

Abstract
Approximately 70% of invasive breast cancers have some degree of dependence on the estrogen hormone for cell proliferation and growth. These tumors have estrogen and/or progresterone receptors (ER/PR+), or generally referred to as hormone receptor positive (HR+) tumors, as indicated by the presence of positive staining and varying intensity levels of estrogen and/or progesterone receptors on immunohistochemistry. Therapies that inhibit ER signaling pathways, such as aromatase inhibitors (letrozole, anastrazole, exemestane), selective ER modulators (tamoxifen), and ER down-regulators (fulvestrant), are the mainstays of treatment for hormone receptor positive breast cancers. However, de novo or acquired resistance to ER targeted therapies is present in many tumors leading to disease progression. The PI3K/AKT/mTOR pathway is implicated in sustaining endocrine resistance and has become the target of many new drugs for ER+ breast cancer. This article will review the function of the phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway and the various classes of PI3K pathway inhibitors that have been developed to disrupt this pathway signaling for the treatment of hormone receptor positive breast cancer.
1. Function of the PI3K/AKT/mTOR pathway and role in tumorigenesis
The PI3K/AKT/mTOR pathway (Figure 1) controls many important cellular functions including metabolism, growth, survival, and proliferation. This pathway transmits a wide variety of extracellular stimuli through a signaling cascade via phosphatidylinositol 3-kinases (PI3Ks).1 PI3Ks are lipid kinases which are divided into three different classes. Class I PI3Ks are heterodimers consisting of a p85 regulatory subunit and a p110 catalytic subunit (p110α, p110β, p110γ or p110δ). This class of PI3Ks is the most studied and is clearly implicated in oncogenesis and tumor growth.1,2 A downstream target of the PI3K pathway is the serine/threonine kinase, AKT, which has 3 isoforms (AKT1, AKT2, and AKT3). This kinase plays a central role in glucose metabolism, cell survival, growth, and proliferation.3 Another important serine/threonine kinase is mTOR, which is composed of two distinct protein complexes. One protein complex, mTORC1, is a rapamycin and nutrient-sensitive multiprotein complex and is downstream AKT.4 mTORC1 stimulates cell growth and progression of the cell cycle in response to amino acids, stress, oxygen, energy, and growth factors. The second protein complex, mTORC2, is also sensitive to growth factors but insensitive to nutrients and rapamycin. mTORC2 regulates the cytoskeleton, cell metabolism, and cell survival in response to growth factors.5 mTORC2 is upstream AKT in the signaling pathway.4
Figure 1:
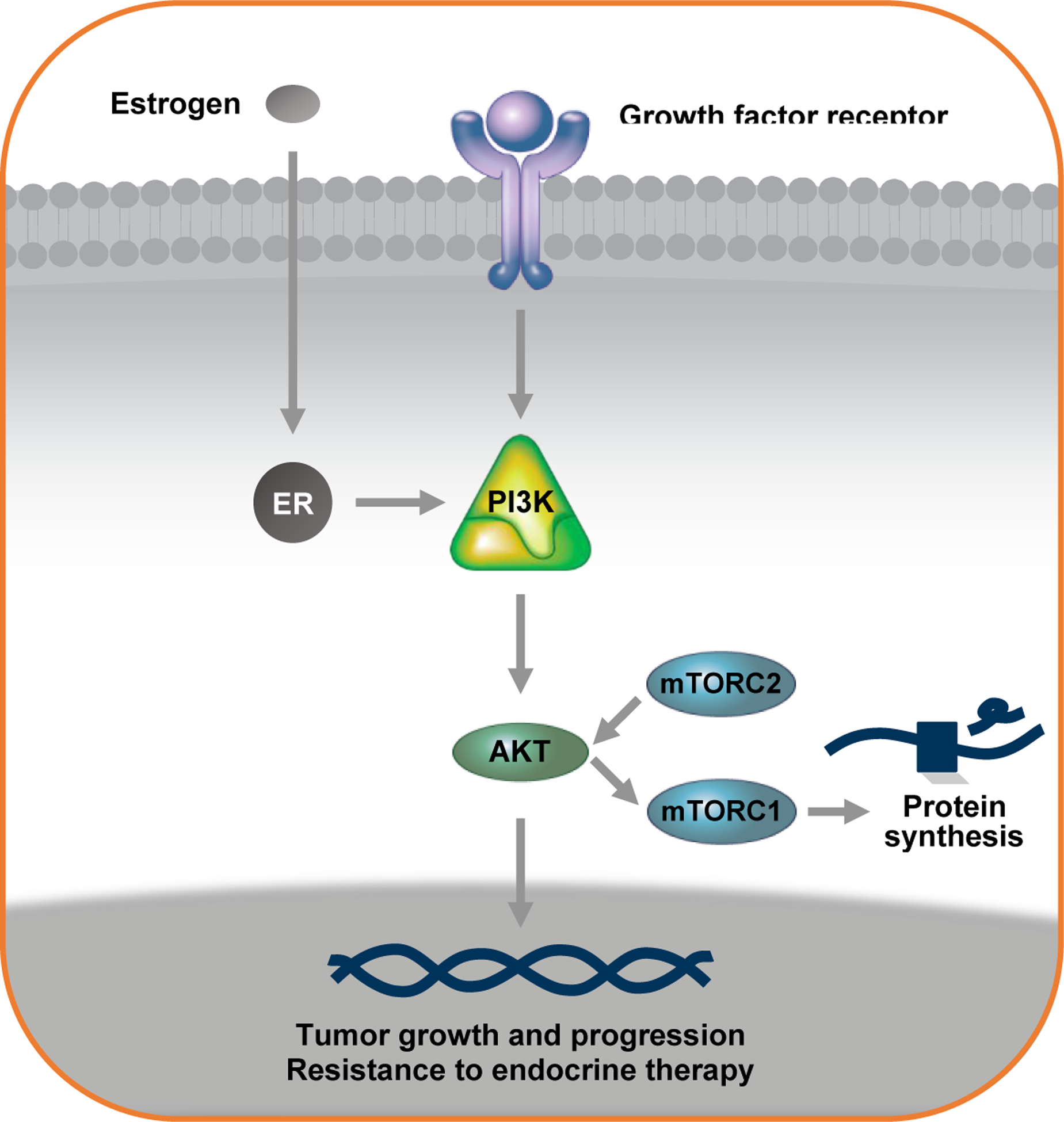
The PI3K/AKT/mTOR pathway
Because of the crucial role that the PI3K/AKT/mTOR pathway has in cell growth, survival, and proliferation, it is not suprising that alterations in this pathway are frequently found in cancer. PI3K/AKT/mTOR pathway alterations are especially common in breast cancer, and it is estimated that up to 70% of tumors have some type of genetic mutation that can render this pathway hyperactivated.6 One of the most common genetic alterations found in breast cancer are mutations in the PIK3CA gene, which encodes the p110α subunit of the PI3K. Approximately 30–40% of advanced ER+ breast cancers have an activating PIK3CA mutation,7 and more than 80% of these mutations cluster within the helical domains (E542K or E545K in exon 9) or the kinase domains (H1047R or H1047L in exon 20) of p110α.8,9 These mutations can either increase the catalytic activity of p110α or increase the retention of p110α respectively.10–13 The helical domain mutations in exon 9 disrupt the intermolecular interaction between p85 and p110α, an interaction which normally inhibits p110α activity.10 The effects of the kinase domain mutations in exon 20 are less clear, but previous studies have shown that these mutations lead to increased retention of p110α at the plasma membrane and constitutive activation of the signaling pathway.11 All of these genetic alterations lead to hyperactivation of the PI3K/AKT/mTOR pathway thereby promoting cell transformation, tumor initiation, and resistance to apoptosis.
2. PI3K/AKT/mTOR pathway and its role in endocrine resistance
Several preclinical studies have demonstrated that PI3K/AKT/mTOR pathway activation is a mechanism of acquired resistance to long-term estrogen deprivation and has subsequently led to further investigation of inhibiting this pathway as a mechanism to bypass estrogen-independent cell survival.14–16 Human breast cancer cell lines after long-term estrogen deprivation showed increased phosphorylation of mTOR substrates as well as the PI3K substrate AKT. Inhibition of mTOR and PI3K induced apoptosis in these cells and prevented the emergence of hormone-independent cells.14 Other preclinical studies as well as retrospective analysis of some clinical trials in the metastatic setting have also suggested that ER+/PIK3CA mutant tumors have a lower response to anti-estrogens compared to ER+/PIK3CA wild-type tumors.17,18 Low levels of estradiol have been shown to rescue ER+/PIK3CA mutant cells from the lethal effect of PI3K inhibitors.19 Furthermore, inhibition of PI3K/AKT leads to upregulation of ERα mRNA and protein transcription suggesting coregulation of the ER and PI3K pathways20–22. Inhibition of both ER and PI3K had synergistic effects against ER+/PIK3CA mutant xenografts.22 These models show that ER+ breast cancer cells can rely on the PI3K/AKT/mTOR pathway for survival and proliferation in the presence of estrogen deprivation and laid the foundation for further investigation in vivo of combined ER and PI3K inhibition as a therapeutic approach for ER+ breast cancers that progress on anti-estrogen treatments.
Mutations in ESR1 have also been well demonstrated to cause resistance to endocrine therapy by rendering the ligand binding domain of estrogen receptors constitutively active.23–25 Molecular modeling studies of two specific ESR1 ligand binding domain mutations,Y537S and D538G, showed that these mutations render the receptor constitutively active even upon antagonist binding.25 Additionally, ESR1 mutations have been shown through transcriptional profiling to upregulate activity of the p53 and mTORC1 signaling pathways thus promoting endocrine resistance and even a metastatic phenotype.26 Furthermore, preclinical and clinical studies have demonstrated that in ER+ metastatic breast cancer, metastatic tumor cells can selectively develop ESR1 mutations while on therapy with aromatase inhibitors.26,27
3. Classes of PI3K pathway inhibitors
A variety of PI3K pathway inhibitors have been developed and can be divided into different classes based on their target(s) in the pathway (Table 1). Several of these agents inhibit all isoforms of class IA PI3Ks. However, these agents are associated with significant off-target effects and toxicities. Consequently, many isoform-specific inhibitors have been developed to selectively inhibit specific PI3Ks with the goal of reducing off-target effects and improving efficacy of the drug by allowing maximal target-inhibitory doses. In addition to targeting the PI3Ks, a vartiety of agents have been developed to inhibit other proteins in the PI3K/AKT/mTOR pathway including mTORC1, mTORC2, and AKT. Many of these drugs when combined with endocrine therapy have shown significant clinical benefit with improved progression free survival (PFS) in ER+ metastatic breast cancers that have progressed on previous lines of endocrine therapy (Table 2).
Table 1:
Classes of PI3K Pathway Inhibitors
| Class | Drug Target(s) | Drugs | Common Toxicities by Class |
|---|---|---|---|
| mTORC1 (mammalian target of rapamycin complex 1) inhibitors | mTORC1 kinase | Everolimus Temsirolimus Deforolimus | Stomatitis Rash Pneumonitis Hyperglycemia Immunosuppression |
| mTORC1/mTORC2 inhibitors | mTORC1/mTORC2 kinases | Sapanisertib (TAK-228) AZD2014 | Hyperglycemia Rash Stomatitis Diarrhea |
| Pan-pi3k inhibitors | All class ia pi3kS | Buparlisib (BKM120) Pilaralisib (XL147) Pictilisib (GDC-0941) GDC-0084 | Hyperglycemia Rash Neutropenia Neuropsychiatric effects (confusion, depression, anxiety) Hepatotoxicity Diarrhea |
| Pan-Akt inhibitors | Three isoforms of Akt (Akt1, 2, and 3) | Capivasertib (AZD5363) Ipatasertib (GDC0068) MK2206 | Rash Hyperglycemia |
| Dual pi3k and mTOR inhibitors | p110 subunit of pi3k and mTOR | Dactolisib (BEZ235) Voxtalisib (XL765) Gedatolisib Bimiralisib (PQR309) | Stomatitis Hyperglycemia Immunosuppression |
| Isoform-specific pi3k inhibitors | pi3k p110α isoform | Alpelisib(BYL719) Taselisib (GDC-0032) Serabelisib (MLN1117) MEN1611 GDC-0077 | Hyperglycemia Rash Diarrhea Pneumonitis |
| pi3k p110δ isoform | Idelalisib (CAL-101) Duvelisib (IPI-145) | Hyperglycemia Rash Diarrhea Hypertension | |
| pi3k p110α and p110δ isoforms | Copanlisib | Hyperglycemia Hypertension Diarrhea Neutropenia |
Table 2:
Summary of Key Clinical Trials assessing PI3K/AKT/mTOR Pathway Inhibitors in HR+ Breast Cancer
| Trial Name | NCT Identifier | Phase | Population | Treatment | Treatment Line | Outcome |
|---|---|---|---|---|---|---|
| Mtorc1 Inhibitors | ||||||
| BOLERO-229 | 00863655 | III | ER+/HER2−mBC after AI n=724 | Exemestane ± everolimus | Subsequent line after progression on AI | Median PFS was 6.9 mos for everolimus plus exemestane vs 2.8 mos for placebo + exemestane (HR for progression or death, 0.43; 95% confidence interval [CI], 0.35 to 0.54; P<0.001) |
| TAMRAD30 | 01298713 | II | ER+/HER2−mBC after AI n= 111 | Tamoxifen ± everolimus | Subsequent line after progression on AI | Primary end point was clinical benefit rate (CBR). 6-month CBR was 61% (95% CI, 47 to 74) with tamoxifen plus everolimus and 42% (95% CI, 29 to 56) with tamoxifen alone. Time to progression (TTP) increased to 8.6 mos with tamoxifen + everolimus from 4.5 mos with tamoxifen alone |
| PrE010231 | 01797120 | II | HR+/HER2−mBC after AI n= 131 | Fulvestrant ± everolimus | Subsequent line After progression on AI | Median PFS was 10.3 mos with everolimus + fulvestrant vs 5.1 mos for fulvestrant + placebo (HR 0.61 [95% CI, 0.40 to 0.92]; stratified log-rank P = .02) |
| e3 (S1207)84 | 01674140 | III | HR+/HER2− high risk, localized BC | Standard adjuvant ET ± one year of everolimus | Adjuvant treatment to prevent disease recurrence | Primary objectives are to assess whether addition of everolimus × 1 year to standard adjuvant ET improves overall survival (OS) and distant recurrence-free survival (DRFS) in this patient population. |
| Pan-PI3K Inhibitors | ||||||
| BELLE-238 | 01610284 | III | ER+/HER2−locally advanced BC or mBC n= 1147 | Fulvestrant ± buparlisib | Subsequent line after progression on AI | Median PFS was 6.9 mos with buparlisib vs 5.0 mos in placebo group (p<0.001); PIK3CA-mut increase in PFS for buparlisib compared to placebo (7 vs 3.2 mos, HR 0.56, p<0.001) |
| BELLE-339 | 01633060 | III | ER+/HER2−locally advanced BC or mBC n= 432 | Fulvestrant ± buparlisib | Subsequent line for progression/relapse after ET + everolimus | Median PFS was 3.9 mos with buparlisib vs 1.8 mos with placebo (HR 0.67, 95% Cl 0.53–0.84, one-sided p=000030); high rates of grade 3–4 adverse events |
| Pan-AKT Inhibitors | ||||||
| FAKTION44 | 01992952 | II | ER+/HER2−locally advanced or mBC n= 140 | Fulvestrant ± capivasertib | Subsequent line for progression/relapse after AI | Median PFS was 10.3 mos (95% CI.5·0–13·2) with capivasertib vs 4.8 mos (3.1–7.7) with placebo [HR 0.58 (95% CI 0.39–0.84); two-sided p=0.0044, one-sided log rank test p=0.0018] |
| NCT0122631646 | 01226316 | I | AKT1E17K-mutant ER+ MBC n=63 | Capivasertib or capivasertib + fulvestrant | Subsequent line for progression on prior therapy | ORR was 20% with capivasertib monotherapy, 36% with fulvestrant + capivasertib in fulvestrant-pretreated patients, 20% with combination in fulvestrant-naive patients |
| Isoform-specific PI3K Inhibitors | ||||||
| SOLAR-155 | 02437318 | III | HR+/HER2−mBC n =572 | Fulvestrant ± alpelisib | Subsequent line for progression/relapse after ET | Patients with PIK3CA-mut PFS was 11.0 mos (95% confidence interval [CI], 7.5 to 14.5) in the alpelisib-fulvestrant group vs 5.7 mos (95% CI, 3.7 to 7.4) in the placebo-fulvestrant group (hazard ratio for progression or death, 0.65; 95% CI, 0.50 to 0.85; P<0.001) |
| NEO-ORB62 | 01923168 | II | HR+/HER2−localized BC n= 257 | Letrozole ± alpelisib | Neoadjuvant therapy | ORR was similar for the alpelisib + letrozole vs. placebo plus letrozole (PIK3CA-mutant, 43% vs. 45%, p= 0.435; PIK3CA-wild-type, 63% vs. 61%, p= 0.611). Low pCR rates in all groups. |
| SANDPIPER67 | 02340221 | III | PIK3CA-mut HR+/HER2−locally advanced BC or mBC n= 516 | Fulvestrant ± taselisib | Subsequent line for progression/relapse after AI | Median PFS was 7.4 mos in taselisib + fulvestrant vs 5.4 mos in fulvestrant alone (HR 0.70, p= 0.0037) |
| LORELEI68 | 02273973 | II | ER+/HER2−localized breast cancer n= 334 | Letrozole ± taselisib | Neoadjuvant therapy | OR was 39% in the placebo group vs 50% in the taselisib group (OR 1.55, 95% CI 1.00–2.38; p=0.049) and in PIK3CA-mut subset 38% vs 56% (OR 2.03, 95% CI 1.06–3.88; p=0.033). No significant differences in pCR between the two groups. |
| BYLIEVE64 | 03056755 | II | PIK3CA-mut HR+/HER2−mBC n= 127 (cohort A) | ET ± alpelisib | Subsequent line for progression/relapse after CKDi | Trial ongoing; Preliminary results of the cohort of patients treated with CDKi + AI immediately prior met primary end point at median follow-up was 11.7 mos: proportion of pts without disease progression at 6 mos was 50.4% (95% CI, 41.2–59.6) |
3.1. mTORC1 (mammalian target of rapamycin complex 1) inhibitors
The mTORC1 (mammalian target of rapamycin complex 1) inhibitors, including sirolimus and its analogs (temsirolimus, everolimus, and deforolimus), are allosteric irreversible inhibitors of mTORC1 kinase28; the mTORC1 or 2 inhibitors block both mTORC1-dependent phosphorylation of S6K1 and mTORC2-dependent phosphorylation of AKT.29 Everolimus is an allosteric inhibitor of mTORC1 but does not affect mTORC2. Results from two randomized trials published in 2012, BOLERO-2 (exemestane with or without the mTOR inhibitor everolimus) and TAMRAD (tamoxifen with or without everolimus),29,30 showed that the addition of everolimus to anti-estrogen therapy in patients with metastatic ER+ breast cancer can mitigate the effect of developing endocrine resistance in vivo. In both trials, patients had been previously treated with AIs then had disease progression after an initial response, indicating acquired resistance to endocrine therapy. The addition of everolimus to anti-estrogen therapy increased the median progression-free survival (PFS) in both studies (BOLERO-2 PFS 7.8 vs 3.2 months, p<0.0001; TAMRAD PFS 8.6 vs 4.5 months, p<0.01). In TAMRAD, randomization was stratified by primary resistance (relapsed disease during or within 6 months of stopping adjuvant aromatase inhibitor treatment or disease progression within 6 months of starting aromatase inhibitor treatment in the metastatic setting) and secondary resistance (relapsing >6 months after stopping adjuvant aromatase inhibitors or responding for ≥6 months to aromatase inhibitors in the metastatic setting). Furthermore, subanalysis showed that the time to progression was longer in patients with secondary endocrine therapy resistance than patients with primary endocrine therapy resistance (14.8 months vs 5.4 months) suggesting a possible adaptive response to long-term estrogen depletion and clinical benefit from adding everolimus to hormone therapy.30 These results led to the FDA and EMA approval of everolimus in combination with endocrine therapy as treatment for metastatic ER+ breast cancer after progression on aromatase inhibitors (AIs). Building on the results of the BOLERO-2 and TAMRAD trials, results of the PrE0102 trial were published in 2018 and showed that the addition of everolimus to fulvestrant, a selective estrogen receptor downregulator, compared to fulvestrant alone improved median progression-free survival from 5.1 to 10.3 months (hazard ratio, 0.61 [95% CI, 0.40 to 0.92]; P = .02) in patients with ER+ metastatic breast cancer resistant to AI-therapy.31 Phase II trials evaluating drugs that inhibit both mTORC1 and mTORC2, AZD2014 and sapanisertib, with the aim of a more complete blockade of mTOR complexes, have been ongoing.32–34 These drugs have also shown activity against everolimus-resistant acquired mutations in the rapamycin-binding domain of mTOR.35,36
3.2. Pan-PI3K inhibitors
Several drugs that work as pan-PI3K inhibitors have been developed but have so far, had low efficacy without much improvement over endocrine therapy alone. These agents block all isoforms of class IA PI3Ks and as a result, are associated with significant off-target effects. They have shown much lower response rates in breast cancer compared to the response rates seen with the inhibition of other oncogenic kinases in other solid tumors (i.e. targeting mutant EGFR and ALK in lung cancer and mutant BRAF in melanoma). Their toxicity precludes adequate dose intensity which could likely explain their relative lack of efficacy. They are represented by several small-molecule drugs including buparlisib (BKM120), pilaralisib (XL147), and pictilisib (GDC-0941).37 The BELLE-2 clinical trial38, a randomized, double-blind, placebo-controlled phase III trial of buparlisib in combination fulvestrant compared to fulvestrant alone in patients with ER+/HER2− locally advanced or metastatic breast cancer that had progressed on AI found a modest improvement in PFS in the cohort treated with buparlisib and fulvestrant (6.9 vs 5.0 months, p<0.001). However for patients with PIK3CA mutations, there was a greater improvement in PFS with the addition of buparlisib to fulvestrant of 3.8 months (7 vs 3.2 months, HR 0.56, p<0.001).38 Results of the BELLE-3 clinical trial39, a phase III study of buparlisib with fulvestrant in patients with ER+/HER2− locally advanced or metastatic breast cancer that had relapsed or progressed on endocrine therapy or mTOR inhibitors, followed and were published in January 2018. Despite displaying a median PFS that was statistically significantly longer in the treatment group vs placebo (3.9 months vs 1.8 months, HR 0.67, p=0.0003), the lack of clinical significance and high rates of grade 3–4 adverse events in the treatment group, including transaminitis, hyperglycemia, dyspnea, pleural effusion, and mood disorders,39 precluded further development of buparlisib in ER+metastatic breast cancer.40–43
3.3. Pan-AKT inhibitors
AKT is a serine/threonine kinase that is a downstream target of PI3K. AKT has three isoforms (AKT1, 2, and 3) which have very similar structures.3 Because of the structural similarities between the three isoforms, isoform-specific inhibitors have proved challenging to develop. Two of these inhibitors, capivasertib (AZD5363) and ipatasertib (GDC0068) are currently in phase III clinical trials (NCT03997123; NCT03337724) for breast cancer in combination with chemotherapy.4 Results of the recently completed FAKTION trial44, a phase II study of capivasertib plus fulvestrant versus fulvestrant plus placebo in patients with ER+/HER2− locally advanced or metastatic breast cancer who had relapsed or progressed on an AI, had promising results and will lead to further investigation of this drug in phase III trials. The addition of capivasertib to fulvestrant resulted in significantly longer PFS of 10.3 months vs 4.8 months in the placebo group (HR 0.58, 95% CI 0.39–0.84; p=0.0018). The most common grade 3–4 adverse events were hypertension, diarrhea, rash, and fatigue.44
AKT1E17K has been identified as the most common somatic mutation in AKT and occurs in approximately 7% of ER+ metastatic breast cancers. This mutation results in a glutamic acid to lysine substitution at amino acid 17 (E17K) in the lipid binding pocket of AKT and ultimately causes constitutive membrane localization and activation of AKT.45 Results of a phase I study of capivasertib as monotherapy or in combination with fulvestrant in heavily pretreated patients with ER+ metastatic breast cancer and the AKT1E17K mutation also had promising results.46 Capivasertib demonstrated clinically meaningful activity and tolerability both as monotherapy and in combination with fulvestrant indicating the potential utility of this drug as a targeted therapy as either single agent or in combination with estrogen blockade in the future.46
3.4. Dual PI3K and mTOR inhibitors
An ongoing area of clinical development are compounds that target both the p110 subunit of PI3K and mTOR. Dual blockade of mTOR and the p110 subunit of PI3K would provide more complete inhibition of the PI3K/AKT/mTOR signaling pathway at multiple points as well as interrupt negative feedback loops and thereby increase clinical efficacy. Multiple dual PI3K and mTOR inhibitors are currently in clinical development. Phase I trials have evaluated dactolisib (BEZ235), voxtalisib (XL765), bimiralisib (PQR309), and gedatolisib.4,47,48 These compounds have a much broader activity profile and could be used to treat a variety of tumors with a range of genetic abnormalities. However as a result of their more broad activity, these agents have more off-target effects and toxicities which has made their development challenging.28 A recent phase 2 trial of voxtalisib in patients with hematologic malignancies showed an acceptable safety profile and promising efficacy.49 No studies with dual inhibitors have been conducted in patients with breast cancer to date.
3.5. Isoform-specific PI3K inhibitors
Isoform-specific inhibitors were developed with the goal of achieving maximal target-inhibitory doses while potentially avoiding the off-target toxicities seen with pan-PI3K inhibitors. A variety of isoform-specific inhibitors have been developed including agents that selectively inhibit the PI3K p110α (e.g. alpelisib [BYL719] and taselisib [GDC-0032]) and the p110β, p110γ, or p110δ (e.g. idelalisib) isoforms.37 Copanlisib selectively inhibits both the p110α and p110δ isoforms and is approved for the treatment of B cell hematologic malignancies. It is also currently being studied in phase I/II trials for metastatic breast cancer.50 The p110α isoform is most commonly mutated in cancer, and studies have shown that selective inhibition of this isoform is enough to block signaling in the PI3K/AKT pathway in response to different growth factors stimuli.51–53 Alpelisib was the first PI3Kα inhibitor studied as a single agent and showed preferential activity against tumors with PIK3CA mutations. Results from phase IB trials53,54 of alpelisib and endocrine therapy showed that treatment with combination endocrine therapy and a PI3K inhibitor provide clinical benefit in ER+ metastatic breast cancers with acquired resistance to endocrine therapy, particularly in the PIK3CA mutated cancers. The large phase III SOLAR-1 trial55 was designed based on these findings and utilized endocrine therapy plus alpelisib in ER+ metastatic breast cancer resistant to endocrine therapy. The SOLAR-1 trial showed prolonged progression-free survival (PFS) in patients with ER+/PIK3CA mutated metastatic breast cancer who had previously been treated with endocrine therapy and led to the FDA approval of alpelisib for patients with ER+/PIK3CA mutated metastatic breast cancer in May 2019. We should note that the vast majority of participants in the SOLAR-1 trial did not have prior exposure to CDK4/6 inhibitors, which are currently approved in combination with endocrine therapies for further-line treatment of ER+ metastatic breast cancer.56–61 Based on the improvement in PFS seen in the metastatic setting with the SOLAR-1 trial, the NEO-ORB trial62 followed to investigate whether the addition of alpelisib to letrozole would also improve resonse rates in the neoadjuvant setting for early ER+ breast cancer. The trial did not meet its primary objectives of improved ORR with the addition of alpelisib to letrozole in either the PIK3CA-mutant tumors or -wild-type tumors after 24 weeks of neoadjuvant treatment, and the pathologic complete response rates were low in all treatment groups.62
The BYLIEVE trial is an ongoing phase II, multicenter, open-label, three-cohort, non-comparative study of alpelisib plus endocrine therapy (either fulvestrant or letrozole) in patients with HR+/HER2− advanced breast cancer with PIK3CA mutation(s) whose disease has progressed on or after prior CDK4/6 inhibitor (CDK4/6i) combination therapy.63 Preliminary results of the cohort of patients treated with CDK4/6i in combination with aromatase inhibitor immediately prior have met the primary endpoint of progression-free survival (PFS) at 6 months, achieved in 50.4% of patients, with median PFS duration of 7.3 months.64 These initial results show clinically meaningful efficacy of alpelisib plus endocrine therapy after prior treatment with CDK4/6 inhibitors.
Taselisib is a β-sparing potent inhibitor of p110α, p110δ, and p110γ, and has a greater selectivity against PIK3CA mutant isoforms than wild-type.65 A phase II trial of taselisib with fulvestrant showed clinical activity regardless of PIK3CA mutation status, but did show a higher objective response rate in patients with PIK3CA mutant tumors compared to wild-type (41% vs 14% respectively).66 Taselisib was further investigated in a randomized phase III study (SANDPIPER trial) in combination with fulvestrant for patients with metastatic or locally advanced ER+ tumors that have progressed during or after AI therapy irrespective of PIK3CA mutation status. The treatment group only had a slight improvement in PFS of 2 months (7.4 vs 5.4 months, p=0.0037).67 The LORELEI trial was a multicenter, randomized, double-blind, placebo-controlled phase II study investigating taselisib with letrozole compared to placebo with letrozole as neoadjuvant treatment in patients with stage I-III, operable, ER/PR+, HER2−negative breast cancer. This study found no significant differences in pathological complete response between the two groups both in the overall population or in patients with PIK3CA-mutated tumors.68 In both the SANDPIPER and LORELEI trials, there were also high rates of grade 3–4 toxicities with taselisib, including diarrhea and hyperglycemia in 17% and 11% of patients respectively, leading to drug discontinuation.67,68 As a result of these two clinically negative trials and the toxicities associated with taselisib, further development of the drug has been stopped.
4. Associated Toxicities
One of the challenges in developing drugs that inhibit the PI3K/mTOR/AKT pathways and in treating patients with these drugs is the associated toxicities. These agents display a wide range of both on-target and off-target effects. Some of the most common side effects seen with PI3K pathway inhibitors are hyperglycemia, dermatitis and rash, stomatitis, diarrhea, nausea, and fatigue69–71. Other less common side effects that are reported include elevation of pancreatic enzymes, elevation of liver enzymes, immune dysfunction/lymphocytopenia, and pneumonitis55,69,72. Autoimmune hepatitis is rare and has been predominantly reported with pan-PI3K inhibitors or combination therapies.40,41 Fortunately, pancreatic enzyme or liver enzyme elevation rarely lead to the development of pancreatitis or liver failure, but they still have significant effects clinically by limiting drug dose and leading to drug discontinuation. Other uncommon toxicities associated with PI3K/AKT inhibitors include opportunistic infections (seen more frequently with mTOR inhibitors), hypertension, and CNS symptoms (seen with buparlisib, a pan-PI3K inhibitor).40,41,69,72 Since these drugs have a short half-life, most side effects are reversible with drug interruption and are manageable with early interventions.
5. Approach to PIK3CA Mutation Testing
Hormone receptors do not remain stable throughout tumor progression, and multiple studies have found a wide range of discordance between HR status from primary tumor to sites of relapse and metastasis.73,74 One review reported a large range of receptor discordance for ER and PR status of 6–40% and 21–41% respectively.74 It has also been well documented that the acquisition of ESR1 mutations during AI therapy in metastatic, ER+ breast cancer is a common mechanism of developing resistance to hormonal therapy.27,75,76 As a result, biopsy of a metastatic site whenever possible in breast cancer patients who develop new metastatic disease is important to confirm concordant ER/PR and HER2 status and to assess for new somatic mutations in the tumor that would impact treatment decisions. Now with the approval of alpelisib for PIK3CA mutant breast cancer and PARP inhibitors for germline BRCA mutant breast cancer, it is mandatory per the NCCN guidelines to perform tumor profiling either through Next generation sequencing (NGS) on tumor tissue or through circulating tumor DNA (ctDNA) from plasma for all patients with metastatic breast cancer, especially ER+ metastatic breast cancer.55,77–79 Additionally, identification of tumors expressing ESR1 mutations is important in guiding therapy since some data suggests that these tumors have improved response to treatment with fulvestrant containint regimens rather than AI regimens.80 In the SoFEA trial, patients with ESR1 mutations had improved PFS with fulvestrant compared to exemestane (n = 18; hazard ratio [HR], 0.52; 95% CI, 0.30 to 0.92; P = .02), versus patients with wild-type ESR1 who had similar PFS with either treatment.80,81 However in the PALOMA-3 trial, there was no difference in PFS observed with treatment of palbociclib plus fulvestrant compared to fulvestrant plus placebo in patients with tumors expressing ESR1 mutations compared to wild-type tumors.60
The ongoing phase III PADA-1 trial82 seeks to answer the question of which endocrine therapy is optimal to combine with CDK4/6 inhibitor and the impact of ESR1 mutations on response to this treatment. This trial includes 1,017 patients with ER+, Her2 negative metastatic breast cancer who had no prior therapy for metastatic disease and no known resistance to AI therapy based on no prior AI treatment or a disease-free interval of more than 12 months from adjuvant treatment with an AI. All patients had cell-free DNA tested for ESR1 mutations at baseline and were on treatment with AI combined with palbociclib at study initiation. At baseline, 33 (3.2%) of patients had an ESR1 mutation, and 135 patients were found to have an emerging ESR1 mutation. These patients were randomized to continue palbociclib and AI or switch to palbociclib combined with fulvestrant. Some initial study results were reported at Virtual ASCO 2020 and found that patients with ESR1 mutation had a worse prognosis with median PFS of 11 months for ESR1 mutants compared to 26.7 months for wild-type at a median follow up of 21.2 months (HR=2.3, p < 0.001). However, clearance of ESR1 mutation after 1 month of treatment improved prognosis with a median PFS of 24.1 months in the patients who cleared the mutation compared to 7.4 months in patients who still had detectable ESR1 mutations.82 Final results from the study comparing patients treated with AI and palbociclib versus fulvestrant and palbociclib have not yet been reported.
In order to be eligible for treatment with alpelisib, patients with advanced or recurrent HR+, HER2− tumors must have PIK3CA mutations detected in tumor or plasma by ctDNA.71 In the SOLAR-1 trial, there were 341 patients enrolled in the cohort with a PIK3CA mutation, and of those 341 patients with a PIK3CA mutation, 336 (99%) patients had one or more PIK3CA mutations confirmed in tumor tissue using the FDA-approved therascreen® PIK3CA RGQ PCR Kit. Nineteen of those patients had no plasma specimen available for testing with therascreen® PIK3CA RGQ PCR Kit. Of the remaining 317 patients with PIK3CA mutations confirmed in tumor tissue, 177 patients (56%) also had PIK3CA mutations identified in plasma, and 140 patients (44%) did not have PIK3CA mutations identified in plasma specimen.55 Based on these findings, if no mutation is detected initially in a plasma specimen, it is recommended to test tumor tissue.71 There are currently two FDA-approved testing modalities for determining presence of PIK3CA mutations. The therascreen® PIK3CA RGQ PCR Kit (QUIAGEN, Germany) is a real-time qualitative PCR test for the detection of 11 mutations in the PIK3CA gene using genomic DNA from formalin-fixed, paraffin-embedded (FFPE) breast tumor tissue or circulating tumor DNA (ctDNA) from plasma.83 Next generation sequencing (NGS) by FoundationOne CDX (Foundation Medicine, Cambridge, MA) for the detection of substitutions, insertion and deletion alterations, and copy number alterations in 324 genes and select gene rearrangements using DNA isolated from FFPE tumor tissue is also FDA-approved.84
6. Conclusion
Alterations in the PI3K/AKT/mTOR pathway are especially common in breast cancer, rendering the pathway hyperactivated and promoting uncontrolled cell proliferation, resistance to apoptosis, and tumorigenesis.6 Specifically, mutations in the PIK3CA gene are the most common activating mutations found in breast cancer and are present in approximately 30% of advanced ER+HER2− breast cancers.7 In recent years, studies have also demonstrated that ER+ breast cancer cells can rely on the PI3K/AKT/mTOR pathway for survival and proliferation as an adaptive mechanism after long-term estrogen deprivation. These discoveries led to the development of combination ER and PI3K/AKT/mTOR pathway inhibition as a therapeutic approach for ER+ breast cancers that progress on anti-estrogen treatments.
There are multiple preferred regimens approved for first-line and subsequent-line therapies in patients with metastatic ER/PR+, HER2− breast cancer, however, optimal sequencing of these various targeted agents or their combinations have not yet been established.79 Randomized clinical trials investigating the optimal sequencing of these targeted agents and their combinations are warranted. The initial targeted therapy that is preferred to combine with endocrine therapy (ET) is a CDK 4/6 inhibitor because their side-effects are generally much more tolerable compared to those of everolimus or alpelisib.56–59 If a patient develops disease progression on ET combined with CDK 4/6 inhibitor, there is not yet data to support subsequent therapy with an alternative CDK 4/6 inhibitor, although several clinical trials are ongoing.79 Patients could next be considered for subsequent-line therapy with ET combined with everolimus or with alpelisib based on the presence of a PIK3CA mutation in the tumor. Disease progression on more than 3 prior lines of ET including combination with a targeted agent as well as the presence of rapidly progressive visceral disease should prompt consideration for transitioning treatment to cytotoxic chemotherapy.
As previously discussed, tumors found to have PIK3CA mutations have improved clinical benefit from alpelisib, and similar improvements in clinical benefit are being seen in early studies with capivasertib in tumors with AKT1E17K mutations. Further discovery of somatic mutations or other biomarkers that can predict which tumors are most dependent on the PI3K/AKT/mTOR pathway and will be most responsive to treatment with pathway inhibitors will allow for improved optimization of risk benefit ratios for patients. Another interesting area of study is whether or not upfront targeting of the PI3K/mTOR/AKT pathway in addition to standard adjuvant ET in high risk disease that has not yet metastasized will improve disease free survival time and prevent late recurrence. Based on the success of the BOLERO-2 trial which showed that the addition of everolimus to examestane more than doubled the average time to disease progression for patients with advanced HR+ positive breast cancer, the Southwest Oncology Group designed the e3 (S1207) trial. The e3 (S1207) Breast Cancer Study is an ongoing phase III, placebo-controlled trial evaluating the use of adjuvant endocrine therapy combined with one year of everolimus compared to adjuvant endocrine therapy alone in women with high-risk HR+/HER2− breast cancer that has not yet metastasized. The goal of this trial is to determine whether the early combination of everolimus with adjuvant endocrine therapy will lengthen disease free survival time compared to adjuvant endocrine therapy alone as well as assess the impact of everolimus on patients’ quality of life.85
In summary, inhibitors of the PI3K/AKT/mTOR pathway are an important part of the current clinical management of ER+ metastatic breast cancer. However, despite significant clinical activity, several challenges for the therapeutic targeting of PI3K/AKT/mTOR remain. Although manageable, on-target toxicities induced by these drugs are not insignificant. Additionally, identifying other targeted drugs that should be used in combination with PI3K pathway inhibitors will require development of rational combinations that have manageable toxicities and better clinical activity than when these targeted agents are used by themselves or sequencing a PI3K pathway inhibitor.
Key Points.
The PI3K/AKT/mTOR pathway plays a central role in cell growth, survival, and proliferation and is implicated in tumorigenesis and the development of endocrine resistance in ER+ breast cancer.
ER+ breast cancer cells can develop genetic mutations, notably mutation of the PIK3CA gene, which cause hyperactivation of the PI3K/AKT/mTOR pathway and allow for cell survival and proliferation despite a state of estrogen deprivation and has led to the investigation of combined ER and PI3K inhibition as a therapeutic approach for ER+ breast cancers that progress on anti-estrogen treatments.
A variety of PI3K pathway inhibitors divided into several classes have been developed and have showed significant clinical benefit with improved PFS in ER+ metastatic breast cancers that have progressed on previous lines of endocrine therapy.
Funding:
No external funding was used in preparation of this manuscript.
Footnotes
Conflicts of Interest: Ingrid Mayer receives institutional research funding from Novartis, Genentech, Pfizer and receives Advisory Board compensation from Novartis, Genentech, Lilly, Astra-Zeneca, GSK, Immunomedics, Macrogenics, Seattle-Genetics. Sara Nunnery has declared no conflicts of interest that might be relevant to the contents of this manuscript.
Ethics approval: Not applicable
Consent to participate: Not applicable
Consent for publication: Not applicable
Availability of data and material: Not applicable
Code availability: Not applicable
References
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Reviews Genetics 2006;7(8):606–619. (In eng). DOI: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene 2008;27(41):5486–5496. (In eng). DOI: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature Reviews Cancer 2009;9(8):550–562. (In eng). DOI: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 4.Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annual Review of Medicine 2016;67:11–28. (In eng). DOI: 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
- 5.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149(2):274–293. (In eng). DOI: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernandez-Aya LF, Gonzalez-Angulo AM. Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. The Oncologist 2011;16(4):404–414. (In eng). DOI: 10.1634/theoncologist.2010-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Network CGA. Comprehensive molecular portraits of human breast tumours. Nature 2012;490(7418):61–70. (In eng). DOI: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciriello G, Gatza ML, Beck AH, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015;163(2):506–19. (In eng). DOI: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004;3(10):1221–4. (In eng). DOI: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 10.Huang CH, Mandelker D, Schmidt-Kittler O, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007;318(5857):1744–8. (In eng). DOI: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 11.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc Natl Acad Sci U S A 2012;109(38):15259–64. (In eng). DOI: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A 2005;102(51):18443–8. (In eng). DOI: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isakoff SJ, Engelman JA, Irie HY, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 2005;65(23):10992–1000. (In eng). DOI: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 14.Miller TW, Hennessy BT, González-Angulo AM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010;120(7):2406–13. (In eng). DOI: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez CG, Ma CX, Crowder RJ, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res 2011;13(2):R21 (In eng). DOI: 10.1186/bcr2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.deGraffenried LA, Friedrichs WE, Russell DH, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res 2004;10(23):8059–67. (In eng). DOI: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 17.Ellis MJ, Lin L, Crowder R, et al. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res Treat 2010;119(2):379–90. (In eng). DOI: 10.1007/s10549-009-0575-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol 2011;29(33):4452–61. (In eng). DOI: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crowder RJ, Phommaly C, Tao Y, et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res 2009;69(9):3955–62. (In eng). DOI: 10.1158/0008-5472.CAN-08-4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox EM, Kuba MG, Miller TW, Davies BR, Arteaga CL. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res 2013;15(4):R55 (In eng). DOI: 10.1186/bcr3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creighton CJ, Fu X, Hennessy BT, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res 2010;12(3):R40 (In eng). DOI: 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosch A, Li Z, Bergamaschi A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med 2015;7(283): 283ra51 (In eng). DOI: 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 1997;57(7):1244–9. (In eng). [PubMed] [Google Scholar]
- 24.Gelsomino L, Gu G, Rechoum Y, et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res Treat 2016;157(2):253–265. (In eng). DOI: 10.1007/s10549-016-3829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45(12):1439–45. (In eng). DOI: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeselsohn R, Bergholz JS, Pun M, et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018;33(2):173–186.e5. (In eng). DOI: 10.1016/j.ccell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu G, Rechoum Y, Gelsomino L, et al. The Y537S ESR1 mutation is a dominant driver of distant ER‐positive breast cancer metastasis Abstract presented at: San Antonio Breast Cancer Symposium 2016; December 6‐10, 2016; San Antonio, TX. [Google Scholar]
- 28.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. The Journal of Clinical Investigation 2011;121(4):1231–1241. (In eng). DOI: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. The New England Journal of Medicine 2012;366(6):520–529. (In eng). DOI: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol 2012;30(22):2718–24. (In eng). DOI: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]
- 31.Kornblum N, Zhao F, Manola J, et al. Randomized Phase II Trial of Fulvestrant Plus Everolimus or Placebo in Postmenopausal Women With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer Resistant to Aromatase Inhibitor Therapy: Results of PrE0102. J Clin Oncol 2018;36(16):1556–1563. (In eng). DOI: 10.1200/JCO.2017.76.9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burris HA, Kurkjian CD, Hart L, et al. TAK-228 (formerly MLN0128), an investigational dual TORC1/2 inhibitor plus paclitaxel, with/without trastuzumab, in patients with advanced solid malignancies. Cancer Chemother Pharmacol 2017;80(2):261–273. (In eng). DOI: 10.1007/s00280-017-3343-4. [DOI] [PubMed] [Google Scholar]
- 33.Gökmen-Polar Y, Liu Y, Toroni RA, et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat 2012;136(3):673–82. (In eng). DOI: 10.1007/s10549-012-2298-8. [DOI] [PubMed] [Google Scholar]
- 34.Guichard SM, Curwen J, Bihani T, et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol Cancer Ther 2015;14(11):2508–18. (In eng). DOI: 10.1158/1535-7163.MCT-15-0365. [DOI] [PubMed] [Google Scholar]
- 35.Wagle N, Grabiner BC, Van Allen EM, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med 2014;371(15):1426–33. (In eng). DOI: 10.1056/NEJMoa1403352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016;534(7606):272–6. (In eng). DOI: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. Journal of Hematology & Oncology 2013;6(1):88 (In eng). DOI: 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baselga J, Im SA, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2−negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18(7):904–916. (In eng). DOI: 10.1016/S1470-2045(17)30376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Leo A, Johnston S, Lee KS, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2−negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2018;19(1):87–100. (In eng). DOI: 10.1016/S1470-2045(17)30688-5. [DOI] [PubMed] [Google Scholar]
- 40.Campone M, Im S-A, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant for postmenopausal, hormone receptor-positive, human epidermal growth factor receptor 2-negative, advanced breast cancer: Overall survival results from BELLE-2. European Journal of Cancer (Oxford, England: 1990) 2018;103:147–154. (In eng). DOI: 10.1016/j.ejca.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Baselga J, Im S-A, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2−negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 2017;18(7):904–916. (In eng). DOI: 10.1016/S1470-2045(17)30376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krop IE, Mayer IA, Ganju V, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Oncology 2016;17(6):811–821. (In eng). DOI: 10.1016/S1470-2045(16)00106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nature Reviews Clinical Oncology 2018;15(5):273–291. (In eng). DOI: 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- 44.Jones RH, Casbard A, Carucci M, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 2020;21(3):345–357. (In eng). DOI: 10.1016/S1470-2045(19)30817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007;448(7152):439–44. (In eng). DOI: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 46.Smyth LM, Tamura K, Oliveira M, et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination With Fulvestrant in Patients With. Clin Cancer Res 2020. (In eng). DOI: 10.1158/1078-0432.CCR-19-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarantelli C, Gaudio E, Arribas AJ, et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin Cancer Res 2018;24(1):120–129. (In eng). DOI: 10.1158/1078-0432.CCR-17-1041. [DOI] [PubMed] [Google Scholar]
- 48.Del Campo JM, Birrer M, Davis C, et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol Oncol 2016;142(1):62–69. (In eng). DOI: 10.1016/j.ygyno.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 49.Brown JR, Hamadani M, Hayslip J, et al. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: an open-label, phase 2 trial. Lancet Haematol 2018;5(4):e170–e180. (In eng). DOI: 10.1016/S2352-3026(18)30030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krause G, Hassenrück F, Hallek M. Copanlisib for treatment of B-cell malignancies: the development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des Devel Ther 2018;12:2577–2590. (In eng). DOI: 10.2147/DDDT.S142406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Utermark T, Rao T, Cheng H, et al. The p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes & Development 2012;26(14):1573–1586. (In eng). DOI: 10.1101/gad.191973.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao JJ, Cheng H, Jia S, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America 2006;103(44):16296–16300. (In eng). DOI: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayer IA, Abramson VG, Formisano L, et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2− Metastatic Breast Cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 2017;23(1):26–34. (In eng). DOI: 10.1158/1078-0432.CCR-16-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juric D, Janku F, Rodón J, et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA oncology 2019;5(2):e184475 (In eng). DOI: 10.1001/jamaoncol.2018.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. The New England Journal of Medicine 2019;380(20):1929–1940. (In eng). DOI: 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 56.Finn RS, Martin M, Rugo HS, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016;375(20):1925–1936. (In eng). DOI: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 57.Hortobagyi GN, Stemmer SM, Burris HA, et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2−negative advanced breast cancer. Ann Oncol 2018;29(7):1541–1547. (In eng). DOI: 10.1093/annonc/mdy155. [DOI] [PubMed] [Google Scholar]
- 58.Goetz MP, Toi M, Campone M, et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol 2017;35(32):3638–3646. (In eng). DOI: 10.1200/JCO.2017.75.6155. [DOI] [PubMed] [Google Scholar]
- 59.Turner NC, Ro J, André F, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med 2015;373(3):209–19. (In eng). DOI: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 60.Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2−negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 2016;17(4):425–439. (In eng). DOI: 10.1016/S1470-2045(15)00613-0. [DOI] [PubMed] [Google Scholar]
- 61.Sledge GW, Toi M, Neven P, et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J Clin Oncol 2017;35(25):2875–2884. (In eng). DOI: 10.1200/JCO.2017.73.7585. [DOI] [PubMed] [Google Scholar]
- 62.Mayer IA, Prat A, Egle D, et al. A Phase II Randomized Study of Neoadjuvant Letrozole Plus Alpelisib for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer (NEO-ORB). Clin Cancer Res 2019;25(10):2975–2987. (In eng). DOI: 10.1158/1078-0432.CCR-18-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rugo HS, Bianchi GV, Chia SKL, Turner NC. BYLieve: A phase II study of alpelisib (ALP) with fulvestrant (FUL) or letrozole (LET) for treatment of PIK3CA mutant, hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2−) advanced breast cancer (aBC) progressing on/after cyclin-dependent kinase 4/6 inhibitor (CDK4/6i) therapy. Journal of Clinical Oncology 2018;36(15). [Google Scholar]
- 64.Rugo HS, Flerebours F, Ciruelos E, et al. Alpelisib (ALP) + Fulvestrant (FUL) in patients (pts) with PIK3CA-mutated (mut) hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2−) advanced breast cancer (ABC) previously treated with cyclin-dependent kinase 4/6 inhibitor (CDKi) + aromatase inhibitor (AI): BYLieve study results. Presented at: 2020 ASCO Virtual Scientific Program; May 29–31, 2020. Abstract 1006. [Google Scholar]
- 65.Ndubaku CO, Heffron TP, Staben ST, et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}−2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem 2013;56(11):4597–610. (In eng). DOI: 10.1021/jm4003632. [DOI] [PubMed] [Google Scholar]
- 66.Dickler MN, Saura C, Richards DA, et al. Phase II Study of Taselisib (GDC-0032) in Combination with Fulvestrant in Patients with HER2−Negative, Hormone Receptor-Positive Advanced Breast Cancer. Clin Cancer Res 2018;24(18):4380–4387. (In eng). DOI: 10.1158/1078-0432.CCR-18-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baselga J, Dent S, Cortes J. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): Primary analysis from SANDPIPER. Journal of Clinical Oncology 2018;36:no. 18_suppl DOI: 10.1200/JCO.2018.36.18_suppl.LBA1006. [DOI] [Google Scholar]
- 68.Saura C, Hlauschek D, Oliveira M, et al. Neoadjuvant letrozole plus taselisib versus letrozole plus placebo in postmenopausal women with oestrogen receptor-positive, HER2−negative, early-stage breast cancer (LORELEI): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 2019;20(9):1226–1238. (In eng). DOI: 10.1016/S1470-2045(19)30334-1. [DOI] [PubMed] [Google Scholar]
- 69.Chia S, Gandhi S, Joy AA, et al. Novel agents and associated toxicities of inhibitors of the pi3k/Akt/mtor pathway for the treatment of breast cancer. Current Oncology 2015;22(1):33–48. DOI: 10.3747/co.22.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Esposito A, Viale G, Curigliano G. Safety, Tolerability, and Management of Toxic Effects of Phosphatidylinositol 3-Kinase Inhibitor Treatment in Patients With Cancer: A Review. JAMA oncology 2019. (In eng). DOI: 10.1001/jamaoncol.2019.0034. [DOI] [PubMed] [Google Scholar]
- 71.Piqray (apelisib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2019. [Google Scholar]
- 72.Brian Greenwell MD I. PI3K Inhibitors: Understanding Toxicity Mechanisms and Management. Cancer Network 2017. (In en). [PubMed] [Google Scholar]
- 73.Lindström LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol 2012;30(21):2601–8. (In eng). DOI: 10.1200/JCO.2011.37.2482. [DOI] [PubMed] [Google Scholar]
- 74.Criscitiello C, André F, Thompson AM, et al. Biopsy confirmation of metastatic sites in breast cancer patients: clinical impact and future perspectives. Breast Cancer Res 2014;16(2):205 (In eng). DOI: 10.1186/bcr3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dustin D, Gu G, Fuqua SAW. ESR1 mutations in breast cancer. Cancer 2019;125(21):3714–3728. (In eng). DOI: 10.1002/cncr.32345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harrod A, Fulton J, Nguyen VTM, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017;36(16):2286–2296. (In eng). DOI: 10.1038/onc.2016.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robson M, Im SA, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med 2017;377(6):523–533. (In eng). DOI: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 78.Litton JK, Rugo HS, Ettl J, et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med 2018;379(8):753–763. (In eng). DOI: 10.1056/NEJMoa1802905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.National Comprehensive Cancer Network. Breast Cancer (Version4.2020). https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed May 15, 2020.
- 80.Fribbens C, O’Leary B, Kilburn L, et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J Clin Oncol 2016;34(25):2961–8. (In eng). DOI: 10.1200/JCO.2016.67.3061. [DOI] [PubMed] [Google Scholar]
- 81.Johnston SR, Kilburn LS, Ellis P, et al. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentre, phase 3 randomised trial. Lancet Oncol 2013;14(10):989–98. (In eng). DOI: 10.1016/S1470-2045(13)70322-X. [DOI] [PubMed] [Google Scholar]
- 82.Bidard FC, Callens C, Dalenc F, et al. : Prognostic impact of ESR1 mutations in ER+ HER2− MBC patients with prior treatment with first-line AI and palbociclib: An exploratory analysis of the PADA-1 trial. ASCO20 Virtual Scientific Program. Abstract 1010. [Google Scholar]
- 83.therascreen PIK3CA RGQ PCR Kit [PMA]. Hilden, Germany: QUIAGEN GMBH. U.S. Food and Drug Administration; website. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P190001 Revised May 2020 Accessed May 16, 2020. . [Google Scholar]
- 84.FoundationOne CDx. Cambridge, MA: Foundation Medicine, Inc. U.S. Food and Drug Administration; website. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=p170019. Revised May 2020 Accessed May 16, 2020. . [Google Scholar]
- 85.Chavez-Macgregor M, Barlow W, Pusztai L, et al. Phase III randomized, placebo-controlled clinical trial evaluating the use of adjuvant endocrine therapy +/− one year of everolimus in patients with high-risk, hormone receptor- (HR) positive and HER2−negative breast cancer: SWOG/NRG/Alliance S1207 (NCT01674140). Journal of Clinical Oncology 33:5s (suppl; abstr TPS637); American Society of Clinical Oncology Annual Meeting (May 29-June 2, 2015, Chicago, IL), poster session. [Google Scholar]