

Abstract
Macromolecular protease inhibitors and camelid single-domain antibodies achieve their enzymic inhibition functions often through protruded structures that directly interact with catalytic centers of targeted proteases. Inspired by this phenomenon, we constructed synthetic human antibody libraries encoding long CDR-H3s, from which highly selective monoclonal antibodies (mAbs) that inhibit multiple proteases were discovered. To elucidate their molecular mechanisms, we performed in-depth biochemical characterizations on a panel of matrix metalloproteinase (MMP)-14 inhibitory mAbs. Assays included affinity and potency measurements, enzymatic kinetics, a competitive enzyme-linked immunosorbent assay, proteolytic stability, and epitope mapping followed by quantitative analysis of binding energy changes. The results collectively indicated that these mAbs of convex paratopes were competitive inhibitors recognizing the vicinity of the active cleft, with their significant epitopes scattered across the north and south rims of the cleft. Remarkably, identified epitopes were the surface loops that were highly diverse among MMPs and predominately located at the prime side of the proteolytic site, shedding light on the mechanisms of target selectivity and proteolytic resistance. Substrate sequence profiling and paratope mutagenesis further suggested that mAb 3A2 bound to the active-site cleft in a canonical (substrate-like) manner, by direct interactions between 100hNLVATP100m of its CDR-H3 and subsites S1–S5′ of MMP-14. Overall, synthetic mAbs carrying convex paratopes can achieve efficient inhibition and thus hold great therapeutic promise for effectively and safely targeting biomedically important proteases.
Graphical Abstract
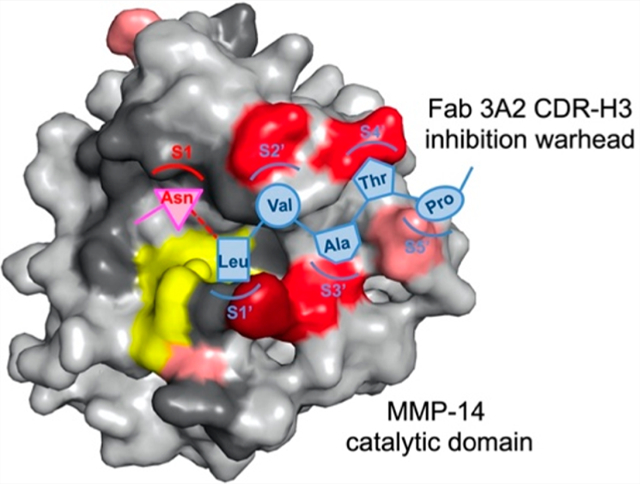
Accounting for ~2% of the human genome, proteases are important signaling molecules that precisely control a wide variety of physiological processes.1–4 To maintain the body’s homeostasis, proteases exist in a delicate balance of networks with their endogenous inhibitors and substrates. Altered protease expression or abnormal substrate proteolysis, therefore, causes many disorders ranging from inflammation,5 cardiac diseases,6 and cancer7 to neuropathy,8 degenerative diseases,9 and osteoporosis.10 In addition, numerous infectious diseases rely on proteases for the pathogen to invade the host cell,11 replicate the viral genome,12 process polyproteins,13 and release progeny virions.14 It is estimated that 5–10% targets for drug development are proteases.15 One apparent therapeutic strategy is to block these abnormal or pathogenic proteolyses by inhibiting their catalytic reactions. Several protease inhibitors have been approved for the treatment of hypertension, coagulation, viral infection, cancer, and diabetes.15 Nevertheless, despite decades of intensive effort, conventional drug discovery has achieved only limited success by targeting a small number of proteases.
Considering the vast proteolytic landscape and the levels of degradome complexity,2,16 specificity is highly desired for any protease inhibition therapy. However, achieving target specificity can be difficult because proteolytic pathways often consist of highly homologous family members that share the same domain folding and catalytic chemistry.17 Indeed, the main reasons why numerous matrix metalloproteinase (MMP) broad spectrum compound inhibitors, e.g., hydroxamates, all failed in clinical trials were a lack of efficacy and severe side effects caused by nonspecific inhibition of other metalloproteinases.18–20 In contrast, monoclonal antibodies (mAbs) provide exquisite specificity capable of distinguishing between closely related protease family members.21–34 Notably, among 569 human proteases identified, 279 (49%) are extracellular or pericellular, and thus can be accessed by therapeutic antibodies.7
Many naturally occurring protease inhibitors exhibit a convex-shaped conformation that inserts into the enzymatic active site and blocks substrate access and/or catalytic function.35,36 However, from a molecular immunology perspective, the likelihood of generating antibodies with convex paratopes is low. In fact, active sites of enzymes have a low antigenicity for murine or human antibodies,37 because the catalytic pocket is often buried inside a major cleft or concave structure and as such is inaccessible and/or incompatible with the antigen-binding surface topography of native human antibodies, often a cavity, groove, or flat surface but rarely a convex conformation. Intriguingly, a large proportion of antibodies isolated from camels and llamas bind the active-site pockets and efficiently inhibit enzymatic reactions.37–39 However, camelid antibodies per se can evoke an immune response when administered to humans. In addition, these animals are usually not available for most researchers. To overcome these limitations, we designed and constructed synthetic human antibody libraries by incorporating enzyme-inhibiting paratopes inspired by camelid antibody repertoires into the human antibody scaffold.33 Using these libraries, panels of nanomolar potent mAbs inhibiting multiple protease targets with high selectivity were identified.33,34 Among these protease targets, MMP-14 is particularly important because of its roles in cancer progression and metastasis,40,41 neuropathic pain,42 and obesity.43
Our previous works have proven the concepts that convex library design and functional selection can facilitate the generation of inhibitory antibodies,33,34 and their proteolytic stability, inhibitory potency, and selectivity can be further engineered.44–46 This study aimed for in-depth characterizations of the inhibition mechanism of isolated anti-MMP14 Fabs (fragments antigen binding) that carry extended CDR-H3s. Numerous biochemical approaches were exploited, including enzymatic kinetics, a competitive enzyme-linked immunosorbent assay (ELISA), proteolytic stability, epitope alanine scanning, and calculation of free binding energy changes. As a result, the types of inhibition and important epitopes were identified for tested Fab inhibitors. Further analysis and paratope mutagenesis on the most potent Fab 3A2 revealed direct recognitions between its CDR-H3 and MMP-14 catalytic subsites. The likely mechanisms for high selectivity over other MMPs and proteolytic stability were also elucidated.
EXPERIMENTAL PROCEDURES
Preparation of Recombinant cdMMP-14 and Its Mutants.
The gene encoding the catalytic domain of MMP-14 (cdMMP-14, Tyr112–Pro290, UniProtKB P50281) was cloned via SfiI sites into a periplasmic expression plasmid pMoPac16.47 The obtained pMoPac16-cdMMP14 carried a Lac promoter, a pelB leader, and a C-terminal polyhistidine tag. Genes of cdMMP-14 site-directed alanine mutants were constructed by overlapping polymerase chain reaction (PCR) with mutagenic primers. Transformed Escherichia coli Jude-I cells [DH10B F′::Tn10 (Tetr)] harboring cdMMP-14 wild type (wt) and mutants were grown in 2×YT medium supplemented with 34 μg/mL chloramphenicol at 30 °C for 16 h without addition of isopropyl β-d-1-thiogalactopyrano-side.48 The cells were harvested and treated with osmotic shocks to recover periplasmic fractions as described previously.49 Briefly, 600 mL cell cultures were pelleted and resuspended in 60 mL of periplasmic buffer [200 mM Tris-HCl (pH 7.5), 20% sucrose, and 30 units/μL lysozyme] for incubation at room temperature for 10 min. The samples were then mixed with 60 mL of ice-cold doubly distilled H2O followed by incubation on ice for 10 min. After centrifugation at 10000g for 30 min at 4 °C, cdMMP-14 wt and mutants were purified from periplasmic preparations by affinity chromatography using Ni-NTA agarose resin (Qiagen). The homogeneities of purified human cdMMP-14 wt and mutants were verified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and their concentrations were measured with a NanoDrop (Thermo Scientific).
Cloning, Expression, and Purification of Fabs and nTIMP-2.
VH and VL genes were cloned into the Fab expression vector containing a PhoA promoter, two STII leaders, and a polyhistidine tag at the C-terminus of the heavy chain.50 VH genes of 3A2 site-directed mutants were assembled by overlapping extension PCR and cloned in the same way. After expression in E. coli BL21 at 30 °C overnight in 2×YT medium, Fabs were purified from the periplasmic fractions by Ni-NTA chromatography and dialyzed against 50 mM HEPES and 150 mM NaCl (pH 6.8). The N-terminal domain of tissue inhibitor metalloproteinase-2 (nTIMP-2, UniProtKB P16035) was produced by E. coli periplasmic expression and affinity purified as described previously.51 Purified proteins were analyzed by SDS–PAGE, and their concentrations were determined.
Biolayer Interferometry.
Purified cdMMP-14 wt was biotinylated by using the EZ-Link Sulfo-NHS-LC kit and then purified following the manufacturer’s instructions (Pierce). Biotinylated cdMMP-14 (100 nM) was loaded onto streptavidin biosensors (ForteBio) for 120 s. After being washed in 50 mM HEPES and 150 mM NaCl (pH 7.5) for 30 s to establish baselines, 50–200 nM Fabs were associated with cdMMP-14-loaded biosensors for 120 s and then dissociated into 50 mM HEPES and 150 mM NaCl (pH 7.5) for 120 s. Averages of measured association constant kon and dissociation constant koff values were determined for KD calculations.
Direct and Competitive ELISA.
Microtiter plates were coated with 5 μg/mL streptavidin and blocked with 0.5% gelatin in assay buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM CaCl2, and 100 μM ZnCl2]. In a direct ELISA, 5 μg/mL biotinylated cdMMP-14 wt in assay buffer was added to streptavidin-coated wells for a 20 min incubation. Purified Fab was serially diluted to 1–1000 nM and incubated with immobilized cdMMP-14 at ambient temperature for 1 h. Bound Fabs were detected with an anti-Fab-HRP conjugate, and signals were developed with a TMB substrate (Thermo Fisher). The half-maximal effective concentration (EC50) was calculated from a four-parameter logistic curve-fitting analysis and set as the subsaturating concentration for each tested Fab. In a competitive ELISA over cdMMP-14 mutants, Fabs at their subsaturating concentrations (e.g., 10 nM Fab 3A2) were first incubated with 0–2000 nM cdMMP-14 mutants for 2 h and then transferred to streptavidin wells coated with biotinylated cdMMP-14 wt for a 15 min incubation. Captured Fabs were measured by using an anti-Fab-HRP and a TMB substrate. For all Fab and cdMMP-14 mutant combinations, IC50 values were determined and Gibbs free energy changes on binding were calculated using eq 1
| (1) |
where R = 1.987 cal mol−1 K−1 and T = 298 K. In a competitive ELISA with nTIMP-2 or GM6001, serially diluted nTIMP-2 or GM6001 at 3 nM to 3 μM was mixed with fixed subsaturating concentrations of Fabs and incubated with biotinylated cdMMP-14 wt immobilized in streptavidin-coated wells, and bound Fabs were detected.
MMP-14 Enzymatic and Inhibition Assays.
Activities of cdMMP-14 wt and mutants were measured at 37 °C by monitoring the hydrolysis of fluorogenic peptide Mca-Lys-Pro-Leu-Gly-Leu-Dap(Dnp)-Ala-Arg-NH2 (M-2350, Bachem) using a fluorescence microplate reader Synergy H4 (BioTek). Typically, 1 μM M-2350 was added to 30 nM cdMMP-14 in assay buffer and the initial slopes of fluorescence signals with an excitation wavelength at 328 nm and an emission wavelength at 393 nm were measured for kinetic calculation. In inhibition assays, 0–1 μM Fabs were incubated with 30 nM cdMMP-14 in assay buffer for 1 h at 37 °C, and then the reaction was initiated by adding 1 μM M-2350. The fluorescence was recorded continuously for 30 min, and the initial reaction rates and inhibition constants were calculated by fitting the data to eq 2
| (2) |
where Vi is the initial velocity in the presence of the inhibitor, Vo is the initial velocity in the absence of the inhibitor, and [I] is the inhibitor concentration. To determine the type of inhibition, the initial velocity of cdMMP-14 wt as a function of substrate concentration (1–40 μM) was measured in the presence of concentrations of Fab (125–5500 nM). The values of apparent Km and Vmax were derived by linearization according to the Lineweaver–Burk equation.
Proteolytic Stability Test.
Purified Fabs (2 μM) were incubated with 2 μM cdMMP-14 wt in 50 mM HEPES and 150 mM NaCl (pH 7.5) at 37 °C for 4 h. Samples were analyzed by using 12% nonreducing SDS–PAGE and stained with Coomassie blue. Densitometric interpretation was conducted using a ChemiDoc imager (Bio-Rad).
RESULTS
Long CDR-H3 Potent Fabs Exhibited Competitive Inhibition.
Inspired by camelid repertoires, synthetic human antibody libraries carrying convex paratopes encoded by CDR-H3 of 23, 25, or 27 amino acids were constructed and applied for the isolation of Fabs inhibiting the catalytic domain (cd) of MMP-14.33 To understand their inhibition mechanisms, five representative Fabs of different potencies and/or postselection abundancies were chosen in this study.52 Four of them have CDR-H3s of 27 amino acids, and Fab 3E9 has a 25-amino acid CDR-H3 (Table 1). These Fabs also have distinct light chains especially on their CDR-L3 sequences. Fabs were produced from E. coli periplasm with typical yields of 0.5–2 mg of purified Fabs per liter of culture (Figure S1). Dissociation constants (KD) measured by biolayer interferometry indicated that Fabs 3E2, 3D9, 2B5, and 3E9 exhibited affinities of 18–45 nM for cdMMP-14, while Fab 3A2 was the most efficient binder with a KD of 7.5 nM (Figure S2). Fab 3A2 also gave a high inhibitory potency (KI) of 8.7 nM, and Fabs 3E2, 3D9, and 2B5 had KI values of 38–220 nM.33 Interestingly, as a highly enriched clone after phage panning, Fab 3E9 displayed only weak inhibition with a KI of 5.4 μM.33 A potent inhibitory Fab DX-2400 (KD = 1.9 nM; KI = 4.3 nM) carrying a short CDR-H3 was characterized in this study, as well.23,53 To determine their inhibition types (except 3E9 due to its weak potency), we measured cdMMP-14 proteolytic kinetics with a FRET peptide substrate in the presence of inhibitory Fabs at varied concentrations. The results clearly showed that for potent inhibitors 3A2, 3E2, and DX-2400, cdMMP-14 showed an unchanged maximum velocity (Vmax) and an increased Michaelis constant (Km) with an increasing concentration of Fabs, indicating that these Fabs inhibited cdMMP-14 proteolytic activity in a substrate competitive manner (Figure 1). For Fabs 2B5 and 3D9 of moderate potency (KI = 220 and 55 nM, respectively), the kinetics of cdMMP-14 represented decreases in both Vmax and Km values, with unparallel Lineweaver–Burk plots suggesting a mixed inhibition type; i.e., 2B5 and 3D9 interfered with both cdMMP-14 as a competitive inhibitor and a cdMMP14–substrate complex as an uncompetitive inhibitor. Overall, from the synthetic human antibody libraries carrying extended CDR-H3s, all isolated Fabs exhibited at least partially competitive inhibition models, and the highly potent Fabs were exclusively competitive inhibitors (Table 1).
Table 1.
Characterization of MMP-14 Inhibitory Fabs
| binding | inhibition | competitive ELISA | ||||||
|---|---|---|---|---|---|---|---|---|
| Faba | CDR-H3 (length) | kon (×104 M−1 s−1) | koff (×10−3 s−1) | KD (nM) | KI (nM) | mode | with nTIMP-2 | with GM6001 |
| 3A2 | VKLQKDKSHQWIRNLVATPYGRYVMDY(27) | 58 ± 2.2 | 4.3 ± 0.2 | 7.5 ± 0.7 | 8.7 | competitive | competitive | not competitive |
| 3E2 | GIKGLVFTGSQMKMLRRGNYNWYVMDY(27) | 29 ± 1.4 | 7.5 ± 0.3 | 26 ± 2.3 | 38 | competitive | competitive | not competitive |
| 3D9 | RLMAYHGSCSSRLCQTAISPQRYAMDY(27) | 68 ± 2.8 | 12 ± 0.4 | 18 ± 1.6 | 55 | mixed | competitive | not competitive |
| 2B5 | IGVNAWAVKMSQRMLATRGSGWYVMDY(27) | 13 ± 1.1 | 6.1 ± 0.3 | 45 ± 8.8 | 220 | mixed | competitive | not competitive |
| 3E9 | NGRYPGFLKRAHKRLLNFKAYVMDY(25) | 14 ± 1.4 | 6.1 ± 0.2 | 44 ± 5.7 | 5400 | ndc | competitive | not competitive |
| DX-2400b | GRAFDI(6) | ndc | ndc | 1.9d | 4.3 | competitive | competitive | not competitive |
Figure 1.
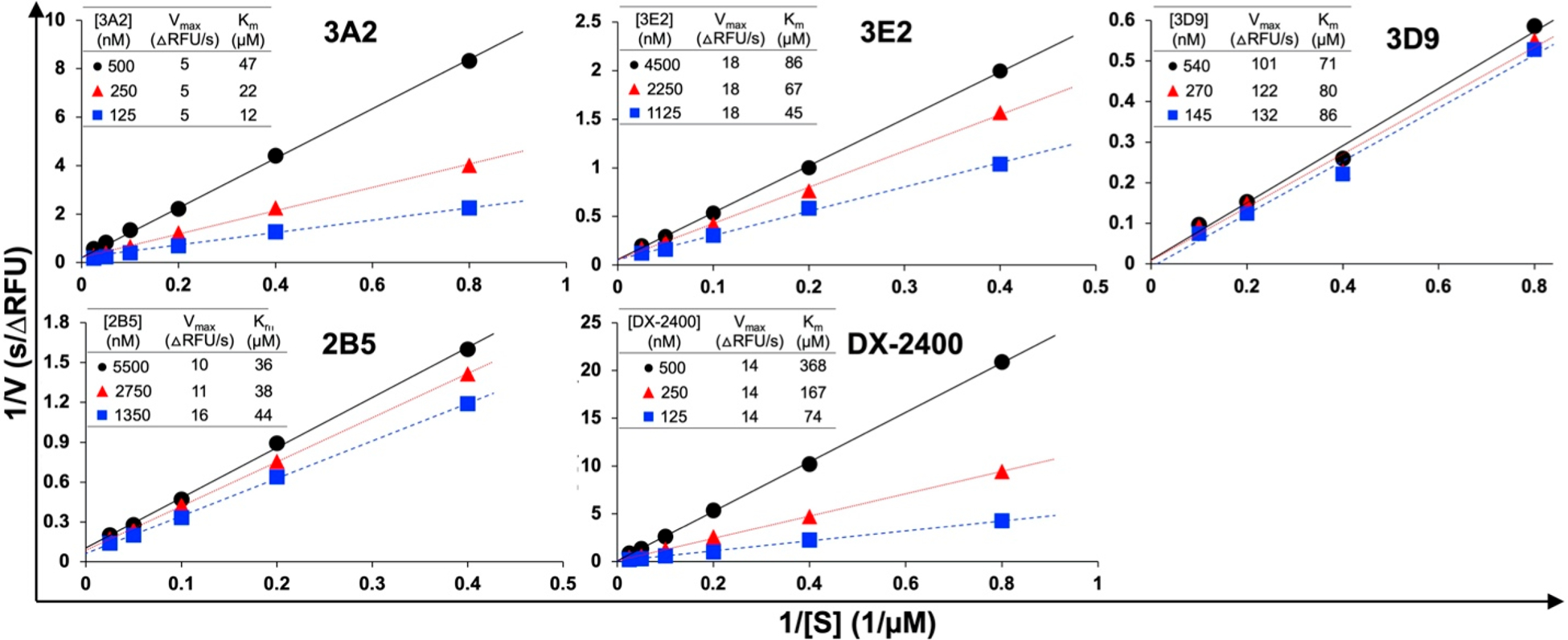
Mode of inhibition determined by kinetics. Reaction rates were measured with 30 nM cdMMP-14 and 1–40 μM FRET peptide substrate M-2350 in the presence of Fabs at various concentrations. With unchanged Vmax and increased apparent Km values, 3A2, 3E2, and DX-2400 exhibited competitive inhibition, while 2B5 and 3D9 were mixed model inhibitors (competitive and uncompetitive).
Inhibitory Fabs Competed with nTIMP for Binding.
The reaction center of MMP-14 and the flanking subsites form a cleftlike structure on the protease surface, which accommodates polypeptide substrates for proteolysis (Figure S3). As endogenous inhibitors of MMPs, tissue inhibitors of metalloproteinases (TIMPs) accomplish their inhibitory activities by directly binding at the active-site clefts and vicinities,36 and thus, their competitive ELISA with antibodies can provide perceptions of antibody’s binding sites. Competitive ELISAs in which each Fab at a fixed subsaturating concentration was mixed with increased amounts of nTIMP-2 were conducted, and the Fabs captured on immobilized cdMMP-14 were detected by anti-Fab-HRP for signal development. The results showed that increasing concentrations of nTIMP-2 reduced the level of binding of all six tested Fabs on immobilized cdMMP-14 (Figure 2), implying that overlaps existed between the epitopes of these Fabs and that of nTIMP-2. Notably, 3A2 displayed a sharp sigmoid curve responding to nTIMP-2, suggesting significant interference of nTIMP-2 on 3A2’s binding to cdMMP-14. As nTIMP-2 makes contacts with numerous residues on surface loops surrounding the active site of MMP-14 (Figure S4),36 some of these residue positions were considered for alanine substitutions in the following epitope mapping. In addition, results of a similar competitive ELISA with a broad spectrum reversible hydroxamate inhibitor GM6001 indicated that the bindings of all tested Fabs on cdMMP-14 were not affected by this potent inhibitor even at high concentrations of ≤3 μM (Figure S5), indicating that the MMP-14 active site is accessible for GM6001 in the presence of Fabs presumably because Fabs did not intimately contact the catalytic center.
Figure 2.
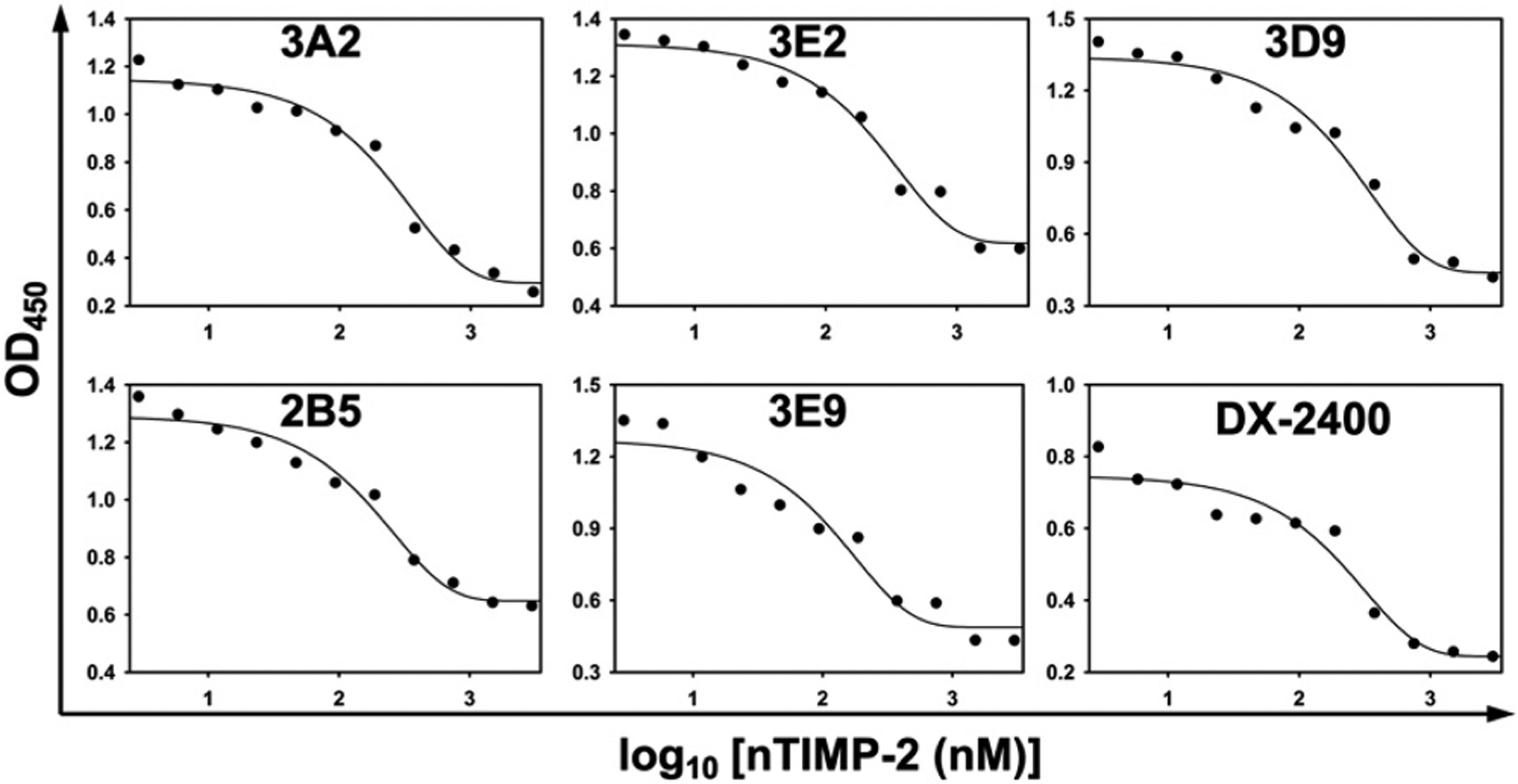
Competitive ELISA with nTIMP-2. Immobilized cdMMP-14 was incubated with Fabs (3A2, 3D9, and DX-2400 at 10 nM; 3E2, 2B5, and 3E9 at 20 nM) in the presence of 3 nM to 3 μM nTIMP-2. Anti-Fab-HRP was used to detect the captured Fabs.
Design and Preparation of cdMMP-14 Alanine Mutations.
To identify the binding epitopes of inhibitory Fabs, site-directed mutagenesis to alanine was used to footprint on the surface of cdMMP-14 (Figure 3). The positions for alanine scanning were selected on the basis of the cdMMP-14 structure. Considerations are (i) on the surface loops flanking its active cleft, (ii) within 15 Å of the catalytic Zn2+, (iii) exhibiting an exposed respective side chain, (iv) overlapped with an epitope of nTIMP-2 (Figure S4), and (v) unique to membrane-type (MT) MMPs but not found in soluble MMPs. In particular, the 200s and 230s loops are located at the nonprime and prime sides of catalytic center, respectively. Loop 260s forms the south rim of the active cleft at the prime side, while loop 190s is at the north rim across both nonprime and prime subsites. Three positions in the MT loop (160s), displaying a high degree of sequence divergence among MMPs, were also selected for Ala substitution. A total of 20 single-point alanine mutants of cdMMP-14 were cloned and expressed in the periplasmic space of E. coli with yields of 60 μg to 5.1 mg of purified proteins per liter of culture (Figure S6). Notably, mutations around the active cleft led to a significantly reduced, albeit still substantial, specific activity of 0.4–8.6% relative to cdMMP-14 wt. Unsurprisingly, these mutations away from the reaction center, e.g., Y166A and E169A, or at far end of the nonprime subsites, e.g., S189A and I209A, did not drastically affect the activity. Prepared in their soluble and active form without refolding, these alanine mutants of cdMMP-14 facilitated epitope mapping with isolated antibodies.
Figure 3.
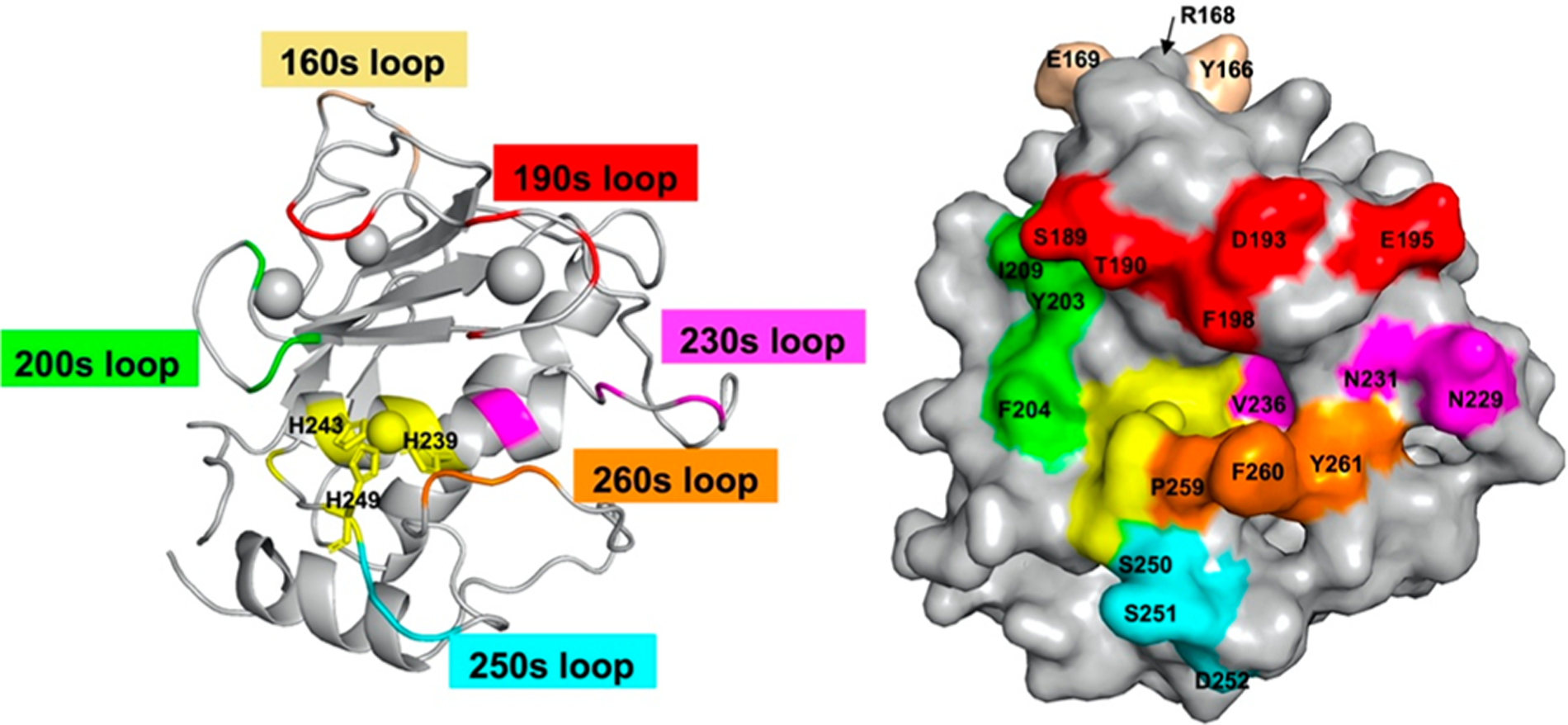
Design of cdMMP-14 alanine point mutants. Structures of cdMMP-14 displayed as a cartoon (left) and in a surface mode (right) show the loops and residues selected for alanine point mutations. The catalytic zinc and its coordinating histidines (sticks) are colored yellow.
Quantitative Mapping of Inhibitory Fab Epitopes.
At their predetermined subsaturating concentrations, Fabs bound to immobilized cdMMP-14 wt were measured in the presence of increasing amounts of cdMMP-14 alanine mutants. Such competitive ELISAs were also performed with soluble cdMMP-14 wt to obtain IC50(wt) values for each Fab. Compared to IC50(wt), the sigmoid curves associated with cdMMP-14 alanine mutants exhibited four scenarios (Figure 4): (a) abolished, e.g., Fab 3A2 completely abandoned its binding to mutant F260A; (b) weakened, e.g., Fab 3E2 showed a decreased affinity for F198A compared to cdMMP-14 wt; (c) unchanged, e.g., Fab DX-2400 had similar binding profiles with respect to D252A and wt; and (d) enhanced, e.g., Fab 3E9 bound actually better with V236A than wt. Like the additional exemplary results shown in Figure S7, 120 total competitive ELISAs for all of the combinations of 6 Fabs with 20 cdMMP-14 alanine mutants were performed for the associated IC50(mut) determination (Figure 5). For a direct comparison disregarding the disparity of the absolute affinity between Fabs, the effect of each alanine substitution was calculated as a change in Gibbs free energy of binding relative to that of wt: ΔΔG(mut‑wt) = ΔG(mut) − ΔG(wt) = RT ln[IC50(mut)/IC50(wt)].54 Specifically, a significant positive value of ΔΔG(mut‑wt), i.e., >0.5 kcal/mol, indicates a disruptive alanine substitution that leads to a reduced binding strength, while a significant negative ΔΔG(mut‑wt) value, i.e., less than −0.5 kcal/mol, indicates that the associated alanine substitution in fact enhances the binding strength. When the absolute value |ΔΔG(mut‑wt)| is <0.5 kcal/mol, equivalent to a <2.3-fold difference in IC50, the change is considered as nonsignificant. Among all tested alanine substitutions, the most disruptive ones were at the 190s and 260s loops, e.g., F260 for 3A2, F198 and P259 for 3E2, and F260 for 3E9, with their associated ΔΔG(mut‑wt) values being >2.0 kcal/mol (corresponding to >28-fold increases in IC50), suggesting that the side chains of these residues significantly contributed to binding. In addition, all Fabs except 3E9 exhibited moderate interactions with D193 and E195 on the 190s loop with ΔΔG(mut‑wt) values of 0.5–2.0 kcal/mol (Figure 5). Interactions with these acidic residues can be possibly mediated with the basic residues in CDR-H3s of tested Fabs (Table 1). Notably, for inhibitory Fabs with high potencies (like 3A2, 3E2, and DX-2400), alanine substitutions mainly resulted in reduced affinities [positive ΔΔG(mut‑wt) values]. In contrast, for low-potency Fabs 2B5 and 3E9, many alanine substitutions gave negative energy changes, e.g., 9 of the 10 significant ΔΔG(mut‑wt) of 3E9 were less than −0.5 kcal/mol, indicating these alanine mutations improved the associated bindings.
Figure 4.
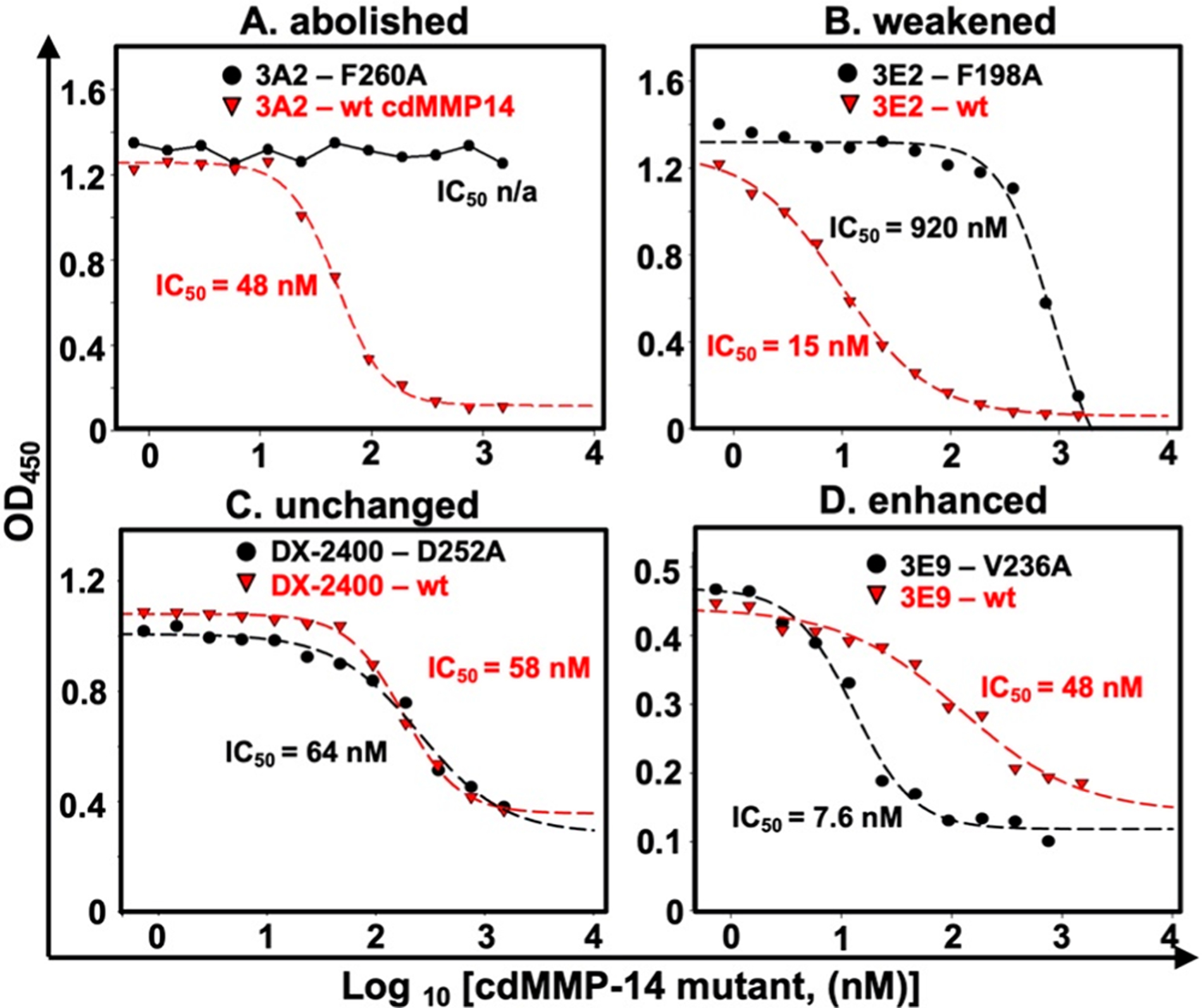
Exemplary results of a competitive ELISA with wt and alanine-substituted cdMMP-14. Immobilized cdMMP-14 wt was incubated with Fabs (3A2, 3D9, and DX-2400 at 10 nM; 3E2, 2B5, and 3E9 at 20 nM) in the presence of 1 nM to 3 μM cdMMP-14 mutants. Captured Fabs were detected with anti-Fab-HRP for signal development. Four scenarios were observed. (A) Binding was abolished with the cdMMP-14 mutant. (B) The binding affinity was weakened with the mutant compared to cdMMP-14 wt. (C) The binding affinity was unchanged. (D) The binding affinity was enhanced with the cdMMP-14 mutant.
Figure 5.
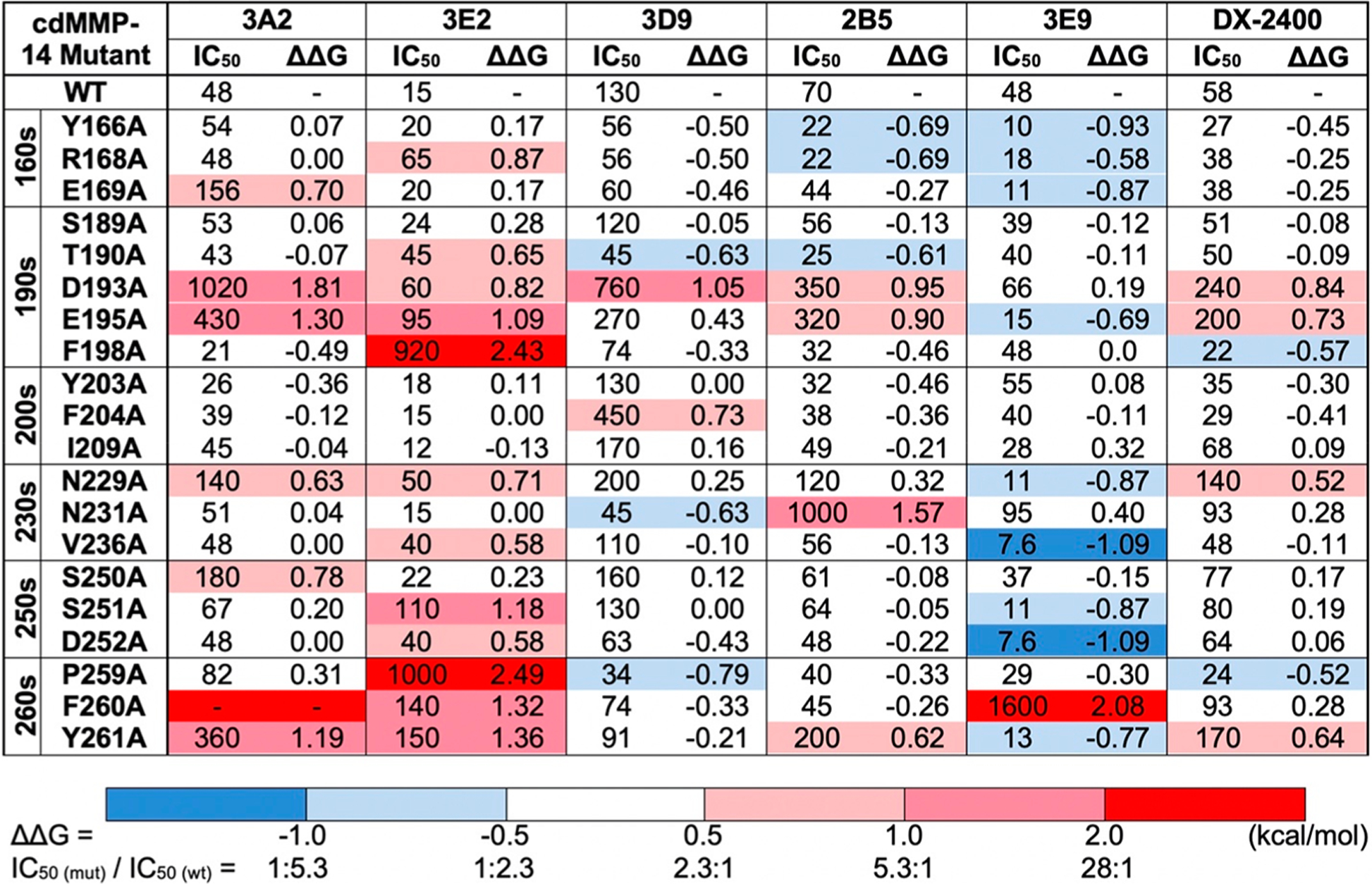
Gibbs free energy changes (ΔΔG) upon binding with mutant cdMMP-14 over wt. Values of IC50 (nanomolar) were measured by a competitive ELISA, and ΔΔG(mut-wt) (kilocalories per mole) values were calculated by the equation , where R = 1.987 cal K−1 mol−1 and T = 298 K. A positive ΔΔG (>0.5 kcal/mol, red) indicates that the binding affinity is weakened with the mutant; a negative ΔΔG (less than −0.5 kcal/mol, blue) indicates that the binding affinity is enhanced with the mutant, and a |ΔΔG| of <0.5 kcal/mol (white) suggests a nonsignificant change.
Residues Surrounding the Active Cleft at the Prime Side Were Important Epitopes.
3A2.
Visualization of alanine scanning data on the cdMMP-14 structure suggested that 3A2 made strong contacts with residues on the 190s and 260s loops in the vicinity of the active site (Figure 6). Interactions of 3A2 with D193, E195, and Y261 side chains accounted for a binding energy of 1.2–1.8 kcal/mol each, corresponding to 7.5–21-fold decreases in affinity when these residues were mutated to alanine. The strongest interaction between 3A2 and cdMMP-14 was mediated by F260, because alanine mutation at this position eliminated binding activity completely (Figure 4A). Weak interactions also occurred with the side chains of E169, N229, and S250, with decreases in the free energy of binding of 0.6–0.8 kcal/mol. Notably, residues that strongly interact with 3A2 are all located at the prime side of the cdMMP-14 active center on the north and south rims of the reaction cleft. In contrast, alanine substitutions at the nonprime side of cdMMP-14, including Y203, F204, and I209 on the 200s loop and S189 and T190 on the 190s loop, did not cause significant changes in ΔΔG(mut‑wt), suggesting these residues made little contribution to 3A2’s recognition. In addition, alanine mutations at N231 and V236, which lie in the bottom of the catalytic cleft, also did not affect the binding affinity, suggesting that Fab 3A2 did not tightly contact with the cleft bottom of cdMMP-14, consistent with the results of the competitive ELISA with GM6001 (Figure S5). Overall, 3A2 recognizes the prime side surface loops of cdMMP-14 in the vicinity of its active cleft.
Figure 6.
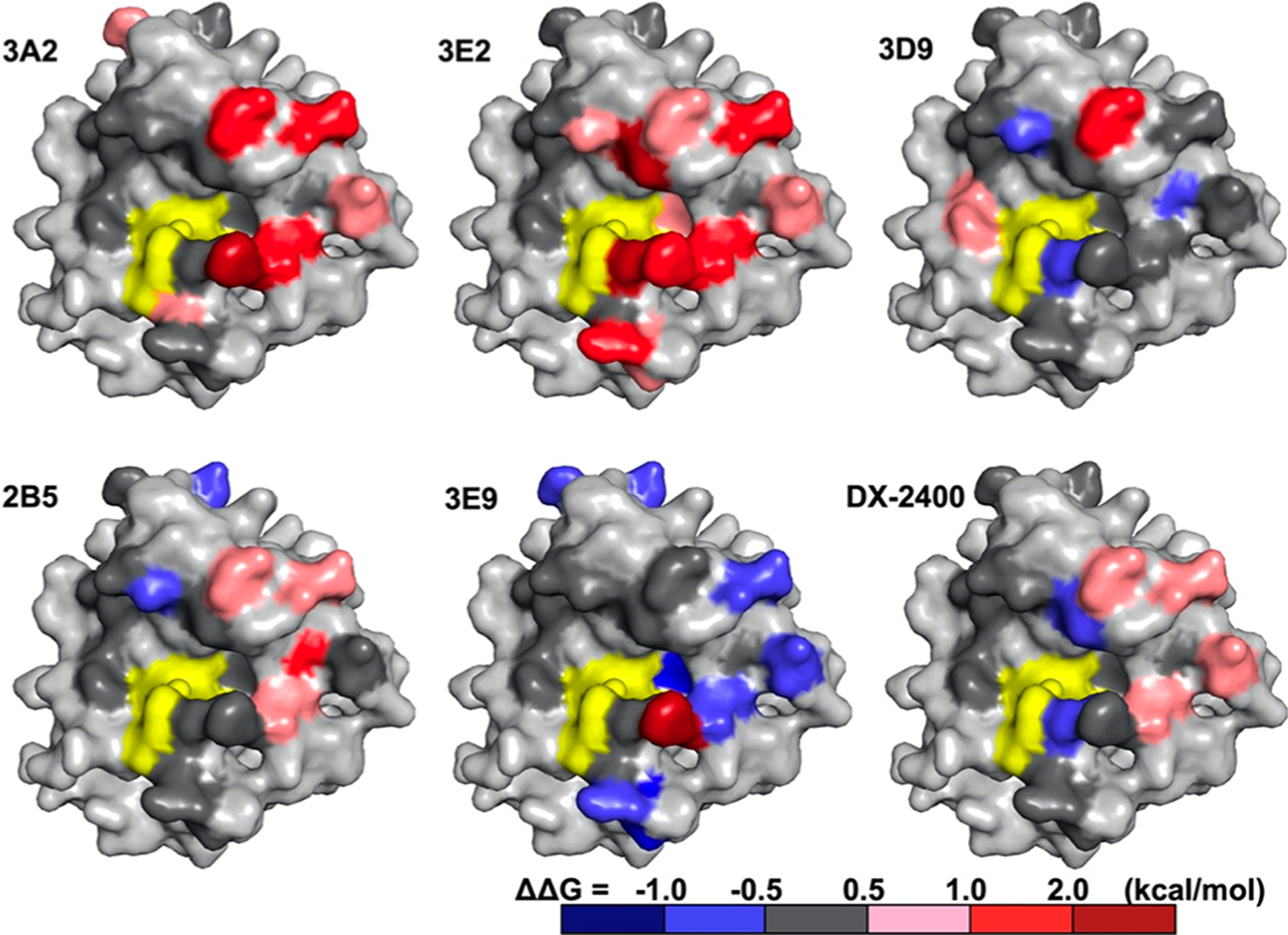
Epitope mapping by alanine scanning mutagenesis. Point mutants that had a positive ΔΔG(mut‑wt) (increased IC50) are colored pink (0.5–1.0 kcal/mol), red (1.0–2.0 kcal/mol), and brown (>2.0 kcal/mol), and mutants that had a negative ΔΔG(mut‑wt) (decreased IC50) are colored blue. Point mutants that had a minimal effect on IC50 [|ΔΔG(mut‑wt)| < 0.5 kcal/mol] are colored gray. The space-filling models are oriented in the same manner as in Figure 3, with the catalytic center colored yellow.
3E2.
Epitope mapping with Fab 3E2 also demonstrated the importance of the prime side (Figure 6). 3E2 exhibited strong interactions with F198 and P259; both are close to the S1′ subsite located on the north and south rims, respectively. Their alanine substitutions increased the IC50 values to >0.9 μM, corresponding to substantial changes in binding energy of >2.4 kcal/mol (Figure 5). In addition, alanine mutations of numerous prime side residues, including D193 and E195 on the 190s loop, N229 and V236 on the 230s loop, and F260 and Y261 on the 260s loop, all had significant effects on 3E2 binding (0.5–2.0 kcal/mol each). In contrast, S189 and the 200s loop, located at the nonprime side, had no significant contributions. Different from 3A2, 3E2 also formed moderate interactions with S251 and D252 on the 250s loop (1.7 kcal/mol collectively) and a weak interaction with T190 close to the S1 subsite, indicating a large binding footprint compared to that of 3A2, though their epitopes were mainly overlapped at the prime side of the cleft rims.
2B5 and DX-2400.
These Fabs shared their epitopes on the prime side surface loops of 190s (D193 and E195 for both), 230s (229N for DX-2400 and 231N for 2B5), and 260s (F261 for both) mostly through combinations of these minor interactions (0.5–1.0 kcal/mol each) (Figure 5). The only exception was the N231A variant that generated a moderate change in free energy on 2B5 binding (1.6 kcal/mol, equivalent to a 14-fold weakened IC50). In addition, neither 2B5 nor DX-2400 made contact with nonprime side residues S189, Y203, F204, and I209 or the 250s loop (Figure 6). Notably, several alanine substitutions on cdMMP-14 improved their recognition by the Fabs, e.g., Y166, R168, and T190 for 2B5 and F198 and P259 for DX-2400. These modest decreases in binding energy (0.5–0.7 kcal/mol) suggested that interactions via the side chains of these residues were suboptimal. As a potent inhibitor with a high binding affinity of 1.9 nM, Fab DX-2400 surprisingly made only four contacts of modest strength among the tested positions, implying that an epitope other than the mapped area may exist for DX-2400.
3E9 and 3D9.
3E9 gained much of its binding energy from a strong interaction with the side chain of F260, as an alanine mutation at this position led to a 33-fold increase in IC50 (2.1 kcal/mol) (Figure 5). Centered at F260, low-affinity Fab 3E9 also made suboptimal contacts [negative ΔΔG(mut‑wt) values] with nine residues scattered on the 160s, 190s, 230s, 250s, and 260s loops (Figure 6). Markedly, a majority of these residues and F260 were located at the prime side related to the cdMMP-14 active site. In contrast, 3D9 formed significant interactions with the side chains of two residues, D193 (1.1 kcal/mol) at the prime side and F204 (0.7 kcal/mol) at the nonprime side (Figure 5). Notably, 3D9 also exhibited a negative ΔΔG(mut‑wt) with T190 at the prime side and weak interactions with N231 and P259 at the nonprime side (Figure 6). All of these observations made 3D9 the only exception that had epitopes across both sides of the reaction center, different from all other tested Fabs.
Contribution of 3A2 CDR-H3 to Inhibition and Stability.
The mechanism of inhibition of highly potent Fab 3A2 was further characterized by paratope alanine mutations. Previous study has suggested that although ineffective as a substrate, Fab 3A2 can be slowly cleaved by MMP-14 with the scissile peptide bond between residues N100h (P1) and L100i (P1′) within its CDR-H3 (Figure 7A).44 Large-scale substrate profiling has identified MMP-14’s subsite amino acid preference:55 Ala at P2, Gly/Asn/Ala at P1, Leu/Ile/Val at P1′, Val/Ile/Leu at P2′, Gly/His/Ala at P3′, and Pro/Asp/Val at P5′. Remarkably, these underlined residues are well matched with the central portion of the 3A2 CDR-H3 sequence (100hNLVATP100m). These observations together with epitope mapping results (Figure 6) imply that this portion of CDR-H3 of 3A2 complements MMP-14 subsites S1–S5′ and thus encourage us to introduce alanine substitutions into 3A2 CDR-H3. More specifically, two positions at the C-terminus of the scissile bond, L100i and T100l, correspond to the prime substrate residues P1′ and P4′, respectively, and two residues, R100g and W100e, at the nonprime (N-terminal side of the scissile bond) P2 and P4 positions were selected for alanine mutations (shaded in Figure 7A). Notably, mutant R100gA flipped P2 from an under-represented substrate residue of MMP-14 to an over-represented one,55 and in contrast, mutant L100iA changed the P1′ residue from MMP-14 preferred to disfavored. These four Fabs of 3A2 alanine mutants were produced (Figure S8). Biolayer interferometry indicated that 3A2 mutants R100gA(P2) and W100eA(P4) kept their binding affinities for cdMMP-14 with KD values of 5.7 and 6.6 nM, respectively (Figure S9), similar to that of 3A2 wt. Assays with a FRET peptide substrate suggested that these two nonprime paratope mutants also effectively retained their inhibitory potency (Figure 7B). In contrast, alanine substitutions at two prime paratope sites, L100iA and T100lA, exhibited reduced affinities with KD values of 21 and 27 nM, respectively (Figure S9 and Figure 7B). In addition, mutant L100iA at position P1′ dramatically decreased its inhibitory potency 290-fold, and mutant T100lA at P4′, distant from the cleavage site, compromised the inhibitory function to a lesser but still significant degree with a 3.3-fold weakened KI (Figure 7C). These results not only echoed the importance of prime subsites as binding epitopes (Figure 6) but also suggested a direct contribution of the 3A2 CDR-H3 prime portion for inhibition function. To understand the possible role of the nonprime portion of 3A2 CDR-H3, we further measured the proteolytic liability of Fabs 3A2 wt, W100eA, and R100gA. After incubation of 2 μM Fab (3A2 wt or its alanine mutants) with 2 μM cdMMP-14 for 4 h at 37 °C and pH 7.5, densitometric analysis by nonreducing SDS–PAGE revealed that compared to 3A2 wt, W100eA and R100gA resulted in significantly more truncated Fab fragments, i.e., 1.9- and 1.6-fold increases for LC-CH1 and 3.0- and 2.0-fold increases for VH, respectively (Figure 7D), suggesting the nonprime portion of CDR-H3 was important for proteolytic stability.
Figure 7.
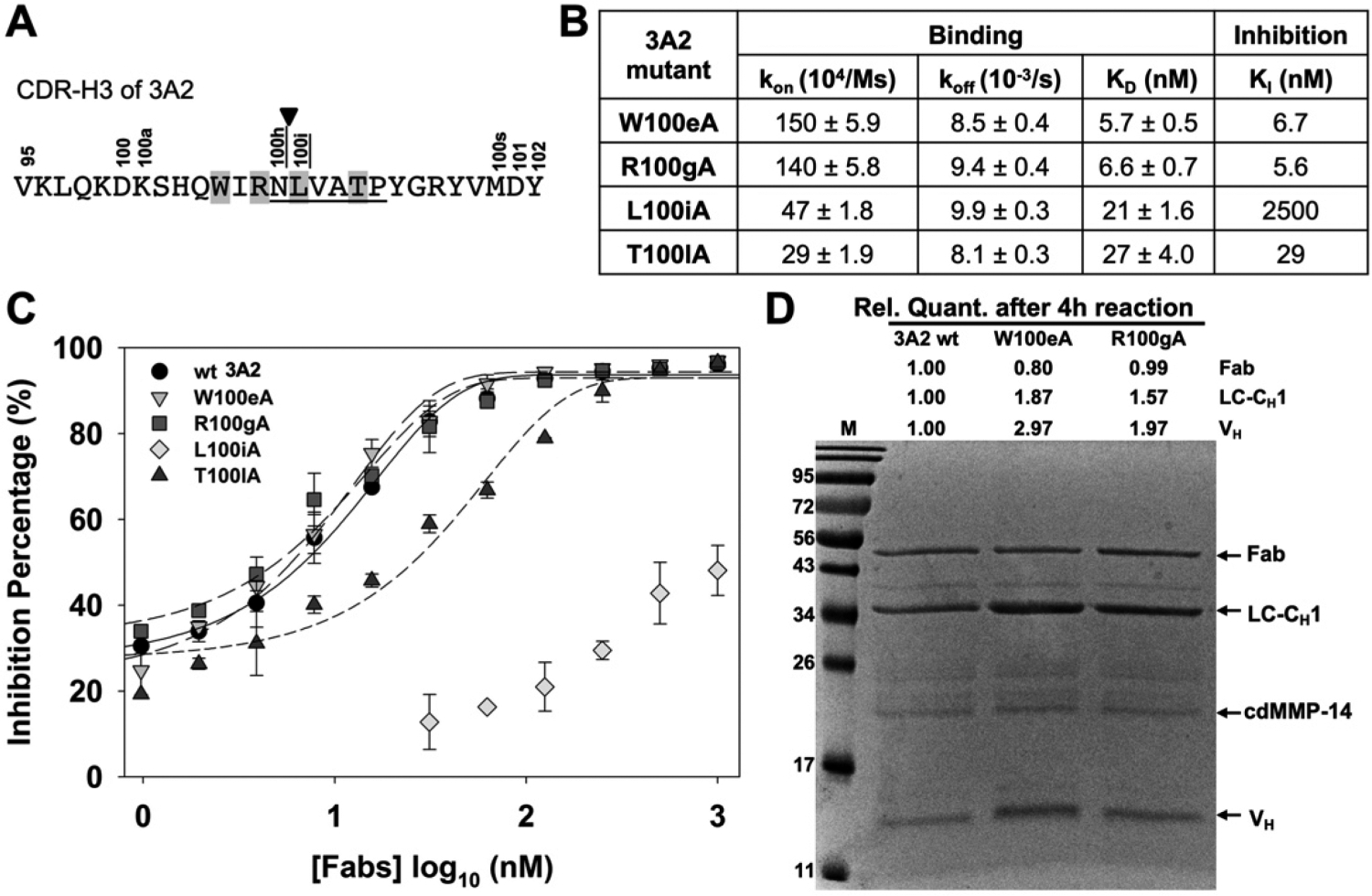
Characterizations of Fab 3A2 alanine point mutants. (A) CDR-H3 amino acid sequence of 3A2, showing the residues that match with the MMP-14 preferred substrate sequences (underlined, positions P1–P5′), cleavage site (arrowhead), and paratope positions chosen for alanine substitution (highlighted). (B) Binding kinetics and inhibitory potency data of 3A2 mutants. (C) Dose–response curves of inhibition measured with 30 nM cdMMP-14 and 1 μM FRET peptide substrate M-2350. (D) Fab stability assays with 3A2 wt and mutants W100eA and R100gA. Purified Fabs (2 μM) were incubated with 2 μM cdMMP-14 in 50 mM HEPES and 150 mM NaCl (pH 7.5) at 37 °C for 4 h and then analyzed by SDS–PAGE.
DISCUSSION
This study has identified the molecular mechanisms by which Fabs of convex paratopes, isolated from synthetic human antibody libraries carrying long CDR-H3s, inhibit proteolytic activity of MMP-14. In addition to enzyme kinetics and a competitive ELISA over physiological or synthetic inhibitors, binding landscapes have been quantitatively mapped by using series of cdMMP-14 alanine mutants. Collectively, the results indicate that these Fabs of extended H3s are competitive inhibitors recognizing the vicinity of the active cleft especially on the prime side of the active site. The most significant epitopes are scattered across the north and south rims of the cleft, i.e., 190s and 260s loops, respectively (Figure 6), but less pronounced at the bottom of the cleft. These results are consistent with observations that nTIMP-2 but not GM6001 can replace Fabs on binding to cdMMP-14 (Figure 2 and Figure S5). Indeed, Fab epitopes overlap with that of nTIMP-2 but cannot block potent GM6001 to access the active site, implying that a void may exist between Fabs and cdMMP-14 in their complexes. Nevertheless, Fab 3A2 can be inefficiently and slowly cleaved at its CDR-H3 by cdMMP-14 (Figure 7D),44 suggesting that its H3 loop must possess a certain flexibility to be able to approach the catalytic center. Analysis of the scissile bond on Fab 3A2 and profiles of the MMP-14 substrate preference44,55 implies that the middle part of 3A2 CDR-H3, 100hNLVATP100m, complements MMP-14 subsites S1–S5′ (Figure 8A). Although recognized by MMP-14 in a substrate-like manner, Fab 3A2 behaves as a strong inhibitor and/or poor substrate, presumably because its interactions with cleft rims are limited primarily to one side of the catalytic zinc, unlike real substrates with intimate contacts with subsites at the bottom of the cleft on both sides. This understanding of 3A2 CDR-H3 function is further supported by paratope mutagenesis studies. Changing P1′ to an unfavorable residue, L100iA, causes the P1′ residue to detach from subsite S1′ (Figure 8B) and thus exposes the catalytic center for substrate access and dramatically decreases the inhibitory potency (Figure 7C). On the contrary, paratope mutation R100gA changes P2 to a MMP-14 preferred substrate residue and thus promotes subsite occupation at S2 and extends the recognition covering S2–S5′ (Figure 8C). Like a substrate, R100gA binds both prime and nonprime subsites and thus is more vulnerable to cdMMP-14 cleavage than 3A2 wt (Figure 7D). Overall, Fab 3A2 is a canonical (substrate-like) inhibitor, achieving its function via direct interactions with the active-site cleft of MMP-14 by using its 27-amino acid extended CDR-H3 likely forming a convex-shaped paratope. For other Fabs of long CDR-H3s tested in this study, however, no clear substrate patterns can be identified within their CDRs.
Figure 8.
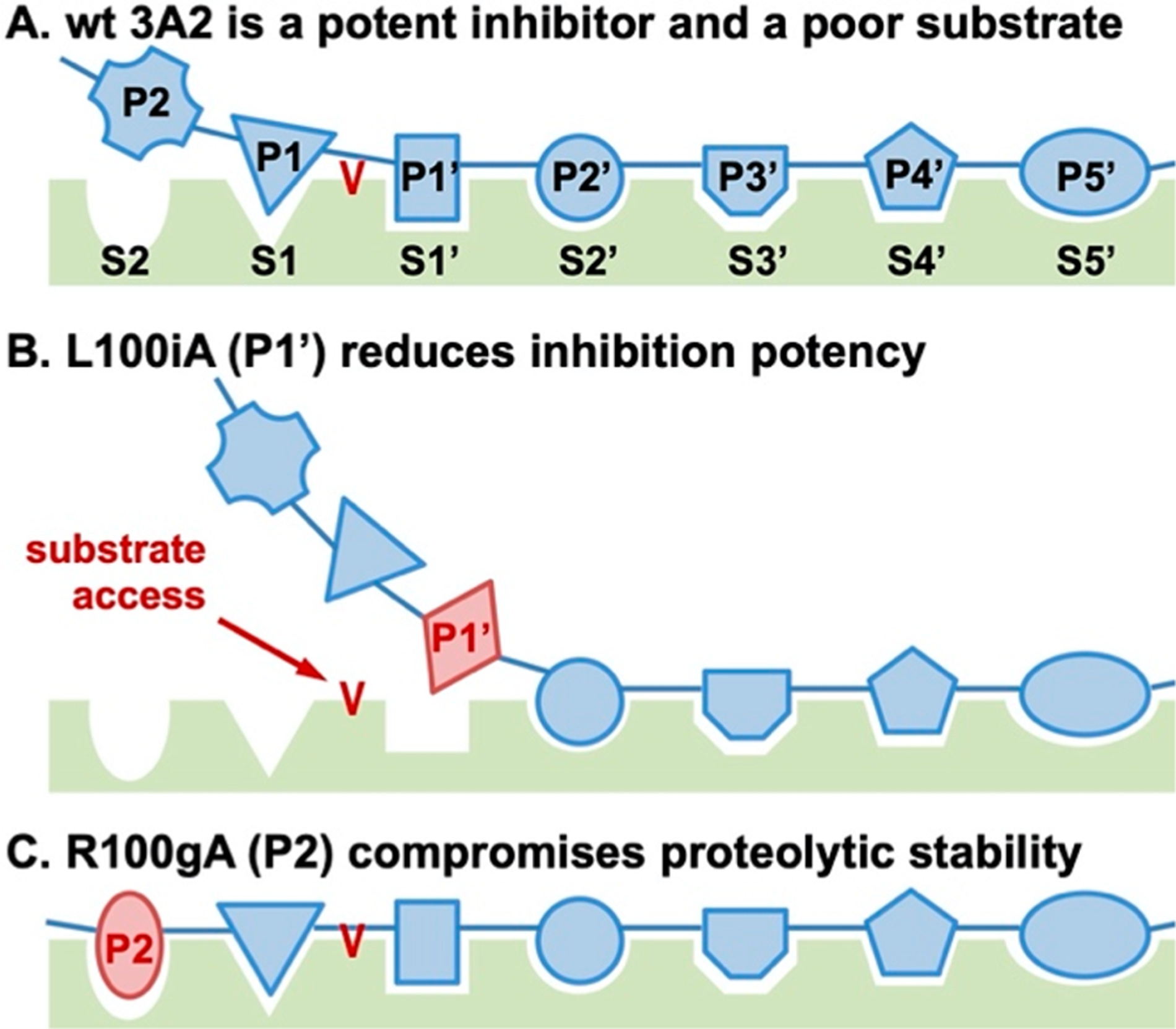
Schematic drawing of the mechanism of inhibition of 3A2. (A) 3A2 is a canonical inhibitor recognizing MMP-14 subsites mainly via the prime portion with its CDR-H3. (B) Paratope alanine substitutions at the prime side, e.g., L100iA at P1′, weaken the inhibitory potency. (C) Paratope alanine substitutions at the nonprime side, e.g., R100gA at P2, compromise proteolytic stability.
Numerous mAbs targeting a variety of proteases have been biochemically and/or structurally elucidated. On the basis of their inhibition mechanisms, a majority of these mAbs can be classified as active-site or exosite inhibitors.56,57 Binding to epitopes other than the catalytic center, an exosite inhibitor usually triggers conformational changes at or around the active cleft and thus interferes with access to the substrate.21,25,58 Interestingly, such allosteric effects are also feasible for accelerating proteolytic reactions for certain substrates.59,60 In contrast, an active-site inhibitor forms intimate contacts with the protease reaction center that is located in the middle of the active cleft.35,56 To reach the reaction pockets, prolate structures are often observed among the paratopes of active-site inhibitors.35 Isolated from synthetic libraries, anti-matriptase mAbs E2, A11, and S422,27 and anti-pKal DX-293028 all carry extended length CDR-H3s that form protruded conformations to insert into the protease active sites. On the contrary, anti-FXIa DEF61 and anti-MMP9 SDS3,24 isolated from naïve and immunized libraries, respectively, have short H3s but intriguingly use their L1 or H2 on the edge of their overall concave paratopes for active-site penetration. Alternatively, substrate access can be blocked without intimate contacts at the active site, as demonstrated by anti-HGFA Ab58, for instance.21 In this study, inhibitory Fabs of long H3s exhibit their main epitopes in the vicinity of the active cleft; e.g., 3E2 strongly interacts with subsite S1′, and 3D9 has epitopes across both prime and nonprime sides of the cleft (Figures 5 and 6). Considering their 25- or 27-amino acid extended CDR-H3s, convex paratopes are probably formed for direct interactions with the active cleft. However, these interpretations can be confirmed only by determining the structures of the Fab–cdMMP-14 complex.
Various strategies for decreasing the proteolytic liability have been exploited by active-site inhibitors: (i) a cleavage–resynthesis equilibrium, e.g., bovine pancreatic trypsin inhibitor,62 (ii) reverse orientation of inserted loops, e.g., anti-matriptase A11 and S4,27 and anti-FXIa DEF,61 and (iii) occupation of one but not both sides of the catalytic center, e.g., anti-pKal DX-2930.28 More specifically, CDR-H3 of DX-2930 binds S1–S3 and abruptly turns away from the prime subsites, thereby preventing cleavage by the catalytic serine of pKal. In contrast, camelid anti-uPA Nb4 inserts its CDR-H3 into the entire S4–S3′ substrate-binding pocket, leading to substantial proteolytic cleavage.63 This study indicates that most tested Fabs of MMP-14 inhibition dominate their recognition on the prime side of the cleft (Figure 6). In addition, binding of Fab 3A2 to cdMMP-14 is unaffected even with high concentrations of GM6001 (Figure S5), implying an unintimate contact between its H3 and the catalytic site. Presumably, the two reasons mentioned above cause Fab 3A2 to be an inefficient substrate. Paratope mutagenesis supports this notion as extending subsite occupation to S2 makes Fab 3A2 mutant R100gA more sensitive to proteolysis (Figure 7D). On the contrary, converting P1 and/or P3′ to MMP-14 unfavored residues significantly improves the stability without compromising the inhibitory potency.44 In addition to proteolytic stability, specificity is another required property for protease inhibition-based therapies. The catalytic domains of MMP family members share a homologous protein folding and secondary structures.64 Despite their highly conserved catalytic Zn2+ and associated motifs, sequential and conformational diversity does exist among MMPs, especially at their surface loops surrounding the active cleft to determine their substrate specificity.65 This study indicates that these surface loops are important epitopes for all tested Fab inhibitors (Figure 6), explaining at least partially why these Fabs are highly selective as reported previously.33
Because of their pivotal roles in extracellular matrix remodeling, MMPs have been regarded as one of the most important regulatory enzymes for a variety of conditions (e.g., tumor invasion and metastasis, development of obesity, and neuropathy),41–43 and thus striking therapeutic targets for associated treatments.66,67 Isolated from synthetic libraries carrying convex paratopes, MMP inhibitory mAbs have demonstrated significant therapeutic efficacies in mouse models. (i) Fab 3A2 reduced both the frequency and the size of melanoma metastatic nodules.40 (ii) IgG 3A2 markedly inhibited growth of the primary breast tumor and more importantly reduced the metastatic spread to the lungs and liver by >90%.41 (iii) Treatment with IgG 3A2 improved glucose intolerance and decreased the body weight in both mice with obesity induced by a high-fat diet and ob/ob mice (unpublished). (iv) Anti-MMP-9 IgG L13 exhibited neuropathic pain attenuation efficacy in chemotherapy-induced pain34 and diabetic neuropathy (unpublished). In addition, pharmacokinetic analysis further suggested that IgG 3A2 had an in vivo half-life of ~4.8 days,41 similar to that of serum IgGs in adult mice.
CONCLUSIONS
In this study, MMP-14 inhibitory Fabs carrying 25- or 27-amino acid CDR-H3s have been biochemically characterized. The results indicate that these Fabs recognize the highly diverse region among MMPs and dominantly target the prime side of the active cleft and its vicinity. The canonical (substrate-like) mechanism of Fab 3A2 and the exact contribution of its CDR-H3 to inhibition have also been revealed. These findings suggest the underlying principles for selectivity and proteolytic resistance of these Fabs. Understanding the molecular mechanism and previous preclinical results mutually support the idea that synthetic antibody libraries encoding extended CDR-H3s are a valuable resource, from which isolated human mAbs hold great therapeutic promise to effectively and selectively inhibit biomedically important proteases.
Supplementary Material
ACKNOWLEDGMENTS
The authors are thankful for UCR Graduate Division’s Dissertation Year Program (DYP) Awards to D.H.N. and K.B.L. and the Mogam Science Scholarship Foundation Fellowship to K.B.L.
Funding
This work was supported by National Institutes of Health Grant 1R01GM115672 and National Science Foundation CAREER Grant 1453645.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00690.
SDS–PAGE of purified Fabs (Figure S1), kinetics of binding of Fabs on cdMMP-14 (Figure S2), active cleft of MMP-14 (Figure S3), nTIMP-2-binding surface on cdMMP-14 (Figure S4), a competitive ELISA with GM6001 (Figure S5), production of cdMMP-14 alanine point mutants (Figure S6), additional exemplary results of a competitive ELISA (Figure S7), production of Fab 3A2 mutants (Figure S8), and biolayer interferometry results of Fab 3A2 mutants (Figure S9) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Turk B, Turk D, and Turk V (2012) Protease signalling: the cutting edge. EMBO J. 31, 1630–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).López-Otín C, and Bond JS (2008) Proteases: multifunctional enzymes in life and disease. J. Biol. Chem 283, 30433–30437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Deu E, Verdoes M, and Bogyo M (2012) New approaches for dissecting protease functions to improve probe development and drug discovery. Nat. Struct. Mol. Biol 19, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Docherty AJ, Crabbe T, O’Connell JP, and Groom CR (2003) Proteases as drug targets. Biochem. Soc. Symp 70, 147–161. [DOI] [PubMed] [Google Scholar]
- (5).Prassas I, Eissa A, Poda G, and Diamandis EP (2015) Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nat. Rev. Drug Discovery 14, 183–202. [DOI] [PubMed] [Google Scholar]
- (6).Singh RB, Dandekar SP, Elimban V, Gupta SK, and Dhalla NS (2004) Role of proteases in the pathophysiology of cardiac disease. Mol. Cell. Biochem 263, 241–256. [DOI] [PubMed] [Google Scholar]
- (7).López-Otín C, and Matrisian LM (2007) Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808. [DOI] [PubMed] [Google Scholar]
- (8).Ji RR, Xu ZZ, Wang X, and Lo EH (2009) Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol. Sci 30, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).De Strooper B (2010) Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev 90, 465–494. [DOI] [PubMed] [Google Scholar]
- (10).Troeberg L, and Nagase H (2012) Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta, Proteins Proteomics 1824, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Greenbaum DC, Baruch A, Grainger M, Bozdech Z, Medzihradszky KF, Engel J, DeRisi J, Holder AA, and Bogyo M (2002) A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science (Washington, DC, U. S.) 298, 2002–2006. [DOI] [PubMed] [Google Scholar]
- (12).Lorenz IC, Marcotrigiano J, Dentzer TG, and Rice CM (2006) Structure of the catalytic domain of the hepatitis C virus NS2–3 protease. Nature 442, 831–835. [DOI] [PubMed] [Google Scholar]
- (13).Wensing AM, van Maarseveen NM, and Nijhuis M (2010) Fifteen years of HIV Protease Inhibitors: raising the barrier to resistance. Antiviral Res. 85, 59–74. [DOI] [PubMed] [Google Scholar]
- (14).Böttcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, and Matrosovich M (2006) Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol 80, 9896–9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Drag M, and Salvesen GS (2010) Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discovery 9, 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Puente XS, Sánchez LM, Overall CM, and López-Otín C (2003) Human and mouse proteases: a comparative genomic approach. Nat. Rev. Genet 4, 544–558. [DOI] [PubMed] [Google Scholar]
- (17).Tallant C, Marrero A, and Gomis-Rüth FX (2010) Matrix metalloproteinases: fold and function of their catalytic domains. Biochim. Biophys. Acta, Mol. Cell Res 1803, 20–28. [DOI] [PubMed] [Google Scholar]
- (18).Overall CM, and Kleifeld O (2006) Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 94, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kessenbrock K, Plaks V, and Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zucker S, and Cao J (2009) Selective matrix metalloproteinase (MMP) inhibitors in cancer therapy: ready for prime time? Cancer Biol. Ther 8, 2371–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wu Y, Eigenbrot C, Liang WC, Stawicki S, Shia S, Fan B, Ganesan R, Lipari MT, and Kirchhofer D (2007) Structural insight into distinct mechanisms of protease inhibition by antibodies. Proc. Natl. Acad. Sci. U. S. A 104, 19784–19789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Farady CJ, Egea PF, Schneider EL, Darragh MR, and Craik CS (2008) Structure of an Fab-protease complex reveals a highly specific non-canonical mechanism of inhibition. J. Mol. Biol 380, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Devy L, Huang L, Naa L, Yanamandra N, Pieters H, Frans N, Chang E, Tao Q, Vanhove M, Lejeune A, van Gool R, Sexton DJ, Kuang G, Rank D, Hogan S, Pazmany C, Ma YL, Schoonbroodt S, Nixon AE, Ladner RC, Hoet R, Henderikx P, Tenhoor C, Rabbani SA, Valentino ML, Wood CR, and Dransfield DT (2009) Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 69, 1517–1526. [DOI] [PubMed] [Google Scholar]
- (24).Sela-Passwell N, Kikkeri R, Dym O, Rozenberg H, Margalit R, Arad-Yellin R, Eisenstein M, Brenner O, Shoham T, Danon T, Shanzer A, and Sagi I (2012) Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med 18, 143–147. [DOI] [PubMed] [Google Scholar]
- (25).Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, Heise CE, Hoyte K, Luk W, Lu Y, Peng K, Wu P, Rouge L, Zhang Y, Lazarus RA, Scearce-Levie K, Wang W, Wu Y, Tessier-Lavigne M, and Watts RJ (2011) A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci. Transl. Med 3, 84ra43. [DOI] [PubMed] [Google Scholar]
- (26).Naito S, Takahashi T, Onoda J, Yamauchi A, Kawai T, Kishino J, Yamane S, Fujii I, Fukui N, and Numata Y (2012) Development of a neutralizing antibody specific for the active form of matrix metalloproteinase-13. Biochemistry 51, 8877–8884. [DOI] [PubMed] [Google Scholar]
- (27).Schneider EL, Lee MS, Baharuddin A, Goetz DH, Farady CJ, Ward M, Wang CI, and Craik CS (2012) A reverse binding motif that contributes to specific protease inhibition by antibodies. J. Mol. Biol 415, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kenniston JA, Faucette RR, Martik D, Comeau SR, Lindberg AP, Kopacz KJ, Conley GP, Chen J, Viswanathan M, Kastrapeli N, Cosic J, Mason S, DiLeo M, Abendroth J, Kuzmic P, Ladner RC, Edwards TE, TenHoor C, Adelman BA, Nixon AE, and Sexton DJ (2014) Inhibition of plasma kallikrein by a highly specific active site blocking antibody. J. Biol. Chem 289, 23596–23608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Appleby TC, Greenstein AE, Hung M, Liclican A, Velasquez M, Villaseñor AG, Wang R, Wong MH, Liu X, Papalia GA, Schultz BE, Sakowicz R, Smith V, and Kwon HJ (2017) Biochemical characterization and structure determination of a potent, selective antibody inhibitor of human MMP9. J. Biol. Chem 292, 6810–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).David T, Kim YC, Ely LK, Rondon I, Gao H, O’Brien P, Bolt MW, Coyle AJ, Garcia JL, Flounders EA, Mikita T, and Coughlin SR (2016) Factor XIa-specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Sci. Transl. Med 8, 353ra112. [DOI] [PubMed] [Google Scholar]
- (31).Ling B, Watt K, Banerjee S, Newsted D, Truesdell P, Adams J, Sidhu SS, and Craig AWB (2017) A novel immunotherapy targeting MMP-14 limits hypoxia, immune suppression and metastasis in triple-negative breast cancer models. Oncotarget 8, 58372–58385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Razai AS, Eckelman BP, and Salvesen GS (2020) Selective inhibition of matrix metalloproteinase 10 (MMP10) with a single-domain antibody. J. Biol. Chem 295, 2464–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nam DH, Rodriguez C, Remacle AG, Strongin AY, and Ge X (2016) Active-site MMP-selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc. Natl. Acad. Sci. U. S. A 113, 14970–14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lopez T, Mustafa Z, Chen C, Lee KB, Ramirez A, Benitez C, Luo X, Ji RR, and Ge X (2019) Functional selection of protease inhibitory antibodies. Proc. Natl. Acad. Sci. U. S. A 116, 16314–16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Farady CJ, and Craik CS (2010) Mechanisms of macromolecular protease inhibitors. ChemBioChem 11, 2341–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Fernandez-Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H, and Maskos K (1998) Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. Embo j 17, 5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lauwereys M, Arbabi Ghahroudi M, Desmyter A, Kinne J, Hölzer W, De Genst E, Wyns L, and Muyldermans S (1998) Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. Embo j 17, 3512–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Desmyter A, Transue TR, Ghahroudi MA, Dao Thi MH, Poortmans F, Hamers R, Muyldermans S, and Wyns L (1996) Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Mol. Biol 3, 803–811. [DOI] [PubMed] [Google Scholar]
- (39).De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, Muyldermans S, and Wyns L (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U. S. A 103, 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Remacle AG, Cieplak P, Nam DH, Shiryaev SA, Ge X, and Strongin AY (2017) Selective function-blocking monoclonal human antibody highlights the important role of membrane type-1 matrix metalloproteinase (MT1-MMP) in metastasis. Oncotarget 8, 2781–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen KE, Chen C, Lopez T, Radecki KC, Bustamante K, Lorenson MY, Ge X, and Walker AM (2018) Use of a novel camelid-inspired human antibody demonstrates the importance of MMP-14 to cancer stem cell function in the metastatic process. Oncotarget 9, 29431–29444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ji RR, Xu ZZ, Wang X, and Lo EH (2009) Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol. Sci 30, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Li X, Zhao Y, Chen C, Yang L, Lee HH, Wang Z, Zhang N, Kolonin MG, An Z, Ge X, Scherer PE, and Sun K (2020) Critical Role of Matrix Metalloproteinase 14 in Adipose Tissue Remodeling during Obesity. Mol. Cell. Biol 40, No. e00564–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lee KB, Dunn Z, and Ge X (2019) Reducing proteolytic liability of a MMP-14 inhibitory antibody by site-saturation mutagenesis. Protein Sci. 28, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lopez T, Ramirez A, Benitez C, Mustafa Z, Pham H, Sanchez R, and Ge X (2018) Selectivity Conversion of Protease Inhibitory Antibodies. Antibody Ther. 1, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lopez T, Chuan C, Ramirez A, Chen KE, Lorenson MY, Benitez C, Mustafa Z, Pham H, Sanchez R, Walker AM, and Ge X (2018) Epitope-specific affinity maturation improved stability of potent protease inhibitory antibodies. Biotechnol. Bioeng 115, 2673–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hayhurst A, Happe S, Mabry R, Koch Z, Iverson BL, and Georgiou G (2003) Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. J. Immunol. Methods 276, 185–196. [DOI] [PubMed] [Google Scholar]
- (48).Nam DH, and Ge X (2016) Direct production of functional matrix metalloproteinase–14 without refolding or activation and its application for in vitro inhibition assays. Biotechnol. Bioeng 113, 717–723. [DOI] [PubMed] [Google Scholar]
- (49).Nam DH, Lee KB, and Ge X (2018) Functional Production of Catalytic Domains of Human MMPs in Escherichia coli Periplasm. Methods Mol. Biol. (N. Y., NY, U. S.) 1731, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Nam DH, and Ge X (2018) Generation of Highly Selective MMP Antibody Inhibitors. Methods Mol. Biol. (N. Y., NY, U. S.) 1731, 307–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lee KB, Nam DH, Nuhn JAM, Wang J, Schneider IC, and Ge X (2017) Direct expression of active human tissue inhibitors of metalloproteinases by periplasmic secretion in Escherichia coli. Microb. Cell Fact 16, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lopez T, Nam DH, Kaihara E, Mustafa Z, and Ge X (2017) Identification of highly selective MMP-14 inhibitory Fabs by deep sequencing. Biotechnol. Bioeng 114, 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Muruganandam A, Tenhoor C, and Devy L (2009) Methods and compositions comprising anti-idiotypic antibodies to anti-mmp-14 antibodies. WO 2009132251 A2. [Google Scholar]
- (54).Cunningham BC, and Wells JA (1993) Comparison of a structural and a functional epitope. J. Mol. Biol 234, 554–563. [DOI] [PubMed] [Google Scholar]
- (55).Eckhard U, Huesgen PF, Schilling O, Bellac CL, Butler GS, Cox JH, Dufour A, Goebeler V, Kappelhoff R, Keller UAD, Klein T, Lange PF, Marino G, Morrison CJ, Prudova A, Rodriguez D, Starr AE, Wang Y, and Overall CM (2016) Active site specificity profiling of the matrix metalloproteinase family: Proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol. 49, 37–60. [DOI] [PubMed] [Google Scholar]
- (56).Ganesan R, Eigenbrot C, and Kirchhofer D (2010) Structural and mechanistic insight into how antibodies inhibit serine proteases. Biochem. J 430, 179–189. [DOI] [PubMed] [Google Scholar]
- (57).Wang W, Liu Y, and Lazarus RA (2013) Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr. Opin. Struct. Biol 23, 797–805. [DOI] [PubMed] [Google Scholar]
- (58).Udi Y, Grossman M, Solomonov I, Dym O, Rozenberg H, Moreno V, Cuniasse P, Dive V, Arroyo AG, and Sagi I (2015) Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure (Oxford, U. K.) 23, 104–115. [DOI] [PubMed] [Google Scholar]
- (59).Zhou L, Chávez-Gutierréz L, Bockstael K, Sannerud R, Annaert W, May PC, Karran E, and De Strooper B (2011) Inhibition of beta-secretase in vivo via antibody binding to unique loops (D and F) of BACE1. J. Biol. Chem 286, 8677–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Petersen HH, Hansen M, Schousboe SL, and Andreasen PA (2001) Localization of epitopes for monoclonal antibodies to urokinase-type plasminogen activator: relationship between epitope localization and effects of antibodies on molecular interactions of the enzyme. Eur. J. Biochem 268, 4430–4439. [DOI] [PubMed] [Google Scholar]
- (61).Ely LK, Lolicato M, David T, Lowe K, Kim YC, Samuel D, Bessette P, Garcia JL, Mikita T, Minor DL Jr., and Coughlin SR (2018) Structural Basis for Activity and Specificity of an Anticoagulant Anti-FXIa Monoclonal Antibody and a Reversal Agent. Structure (Oxford, U. K.) 26, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zakharova E, Horvath MP, and Goldenberg DP (2009) Structure of a serine protease poised to resynthesize a peptide bond. Proc. Natl. Acad. Sci. U. S. A 106, 11034–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kromann-Hansen T, Oldenburg E, Yung KW, Ghassabeh GH, Muyldermans S, Declerck PJ, Huang M, Andreasen PA, and Ngo JC (2016) A Camelid-derived Antibody Fragment Targeting the Active Site of a Serine Protease Balances between Inhibitor and Substrate Behavior. J. Biol. Chem 291, 15156–15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gomis-Rüth FX (2009) Catalytic domain architecture of metzincin metalloproteases. J. Biol. Chem 284, 15353–15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nagase H (2001) Substrate Specificity of MMPs In Matrix Metalloproteinase Inhibitors in Cancer Therapy (Clendeninn NJ, and Appelt K, Eds.) pp 39–66, Humana Press, Totowa, NJ. [Google Scholar]
- (66).Roy R, Yang J, and Moses MA (2009) Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J. Clin. Oncol 27, 5287–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Fingleton B (2007) Matrix metalloproteinases as valid clinical targets. Curr. Pharm. Des 13, 333–346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.