

Abstract
Biosynthesis and degradation of heme, an iron-bound protoporphyrin molecule utilized by a wide variety of metabolic processes, is tightly regulated. Two closely related enzymes, heme oxygenase 1 (HMOX1) and heme oxygenase 2 (HMOX2), degrade free heme to produce carbon monoxide, Fe2+ and biliverdin. HMOX1 expression is controlled via the transcriptional activator, NFE2L2, and the transcriptional repressor, Bach1. Transcription of HMOX1 and other NFE2L2-dependent genes is increased in response to electrophilic and reactive oxygen species. Many tumor-derived cell lines have elevated levels of NFE2L2. Elevated expression of NFE2L2-dependent genes contributes to tumor growth and acquired resistance to therapies. Here we report a novel role for heme oxygenase activity in melanosphere formation by human melanoma-derived cell lines. Transcriptional induction of HMOX1 through derepression of Bach1 or transcriptional activation of HMOX2 by oncogenic B-RafV600E results in increased melanosphere formation. Genetic ablation of HMOX1 diminishes melanosphere formation. Further, inhibition of heme oxygenase activity with tin protoporphyrin markedly reduces melanosphere formation driven by either Bach1 derepression or B-RafV600E expression. Global transcriptome analysis implicates genes involved in focal adhesion and extracellular matrix interactions in melanosphere formation.
Keywords: Melanoma, Reactive Oxygen Species, Proto-oncogene proteins B-Raf, Heme Oxygenase-1, Neoplastic Stem Cells, Bach1, Antioxidant, NFE2L2, Extracellular Matrix Proteins, Melanosphere
Introduction
Reactive oxygen species (ROS) function as signaling molecules under physiological conditions, but excessive production of ROS leads to oxidative stress (Alfadda and Sallam, 2012, Benhar et al., 2002, Nishigori et al., 2004, Ray et al., 2012, Sorbara and Girardin, 2011). Due to increased metabolic activity, elevated ROS production is characteristic of neoplastic growth. Oxidative stress is involved in multiple stages of cancer development, including oncogene activation (Maynard et al., 2009, Lander et al., 1997, Sun and Kemble, 2009), increased cell proliferation (Burdon et al., 1990, Burdon, 1995), cell motility and metastasis (Pelicano et al., 2009, Kundu et al., 1995), metabolic reprogramming (Lee et al., 2018, Hayes and Dinkova-Kostova, 2014) and maintenance of a cancer stem cell population (Tanno and Matsui, 2011, Luanpitpong et al., 2018).
The Nuclear Factor Erythroid 2-Like 2 (NFE2L2; Nrf2) is the master transcriptional activator of the oxidative stress response. Nrf2 binds to antioxidant response elements (AREs) in DNA and regulates a cytoprotective transcriptional program (Kaspar et al., 2009, Zhang, 2006, Malhotra et al., 2010). The Kelch-like ECH-associated protein 1 (Keap1) is a component of a Cul3-dependent E3-ubiquitin ligase that ubiquitinates multiple lysine residues within the N-terminal Neh2 domain of Nrf2 and targets Nrf2 for proteasomal degradation under basal conditions (Itoh et al., 1999, Zhang et al., 2004, Kobayashi et al., 2004, Cullinan et al., 2004). Reactive molecules, including ROS, modify cysteine residues within Keap1 and abolish its ability to form a functional E3-ubiquitin ligase (Dinkova-Kostova et al., 2002, Zhang and Hannink, 2003, Cleasby et al., 2014). Consequently, Nrf2 accumulates in the nucleus and activates gene transcription.
Nrf2 target genes encode phase II detoxifying enzymes (Hayes and McMahon, 2009, Itoh et al., 1997), drug transporters (Maher et al., 2007), anti-apoptotic proteins (Niture and Jaiswal, 2012) and components of the proteasome (Kwak et al., 2003). Induction of Nrf2 is thought to inhibit cancer progression by facilitating the removal of ROS and damaged proteins (Hayes and McMahon, 2009). However, increased expression of Nrf2 target genes have been implicated in multiple cancer hallmarks – tumorigenesis, aberrant survival, invasion, metastasis, metabolic reprogramming and drug resistance – in a variety of cancer types (Lau et al., 2008, Wang et al., 2008b, Homma et al., 2009, Kensler and Wakabayashi, 2010, Bauer et al., 2011, Hayes and McMahon, 2009, Kang et al., 2010, Mitsuishi et al., 2012, Niture and Jaiswal, 2012, Rojo et al., 2014, Singh et al., 2008, Zhang et al., 2010, Rojo de la Vega et al., 2018). Mutations in either KEAP1 or NFE2L2 that stabilize Nrf2, as well as epigenetic silencing of KEAP1, have been identified in a number of different tumor types (Hayes and McMahon, 2009, Muscarella et al., 2011, Shibata et al., 2008, Wang et al., 2008a, Kim et al., 2010, Solis et al., 2010, Singh et al., 2006, Rotblat et al., 2012). For example, somatic mutation of the KEAP1 gene in malignant melanoma confers resistance against cisplatin and dacarbazine (Miura et al., 2014). Dysregulation of other signaling pathways that regulate Nrf2 expression, stability or function can also contribute to increased expression of Nrf2-dependent target genes in tumor cells (Rada et al., 2011, DeNicola et al., 2011). Oncogenic forms of both Ras and B-Raf have been reported to increase transcription of the NFE2L2 gene, leading to increased expression of Nrf2-dependent target genes (DeNicola et al., 2011, Chen et al., 2017).
The BTB and CNC Homology 1 (Bach1) protein competes with Nrf2 for binding to ARE motifs and represses many Nrf2 target genes (Ogawa et al., 2001, Warnatz et al., 2011). The heme oxygenase-1 (HMOX1) gene is the best-characterized target for Bach1 repression (Ishikawa et al., 2005, Warnatz et al., 2011). Under basal conditions, Bach1 is bound to several ARE enhancer motifs that regulate HMOX1 expression (Reichard et al., 2007). However, either oxidative stress or an excess heme level results in the removal of Bach1 from DNA and its translocation to the cytoplasm, where it is targeted for proteosomal degradation. Candidate ubiquitin ligases for Bach1 include HOIL (Zenke-Kawasaki et al., 2007) and FBXL17 (Tan et al., 2013). Loss of Bach1-mediated repression enables transcriptional activation of HMOX1 by Nrf2. HMOX1 catalyzes the degradation of heme into carbon monoxide (CO), iron (Fe2+) and biliverdin (BV) (Otterbein et al., 2003). These three products of heme degradation play anti-inflammatory, anti-apoptotic and anti-proliferative roles (Bilban et al., 2008, Otterbein et al., 2003, Tauber et al., 2010, Ryter and Tyrrell, 2000, Vile and Tyrrell, 1993). HMOX1 prevents cancer initiation by inhibiting ROS-induced damage (Jansen et al., 2010) and by promoting DNA repair (Otterbein et al., 2011). However, HMOX1 is overexpressed in a number of cancer types (Berberat et al., 2005, Busserolles et al., 2006, Maines and Abrahamsson, 1996, Mayerhofer et al., 2004, Nishie et al., 1999, Nuhn et al., 2009, Torisu-Itakura et al., 2000, Degese et al., 2012, Noh et al., 2013) and HMOX1 overexpression has been implicated in promoting metastatic tumor growth, evasion of apoptosis and angiogenesis in a variety of cancers (Jozkowicz et al., 2007, Chau, 2015b, Lin et al., 2013, Sunamura et al., 2003, Was et al., 2006). It has recently been reported that loss of Keap1 and thus Nrf2 activation promotes metastasis of lung adenocarcinoma by leading to the accumulation of Bach1 (Lignitto et al., 2019). Further, this group showed that in vivo inhibition of HMOX1 by treatment with zinc protoporphyrin (ZnPPIX) reduced metastasis (Lignitto et al., 2019). While transcription of the HMOX1 gene is tightly regulated by multiple signals, including heme, oxidative stress, cytokines, heavy metals and bacterial endotoxin, the HMOX2 gene is constitutively expressed (Wegiel et al., 2014, Bilban et al., 2008). A role for HMOX2 in cancer has not been evaluated.
The ability of melanoma cells to form spheroids – or melanospheres – when cultured in non-adherent conditions in vitro correlates with their tumorigenic capacity in vivo (Perego et al., 2010, Sette et al., 2012, 2013, Eramo et al., 2006, 2008, Marzagalli et al., 2018). In this report, we examined the role of Nrf2 and HMOX1 in melanosphere formation of human melanoma cell lines. We demonstrate that silencing of either Nrf2 or HMOX1 diminishes melanosphere formation in B-RafV600E –driven SK-Mel-5 and SK-Mel-28 melanoma cell lines. We further provide evidence that heme oxygenase enzymatic activity, either as a result of HMOX1 or HMOX2 function, is required for melanosphere formation. Global transcriptome analyses of melanospheres induced by B-RafV600E expression or derepression of Bach1 target genes revealed enrichment for genes involved in focal adhesion and extracellular matrix (ECM)-receptor interactions. Taken together with recent evidence implicating HMOX1 in melanoma invasion, migration and drug resistance, our data suggest HMOX1 may provide a suitable target for the treatment of metastatic melanoma. Metalloporphyrins, such as tin protoporphyrin (SnPP) and Stannsoporfin (SnMP), are competitive heme oxygenase inhibitors that have long been studied for the treatment of neonatal hyperbilirubinemia (Schulz et al., 2012). Additionally, SnPP is a clinically relevant compound utilized for the treatment of hepatic porphyria (Dover et al., 1991, Dover et al., 1993), and is currently being investigated in clinical trials for the treatment of chronic kidney disease (clinicaltrials.gov). Thus, current therapeutic metalloporphyrins may be repurposed for the treatment of metastatic melanoma.
Materials And Methods
Cell Culture
Normal human embryonic melanocytes (NHEM) isolated from the juvenile foreskin were purchased from PromoCell and cultured in Melanocyte Growth Medium (PromoCell, Heidelberg, Germany). SK-Mel-2 cells were obtained from the National Cancer Institute (NCI) DCTD Tumor Repository. All other human melanoma cell lines were a gift of Dr. Thomas Quinn (University of Missouri) and were cultured in DMEM (Corning, Corning, NY) containing 10% (v/v) FBS (Atlanta Biologicals, Flowery Branch, GA) and 2 mM L-glutamine. HEK293FT cells were cultured in the presence of geneticin (500 μg/ml).
Sequencing
RNA was isolated using a PureLink RNA mini kit (Thermo Scientific, Grand Island, NY). cDNA was synthesized using a Superscript™ III First Strand Synthesis Kit (Thermo Scientific). DNA was amplified by PCR and purified using a GeneJet PCR Purification Kit (Thermo Scientific). The DNA Core at the University of Missouri carried out sequencing using a 3730xl 96-capillary DNA Analyzer with Applied Biosystems Big Dye Terminator. Primers were purchased from Sigma Aldrich (St. Louis, MO). Reference sequences were obtained from the National Center for Biotechnology Information using the following gene accession numbers: N-Ras (NM_002524) and B-Raf (NM_004333).
Silencing Hairpin RNA Design and Synthesis
Target sequences were identified using both pSicoOligomaker 1.5 and siRNA at Whitehead software (http://sirna.wi.mit.edu). Oligo design and cloning into the pSICOR PGK vector (Plasmid 12084, Addgene, Cambridge, MA) were completed according to the Jack’s Lab protocol (http://jacks-lab.mit.edu/protocols/psico).
Retroviral and Lentiviral Preparation
HEK293FT cells were transfected using LipoD293™ DNA in vitro transfection reagent (SignaGen, Gaithersburg, MD). For retroviral particle generation, pHCMV-10A1 (Addgene plasmid #15805) and pGag-Pol (Addgene plasmid #14887) were transfected with either pBABE-Empty Vector (gift of Dr. Alan Diehl, Case Western Reserve University) or pBABE-B-RafV600E (Addgene plasmid #17544). For lentiviral particle generation, psPAX2 (Addgene plasmid #12260) and pMD2.G (Addgene plasmid #12259) were transfected with pSICOR constructs (described above). Supernatant was collected at both 24 and 48 hours, combined, and filtered through a 0.45 μm filter unit (Millipore, Billerica, MA). Viral-containing supernatant was supplemented with 8 μg/ml polybrene for infection.
Nrf2 and HMOX1 Ablation
SK-Mel-5 and SK-Mel-28 cells were grown until 70% confluent in 75 cm flasks and then transfected with 20 μg of CRISPR/Cas9 constructs containing guide RNAs targeting HMOX1 or NFE2L2 (Horizon Discovery DNA 2.0) along with a plasmid expressing eGFP using Lipofectamine 2000 transfection reagent according to manufacturer’s instructions. Twenty-four hours after transfection, the cells were sorted by eGFP expression and plated at clonal density on 96-well plates. Individual clones were screened for Nrf2 or HMOX1 expression by immunoblot analysis following MG132 inhibitor or CoPP treatment, respectively. Each cell line was derived from a single clonal expansion.
Immunoblot Analysis
Cells were lysed in High Salt ELB as described previously (Attucks et al., 2014) and electrophoresed through a Genscript ExpressPlus™ PAGE Precast gel and transferred onto 0.45 μm nitrocellulose membrane (BioRad, Hercules, CA). The membrane was incubated for 2 hours with 5% (w/v) non-fat dry milk in PBS/0.1% (v/v) Tween-20 (Fischer Scientific) and then incubated overnight with the appropriate primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, 1:000 dilution): anti-Nrf2(H-300), anti-HMOX1(A-3), anti-HMOX2(B-3), anti-Bach1(F-9) and anti-Raf-B(F-7). Anti-β-tubulin(E7) was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA) and used as a loading control (1:1000). Then, membranes were incubated for one hour with the appropriate horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, 1:2000 dilution). Immunodetection was achieved using SuperSignal™ West Pico Chemiluminescent Substrate and developed in a FujiFilm Intelligent Dark Box using LAS-3000 software or a UVP ChemiDoc. For quantitation, band intensity was measured using MultiGauge or ImageJ software, normalized to β-tubulin expression.
Melanosphere Assay
Melanosphere assays were conducted in Corning Ultra-Low Attachment 6-well plates (Corning). Cells (20,000) were plated per well in 4 ml of stem cell medium (DMEM/F12 (1:1)) supplemented with B27 Serum-Free Supplement, 20 ng/ml Recombinant Human FGF, 20 ng/ml Recombinant Human EGF and 2.5 μg/ml Amphotericin B) (Thermo Scientific). After 10 days, melanospheres were imaged and those that measured at least 100 μM in diameter were counted.
Real-Time RT-PCR
RNA was extracted from 1 × 106 cells per sample using a Qiagen RNeasy mini kit (Valencia, CA). cDNA was synthesized using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY). Quantitative PCR was carried out for 40 cycles using Thermo Scientific Maxima SYBR Green/ROX qPCR Master Mix, 7 μl of purified cDNA (representing approximately 5 × 104 cells) and a primer concentration of 0.15 μM. All PCR reactions were conducted in triplicate. Expression levels were determined using the Comparative CT Method for Quantitative RT-PCR (Schmittgen and Livak, 2008). Briefly, expression is normalized to actin and then calculated as the fold-change over an untreated sample.
RNA-Seq Analysis and Functional Annotation Clustering
Cells were grown in non-adherent conditions for 5 days. Each condition was done in triplicate. RNA was isolated from 6 × 105 cells per sample using a Qiagen RNeasy mini kit. Then, 2.5 μg of RNA were diluted to 100 ng/μl in RNAse-free water and submitted to the DNA Core at the University of Missouri for RNA-seq analysis using an Illumina HiSeq 2000 with a 1 × 50 run type, returning approximately 34 million reads per sample. Pairwise comparisons were done using both DESeq and EdgeR Bioconductor packages as described previously (Li et al., 2015). Those genes found to be differentially expressed by a minimum of a 2-fold increase by both packages were used for DAVID functional annotation clustering (http://david.abcc.ncifcrf.gov). Significance for DAVID functional annotation clustering was calculated using a Fisher’s Exact Test (EASE Score). For Nrf2-target gene expression analysis, the DESeq Bioconductor package was used.
Pharmacological Inhibitors
Tin protoporphyrin IX dichloride (SnPP) (Tocris Biosciences, Bristol, UK), protoporphyrin IX cobalt dichloride (CoPP) (Sigma-Aldrich) and MG132 were used at 10 μM. For melanosphere assays, SnPP-treated wells were re-treated with an additional 10 μM every 48 hours. PLX-4032 (Selleck Chemicals, Houston, TX) was used at 5 μM.
Bilirubin Quantitation
Twenty-four hours after seeding 2.5 × 105 cells per plate, CoPP was added to the appropriate plates overnight. Cells were lysed in High Salt ELB supplemented with protease and phosphatase inhibitors and immediately collected into a dark tube to avoid light exposure. A Bilirubin Assay Kit (MAK126, Sigma-Aldrich) was used to measure bilirubin. Absorbance was measured at 530 nm. Total bilirubin was calculated as described by the manufacturer.
MTT Assay
Ten thousand cells were seeded per well of a 96-well plate and treated with inhibitors (CoPP, SnPP, PLX-4032) as indicated. After 48 hours, the medium was replaced with 100 μl of phenol-red free medium and 10 μl of 12 mM MTT (Molecular Probes, Life Technologies) was added to each well. After a four hour incubation at 37°C, 75 μl of media was removed and 50 μl of DMSO was added. After a 10 minute incubation at 37°C, the absorbance was measured at 540 nm. Data was normalized to the absorbance of untreated SK-Mel-5 or SK-Mel-28 cells.
Statistics
Except where otherwise noted, data represent the mean ± SEM of at least three biological replicates. Significance was determined using the standard Student’s t-test.
Results
Melanoma cell lines which harbor the activating mutation, B-RafV600E, form melanospheres when grown in non-adherent culture conditions.
NHEM and a panel of six human melanoma cell lines were plated in ultra-low attachment plates in defined stem cell medium. After ten days, NHEM did not survive the non-adherent culture conditions while all melanoma cell lines remained viable (data not shown). Four melanoma cell lines formed melanospheres of at least 100 μM in size (SK-Mel-2, M14, SK-Mel-5, SK-Mel-28), while two did not (Hs936T, TXM13) (Fig. 1A). Over the 10-day period, SK-Mel-2, Hs936T and TXM13 cells did not proliferate while M14, SK-Mel-5, and SK-Mel-28 cells increased in cell number (data not shown). SK-Mel-5 cells formed the largest number of melanospheres: 722 ± 62 as compared to 100 ± 11 (M14), 49 ± 3 (SK-Mel-28), and 17 ± 5 (SK-Mel-2) for the other three sphere-forming lines (Fig. 1B).
Figure 1. Melanoma cell lines harboring the B-RafV600E mutation form melanospheres.
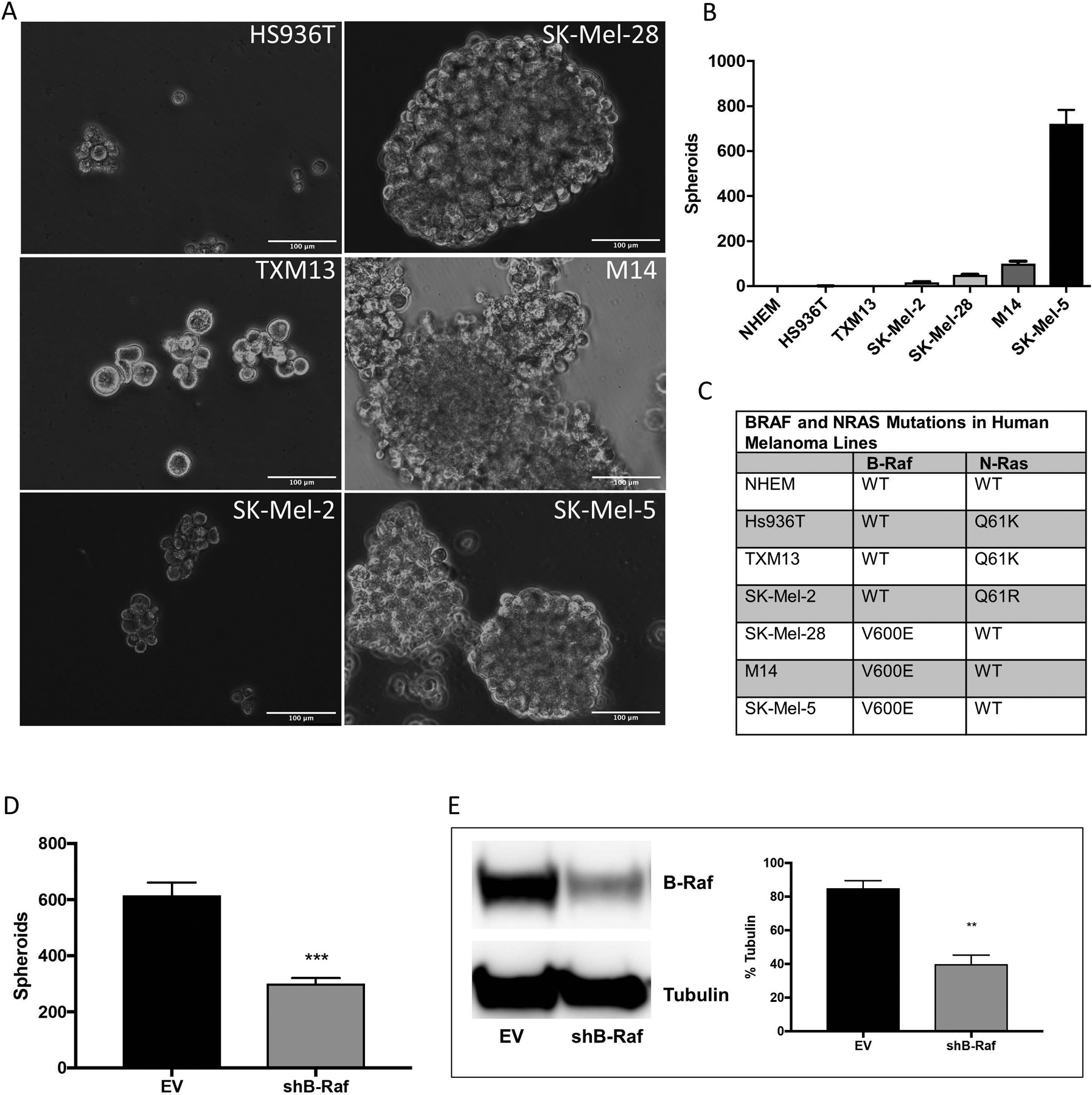
Six human melanoma cell lines and primary normal human embryonic melanocytes (NHEM) were plated for melanosphere formation assays as described in Materials and Methods. After 10 days, the spheroids were imaged (A) and counted (B). (C) cDNA synthesized from RNA isolated from melanoma cell lines and NHEM was amplified by PCR and then N-Ras (NM_002524) and B-Raf (NM_004333) were sequenced. (D) SK-Mel-5 cells stably expressing either an empty pSicoR vector (EV) or a silencing hairpin RNA (shRNA) targeting B-Raf (shB-Raf) were plated for melanosphere assays. (E) B-Raf protein levels were assessed by immunoblot analysis using β-tubulin as a loading control. Error bars represent the standard error of the mean (SEM) of three biological replicates for immunoblot quantitation and four for melanosphere assays. Statistical significance was determined using the standard Student’s t-test. **P < 0.01, ***P < 0.001.
To determine whether the melanoma cell lines harbored B-Raf and/or N-Ras mutations, the two most commonly mutated genes in melanoma (Reddy et al., 2017), cDNA was synthesized from RNA isolated from each of the cell lines. Sequencing of PCR-amplified cDNA revealed that the melanoma lines that formed the largest number of melanospheres (SK-Mel5, M14, SK-Mel-28) harbored the V600E mutation in B-Raf while the other three lines harbored mutations in the N-Ras gene (Q61R for SK-Mel-2 and Q61K for Hs936T and TXM13) (Fig. 1C). None of the lines harbored mutations in either the Nrf2 or Keap1 genes (data not shown). Knockdown of B-Raf by infection with a silencing hairpin RNA (shRNA) targeting B-Raf (shB-Raf) showed a reduction in melanosphere formation in SK-Mel-5 cells (Fig. 1D) from 616 ± 110 to 300 ± 50 spheroids. Infection with shB-Raf decreased B-Raf protein levels by 50% (Fig. 1E).
Nrf2 and HMOX1 are important for melanosphere formation in B-Raf-active melanoma.
To determine whether Nrf2 contributes to melanosphere formation in B-RafV600E–containing melanoma cell lines, SK-Mel-5 and SK-Mel-28 cells were transfected with a CRISPR/Cas9 construct to ablate Nrf2 with MG132 added to stabilize Nrf2 protein levels. Loss of Nrf2 in clonally expanded cell lines was verified by western blot analysis (Fig. 2A). Loss of Nrf2 in SK-Mel-5 and SK-Mel-28 cells reduced melanosphere formation in both lines (Fig. 2B) from 813 ± 113 to 279 ± 75 and 79 ± 20 to 40 ± 7, respectively. Loss of Nrf2 had a modest effect on cell viability of SK-Mel-5 cells but did not affect SK-Mel-28 cells (Fig. 2C) as determined by MTT assay. All lines were sensitive to the B-RafV600E inhibitor, Vemurafanib (PLX-4032), demonstrating that loss of Nrf2 did not disrupt B-RafV600E-driven cell proliferation (Fig. 2C). A reduction in melanosphere formation was also observed in SK-Mel-5 cells in which levels of Nrf2 were reduced by shRNA (Supplementary Fig. S1A and S1B). Taken together, our data suggest that Nrf2 plays a role in melanosphere formation observed in B-RafV600E–expressing melanoma cell lines.
Figure 2. Loss of Nrf2 diminishes melanosphere formation.
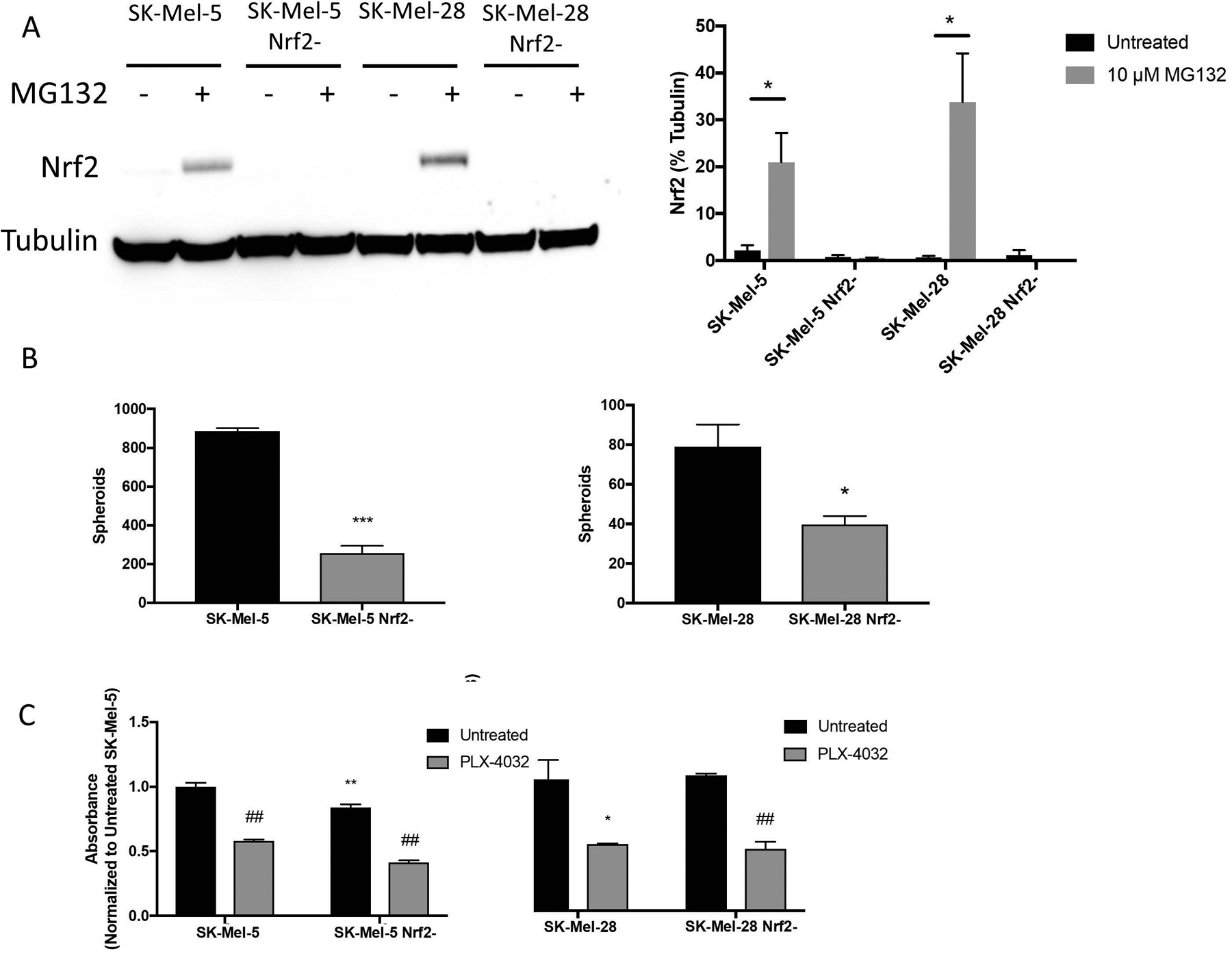
SK-Mel-5 and SK-Mel-28 cells were transfected with CRISPR/Cas9 constructs to ablate Nrf2 expression. Transfected cells were sorted by GFP expression and individual clones were expanded. (A) Complete loss of Nrf2 was validated by immunoblot analysis following MG132 treatment (10 μM for 6 hours) to stabilize Nrf2 protein levels. Representative blot shown in the left panel. In the right panel, band intensity of three blots was measured and normalized to tubulin. (B) Melanosphere formation was assessed for SK-Mel-5, SK-Mel-28 and their Nrf2-null counterparts. (C) For the MTT assay, 10,000 cells were plated per well of a 96-well plate and treated with or without 5 μM PLX-4032 to inhibit B-RafV600E. After 48 hours, the MTT assay was conducted as described in Materials and Methods. Comparisons between wild-type and Nrf2-null lines are noted with asterisks. Comparisons between treated and untreated cells are noted by ## (P < 0.01). Error bars represent the standard error of the mean (SEM) of three biological replicates for immunoblot quantitation, four for the melanosphere assay and six for the MTT assay. Statistical significance was determined using the standard Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Increased expression of one Nrf2 target gene, heme oxygenase-1 (HMOX1), has been implicated in promoting metastatic growth in a number of cancer types (Lignitto et al., 2019, Jozkowicz et al., 2007, Chau, 2015a), including melanoma (Was et al., 2006, Okamoto et al., 2006). To investigate whether HMOX1 may be involved in melanosphere formation, we first increased expression of HMOX1 in SK-Mel-5 cells by treating cells with cobalt protoporphyrin IX (CoPP), a heme mimetic. When heme or CoPP bind Bach1, the repressor is exported from the nucleus and degraded in the cytoplasm, allowing Nrf2-dependent expression of genes normally repressed by Bach1 (Zenke-Kawasaki et al., 2007, Suzuki et al., 2004). As expected, CoPP treatment resulted in increased HMOX1 expression in SK-Mel-5 cells (Fig. 3C). Melanosphere formation in SK-Mel-5 cells was increased following CoPP treatment, from 788 ± 17 to 1192 ± 107 (Fig. 3A), despite a decrease in cell number as compared to untreated SK-Mel-5 cells (Fig. 3B).
Figure 3. CoPP treatment enhances melanosphere formation.
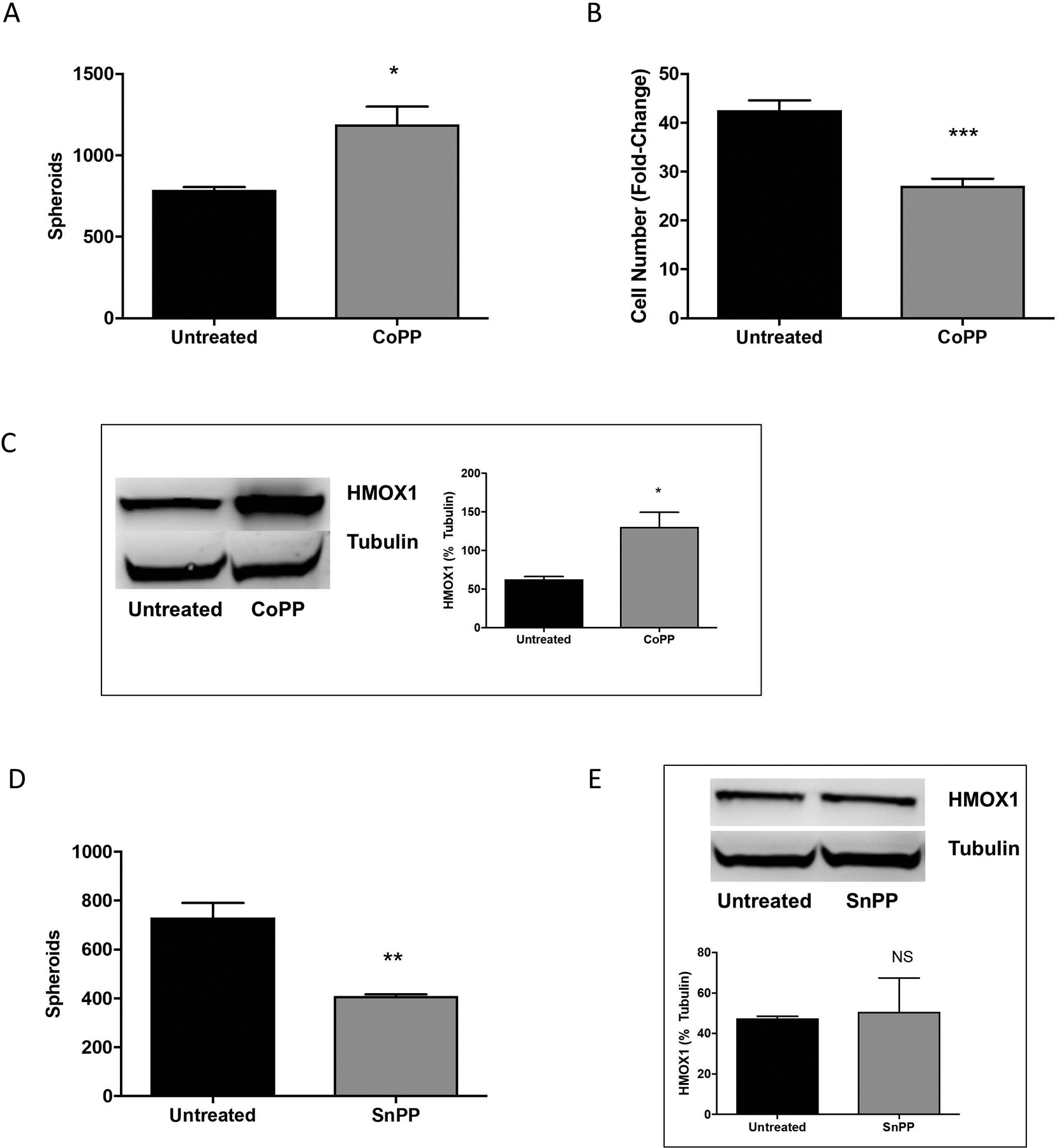
(A) SK-Mel-5 cells were treated with 10 μM cobalt protoporphyrin IX (CoPP) and melanosphere formation was assessed. Error bars represent the SEM of four biological replicates of each melanosphere assay. (B) Following the melanosphere assay, all cells from each well were collected independently, melanospheres were dissociated by incubating with TrypLE and the cells were counted. Error bars represent the SEM of four biological replicates. (C) SK-Mel-5 cells were grown in adherent conditions and treated with 10 μM CoPP overnight. HMOX1 protein levels were assessed by immunoblot analysis and band intensity measured and normalized to tubulin. Error bars represent the SEM of three biological replicates. (D) SK-Mel-5 cells were treated with 10 μM tin protoporphyrin IX dichloride (SnPP) every 48 hours and melanosphere formation assessed over a 10-day melanosphere assay. Error bars represent the SEM of four biological replicates. (E) HMOX1 protein levels in SK-Mel-5 cells grown in adherent conditions and treated with 10 μM SnPP overnight were assessed by immunoblot analysis and band intensity measured and normalized to tubulin. Error bars represent the SEM of three biological replicates for immunoblot analysis and all statistical significance was determined using the standard Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, NS is not significant.
Independent of its enzymatic function, HMOX1 promotes gene expression changes in the nucleus by activating other transcription factors (Lin et al., 2007). Under basal conditions, HMOX1 is located in the ER membrane, tethered by its C-terminus (Lin et al., 2007). However, hypoxic exposure or hemin can induce cleavage of this C-terminal region, releasing HMOX1 from the endoplasmic reticulum and allowing localization of HMOX1 to the nucleus, where it can modulate transcription (Lin et al., 2007). To determine whether the role of HMOX1 in melanosphere formation was a result of its enzymatic or transcriptional roles, activity was inhibited with tin protoporphyrin (SnPP), a metalloporphyrin that acts as a competitive inhibitor of heme oxygenase. SnPP treatment decreased melanosphere formation in SK-Mel-5 cells (Fig. 3D) from 732 ± 59 to 410 ± 7 spheroids, providing evidence that heme oxygenase activity is involved in melanosphere formation in B-Raf-active melanoma. SnPP treatment did not affect HMOX1 protein levels (Fig. 3E). It is worth noting that some have reported a compensatory mechanism leading to the upregulation of HMOX1 following inhibition with SnPP or similar metalloporphyrins (Sardana and Kappas, 1987, Xia et al., 2006, Kwok, 2013). However, others have reported no impact of metalloporphyrins on HMOX1 protein levels in human breast cancer cells in vitro (Lin et al., 2008) or DBA/1J mice in vivo (Devesa et al., 2005). Additionally, ZnPP, but not SnPP, has been reported to cause a compensatory upregulation (mRNA and protein) of HMOX1 in human pancreatic cancer cells (Mohammad et al., 2019) and hamster fibroblasts (Yang et al., 2001). Taken together, the compensatory effect of metalloporphyrins on HMOX1 expression may be cell line-, tissue- or species-dependent and there is some evidence that SnPP may be a more modest inducer of HMOX1 expression than ZnPP.
To further assess the role of HMOX1 in B-RafV600E–containing melanoma cells, HMOX1 expression was ablated in both SK-Mel-5 and SK-Mel-28 cells by CRISPR/Cas9 (Fig. 4A) and clonal cell lines were acquired. Ablation of HMOX1 significantly decreased melanosphere formation in both SK-Mel-5 and SK-Mel-28 cells (Fig. 4B). SK-Mel-5 cells formed 550 ± 94 melanospheres, while the HMOX1-null lines, SK-Mel-5 (5KO1) and SK-Mel-5 (5KO2), produced 1 ± 1 and 31 ± 15 melanospheres, respectively. SK-Mel-28 cells formed 131 ± 22 melanospheres, while the HMOX1-null line, SK-Mel-28 (28KO1), produced 37 ± 5 melanospheres (Fig. 4B). An MTT assay showed that treatment with CoPP or SnPP did not affect the cell viability of SK-Mel-5, SK-Mel-28 or any of the HMOX1-null cell lines (Fig. 4C). All lines remained sensitive to treatment with PLX-4032, demonstrating that loss of HMOX1 did not disrupt B-RafV600E-driven proliferation. A reduction in melanosphere formation was also observed in SK-Mel-5 cells in which levels of HMOX1 were reduced by shRNA (Supplementary Fig. S1C and S1D). Together our data demonstrate that loss of HMOX1 significantly diminishes melanosphere formation, without affecting proliferative capability. Use of CRISPR/Cas9 to knock out HMOX1 in two B-Raf active melanoma cell lines suggests that HMOX1 dependence is not cell line-specific.
Figure 4. Ablation of HMOX1 abrogates melanosphere formation.
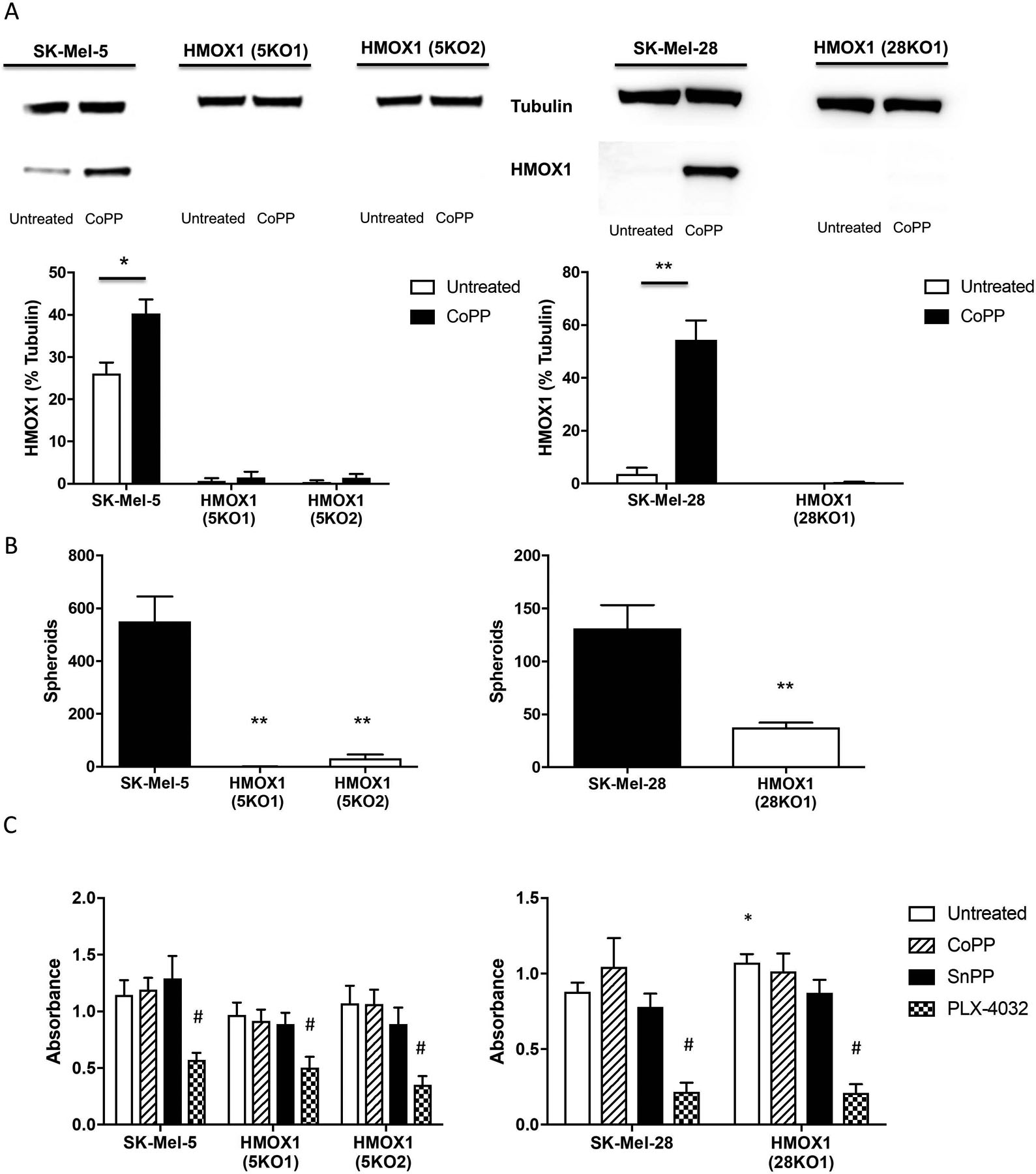
SK-Mel-5 and SK-Mel-28 cells were transfected with CRISPR/Cas9 constructs to ablate HMOX1 expression. Transfected cells were sorted by GFP expression and individual clones were expanded. (A) Complete loss of HMOX1 was validated by immunoblot analysis following overnight CoPP (10 μM) treatment to upregulate HMOX1. Quantitation of three blots is shown in the lower panels. (B) SK-Mel-5, SK-Mel-28 and their HMOX1-null counterparts, SK-Mel-5HMOX1- (5KO1), SK-Mel-5HMOX1- (5KO2) and SK-Mel-28HMOX1- (28KO1) were plated and used for melanosphere assay. (C) For MTT assay, 10,000 cells were plated per well of a 96-well plate and untreated or treated with 10 μM CoPP, 10 μM SnPP or 5 μM PLX-4032. Comparisons between wild-type and HMOX1-null lines are noted with asterisks. Comparisons amongst the various treatments are noted by # (#P < 0.001). Error bars represent the standard error of the mean (SEM) of three biological replicates for immunoblot quantitation, four for melanosphere assay and six for MTT assay. Statistical significance was determined using the standard Student’s t-test. *P < 0.05, **P < 0.01. Symbols were omitted where no significance was found.
Oncogenic B-RafV600E induces Nrf2 expression and melanosphere formation in Hs936T cells
In the human melanoma cell lines tested, the presence of mutant B-RafV600E correlated with melanosphere formation (Fig. 1). To test whether expression of oncogenic B-RafV600E was sufficient to induce melanosphere formation, Hs936T cells were infected with a retroviral vector encoding oncogenic B-RafV600E. Hs936T cells were chosen because they do not harbor mutant B-RafV600E and do not form melanospheres (Fig. 1). In contrast to wild-type Hs936T cells, Hs936T cells that stably express B-RafV600E readily formed melanospheres (324 ± 34 spheroids) (Fig. 5A), suggesting that transformation by oncogenic B-RafV600E alone is sufficient for melanosphere formation. Melanosphere formation was markedly reduced by PLX-4032 treatment (35 ± 4 spheroids) (Fig. 5A). Hs936T cells were counted following the assay to assess the proliferation of each cell line during the ten-day period. Hs936T cells infected with an empty vector (EV) showed a slight increase in cell number over the 10-day period (1.35-fold increase) while those infected with B-RafV600E showed a robust increase in cell number (100-fold increase). This increase in cell number was markedly diminished to levels near EV-expressing controls following treatment with PLX-4032 (4.19-fold) (Fig. 5B).
Figure 5. Oncogenic B-Raf promotes melanosphere formation.
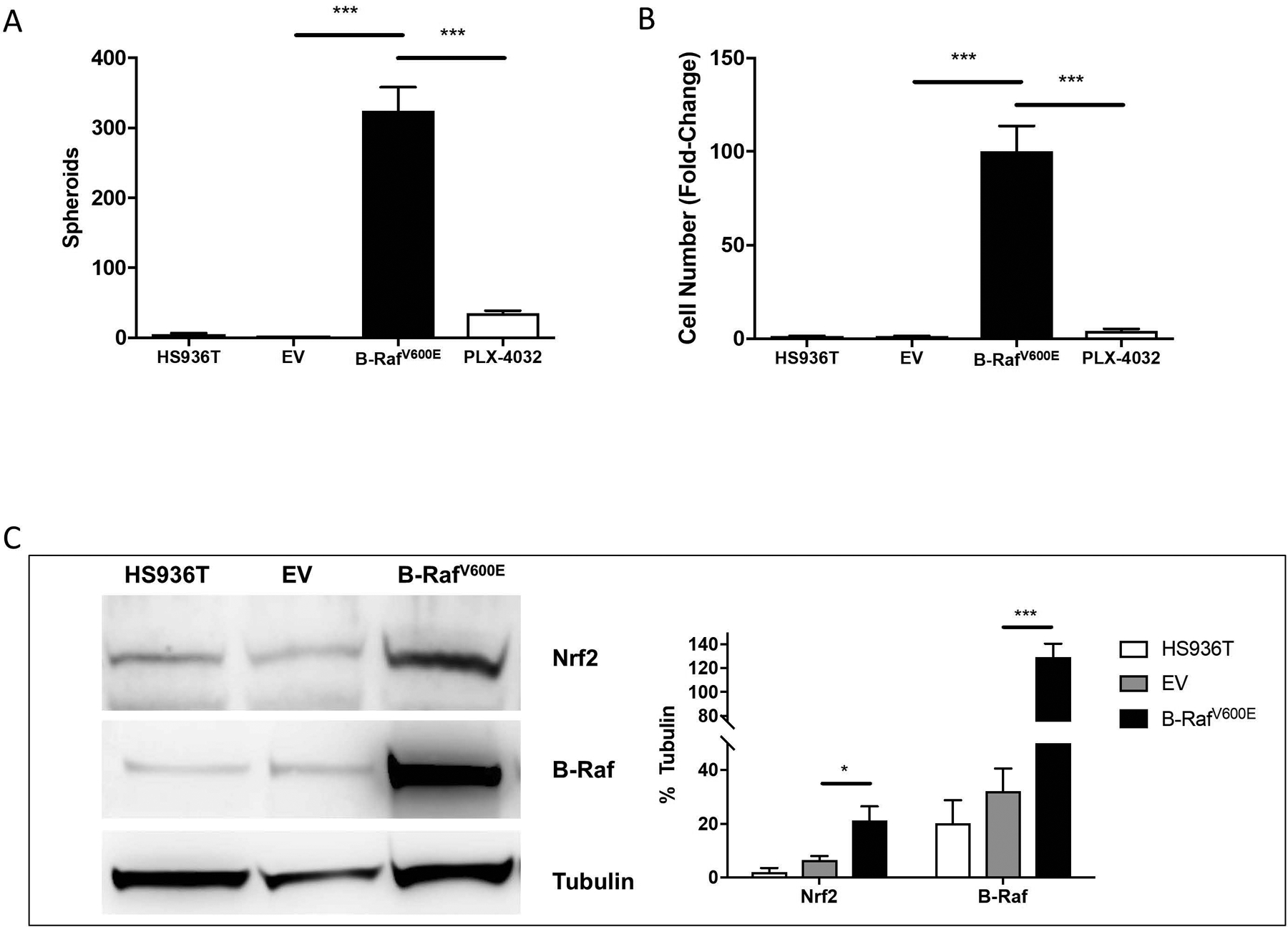
(A) Melanosphere formation was assessed for Hs936T cells, Hs936T cells stably expressing either an empty pBABE vector (EV) or pBABE vector containing oncogenic B-RafV600E, and Hs936T cells stably expressing pBABE-B-RafV600E and treated with 5 μM PLX-4032. (B) Following the 10-day melanosphere assay, cells were collected and melanospheres dissociated by incubation with TrypLE and counted. Error bars represent the SEM of four biological replicates of each melanosphere assay. (C) Cells were grown in adherent conditions and protein levels for Nrf2 and B-Raf were assessed by immunoblot analysis. Band intensities were measured and normalized to tubulin. Error bars represent the SEM of the three biological replicates used for immunoblot analysis. Statistical significance was determined using the standard Student’s t-test. *P < 0.05, ***P < 0.001.
A marked increase (7.5-fold) in Nrf2 protein levels was observed in Hs936T cells upon stable expression of oncogenic B-RafV600E (Figs. 5C, 6A and 6B). Since Nrf2 protein levels were elevated, we expected to find an increase in the expression of Nrf2 target genes, such as HMOX1. However, HMOX1 protein levels significantly decreased in B-RafV600E-expressing cells compared to EV-expressing cells (Figs. 6A and 6B). Bach1 mRNA levels were significantly increased, providing a likely explanation for the observed decrease in HMOX1 levels in Hs936T cells that stably express B-RafV600E. However, Bach1 protein levels had a trend toward elevation that was not statistically significant. While expression of HMOX1 was decreased, HMOX2 levels were increased in B-RafV600E-expressing cells (Fig. 6A and 6B). Quantitative RT-PCR (qRT-PCR) revealed an increase in Nrf2, Bach1 and HMOX2 mRNA levels with B-RafV600E-expression (Fig. 6C). Together, our data are consistent with the suggestion that Nrf2 is a target gene of the B-RafV600E signal transduction pathway (DeNicola et al., 2011). However, increased expression of Nrf2 is insufficient to drive HMOX1 expression due to a simultaneous increase in Bach1. Instead, we show elevated expression of HMOX2 following expression of B-RafV600E in Hs936T cells.
Figure 6. Oncogenic pBABE-B-RafV600E transformed Hs936T cells demonstrate an increase in HMOX2 mRNA and protein levels.
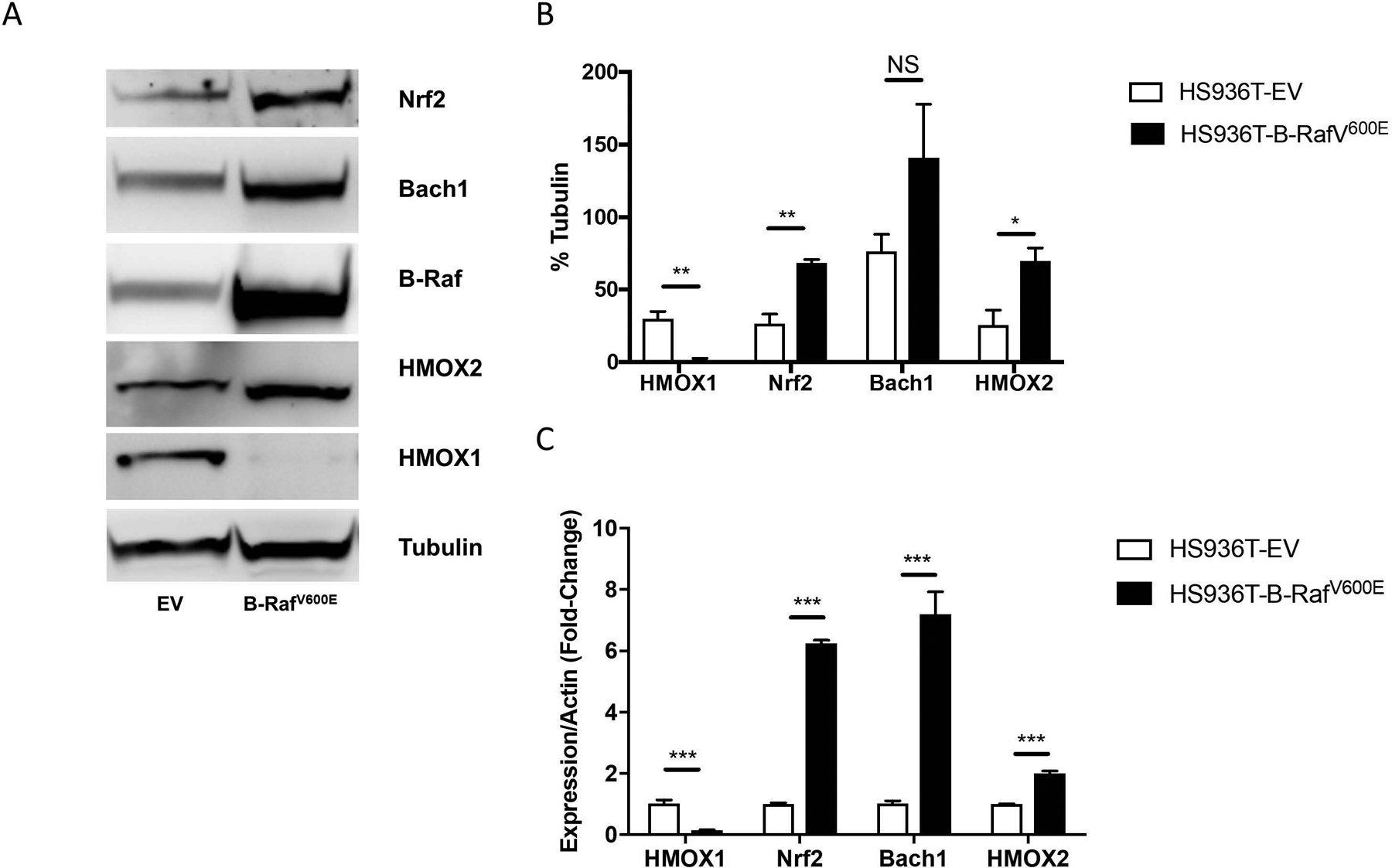
(A) Hs936T cells stably expressing either an empty pBABE vector (EV) or a pBABE vector containing oncogenic B-RafV600E were grown in adherent conditions. Immunoblot analysis was done to assess the protein levels of HMOX1, HMOX2, B-Raf, Bach1, and Nrf2. (B) Band intensities were measured and normalized to tubulin. Error bars represent the SEM of three biological replicates. Statistical significance was determined using the standard Student’s t-test. *P < 0.05, **P < 0.01, NS is not significant. (C) Expression levels of HMOX1, Nrf2, Bach1, HMOX2 and actin mRNA were determined by qRT-PCR. Error bars represent the SEM of three biological replicates. Statistical significance was determined using the standard Student’s t-test. ***P < 0.0001 for all genes in real-time PCR.
Heme oxygenase activity is sufficient for melanosphere formation
We next determined if induction of HMOX1 in Hs936T cells by CoPP treatment could contribute to melanosphere formation in Hs936T cells. Indeed, Hs936T cells treated with CoPP formed melanospheres (630 ± 128 spheroids) compared to untreated Hs936T cells (5 ± 2 spheroids) (Fig. 7A). There was no change in cell number between untreated and CoPP-treated Hs936T cells, indicating that melanosphere formation was independent of proliferation (Fig. 7B). Steady-state levels of HMOX1 but not HMOX2 protein and mRNA were markedly elevated in CoPP-treated Hs936T cells (Fig. 7C and 7D). Thus, derepression of Bach1 via CoPP and subsequent induction of HMOX1 is sufficient to promote melanosphere formation by a proliferation-independent mechanism.
Figure 7. CoPP treatment facilitates melanosphere formation in Hs936T cells.
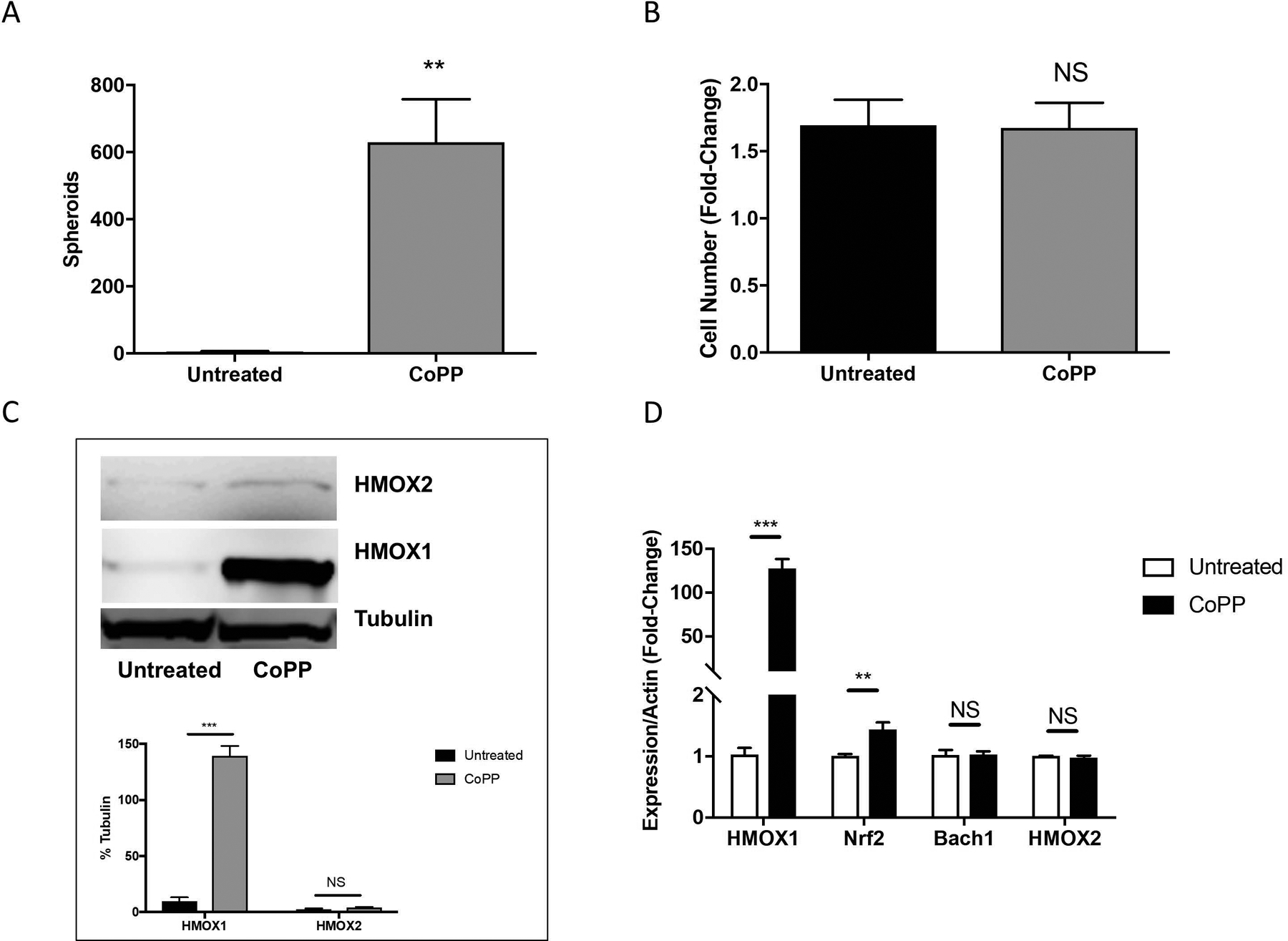
(A) Melanosphere formation was assessed in Hs936T cells treated with or without 10 μM CoPP. Error bars represent the SEM of four biological replicates. (B) Following the 10-day melanosphere assay, all cells were collected and melanospheres were dissociated by incubation with TrypLE and cells were counted. (C) Protein levels for HMOX1 and HMOX2 were assessed by immunoblot analysis for Hs936T cells grown in adherent conditions and either left untreated or treated with 10 μM CoPP overnight. Band intensities were measured and normalized to tubulin. Error bars represent the SEM of three biological replicates. (D) Expression levels of HMOX1, HMOX2, Nrf2, Bach1 and actin mRNA were assessed in Hs936T cells using qRT-PCR. Error bars represent the SEM of three biological replicates. Statistical significance was determined using the standard Student’s t-test. **P < 0.01, ***P < 0.001, NS is not significant.
To test the effect of HMOX activity on melanosphere formation in Hs936T cells expressing B-RafV600E, cells were treated with SnPP, which blocks the activity of both HMOX1 and HMOX2. The ability of Hs936T cells expressing oncogenic B-RafV600E to form melanospheres (325 ± 34 spheroids) was diminished upon treatment with SnPP (99 ± 34 spheroids) (Fig. 8A). Likewise, melanosphere formation in Hs936T cells treated with CoPP was reduced upon treatment with SnPP, from 607 ± 44 spheroids in CoPP-treated cells to 129 ± 16 spheroids in Hs936T cells treated with both CoPP and SnPP (Fig. 8D). SnPP treatment did not affect HMOX1 or HMOX2 protein levels in HS936T cells (Fig. 8C). Neither SnPP treatment in B-RafV600E-expressing Hs936T cells nor SnPP + CoPP treatment in Hs936T cells affected proliferation (Fig. 8B and 8E). Together, these data support a role for heme oxygenase activity in melanosphere formation, independent of cell proliferation. Furthermore, these results indicate that increased expression of either HMOX1 or HMOX2 is sufficient to drive melanosphere formation. To determine whether HMOX1 or HMOX2 levels differed among human melanoma cell lines, we performed western blot analysis for both HMOX1 and HMOX2 in our panel of cell lines (Supplementary Fig. S2). CoPP-induced HMOX1 levels were much higher in the B-RafV600E-expressing cell lines, SK-Mel-5 and SK-Mel-28 than in any of the other cell lines.
Figure 8. Pharmacological inhibition of HMOX1 and HMOX2 diminishes melanosphere formation in transformed Hs936T cells.
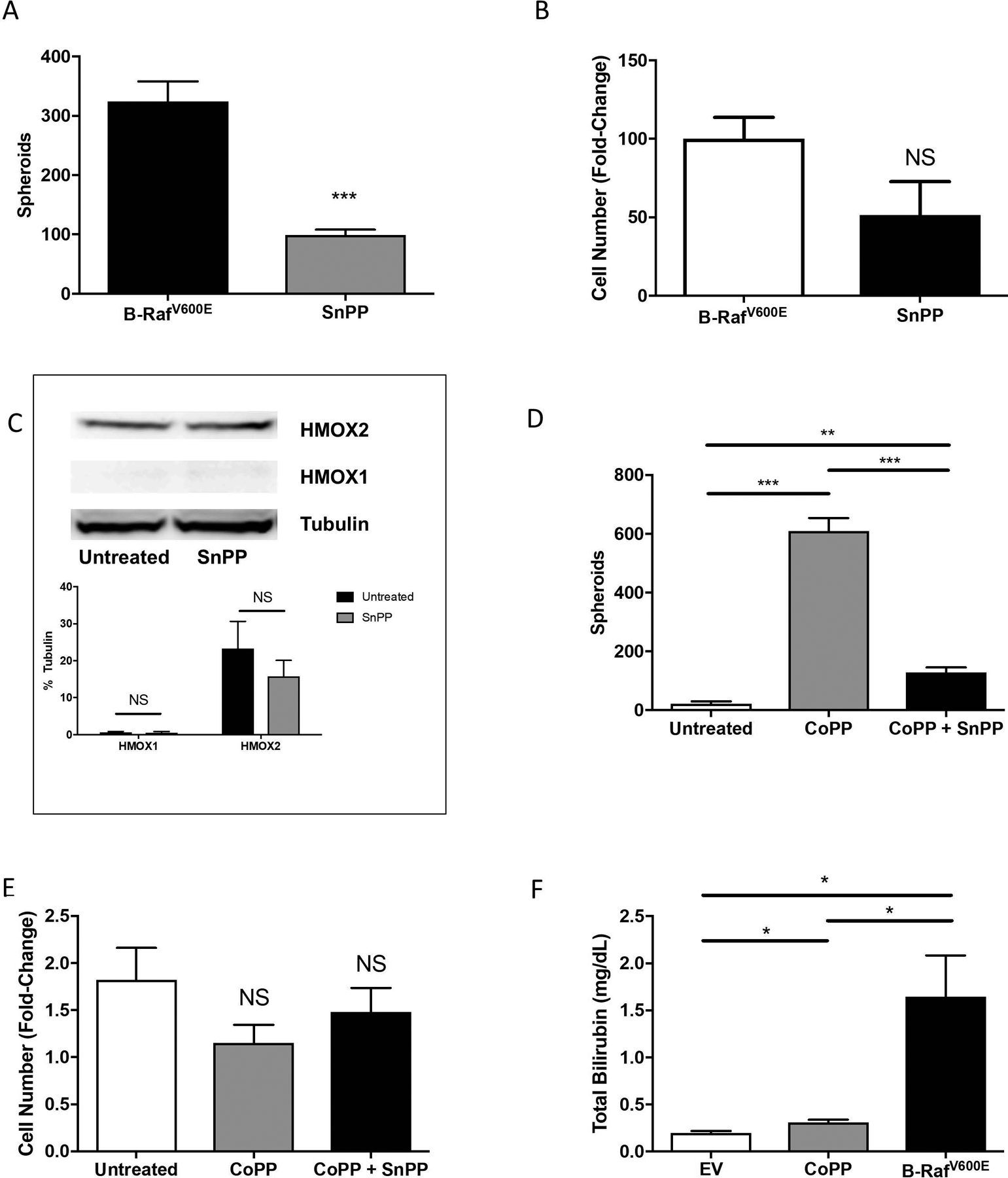
(A) Transformed Hs936T cells stably expressing B-RafV600E were either untreated or treated with 10 μM tin protoporphyrin IX dichloride (SnPP) every 48 hours to inhibit both HMOX1 and HMOX2 activity. Melanosphere formation was assessed as compared to untreated cells. (B) Following the 10-day melanosphere assay, all cells were collected, melanospheres were dissociated by incubation with TrypLE and cells were counted. Error bars represent the SEM of four biological replicates for each melanosphere assay. (C) Immunoblot analysis for Hs936T cells stably expressing B-RafV600E either untreated or treated with SnPP overnight was done to assess protein levels of HMOX1 and HMOX2. Band intensities were measured and normalized to tubulin. Error bars represent the SEM of three biological replicates. (D) Hs936T cells were either untreated, treated with 10 μM CoPP or treated with 10 μM CoPP plus 10 μM SnPP and melanosphere formation assessed. SnPP was added every 48 hours while CoPP was only added on day one of the melanosphere assay. (E) Melanospheres were dissociated and cells counted as described for (B). (F) Heme degradation levels were assessed by measuring total bilirubin in Hs936T cells stably expressing an empty pBABE vector (EV) and either untreated or treated with 10 μM CoPP overnight and in Hs936T cells stably expressing B-RafV600E. Error bars represent the SEM of three biological replicates. All statistical significance was determined using a standard Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, NS is not significant.
Following heme degradation, biliverdin is rapidly converted to bilirubin (Ryter and Tyrrell, 2000). To measure heme oxygenase activity, we measured bilirubin levels in Hs936T cells compared to CoPP-treated Hs936T or B-RafV600E-expressing Hs936T cells (Fig. 8F). Bilirubin levels were increased in both CoPP-treated Hs936T and B-RafV600E-expressing Hs936T cell lysates (Fig. 8F).
CoPP-treated and B-RafV600E-expressing Hs936T cells show increased expression levels of genes involved in focal adhesion and ECM-receptor interactions.
Both CoPP-induced overexpression of HMOX1 as well as oncogenic transformation by expression of B-RafV600E enabled Hs936T cells to produce melanospheres, suggesting that a mechanism common to both changes was responsible for this effect. To elucidate an underlying mechanism for increased melanosphere formation by CoPP and B-RafV600E, mRNA was isolated from Hs936T cells infected with an empty vector (EV) and either untreated or treated with CoPP and from Hs936T cells stably expressing B-RafV600E. Prior to RNA isolation, the cells were grown for five days in non-adherent conditions, a time at which melanosphere formation was just beginning to be visible. Under these conditions, no aggregation of untreated cells was observed. Since both CoPP-treatment and B-RafV600E expression promote melanosphere formation in Hs936T cells, our interest was predominantly focused on where the transcriptome signatures for these two conditions overlapped. Two R Bioconductor packages, DESeq and EdgeR (Li et al., 2015), were used to construct a consensus list of differentially expressed transcripts (Fig. 9). This list contained transcripts that were up- or down-regulated by a minimum of two-fold following either stable expression of B-RafV600E (14,411 transcripts) or treatment with CoPP (1,246 transcripts) (Fig. 9A). The two groups shared 901 differentially expressed transcripts (Fig. 9A). Those transcripts which were up-regulated by a minimum of two-fold were used for DAVID functional annotation clustering. After sorting by P-values, the three most significantly enriched functional groups are shown in Figure 9B. Both conditions demonstrated enrichment for genes involved in focal adhesion and ECM-receptor interactions, including several integrin alpha genes, laminin beta 1 and growth factors (Table 1). Hs936T cells infected with B-RafV600E showed robust increases in mRNA expression of a subset of Nrf2-target genes, notably malic enzyme 1 (ME1) (163-fold, P = 4.02E-69), extracellular superoxide dismutase 3 (SOD3) (402-fold, P = 4.35E-30) and glutathione S-transferase mu 1 (GSTM1) (59-fold, P = 4.30E-22) (Table 2). As previously discussed, HMOX1 was markedly repressed in B-RafV600E-expressing cells (P = 6.01E-185) but increased 14-fold in CoPP-treated cells (P = 1.10E-183) (Table 2).
Figure 9. Hs936T cells treated with CoPP or stably expressing the active form of B-Raf, B-RafV600E, have up-regulated expression of genes involved in focal adhesion and ECM-receptor interaction.
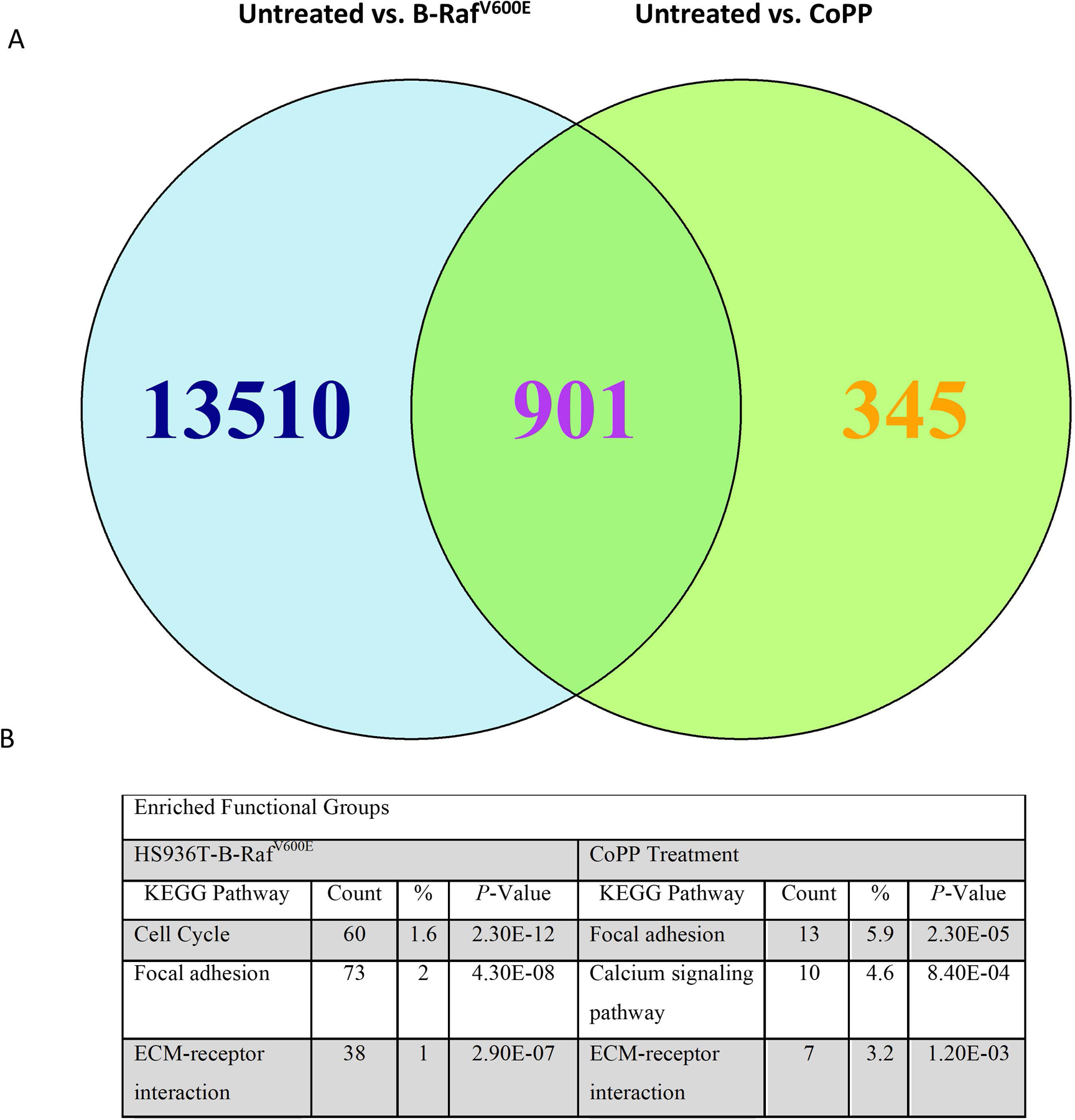
Cells grown in suspension for 4 days were collected and mRNA isolated for high throughput RNA sequencing. Triplicate samples for each treatment group were sequenced. Since CoPP treatment and Hs936T cells stably expressing B-RafV600E both cause melanosphere formation, we were interested in where the global transcriptome signatures overlapped. (A) DESeq and EdgeR Bioconductor packages were used to identify differentially expressed transcripts that were up- or down-regulated at least 2-fold. Results indicate that 901 differentially expressed transcripts were similar between the B-RafV600E-expressing Hs936T cells and Hs936T cells treated with CoPP as compared to the untreated empty vector-expressing Hs936T cells. (B) DESeq and EdgeR identified 6,997 transcripts and 357 transcripts for B-RafV600E-expressing Hs936T cells and CoPP-treated Hs936T cells, respectively, which had at least a 2-fold increase in expression as compared to untreated Hs936T cells. These genes were used for DAVID functional annotation clustering. The top three enriched functional groups, sorted by P-value, are shown above. P-values are from a modified Fisher’s Exact Test (EASE Score). “Count” = the number of genes from the total list of up-regulated genes that are represented in this functional group; “%” = the percentage of genes from the total list of up-regulated genes that are represented in that functional group.
Table 1. Genes enriched in both B-RafV600E-expressing and CoPP-treated Hs936T cells by functional group.
Statistically significant genes were identified using both DESeq and EdgeR Bioconductor packages. Genes enriched by greater than 2-fold were used to identify enriched functional groups using DAVID functional annotation clustering. Of the top three enriched functional groups identified for both B-RafV600E-expressing or CoPP-treated Hs936T cells (Fig. 9), two were shared as shown in the table. Each of these functional groups is comprised of a number of genes. Genes enriched in both groups are shown.
| Focal Adhesion | ECM-Receptor Interaction | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | ID | Hs936T B-RafV600E | CoPP-Treated | Gene | ID | Hs936T B-RafV600E | CoPP-Treated | ||||
| Fold-Change | P-Value | Fold-Change | P-Value | Fold-Change | P-Value | Fold-Change | P-Value | ||||
| ACTN2 | NM_001103 | 2.27 | 6.30E-07 | 2.14 | 4.66E-08 | ITGA1 | NM_181501 | 10.46 | 1.85E-77 | 2.57 | 1.43E-29 |
| CCND1 | NM_053056 | 4.71 | 3.14E-45 | 2.46 | 2.99E-71 | ITGA10 | NM_003637 | 9.5 | 1.09E-60 | 2.59 | 3.38E-16 |
| EGFR | NM_005228 | 3.06 | 3.34E-18 | 2.06 | 7.07E-15 | ITGA5 | NM_002205 | 54.57 | 7.75E-185 | 3.00 | 5.57E-32 |
| ITGA1 | NM_181501 | 10.46 | 1.85E-77 | 2.57 | 1.43E-29 | LAMB1 | NM_002291 | 19.81 | 2.33E-136 | 2.02 | 8.42E-36 |
| ITGA10 | NM_003637 | 9.50 | 1.09E-60 | 2.59 | 3.38E-16 | SDC2 | NM_002998 | 2.46 | 2.62E-14 | 2.34 | 1.26E-32 |
| ITGA5 | NM_002205 | 54.57 | 7.75E-185 | 3.00 | 5.57E-32 | ||||||
| LAMB1 | NM_002291 | 19.81 | 2.33E-136 | 2.02 | 8.42E-36 | ||||||
| MYLK | NM_053025 | 3.30 | 1.56E-21 | 2.24 | 8.26E-21 | ||||||
| PDGFA | NM_002607 | 10.84 | 1.19E-66 | 2.24 | 1.82E-12 | ||||||
| PDGFA | NM_033023 | 10.99 | 3.93E-67 | 2.26 | 1.03E-12 | ||||||
Table 2. Nrf2-target gene expression changes in CoPP-treated and B-RafV600E-transformed Hs936T cells.
Statistically significant genes were identified using the DESeq R Bioconductor package.
| Hs936T-B-RafV600E | CoPP Treatment | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | ID | Fold change | P-value | Qvalue | Significant | Fold change | P-value | Qvalue | Significant | |
| ARE Regulation | ||||||||||
| NFE2L2 | NM_006164 | 0.65 | 4.18E-11 | 1.40E-10 | yes | 1.14 | 2.21E-03 | 8.38E-03 | yes | |
| BACH1 | NM_206866 | 1.45 | 2.88E-02 | 4.36E-02 | yes | 1.02 | 7.23E-01 | 8.22E-01 | no | |
| MAFK | NM_002360 | 1.17 | 2.24E-01 | 2.83E-01 | no | 0.82 | 3.37E-03 | 1.21E-02 | yes | |
| MAFF | NM_001161572 | 0.37 | 2.80E-21 | 1.57E-20 | yes | 1.52 | 2.36E-15 | 7.46E-14 | yes | |
| MAFG | NM_032711 | 0.31 | 5.30E-60 | 1.00E-58 | yes | 1.05 | 3.12E-01 | 4.65E-01 | no | |
| Heme Degradation | ||||||||||
| HMOX1 | NM_002133 | 0.07 | 6.01E-185 | 8.35E-183 | yes | 14.20 | 1.10E-183 | 1.35E-179 | yes | |
| FTH1 | NM_002032 | 0.35 | 4.79E-44 | 6.09E-43 | yes | 1.58 | 9.68E-20 | 5.24E-18 | yes | |
| FTL | NM_000146 | 0.15 | 1.73E-108 | 8.65E-107 | yes | 0.98 | 6.95E-01 | 8.01E-01 | no | |
| ME1 | NM_002395 | 162.97 | 4.02E-69 | 9.28E-68 | yes | 1.23 | 2.23E-01 | NA | no | |
| Detoxifying Enzymes | ||||||||||
| NQO1 | NM_000903 | 0.24 | 1.29E-74 | 3.33E-73 | yes | 2.32 | 9.50E-82 | 1.30E-78 | yes | |
| NQO2 | NM_000904 | 0.34 | 2.44E-34 | 2.29E-33 | yes | 1.37 | 2.34E-08 | 2.75E-07 | yes | |
| GSTM1 | NM_000561 | 59.24 | 4.30E-22 | 2.51E-21 | yes | 1.02 | 8.52E-01 | NA | no | |
| Glutathione Metabolism | ||||||||||
| GCLC | NM_001498 | 0.75 | 6.26E-03 | 1.05E-02 | yes | 0.99 | 8.64E-01 | 9.16E-01 | no | |
| GCLM | NM_002061 | 0.56 | 6.50E-05 | 1.36E-04 | yes | 2.23 | 1.68E-32 | 2.51E-30 | yes | |
| SLC7A11 | NM_014331 | 0.14 | 2.25E-80 | 6.60E-79 | yes | 1.83 | 8.28E-26 | 7.62E-24 | yes | |
| G6PD | NM_000402 | 0.62 | 5.02E-09 | 1.47E-08 | yes | 1.77 | 1.28E-39 | 3.14E-37 | yes | |
| Cell Cycle and Apoptosis | ||||||||||
| SQSTM1 | NM_003900 | 0.10 | 1.44E-127 | 1.01E-125 | yes | 0.93 | 6.75E-02 | 1.45E-01 | no | |
| TFE3 | NM_006521 | 0.73 | 1.90E-03 | 3.40E-03 | yes | 0.94 | 2.18E-01 | 3.59E-01 | no | |
| BCL2L11 | NM_138621 | 0.45 | 3.06E-09 | 9.12E-09 | yes | 0.82 | 7.13E-03 | 2.28E-02 | yes | |
| Additional Antioxidants | ||||||||||
| TXNRD1 | NM_001093771 | 0.33 | 5.02E-69 | 1.15E-67 | yes | 1.33 | 7.32E-11 | 1.25E-09 | yes | |
| SOD3 | NM_003102 | 402.13 | 4.35E-30 | 3.42E-29 | yes | 0.95 | 3.47E-01 | NA | no | |
| Growth Factors | ||||||||||
| TGFB2 | NM_001135599 | 4.61 | 2.11E-03 | 3.74E-03 | yes | 0.98 | 8.72E-01 | NA | no | |
| TGFB1 | NM_000660 | 7.44 | 5.80E-110 | 3.04E-108 | yes | 1.59 | 2.31E-07 | 2.28E-06 | yes | |
| FGF13 | NM_004114 | 1.96 | 2.11E-11 | 7.23E-11 | yes | 1.82 | 5.57E-16 | 1.91E-14 | yes | |
| Proteosome | ||||||||||
| PSMB3 | NM_002795 | 1.43 | 5.84E-05 | 1.23E-04 | yes | 0.89 | 6.29E-02 | 1.37E-01 | no | |
| PSMA4 | NM_002789 | 2.53 | 2.03E-30 | 1.62E-29 | yes | 1.14 | 3.62E-02 | 8.79E-02 | yes | |
| PSMA1 | NM_148976 | 1.66 | 5.80E-11 | 1.93E-10 | yes | 1.17 | 6.95E-03 | 2.23E-02 | yes | |
| PSMB6 | NM_002798 | 1.84 | 7.85E-10 | 2.43E-09 | yes | 0.86 | 3.61E-02 | 8.77E-02 | yes | |
Discussion
Mutually exclusive mutations in either N-Ras or in B-Raf are found in a majority of melanoma tumors (Colombino et al., 2012, Reddy et al., 2017). We find that melanoma cell lines harboring the B-RafV600E mutation were capable of melanosphere formation, while melanosphere formation was markedly reduced in melanoma cell lines that contained mutant N-RASQ61K/R (Fig. 1). Furthermore, ectopic expression of B-RafV600E in the N-RAS-mutant cell line, Hs936T, markedly increased melanosphere formation (Fig. 5). The experiments reported in this manuscript, which indicate an important role for heme oxygenase activity in melanosphere formation, provide novel insight into the mechanism of melanosphere formation. Inhibition of HMOX1 protein expression by shRNA (Supplementary Fig. S1), ablation of HMOX1 protein expression by CRISPR/Cas9 (Fig. 4) or inhibition of HMOX activity by SnPP (Figs. 3 and 8) diminishes the ability of BRafV600E to promote melanosphere formation. Derepression of Bach1 with CoPP, leading to increased expression of HMOX1, promotes melanosphere formation (Figs. 3, 7 and 8) and CoPP-induced melanosphere formation is abolished in HMOX1-null cells (Fig. 4). Ectopic expression of B-RafV600E or treatment with CoPP increased expression of genes involved in focal adhesion and ECM-receptor signaling pathways (Fig. 9 and Table 2), suggesting that these signaling pathways contribute to melanosphere formation.
Melanosphere formation is influenced by a number of processes including anchorage-independent proliferation, anchorage-independent survival and the ability to form cell-cell adhesions that facilitate multicellular spheroid formation. Our results indicate that B-RafV600E promotes both anchorage-independent proliferation and the formation of multicellular spheroids. In contrast, CoPP treatment increased spheroid formation without increasing cell proliferation. To identify genes specifically involved in melanosphere formation, we carried out an RNA-seq analysis of Hs936T cells maintained in non-adherent conditions. We used non-adherent growth conditions for the RNA-seq analysis, as a previous study showed a unique transcriptome signature for melanoma cells grown as melanospheres (Hartman et al., 2014). Culture conditions can significantly influence gene expression. In some instances, the expression of the smaller subset of genes we measured by qRT-PCR in adherent cells (Fig. 6) differed from the results of our transcriptome analysis. For example, when grown in adherent conditions Hs936T cells expressing B-RafV600E had elevated expression of Nrf2 and HMOX2. When grown in melanospheres, these same cells showed a decrease in Nrf2 (Table 2) and no significant change in HMOX2. Bach1 expression was increased in both conditions with a subsequent decrease in HMOX1 expression in both conditions. Our analysis revealed a robust effect of B-RafV600E on gene transcription (Fig. 9), including a strong cell cycle-related gene signature, likely due to activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) by B-RafV600E. The major downstream mediators of B-Raf-dependent gene expression are ERK1 and ERK2, which phosphorylate Ets family transcription factors in the nucleus (Yordy and Muise-Helmericks, 2000). Consistent with the absence of CoPP-induced anchorage-independent proliferation, a cell-cycle signature was not observed in the RNA-seq analysis of CoPP-treated cells. Rather, the top three KEGG pathways that were altered in CoPP-treated cells were focal adhesion, calcium signaling and ECM-receptor interactions. Notably, both the focal adhesion and ECM-receptor interactions were also altered by stable expression of B-RafV600E. As HMOX activity is required for melanosphere formation by both B-RafV600E-expressing and CoPP-treated cells, our results indicate that HMOX promotes the generation of cell-cell contacts that facilitate spheroid formation.
Our results suggest that either HMOX1 or HMOX2 can drive spheroid formation, as HMOX1 is increased by CoPP treatment while HMOX2 levels are increased in B-RafV600E-expressing Hs936T cells. Stable expression of B-RafV600E in Hs936T cells led to an increase in Nrf2 mRNA and protein levels (Fig. 5 and 6), consistent with what others have reported (DeNicola et al., 2011). Increased steady-state levels of Nrf2 typically lead to an increase in the expression of target genes, including HMOX1. Thus, we were surprised to find that HMOX1 expression levels are markedly reduced by stable B-RafV600E expression in Hs936T cells (Fig. 6). Since expression of Bach1 mRNA is also increased by B-RafV600E expression in Hs936T cells, it is likely that increased expression of Bach1 is responsible for repression of HMOX1 expression. Notably, not all ARE-dependent genes responded the same way to stable B-RafV600E expression in Hs936T cells. Expression of several Nrf2-dependent genes, including malic enzyme 1 (ME1), superoxide dismutase 3 (SOD3) and glutathione S-transferase mu 1 (GSTM1), were increased by B-RafV600E expression in Hs936T cells while expression of NAD(P)H quinone dehydrogenase 1 (NQO1) was decreased (Table 2). It is likely that, in addition to Nrf2 and Bach1, other transcriptional mediators also contribute to regulation of oxidative stress-responsive genes. For example, GSTM1 is regulated, in part, by the Myb transcription factor (Bartley et al., 2003). Our RNA-seq analysis revealed a 41-fold increase in MYB expression (P = 2.00E-21) (Data not shown) following B-RafV600E expression in Hs936T cells, suggesting that up-regulation of GSTM1, but not the NQO1 and NQO2, is the result of increased Myb-dependent transcription. Transcriptional regulation of the large cohort of oxidative-stress response genes is more complex than simple competition between Nrf2 and Bach1 for ARE binding.
Our work adds to a growing body of evidence implicating HMOX1 in melanoma tumor growth, migration, invasion and Vemurafenib (PLX-4032) resistance. A recent publication demonstrated that down-regulation of HMOX1 improved sensitivity to the B-RafV600E kinase inhibitor, PLX-4032, in human melanoma cells harboring the B-RafV600E mutation (Furfaro et al., 2020). As acquired resistance remains a significant problem for melanoma patients (Welsh et al., 2016), targeting of HMOX1 may provide a means to sensitize melanomas to Vemurafenib treatment. Knockdown of Nrf2, which results in a reduction in HMOX1 expression, has been shown to inhibit migration and invasion of B16-F10 mouse melanoma cells during ionizing radiation (Gao et al., 2018). Another group recently reported a direct interaction between HMOX1 and B-Raf in A375 human melanoma cells (Liu et al., 2019). They also demonstrate that HMOX1 overexpression in A375 cells leads to an increased tumor volume while knockout of HMOX1 reduces tumor volumes in a mouse tumor xenograft model (Liu et al., 2019). Finally, they propose a mechanism by which HMOX1 promotes cellular proliferation by upregulation of cyclin E and CDK2 in a B-Raf-dependent manner (Liu et al., 2019).
Taken together, the pharmacological and genetic approaches that we used to investigate the role of heme oxygenase activity in melanosphere formation support a model in which the enzymatic activity of heme oxygenase is required for melanosphere formation. We propose that the underlying mechanism by which HMOX enzymatic activity promotes melanosphere formation involves gene expression changes that lead to modulation in focal adhesion and ECM-receptor signaling pathways. Heme degradation by either HMOX1 or HMOX2 results in the production of CO, free iron and biliverdin/bilirubin. Several studies have reported that CO influences gene expression (Dulak and Jozkowicz, 2003, Oliveira et al., 2015, Chi et al., 2015). Thus, production of CO could be responsible for increased expression of genes involved in ECM dynamics that were identified in our RNA-seq data. Dysregulation of ECM dynamics is a hallmark of metastasis (Lu et al., 2012), allowing cancer cells to alter the way in which they interact with surrounding tissues. In a meta-profiling analysis of expression data from 190 human tumors, HMOX1 expression correlated with the expression of a number of genes involved in extracellular matrix (ECM) remodeling (Tauber et al., 2010). One of the genes identified in this analysis, peroxidasin (PXDN), encodes a peroxidase enzyme that contributes to collagen cross-linking (Bhave et al., 2012). Expression of PXDN was dependent on HMOX1 expression in both BeWo choriocarcinoma cells and 607B melanoma cells (Tauber et al., 2010). Loss of PXDN inhibited invasion and cell growth (Tauber et al., 2010). HMOX1 has been shown to play a unique role in lung tumor colonization in mice (Dey et al., 2015). In normal cells, ECM detachment leads to a specialized type of apoptosis called anoikis. Following ECM detachment in human fibrosarcoma cells, the transcription factor ATF4 is increased which promotes resistance to anoikis. Following ECM detachment, both Nrf2 and ATF4 bind the HMOX1 promoter and promote gene expression (Dey et al., 2015). When injected into mice, ATF4-deficient human fibrosarcoma cells were unable to colonize the lung. However, lung tumor colonization was subsequently rescued by overexpression of HMOX1 (Dey et al., 2015). Together with our data, these studies support a role for heme oxygenase activity in modulation of ECM dynamics.
Our data suggest an HMOX-dependent mechanism for B-Raf-driven melanosphere formation. While melanosphere formation has been shown to correlate with tumorigenic potential, the role of HMOX activity in tumor growth and metastasis is not fully understood. Further studies directed towards understanding the role of HMOX activity in tumor biology may provide insight into how therapeutic metalloporphyrins, such as SnPP and SnMP, might be repurposed for treatment of metastatic melanoma.
Supplementary Material
Supplementary Figure S1. Inhibition of either Nrf2 or HMOX1 diminishes melanosphere formation. (A) SK-Mel-5 cells stably expressing a pSicoR vector containing either silencing hairpin RNA (shRNA) targeting Nrf2 or a scrambled shRNA were plated for melanosphere assays. Error bars represent the SEM of four biological replicates of each melanosphere assay. (B) Nrf2 protein levels in Sk-Mel-5 cells grown in adherent conditions were assessed by immunoblot analysis and band intensity was measured and normalized to tubulin. Error bars represent the SEM of three biological replicates. (C) SK-Mel-5 cells stably expressing a pSicoR vector containing either a scrambled shRNA or an shRNA targeting HMOX1 were used for melanosphere assays. Error bars represent the SEM of four biological replicates of each melanosphere assay. (D) HMOX1 protein levels in SK-Mel-5 cells stably expressing scrambled shRNA or shRNA targeting HMOX1 and grown in adherent conditions were assessed by immunoblot analysis and band intensity measured and normalized to tubulin. Error bars represent the SEM of three biological replicates for western blot quantitation. Statistical significance was determined by the standard Student’s t-test. *P < 0.05, **P < 0.01.
Supplementary Figure S2. Heme Oxygenase I and II protein expression in human melanoma lines. (A) Immunoblot analysis for six human melanoma cell lines with or without 10 μM CoPP treatment overnight to assess protein levels of HMOX1 and HMOX2. (B) For HMOX1, band intensities were measured and normalized to tubulin. Error bars represent the SEM of three biological replicates.
Significance:
Melanoma is the fifth most common malignancy in the US. While the overall 5-year survival rate has improved in the past decade the 5-year survival rate for melanoma patients that are diagnosed with metastatic disease is only 23%, due in part to acquired drug resistance to both targeted therapies (i.e. B-Raf inhibitors) and immunotherapies. Our results suggest heme oxygenase activity as a novel target for drug development. Importantly, current therapeutic metalloporphoryins (e.g. SnMP, SnPP) may be repurposed for the treatment of metastatic melanoma. This report provides the first evidence implicating heme oxygenase in a B-Raf-dependent mechanism for promoting melanoma progression.
Acknowledgements
This work was supported by a grant from the National Institutes of Health, NIH 1 P50 AT006273 and a grant from the University of Missouri Research Board. Support for Kimberly Jasmer was provided by a grant from the Department of Education, GAANN IX P200A100178.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References:
- ALFADDA AA & SALLAM RM 2012. Reactive oxygen species in health and disease. J Biomed Biotechnol, 2012, 936486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATTUCKS OC, JASMER KJ, HANNINK M, KASSIS J, ZHONG Z, GUPTA S, VICTORY SF, GUZEL M, POLISETTI DR, ANDREWS R, MJALLI AM & KOSTURA MJ 2014. Induction of heme oxygenase I (HMOX1) by HPP-4382: a novel modulator of Bach1 activity. PLoS One, 9, e101044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARTLEY PA, KEOUGH RA, LUTWYCHE JK & GONDA TJ 2003. Regulation of the gene encoding glutathione S-transferase M1 (GSTM1) by the Myb oncoprotein. Oncogene, 22, 7570–5. [DOI] [PubMed] [Google Scholar]
- BAUER AK, CHO HY, MILLER-DEGRAFF L, WALKER C, HELMS K, FOSTEL J, YAMAMOTO M & KLEEBERGER SR 2011. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS One, 6, e26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENHAR M, ENGELBERG D & LEVITZKI A 2002. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep, 3, 420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERBERAT PO, DAMBRAUSKAS Z, GULBINAS A, GIESE T, GIESE N, KUNZLI B, AUTSCHBACH F, MEUER S, BUCHLER MW & FRIESS H 2005. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res, 11, 3790–8. [DOI] [PubMed] [Google Scholar]
- BHAVE G, CUMMINGS CF, VANACORE RM, KUMAGAI-CRESSE C, ERO-TOLLIVER IA, RAFI M, KANG JS, PEDCHENKO V, FESSLER LI, FESSLER JH & HUDSON BG 2012. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat Chem Biol, 8, 784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BILBAN M, HASCHEMI A, WEGIEL B, CHIN BY, WAGNER O & OTTERBEIN LE 2008. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J Mol Med (Berl), 86, 267–79. [DOI] [PubMed] [Google Scholar]
- BURDON RH 1995. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic Biol Med, 18, 775–94. [DOI] [PubMed] [Google Scholar]
- BURDON RH, GILL V & RICE-EVANS C 1990. Oxidative stress and tumour cell proliferation. Free Radic Res Commun, 11, 65–76. [DOI] [PubMed] [Google Scholar]
- BUSSEROLLES J, MEGIAS J, TERENCIO MC & ALCARAZ MJ 2006. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int J Biochem Cell Biol, 38, 1510–7. [DOI] [PubMed] [Google Scholar]
- CHAU LY 2015a. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci, 22, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAU LY 2015b. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci, 22, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN HH, CHANG HH, CHANG JY, TANG YC, CHENG YC, LIN LM, CHENG SY, HUANG CH, SUN MW, CHEN CT & KUO CC 2017. Enhanced B-Raf-mediated NRF2 gene transcription and HATs-mediated NRF2 protein acetylation contributes to ABCC1-mediated chemoresistance and glutathione-mediated survival in acquired topoisomerase II poison-resistant cancer cells. Free Radic Biol Med, 113, 505–518. [DOI] [PubMed] [Google Scholar]
- CHI PL, LIN CC, CHEN YW, HSIAO LD & YANG CM 2015. CO Induces Nrf2-Dependent Heme Oxygenase-1 Transcription by Cooperating with Sp1 and c-Jun in Rat Brain Astrocytes. Mol Neurobiol, 52, 277–92. [DOI] [PubMed] [Google Scholar]
- CLEASBY A, YON J, DAY PJ, RICHARDSON C, TICKLE IJ, WILLIAMS PA, CALLAHAN JF, CARR R, CONCHA N, KERNS JK, QI H, SWEITZER T, WARD P & DAVIES TG 2014. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One, 9, e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLOMBINO M, CAPONE M, LISSIA A, COSSU A, RUBINO C, DE GIORGI V, MASSI D, FONSATTI E, STAIBANO S, NAPPI O, PAGANI E, CASULA M, MANCA A, SINI M, FRANCO R, BOTTI G, CARACO C, MOZZILLO N, ASCIERTO PA & PALMIERI G 2012. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol, 30, 2522–9. [DOI] [PubMed] [Google Scholar]
- CULLINAN SB, GORDAN JD, JIN J, HARPER JW & DIEHL JA 2004. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol, 24, 8477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEGESE MS, MENDIZABAL JE, GANDINI NA, GUTKIND JS, MOLINOLO A, HEWITT SM, CURINO AC, COSO OA & FACCHINETTI MM 2012. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer, 77, 168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENICOLA GM, KARRETH FA, HUMPTON TJ, GOPINATHAN A, WEI C, FRESE K, MANGAL D, YU KH, YEO CJ, CALHOUN ES, SCRIMIERI F, WINTER JM, HRUBAN RH, IACOBUZIO-DONAHUE C, KERN SE, BLAIR IA & TUVESON DA 2011. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature, 475, 106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVESA I, FERRANDIZ ML, TERENCIO MC, JOOSTEN LA, VAN DEN BERG WB & ALCARAZ MJ 2005. Influence of heme oxygenase 1 modulation on the progression of murine collagen-induced arthritis. Arthritis Rheum, 52, 3230–8. [DOI] [PubMed] [Google Scholar]
- DEY S, SAYERS CM, VERGINADIS II, LEHMAN SL, CHENG Y, CERNIGLIA GJ, TUTTLE SW, FELDMAN MD, ZHANG PJ, FUCHS SY, DIEHL JA & KOUMENIS C 2015. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest, 125, 2592–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DINKOVA-KOSTOVA AT, HOLTZCLAW WD, COLE RN, ITOH K, WAKABAYASHI N, KATOH Y, YAMAMOTO M & TALALAY P 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A, 99, 11908–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOVER SB, GRAHAM A, FITZSIMONS E, MOORE MR & MCCOLL KE 1991. Haem-arginate plus tin-protoporphyrin for acute hepatic porphyria. Lancet, 338, 263. [DOI] [PubMed] [Google Scholar]
- DOVER SB, MOORE MR, FITZSIMMONS EJ, GRAHAM A & MCCOLL KE 1993. Tin protoporphyrin prolongs the biochemical remission produced by heme arginate in acute hepatic porphyria. Gastroenterology, 105, 500–6. [DOI] [PubMed] [Google Scholar]
- DULAK J & JOZKOWICZ A 2003. Carbon monoxide -- a “new” gaseous modulator of gene expression. Acta Biochim Pol, 50, 31–47. [PubMed] [Google Scholar]
- ERAMO A, LOTTI F, SETTE G, PILOZZI E, BIFFONI M, DI VIRGILIO A, CONTICELLO C, RUCO L, PESCHLE C & DE MARIA R 2008. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ, 15, 504–14. [DOI] [PubMed] [Google Scholar]
- ERAMO A, RICCI-VITIANI L, ZEUNER A, PALLINI R, LOTTI F, SETTE G, PILOZZI E, LAROCCA LM, PESCHLE C & DE MARIA R 2006. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ, 13, 1238–41. [DOI] [PubMed] [Google Scholar]
- FURFARO AL, OTTONELLO S, LOI G, COSSU I, PIRAS S, SPAGNOLO F, QUEIROLO P, MARINARI UM, MORETTA L, PRONZATO MA, MINGARI MC, PIETRA G & NITTI M 2020. HO-1 downregulation favors BRAF(V600) melanoma cell death induced by Vemurafenib/PLX4032 and increases NK recognition. Int J Cancer, 146, 1950–1962. [DOI] [PubMed] [Google Scholar]
- GAO Y, ZHAO Z, MENG X, CHEN H & FU G 2018. Migration and invasion in B16-F10 mouse melanoma cells are regulated by Nrf2 inhibition during treatment with ionizing radiation. Oncol Lett, 16, 1959–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARTMAN ML, TALAR B, NOMAN MZ, GAJOS-MICHNIEWICZ A, CHOUAIB S & CZYZ M 2014. Gene expression profiling identifies microphthalmia-associated transcription factor (MITF) and Dickkopf-1 (DKK1) as regulators of microenvironment-driven alterations in melanoma phenotype. PLoS One, 9, e95157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYES JD & DINKOVA-KOSTOVA AT 2014. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci, 39, 199–218. [DOI] [PubMed] [Google Scholar]
- HAYES JD & MCMAHON M 2009. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci, 34, 176–88. [DOI] [PubMed] [Google Scholar]
- HOMMA S, ISHII Y, MORISHIMA Y, YAMADORI T, MATSUNO Y, HARAGUCHI N, KIKUCHI N, SATOH H, SAKAMOTO T, HIZAWA N, ITOH K & YAMAMOTO M 2009. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin Cancer Res, 15, 3423–32. [DOI] [PubMed] [Google Scholar]
- ISHIKAWA M, NUMAZAWA S & YOSHIDA T 2005. Redox regulation of the transcriptional repressor Bach1. Free Radic Biol Med, 38, 1344–52. [DOI] [PubMed] [Google Scholar]
- ITOH K, CHIBA T, TAKAHASHI S, ISHII T, IGARASHI K, KATOH Y, OYAKE T, HAYASHI N, SATOH K, HATAYAMA I, YAMAMOTO M & NABESHIMA Y 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun, 236, 313–22. [DOI] [PubMed] [Google Scholar]
- ITOH K, WAKABAYASHI N, KATOH Y, ISHII T, IGARASHI K, ENGEL JD & YAMAMOTO M 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev, 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANSEN T, HORTMANN M, OELZE M, OPITZ B, STEVEN S, SCHELL R, KNORR M, KARBACH S, SCHUHMACHER S, WENZEL P, MUNZEL T & DAIBER A 2010. Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J Mol Cell Cardiol, 49, 186–95. [DOI] [PubMed] [Google Scholar]
- JOZKOWICZ A, WAS H & DULAK J 2007. Heme oxygenase-1 in tumors: is it a false friend? Antioxid Redox Signal, 9, 2099–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG HJ, HONG YB, KIM HJ & BAE I 2010. CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2 (NRF2) protein stability by proteasome-mediated degradation. J Biol Chem, 285, 21258–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KASPAR JW, NITURE SK & JAISWAL AK 2009. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med, 47, 1304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENSLER TW & WAKABAYASHI N 2010. Nrf2: friend or foe for chemoprevention? Carcinogenesis, 31, 90–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM YR, OH JE, KIM MS, KANG MR, PARK SW, HAN JY, EOM HS, YOO NJ & LEE SH 2010. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol, 220, 446–51. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI A, KANG MI, OKAWA H, OHTSUJI M, ZENKE Y, CHIBA T, IGARASHI K & YAMAMOTO M 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol, 24, 7130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUNDU N, ZHANG S & FULTON AM 1995. Sublethal oxidative stress inhibits tumor cell adhesion and enhances experimental metastasis of murine mammary carcinoma. Clin Exp Metastasis, 13, 16–22. [DOI] [PubMed] [Google Scholar]
- KWAK MK, WAKABAYASHI N, GREENLAW JL, YAMAMOTO M & KENSLER TW 2003. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol, 23, 8786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWOK SC 2013. Zinc Protoporphyrin Upregulates Heme Oxygenase-1 in PC-3 Cells via the Stress Response Pathway. Int J Cell Biol, 2013, 162094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANDER HM, HAJJAR DP, HEMPSTEAD BL, MIRZA UA, CHAIT BT, CAMPBELL S & QUILLIAM LA 1997. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem, 272, 4323–6. [DOI] [PubMed] [Google Scholar]
- LAU A, VILLENEUVE NF, SUN Z, WONG PK & ZHANG DD 2008. Dual roles of Nrf2 in cancer. Pharmacol Res, 58, 262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE SB, SELLERS BN & DENICOLA GM 2018. The Regulation of NRF2 by Nutrient-Responsive Signaling and Its Role in Anabolic Cancer Metabolism. Antioxid Redox Signal, 29, 1774–1791. [DOI] [PubMed] [Google Scholar]
- LI J, HOU J, SUN L, WILKINS JM, LU Y, NIEDERHUTH CE, MERIDETH BR, MAWHINNEY TP, MOSSINE VV, GREENLIEF CM, WALKER JC, FOLK WR, HANNINK M, LUBAHN DB, BIRCHLER JA & CHENG J 2015. From Gigabyte to Kilobyte: A Bioinformatics Protocol for Mining Large RNA-Seq Transcriptomics Data. PLoS One, 10, e0125000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIGNITTO L, LEBOEUF SE, HOMER H, JIANG S, ASKENAZI M, KARAKOUSI TR, PASS HI, BHUTKAR AJ, TSIRIGOS A, UEBERHEIDE B, SAYIN VI, PAPAGIANNAKOPOULOS T & PAGANO M 2019. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell, 178, 316–329 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN CW, SHEN SC, HOU WC, YANG LY & CHEN YC 2008. Heme oxygenase-1 inhibits breast cancer invasion via suppressing the expression of matrix metalloproteinase-9. Mol Cancer Ther, 7, 1195–206. [DOI] [PubMed] [Google Scholar]
- LIN PH, LAN WM & CHAU LY 2013. TRC8 suppresses tumorigenesis through targeting heme oxygenase-1 for ubiquitination and degradation. Oncogene, 32, 2325–34. [DOI] [PubMed] [Google Scholar]
- LIN Q, WEIS S, YANG G, WENG YH, HELSTON R, RISH K, SMITH A, BORDNER J, POLTE T, GAUNITZ F & DENNERY PA 2007. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem, 282, 20621–33. [DOI] [PubMed] [Google Scholar]
- LIU L, WU Y, BIAN C, NISAR MF, WANG M, HU X, DIAO Q, NIAN W, WANG E, XU W & ZHONG JL 2019. Heme oxygenase 1 facilitates cell proliferation via the B-Raf-ERK signaling pathway in melanoma. Cell Commun Signal, 17, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU P, WEAVER VM & WERB Z 2012. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol, 196, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUANPITPONG S, POOHADSUAN J, SAMART P, KIRATIPAIBOON C, ROJANASAKUL Y & ISSARAGRISIL S 2018. Reactive oxygen species mediate cancer stem-like cells and determine bortezomib sensitivity via Mcl-1 and Zeb-1 in mantle cell lymphoma. Biochim Biophys Acta Mol Basis Dis, 1864, 3739–3753. [DOI] [PubMed] [Google Scholar]
- MAHER JM, DIETER MZ, ALEKSUNES LM, SLITT AL, GUO G, TANAKA Y, SCHEFFER GL, CHAN JY, MANAUTOU JE, CHEN Y, DALTON TP, YAMAMOTO M & KLAASSEN CD 2007. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology, 46, 1597–610. [DOI] [PubMed] [Google Scholar]
- MAINES MD & ABRAHAMSSON PA 1996. Expression of heme oxygenase-1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology, 47, 727–33. [DOI] [PubMed] [Google Scholar]
- MALHOTRA D, PORTALES-CASAMAR E, SINGH A, SRIVASTAVA S, ARENILLAS D, HAPPEL C, SHYR C, WAKABAYASHI N, KENSLER TW, WASSERMAN WW & BISWAL S 2010. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res, 38, 5718–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARZAGALLI M, MORETTI RM, MESSI E, MARELLI MM, FONTANA F, ANASTASIA A, BANI MR, BERETTA G & LIMONTA P 2018. Targeting melanoma stem cells with the Vitamin E derivative delta-tocotrienol. Sci Rep, 8, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAYERHOFER M, FLORIAN S, KRAUTH MT, AICHBERGER KJ, BILBAN M, MARCULESCU R, PRINTZ D, FRITSCH G, WAGNER O, SELZER E, SPERR WR, VALENT P & SILLABER C 2004. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res, 64, 3148–54. [DOI] [PubMed] [Google Scholar]
- MAYNARD S, SCHURMAN SH, HARBOE C, DE SOUZA-PINTO NC & BOHR VA 2009. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis, 30, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITSUISHI Y, TAGUCHI K, KAWATANI Y, SHIBATA T, NUKIWA T, ABURATANI H, YAMAMOTO M & MOTOHASHI H 2012. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell, 22, 66–79. [DOI] [PubMed] [Google Scholar]
- MIURA S, SHIBAZAKI M, KASAI S, YASUHIRA S, WATANABE A, INOUE T, KAGESHITA Y, TSUNODA K, TAKAHASHI K, AKASAKA T, MASUDA T & MAESAWA C 2014. A somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. J Invest Dermatol, 134, 553–6. [DOI] [PubMed] [Google Scholar]
- MOHAMMAD J, SINGH RR, RIGGLE C, HAUGRUD B, ABDALLA MY & REINDL KM 2019. JNK inhibition blocks piperlongumine-induced cell death and transcriptional activation of heme oxygenase-1 in pancreatic cancer cells. Apoptosis, 24, 730–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUSCARELLA LA, PARRELLA P, D’ALESSANDRO V, LA TORRE A, BARBANO R, FONTANA A, TANCREDI A, GUARNIERI V, BALSAMO T, COCO M, COPETTI M, PELLEGRINI F, DE BONIS P, BISCEGLIA M, SCARAMUZZI G, MAIELLO E, VALORI VM, MERLA G, VENDEMIALE G & FAZIO VM 2011. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics, 6, 710–9. [DOI] [PubMed] [Google Scholar]
- NISHIE A, ONO M, SHONO T, FUKUSHI J, OTSUBO M, ONOUE H, ITO Y, INAMURA T, IKEZAKI K, FUKUI M, IWAKI T & KUWANO M 1999. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res, 5, 1107–13. [PubMed] [Google Scholar]
- NISHIGORI C, HATTORI Y & TOYOKUNI S 2004. Role of reactive oxygen species in skin carcinogenesis. Antioxid Redox Signal, 6, 561–70. [DOI] [PubMed] [Google Scholar]
- NITURE SK & JAISWAL AK 2012. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem, 287, 9873–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOH SJ, BAE JS, JAMIYANDORJ U, PARK HS, KWON KS, JUNG SH, YOUN HJ, LEE H, PARK BH, CHUNG MJ, MOON WS, KANG MJ & JANG KY 2013. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer, 13, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUHN P, KUNZLI BM, HENNIG R, MITKUS T, RAMANAUSKAS T, NOBILING R, MEUER SC, FRIESS H & BERBERAT PO 2009. Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Mol Cancer, 8, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGAWA K, SUN J, TAKETANI S, NAKAJIMA O, NISHITANI C, SASSA S, HAYASHI N, YAMAMOTO M, SHIBAHARA S, FUJITA H & IGARASHI K 2001. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J, 20, 2835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKAMOTO I, KROGLER J, ENDLER G, KAUFMANN S, MUSTAFA S, EXNER M, MANNHALTER C, WAGNER O & PEHAMBERGER H 2006. A microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with risk for melanoma. Int J Cancer, 119, 1312–5. [DOI] [PubMed] [Google Scholar]
- OLIVEIRA SR, VIEIRA HL & DUARTE CB 2015. Effect of Carbon Monoxide on gene expression in cerebrocortical astrocytes: validation of reference genes for quantitative Real-time PCR. Nitric Oxide. [DOI] [PubMed] [Google Scholar]
- OTTERBEIN LE, HEDBLOM A, HARRIS C, CSIZMADIA E, GALLO D & WEGIEL B 2011. Heme oxygenase-1 and carbon monoxide modulate DNA repair through ataxia-telangiectasia mutated (ATM) protein. Proc Natl Acad Sci U S A, 108, 14491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OTTERBEIN LE, SOARES MP, YAMASHITA K & BACH FH 2003. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol, 24, 449–55. [DOI] [PubMed] [Google Scholar]
- PELICANO H, LU W, ZHOU Y, ZHANG W, CHEN Z, HU Y & HUANG P 2009. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res, 69, 2375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEREGO M, TORTORETO M, TRAGNI G, MARIANI L, DEHO P, CARBONE A, SANTINAMI M, PATUZZO R, MINA PD, VILLA A, PRATESI G, COSSA G, PEREGO P, DAIDONE MG, ALISON MR, PARMIANI G, RIVOLTINI L & CASTELLI C 2010. Heterogeneous phenotype of human melanoma cells with in vitro and in vivo features of tumor-initiating cells. J Invest Dermatol, 130, 1877–86. [DOI] [PubMed] [Google Scholar]
- RADA P, ROJO AI, CHOWDHRY S, MCMAHON M, HAYES JD & CUADRADO A 2011. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol, 31, 1121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAY PD, HUANG BW & TSUJI Y 2012. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal, 24, 981–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REDDY BY, MILLER DM & TSAO H 2017. Somatic driver mutations in melanoma. Cancer, 123, 2104–2117. [DOI] [PubMed] [Google Scholar]
- REICHARD JF, MOTZ GT & PUGA A 2007. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res, 35, 7074–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROJO AI, RADA P, MENDIOLA M, ORTEGA-MOLINA A, WOJDYLA K, ROGOWSKA-WRZESINSKA A, HARDISSON D, SERRANO M & CUADRADO A 2014. The PTEN/NRF2 axis promotes human carcinogenesis. Antioxid Redox Signal, 21, 2498–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROJO DE LA VEGA M, CHAPMAN E & ZHANG DD 2018. NRF2 and the Hallmarks of Cancer. Cancer Cell, 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTBLAT B, MELINO G & KNIGHT RA 2012. NRF2 and p53: Januses in cancer? Oncotarget, 3, 1272–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RYTER SW & TYRRELL RM 2000. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med, 28, 289–309. [DOI] [PubMed] [Google Scholar]
- SARDANA MK & KAPPAS A 1987. Dual control mechanism for heme oxygenase: tin(IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc Natl Acad Sci U S A, 84, 2464–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMITTGEN TD & LIVAK KJ 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc, 3, 1101–8. [DOI] [PubMed] [Google Scholar]
- SCHULZ S, WONG RJ, VREMAN HJ & STEVENSON DK 2012. Metalloporphyrins - an update. Front Pharmacol, 3, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SETTE G, FECCHI K, SALVATI V, LOTTI F, PILOZZI E, DURANTI E, BIFFONI M, PAGLIUCA A, MARTINETTI D, MEMEO L, MILELLA M, DE MARIA R & ERAMO A 2013. Mek inhibition results in marked antitumor activity against metastatic melanoma patient-derived melanospheres and in melanosphere-generated xenografts. J Exp Clin Cancer Res, 32, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SETTE G, SALVATI V, MEMEO L, FECCHI K, COLAROSSI C, DI MATTEO P, SIGNORE M, BIFFONI M, D’ANDREA V, DE ANTONI E, CANZONIERI V, DE MARIA R & ERAMO A 2012. EGFR inhibition abrogates leiomyosarcoma cell chemoresistance through inactivation of survival pathways and impairment of CSC potential. PLoS One, 7, e46891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBATA T, OHTA T, TONG KI, KOKUBU A, ODOGAWA R, TSUTA K, ASAMURA H, YAMAMOTO M & HIROHASHI S 2008. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A, 105, 13568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH A, BOLDIN-ADAMSKY S, THIMMULAPPA RK, RATH SK, ASHUSH H, COULTER J, BLACKFORD A, GOODMAN SN, BUNZ F, WATSON WH, GABRIELSON E, FEINSTEIN E & BISWAL S 2008. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res, 68, 7975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH A, MISRA V, THIMMULAPPA RK, LEE H, AMES S, HOQUE MO, HERMAN JG, BAYLIN SB, SIDRANSKY D, GABRIELSON E, BROCK MV & BISWAL S 2006. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med, 3, e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOLIS LM, BEHRENS C, DONG W, SURAOKAR M, OZBURN NC, MORAN CA, CORVALAN AH, BISWAL S, SWISHER SG, BEKELE BN, MINNA JD, STEWART DJ & WISTUBA II 2010. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res, 16, 3743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SORBARA MT & GIRARDIN SE 2011. Mitochondrial ROS fuel the inflammasome. Cell Res, 21, 558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN G & KEMBLE DJ 2009. To C or not to C: direct and indirect redox regulation of Src protein tyrosine kinase. Cell Cycle, 8, 2353–5. [DOI] [PubMed] [Google Scholar]
- SUNAMURA M, DUDA DG, GHATTAS MH, LOZONSCHI L, MOTOI F, YAMAUCHI J, MATSUNO S, SHIBAHARA S & ABRAHAM NG 2003. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis, 6, 15–24. [DOI] [PubMed] [Google Scholar]
- SUZUKI H, TASHIRO S, HIRA S, SUN J, YAMAZAKI C, ZENKE Y, IKEDA-SAITO M, YOSHIDA M & IGARASHI K 2004. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J, 23, 2544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN MK, LIM HJ, BENNETT EJ, SHI Y & HARPER JW 2013. Parallel SCF adaptor capture proteomics reveals a role for SCFFBXL17 in NRF2 activation via BACH1 repressor turnover. Mol Cell, 52, 9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANNO T & MATSUI W 2011. Development and maintenance of cancer stem cells under chronic inflammation. J Nippon Med Sch, 78, 138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAUBER S, JAIS A, JEITLER M, HAIDER S, HUSA J, LINDROOS J, KNOFLER M, MAYERHOFER M, PEHAMBERGER H, WAGNER O & BILBAN M 2010. Transcriptome analysis of human cancer reveals a functional role of heme oxygenase-1 in tumor cell adhesion. Mol Cancer, 9, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORISU-ITAKURA H, FURUE M, KUWANO M & ONO M 2000. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn J Cancer Res, 91, 906–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILE GF & TYRRELL RM 1993. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J Biol Chem, 268, 14678–81. [PubMed] [Google Scholar]
- WANG R, AN J, JI F, JIAO H, SUN H & ZHOU D 2008a. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun, 373, 151–4. [DOI] [PubMed] [Google Scholar]
- WANG XJ, SUN Z, VILLENEUVE NF, ZHANG S, ZHAO F, LI Y, CHEN W, YI X, ZHENG W, WONDRAK GT, WONG PK & ZHANG DD 2008b. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis, 29, 1235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARNATZ HJ, SCHMIDT D, MANKE T, PICCINI I, SULTAN M, BORODINA T, BALZEREIT D, WRUCK W, SOLDATOV A, VINGRON M, LEHRACH H & YASPO ML 2011. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J Biol Chem, 286, 23521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAS H, CICHON T, SMOLARCZYK R, RUDNICKA D, STOPA M, CHEVALIER C, LEGER JJ, LACKOWSKA B, GROCHOT A, BOJKOWSKA K, RATAJSKA A, KIEDA C, SZALA S, DULAK J & JOZKOWICZ A 2006. Overexpression of heme oxygenase-1 in murine melanoma: increased proliferation and viability of tumor cells, decreased survival of mice. Am J Pathol, 169, 2181–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEGIEL B, NEMETH Z, CORREA-COSTA M, BULMER AC & OTTERBEIN LE 2014. Heme oxygenase-1: a metabolic nike. Antioxid Redox Signal, 20, 1709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WELSH SJ, RIZOS H, SCOLYER RA & LONG GV 2016. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur J Cancer, 62, 76–85. [DOI] [PubMed] [Google Scholar]
- XIA ZW, ZHONG WW, XU LQ, SUN JL, SHEN QX, WANG JG, SHAO J, LI YZ & YU SC 2006. Heme oxygenase-1-mediated CD4+CD25high regulatory T cells suppress allergic airway inflammation. J Immunol, 177, 5936–45. [DOI] [PubMed] [Google Scholar]
- YANG G, NGUYEN X, OU J, REKULAPELLI P, STEVENSON DK & DENNERY PA 2001. Unique effects of zinc protoporphyrin on HO-1 induction and apoptosis. Blood, 97, 1306–13. [DOI] [PubMed] [Google Scholar]
- YORDY JS & MUISE-HELMERICKS RC 2000. Signal transduction and the Ets family of transcription factors. Oncogene, 19, 6503–13. [DOI] [PubMed] [Google Scholar]
- ZENKE-KAWASAKI Y, DOHI Y, KATOH Y, IKURA T, IKURA M, ASAHARA T, TOKUNAGA F, IWAI K & IGARASHI K 2007. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol, 27, 6962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG DD 2006. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev, 38, 769–89. [DOI] [PubMed] [Google Scholar]
- ZHANG DD & HANNINK M 2003. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol, 23, 8137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG DD, LO SC, CROSS JV, TEMPLETON DJ & HANNINK M 2004. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol, 24, 10941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG P, SINGH A, YEGNASUBRAMANIAN S, ESOPI D, KOMBAIRAJU P, BODAS M, WU H, BOVA SG & BISWAL S 2010. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther, 9, 336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Inhibition of either Nrf2 or HMOX1 diminishes melanosphere formation. (A) SK-Mel-5 cells stably expressing a pSicoR vector containing either silencing hairpin RNA (shRNA) targeting Nrf2 or a scrambled shRNA were plated for melanosphere assays. Error bars represent the SEM of four biological replicates of each melanosphere assay. (B) Nrf2 protein levels in Sk-Mel-5 cells grown in adherent conditions were assessed by immunoblot analysis and band intensity was measured and normalized to tubulin. Error bars represent the SEM of three biological replicates. (C) SK-Mel-5 cells stably expressing a pSicoR vector containing either a scrambled shRNA or an shRNA targeting HMOX1 were used for melanosphere assays. Error bars represent the SEM of four biological replicates of each melanosphere assay. (D) HMOX1 protein levels in SK-Mel-5 cells stably expressing scrambled shRNA or shRNA targeting HMOX1 and grown in adherent conditions were assessed by immunoblot analysis and band intensity measured and normalized to tubulin. Error bars represent the SEM of three biological replicates for western blot quantitation. Statistical significance was determined by the standard Student’s t-test. *P < 0.05, **P < 0.01.
Supplementary Figure S2. Heme Oxygenase I and II protein expression in human melanoma lines. (A) Immunoblot analysis for six human melanoma cell lines with or without 10 μM CoPP treatment overnight to assess protein levels of HMOX1 and HMOX2. (B) For HMOX1, band intensities were measured and normalized to tubulin. Error bars represent the SEM of three biological replicates.