

Abstract
Predicting the poses of small-molecule ligands in protein binding sites is often done by virtual screening algorithms like DOCK. In principle, Molecular Dynamics (MD) using atomistic force fields could give better free-energy-based pose selection, but MD is computationally expensive. Here, we ask if MELD-accelerated MD (MELD × MD) can pick out the best DOCK poses taken as input. We study 30 different ligand-protein pairs. MELD × MD finds native poses, based on best free energies, in 23 out of the 30 cases, twenty of which were previously known DOCK failures. We conclude that MELD × MD can add value for predicting accurate poses of small-molecules bound to proteins.
Introduction
To assist in drug discovery, it can be useful to compute the native poses of small-molecule ligands bound to proteins.1,2 A widely used algorithm for this is DOCK3 and variants of it.4–7 Docking methods have become workhorses of virtual screening because they are fast to compute.2,8,9 But speed comes at the cost of accuracy: (i) DOCK scoring functions use simplified physics, usually the non-bonded terms of molecular mechanics force field3 (ii) DOCK treats proteins as rigid, yet flexibility is important.10,11 Various methods incorporate receptor flexibility,12,13 but more improvement is needed.14
Alternatively, ligand poses in protein sites can be found by Molecular Dynamics (MD) simulations using atomistic force fields.15,16 Such atomistic modeling can capture more of the physics, treat water explicitly, and sample the degrees of freedom more extensively. An important virtue of MD methods is that they give relative populations, and thus free energies, of different states, for example of binding. Using conventional MD even on special-purpose computers17,18 is too computationally demanding to sample the vast conformational spaces of binding/unbinding and obtaining good statistics. Enhanced sampling approaches have been developed to accelerate small-molecule binding, either using general ensemble approaches like Replica Exchange (REMD)19 or using collective variables with biasing potentials.20–25 For a recent review, see Limongelli et al.26
Here, we apply an accelerator of MD, called MELD (Modeling Employing Limited Data), to improve upon DOCK models of ligands bound to proteins.27,28 MELD × MD accelerates MD simulations when some target goal can be specified, even vaguely or incompletely. Using MELD × MD, we ask: (1) Can we rescue DOCK scoring failures (i.e incorrect rankings of dock poses) using a current atomistic force field and explicit solvent? (2) Can we speed up the atomistic MD modeling using the MELD accelerator, guided by contacts (extracted from DOCK poses) to give the MD method some starting points for sampling? We’re asking here if MELD × MD is su ciently fast and accurate, in the limited tests here, to improve upon DOCK poses using free energy criteria. We tested 30 different ligand-protein complexes from the DOCK SB2012 dataset,29 including some cases where DOCK is already successful. For each complex, we take the 30 best poses according to the DOCK scoring function, extract binding pose information as input, model ligand binding from completely unbounded state with MELD × MD in the TIP3P30 explicit-solvent model. We compute the populations of sampled binding poses, to see if the atomistic modeling correctly identifies the native poses. We find that MELD × MD commonly (but not always) rescues the scoring failures of DOCK, and that it is much faster than brute-force MD.
Methods
MELD is an accelerator of MD when some guiding directives are available
MELD (Modeling Employing Limited Data) is a computational method that accelerates MD simulations by ‘melding’ in external directive information of some kind.27,28,31 MELD is useful in many different situations: (i) in determining protein structures from experimental data that is limited in some way or noisy,32 (ii) in computing protein 3D native structures from amino acid sequences when given vague directives (such as: “make a hydrophobic core” or “make good secondary structures”),33 (iii) in computing protein-peptide34 or protein-protein35 binding structures, (iv) in computing relative binding affinity between two peptide binders to a protein.36 The power of MELD is its ability to use vague or combinatoric information or to choose options among choices; it does this through a Bayesian ‘sub-haystacking’ procedure.
Here is how we employ the MELD accelerator in the present work. DOCK serves as a front-end to provide sets of contacts in the form of ligand binding poses. Those poses are identified as the top 30 ones based on DOCK6 socring function. We accept that DOCK may not correctly rank order those poses. We do require, however, that the true native pose be within the vicinity of DOCK best-scored N poses that is our input. Then, starting from the ligand unbound state, MELD × MD samples the ligand in the vicinity of subsets of the N poses, and seeks the best pose based on the atomistic potential function. The ligand unbound state is prepared by only a translational move from the crystal pose along the vector defined by the protein and the ligand center-of-mass, until it is 25Å away from protein. The original orientation is rapidly lost as the ligand diffuses through the water box during binding simulations.
Chosing appropriate ligand-protein systems
Among the 1024 ligand-protein complexes in the SB2012 dataset,29 we applied the following criteria for selecting ligand-protein pairs to test: A) The receptor must be smaller than 250 amino acids, to be within our current computational capabilities. B) It contains no co-factors or ions (to avoid possible force field deficiencies). Filtered in this way, the dataset contains 371 complexes. C) To further eliminate complexes that are incompatible with the physics model, we performed a stability test (see Simulation Protocols) starting from the native pose. We further eliminated those protein-ligand pairs having an average LRMSD value larger than 3Å. The final dataset contains 268 ligand-protein complexes. From those, 20 DOCK failures and 10 DOCK successes were selected as our MELD × MD test set.
Simulation Protocols
For all our simulations here, we used the AMBER ff14SB force field37 to describe the protein, and the GAFF force field38 for the ligand molecule. The atomic partial charges of the ligand are assigned using the AM1-BCC method.39 We used a TIP3P30 equilibrated water box with 10Å buffer size for each system. Equilibration of the solvated complex is following this protocol.40 We did MD production runs to test stabilities, using the AMBER16 software41 with Cartesian restraints using a force constant of 100.0kJ mol−1nm−2 applied to the protein heavy atoms. MELD × MD simulations were run using MELD, a plugin for the OpenMM MD software.42 Two types of information are input into MELD as restraints. (1) The protein receptor was not allowed to completely unfold during the simulations, but was allowed to rearrange upon ligand binding. To this end backbone atoms are restrained using flat-bottomed Cartesian restraints. No force is applied to backbone atoms inside a 3.5Å radius around their initial crystal position, and a harmonic potential with a 250.0kJ mol−1nm−2 force constant is applied to atoms that move outside this radius. This force constant gives ~ kBT of energy when the atom deviates 1.0Å from the flat-bottom region. The strength of these restraints is kept constant across the replica ladder. (2) We use contacts extracted by DOCK prediction to drive the docking of the ligand onto the protein (see SI for details). DOCK poses are clustered according to the protocol described in SI. MELD restraints driving the system to different pose clusters are slowly turned on while the system descends the replica ladder using a MELD nonlinear scaler (α=1, force constant=0.0kJ mol−1nm−2, α=0, force constant=100.0kJ mol−1nm−2).
To accelerate the conformational sampling in the explicit solvent, we use Replica Exchange with Solute Tempering (REST2).43 The effective temperature of each replica, which correlates with the strength of the non-solvent nonbonded and torsion interactions, varies geometrically from 300K at α=0.0 to 500K at α=1.0.27 To further accelerate ligand transi-tioning among different poses, step-wise pushing restraints are turned on as the ligand moves up the replica ladder (see SI). The pushing restraints coupled with temperature scaling accelerate the ligand unbinding as the system moves up the replica ladder. The H, T-REMD simulations are run using 30 replicas. Exchanges of configurations between replicas are attempted every 50ps. The total simulation time of a single replica is at least 500ns for each system. Initially parallel-simulations are evenly spaced along the replica ladder. Their position in replica index space are being optimized by MELD on-the-fly to maximize the exchange probability. The initial conformation of all the 30 replicas is the ligand-unbound state.
Criteria to evaluate MELD × MD success or failure
In rigid docking studies, the quality of a docking prediction compared to the reference crystal pose can be effectively described using ligand root-mean-square deviation (LRMSD). LRMSD measures the deviation of ligand heavy atoms position between two poses after aligning the protein main chains. In most docking studies the protein main chain is rigid. In these cases, LRMSD values reflect only discrepancies in ligand positions. When protein structures are flexible, like in our MELD × MD approach, LRMSD reflects both discrepancies in the position of the ligand and uncertainties in the alignment of the protein structures. This has two implications: (i) we need to use a different LRMSD cut-off as a measure of success between rigid and flexible docking studies, and (ii) we need to identify other measures, less affected by protein conformations, to help describing the quality of predicted pose in flexible systems.
To identify a LRMSD cut-off value that accounts for protein flexibility, we need an estimation of the contribution of receptor flexibility to the standard LRMSD measurement. Thus, we performed MD simulations in AMBER, in which we applied Cartesian restraints to the positions of heavy atoms of the protein residues within 5 Å of any ligand heavy atoms and to all ligand heavy atoms. The restraint strength was 100 kcal mol−1 Å−2. After minimization and equilibration, each system was run for 100 ns. The LRMSD of each frame in the trajectory was computed by aligning receptor Cα atoms against the native crystal structure. The value corresponds to two standard deviation of the LRMSD distribution was the receptor flexibility contribution to the standard LRMSD measurement. Finally, this value (1Å, see Fig. S1) plus the standard cut-off value used by DOCK community (2Å) was the cut-off to evaluate MELD × MD prediction.
To describe the quality of the structures from MELD × MD predictions, we also evaluate the fraction of native contacts between the ligand and the receptor. Contacts are computed following the protocol described in SI. As a rule of thumb, we consider our prediction to be successful when it captures 60% of the native contacts. In general this means that we identify important contacts between the ligand and the protein (e.g. hydrogen bond or π stacking interactions), these are important contacts that are typically maintained in the lead optimization stages of drug discovery. Nonetheless, not all contacts are created equal. For example identifying an hydrogen bond between specific ligand and protein atoms is arguably more informative than a “promiscuous” hydrophobic contacts. Therefore, while satisfying the 60% cut-off is good enough to denote success, there are cases when these criteria are not met that warrant special consideration.
Results and discussion
Figure 1 compares the MELD × MD-refined ligand conformations to the experimental native structures.
Figure 1: Docking by MELD × MD successfully predicts bound structures in 23/30 cases.
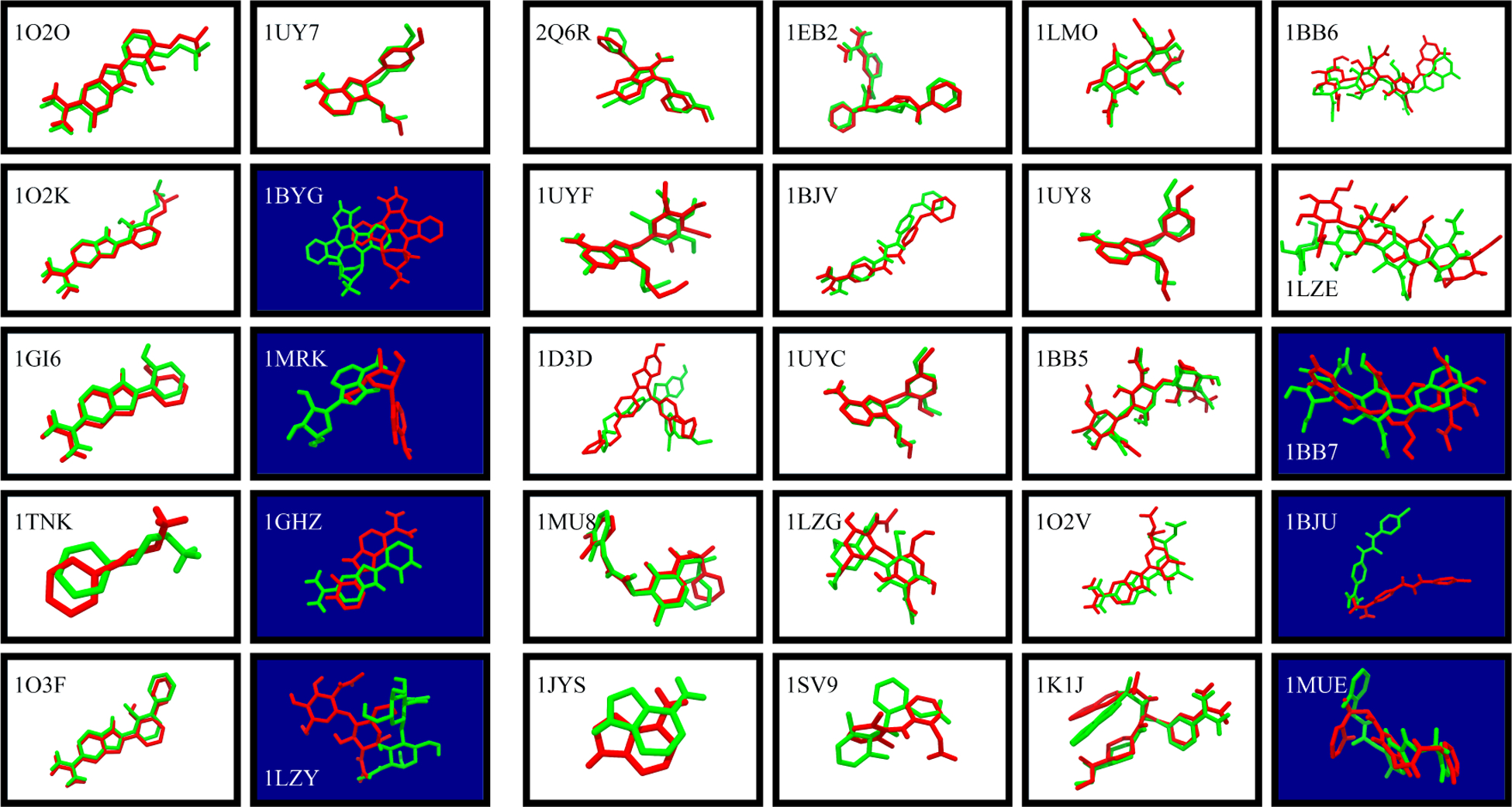
White panels show successful TOP1 predictions. Dark blue ones are not successes at the TOP1 level. (Green) Native ligand pose, superimposed on receptor Cα atom. (Red) MELD × MD predicted ligand poses. (Left 10 panels): DOCK by itself produces a native-like pose and rank it as TOP1, even without MELD × MD. (Right 20 panels): DOCK by itself fails to rank a native-like pose as TOP1.
Here is a summary of our results: (i) Reference tests. The left two columns show that MELD × MD succeeds in 6 of the 10 cases where DOCK itself already successfully predicted a native-like pose and ranked it first. This was just a reference check that MD could give good predictions when the DOCK input was already good. (ii) Rescuing DOCK scoring failures. The right four columns show that out of the 20 DOCK failures, MELD rescues 17 of them. This test was the main point of the present work.
One virtue of MELD × MD is the ability to drive the ligand to different poses from completely unbound state and unbiasedly sample them based on free energy. By enforcing “fuzzy” intermolecular ligand-protein contacts compatible with multiple microstates, MELD can identify and refine the best DOCK pose, and sometimes go beyond input DOCK poses and find novel ones (Fig. S3). Meantime, leveraging the sampling power of MELD × MD and flat-bottom MELD distance restraints, those poses can be sampled unbiasedly and more extensively, which leads to more accurate evaluation of the relative free energy of different poses, results to the better ranking of native poses (Fig. S5) and more successes at the TOP1 level (Fig. S4).
Assessing MELD × MD prediction failures and successes
We discuss the quality of our TOP1 predictions in terms of LRMSD and fraction of native contact of our predictions against the reference crystal structure. According to the cut-off presented in section “Criteria to evaluate MELD × MD success or failure”, we partition MELD × MD predictions into the four sectors highlighted in the graph of Figure 2. The green top-left region identifies accurate predictions with low value of LRMSD and high fraction of true contacts. The bottom-right red region identifies failed predictions with high values of LRMSD and low fraction of true contacts. Predictions falling in the two other areas need an accurate system-by-system discussion. The bottom-left blue region represents predictions with a relatively low fraction of true contacts but with a low value of LRMSD. In the 1TNK system the ligand establishes only 2 contacts with the protein -according to our definition of contacts. Assessing the quality of this prediction according to the fraction of native contacts is therefore not particularly meaningful. Considering the low value of LRMSD of the MELD TOP1 prediction (2.0Å), we define this prediction a success. In the 1D3D system, significant sidechain rearrangements prevent key contacts to be formed. Rearrangement of the TRP220 sidechain prevents the benzothiophene group of the ligand to sit in its crystal position and therefore to form hydrogen bonds between the hydroxyl oxygen and protein residue ASP192 and ALA193 (Fig. S6). Similarly the rearrangement of the TRP50 sidechain hinders the tertiary amine of the ligand from forming pi-cation interactions with the indole sidechain of tryptophan. A well-known property of the DBSCAN for discovering arbitrary shaped cluster44 means that any cluster member is in principle as representative as the centroid. Our choice of centroid was just meant to be systematic, but in no way the best. We observed that there are better structures in the MELD TOP1 ensemble (an example is shown in Fig. S7). Moreover, one advantage of a physics based method like MELD × MD is that there is a whole ensemble of structures associated with out TOP1 prediction -i.e. all the structures that belong to the same cluster. Analyzing this ensemble all native contacts can be identified (Fig. S8). Considering the low value of LRMSD of our prediction and the existence of high quality structures in the TOP1 ensemble of structures that allow to identify all native contacts, we consider this prediction successful. Finally the top-right orange region captures predictions with high value of LRMSD, but that also satisfy a high fraction of of native contacts. Our 1LZE prediction captures 6 out of 8 native contacts, that include 4 out of 6 native hydrogen bonds. Figure S9 shows the captured contacts and the overall good binding pose. The LRMSD value of 3.6Å is due to the displacement of the flexible loop backbone region between residues ASP101 and ASN103 that “pushes” one side of ligand away from the crystal pose (Fig. S10). Further investigation shows that structures with significantly lower value of LRMSD can be found inside the TOP1 ensemble (Fig. S11). Considering that we capture most of the hydrogen bond interactions, and the presence of good structures in the ensemble, we can consider 1LZE a successful prediction. The other two systems in this orange region, 1MRK and 1BYG, are two failures due to the overall different binding modes compared to native (Fig. 1). For these two systems, our simplified contact representation lead to a significant fraction of false positive contacts.
Figure 2: Assessing MELD × MD predictions for all 30 complexes.
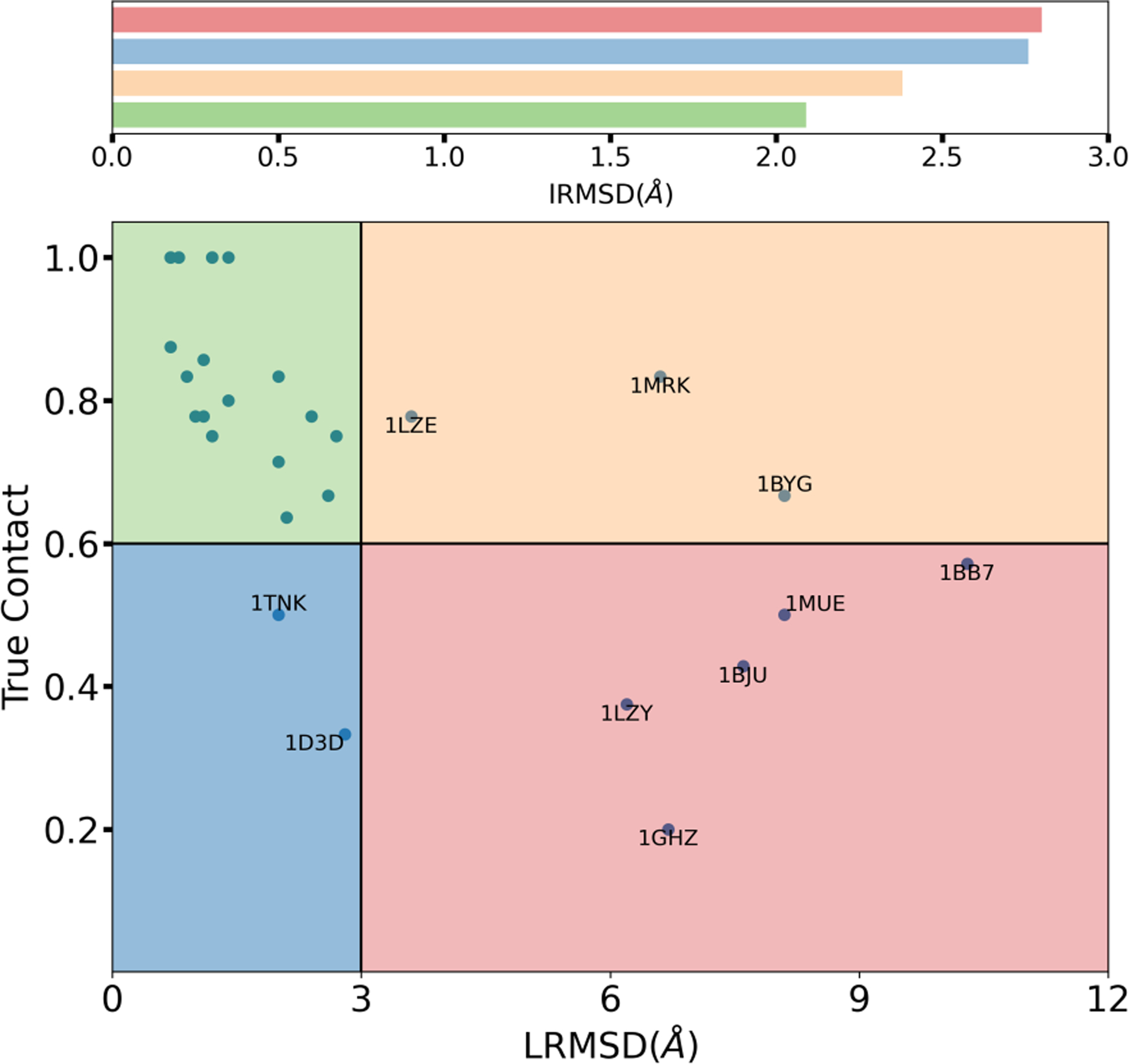
MELD × MD TOP1 predictions for all 30 complexes are partitioned into 4 different groups based the fraction of true contact and ligand root-mean-square distance(LRMSD). The average binding pocket flexibility across all complexes in each groups are accessed using interface root-mean-square distance (IRMSD) and the corresponding bar plot is shown on top.
MELD simulations converge relatively quickly to the native pose
When MELD simulation converged to the native pose, how long does it take and what is the indicator? Figure 3 shows results of clustering trajectory over different time intervals. The ratio of the most populated native one (P(c0)) and most populated non-native cluster’s population (P(c1) indicates the reliability of our prediction. When this ratio keeps above 1, it indicates that our MELD prediction converged to the native pose.
Figure 3:
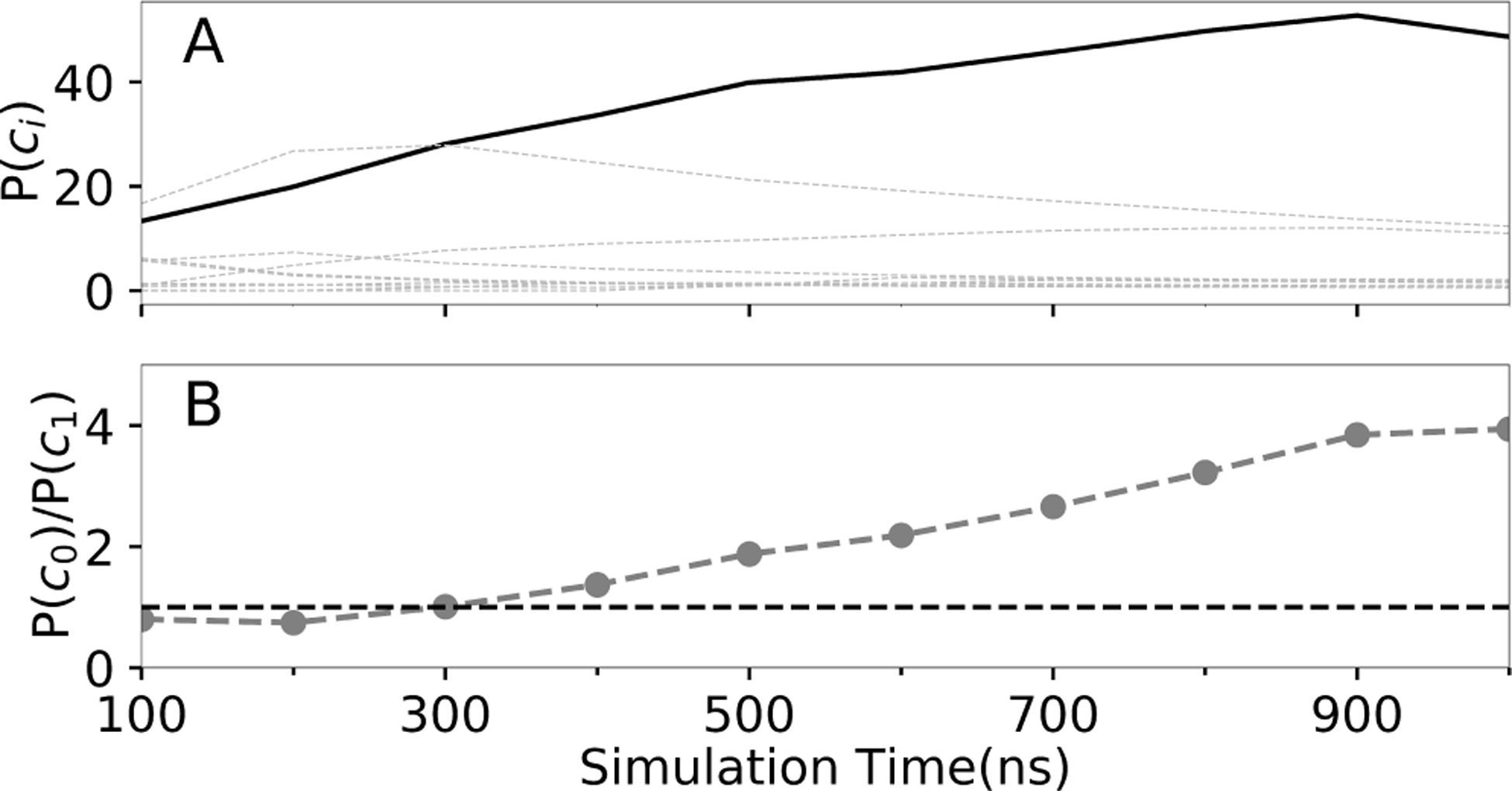
(A) Convergence of cluster populations for 1LMO. The bold line represents the native cluster, c0. (B) Emergence of the dominant cluster population for 1LMO, c0 relative to the second best, c1.
Figure 3 shows convergence to native, on average, after about 300ns for 1LMO. Similar results are shown below for all 23 MELD × MD successes. For most MELD × MD successes (18/23, ~80%), the MELD × MD simulation converged to native in ~300ns. Simulating a protein of less than 250 amino acids with a ligand in explicit TIP3P water can be done within a day on modern GPUs (Table. S2). So, it may be useful for practical structure-based drug design.
But, some simulations converged slowly. For 1 out of 5 slowly converging complexes, our clustering protocol fails to separate populations (Fig. S12). For the rest, we did not find any clustering issues. Slow convergence might due to the existence of multiple native binding modes or alternative non-native poses with similar binding free energies as the native ones in the simulated protein-ligand complexes.45
MELD × MD failures are from force field flaws or protocol limitations
Here, we looked at the sources of error when our modeling failed. In some cases, the active site has floppy loops. In those cases, the flexibility allowed by MELD × MD makes it harder for the method to find the binding mode. So, we experimented with reducing the flexibility by narrowing the flat-bottom region from 3.5Å to 1.8Å. That gave improved predictions for four systems (1GHZ, 1BJU, 1MUE, 1MRK, see Table. S3). It did not help, however, for 1BYG. 1BYG has a deep cavity, so reducing the flexibility makes it harder for the ligand to diffuse into the cavity (see Fig. S15). We regard both types as a sampling failure of the MELD protocol: too much sampling distorts the cavity and restricts binding; and too little sampling doesn’t sufficiently populate the native state. If we know such information about the cavity in advance, it may allow for improvements in our modeling. In the failure of 1BB7, the input contacts were not sufficiently directive to native (Fig. S16), probably causing slow convergence. For 1LZY, the native state is sampled, but not preferred; this is a forcefield error (Fig. S14).
MELD × MD adds values to rigid and flexible docking
To assess whether the more expensive explicit solvent MELD × MD simulations add value and how other docking software performs on the same testing systems, we did some rigid/flexible docking simulations using AutoDockFR.46 We used default “vanilla” parameters as outlined in the SI. Figure. 5 shows the results. All three of the docking algorithms gave about the similar results, and MELD × MD free energies add considerable value in picking out the single most native-like ligand pose. The simulation cost of each method is given in Table. S2.
Figure 5: Comparing predictions among 4 different methods.
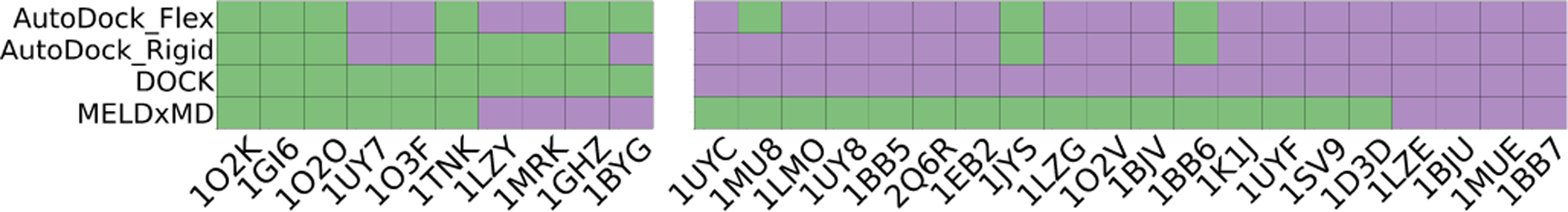
Predicted TOP1 poses for all 30 complexes using AutoDock rigid(AutoDock_Rigid) and flexible receptor (AutoDock_Flex), DOCK and MELD×MD are compared. The success and failure are colored as green and purple respectively. The left-most 10 cases are DOCK successes while the right-most 20 ones are DOCK failures.
Conclusions
We addressed two questions here. (1) Can DOCK scoring failures be rescued by Molecular Dynamics simulations, which presumably have two advantages that: (i) they represent the atomic interaction physics and solvation more accurately, and (ii) they sample the protein degrees of freedom more fully. And (2) Can MELD give sufficient acceleration to MD, when fed DOCK poses as input, to be a practical tool for finding ligand poses bound to proteins? MELD × MD was largely successful on both counts. We tested 30 protein-ligand complexes. Our protocol predicts the correct native ligand binding pose for 23/30 complexes and rescues 17/20 DOCK scoring failures. In general, where extra computational expense is warranted, MELD × MD can add value to DOCKing.
Supplementary Material
Figure 4: ROC curve of simulation time needed to converge to native.
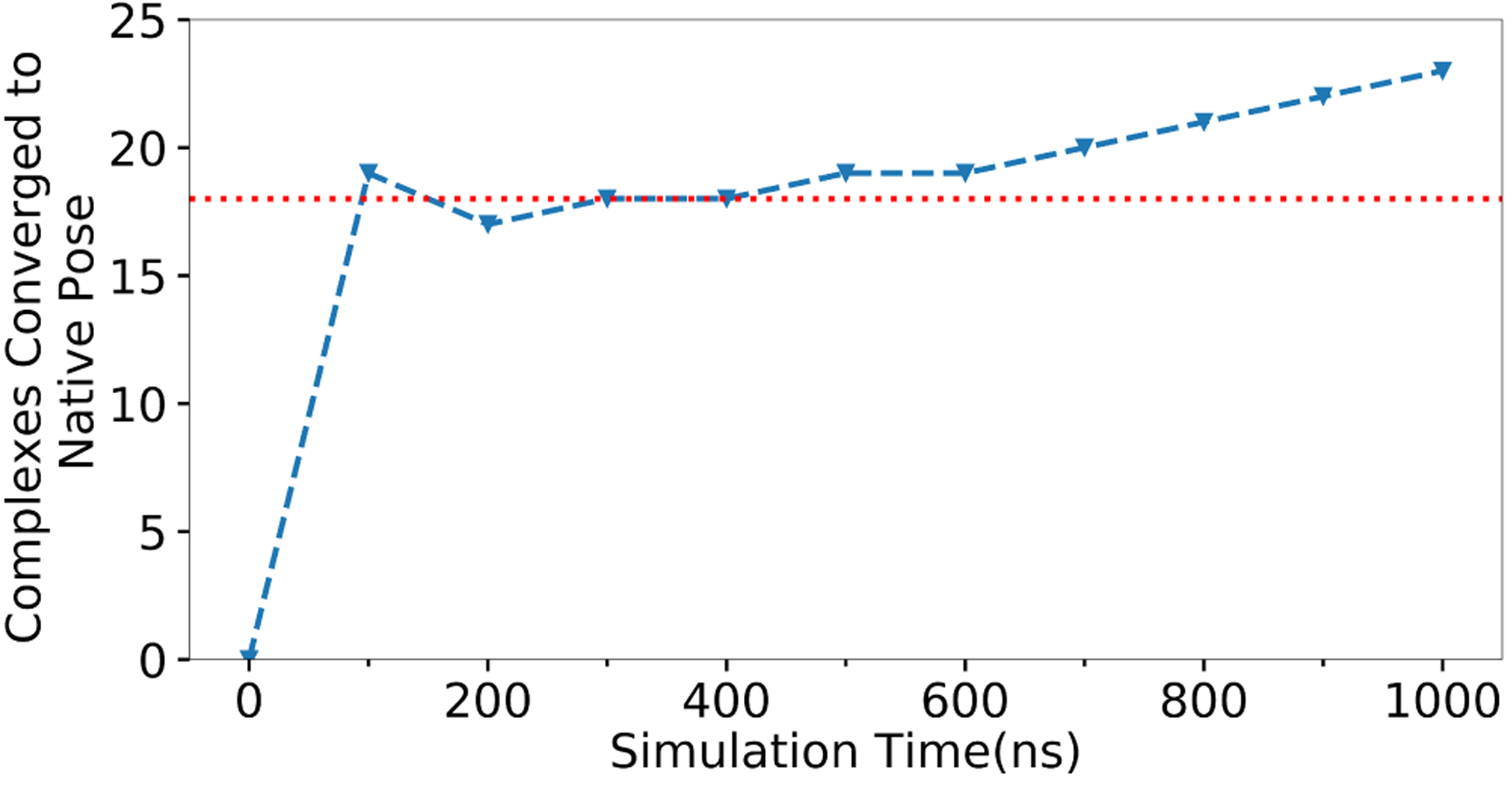
The percentage of MELD×MD successes that converged to native poses after simulation times given on the x-axis.
Acknowledgement
The author thanks the support of the Blue Waters sustained-petascale computing project and NSF PRAC award ACI1514873. Blue Waters is supported by the National Science Foundation (Awards OCI-0725070 and ACI-1238993) and the State of Illinois. We also appreciate funding from NIH Grant GM125813, the Laufer Center and University of Florida start-up resources. The author also thanks Dr. Jiaye Guo for providing the SB2012 dataset, Dr. Robert C. Rizzo and Lauren Prentis for helpful discussions.
Footnotes
Supporting Information Available
Detailed information about the following: how we cluster DOCK ensemble to extract contacts, how we set up unbinding restraints in MELD simulation, how we quantify the effect of receptor flexibility on the ligand root-mean-square distance, the protocol to compute contacts using protein-ligand interaction profiler (PLIP), the protocol to run AutoDockFR simulations, the convergence of the simulation and the computational cost of different methods reported in the paper.
References
- (1).Kuntz ID Structure-based strategies for drug design and discovery. Science 1992, 257, 1078–1082. [DOI] [PubMed] [Google Scholar]
- (2).Jorgensen WL The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [DOI] [PubMed] [Google Scholar]
- (3).Allen WJ; Balius TE; Mukherjee S; Brozell SR; Moustakas DT; Lang PT; Case DA; Kuntz ID; Rizzo RC DOCK 6: Impact of new features and current docking performance. J. Comput. Chem 2015, 36, 1132–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- (5).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (6).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pagadala NS; Syed K; Tuszynski J Software for molecular docking: a review. Biophys. J 2017, 9, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kitchen DB; Decornez H; Furr JR; Bajorath J Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discovery 2004, 3, 935. [DOI] [PubMed] [Google Scholar]
- (9).Shoichet BK Virtual screening of chemical libraries. Nature 2004, 432, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mobley DL; Graves AP; Chodera JD; McReynolds AC; Shoichet BK; Dill KA Predicting absolute ligand binding free energies to a simple model site. J. Mol. Biol 2007, 371, 1118–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Totrov M; Abagyan R Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr. Opin. Struct. Biol 2008, 18, 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Leach AR Ligand docking to proteins with discrete side-chain flexibility. J. Mol. Biol 1994, 235, 345–356. [DOI] [PubMed] [Google Scholar]
- (13).Korb O; Olsson TS; Bowden SJ; Hall RJ; Verdonk ML; Liebeschuetz JW; Cole JC Potential and limitations of ensemble docking. J. Chem. Inf. Model 2012, 52, 1262–1274. [DOI] [PubMed] [Google Scholar]
- (14).Guedes IA; de Magalhães CS; Dardenne LE Receptor–ligand molecular docking. Biophys. Rev 2014, 6, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shan Y; Kim ET; Eastwood MP; Dror RO; Seeliger MA; Shaw DE How does a drug molecule find its target binding site? J. Am. Chem. Soc 2011, 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dror RO; Pan AC; Arlow DH; Borhani DW; Maragakis P; Shan Y; Xu H; Shaw DE Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bokoch MP; Jo H; Valcourt JR; Srinivasan Y; Pan AC; Capponi S; Grabe M; Dror RO; Shaw DE; DeGrado WF, et al. Entry from the lipid bilayer: a possible pathway for inhibition of a peptide G protein-coupled receptor by a lipophilic small molecule. Biochemistry 2018, 57, 5748–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Song X; Jensen MØ; Jogini V; Stein RA; Lee C-H; Mchaourab HS; Shaw DE; Gouaux E Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature 2018, 556, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang K; Chodera JD; Yang Y; Shirts MR Identifying ligand binding sites and poses using GPU-accelerated Hamiltonian replica exchange molecular dynamics. J. Comput.-Aided Mol. Des 2013, 27, 989–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Torrie GM; Valleau JP Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys 1977, 23, 187–199. [Google Scholar]
- (21).Grubmüller H; Heymann B; Tavan P Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science 1996, 271, 997–999. [DOI] [PubMed] [Google Scholar]
- (22).Izrailev S; Stepaniants S; Balsera M; Oono Y; Schulten K Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J 1997, 72, 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Laio A; Parrinello M Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hamelberg D; Mongan J; McCammon JA Accelerated molecular dynamics: a promising and effcient simulation method for biomolecules. J. Chem. Phys 2004, 120, 11919–11929. [DOI] [PubMed] [Google Scholar]
- (25).Limongelli V; Bonomi M; Parrinello M Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 6358–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Limongelli V Ligand binding free energy and kinetics calculation in 2020. Wiley Interdiscip. Rev. Comput. Mol. Sci 2020, e1455. [Google Scholar]
- (27).MacCallum JL; Perez A; Dill KA Determining protein structures by combining semireliable data with atomistic physical models by Bayesian inference. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Perez A; MacCallum JL; Dill KA Accelerating molecular simulations of proteins using Bayesian inference on weak information. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 11846–11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mukherjee S; Balius TE; Rizzo RC Docking validation resources: protein family and ligand flexibility experiments. J. Chem. Inf. Model 2010, 50, 1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (31).Perez A; Morrone JA; Dill KA Accelerating physical simulations of proteins by leveraging external knowledge. Wiley Interdiscip. Rev. Comput. Mol. Sci 2017, 7, e1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Perez A; Morrone JA; Brini E; MacCallum JL; Dill KA Blind protein structure prediction using accelerated free-energy simulations. Sci. Adv 2016, 2, e1601274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Robertson JC; Perez A; Dill KA MELD× MD Folds Nonthreadables, Giving Native Structures and Populations. J. Chem. Theory Comput 2018, 14, 6734–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morrone JA; Perez A; MacCallum J; Dill KA Computed binding of peptides to proteins with MELD-accelerated molecular dynamics. J. Chem. Theory Comput 2017, 13, 870–876. [DOI] [PubMed] [Google Scholar]
- (35).Brini E; Kozakov D; Dill KA Predicting Protein Dimer Structures Using MELD× MD. J. Chem. Theory Comput 2019, 15, 3381–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Morrone JA; Perez A; Deng Q; Ha SN; Holloway MK; Sawyer TK; Sherborne BS; Brown FK; Dill KA Molecular simulations identify binding poses and approximate a nities of stapled α-helical peptides to MDM2 and MDMX. J. Chem. Theory Comput 2017, 13, 863–869. [DOI] [PubMed] [Google Scholar]
- (37).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (39).Jakalian A; Bush BL; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem 2000, 21, 132–146. [DOI] [PubMed] [Google Scholar]
- (40).Hornak V; Okur A; Rizzo RC; Simmerling C HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Case D; Betz R; Cerutti D; Cheatham T; Darden T; Duke R, et al. AMBER 16. 2016. San Francisco [Google Scholar]
- (42).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang L-P; Simmonett AC; Harrigan MP; Stern CD, et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol 2017, 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang L; Friesner RA; Berne B Replica exchange with solute scaling: a more efficient version of replica exchange with solute tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ester M; Kriegel H-P; Sander J; Xu X, et al. A density-based algorithm for discovering clusters in large spatial databases with noise 1996, 96, 226–231. [Google Scholar]
- (45).Mobley DL; Dill KA Binding of small-molecule ligands to proteins:“what you see” is not always “what you get”. Structure 2009, 17, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ravindranath PA; Forli S; Goodsell DS; Olson AJ; Sanner MF AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol 2015, 11, e1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Thompson TM From error-correcting codes through sphere packings to simple groups; American Mathematical Soc., 1983; Vol. 21. [Google Scholar]
- (48).Salentin S; Schreiber S; Haupt VJ; Adasme MF; Schroeder M PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res 2015, 43, W443–W447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Roe DR; Cheatham III TE PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (50).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (51).Zhao Y; Stoffler D; Sanner M Hierarchical and multi-resolution representation of protein flexibility. Bioinformatics 2006, 22, 2768–2774. [DOI] [PubMed] [Google Scholar]
- (52).Huey R; Morris GM; Olson AJ; Goodsell DS A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem 2007, 28, 1145–1152. [DOI] [PubMed] [Google Scholar]
- (53).Spyrakis F; Ahmed MH; Bayden AS; Cozzini P; Mozzarelli A; Kellogg GE The roles of water in the protein matrix: a largely untapped resource for drug discovery. J. Med. Chem 2017, 60, 6781–6827. [DOI] [PubMed] [Google Scholar]
- (54).Nguyen H; Roe DR; Simmerling C Improved generalized born solvent model parameters for protein simulations. J. Chem. Theory Comput 2013, 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.