

Abstract
Cardiovascular disease remains a worldwide public health concern, despite decades of research and the availability of numerous targeted therapies. While the intrinsic physiological mechanisms regulating cardiovascular function are similar between males and females, marked sex differences are established in terms of cardiovascular disease onset, pathophysiology, manifestation, susceptibility, prevalence, treatment responses and outcomes in animal models and clinical populations. Premenopausal females are generally protected from cardiovascular disease when compared to men of similar age, and tend to develop cardiovascular complications later in life following menopause. Emerging evidence suggests this cardioprotection in females is, in part, attributed to sex differences in hormonal regulators such as the renin-angiotensin system. To date, research has largely focused on canonical renin-angiotensin system pathways, and shown that premenopausal females are protected from cardiovascular derangements produced by activation of angiotensin II pathways. More recently, a vasodilatory arm of the renin-angiotensin system has emerged that is characterized by angiotensin-(1–7), angiotensin-converting enzyme 2, and Mas receptors. Emerging studies provide evidence for a shift towards these cardioprotective angiotensin-(1–7) pathways in females, with effects modulated by interactions with estrogen. Despite well-established sex differences, female comparison studies on cardiovascular outcomes are lacking at both the preclinical and clinical levels. Furthermore, there are no specific guidelines in place for treatment of cardiovascular disease in men versus women, including for therapies targeting the renin-angiotensin system. This review summarizes current knowledge of sex differences in cardiovascular actions of the renin-angiotensin system, focusing on interactions with gonadal hormones, emerging data for protective angiotensin-(1–7) pathways, and potential clinical implications for established and novel therapies.
Keywords: angiotensin, blood pressure, hypertension, sex, estrogen, clinical, animal models
Introduction
Cardiovascular disease (CVD) remains the leading cause of death for men and women worldwide, despite the availability of numerous targeted treatment approaches [1]. Hypertension is a leading independent risk factor for developing cardiovascular-related diseases including atherosclerosis, myocardial infarction, stroke, heart failure, end-stage renal disease, and peripheral arterial disease [2]. Importantly, there are recognized sex differences in blood pressure regulation as well as CVD susceptibility, onset, prevalence, clinical presentation, pathophysiology, treatment responses and outcomes in animal models and clinical populations [3]. For example, premenopausal women generally have a lower incidence and severity of hypertension compared with age-matched men [3]. Despite this, following a cardiovascular event, women do not respond as well to treatment and have higher risk of death [4]. This cardioprotection in premenopausal women is generally lost with obesity and type II diabetes and during menopause, with a greater incidence of CVD and more vascular and myocardial stiffness in hypertensive older women [1]. The manifestation of ischemic heart disease also exhibits sex-specific features, with men typically having obstructive dysfunction, whereas women suffer from non-obstructive or microvascular phenotypes [3].
The mechanisms underlying sex differences in CVD remain poorly understood, with females being underrepresented in preclinical and clinical research. Several mechanisms have been proposed including lifestyle and behavioral factors, functioning of cardiovascular end organs, sex hormones, and physiological regulatory systems including sympathetic and immune activation and vasoactive hormones such as nitric oxide (NO) and endothelin [4]. Emerging evidence suggests that differential activation of the renin-angiotensin system (RAS) also contributes to sex differences in CVD [5,4]. While the RAS intrinsically regulates blood pressure and cardiovascular end organ function in both sexes, premenopausal females exhibit a shift in this hormone system towards cardioprotective counter-regulatory pathways mediated by the hormone angiotensin (Ang)-(1–7) [4]. This protective role of Ang-(1–7) pathways in females appears to be modulated, in part, by interactions with estrogen. This review will highlight current knowledge of sex differences in cardiovascular actions of the RAS, including interactions with gonadal hormones, emerging data for protective Ang-(1–7) pathways, and therapeutic implications for established and novel therapies in clinical populations. In addition to hypertension, this review will describe evidence for sex differences in RAS actions in heart failure as well as diseases associated with vessel wall remodeling such as atherosclerosis and abdominal aortic aneurysm. While not a focus of this review, sex differences in the RAS have also been described in animal models and clinical populations for other CVD-related conditions including obesity, aging, and developmental programming [6–8].
RAS Signaling Pathways in Cardiovascular Regulation
The RAS has been established as a critical contributor to blood pressure regulation, and the pathogenesis of CVD, for over a century. This system consists of a series of enzymes and bioactive peptides regulating cardiovascular function, with a simplified overview provided in Figure 1. In the canonical RAS, the enzyme renin (an aspartyl protease) is secreted from renal juxtaglomerular cells in response to various stimuli such as decreased perfusion in the afferent renal arterioles, locally acting prostanoids and NO, increased sympathetic activity, or low sodium chloride concentration in the macula densa [9]. Renin cleaves angiotensinogen to produce Ang I, which is hydrolyzed by Ang-converting enzyme (ACE) to form Ang II, the main effector of the canonical pathway [10]. Ang II acts at cell surface type I G protein-coupled receptors (AT1R) to elevate blood pressure via multiple mechanisms including vasoconstriction, sodium reabsorption, sympathetic and immune activation, impairment of arterial baroreceptor reflex sensitivity, and increases in aldosterone, oxidative stress, fibrosis, and inflammation [10]. Uncontrolled Ang II activation is a contributing factor to hypertension as well as cardiac and vascular dysfunction and fibrosis, to promote development of CVD [10]. To counteract AT1R actions, Ang II binds cell surface type II receptors (AT2R) to increase arterial baroreflex sensitivity and promote vasodilation, natriuresis, and NO production [11]. The precise mechanisms responsible for AT2R actions are incompletely understood, however, with activity of this receptor limited in adults due to low affinity and tissue expression [11].
Fig. 1.
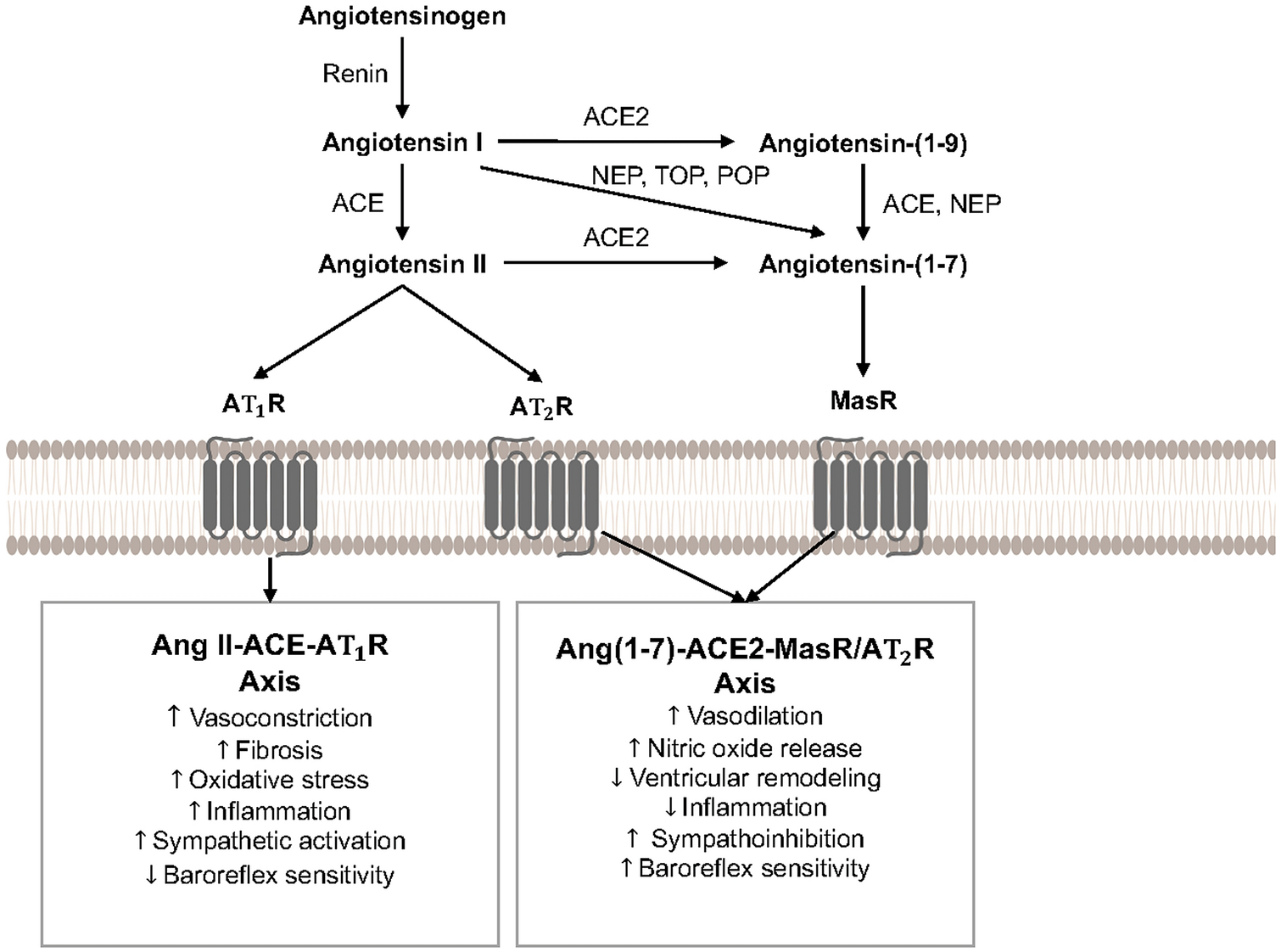
Simplified overview of the renin-angiotensin system, including primary cardiovascular effects elicited by activation of angiotensin II and angiotensin-(1–7) signaling pathways. ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme 2; Ang, angiotensin; AT1R, angiotensin II type 1 receptor; AT2R; angiotensin II type 2 receptor; MasR, angiotensin-(1–7) mas receptor; NEP, neprilysin; POP, prolyl oligopeptidase; TOP, thimet oligopeptidase.
More recently, a noncanonical counter-regulatory arm of the RAS has been discovered, which is characterized by the biologically active heptapeptide Ang-(1–7) [12]. As shown in Figure 1, Ang-(1–7) results from degradation of Ang I by endopeptidases (e.g., neprilysin, prolyl oligopeptidase, thimet oligopeptidase), or degradation of Ang II by carboxypeptidases such as ACE2. Ang I can also be converted to Ang-(1–9) by ACE2, which is then subsequently cleaved by ACE or neprilysin to form Ang-(1–7); however, this formation pathway has lower catalytic efficiency [13]. Ang-(1–7) elicits physiological effects by binding Mas G protein-coupled receptors (MasR), which are found in tissues pivotal to cardiovascular control including the heart, vasculature, kidneys, and brain [12]. The actions of Ang-(1–7) at cell surface MasR promotes vasodilation, NO release, and improved arterial baroreflex sensitivity [12]. Ang-(1–7) also has antihypertensive, sympathoinhibitory, antihypertrophic, antifibrotic, antiarrhythmogenic, and antithrombotic properties in animal models of CVD [13,12]. While most evidence suggests that in vivo actions of Ang-(1–7) require MasR, a few recent studies have shown potential heterodimerization and functional interactions between MasR and AT1R or AT2R [13]. Overall, Ang-(1–7)-ACE2-MasR pathways generally opposes the deleterious cardiovascular actions elicited by activation of the Ang II-ACE-AT1R axis.
As recently described [6], in addition to these Ang II and Ang-(1–7) pathways, research continues to reveal further layers of complexity in the RAS with more recent discoveries including: the renin-independent precursor angiotensin-(1–12); prorenin, and the prorenin receptor; localization of RAS components in tissues; intracellular RAS components; ACE-independent enzymatic pathways for Ang II formation; and binding of the endogenous heptapeptide alamandine to mas-related G protein-coupled receptor D to elicit cardiovascular actions similar to Ang-(1–7). These alternate pathways will not be a focus of this review, as there is currently limited or non-existent data related to sex differences in cardiovascular outcomes.
Effects of Sex Hormones on RAS Pathways
Accumulating evidence exists for profound effects of gonadal hormones such as estrogen and testosterone (as well as hormone replacement therapies) on expression and activity of RAS components, to contribute to sex differences in cardiovascular outcomes and pathophysiology. An overview of interactions of the RAS with gonadal hormones for cardiovascular control is described below, with findings for estrogen and testosterone summarized in Figure 2.
Fig. 2.
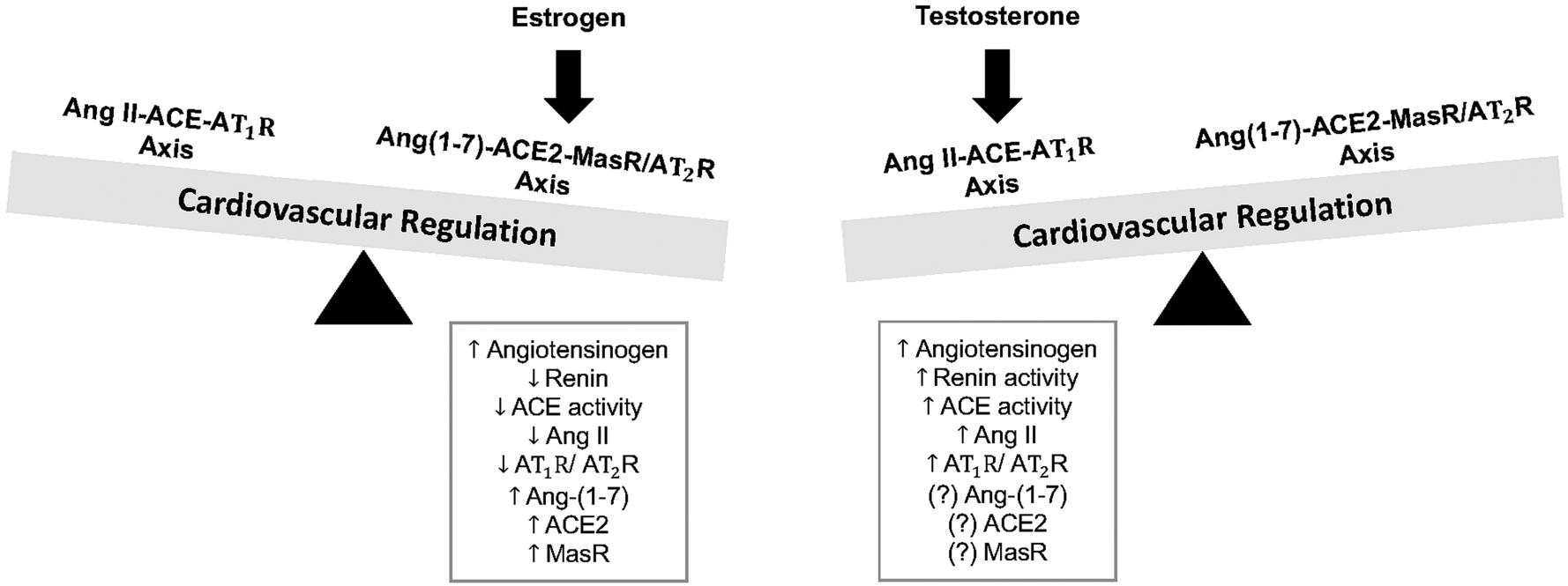
Interactions between sex hormones and components of the renin-angiotensin system for cardiovascular regulation. Estrogen shifts the balance of the renin-angiotensin system towards angiotensin-(1–7)-ACE2-mas receptor-AT2 receptor pathways to elicit cardiovascular protection. In contrast, testosterone shifts the balance of the renin-angiotensin system towards angiotensin II-ACE-AT1 receptor pathways to elicit deleterious cardiovascular actions. ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme 2; Ang, angiotensin; AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; MasR, mas receptor.
RAS-Estrogen Interactions:
Estrogen is primarily produced by theca and granulosa cells in the ovaries, but can also be synthesized in extragonadal tissues such as adipose, brain, heart and vasculature [14]. The majority of circulating estrogen is in the form of 17β-estradiol, but other naturally occurring and less potent estrogens include estrone and estriol. Levels of circulating angiotensinogen are generally higher in premenopausal women compared to men. This trend has a biological age feature with postmenopausal women having lower angiotensinogen levels compared with premenopausal women [5,15]. Oral estrogen replacement increases angiotensinogen secretion in postmenopausal women, irrespective of hypertensive status [16,5]. These overall findings suggest that estrogen increases synthesis of angiotensinogen, which is likely due to the presence of an estrogen response element in its gene promoter [5,15].
While increasing angiotensinogen production, most studies support that estrogen reduces plasma renin concentrations. This reflects, in part, estrogen-mediated reductions in sympathetic activity, to suppress renin secretion from renal juxtaglomerular cells [5]. For example, plasma renin concentration is lower in premenopausal women and postmenopausal women on estrogen replacement therapy when compared to either postmenopausal women without hormonal therapy or men [17,18]. Estrogen replacement can increase plasma renin activity in postmenopausal women, however, perhaps reflecting more angiotensinogen availability [15]. In addition, oral versus transdermal estrogen replacement can produce similar or differential effects on renin levels in postmenopausal women [15,5]. Despite this, estrogen consistently reduces serum and tissue ACE expression and activity, AT1R expression and signaling in tissues (e.g. kidney, adrenal cortex, vasculature), and aldosterone production in animal models [19,5,15]. The ability of estrogen to downregulate AT1R may result from direct interference with gene transcription via enhanced NO production as well as with translation by blocking ribosomal scanning [20,21].
There are varying effects of estrogen replacement on Ang II levels with one study showing a decrease in young female hypertensive rats, and another showing an increase in ovariectomized female rats [22,23]. A decrease in Ang II levels with estrogen is thought to result from reduced formation due to suppression of renin and ACE activity [5]. Despite controversy for modifying hormone levels, pressor responses to Ang II are attenuated following estrogen replacement in young female hypertensive rodents [15]. Furthermore, treatment with 17β-estradiol inhibits Ang II-induced cellular proliferation and oxidative stress in cardiac fibroblasts obtained from neonatal rats [24]. Clinically, oral estrogen replacement increases plasma renin activity, decreases circulating ACE, and increases Ang II in postmenopausal women, with no impact of transdermal estrogen replacement on these RAS components [15,5,25]. These divergent effects likely reflect that transdermal administration avoids hepatic metabolism (first pass effect), which can produce non-physiological elevations in estrone as well as renin substrate to increase Ang II formation [25]. Overall, while some conflicting evidence exists depending on route of administration, estrogen is generally accepted to reduce activation of Ang II-ACE-AT1R pathways [15,5].
In addition to inhibiting Ang II pathways, evidence from preclinical models indicates that estrogen upregulates protective Ang-(1–7)-ACE2-MasR-AT2R pathways (Figure 2). Circulating Ang-(1–7) levels are higher in obese young female mice and healthy premenopausal women when compared with their male counterparts [26,27]. In human endothelial cells, estradiol increases local Ang-(1–7) production through an estrogen receptor α (ERα) dependent mechanism [28]. Similarly, estrogen increases ACE2 gene expression in cultured 3T3-L1 adipocytes [26], and increases ACE2 and MasR expression in human atrial tissue from elderly men [29]. Administration of 17β-estradiol to young female ovariectomized rats lowers blood pressure and attenuates Ang II-induced pressor responses, effects associated with reduced plasma ACE and Ang II and increased Ang-(1–7) levels [22]. In young obese female mice, loss of estrogen via ovariectomy increases resting blood pressure and decreases adipose ACE2 and plasma Ang-(1–7) levels [26]. Furthermore, young female rats are protected from renovascular hypertension, due to an estrogen-regulated increase in intratubular Ang-(1–7)-ACE2-MasR pathways [30]. Estrogen also upregulates AT2R expression and binding in cardiovascular tissues from young male and female rats [31,32], and reciprocally AT2R agonism increases estrogen production in ovaries from young female rabbits [33]. These protective estrogen-RAS interactions appear diminished during aging. A recent study showed a decline in circulating Ang-(1–7) levels during aging in clinical populations, which was particularly evident in overweight and obese individuals [34]. Furthermore, loss of vasodilatory responses to Ang-(1–7) is observed in older female mice, with vascular responsiveness and NO levels restored following estradiol replacement [35].
Overall, estrogen provides cardioprotection in premenopausal females by shifting the balance of the RAS towards Ang-(1–7)-ACE2-MasR-AT2R pathways. The increased production of Ang-(1–7) with estrogen is mediated by ERα to increase ACE2 activity and expression in a concentration-dependent manner [36]. In addition, estrogen promotes NO-dependent vasodilation through a mechanism involving crosstalk with MasR [36]. In premenopausal women, loss of estrogen to modulate these protective RAS pathways may contribute to increased cardiovascular risk in this population [4]. The precise mechanisms by which estrogen interacts with the RAS to induce cardioprotective properties are still incompletely understood, but appear dependent on the type of estrogen administered, the experimental context, and use of preclinical versus clinical models.
RAS-Testosterone Interactions
In young male rats, renal and hepatic angiotensinogen gene expression are increased following testosterone administration, and conversely decreased following castration [5,37]. Testosterone administration to either castrated male or ovariectomized female rodents increases plasma renin activity suggesting a positive regulatory relationship [5,15]. Conversely, anti-androgen therapies decrease plasma renin in young male and female hypertensive rats [38,39]. Testosterone exposure also increases ACE activity and tissue AT1R expression and plays an essential role in the development and maintenance of Ang-II induced vascular dysfunction, hypertension, and cardiac hypertrophy in pregnant rats and in young male mice and rats [40–42]. Ang-II induced hypertensive and vasoconstrictor responses are attenuated in young castrated male rats and associated with a reduced vascular AT1R/AT2R ratio, with effects reversed by testosterone replacement [43]. Thus, in contrast to estrogen, sex-specific differences in CVD pathophysiology are amplified by androgens such as testosterone through stimulation of the Ang II-ACE-AT1R axis (Figure 2).
While increasing Ang II pathways, much less is known about the effects of testosterone on counter-regulatory pathways of the RAS. Testosterone downregulates AT2R expression in aorta from young male and female rats [44], with no effect on ACE2 expression in cultured 3T3-L1 adipocytes or testicular ACE2 activity in young male rats [26,45]. Conversely, AT2R expression is increased in young male castrated rats with heart failure, with effects prevented by testosterone replacement [46]. Taken together, testosterone shifts the balance of the RAS towards Ang II-ACE-AT1R pathways to induce vasoconstriction, vascular dysfunction, and cardiac hypertrophy and fibrosis. Further research is needed to better understand how testosterone, and anti-androgen therapies, interacts with protective Ang-(1–7)-ACE2-MasR-AT2R pathways to influence cardiovascular control.
RAS Interactions with Other Sex-Related Hormones
In addition to estrogen and testosterone, there is some evidence the RAS interacts with other sex-related hormones for cardiovascular control. Progesterone is a hormone primarily produced by the ovaries, which plays a critical role in reproductive functions including the menstrual cycle, pregnancy, and embryonic development. Progesterone receptors are also widely expressed in the vasculature and can promote either vasodilation or vasoconstriction in humans depending on the vascular bed studied, dose administered, and if co-administered with estrogen [47]. Premenopausal women taking oral contraceptives containing both estrogen and progesterone have elevated circulating Ang II levels when compared with premenopausal women not taking oral contraceptives and men [48,49]. Progesterone increases AT1R gene and protein expression in vascular smooth muscle cells from thoracic aorta of young female rats [50], and increases AT1R binding in the arcuate nucleus of the hypothalamus in young ovariectomized female rats [51,52]. Despite potentially elevating Ang II and AT1R levels, progesterone does not appear to functionally affect pressor responses to Ang II in women [53]. Ang II can conversely produce dose- and time-dependent stimulatory effects on progesterone secretion in human granulosa and rat zona glomerulosa cells [54,55].
Progesterone is also a competitive inhibitor of the mineralocorticoid receptor, resulting in increased compensatory aldosterone production to maintain sodium homeostasis [56]. In support of this concept, aldosterone is higher in the luteal phase in cycling women in high-sodium balance, and progesterone increases aldosterone production in isolated rat zona glomerulosa cells [57]. This ability of progesterone to increase adrenal aldosterone production appears independent of changes in plasma renin activity or Ang II levels suggesting RAS-independent mechanisms [57,58]. More recently, it has been shown that progesterone upregulates endothelial mineralocorticoid receptor expression to contribute to leptin-induced endothelial dysfunction in young obese female mice [59]. Overall, progesterone appears to increase Ang II and aldosterone pathways, although there are limited data on the functional importance of these interactions for cardiovascular outcomes.
Much less is known about interactions of progesterone with Ang-(1–7) pathways. Ang-(1–7) attenuates the ability of Ang II to increase progesterone secretion in rat zona glomerulosa cells [55]. In addition, female rats lacking either estradiol or progesterone have decreased Ang-(1–7) levels and MasR gene expression in the cerebral cortex and hippocampus [60]. While less studied, Ang II interactions with relaxin and oxytocin have also been reported in rat models of cardiac fibrosis and hypertension [61,62]. Given this limited research, studies are needed to determine how these additional sex hormones interact with RAS components to contribute to cardiovascular control, with a major gap in knowledge existing related to their influence on cardioprotective Ang-(1–7) pathways.
Sex Differences in Cardiovascular Actions of the RAS
The development and progression of CVD are enhanced by uncontrolled RAS activation and display sex-specific features. While blood pressure and cardiovascular end organ function are controlled by the same intrinsic mechanisms in males and females, differential activation of the vasoconstrictor versus vasodilatory arms of the RAS contributes to sex differences in cardiovascular control [4]. In males, activation of Ang II-ACE-AT1R pathways is well recognized to contribute to the etiology of CVD. In contrast, premenopausal women and animal models are generally protected from cardiovascular complications due to upregulation of counter-regulatory Ang-(1–7)-ACE2-MasR-AT2R pathways. This protection is diminished in postmenopausal women and ovariectomized animal models following loss of estrogen, with evidence for decreased circulating Ang-(1–7) levels as well as reduced responsiveness to the vasodilatory effects of exogenous Ang-(1–7) administration with aging to increase cardiovascular risk (Figure 3) [35,34]. Importantly, this differential RAS activation is multifaceted and affects cardiovascular control at multiple levels resulting in sex differences in CVD presentation, pathophysiology, and outcomes. This section will describe known sex differences in cardiovascular actions of RAS components, with a focus on hypertension, atherosclerosis, aneurysm formation, and heart failure.
Fig. 3.
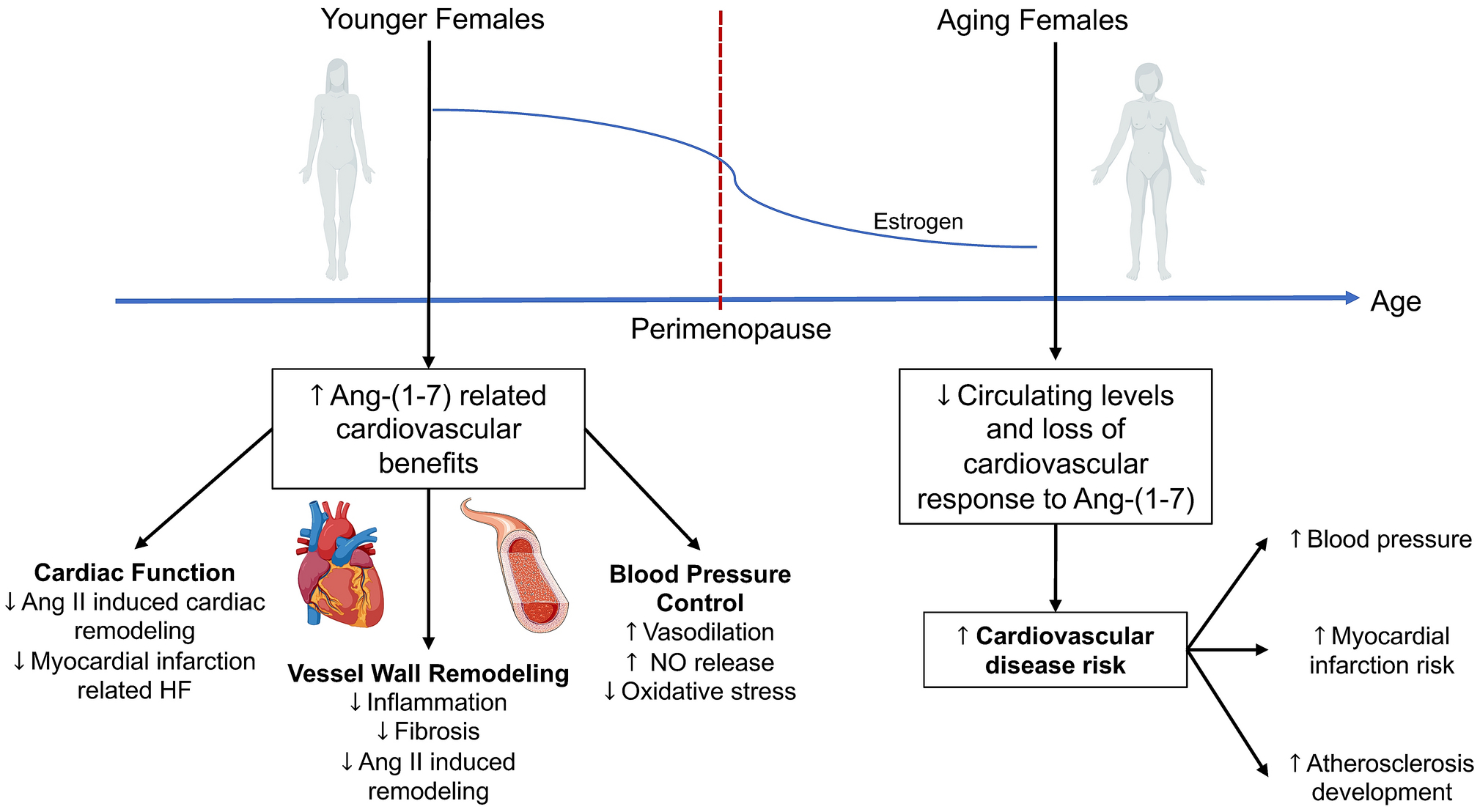
Biological aging feature of angiotensin-(1–7) pathways and effects on the cardiovascular system. In premenopausal females, angiotensin-(1–7) elicits protective effects on cardiac function, vessel wall remodeling, and blood pressure control. These protective responses to angiotensin-(1–7) appear reduced in postmenopausal women following loss of estrogen, to potentially increase risk for developing cardiovascular diseases such as hypertension, myocardial infarction, and atherosclerosis. Ang, angiotensin; HF, heart failure; NO, nitric oxide.
Blood Pressure Regulation and Hypertension
Hypertension affects over 85 million adults in the United States alone, and is a leading predictor for cardiovascular morbidity and mortality [1]. Sex differences in hypertension are well recognized, with premenopausal women generally having lower incidence and severity of hypertension compared with age-matched men [4]. This protection declines after menopause with percentage of hypertensive women outweighing the number of men in elderly populations [4,3]. Hypertensive women are also more likely to develop vascular and myocardial stiffness, as well as isolated systolic hypertension, during aging when compared with men [3]. These sex differences in hypertension have been, in part, attributed to differential levels and activity of RAS components in males versus females.
The main precursor of the RAS, angiotensinogen, is primarily produced in liver but is also expressed in cardiovascular-regulatory tissues such as heart, kidney, vasculature, and adipose [5]. A handful of recent studies have focused on adipose tissue, as adipose-derived angiotensinogen is reported to contribute to up to 30% of total levels in mice. In young male mice, adipose over-expression of angiotensinogen elicits hypertension, whereas adipose-specific deletion lowers resting blood pressure and attenuates development of obesity hypertension [63,64]. Similar to males, adipose-specific deletion of angiotensinogen lowers resting blood pressure in both younger and older female mice [65]. As recently described [6], sex differences have been reported in stimuli influencing renin release, with females having reduced sympathetic activation, increased renal nitric oxide synthesis, and a differential pattern of renal transporters. Despite this, young male and female rats with global deletion of the renin gene exhibit similar reductions in resting blood pressure [66]. In addition, direct renin inhibition with aliskiren similarly lowers blood pressure in men and women with mild-to-moderate hypertension [67]. Adipose deletion of the prorenin receptor also exacerbates obesity hypertension in young female mice via an intra-renal Ang II-dependent mechanism [68]. Therefore, there does not appear to be major sex differences in the blood pressure regulatory actions of angiotensinogen and renin, particularly when targeting these components in adipose tissue.
In terms of Ang II pathways, elevated ACE activity is observed in the circulation, heart, and kidney of young male hypertensive rodents [5]. In humans, serum ACE activity is generally higher in men versus women, but declines during healthy aging in men [7]. In young mice, global ACE knockout lowers blood pressure in males but not females, despite similar reductions in serum ACE activity between sexes [69]. While plasma and tissue Ang II is higher in young male rodents compared with females, consistent sex differences for circulating levels of this hormone have not been observed clinically [5]. Despite this, males exhibit greater sensitivity to Ang II-induced cardiovascular effects in both animal models and clinical populations. One study showed that Ang II produces greater pressor and renal vasoconstrictor responses in healthy young men versus women [70]. Chronic Ang II infusion results in a greater pressor response in young male compared with female rodents, and appears androgen-dependent and associated with increased sympathetic activation as well as higher AT1R and lower AT2R gene expression in the vasculature and kidneys [71–74]. This protection from hypertension in female rodents appears mediated by AT2R as well as estrogen actions at centrally expressed ERα to inhibit Ang II-induced sympathetic activation and oxidative stress [75–77]. Similarly, young female rats are protected from Ang II-induced sensitization of hypertension via central estrogenic mechanisms [78]. Furthermore, obesity hypertension in young male mice is associated with increased circulating Ang II levels and is mediated by AT1R activation [26]. In contrast, young female mice do not exhibit elevated Ang II and are protected from obesity hypertension [26]. Similarly, the elevated blood pressure in young male fructose fed rats is associated with increased cardiac ACE and AT1R expression, with females protected from this cardiovascular phenotype [79]. As a potential mechanism underlying these sex differences, recent studies have shown that chronic Ang II infusion increases renal pro-inflammatory T cells in young male rats, while increasing anti-inflammatory T regulatory cells in females [80]. These data suggest females are protected from hypertension resulting from activation of Ang II-ACE-AT1R pathways of the RAS.
In clinical populations, a few studies have shown that plasma and urinary Ang-(1–7) levels are higher in young healthy women versus men, and positively correlate with blood pressure only in women [27,81]. Another study, however, showed higher plasma Ang-(1–7) in young healthy men [82]. These disparate findings could reflect heterogeneity in blood sample collection and patient demographics. In addition, no differences in Ang-(1–7)-forming enzyme activities (e.g. ACE2, neprilysin) are observed between men and women during healthy aging [7]. In young hypertensive rats, circulating and renal Ang-(1–7) are higher in females versus males [83]. Similarly, young obese female mice have higher circulating Ang-(1–7) and adipose ACE2 and are protected from hypertension [26]. This protection from obesity hypertension in female mice is mediated by endogenous Ang-(1–7) actions at MasR and interactions with estrogen [26]. In addition, the lower blood pressure and increased Ang-(1–7) levels in young female hypertensive rats is associated with more anti-inflammatory T regulatory cells and cytokines compared with males [84].
In addition to elevated Ang-(1–7) levels, endogenous activation of ACE2, MasR and AT2R induces cardioprotective effects, particularly in females. Endogenous brain Ang-(1–7) pathways are upregulated via estrogen -dependent mechanisms to protect against hypertension development in young female rats [85]. Adipocyte ACE2 deficiency increases resting blood pressure as well as pressor responses to Ang II in young obese female but not male mice [86]. In Chinese individuals with untreated essential hypertension, the ACE2 alleles rs2074192 and rs2106809 are associated with reduced circulating Ang-(1–7) levels, which correlates with increased blood pressure only in women [87]. Furthermore, global MasR deletion in young mice increases sympathetic tone in both sexes, but only reduces heart rate variability in females [88]. In young obese female mice, global MasR deletion increases blood pressure compared to lean within genotype controls, suggesting endogenous MasR protect females from obesity-related hypertension [89]. In contrast to females, young obese male MasR knockout mice have reduced blood pressure, which is attributed to chronically elevated Ang II levels to impair cardiac function. The depressor and natriuretic effects elicited by activation of AT2R are also greater in females, with levels of this receptor modulated by estrogen [90]. These overall findings suggest that in females, activation of Ang-(1–7)-ACE2-MasR-AT2R pathways promotes blood pressure lowering to protect against development and progression of hypertension.
Vessel wall remodeling
Changes in vessel wall structure are implicated in numerous CVD including atherosclerosis, abdominal aortic aneurysm (AAA), myocardial infarction, and peripheral arterial disease. Sex differences in these diseases are established, with men dying from atherosclerosis and myocardial infarction at a younger age compared with women [3]. The estrogenic effect in the vasculature is claimed as one of the main reasons for this beneficial cardiovascular profile in premenopausal women, with more women developing atherosclerosis following menopause [91]. In addition, there is a 4- to 5-fold higher incidence of AAA in men compared to age-matched women, as well as higher incidence in males in the Ang II-infused mouse model of AAA [92]. Interactions of the RAS with testosterone are implicated in these sex differences in aneurysm formation. Testosterone upregulates ACE activity, with older healthy men having lower ACE activity compared with younger healthy men [7]. Furthermore, the higher prevalence of AAA in Ang II-infused male mice is associated with androgen-mediated upregulation of AT1R in the abdominal aorta [93]. Despite occurring less frequently, postmenopausal women exhibit rapid growth rates of AAA and rupture at smaller sizes [94]. This has been attributed to sex hormones, with exogenous estrogen administration reducing progression and severity of Ang II-induced AAA in ovariectomized female mice [95]. Similar to males, AT1R blockade attenuates Ang II-induced AAA formation in young hypercholesterolemic female mice [96].
Sex differences in protective RAS pathways in diseases associated with vessel wall remodeling are less understood. In severe coronary atherosclerosis, there is a high Ang II to Ang-(1–7) ratio in both sexes, which may increase vascular proliferation and inflammation to promote plaque development [97]. In young and middle aged male hypercholesterolemic mice, global knockout of either ACE2 or MasR exacerbates Ang II-induced atherosclerosis and aortic aneurysm ruptures, and conversely pharmacologic ACE2 activation reduces incidence of AAA [98,99], with no information in females. In human brain vascular smooth muscle cells and young mice, Ang-(1–7) counteracts Ang-II induced vascular remodeling by attenuating proinflammatory pathways [100]. In young female hypercholesterolemic mice, AT2R antagonism increases incidence and severity of AAA and atherosclerosis, providing evidence for a protective role of AT2R against Ang II-mediated vascular pathophysiology [96]. Thus, while some evidence exists for a protective role of Ang-(1–7)-ACE2-MasR-AT2R pathways against atherosclerosis and AAA, sex differences in these mechanisms have yet to be defined. Further, while emerging literature suggests a potential role for sex chromosomes to regulate atherosclerosis and AAA development [92], whether this involves interactions with the RAS is unclear. Given these known sex disparities, guidelines were recently established for the design and reporting of sex as a biological variable in preclinical studies on atherosclerosis, aneurysms, and peripheral arterial disease [92].
Heart failure
Emerging evidence suggests the epidemiology and clinical characteristics of heart failure, as well as response to heart failure therapies including those inhibiting the RAS, are sex dependent. The RAS is a major contributor to heart failure with chronic Ang II exposure promoting cardiac hypertrophy via activation of growth factors and increased mitochondrial oxidative stress [101]. In addition, Ang II-mediated activation of AT1R induces structural remodeling of the ventricular wall including proliferation of cardiac fibroblasts, deposition of extracellular matrix proteins such as collagen, and hypertrophic growth of cardiomyocytes [101]. Indeed, cardiac Ang II levels increase with age and are linked to hypertrophy, fibrosis, and diastolic dysfunction [102]. Sex differences in RAS mechanisms involved in cardiac dysfunction have been reported, particularly related to aging and heart failure. In a mouse model of chronically elevated cardiac Ang II, age-related deterioration of cardiomyocyte function is sex-dependent with females more susceptible to contractile deficits but having low spontaneous activity, and males maintaining contractile function but having desensitization of myofilaments to calcium and heightened vulnerability to arrhythmias [102]. Over-expression of AT1R in cardiomyocytes results in heart failure in both male and female mice, but with females having increased susceptibility to dilated cardiomyopathy and higher mortality following cardiac Ang II overstimulation [103]. In addition, young female rats at the onset of the decompensated phase of congestive heart failure exhibit higher mortality, associated with increased renal vascular responsiveness to norepinephrine and Ang II [104]. These findings suggest female rodents are more sensitive to Ang II in terms of susceptibility to heart failure, although the precise mechanisms underlying this finding are not fully understood. This is consistent with clinical findings showing that women are more susceptible to developing heart failure in response to hypertension compared with men [105].
Clinically, pressure overload-induced hypertrophy is associated with a smaller left ventricular chamber and larger wall thickness in women than in men [106,107]. These findings are in agreement with animal studies showing that female mice develop concentric myocardial hypertrophy whereas males develop eccentric myocardial hypertrophy [3]. This may be explained by a reduced number of ventricular myocytes with age via apoptosis in men but not women [108]. These findings could help explain why older men are more likely than age-matched women to experience heart failure with reduced ejection fraction [102]. Interestingly, hypertensive women do not respond as well to long-term RAS inhibition with the angiotensin receptor blocker (ARB) losartan in terms of left ventricular structure and function when compared to men [109]. Despite this, ARBs appear more effective to improve survival in women with congestive heart failure compared with ACE inhibitors irrespective of hypertensive status [110]. These collective data suggest that prolonged activation of Ang II pathways plays a pathophysiological role in cardiac remodeling, and that sex differences exist in terms of susceptibility for developing myocardial hypertrophy and heart failure as well as responsiveness to therapies targeting Ang II for treatment of hypertensive cardiac abnormalities.
In contrast to Ang II, emerging evidence suggests that activation of non-canonical RAS pathways involving Ang-(1–7) provides protection against heart failure [111,112]. In support of this, in rodent models: Ang-(1–7) prevents Ang II-induced cardiac remodeling in young male rats; the MasR agonist AVE-0991 attenuates myocardial infarction-induced heart failure in young male rats; and genetic deletion of ACE2 accelerates hypertension-induced cardiac dysfunction in young male mice [111–113]. Unfortunately, these preclinical studies showing protective effects of Ang-(1–7) pathways in heart failure have been limited to males. In heart failure patients, ACE2 is upregulated in the myocardium and associated with the degree of left ventricular ejection fraction in both men and women [114]. This ACE2 upregulation is suggested as an adaptive compensatory mechanism to prevent myocardial remodeling in both sexes. While similar cardiac ACE2 has been observed, a recent study suggests plasma ACE2 concentrations are higher in men versus women with heart failure, and not associated with ACE inhibitor or ARB treatment [115]. Further research is needed to determine the importance of these sex differences in ACE2 levels to outcomes, as well as the effectiveness of targeting Ang II versus Ang-(1–7) pathways in men versus women, in heart failure.
Sex Differences in Cardiovascular Therapeutic Formulations Targeting the RAS
Despite well-established sex differences in CVD, preclinical and clinical research efforts have largely focused on males. Even with recent efforts to include females equally in CVD research, women still only represent ~37% of study participants in controlled NIH-funded trials, with only 35% of published cardiovascular clinical studies analyzing outcomes according to sex [116]. As a result, sex-specific guidelines have not been developed for hypertension or CVD treatment, including for therapies targeting the RAS. The current knowledge of sex differences in cardiovascular outcomes for RAS therapies is shown in Table 1.
Table 1.
Sex-specific responses to established and novel pharmacological therapeutic formulations targeting the renin-angiotensin system in clinical populations.
| Therapeutic Agent | Mechanism of Action | Sex-Specific Responses |
|---|---|---|
| ACE inhibitors | Decrease Ang II formation | Men demonstrate better blood pressure control [117], with women having increased susceptibility to side effects [119,120]. |
| Angiotensin receptor blockers (ARBs) | Block Ang II effects at AT1R | Women exhibit greater antihypertensive effect and require lower drug dosage [121,122,110]. Improved survival in women with CHF when compared to ACE inhibitors [110]. |
| Mineralocorticoid antagonists | Block aldosterone effects at mineralocorticoid receptors | More effective to lower blood pressure in women and in obesity-related hypertension [123,124]. |
| Direct Renin Inhibitors | Antagonize renin to prevent downstream activation of the RAS | Similar blood pressure lowering efficacy in hypertensive men and women treated with aliskiren [67]. |
| MasR Agonists | Increase activity of Ang-(1–7) mas receptors | Unknown |
| ACE2 Activators | Decrease Ang II and increase Ang-(1–7) levels | Unknown |
| Recombinant human ACE2 | Decrease Ang II and increase Ang-(1–7) levels | Unknown |
ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme 2; Ang, angiotensin; AT1R, angiotensin II type 1 receptor; CHF, congestive heart failure; MasR, angiotensin-(1–7) mas receptor; RAS, renin-angiotensin system.
RAS inhibition has been one of the most common treatment approaches for CVD for decades. Pharmacological agents such as direct renin inhibitors, ACE inhibitors, ARBs, and mineralocorticoid antagonists have helped to convert what was once a lethal condition into a chronic one. These therapies all elicit antihypertensive, antifibrotic, and anti-remodeling effects and are generally safe and efficacious across diverse clinical populations. For direct renin inhibition, a retrospective meta-analysis of eight clinical trials showed no differences in blood pressure lowering efficacy of aliskiren in hypertensive men versus women [67]. In contrast, evidence suggests potential dimorphic responses to therapeutics blocking Ang II activity based on sex. In 2008, a systematic review was published on sex-based analysis of controlled clinical trials using ACE inhibitors or ARBs for hypertension treatment [117]. Sex-specific results were only published in 43% of the trials included, with data suggesting both of these therapies are slightly more effective in men versus women. Additional literature, however, suggests women respond more favorably to ARBs in terms of cardiovascular outcomes, whereas ACE inhibitors appear more effective in men. Women treated with ACE inhibitors are more likely to develop adverse reactions such as cough and angioedema, with effectiveness potentially decreasing over time and offering less benefit for total mortality [118–120]. A lower dose of the ARB irbesartan is required for young healthy women to inhibit Ang-II mediated reduction in renal blood flow compared to men [121]. Women also have a greater antihypertensive response to treatment with the ARB valsartan combined with amlodipine, a calcium channel blocker, when compared with men treated with the same agents [122]. Another study described better outcomes in women with congestive heart failure treated with an ARB versus an ACE inhibitor, while no differences in outcomes were observed in men between these therapeutics [110]. Additionally, women appear more responsive to blockade of aldosterone actions with mineralocorticoid receptor antagonists. This is particularly evident in obese women where increased aldosterone correlates with visceral adiposity, body mass index, and blood pressure [123,124]. Women also appear to have greater salt sensitivity of blood pressure compared with men, irrespective of age or hypertensive status, due to increased adrenal aldosterone production [125]. These findings highlight emerging data for sex differences in therapeutic effectiveness to established RAS therapies, with women potentially responding more favorably to ARBs and mineralocorticoid receptor antagonists.
In addition to blockade of Ang II pathways, direct targeting of Ang-(1–7) has been explored for treatment hypertension and CVD. Unfortunately, Ang-(1–7) has unfavorable pharmacokinetic properties including a short half-life [13]. Therefore, novel therapeutics are in development to increase Ang-(1–7) levels or actions such as oral formulations, stable analogs, MasR agonists, ACE2 activators, and recombinant human ACE2 [12]. These therapies reduce blood pressure and attenuate cardiovascular damage in animal models, making them attractive for clinical development [12]. There are limited published or ongoing trials targeting Ang-(1–7) pathways in humans, however, with none designed to identify sex-specific differences in cardiovascular outcomes. Importantly, targeting Ang-(1–7) pathways may provide advantage over use of ACE inhibitors and ARBs. ACE inhibitors are limited in up to 11% of patients by cough due to bradykinin production [126]. Furthermore, both ACE inhibitors and ARBs increase endogenous Ang-(1–7) generation, which contributes to the beneficial cardiovascular and metabolic effects of these therapies in young male rodent models of hypertension and obesity [127–129]. Direct targeting of Ang-(1–7) pathways may therefore provide cardiovascular benefit while avoiding potentially limiting side effects of therapies blocking Ang II activity. Overall, these limited data illustrate the critical need for additional preclinical and clinical studies to determine the impact of sex on cardiovascular effects of RAS inhibition, for both established therapies targeting Ang II pathways and more novel therapies targeting Ang-(1–7) pathways.
Conclusions
The identification of sex-specific mechanisms by which the RAS influences cardiovascular control, including potential interactions with gonadal hormones, remains an active area of research. There are known sex differences in expression and activity of several RAS components, with estrogen upregulating protective Ang-(1–7)-ACE2-MasR-AT2R pathways [4]. On the other hand, testosterone favors activation of Ang-II-ACE-AT1R pathways, thus enhancing the vasoconstrictive arm of the RAS [15,5]. These variations in RAS activity can contribute to differential CVD clinical presentation, pathophysiology, and treatment responses and outcomes between men and women. The role of the RAS in regulating cardiovascular function also exhibits a biological age pattern, offering cardioprotection to premenopausal women with effects decreasing in menopause following loss of estrogen [35]. There remain several gaps in our knowledge of sex differences in RAS mechanisms regulating cardiovascular function that require future research, as summarized below and in Table 2. While interactions with estrogen and testosterone are well described, there are limited data on how other sex-related hormones (e.g. progesterone, relaxin, oxytocin) or newer forms of contraception influence RAS components and activity. Importantly, we still do not fully understand sex differences in therapies targeting the RAS, due to underrepresentation of females in clinical trials. Furthermore, therapeutic management of CVD is similar between men and women, despite initial evidence for sex-specific outcomes in therapies blocking Ang II activity. In particular, women may respond better to ARBs and tend to experience increased side effects with ACE inhibitors [120,118]. While targeting of Ang-(1–7) and other components of the vasodilatory arm of the RAS seems to be a promising therapeutic target, there is currently a paucity of clinical data. Preclinical studies have shown that novel therapeutic formulations targeting Ang-(1–7) pathways are protective in CVD, but the cellular mechanisms underlying these effects are not fully understood [12]. Additionally, further studies are needed to determine the effects of Ang-(1–7) on cardiovascular outcomes in clinical populations with hypertension and CVD. Overall, there is crucial need for additional research to clarify the impact of sex-specific RAS differences on the cardiovascular system, to aid in the development of novel cardioprotective agents.
Table 2.
Potential Questions for Future Research on Sex Differences in Cardiovascular Actions of the Renin-Angiotensin System (RAS)
| Effects of Sex Hormones on RAS Pathways |
|
| Sex Differences in Cardiovascular Actions of the RAS |
|
| Sex Differences in RAS Therapies |
|
Acknowledgements:
ACA is supported by NIH grants R00HL122507 and UL1TR002014.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References Cited
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics C, Stroke Statistics S (2017) Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 135 (10):e146–e603. doi: 10.1161/CIR.0000000000000485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kjeldsen SE (2018) Hypertension and cardiovascular risk: General aspects. Pharmacol Res 129:95–99. doi: 10.1016/j.phrs.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 3.Regitz-Zagrosek V, Kararigas G (2017) Mechanistic Pathways of Sex Differences in Cardiovascular Disease. Physiol Rev 97 (1):1–37. doi: 10.1152/physrev.00021.2015 [DOI] [PubMed] [Google Scholar]
- 4.Colafella KMM, Denton KM (2018) Sex-specific differences in hypertension and associated cardiovascular disease. Nat Rev Nephrol 14 (3):185–201. doi: 10.1038/nrneph.2017.189 [DOI] [PubMed] [Google Scholar]
- 5.Komukai K, Mochizuki S, Yoshimura M (2010) Gender and the renin-angiotensin-aldosterone system. Fundam Clin Pharmacol 24 (6):687–698. doi: 10.1111/j.1472-8206.2010.00854.x [DOI] [PubMed] [Google Scholar]
- 6.White MC, Fleeman R, Arnold AC (2019) Sex differences in the metabolic effects of the renin-angiotensin system. Biol Sex Differ 10 (1):31. doi: 10.1186/s13293-019-0247-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Atucha A, Izagirre A, Fraile-Bermudez AB, Kortajarena M, Larrinaga G, Martinez-Lage P, Echevarria E, Gil J (2017) Sex differences in the aging pattern of renin-angiotensin system serum peptidases. Biol Sex Differ 8:5. doi: 10.1186/s13293-017-0128-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dasinger JH, Alexander BT (2016) Gender differences in developmental programming of cardiovascular diseases. Clin Sci (Lond) 130 (5):337–348. doi: 10.1042/CS20150611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurtz A (2011) Renin release: sites, mechanisms, and control. Annu Rev Physiol 73:377–399. doi: 10.1146/annurev-physiol-012110-142238 [DOI] [PubMed] [Google Scholar]
- 10.Miller AJ, Arnold AC (2019) The renin-angiotensin system in cardiovascular autonomic control: recent developments and clinical implications. Clin Auton Res 29 (2):231–243. doi: 10.1007/s10286-018-0572-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemarie CA, Schiffrin EL (2010) The angiotensin II type 2 receptor in cardiovascular disease. J Renin Angiotensin Aldosterone Syst 11 (1):19–31. doi: 10.1177/1470320309347785 [DOI] [PubMed] [Google Scholar]
- 12.Medina D, Arnold AC (2019) Angiotensin-(1–7): Translational Avenues in Cardiovascular Control. Am J Hypertens 32 (12):1133–1142. doi: 10.1093/ajh/hpz146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, Campagnole-Santos MJ (2018) The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol Rev 98 (1):505–553. doi: 10.1152/physrev.00023.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stocco C (2012) Tissue physiology and pathology of aromatase. Steroids 77 (1–2):27–35. doi: 10.1016/j.steroids.2011.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer M, Baessler A, Schunkert H (2002) Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res 53 (3):672–677. doi: 10.1016/s0008-6363(01)00479-5 [DOI] [PubMed] [Google Scholar]
- 16.Schunkert H, Danser AH, Hense HW, Derkx FH, Kurzinger S, Riegger GA (1997) Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation 95 (1):39–45. doi: 10.1161/01.cir.95.1.39 [DOI] [PubMed] [Google Scholar]
- 17.De Lignieres B, Basdevant A, Thomas G, Thalabard JC, Mercier-Bodard C, Conard J, Guyene TT, Mairon N, Corvol P, Guy-Grand B, et al. (1986) Biological effects of estradiol-17 beta in postmenopausal women: oral versus percutaneous administration. J Clin Endocrinol Metab 62 (3):536–541. doi: 10.1210/jcem-62-3-536 [DOI] [PubMed] [Google Scholar]
- 18.Hassager C, Riis BJ, Strom V, Guyene TT, Christiansen C (1987) The long-term effect of oral and percutaneous estradiol on plasma renin substrate and blood pressure. Circulation 76 (4):753–758. doi: 10.1161/01.cir.76.4.753 [DOI] [PubMed] [Google Scholar]
- 19.Roesch DM, Tian Y, Zheng W, Shi M, Verbalis JG, Sandberg K (2000) Estradiol attenuates angiotensin-induced aldosterone secretion in ovariectomized rats. Endocrinology 141 (12):4629–4636. doi: 10.1210/endo.141.12.7822 [DOI] [PubMed] [Google Scholar]
- 20.Wu Z, Maric C, Roesch DM, Zheng W, Verbalis JG, Sandberg K (2003) Estrogen regulates adrenal angiotensin AT1 receptors by modulating AT1 receptor translation. Endocrinology 144 (7):3251–3261. doi: 10.1210/en.2003-0015 [DOI] [PubMed] [Google Scholar]
- 21.Ichiki T, Usui M, Kato M, Funakoshi Y, Ito K, Egashira K, Takeshita A (1998) Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension 31 (1 Pt 2):342–348. doi: 10.1161/01.hyp.31.1.342 [DOI] [PubMed] [Google Scholar]
- 22.Brosnihan KB, Li P, Ganten D, Ferrario CM (1997) Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS. Am J Physiol 273 (6):R1908–1915. doi: 10.1152/ajpregu.1997.273.6.R1908 [DOI] [PubMed] [Google Scholar]
- 23.Xu X, Xiao JC, Luo LF, Wang S, Zhang JP, Huang JJ, Liu ML, Liu CG, Xu KQ, Li YJ, Song HP (2008) Effects of ovariectomy and 17beta-estradiol treatment on the renin-angiotensin system, blood pressure, and endothelial ultrastructure. Int J Cardiol 130 (2):196–204. doi: 10.1016/j.ijcard.2007.08.041 [DOI] [PubMed] [Google Scholar]
- 24.Chao HH, Chen JJ, Chen CH, Lin H, Cheng CF, Lian WS, Chen YL, Juan SH, Liu JC, Liou JY, Chan P, Cheng TH (2005) Inhibition of angiotensin II induced endothelin-1 gene expression by 17-beta-oestradiol in rat cardiac fibroblasts. Heart 91 (5):664–669. doi: 10.1136/hrt.2003.031898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichikawa J, Sumino H, Ichikawa S, Ozaki M (2006) Different effects of transdermal and oral hormone replacement therapy on the renin-angiotensin system, plasma bradykinin level, and blood pressure of normotensive postmenopausal women. Am J Hypertens 19 (7):744–749. doi: 10.1016/j.amjhyper.2005.10.006 [DOI] [PubMed] [Google Scholar]
- 26.Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, Karounos M, Cassis LA (2012) Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol 32 (6):1392–1399. doi: 10.1161/ATVBAHA.112.248559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sullivan JC, Rodriguez-Miguelez P, Zimmerman MA, Harris RA (2015) Differences in angiotensin (1–7) between men and women. Am J Physiol Heart Circ Physiol 308 (9):H1171–1176. doi: 10.1152/ajpheart.00897.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mompeon A, Lazaro-Franco M, Bueno-Beti C, Perez-Cremades D, Vidal-Gomez X, Monsalve E, Gironacci MM, Hermenegildo C, Novella S (2016) Estradiol, acting through ERalpha, induces endothelial non-classic renin-angiotensin system increasing angiotensin 1–7 production. Mol Cell Endocrinol 422:1–8. doi: 10.1016/j.mce.2015.11.004 [DOI] [PubMed] [Google Scholar]
- 29.Bukowska A, Spiller L, Wolke C, Lendeckel U, Weinert S, Hoffmann J, Bornfleth P, Kutschka I, Gardemann A, Isermann B, Goette A (2017) Protective regulation of the ACE2/ACE gene expression by estrogen in human atrial tissue from elderly men. Exp Biol Med (Maywood) 242 (14):1412–1423. doi: 10.1177/1535370217718808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SH, Lee YH, Jung SW, Kim DJ, Park SH, Song SJ, Jeong KH, Moon JY, Ihm CG, Lee TW, Kim JS, Sohn IS, Lee SY, Kim DO, Kim YG (2019) Sex-related differences in the intratubular renin-angiotensin system in two-kidney, one-clip hypertensive rats. Am J Physiol Renal Physiol 317 (3):F670–F682. doi: 10.1152/ajprenal.00451.2018 [DOI] [PubMed] [Google Scholar]
- 31.Baiardi G, Macova M, Armando I, Ando H, Tyurmin D, Saavedra JM (2005) Estrogen upregulates renal angiotensin II AT1 and AT2 receptors in the rat. Regul Pept 124 (1–3):7–17. doi: 10.1016/j.regpep.2004.06.021 [DOI] [PubMed] [Google Scholar]
- 32.Macova M, Armando I, Zhou J, Baiardi G, Tyurmin D, Larrayoz-Roldan IM, Saavedra JM (2008) Estrogen reduces aldosterone, upregulates adrenal angiotensin II AT2 receptors and normalizes adrenomedullary Fra-2 in ovariectomized rats. Neuroendocrinology 88 (4):276–286. doi: 10.1159/000150977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshimura Y, Karube M, Aoki H, Oda T, Koyama N, Nagai A, Akimoto Y, Hirano H, Nakamura Y (1996) Angiotensin II induces ovulation and oocyte maturation in rabbit ovaries via the AT2 receptor subtype. Endocrinology 137 (4):1204–1211. doi: 10.1210/endo.137.4.8625890 [DOI] [PubMed] [Google Scholar]
- 34.Vargas-Castillo A, Tobon-Cornejo S, Del Valle-Mondragon L, Torre-Villalvazo I, Schcolnik-Cabrera A, Guevara-Cruz M, Pichardo-Ontiveros E, Fuentes-Romero R, Bader M, Alenina N, Vidal-Puig A, Hong E, Torres N, Tovar AR (2019) Angiotensin-(1–7) induces beige fat thermogenesis through the Mas receptor. Metabolism 103:154048. doi: 10.1016/j.metabol.2019.154048 [DOI] [PubMed] [Google Scholar]
- 35.Costa-Fraga FP, Goncalves GK, Souza-Neto FP, Reis AM, Capettini LA, Santos RA, Fraga-Silva RA, Stergiopulos N, da Silva RF (2018) Age-related changes in vascular responses to angiotensin-(1–7) in female mice. J Renin Angiotensin Aldosterone Syst 19 (3):1470320318789332. doi: 10.1177/1470320318789332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Novella S, Perez-Cremades D, Mompeon A, Hermenegildo C (2019) Mechanisms underlying the influence of oestrogen on cardiovascular physiology in women. J Physiol 597 (19):4873–4886. doi: 10.1113/JP278063 [DOI] [PubMed] [Google Scholar]
- 37.Ellison KE, Ingelfinger JR, Pivor M, Dzau VJ (1989) Androgen regulation of rat renal angiotensinogen messenger RNA expression. J Clin Invest 83 (6):1941–1945. doi: 10.1172/JCI114102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YF, Naftilan AJ, Oparil S (1992) Androgen-dependent angiotensinogen and renin messenger RNA expression in hypertensive rats. Hypertension 19 (5):456–463. doi: 10.1161/01.hyp.19.5.456 [DOI] [PubMed] [Google Scholar]
- 39.Baltatu O, Cayla C, Iliescu R, Andreev D, Bader M (2003) Abolition of end-organ damage by antiandrogen treatment in female hypertensive transgenic rats. Hypertension 41 (3 Pt 2):830–833. doi: 10.1161/01.HYP.0000048702.55183.89 [DOI] [PubMed] [Google Scholar]
- 40.Chinnathambi V, More AS, Hankins GD, Yallampalli C, Sathishkumar K (2014) Gestational exposure to elevated testosterone levels induces hypertension via heightened vascular angiotensin II type 1 receptor signaling in rats. Biol Reprod 91 (1):6. doi: 10.1095/biolreprod.114.118968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freshour JR, Chase SE, Vikstrom KL (2002) Gender differences in cardiac ACE expression are normalized in androgen-deprived male mice. Am J Physiol Heart Circ Physiol 283 (5):H1997–2003. doi: 10.1152/ajpheart.01054.2001 [DOI] [PubMed] [Google Scholar]
- 42.Leung PS, Wong TP, Chung YW, Chan HC (2002) Androgen dependent expression of AT1 receptor and its regulation of anion secretion in rat epididymis. Cell Biol Int 26 (1):117–122. doi: 10.1006/cbir.2001.0830 [DOI] [PubMed] [Google Scholar]
- 43.Mishra JS, More AS, Gopalakrishnan K, Kumar S (2019) Testosterone plays a permissive role in angiotensin II-induced hypertension and cardiac hypertrophy in male rats. Biol Reprod 100 (1):139–148. doi: 10.1093/biolre/ioy179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mishra JS, Hankins GD, Kumar S (2016) Testosterone downregulates angiotensin II type-2 receptor via androgen receptor-mediated ERK1/2 MAP kinase pathway in rat aorta. J Renin Angiotensin Aldosterone Syst 17 (4). doi: 10.1177/1470320316674875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Douglas GC, O’Bryan MK, Hedger MP, Lee DK, Yarski MA, Smith AI, Lew RA (2004) The novel angiotensin-converting enzyme (ACE) homolog, ACE2, is selectively expressed by adult Leydig cells of the testis. Endocrinology 145 (10):4703–4711. doi: 10.1210/en.2004-0443 [DOI] [PubMed] [Google Scholar]
- 46.Kang NN, Fu L, Xu J, Han Y, Cao JX, Sun JF, Zheng M (2012) Testosterone improves cardiac function and alters angiotensin II receptors in isoproterenol-induced heart failure. Arch Cardiovasc Dis 105 (2):68–76. doi: 10.1016/j.acvd.2011.12.002 [DOI] [PubMed] [Google Scholar]
- 47.dos Santos RL, da Silva FB, Ribeiro RF Jr., Stefanon I (2014) Sex hormones in the cardiovascular system. Horm Mol Biol Clin Investig 18 (2):89–103. doi: 10.1515/hmbci-2013-0048 [DOI] [PubMed] [Google Scholar]
- 48.Kang AK, Duncan JA, Cattran DC, Floras JS, Lai V, Scholey JW, Miller JA (2001) Effect of oral contraceptives on the renin angiotensin system and renal function. Am J Physiol Regul Integr Comp Physiol 280 (3):R807–813. doi: 10.1152/ajpregu.2001.280.3.R807 [DOI] [PubMed] [Google Scholar]
- 49.Zakheim RM, Molteni A, Mattioli L, Mullis KB (1976) Angiotensin I-converting enzyme and angiotensin II levels in women receiving an oral contraceptive. J Clin Endocrinol Metab 42 (3):588–589. doi: 10.1210/jcem-42-3-588 [DOI] [PubMed] [Google Scholar]
- 50.Nickenig G, Strehlow K, Wassmann S, Baumer AT, Albory K, Sauer H, Bohm M (2000) Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation 102 (15):1828–1833. doi: 10.1161/01.cir.102.15.1828 [DOI] [PubMed] [Google Scholar]
- 51.Donadio MV, Gomes CM, Sagae SC, Franci CR, Anselmo-Franci JA, Lucion AB, Sanvitto GL (2006) Estradiol and progesterone modulation of angiotensin II receptors in the arcuate nucleus of ovariectomized and lactating rats. Brain Res 1083 (1):103–109. doi: 10.1016/j.brainres.2006.02.018 [DOI] [PubMed] [Google Scholar]
- 52.Seltzer A, Tsutsumi K, Shigematsu K, Saavedra JM (1993) Reproductive hormones modulate angiotensin II AT1 receptors in the dorsomedial arcuate nucleus of the female rat. Endocrinology 133 (2):939–941. doi: 10.1210/endo.133.2.8344227 [DOI] [PubMed] [Google Scholar]
- 53.Chesley LC, Tepper IH (1967) Effects of progesterone and estrogen on the sensitivity to angiotensin II. J Clin Endocrinol Metab 27 (4):576–581. doi: 10.1210/jcem-27-4-576 [DOI] [PubMed] [Google Scholar]
- 54.Johnson MC, Vega M, Vantman D, Troncoso JL, Devoto L (1997) Regulatory role of angiotensin II on progesterone production by cultured human granulosa cells. Expression of angiotensin II type-2 receptor. Mol Hum Reprod 3 (8):663–668. doi: 10.1093/molehr/3.8.663 [DOI] [PubMed] [Google Scholar]
- 55.Shefer G, Marcus Y, Knoll E, Dolkart O, Foichtwanger S, Nevo N, Limor R, Stern N (2016) Angiotensin 1–7 Is a Negative Modulator of Aldosterone Secretion In Vitro and In Vivo. Hypertension 68 (2):378–384. doi: 10.1161/HYPERTENSIONAHA.116.07088 [DOI] [PubMed] [Google Scholar]
- 56.Funder JW (2013) Mineralocorticoid receptor antagonists: emerging roles in cardiovascular medicine. Integr Blood Press Control 6:129–138. doi: 10.2147/IBPC.S13783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szmuilowicz ED, Adler GK, Williams JS, Green DE, Yao TM, Hopkins PN, Seely EW (2006) Relationship between aldosterone and progesterone in the human menstrual cycle. J Clin Endocrinol Metab 91 (10):3981–3987. doi: 10.1210/jc.2006-1154 [DOI] [PubMed] [Google Scholar]
- 58.Stachenfeld NS, Taylor HS (2005) Progesterone increases plasma volume independent of estradiol. J Appl Physiol (1985) 98 (6):1991–1997. doi: 10.1152/japplphysiol.00031.2005 [DOI] [PubMed] [Google Scholar]
- 59.Faulkner JL, Kennard S, Huby AC, Antonova G, Lu Q, Jaffe IZ, Patel VS, Fulton DJR, Belin de Chantemele EJ (2019) Progesterone Predisposes Females to Obesity-Associated Leptin-Mediated Endothelial Dysfunction via Upregulating Endothelial MR (Mineralocorticoid Receptor) Expression. Hypertension 74 (3):678–686. doi: 10.1161/HYPERTENSIONAHA.119.12802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng Y, Li Q, Zhang Y, Wen Q, Zhao J (2015) Effects of female sex hormones on expression of the Ang-(1–7)/Mas-R/nNOS pathways in rat brain. Can J Physiol Pharmacol 93 (11):993–998. doi: 10.1139/cjpp-2015-0087 [DOI] [PubMed] [Google Scholar]
- 61.Samuel CS, Unemori EN, Mookerjee I, Bathgate RA, Layfield SL, Mak J, Tregear GW, Du XJ (2004) Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology 145 (9):4125–4133. doi: 10.1210/en.2004-0209 [DOI] [PubMed] [Google Scholar]
- 62.Phie J, Haleagrahara N, Newton P, Constantinoiu C, Sarnyai Z, Chilton L, Kinobe R (2015) Prolonged Subcutaneous Administration of Oxytocin Accelerates Angiotensin II-Induced Hypertension and Renal Damage in Male Rats. PLoS One 10 (9):e0138048. doi: 10.1371/journal.pone.0138048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Massiera F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulange A, Negrel R, Ailhaud G, Seydoux J, Meneton P, Teboul M (2001) Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J 15 (14):2727–2729. doi: 10.1096/fj.01-0457fje [DOI] [PubMed] [Google Scholar]
- 64.Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, Daugherty A, Cassis LA (2012) Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension 60 (6):1524–1530. doi: 10.1161/HYPERTENSIONAHA.112.192690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A, Cassis LA (2012) Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol 302 (2):R244–251. doi: 10.1152/ajpregu.00323.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moreno C, Hoffman M, Stodola TJ, Didier DN, Lazar J, Geurts AM, North PE, Jacob HJ, Greene AS (2011) Creation and characterization of a renin knockout rat. Hypertension 57 (3):614–619. doi: 10.1161/HYPERTENSIONAHA.110.163840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gradman AH, Weir MR, Wright M, Bush CA, Keefe DL (2010) Efficacy, safety and tolerability of aliskiren, a direct renin inhibitor, in women with hypertension: a pooled analysis of eight studies. J Hum Hypertens 24 (11):721–729. doi: 10.1038/jhh.2010.11 [DOI] [PubMed] [Google Scholar]
- 68.Gatineau E, Cohn DM, Poglitsch M, Loria AS, Gong M, Yiannikouris F (2019) Losartan prevents the elevation of blood pressure in adipose-PRR deficient female mice while elevated circulating sPRR activates the renin-angiotensin system. Am J Physiol Heart Circ Physiol 316 (3):H506–H515. doi: 10.1152/ajpheart.00473.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krege JH, John SW, Langenbach LL, Hodgin JB, Hagaman JR, Bachman ES, Jennette JC, O’Brien DA, Smithies O (1995) Male-female differences in fertility and blood pressure in ACE-deficient mice. Nature 375 (6527):146–148. doi: 10.1038/375146a0 [DOI] [PubMed] [Google Scholar]
- 70.Toering TJ, van der Graaf AM, Visser FW, Buikema H, Navis G, Faas MM, Lely AT (2015) Gender differences in response to acute and chronic angiotensin II infusion: a translational approach. Physiol Rep 3 (7). doi: 10.14814/phy2.12434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sampson AK, Moritz KM, Denton KM (2012) Postnatal ontogeny of angiotensin receptors and ACE2 in male and female rats. Gend Med 9 (1):21–32. doi: 10.1016/j.genm.2011.12.003 [DOI] [PubMed] [Google Scholar]
- 72.Xue B, Pamidimukkala J, Hay M (2005) Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol 288 (5):H2177–2184. doi: 10.1152/ajpheart.00969.2004 [DOI] [PubMed] [Google Scholar]
- 73.Brosnihan KB, Hodgin JB, Smithies O, Maeda N, Gallagher P (2008) Tissue-specific regulation of ACE/ACE2 and AT1/AT2 receptor gene expression by oestrogen in apolipoprotein E/oestrogen receptor-alpha knock-out mice. Exp Physiol 93 (5):658–664. doi: 10.1113/expphysiol.2007.041806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Silva-Antonialli MM, Tostes RC, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MH, Fortes ZB, Nigro D (2004) A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res 62 (3):587–593. doi: 10.1016/j.cardiores.2004.01.020 [DOI] [PubMed] [Google Scholar]
- 75.Sampson AK, Hilliard LM, Moritz KM, Thomas MC, Tikellis C, Widdop RE, Denton KM (2012) The arterial depressor response to chronic low-dose angiotensin II infusion in female rats is estrogen dependent. Am J Physiol Regul Integr Comp Physiol 302 (1):R159–165. doi: 10.1152/ajpregu.00256.2011 [DOI] [PubMed] [Google Scholar]
- 76.Xue B, Zhao Y, Johnson AK, Hay M (2008) Central estrogen inhibition of angiotensin II-induced hypertension in male mice and the role of reactive oxygen species. Am J Physiol Heart Circ Physiol 295 (3):H1025–H1032. doi: 10.1152/ajpheart.00021.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xue B, Pamidimukkala J, Lubahn DB, Hay M (2007) Estrogen receptor-alpha mediates estrogen protection from angiotensin II-induced hypertension in conscious female mice. Am J Physiol Heart Circ Physiol 292 (4):H1770–1776. doi: 10.1152/ajpheart.01011.2005 [DOI] [PubMed] [Google Scholar]
- 78.Xue B, Zhang Z, Beltz TG, Guo F, Hay M, Johnson AK (2014) Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am J Physiol Heart Circ Physiol 307 (2):H191–198. doi: 10.1152/ajpheart.01012.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bundalo MM, Zivkovic MD, Romic S, Tepavcevic SN, Koricanac GB, Djuric TM, Stankovic AD (2016) Fructose-rich diet induces gender-specific changes in expression of the renin-angiotensin system in rat heart and upregulates the ACE/AT1R axis in the male rat aorta. J Renin Angiotensin Aldosterone Syst 17 (2):1470320316642915. doi: 10.1177/1470320316642915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ramirez LA, Sullivan JC (2018) Sex Differences in Hypertension: Where We Have Been and Where We Are Going. Am J Hypertens 31 (12):1247–1254. doi: 10.1093/ajh/hpy148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cohall DH, Scantlebury-Manning T, James S, Hall K, Ferrario CM (2015) Renin-angiotensin-aldosterone system gender differences in an Afro-Caribbean population. J Renin Angiotensin Aldosterone Syst 16 (3):539–546. doi: 10.1177/1470320314523659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Reyes-Engel A, Morcillo L, Aranda FJ, Ruiz M, Gaitan MJ, Mayor-Olea A, Aranda P, Ferrario CM (2006) Influence of gender and genetic variability on plasma angiotensin peptides. J Renin Angiotensin Aldosterone Syst 7 (2):92–97. doi: 10.3317/jraas.2006.015 [DOI] [PubMed] [Google Scholar]
- 83.Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI (2014) Update on the Angiotensin converting enzyme 2-Angiotensin (1–7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne) 4:201. doi: 10.3389/fendo.2013.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Taylor LE, Sullivan JC (2016) Sex differences in obesity-induced hypertension and vascular dysfunction: a protective role for estrogen in adipose tissue inflammation? Am J Physiol Regul Integr Comp Physiol 311 (4):R714–R720. doi: 10.1152/ajpregu.00202.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xue B, Zhang Z, Johnson RF, Guo F, Hay M, Johnson AK (2013) Central endogenous angiotensin-(1–7) protects against aldosterone/NaCl-induced hypertension in female rats. Am J Physiol Heart Circ Physiol 305 (5):H699–705. doi: 10.1152/ajpheart.00193.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shoemaker R, Tannock LR, Su W, Gong M, Gurley SB, Thatcher SE, Yiannikouris F, Ensor CM, Cassis LA (2019) Adipocyte deficiency of ACE2 increases systolic blood pressures of obese female C57BL/6 mice. Biol Sex Differ 10 (1):45. doi: 10.1186/s13293-019-0260-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen YY, Zhang P, Zhou XM, Liu D, Zhong JC, Zhang CJ, Jin LJ, Yu HM (2018) Relationship between genetic variants of ACE2 gene and circulating levels of ACE2 and its metabolites. J Clin Pharm Ther 43 (2):189–195. doi: 10.1111/jcpt.12625 [DOI] [PubMed] [Google Scholar]
- 88.Walther T, Wessel N, Kang N, Sander A, Tschope C, Malberg H, Bader M, Voss A (2000) Altered heart rate and blood pressure variability in mice lacking the Mas protooncogene. Braz J Med Biol Res 33 (1):1–9. doi: [DOI] [PubMed] [Google Scholar]
- 89.Wang Y, Shoemaker R, Powell D, Su W, Thatcher S, Cassis L (2017) Differential effects of Mas receptor deficiency on cardiac function and blood pressure in obese male and female mice. Am J Physiol Heart Circ Physiol 312 (3):H459–H468. doi: 10.1152/ajpheart.00498.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hilliard LM, Mirabito KM, Denton KM (2013) Unmasking the potential of the angiotensin AT2 receptor as a therapeutic target in hypertension in men and women: what we know and what we still need to find out. Clin Exp Pharmacol Physiol 40 (8):542–550. doi: 10.1111/1440-1681.12067 [DOI] [PubMed] [Google Scholar]
- 91.Prabhushankar R, Krueger C, Manrique C (2014) Membrane estrogen receptors: their role in blood pressure regulation and cardiovascular disease. Curr Hypertens Rep 16 (1):408. doi: 10.1007/s11906-013-0408-6 [DOI] [PubMed] [Google Scholar]
- 92.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD (2018) Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies-Statement From ATVB Council. Arterioscler Thromb Vasc Biol 38 (2):292–303. doi: 10.1161/ATVBAHA.117.309524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Henriques T, Zhang X, Yiannikouris FB, Daugherty A, Cassis LA (2008) Androgen increases AT1a receptor expression in abdominal aortas to promote angiotensin II-induced AAAs in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 28 (7):1251–1256. doi: 10.1161/ATVBAHA.107.160382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lo RC, Schermerhorn ML (2016) Abdominal aortic aneurysms in women. J Vasc Surg 63 (3):839–844. doi: 10.1016/j.jvs.2015.10.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thatcher SE, Zhang X, Woody S, Wang Y, Alsiraj Y, Charnigo R, Daugherty A, Cassis LA (2015) Exogenous 17-beta estradiol administration blunts progression of established angiotensin II-induced abdominal aortic aneurysms in female ovariectomized mice. Biol Sex Differ 6:12. doi: 10.1186/s13293-015-0030-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Daugherty A, Manning MW, Cassis LA (2001) Antagonism of AT2 receptors augments angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol 134 (4):865–870. doi: 10.1038/sj.bjp.0704331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li W, Li J, Hao P, Chen W, Meng X, Li H, Zhang Y, Zhang C, Yang J (2016) Imbalance between angiotensin II and angiotensin-(1–7) in human coronary atherosclerosis. J Renin Angiotensin Aldosterone Syst 17 (3). doi: 10.1177/1470320316659618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thatcher SE, Zhang X, Howatt DA, Yiannikouris F, Gurley SB, Ennis T, Curci JA, Daugherty A, Cassis LA (2014) Angiotensin-converting enzyme 2 decreases formation and severity of angiotensin II-induced abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 34 (12):2617–2623. doi: 10.1161/ATVBAHA.114.304613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stegbauer J, Thatcher SE, Yang G, Bottermann K, Rump LC, Daugherty A, Cassis LA (2019) Mas receptor deficiency augments angiotensin II-induced atherosclerosis and aortic aneurysm ruptures in hypercholesterolemic male mice. J Vasc Surg 70 (5):1658–1668 e1651. doi: 10.1016/j.jvs.2018.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bihl JC, Zhang C, Zhao Y, Xiao X, Ma X, Chen Y, Chen S, Zhao B, Chen Y (2015) Angiotensin-(1–7) counteracts the effects of Ang II on vascular smooth muscle cells, vascular remodeling and hemorrhagic stroke: Role of the NFsmall ka, CyrillicB inflammatory pathway. Vascul Pharmacol 73:115–123. doi: 10.1016/j.vph.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rosenkranz S (2004) TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res 63 (3):423–432. doi: 10.1016/j.cardiores.2004.04.030 [DOI] [PubMed] [Google Scholar]
- 102.Keller KM, Howlett SE (2016) Sex Differences in the Biology and Pathology of the Aging Heart. Can J Cardiol 32 (9):1065–1073. doi: 10.1016/j.cjca.2016.03.017 [DOI] [PubMed] [Google Scholar]
- 103.Mathieu S, El Khoury N, Rivard K, Paradis P, Nemer M, Fiset C (2018) Angiotensin II Overstimulation Leads to an Increased Susceptibility to Dilated Cardiomyopathy and Higher Mortality in Female Mice. Scientific reports 8 (1):952. doi: 10.1038/s41598-018-19436-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kratky V, Kikerlova S, Huskova Z, Sadowski J, Kolar F, Cervenka L (2019) Enhanced Renal Vascular Responsiveness to Angiotensin II and Norepinephrine: A Unique Feature of Female Rats with Congestive Heart Failure. Kidney Blood Press Res 44 (5):1128–1141. doi: 10.1159/000502379 [DOI] [PubMed] [Google Scholar]
- 105.Jessup M, Brozena S (2003) Heart failure. N Engl J Med 348 (20):2007–2018. doi: 10.1056/NEJMra021498 [DOI] [PubMed] [Google Scholar]
- 106.Aurigemma GP, Silver KH, McLaughlin M, Mauser J, Gaasch WH (1994) Impact of chamber geometry and gender on left ventricular systolic function in patients > 60 years of age with aortic stenosis. Am J Cardiol 74 (8):794–798. doi: 10.1016/0002-9149(94)90437-5 [DOI] [PubMed] [Google Scholar]
- 107.Cramariuc D, Rogge BP, Lonnebakken MT, Boman K, Bahlmann E, Gohlke-Barwolf C, Chambers JB, Pedersen TR, Gerdts E (2015) Sex differences in cardiovascular outcome during progression of aortic valve stenosis. Heart 101 (3):209–214. doi: 10.1136/heartjnl-2014-306078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Olivetti G, Melissari M, Capasso JM, Anversa P (1991) Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res 68 (6):1560–1568. doi: 10.1161/01.res.68.6.1560 [DOI] [PubMed] [Google Scholar]
- 109.Gerdts E, Okin PM, de Simone G, Cramariuc D, Wachtell K, Boman K, Devereux RB (2008) Gender differences in left ventricular structure and function during antihypertensive treatment: the Losartan Intervention for Endpoint Reduction in Hypertension Study. Hypertension 51 (4):1109–1114. doi: 10.1161/HYPERTENSIONAHA.107.107474 [DOI] [PubMed] [Google Scholar]
- 110.Hudson M, Rahme E, Behlouli H, Sheppard R, Pilote L (2007) Sex differences in the effectiveness of angiotensin receptor blockers and angiotensin converting enzyme inhibitors in patients with congestive heart failure--a population study. Eur J Heart Fail 9 (6–7):602–609. doi: 10.1016/j.ejheart.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 111.Ferreira AJ, Jacoby BA, Araujo CA, Macedo FA, Silva GA, Almeida AP, Caliari MV, Santos RA (2007) The nonpeptide angiotensin-(1–7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am J Physiol Heart Circ Physiol 292 (2):H1113–1119. doi: 10.1152/ajpheart.00828.2006 [DOI] [PubMed] [Google Scholar]
- 112.Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, Speth RC, Raizada MK, Katovich MJ (2007) Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7). Am J Physiol Heart Circ Physiol 292 (2):H736–742. doi: 10.1152/ajpheart.00937.2006 [DOI] [PubMed] [Google Scholar]
- 113.Yamamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M, Tatara Y, Shiota A, Sugano S, Takeda S, Rakugi H, Ogihara T (2006) Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension 47 (4):718–726. doi: 10.1161/01.HYP.0000205833.89478.5b [DOI] [PubMed] [Google Scholar]
- 114.Wang J, Li N, Gao F, Song R, Zhu S, Geng Z (2015) Balance between angiotensin converting enzyme and angiotensin converting enzyme 2 in patients with chronic heart failure. J Renin Angiotensin Aldosterone Syst 16 (3):553–558. doi: 10.1177/1470320315576257 [DOI] [PubMed] [Google Scholar]
- 115.Sama IE, Ravera A, Santema BT, van Goor H, Ter Maaten JM, Cleland JGF, Rienstra M, Friedrich AW, Samani NJ, Ng LL, Dickstein K, Lang CC, Filippatos G, Anker SD, Ponikowski P, Metra M, van Veldhuisen DJ, Voors AA (2020) Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur Heart J 41 (19):1810–1817. doi: 10.1093/eurheartj/ehaa373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mosca L, Barrett-Connor E, Wenger NK (2011) Sex/gender differences in cardiovascular disease prevention: what a difference a decade makes. Circulation 124 (19):2145–2154. doi: 10.1161/CIRCULATIONAHA.110.968792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rabi DM, Khan N, Vallee M, Hladunewich MA, Tobe SW, Pilote L (2008) Reporting on sex-based analysis in clinical trials of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker efficacy. Can J Cardiol 24 (6):491–496. doi: 10.1016/s0828-282x(08)70624-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sullivan JC (2008) Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol 294 (4):R1220–1226. doi: 10.1152/ajpregu.00864.2007 [DOI] [PubMed] [Google Scholar]
- 119.Kostis WJ, Shetty M, Chowdhury YS, Kostis JB (2018) ACE Inhibitor-Induced Angioedema: a Review. Curr Hypertens Rep 20 (7):55. doi: 10.1007/s11906-018-0859-x [DOI] [PubMed] [Google Scholar]
- 120.Song JJ, Ma Z, Wang J, Chen LX, Zhong JC (2020) Gender Differences in Hypertension. J Cardiovasc Transl Res 13 (1):47–54. doi: 10.1007/s12265-019-09888-z [DOI] [PubMed] [Google Scholar]
- 121.Miller JA, Cherney DZ, Duncan JA, Lai V, Burns KD, Kennedy CR, Zimpelmann J, Gao W, Cattran DC, Scholey JW (2006) Gender differences in the renal response to renin-angiotensin system blockade. J Am Soc Nephrol 17 (9):2554–2560. doi: 10.1681/ASN.2005101095 [DOI] [PubMed] [Google Scholar]
- 122.Wang H, Chen H (2016) Gender difference in the response to valsartan/amlodipine single-pill combination in essential hypertension (China Status II): An observational study. J Renin Angiotensin Aldosterone Syst 17 (2):1470320316643903. doi: 10.1177/1470320316643903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Calhoun DA, Sharma K (2010) The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiol Clin 28 (3):517–527. doi: 10.1016/j.ccl.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ (1999) Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res 7 (4):355–362. doi: 10.1002/j.1550-8528.1999.tb00418.x [DOI] [PubMed] [Google Scholar]
- 125.Shukri MZ, Tan JW, Manosroi W, Pojoga LH, Rivera A, Williams JS, Seely EW, Adler GK, Jaffe IZ, Karas RH, Williams GH, Romero JR (2018) Biological Sex Modulates the Adrenal and Blood Pressure Responses to Angiotensin II. Hypertension 71 (6):1083–1090. doi: 10.1161/HYPERTENSIONAHA.117.11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bangalore S, Kumar S, Messerli FH (2010) Angiotensin-converting enzyme inhibitor associated cough: deceptive information from the Physicians’ Desk Reference. Am J Med 123 (11):1016–1030. doi: 10.1016/j.amjmed.2010.06.014 [DOI] [PubMed] [Google Scholar]
- 127.Loloi J, Miller AJ, Bingaman SS, Silberman Y, Arnold AC (2018) Angiotensin-(1–7) contributes to insulin-sensitizing effects of angiotensin-converting enzyme inhibition in obese mice. Am J Physiol Endocrinol Metab 315 (6):E1204–E1211. doi: 10.1152/ajpendo.00281.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schuchard J, Winkler M, Stolting I, Schuster F, Vogt FM, Barkhausen J, Thorns C, Santos RA, Bader M, Raasch W (2015) Lack of weight gain after angiotensin AT1 receptor blockade in diet-induced obesity is partly mediated by an angiotensin-(1–7)/Mas-dependent pathway. Br J Pharmacol 172 (15):3764–3778. doi: 10.1111/bph.13172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Benter IF, Yousif MH, Anim JT, Cojocel C, Diz DI (2006) Angiotensin-(1–7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am J Physiol Heart Circ Physiol 290 (2):H684–691. doi: 10.1152/ajpheart.00632.2005 [DOI] [PubMed] [Google Scholar]