

SUMMARY
Discovering potent human monoclonal antibodies (mAbs) targeting the Plasmodium falciparum circumsporozoite protein (PfCSP) on sporozoites and elucidating their mechanisms of neutralization will facilitate translation for passive prophylaxis and aid next-generation vaccine development. Here, we isolated a neutralizing human mAb, L9, that preferentially bound NVDP minor repeats of PfCSP with high affinity while cross-reacting with NANP major repeats. L9 was more potent than six published neutralizing human PfCSP mAbs at mediating protection against mosquito bite challenge in mice. Isothermal titration calorimetry and multiphoton microscopy showed that L9 and the other most protective mAbs bound PfCSP with two binding events and mediated protection by killing sporozoites in the liver and by preventing their egress from sinusoids and traversal of hepatocytes. This study defines the subdominant PfCSP minor repeats as neutralizing epitopes, identifies an in vitro biophysical correlate of SPZ neutralization, and demonstrates that the liver is an important site for antibodies to prevent malaria.
Graphical Abstract

In Brief (eTOC Blurb)
Wang et al. isolate a potent neutralizing human mAb, L9, that preferentially binds NVDP minor repeats of Plasmodium falciparum circumsporozoite protein (PfCSP) on sporozoites with high affinity while cross-reacting with NANP major repeats. Their findings define the subdominant PfCSP minor repeats as neutralizing epitopes and demonstrate that the liver is an important site for antibodies to prevent malaria.
INTRODUCTION
Malaria is a mosquito-borne parasitic disease affecting ~200–400 million people leading to ~400,000 deaths annually, primarily in children in sub-Saharan Africa (WHO, 2018). Antimalarial drugs, insecticide-treated nets, and other public health interventions contributed to a 50–75% reduction in global malaria cases between 2000–2015 (WHO, 2015). Despite these efforts, malaria incidence has increased in many areas since 2015 (WHO, 2018). These data highlight the need for additional interventions to control and eliminate malaria (Cockburn and Seder, 2018).
A long-sought goal for preventing malaria is the development of an effective vaccine. RTS,S, a protein subunit vaccine administered with the adjuvant AS01, is the most clinically advanced vaccine against Plasmodium falciparum (Pf), the species that accounts for most malaria-associated mortality (Kester et al., 2009; Olotu et al., 2011). In phase III clinical trials, three vaccinations with RTS,S/AS01 conferred ~50% protection against clinical disease at one year and ~30% protection over four years in 5–17 month-old infants (RTS,S Clinical Trials Partnership, 2015). High antibody titers are associated with protection but wane over time and require further vaccine boosting (Bejon et al., 2013; White et al., 2014, 2015). An alternative approach that may mediate higher levels of protection for defined periods of time is passive immunization with potent monoclonal antibodies (mAbs).
Antibodies can prevent malaria by neutralizing sporozoites (SPZ; the infectious form of Plasmodium parasites deposited into the skin when a mosquito bites) before they infect hepatocytes in the liver (Julien and Wardemann, 2019). The major target of anti-PfSPZ antibodies is the Pf circumsporozoite protein (PfCSP). PfCSP is the most abundant SPZ surface protein and is essential for their motility and invasion of hepatocytes (Cerami et al., 1992; Tewari et al., 2002). PfCSP has three domains: an N-terminus, a central region composed of repeating tetrapeptides, and a C-terminus. In the Pf reference isolate 3D7 (PfCSP_3D7), the “junctional region” at the end of the N-terminus and start of the tetrapeptide repeats begins with NPDP followed by 3 interspersed NANP and NVDP repeats. This junctional region is followed by 35 NANP repeats, with a fourth NVDP inserted after the twentieth NANP (Cockburn and Seder, 2018). Structural studies indicate that anti-repeat antibody binding motifs are actually DPNA, NPNV, and NPNA, derived from the joining of major and minor repeats (Dyson et al., 1990; Ghasparian et al., 2006; Oyen et al., 2017, 2018; Plassmeyer et al., 2009). Importantly, RTS,S includes a truncated form of PfCSP with 19 NANP repeats and the C-terminus and so does not contain the N-terminus, NPDP or NVDP repeats (Stoute et al., 1997).
Mouse and human mAbs have been characterized against all domains of PfCSP (Julien and Wardemann, 2019). One N-terminal mAb mediates some protection against SPZ challenge in mice (Espinosa et al., 2015), while no protective C-terminal mAbs are known (Scally et al., 2018). Most neutralizing mAbs bind the repeat region, particularly the immunodominant NANP repeats (Imkeller et al., 2018; Oyen et al., 2017; Triller et al., 2017; Zavala et al., 1983). The isolation of potent human mAbs exhibiting dual specificity for NANP repeats and the unique tetrapeptide, NPDP, at the junction of the N-terminus and repeat region identify this subdominant “junctional epitope” as a site of vulnerability (Kisalu et al., 2018; Tan et al., 2018). These data have led to ongoing efforts to isolate more PfCSP mAbs against epitopes in the junctional region (Oyen et al., 2020).
Here, to discover additional human mAbs against neutralizing epitopes in the junctional region of PfCSP, a junctional probe was used to isolate PfCSP mAbs from a subject immunized with radiation-attenuated PfSPZ. One of these mAbs, L9, preferentially bound NPNV motifs associated with NVDP minor repeats of PfCSP. When compared to a published panel of protective human PfCSP mAbs, L9 potently protected mice against intravenous and mosquito bite SPZ challenge. To correlate the mAb panel’s binding and functional characteristics, isothermal titration calorimetry and multiphoton microscopy were respectively used to define mAb binding to the PfCSP repeat region and visualize mAb-mediated SPZ neutralization in the livers of mice. Collectively, these data classify the complex binding properties of highly protective human PfCSP repeat mAbs, define the in vivo mechanisms by which repeat mAbs neutralize SPZ in the liver, and identify L9 as a promising clinical candidate for passive malaria prophylaxis.
RESULTS
Isolation of a neutralizing human mAb that preferentially binds the PfCSP minor repeats
CIS43, a human mAb targeting the junctional epitope of PfCSP induced by vaccination with radiation-attenuated PfSPZ (Seder et al., 2013), protects mice from SPZ challenge (Kisalu et al., 2018). To isolate additional and potentially more potent junctional mAbs, sera from subjects immunized with higher doses of radiation-attenuated PfSPZ were tested for reactivity against S02, a junctional epitope mimic designed to select for CIS43-like mAbs. One subject with the highest anti-S02 titers that was protected following controlled human malaria infection (CHMI) was selected for mAb isolation. PfCSP-reactive memory B cells were sorted from peripheral blood mononuclear cells collected from this subject using recombinant full-length PfCSP (rPfCSP_FL) and S02 probes. 28 mAbs were cloned that bound rPfCSP.
These 28 PfCSP mAbs were assessed for their ability to reduce parasite liver burden in mice challenged intravenously (IV) with transgenic P. berghei SPZ expressing PfCSP and a green fluorescent protein/luciferase fusion protein (Pb-PfCSP-GFP/Luc-SPZ; hereafter Pb-PfCSP-SPZ) 2 hours after passive transfer, a model used to screen mAbs for in vivo protection against SPZ liver invasion (Raghunandan et al., 2020). All mAbs were benchmarked against CIS43 at a dose of 300 µg/mouse, which confers maximum protection in this model (Kisalu et al., 2018). Only one of these 28 mAbs, L9, reduced liver burden comparably to CIS43 at 300 µg/mouse. At a more limiting dose of 100 µg/mouse, L9 trended towards greater liver burden reduction than CIS43 (P=0.06) at similar serum mAb titers (Figure 1A), suggesting that L9 might be more potent than CIS43.
Figure 1. L9 is a neutralizing human mAb that binds the NPNV motif of PfCSP.
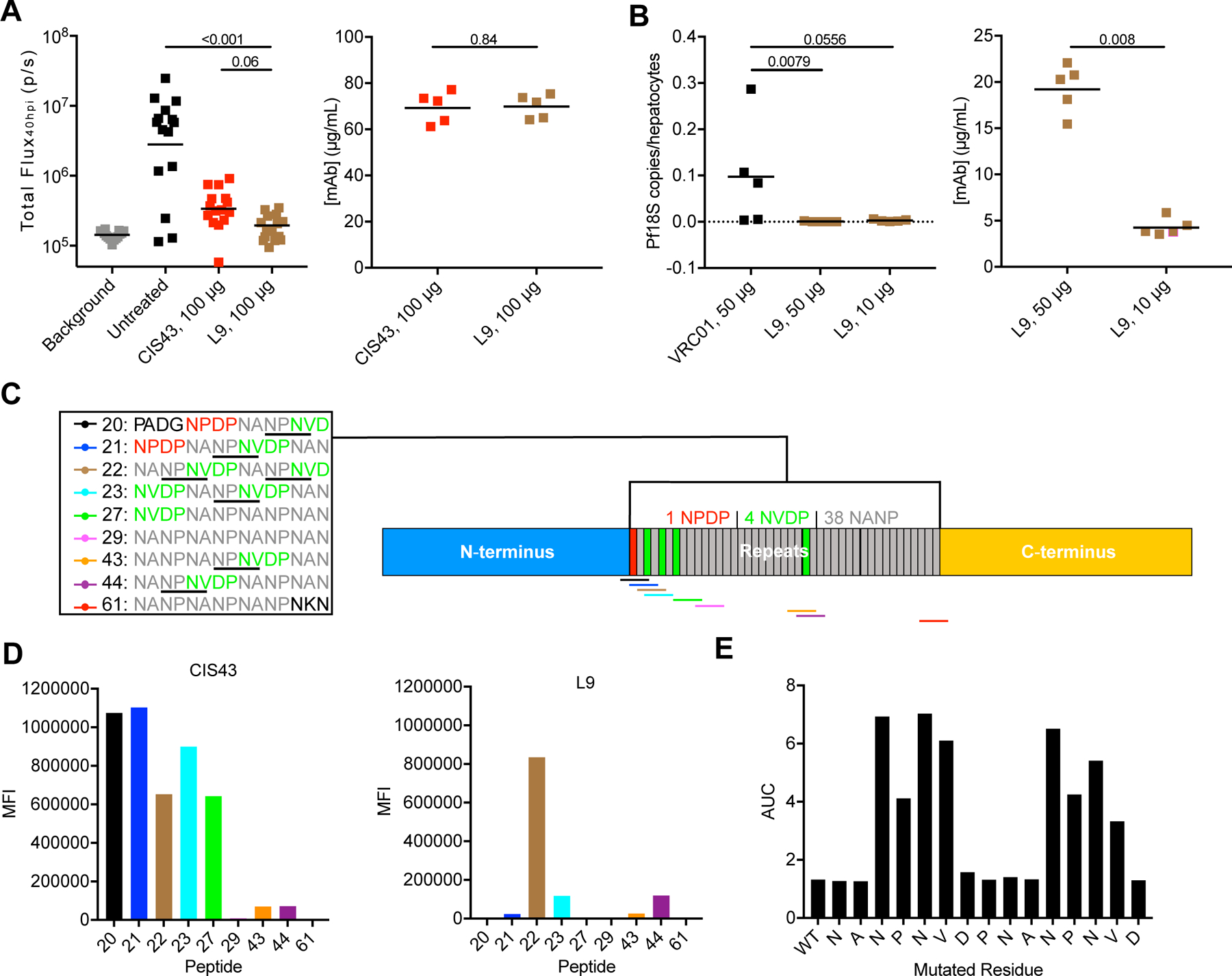
(A) Left: liver burden reduction (bioluminescence; total flux, photons/sec) in B6-albino mice 40 hrs post-infection (hpi; n=15/group; data pooled from three independent experiments) mediated by 100 µg CIS43 or L9 administered 2 hours before IV challenge with 2,000 Pb-PfCSP-GFP/Luc-SPZ. P-values were determined by comparing L9 to CIS43 and untreated control using the Kruskal-Wallis test with Dunn’s post-hoc correction. Right: serum mAb titers 2 hours after administration of 100 µg CIS43 or L9 in separate mice (n=5/group) determined through ELISA. Differences between CIS43 and L9 were determined using the two-tailed Mann–Whitney test. (B) Left: liver burden reduction (Pf 18S rRNA normalized to number of human hepatocytes) in FRG-huHep mice (n=5/group) administered 50 or 10 µg L9 and challenged IV with 100,000 PfSPZ. Right: serum mAb titers in challenged FRG-huHep mice. Differences between VRC01 (anti-HIV-1 isotype control IgG) and L9 (50 vs. 10 µg) were determined using the two-tailed Mann–Whitney test. (C) Schematic of PfCSP_3D7 depicting the N-terminus, repeat region (with color-coded overlapping 15mer peptides 20–61), and C-terminus. Every NPNV motif in each peptide is underlined. (D) ELISA (MFI, median fluorescence intensity) of CIS43 and L9 binding to peptides 20–61. (E) Competition ELISA (AUC, area under the curve) of L9 binding to rPfCSP_FL in the presence of varying concentrations of peptide 22 (WT, leftmost bar) or variant peptides (subsequent bars) where the indicated residue was mutated to alanine or serine. A-B: lines represent geometric mean. See also Figure S1.
To extend these findings and demonstrate that L9 is protective against natural PfSPZ challenge, we assessed whether L9 reduced liver burden in human liver-chimeric (FRG-huHep) mice challenged IV with PfSPZ (Foquet et al., 2018). Remarkably, only 50 or 10 µg of L9 (which resulted in serum titers as low as ~5 µg/mL) reduced liver burden to undetectable levels (Figure 1B). Collectively, these data show that L9 mediates high-level protection against SPZ challenge in two complementary mouse models of Pf malaria infection.
To define the epitope(s) recognized by L9, mapping was done using a series of overlapping peptides spanning the repeat region of PfCSP (peptides 20–61; Figure 1C) and compared to CIS43 as a benchmark (Figure 1D). Consistent with our previous report (Kisalu et al., 2018), CIS43 preferentially bound DPNA-containing peptides 20 (PADGNPDPNANPNVD) and 21 (NPDPNANPNVDPNAN). Conversely, L9 preferentially bound peptide 22 (NANPNVDPNANPNVD). Both mAbs had undetectable binding to peptide 29 (NANPNANPNANPNAN), which contains only NPNA. Alanine scanning of peptide 22 demonstrated that its two NPNV motifs were critical for L9 binding (Figure 1E). Peptide 22 is the only peptide that contains two NPNV motifs (Figure 1C), likely accounting for its strong recognition by L9. These data demonstrate that L9 preferentially binds NPNV minor repeat motifs and exhibits undetectable affinity for the 3 NPNA major repeat motifs in peptide 29.
CIS43 and L9 preferentially bind peptides in the junctional region of PfCSP
Having established L9 as an neutralizing PfCSP mAb, we next compared its binding characteristics to six of the most potent human PfCSP mAbs published to date [CIS43, mAb10 (Kisalu et al., 2018), MGU12 (Tan et al., 2018), 1210 (Imkeller et al., 2018), 311 and 317 (Oyen et al., 2017)]. All mAbs were expressed in the same IgG1 vector and bound comparably to rPfCSP_FL (Figure S1A) and Pb-PfCSP-SPZ (Figure S1B).
The mAb panel’s specificities were mapped by competition ELISA using peptides 20–61 (Figure S1C). All mAbs except CIS43 and L9 preferentially bound peptides with ≥2 NPNA repeats (i.e., peptides 27, 29, 61). Conversely, CIS43 and L9 preferentially bound peptide 21 and 22, respectively, and exhibited no binding to peptide 29. The mAbs’ peptide preferences were substantiated using biolayer interferometry to directly measure their binding response and apparent avidity to peptides 21, 22, and 29 (Figure S1D–F).
Additionally, (NANP)1 to (NANP)9 peptides were used to determine the minimal number of NPNA motifs required for binding (Figure S1G). Consistent with previous reports that the minimum epitope of NPNA-preferring mAbs is 2 NPNA repeats (Imkeller et al., 2018; Oyen et al., 2017, 2018), mAb10, MGU12, 1210, 311, and 317 bound peptides with ≥2 NPNA repeats [(NANP)3–9] with greater affinity than CIS43 and L9. Interestingly, L9 binding increased from nearly undetectable for (NANP)3 to (NANP)9 binding that was comparable to CIS43, though still less than the other mAbs. Together, these data show that all mAbs except CIS43 and L9 prefer NPNA motifs and that, although CIS43 and L9 respectively prefer DPNA and NPNV motifs, they could bind NPNA when these motifs were sufficiently concatenated to form long peptides.
PfCSP mAbs with high junctional affinity bind rPfCSP in two distinct steps
To correlate the mAb panel’s peptide specificities to their recognition of full-length protein, we used isothermal titration calorimetry (ITC) to characterize the affinity and stoichiometry (i.e., valency) of their binding to rPfCSP. CIS43 IgG binds rPfCSP_FL in two sequential high-affinity events by ITC (termed “two-step binding”): the first binding event involves ~1 binding site to NPDP and the second involves ~5 binding sites to NANP repeats (Kisalu et al., 2018).
Remarkably, L9 IgG also showed two high-affinity binding events to rPfCSP_FL: the first event involved ~2 binding sites (dissociation constant KD1 = 22 nM) and the second event involved ~9 binding sites (KD2 = 47 nM) (Figure 2A). Based on L9’s preference for NPNV, we assessed whether the first binding event targeted this motif. The four NVDP repeats in rPfCSP_FL (denoted A, B, C, D) were mutated to NANP in all possible combinations (Figure S2A). L9 binding was reduced to a single event only when all four NVDP were mutated to NANP (rPfCSP_∆ABCD), suggesting that the first high-affinity binding event targeted NPNV motifs and required at least one NPNV (Figure 2A). Furthermore, the affinity of L9 for rPfCSP_∆ABCD decreased 3-fold compared to wild-type (KD1 = 73 nM vs. 22 nM, respectively) while the total stoichiometry (N) was not appreciably perturbed (N=12.7 vs. 11.2), substantiating that L9 could bind concatenated NPNA motifs. L9 demonstrated two-step binding, as well as similar affinity and stoichiometry, for all other NVDP mutants (Figure S2A). As controls, the affinity and stoichiometry of mAb10 and 317 for rPfCSP_∆ABCD were similar (Figure S2B), suggesting that mutating all NVDP to NANP repeats in rPfCSP did not perturb the binding of these NPNA-preferring mAbs. Together, these data show that L9 binds rPfCSP in two steps: the first high-affinity step targets NPNV motifs and the second lower-affinity step involves NPNA motifs.
Figure 2. mAb two-step binding to rPfCSP is associated with high junctional affinity.
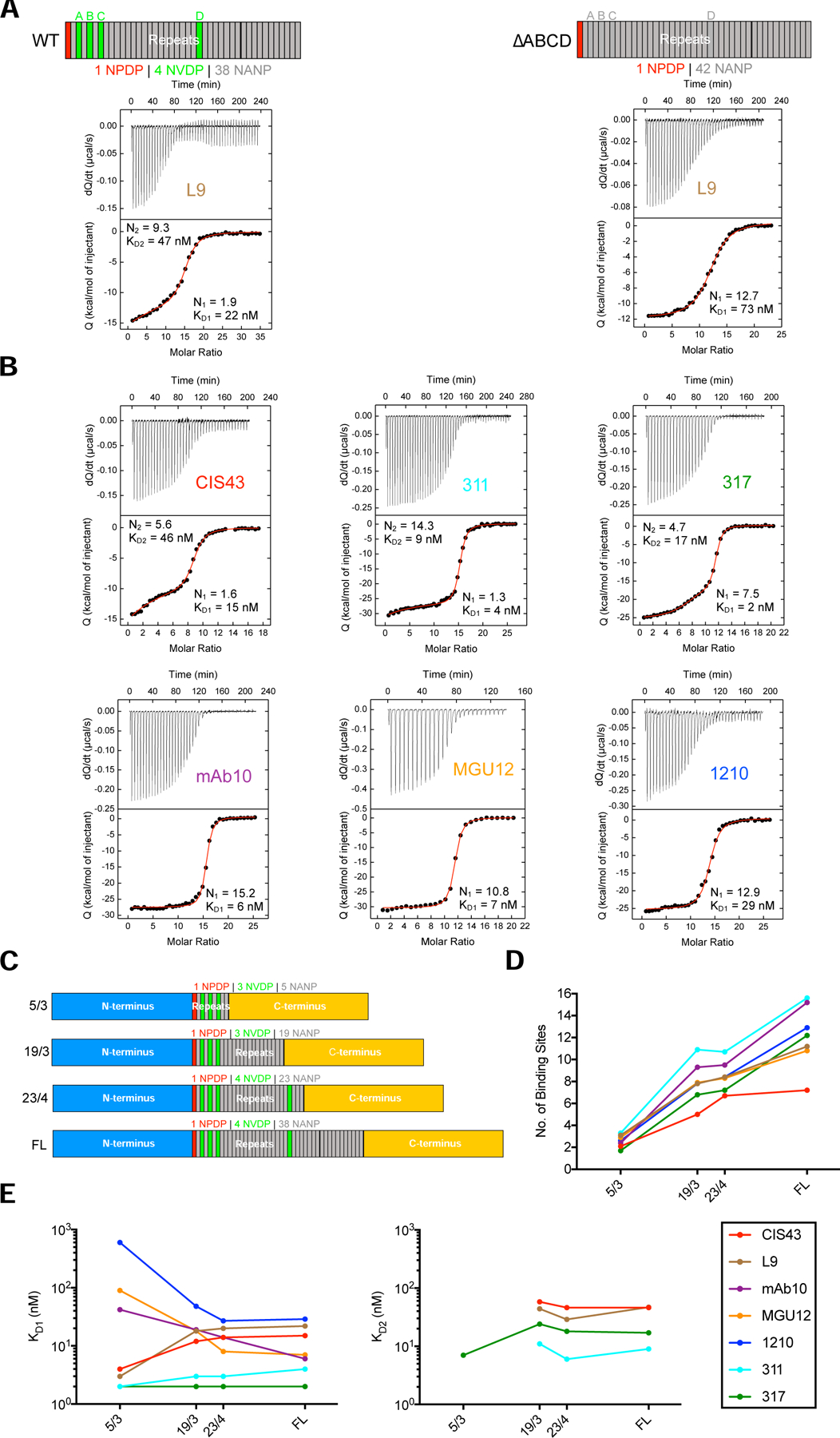
(A) ITC analyses of L9 IgG binding to wild-type rPfCSP (rPfCSP_WT or FL) and rPfCSP with all four NVDP mutated to NANP (rPfCSP_∆ABCD); N- and C-termini in schematics were omitted. Top, dQ/dt (heat flow, Q, as a function of time). Bottom, the integrated heat associated with each IgG injection shown as a function of the molar ratio between IgG antigen binding sites and rPfCSP in the calorimetric cell. The red line represents the result from best nonlinear least squares fit of the data. Dissociation constant (KD) and stoichiometry (N) of binding are shown for binding events 1 and 2. ITC data was fit to a two-step binding model if the IgG titrant bound to two sets of sites with different affinity values. The first set of high-affinity sites is saturated at lower IgG concentrations before the second set of lower-affinity sites. (B) ITC analyses of CIS43, 311, 317, 1210, mAb10, and MGU12 IgG binding to rPfCSP_WT (or FL). (C) Schematics of rPfCSP_FL and rPfCSP mutants with truncated repeat regions (23/4, 19/3, 5/3 NANP/NVDP repeats) and identical N- and C-termini. (D) Aggregate stoichiometry (binding events 1+2; no. of antigen binding sites) of mAb binding to 5/3, 19/3, 23/4, and rPfCSP_FL determined through ITC. (E) Affinity (binding events 1 vs. 2; KD, nM) of mAb binding to 5/3, 19/3, 23/4, and rPfCSP_FL determined through ITC. All ITC plots are representative of 2–3 independent experiments; D-E reflect an average of these experiments. See also Figures S1–2; Tables S1–2, S4.
Since L9 displayed two-step, high-affinity binding to rPfCSP_FL if one NVDP repeat was present, we assessed NVDP conservation in a large number of Pf field isolates (Table S1). While there was considerable variation in both the number and position of NVDP repeats, all isolates contained at least one NVDP and every NVDP was preceded by an NANP or ANP sequence that would give rise to NPNV. Similar to PfCSP_3D7, most (34 of 39, 87%) African isolates contained 4 NVDP repeats. These data show that the NPNV motif is highly conserved, suggesting that L9 should react broadly with circulating Pf strains.
To complete the comparative ITC analyses, the binding affinity and stoichiometry of CIS43, mAb10, MGU12, 1210, 311, and 317 to rPfCSP_FL were determined. All mAbs bound rPfCSP_FL with high affinity (KD1= 2–29 nM) and stoichiometry (~7–16 binding sites) (Figure 2B). Interestingly, 311 and 317 also exhibited two-step binding to rPfCSP_FL while 1210, mAb10, and MGU12 bound in a single step.
To further define the mAbs’ binding affinity and stoichiometry, ITC was performed using three rPfCSP constructs with identical N- and C-termini but truncated repeat regions containing 23/4, 19/3, and 5/3 NANP/NVDP repeats (Figure 2C; Table S2). As the number of repeats were increased, stoichiometry increased linearly for all mAbs except for CIS43, which showed no appreciable binding to the last 15 NANP repeats (Figure 2D), consistent with its lower total stoichiometry (N=7.2 for FL; Figure 2B). This suggests that the binding of every mAb except CIS43 is evenly distributed across the repeat region.
Notably, the stoichiometry of binding event 1 (N1) for CIS43, L9, and 311 was relatively unchanged as the repeat region was truncated (Table S2), suggesting that the epitopes recognized by these mAbs in binding event 1 were located in rPfCSP_5/3. As the repeats were truncated down to 5/3, the KD1 of two-step binding mAbs (CIS43, L9, 311, 317) improved or was unchanged, while the KD1 of single-step binding mAbs (mAb10, MGU12, 1210) worsened (Figure 2E). Overall, these data suggest that the epitope(s) bound by CIS43, L9, and 311 in binding event 1 are located in the junctional region encompassed by rPfCSP_5/3 and that two-step binding is associated with high affinity for rPfCSP_5/3.
CIS43, L9, 311, and 317 mediate the greatest in vivo SPZ neutralization in the liver
Having classified the mAbs based on their binding to PfCSP peptides and proteins, we next evaluated their ability to prevent SPZ invasion of hepatocytes in vitro or in vivo following IV challenge. All mAbs comparably and significantly reduced invasion in vitro (Figure 3A). Conversely, only CIS43, L9, 311, and 317 significantly reduced liver burden following IV challenge (Figure 3B), substantiating that in vitro hepatocyte invasion assays do not predict in vivo liver protection (Kisalu et al., 2018).
Figure 3. Cytotoxic PfCSP mAbs prevent SPZ from accessing and invading hepatocytes.
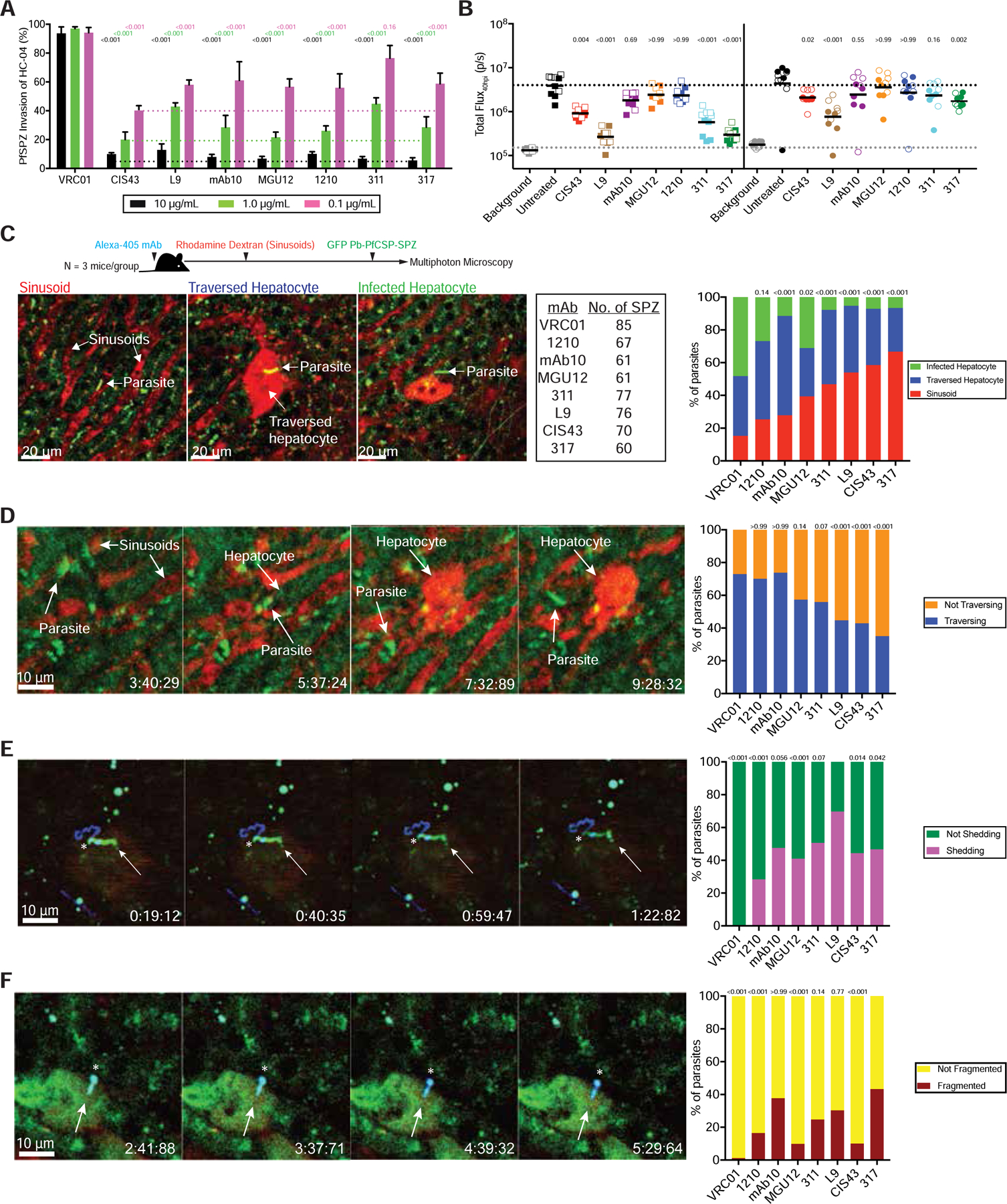
(A) Inhibition of PfSPZ invasion of HC-04 hepatocytes by 10, 1.0, and 0.1 µg/mL PfCSP mAbs. Data were combined from two independent experiments (triplicate wells/mAb). Bars represent the mean ± SEM; dotted lines indicate the highest mAb-mediated inhibition at each concentration. Each mAb was compared to the corresponding concentration of VRC01 using a two-way ANOVA with Bonferroni’s correction. (B) Liver burden reduction in mice 40 hpi (n=10/group; data pooled from two independent experiments, solid vs. open symbols; line represents geometric mean) mediated by 75 (squares) or 25 (circles) µg/mouse mAb administered 2 hours before IV challenge with 2,000 Pb-PfCSP-GFP/Luc-SPZ. Dotted lines indicate the geometric mean of the background and untreated controls. P-values were determined by comparing each mAb to untreated using the Kruskal-Wallis test with Dunn’s post-hoc correction. (C) Left: schema for intravital liver imaging in mice (N=3/group) sequentially administered 30 µg Alexa405-labeled mAb (blinded), 1 µg rhodamine-labeled dextran, and 100,000 Pb-PfCSP-GFP-SPZ. The number of SPZ (No. of SPZ) observed across the 3 independent experiments per mAb were combined for analysis. The locations of individual Pb-PfCSP-SPZ were noted; traversal was detected by dextran uptake into wounded hepatocytes. Representative images depict discrete Pb-PfCSP-SPZ in the sinusoid, traversing a dextran+ hepatocyte, or invading a dextran− hepatocyte. Right: locations of Pb-PfCSP-SPZ in the liver calculated as the percentage of total SPZ observed. (D-F) Left: representative time-lapse images (min:s:ms) of discrete Pb-PfCSP-SPZ (D) exiting a sinusoid and traversing a hepatocyte, (E) shedding a mAb-bound PfCSP tail in a CSPR, and (F) undergoing dotty death. Right: percentages of Pb-PfCSP-SPZ that (D) initiated traversal (i.e., traversed ≥1 hepatocyte), (E) underwent a CSPR, and (F) underwent dotty death. C-F: P-values were determined by comparing each mAb to VRC01, L9, or 317 using the chi-squared test with Bonferroni’s correction. E-F: arrow and star respectively indicate anterior and posterior ends of SPZ. See also Figure S3; Table S4.
To determine the potential in vivo mechanisms contributing to the differences in liver burden reduction meditated by the mAb panel, we developed a multiphoton microscopy assay to quantify SPZ neutralization in the liver following IV inoculation of Alexa 405-labeled mAbs, rhodamine-labeled dextran, and Pb-PfCSP-SPZ (Figure 3C). SPZ were imaged as they exited sinusoids, traversed hepatocytes, and invaded a hepatocyte. Complete prevention of hepatocyte infection was not expected due to the high SPZ inoculum required for these imaging studies.
Consistent with the IV challenge data (Figure 3B), mice that received 311, L9, CIS43, and 317 had the highest percentages (47–67%) of Pb-PfCSP-SPZ in the sinusoids and the lowest percentages (5–8%) of SPZ infecting hepatocytes (Figures 3C, S3A–B). SPZ traversed several hepatocytes before productively invading a hepatocyte, and traversal was detected by dextran uptake into hepatocytes with compromised membranes (Mota et al., 2001; Yang et al., 2017). Only L9, CIS43, and 317 significantly prevented Pb-PfCSP-SPZ from initiating traversal (35–45% traversing versus 73% in control), while 311 trended towards traversal inhibition (P=0.07) (Figures 3D, S3C). Furthermore, CIS43 and L9 significantly lowered the average number of hepatocytes traversed by the ~40% of SPZ that initiated traversal (Figure S3D). Together, these data show that the four most protective mAbs against IV challenge (CIS43, L9, 311, 317) limit hepatocyte infection by preventing Pb-PfCSP-SPZ egress from sinusoids and subsequent traversal of hepatocytes.
Anti-CSP repeat antibodies directly kill SPZ in vitro and in the skin in a process called “dotty death,” which manifests as the gradual disappearance of cytosolic GFP accompanied by a persistent fluorescent dot at the parasite’s posterior pole. Dotty death occurs downstream of the circumsporozoite precipitation reaction (CSPR), wherein antibodies crosslink CSP on SPZ and cause the shedding of CSP as thread-like precipitates (Aliprandini et al., 2018; Vanderberg et al., 1969). Since no studies have assessed whether this cytotoxic process occurs in the liver, we quantified the percentage of Pb-PfCSP-SPZ that underwent a CSPR (Figure 3E). L9 induced ~70% of SPZ to undergo a CSPR, which was significantly higher than all mAbs except 311 (51%) and mAb10 (48%) (Figures 3E, S3E). mAbs induced the CSPR primarily while SPZ were migrating through sinusoids or traversing hepatocytes (57% and 36%, respectively) and rarely after infecting hepatocytes (7%) (Figure S3F).
Subsequently, dotty death was quantified by determining the percentage of immobilized, mAb-bound Pb-PfCSP-SPZ that became fragmented and by counting the number of fragments per dead SPZ (Figure 3F). Only 317, L9, mAb10, and 311 significantly induced dotty death (Figures 3F, S3G–H). Like the CSPR, SPZ underwent dotty death while migrating through sinusoids and traversing hepatocytes (50% and 46%, respectively) and rarely after invading hepatocytes (4%) (Figure S3I). Interestingly, we observed a cytotoxic phenomenon distinct from dotty death in which mAb-bound Pb-PfCSP-SPZ rapidly lost membrane integrity and burst, releasing GFP into the surrounding tissue (Figure S3J). This “bursting” was a rare event and only observed with 317 and L9. Collectively, these data demonstrate that PfCSP mAbs induce the CSPR and cytotoxic death of SPZ in the liver and substantiate that most mAb-mediated SPZ neutralization occurs prior to hepatocyte invasion.
L9, 317, and CIS43 mediate potent protection against mosquito bite challenge
The gold standard for assessing the in vivo functional activity of anti-SPZ mAbs are natural transmission studies wherein mice are challenged with infected mosquitos and assessed for parasitemia (Raghunandan et al., 2020). Thus, sterile protection from parasitemia mediated by the mAbs in the panel was assessed across several mAb doses in multiple blinded, independent studies wherein mice were challenged with five mosquito bites 3 days after passive transfer (Figure S4A–B).
Within each dose, L9 was compared to the other mAbs for sterile protection (Figure 4A). At 600 µg/mouse, 94% of L9-treated mice were sterilely protected, which was comparable to 317 (100%) and CIS43 (94%), trended higher than mAb10 (67%) and 311 (50%), and was significantly greater than 1210 (18%) and MGU12 (0%). At 300 µg/mouse, sterile protection by L9 (64%) trended higher than 317 (44%) and was significantly greater than CIS43, mAb10, 1210 (~20%), 311 (13%), and MGU12 (0%). At 100 µg/mouse, L9 (40%) was significantly more protective than CIS43 (10%) and 317 (0%). To determine whether differences in mAb titers may have contributed to differences in protection, serum mAb titers were measured one day prior to challenge in all challenged mice (Figure 4B). Notably, L9 titers were similar to 317 at 300 and 100 µg/mouse despite L9 providing improved protection. Furthermore, mAb10 titers were significantly lower than the other mAbs at 300 µg/mouse.
Figure 4. L9 is more potently protective than several published PfCSP mAbs.
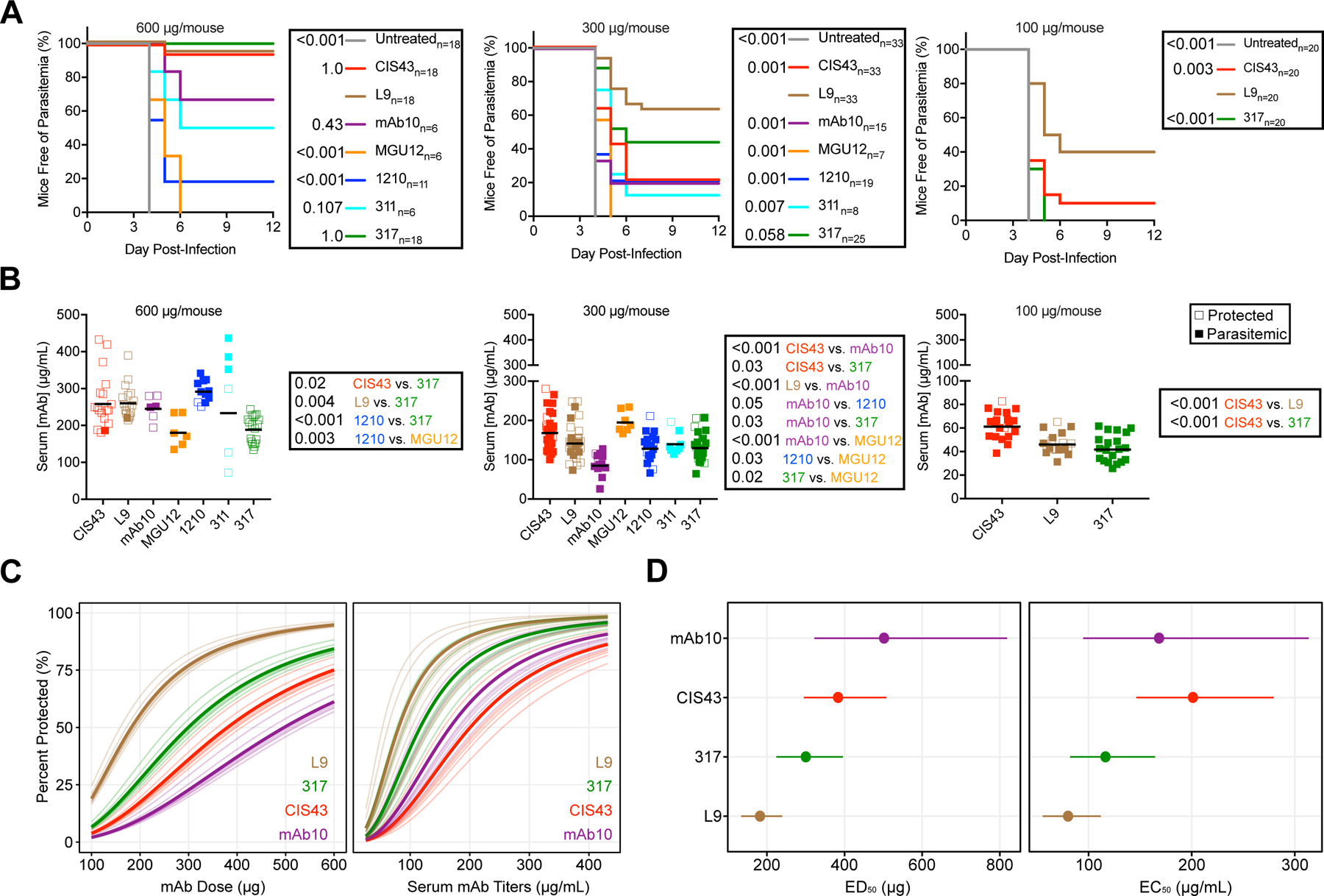
(A) Survival curves of mice challenged with five infected mosquito bites 3 days after passive transfer of 600, 300, or 100 µg PfCSP mAbs (n=number of mice/group; data were combined from seven independent blinded experiments). P-values were determined by comparing L9 to every other mAb and untreated control using the log-rank test. (B) Serum mAb titers one day prior to challenge in mice from A. P-values were determined by comparing mAbs to each other using the Kruskal-Wallis test with Dunn’s post-hoc correction. (C) Dose-response relationship of infection probability (percent protected) versus mAb dose (µg) and pre-challenge serum titers (µg/mL) for L9, 317, CIS43, and mAb10 estimated by a 2PL regression model. Thick line denotes average relationship across all experiments; lighter lines denote individual experiment relationships predicted from the model. (D) ED50 and EC50 with 95% confidence interval (CI) of L9, 317, CIS43, and mAb10 estimated from the 2PL model. See also Figure S4; Tables S3–4.
Based on the mosquito bite protection and mAb titers data (Figure 4A–B), dose-response curves were generated using 2-parameter logistic (2PL) regression models (Figure 4C). From these models, protection elicited by each mAb can be summarized across all doses by the effective dose and serum concentration required for 50% inhibition (ED50 and EC50, respectively). The ED50 and EC50 for MGU12, 1210, and 311 were not estimated, as these mAbs elicited ≤50% protection at the maximum dose of 600 µg/mouse. Additionally, we assessed the odds of protection among mice receiving L9 compared to mice receiving other mAbs, adjusting for either dose or pre-challenge mAb titers.
L9 had the lowest ED50 and EC50 (181.6 µg and 80.3 µg/mL, respectively) among all mAbs in the panel (Figure 4D; Table S3). mAb10 had a higher ED50 but lower EC50 than CIS43 due to its poorer pharmacokinetics (Figure 4B), suggesting that mAb10 was more potent than CIS43 but required higher dosing. Among the three mAbs that elicited ≥80% protection (L9, 317, and CIS43), L9 had the lowest ED80 and EC80 (325.7 µg and 145.1 µg/mL, respectively) (Table S3). Furthermore, the odds of L9-treated mice being protected from challenge were significantly higher than all other mAbs, adjusted across dose or pre-challenge titers (Table S3). Thus, L9 provides more potent protection than all mAbs in the panel against mosquito bite challenge. A thorough classification of the mAbs in the panel based on their gene usage, binding characteristics and in vivo functional activity was compiled in Table S4.
DISCUSSION
This study defined a highly potent human mAb, L9 that preferentially bound the NPNV motif associated with NVDP minor repeats of PfCSP. There are other mAbs that react with NVDP repeats: 2B6, a mouse mAb that recognizes NVDPNANP but was not functionally assessed (Sällberg et al., 2002), and 311, which binds DPNANPNVDPNA and NPNVDPNANPNV in the µM range but has five-fold greater affinity for NANP repeats (Oyen et al., 2017, 2018). Compared to 311, L9 binding to rPfCSP_FL was ~6E5-fold more potently outcompeted by peptide 22 (NANPNVDPNANPNVD). The unique preference of L9 for NPNV motifs was further underscored by the demonstration that every NVDP had to be mutated to NANP to disrupt two-step binding of L9 to rPfCSP. These data suggest that L9 should bind all circulating strains, as 100% of known Pf field isolates have ≥1 NVDP and L9 maintained high-affinity binding to rPfCSP with ≥1 NVDP.
While PfCSP repeat mAbs preferentially target either DPNA/NPNV/NPNA, they can promiscuously bind peptides composed of all three tetrapeptides due to paratopes that accommodate the subtle differences between these motifs (Imkeller et al., 2018; Kisalu et al., 2018; Oyen et al., 2017; Scally and Julien, 2018). For example, while MGU12 has been reported to have dual specificity for the junctional epitope and NPNA repeats (Tan et al., 2018), this study’s peptide mapping classified MGU12 as an NPNA-preferring mAb with minimal junctional affinity. Similarly, a recent study describes two dual-specific mAbs (667 and 668) that demonstrate some affinity for a junctional peptide KQPADGNPDPNANPNV (12 µM and 206 nM, respectively) but far greater affinity for a peptide containing only NPNA (176 nM and 55.6 nM, respectively) (Oyen et al., 2020). Consistent with these and other reports (Imkeller et al., 2018; Kisalu et al., 2018; Oyen et al., 2017), our data classified mAb10, 1210, 311, and 317 as NPNA-preferring mAbs that can bind DPNA- and NPNV-containing peptides, albeit with much lower affinity. In contrast, CIS43 and L9 respectively exhibited ~1.4E4-fold and ~2.5E6-fold greater preference for DPNA-containing peptide 21 and NPNV-containing peptide 22 compared to peptide 29, which contains only NPNA. Given their binding promiscuity, PfCSP repeat mAbs are more accurately classified based on their preferred peptides, which is stringently determined by competition ELISAs with short peptides across a range of concentrations. Finally, the observation that the three most protective mAbs in this study (CIS43, L9, and 317) are respectively DPNA-, NPNV-, and NPNA-preferring mAbs demonstrates that peptide preference may not predict in vivo protection but is useful for classification.
Despite their different peptide preferences, these three mAbs and 311 demonstrated two-step binding to rPfCSP_FL and high-affinity binding to rPfCSP_5/3 by ITC. While CIS43 and L9 were cloned from subjects immunized with radiation-attenuated PfSPZ presenting PfCSP_FL, 311 and 317 are from subjects immunized with RTS,S/AS01 (Oyen et al., 2017), which does not include the junctional region in rPfCSP_5/3. These data suggest that two-step binding is exhibited by promiscuous repeat mAbs that preferentially bind a specific epitope (e.g., DPNA or NPNV) but have achieved significant cross-reactivity to other epitopes (e.g., NPNA), resulting in two binding events to rPfCSP_FL with distinct affinities. Of note, previous ITC analyses of 311 and 317 using peptides did not report two-step binding (Oyen et al., 2017), highlighting the importance of additionally classifying repeat mAbs based on their binding to rPfCSP proteins by ITC.
The most protective mAbs against IV challenge (CIS43, L9, 311, and 317) demonstrated two-step binding to rPfCSP_FL and high affinity for rPfCPS_5/3. In contrast, mAbs with high-affinity, multivalent binding to rPfCSP_FL (mAb10, MGU12, and 1210) showed no protection at limiting doses. These data suggest that high-affinity, multivalent binding to rPfCSP_FL may be necessary, but is not sufficient, for potent in vivo SPZ neutralization in the liver. Notably, a recent analysis also finds that high PfCSP affinity is a prerequisite for SPZ neutralization and that NANP-preferring mAbs with cross-reactivity to junctional peptides are more potent (Murugan et al., 2020). Though further studies are needed to understand the relationship between two-step binding, junctional and NANP region cross-reactivity, and SPZ neutralization, our data suggests that ITC might be useful for down-selecting two-step binding PfCSP mAbs for functional testing in vivo.
In terms of elucidating how antibodies neutralize SPZ in vivo, our study built upon prior mechanistic studies in the skin (i.e., the first site for CSP antibodies to neutralize SPZ) (Aliprandini et al., 2018; Flores-Garcia et al., 2018; Vanderberg and Frevert, 2004) by showing that PfCSP mAbs mediated in vivo protection in the liver by killing SPZ and preventing their egress from sinusoids to traverse and invade hepatocytes. CSP antibodies are known to prevent SPZ traversal and invasion of HC-04 hepatocytes, a commonly used in vitro neutralization assay (Rodríguez-Galán et al., 2017; Tan et al., 2018; Triller et al., 2017). However, this in vitro assay did not predict in vivo protection against IV challenge likely due to its lack of liver sinusoids, which are a major barrier that SPZ must cross to access hepatocytes (Tavares et al., 2013; Yang et al., 2017). Effective SPZ neutralization in the vasculature and liver may be critical to prevent malaria, as a low number of SPZ are directly inoculated into the vasculature following mosquito bites (Krettli and Miller, 2001; Sidjanski and Vanderberg, 1997) and even a single SPZ that invades a hepatocyte can initiate a blood-stage infection (Frischknecht and Matuschewski, 2017).
In conclusion, mAb L9 is a promising clinical candidate for malaria prophylaxis in travelers and aid workers, as well as seasonal control and elimination campaigns. Moreover, the NPNV minor repeat motif of PfCSP presents a neutralizing epitope not included in RTS,S that might improve next-generation malaria vaccines.
Limitations of Study
While this study showed that L9 induced a high degree of CSPR in the liver, the molecular mechanisms for how L9 binding results in increased PfCSP shedding by SPZ and whether this cytopathic process alone accounts for its enhanced potency are not fully understood. Future studies are required to elucidate the structural basis for NPNV recognition by NPNV-preferring mAbs like L9, as well as how two-step binding to rPfCSP and cross-linking of native PfCSP on SPZ contribute to functional inhibition. Furthermore, it remains unclear whether SPZ neutralization by L9 requires cross-recognition of both NPNV and NPNA motifs, or is solely dependent on its preferential targeting of NPNV. In vivo protection studies with transgenic SPZ lacking minor repeats should provide insight into this question. Lastly, this study did not assess whether the addition of NPNV motifs improves current PfCSP-based vaccines.
The finding that high serum mAb titers were required to prevent malaria in mice infected with Pb-PfCSP-SPZ, compared to the substantially lower mAb titers required to mediate protection against PfSPZ in FRG-huHep mice, raises important questions as to which model will be more predictive of protection in humans. Furthermore, the half-life of PfCSP mAbs will be a critical variable for how this approach will be used.
STAR METHODS TEXT
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert Seder (rseder@mail.nih.gov).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The heavy and light chain gene sequences of anti-PfCSP human monoclonal antibodies isolated in this study, along with the sequences of PCR primers used to amplify the antibody genes, were deposited in GenBank (Accession Numbers MT811859–MT811914). Sequences of synthetic rPfCSP constructs created in this study were deposited in GenBank (Accession Numbers MT891160 –MT891178).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human clinical specimens
Clinical specimens were derived from malaria-naive, healthy adults (18–45 years of age) in the VRC 314 clinical trial (https://clinicaltrials.gov/; NCT02015091) after obtaining written informed consent. Briefly, VRC 314 was a multi-institution, phase 1, open-label, dose-escalation trial with controlled human malaria infection (CHMI) that was designed to assess the safety, immunogenicity, and protective efficacy of the Sanaria PfSPZ Vaccine administered by intravenous or intramuscular injection (Lyke et al., 2017). The Sanaria PfSPZ Vaccine is composed of radiation-attenuated, aseptic, purified, cryopreserved Plasmodium falciparum sporozoites derived from the NF54 strain (Seder et al., 2013).
Mice
Female 6- to 8-weeks old B6(Cg)-Tyrc-2J/J albino mice and female 6- to 12-weeks old C57BL/6 mice were obtained from The Jackson Laboratory. Female 6- to 8-weeks old C57BL/6 mice and female 8-weeks old Swiss Webster mice were obtained from Charles River Laboratories. Female 8–9 month old FRG-huHep were obtained from Yecuris Corp. All mouse research was performed according to National Institutes of Health (NIH) guidelines for use and care of live animals approved by the institutional animal care and use ethics committees of the Vaccine Research Center (Animal Study Protocol VRC-17–702), Johns Hopkins University (Approved protocol permit no. MO18H419), Radboud University Medical Center (ADV103002016452), and Australia National University (A2016/17 & A2019/36).
Cell Lines
Expi293 and 293F cells used were from Thermo Fisher Scientific. The human hepatoma cell line HC-04 (MRA-965, deposited by Jetsumon Sattabongkot) (Sattabongkot et al., 2006) was obtained through the Malaria Research and Reference Reagent Resource Center as part of the Biodefense and Emerging Infections Research Resources Repository.
Sporozoites
Transgenic P. berghei (strain ANKA 676m1c11, MRA-868) expressing full-length P. falciparum CSP and a green fluorescent protein/luciferase fusion protein (Pb-PfCSP-GFP/Luc-SPZ) were obtained as previously described (Flores-Garcia et al., 2019). GFP-labeled P. berghei SPZ expressing PfCSP (Pb-PfCSP-GFP-SPZ) were generated by crossing the parental Pb-PfCSP-SPZ line (Espinosa et al., 2017) with the previously described P. berghei-ConF parasite line that expresses GFP under the control of an HSP70 promotor (Amino et al., 2008). Briefly, C57BL/6 mice were coinfected with Pb-PfCSP-SPZ and P. berghei-ConF at a ratio of 10:1. Anopheles stephensi mosquitoes were allowed to feed on the mice, and subsequently, sporozoites dissected from these mosquitoes were used to infect naive animals. Parasites expressing the GFP transgene were sorted from the blood of these mice by use of a FACSAria cell sorter and used to infect mice. Subsequently, the GFP+ progeny was cloned, and the clones screened for the insertion of the PfCSP knock-in via measurement of anti-PfCSP antibody binding to progeny SPZ. P. falciparum NF54 sporozoites were isolated from an individual near Schiphol Airport (The Netherlands) (Delemarre-van de Waal and de Waal, 1981).
METHOD DETAILS
Generation of junctional probe
S02, a 43-residue structurally stabilized peptide mimicking the junctional epitope (MPESSSNPDCNANPNVDPNEDLIKKCEKINVPTEEIKKEIEEKK), was designed using Rosetta (Chevalier et al., 2017) by modifying the junctional peptide 21 with flanking sequences to stabilize the peptide in a conformation that retains binding by the neutralizing junctional antibody CIS43, but which is unfavorable to binding by the poorly neutralizing junctional antibody CIS42 (Kisalu et al., 2018). Sera from 14 protected subjects in group 6 of VRC 314 who received a total of three doses of radiation-attenuated PfSPZ (9.0 x 105) intravenously at weeks 0, 8, and 16 were screened for antibody titers against S02 using ELISA. A subject whose sera demonstrated the highest reactivity against S02 at the week 20 timepoint, 4 weeks after the last immunization, was chosen for memory B cell sorting and mAb isolation.
Production of recombinant PfCSP constructs
The amino acid sequence of PfCSP in the 3D7 clone of the NF54 isolate (PlasmoDB ID: PF3D7_0304600.1) was used to generate a codon-optimized synthetic gene for expression in mammalian cells (GenScript). The DNA construct corresponding to the full-length rPfCSP, in which the leader peptide residues 1–20 were replaced with a mammalian secretory signal peptide derived from the modified bovine prolactin (MDSKGSSQKGSRLLLLLVVSNLLLPQGVLA) and the glycosylphosphatidylinositol (GPI) anchor residues 376–397 were excluded, was cloned into a CMV/R-expression vector with a C-terminal AviTag, HRV3C-processing tag, and a 6X histidine tag. This construct, termed PfCSP_SAmut_C5S, encodes the N-terminal domain (with four amino acid mutations that removed processing sites and prevented dimerization upon solubilization to increase yield and facilitate consistent analyses), the central domain consisting of 38 NANP tandem repeats interspersed with 4 NVDP repeats, and the C-terminal domain. Truncated (Oyen et al., 2018) and NVDP repeat mutant rPfCSP synthetic constructs were created in the same expression vector with identical N- and C-termini to full-length PfCSP_SAmut_C5S, expressed through transient transfection in 293F cells using the Freestyle 293F expression system (Thermo Fisher Scientific) at 37°C, 8% CO2 for 6 days, and purified from culture supernatants through polyhistidine-tag affinity chromatography followed by size-exclusion chromatography on an ÄKTA ™ start (GE Healthcare). Monomer-containing fractions were pooled, concentrated, snap frozen, and stored at −80°C.
rPfCSP and S02 probe generation
For tetramer probe generation, rPfCSP or S02-DsbC (S02 fused to the disulfide bond C protein) were first biotinylated and then respectively conjugated to the fluorophores FITC (fluorescein isothiocyanate) and BV605 (Brilliant™ Violet 605) (BD Biosciences). Biotinylation was performed using ligase Bir A (Avidity) at 30°C for 4 hours prior to buffer exchange with 1X PBS (pH 7.4) over a 30-kDa Centricon Plus-70 Centrifugal Filter (Millipore) to remove excess free biotin. Biotinylated rPfCSP and S02-DsbC were fluorescently labeled through sequential addition of streptavidin conjugated to FITC (SA-FITC) or BV605 (SA-BV605), respectively, in a 4:1 molar ratio.
Isolation of PfCSP-specific memory B cells
Probe-specific memory B cells were isolated from cryopreserved peripheral blood mononuclear cells stained with the following panel: Aqua LIVE/DEAD (Thermo Fisher Scientific), rPfCSP-FITC and S02-BV605 tetramer probes, and antibodies against CD3-APC/Cy7 (BioLegend), CD8-V450 (BD Biosciences), CD14-BV785 (BioLegend), CD20-Alexa Fluor 700/PE (BioLegend), IgM-PE/Cy5 (BD Biosciences) and IgG-APC (BD Biosciences). Cells were sorted using a BD FACS Aria II (BD Immunocytometry Systems), and flow cytometry data were analyzed using FlowJo software (Tree Star). PfCSP-reactive (rPfCSP+ and/or S02+) CD20+CD3−CD14− memory B cells were single-cell sorted into 96-well PCR plates containing lysis buffer (RNase OUT, 5X First-Strand buffer, DTT, IgePAL, and water; SuperScript III First-Strand Synthesis System, Thermo Fisher Scientific).
Production of recombinant immunoglobulins
Immediately following single cell sorting and lysis of probe-specific memory B cells, all RNA transcripts were reverse transcribed to cDNA (SuperScript III First-Strand Synthesis System; Thermo Fisher Scientific). Amplification of the genes encoding the immunoglobulin variable regions heavy chains, as well as kappa or lambda light chains, was performed using a cocktail of primers followed by sequencing (ACGT) and cloning into the pVRC8400 huIgG1, pVRC8400 huIgK, or SBShuLambda expression vectors (GenScript) containing the relevant constant region. Sequence analysis was performed using The International Immunogenetics Information System (IMGT, http://www.imgt.org/). Matched heavy and light chain constructs were co-transfected into Expi293 cells using the ExpiFectamine™ 293 Transfection Kit (Thermo Fisher Scientific) and cultures were incubated at 37°C, 8% CO2 for 6 days. Supernatants were harvested and purified using rProtein A Sepharose Fast Flow resin (GE Healthcare) and buffer exchanged with 1X PBS (pH 7.4) before being concentrated using Amicon Centrifugal Filters (Millipore). Purified mAb concentrations were determined using a Nanodrop spectrophotometer. The sequences of CIS43, mAb10 (Kisalu et al., 2018), MGU12 (Tan et al., 2018), 1210 (Imkeller et al., 2018), 311, and 317 (Oyen et al., 2017) were retrieved from PDB or GenBank and produced and purified as described above.
Fluorescent antibody labeling
Antibodies were conjugated to Alexa Fluor-405 molecules using a SAIVI Antibody Labeling Kit according to the manufacturers’ directions (ThermoFisher Scientific) but using Alexa Fluor-405 NHS ester (ThermoFisher Scientific), which was mixed with each antibody at an 8:1 molar ratio for 1 hr at room temperature. The reaction was then purified over the SAIVI column, with fractions collected to determine the location of the conjugate. Resulting PfCSP mAb conjugates had degree of labeling (DOL) ratios between 1.4–2.2. For determination of concentration and DOL of Alexa Fluor-405 conjugates: absorbance correction factor = 0.7; extinction coefficient (e) = 34,500. Prior to use, conjugate binding was compared to unlabeled mAbs by rPfCSP ELISA and/or Pb-PfCSP-SPZ flow cytometry to confirm binding was not dramatically altered.
ELISA for binding of mAbs to rPfCSP_FL
Immulon 4HBX flat bottom microtiter plates (Thermo Fisher Scientific) were coated with 100 µl per well of antigen (1.0 µg/mL) in bicarbonate buffer overnight at 4°C. Coated plates were blocked with 200 µl of PBS + 10% FBS for 2 hrs at room temperature, followed by incubation for 2 hrs at 37°C with 100 µl of PfCSP or control mAbs at varying concentrations (5x10−7 – 5.0 µg/mL, 10-fold serial dilutions). Plates were incubated with 100 µl/well of 0.1 µg/mL HRP-conjugated goat anti–human IgG (Bethyl Laboratories). Plates were washed six times with PBS-Tween between each step. After a final wash, samples were incubated for 10 min with 1-Step Ultra TMB-ELISA Substrate (Thermo Fisher Scientific). The optical density was read at 450 nm after addition of stopping solution (2N sulfuric acid, 100 µl/well).
ELISA for binding of mAbs to NANP peptides
MSD Gold microtiter plates (Meso Scale Discovery) were blocked with PBS + 5% BSA (20 µl/well). Blocked plates were coated with 10 µl/well of NANP biotinylated peptides (240 pmol, Genscript) in PBS + 1% BSA for 1 hr at room temperature. The coated plates were incubated for 2 hrs at room temperature with 10 µl of PfCSP or control mAbs at varying concentrations (5x10−7 – 5.0 µg/mL, 5-fold serial dilutions). Plates were then incubated with 10 µl/well of 1.0 µg/mL Sulfo-tag goat anti–human IgG (Meso Scale Discovery) for 1 hr at room temperature. Plates were washed six times with PBS-Tween between each step. After a final wash, 35 µl of 1X MSD Read T Buffer (Meso Scale Discovery) was added to each well and plates were analyzed on an MSD Sector Image 2400 instrument.
Epitope mapping and competition ELISAs
Epitope mapping of CIS43 and L9 was performed using PfCSP overlapping peptides (peptides 20–61) that were 15 amino acids in length (GenScript) and overlapped by 11 residues spanning the central repeat region of PfCSP using the MSD U-Plex Assay platform (Meso Scale Discovery) according to the manufacturer’s instructions, with all mAb concentrations at 0.01 µg/mL. Competitive ELISA was also performed using peptides 20–61. Briefly, ELISA plates were coated with 10 µl of rPfCSP (200 ng/mL) for 1 hr at room temperature. After coating, PfCSP-specific monoclonal antibodies (10 ng/mL) preincubated overnight with varying concentrations (0 – 1,000 µg/mL) of selected PfCSP peptides in PBS + 1% BSA were added to the rPfCSP-coated plates, and ELISA was performed on the MSD platform as described above. For the alanine scanning mutagenesis experiments, competitive ELISA was performed as described above using peptide 22 variants where each residue was mutated to an alanine or a serine if the original residue was an alanine (GenScript).
Biolayer interferometry kinetic binding assay
Antibody binding kinetics were measured using biolayer interferometry on an Octet HTX instrument (FortéBio) using streptavidin-capture biosensors (fortéBio). PfCSP mAb solutions were plated in black tilted-bottom 384-well microplates (fortéBio); assays were performed with agitation at 30°C. mAb serial concentrations used are as follow: 1.25, 0.625, 0.3125, and 0.15625 µg/mL. Loading of biotinylated peptides 21, peptide 22, and peptide 29 (GenScript) was performed for 300s, followed by dipping of biosensors into buffer (PBS + 1% BSA) for 60s to assess baseline assay drift. Association with whole IgG (serially diluted from 16.67 to 1.04 µM) was done for 300s, followed by a dissociation step in buffer for 600s. Background subtraction of nonspecific binding was performed through measurement of association in buffer alone. Data analysis and curve fitting were performed using Octet software, version 7.0. Experimental data were fitted with the binding equations describing a 1:1 analyte-ligand interaction. Global analyses of the complete data sets, assuming binding was reversible (full dissociation), were carried out using nonlinear least-squares fitting allowing a single set of binding parameters to be obtained simultaneously for all concentrations of a given mAb dilution series.
Isothermal titration calorimetry
Isothermal titration calorimetry was carried out using a VP-ITC microcalorimeter (Malvern Panalytical). In all titration experiments, the rPfCSP constructs and mAbs were prepared in PBS, pH 7.4. Each antibody solution, prepared at a concentration of ~40 µM (expressed per antigen binding site), was injected in 5 or 7 µl aliquots into the calorimetric cell containing the respective rPfCSP construct at a concentration of ~0.4 µM except for rPfCSP_5/3, which was prepared at 0.8 µM. All titrations were performed at 25°C. The exact concentrations of the reactants in each experiment were determined from the absorbance at 280 nm. The heat evolved upon each injection of antibody was obtained from the integral of the calorimetric signal. The heat associated with binding to the different rPfCSP constructs was obtained by subtracting the heat of dilution from the heat of reaction. The individual heats were plotted against the molar ratio, and the enthalpy change, ∆H, the association constant, Ka (the dissociation constant, Kd =1/Ka) and the stoichiometry (valency of antigen binding sites), N, were obtained by nonlinear regression of the data to a model that takes into account the binding to either one or two sets of sites with different binding affinities. Gibbs energy, ∆G, was calculated from the relation ∆G = − RTlnKa, where R is the universal gas constant, (1.987 cal/(K × mol)) and T the absolute temperature in Kelvin. The entropy contribution to Gibbs energy, -T∆S, was calculated from the known relation ∆G = ∆H − T∆S. The results were expressed per mole of antigen binding sites and the stoichiometry, N, denotes the number of antigen binding sites per mole of the respective rPfCSP construct.
FACS analysis of mAb binding to sporozoites
Freshly isolated Pb-PfCSP-GFP/Luc-SPZ were purified across an Accudenz density gradient (Accurate Chemical) to remove mosquito debris as previously described (Kennedy et al., 2012) and resuspended in PBS containing the protease inhibitor E64 (Sigma-Aldrich) to prevent proteolytic processing of PfCSP. 8,000 SPZ were aliquoted to each well of a 96-well V-bottom plate (50 µl/well) and incubated for 30 min at 4 °C with various concentrations (0.02 – 20 µg/mL) of PfCSP-specific or control mAbs in PBS+E64, washed with 200 µl PBS+E64, and stained for 20 minutes at 4°C with goat anti-human IgG-Alexa Fluor® 647 secondary antibody (Thermo Fisher Scientific) at a 1:1,000 dilution in PBS+E64. After washing with 200 µl PBS+E64 and fixation in 250 µl PBS with 0.5% paraformaldehyde, events were acquired on a modified LSR II (BD Biosciences).
In vitro hepatocyte invasion inhibition assay
The human hepatoma cell line HC-04 was used to evaluate the in vitro capacity of the PfCSP mAbs to block hepatocyte invasion by PfSPZ NF54. HC-04 cells were seeded at 50,000 cells/well in rat tail collagen pre-treated 96 well plates for 16–24 hrs. Salivary gland PfSPZ were pre-mixed with heat-inactivated human serum and varying concentrations of PfCSP mAbs (10, 1 or 0.1 µg/mL) for 30 minutes on ice and then seeded in triplicate (50,000 sporozoites/well) onto the HC-04 cells. Plates were centrifuged for 10 minutes at 3000 xg and incubated for 3 hrs at 37°C with 5% C02. Cells were washed with PBS (Gibco) and treated with trypsin (0.05% Trypsin-ethylenediamine tetra acetic acid (EDTA); Gibco) to generate a single-cell suspension. Subsequently, cells were fixed and impermeabilized (eBioscience) for 30 min at 4°C. The staining was done with a cell viability dye (1:2,000; Fixable viability dye (FVD) eFluor 780, eBioscience, Thermo Fisher Scientific) and a fluorescently-labelled mouse anti-PfCSP antibody (1:400; 3SP2-FITC) for 30 min at 4°C. After washing with 2% FCS/PBS, cells were fixed with 1% paraformaldehyde (PFA) and analyzed by flow cytometry using Gallios (Beckman Coulter) and FlowJo software (version 10.0.8, Tree Star).
IV challenge with Pb-PfCSP-SPZ
To measure mAb neutralization of SPZ and reduction of parasite burden in vivo, specified amounts of PfCSP-specific or control mAbs diluted in sterile filtered 1X PBS (pH 7.4; total volume 200 µl/mouse) were injected into the tail veins of female 6- to 8-week old B6(Cg)-Tyrc-2J/J albino mice (The Jackson Laboratory). Mice were then intravenously challenged in the tail vein with 2,000 freshly harvested Pb-PfCSP-GFP/Luc-SPZ (Flores-Garcia et al., 2019) 2 hours after mAb administration. 40–42 hours post-challenge, mice were injected intraperitoneally with 150 µl of D-Luciferin (30 mg/mL), anesthetized with isoflurane and imaged with the IVIS® Spectrum in vivo imaging system (PerkinElmer) 10 minutes after luciferin injection. Liver burden was quantified by analyzing a region of interest (ROI) in the upper abdominal region and determining the total flux or bioluminescent radiance (photons/sec) expressed by Pb-PfCSP-GFP/Luc-SPZ using the manufacturer’s software (Living Image 4.5, PerkinElmer).
Mosquito bite challenge with Pb-PfCSP-SPZ
Anopheles stephensi female mosquitoes were allowed to feed on female 8-week old Swiss Webster mice (Charles Laboratories) infected with blood-stage Pb-PfCSP-SPZ. Twenty days after infected bloodmeal, the proportion of infected mosquitoes was between 70–80%, as assessed by microscopic observation of 20 salivary glands. Based on this observation, we determined that 6–7 mosquitoes were needed to expose mice to the bites of ~5 infected mosquitoes. 6–8 week old C57BL/6 female mice (Charles Laboratories) were injected IV with PfCSP mAbs (100, 300, or 600 µg mAb/mouse; blinded and in differing orders per experimental replicate) diluted in 1X PBS (pH 7.4) in a total volume of 200 µL. Forty-eight hours after mAb administration, mice were subjected to a small tail vein bleed to ascertain pre-challenge mAb serum titers. Seventy-two hours after mAb administration, test and control mice were anesthetized with 2% Avertin (Alfa Aesar, Ward Hill, MA). Mosquitoes were allowed to feed on mice for 10 minutes. Following feeding, mosquito abdomens were inspected to confirm the blood meal. Mouse parasitemia was assessed daily through Giemsa staining of blood smears starting on day 4 and up to day 10–12 after exposure to infected mosquito bites.
IV challenge with PfSPZ
Female 8–9 month old FRG-huHep mice with engrafted human hepatocytes from two different donors (HHM19027 and HHM13022) were purchased from Yecuris Corp. Repopulation of human hepatocytes were confirmed by the level of serum albumin and ranged between 4000 to 8000 µg/mL, evenly divided between the different experimental groups. Mice were intravenously injected with the indicated dose of VRC01 or PfCSP mAbs (100 µL per mouse) 24 hours before sporozoite challenge. On the day of sporozoite challenge, Anopheles stephensi mosquitoes infected with Pf NF54 (on day 14–18 post blood meal) were harvested into Dutch modified RPMI 1640 media (ThermoFisher). 100,000 sporozoites (in a total volume of 100 µl) were injected IV in the tail vein of each mouse. Six days following challenge, serum was collected via cardiac bleed and livers of each mice were harvested as previously described (Foquet et al., 2013; Yang et al., 2017). Briefly, lobes were pooled and emulsified to obtain single-cell suspensions for subsequent genomic DNA (gDNA) extraction. gDNA were extracted from roughly 25% of the chimeric livers and used to quantify parasite load using oligonucleotides specific for Pf 18S rRNA as previously described (McCall et al., 2017; Yang et al., 2017). qPCR was used to quantify the relative amount of human hepatocytes engrafted onto the mouse liver, as previously described (Alcoser et al., 2011).
ELISA for quantitation of mAb serum titers
ELISA was performed on serum from mice passively transferred human PfCSP mAbs as previously described (Kisalu et al., 2018) using rPfCSP-coated plates (200 ng/mL). A standard curve for each mAb was generated using eight two-fold serial dilutions of mAb starting at 10 ng/mL. Serum samples were applied at various dilutions in dilution/blocking buffer. For datapoints in the linear range of the standard curve, the average of the calculated concentration values was used for each individual sample.
Intravital liver imaging of Pb-PfCSP-SPZ
Female 6- to 12-weeks old C57BL/6 mice (The Jackson Laboratory) received sequential IV injections of Alexa Fluor-405-labeled mAbs (30 µg, blinded), 1 x 105 Pb-PfCSP-GFP-SPZ, and rhodamine-labeled dextran (20 µg/mL, 50µL). Mice were immediately prepared for multiphoton microscopy as previously described (McNamara et al., 2017). Briefly, mice were anaesthetized with a mix of Ketamine (100 mg/kg) and Xylazine (10 mg/kg). The mouse temperature was maintained at 37ºC using a heating mat attached to a feedback probe inserted in the mouse rectum throughout the surgery and imaging procedure. A lateral incision was made over the left lobe of the liver and any vessels cauterized by applying light pressure to the vessel until clotting occurred naturally. The mouse was then placed in a custom-made holder. The liver was exposed and directly adhered to a coverslip that was secured in the holder. Once stable, the preparation was transferred to a Fluoview FVMPE-RS multiphoton microscope system (Olympus) equipped with a XLPLN25XWMP2 objective (25x; NA1.05; water immersion; 2mm working distance). For quantification of parasites, damaged hepatocytes, and shedding of PfCSP, a single 50µm Z-stack (2µm/slice) was acquired using a resonance scanner. Fluorescence of Alexa Fluor-405, GFP and rhodamine were detected using an 860nm wavelength laser. For videos of traversal or shedding, a sequence of between 1,000–2,000 50µm Z-stacks (2µm/slice) was acquired using a resonance scanner. Images were acquired using FV30 software (Olympus) and exported to Imaris (Bitplane) for downstream processing. Sporozoites and shed PfCSP were measured using the measurement function on Imaris. Sporozoites were classified as either sinusoid-bound, traversing hepatocytes, or having infected a hepatocyte based on the following criteria. Any sporozoite located within a sinusoid and not within 40µm (i.e., diameter of a hepatocyte)of a rhodamine+ hepatocyte was considered to be vessel bound in the sinusoid. Any sporozoite within 40µm of at least one rhodamine+ hepatocyte and still in contact with the traversed hepatocyte was considered to be traversing. Any sporozoite within 40µm of at least one rhodamine+ hepatocyte, but not in a sinusoid nor in contact with a rhodamine+ hepatocyte, was considered to have established infection. Additionally, any sporozoite with a discontinuation (i.e., fragmentation) of GFP along its length was classified as undergoing “dotty death”, and any sporozoite with a length of Alexa Fluor-405 not colocalized with GFP was considered to be shedding its mAb-bound PfCSP coat and undergoing a CSPR. Parasite death in any of the resonance videos was characterized as bursting if a sudden loss of membrane integrity and release of GFP into the surrounding tissue was observed.
NVDP conservation in global field isolates
PfCSP sequences and country in which the sequences were isolated were retrieved from GenBank. N- and C-terminal sequences were trimmed in Geneious Prime (Geneious) and the central repeat region sequences was exported into Microsoft Excel. All NPDP tetrapeptides were transformed into 0, NANP repeats into 1, and NVDP repeats into 2; sequences were numerically ordered based on number of NVDP repeats and the geographic region in which they were isolated was indicated.
Heat map classifying the panel of PfCSP mAbs
Numerical values for each parameter describing each of the seven mAbs in the panel were collated and ranked using conditional formatting (Microsoft Excel), with darker colors denoting improved performance and lighter colors indicating poorer performance. All numerical values that varied logarithmically were log-transformed into linear values to facilitate consistent analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests used, exact value of n, what n represents, and precision measures can be found in figure legends. Unless otherwise stated, all mAbs were compared for significance to untreated/isotype control or to each other using the Kruskal-Wallis test with Dunn’s post-hoc test correcting for multiple comparisons. For the PfSPZ challenge and L9 titers in FRG-huHep mice and the comparison of L9 and CIS43 serum titers, the two-tailed Mann-Whitney test was used. For the in vitro hepatocyte invasion assay, two-way ANOVA was used to compare each mAb to the isotype control at each respective concentration and adjusted for multiple comparisons using Bonferroni’s post-hoc test. For the intravital liver imaging data (locations, traversal, CSPR, and dotty death), all mAbs were first compared to each other using an omnibus chi-squared test; if the distribution varied significantly, then each individual mAb was compared to isotype control (or the best performing mAb) and adjusted for multiple comparisons using Bonferroni’s post-hoc test. For measurement of PfCSP mAbs in mouse serum, standard curves were fitted with a hyperbolic parameter curve, and concentration values in the linear range of the standard curve were interpolated. For the ITC stoichiometry data, errors with 95% confidence were estimated from the fits of the data. Unless otherwise indicated, all data were plotted using GraphPad Prism, version 7.0. For the mosquito bite challenge experiments, L9 was tested for superiority against the other mAbs (one-sided tests) and P-values were corrected using the Holm method (Holm, 1979). For the Kaplan–Meier curves, comparisons were tested using the log-rank test; an exact P-value was calculated when the sample size did not exceed 15 in either group (Hothorn et al., 2008). The relationship between dose or circulating serum mAb concentration prior to challenge and protection were modeled using two-parameter logistic (2PL) models with the following functional form: y = 1/1 + (x/ED50)h) (Raghunandan et al., 2020). The ED50 (or EC50 for concentration) is the effective dose that elicits 50% protection and the hill slope, h, determines the steepness of the logistic curve. The 2PL model was fit to the data using logistic regression with a log-transformed dose or concentration covariate. For mAbs eliciting at least 50% protection, a single model was fit assuming a common hill slope and a mixed model was used with a random intercept specified for each experiment. ED50, EC50, ED80, and ED80 with 95% confidence intervals were estimated from these fitted models using parametric simulation. Protection was compared between L9 and the other mAbs using the dose-adjusted or concentration-adjusted odds ratio from these models. For mAbs eliciting less than 50% protection, separate logistic models with small-sample size corrections (Firth-correction) were fit for each paired adjusted odds ratio comparison (Kosmidis and Firth, 2009). P-values of odds ratios were corrected together within dose or concentration comparisons. Protection analyses were conducted in R programming language (CRAN) (R Core Team). Mixed effects logistic regression models were implemented with the lme4 package (Bates et al., 2015), simulations were implemented with the arm package (Gelman et al., 2020), and data manipulation and visualization were implemented with the tidyverse packages (Wickham et al., 2019; Wilke, 2019).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD3 APC/Cy7 (clone OKT3) | BioLegend | Cat#317341 |
| Anti-human CD8 V450 (clone RPA-T8) | BD Biosciences | Cat#560347 |
| Anti-human CD14 BV785 (clone M5E2) | BioLegend | Cat#301839 |
| Anti-human CD20 Alexa Fluor 700/PE (clone 2H7) | BioLegend | Cat#302322 |
| Anti-human IgM PE/Cy5 (clone G20-127) | BD Biosciences | Cat#551079 |
| Anti-human IgG APC (clone G18-145) | BD Biosciences | Cat#550931 |
| Anti–human IgG, HRP conjugated | Bethyl Laboratories | Cat#A80-119P |
| Anti–human IgG, Sulfo-tag | Meso Scale Discovery | Cat#R32AJ-1 |
| Anti-human IgG-Alexa Fluor® 647 | Thermo Fisher Scientific | Cat#A-21445 |
| Anti-PfCSP antibody, 3SP2-FITC | (Verhave et al., 1988) | N/A |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant PfCSP constructs with truncated repeat regions and minor repeat mutations | This Paper | GenBank Accession Numbers MT891160 - MT891178 |
| Fluorescein Isothiocyanate Streptavidin | BD Biosciences | Cat#554060 |
| Brilliant™ Violet 605 Streptavidin | BD Biosciences | Cat#563260 |
| Dextran, Rhodamine B | Thermo Fisher Scientific | Cat#D1824 |
| Bir A | Avidity | Cat#BirA500 |
| RNaseOUT™ Recombinant Ribonuclease Inhibitor | Thermo Fisher Scientific | Cat#10777019 |
| IgePAL | Millipore Sigma | Cat#I8896 |
| Alexa Fluor™ 405 NHS Ester (Succinimidyl Ester) | Thermo Fisher Scientific | Cat#A30000 |
| 1-Step™ Ultra TMB-ELISA | Thermo Fisher Scientific | Cat#34029 |
| Accudenz density gradient | Accurate Chemical | Cat#AN7050 |
| E-64 | Millipore Sigma | Cat#E3132 |
| D-Luciferin | Perkin Elmer | Cat#1227991 |
| eBioscience™ Fixable Viability Dye eFluor™ 506/780 | Thermo Fisher Scientific | Cat#65-2860-40 |
| Avertin | Alfa Aesar | Cat#A18706 |
| Critical Commercial Assays | ||
| SuperScript™ First-Strand Synthesis System for RT-PCR | Thermo Fisher Scientific | Cat#11904018 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34966 |
| Expi293™ Expression System Kit | Thermo Fisher Scientific | Cat#A14635 |
| FreeStyle™ 293 Expression System | Thermo Fisher Scientific | Cat#K900001 |
| rProtein A Sepharose® Fast Flow | Millipore Sigma | Cat#GE17-1279-03 |
| SAIVI™ Alexa Fluor™ 647 Antibody/Protein Labeling Kit | Thermo Fisher Scientific | Cat#S30044 |
| Deposited Data | ||
| Human PfCSP mAb heavy and light chain sequences | This Paper | GenBank Accession Numbers MT811859 - MT811914 |
| Experimental Models: Cell Lines | ||
| Human: HC-04 hepatoma cells | (Sattabongkot et al., 2006) | N/A |
| Human: Expi293 cells | Thermo Fisher Scientific | Cat#A14527 |
| Human: 293F cells | Thermo Fisher Scientific | Cat#R79007 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6(Cg)-Tyrc-2J/J albino | The Jackson Laboratory | Cat #000058 |
| Mouse: C57BL/6 | Charles River Laboratories | Cat#027 |
| Mouse: Swiss Webster | Charles River Laboratories | Cat#024 |
| Mouse: FRG huHep | Yecuris Corp | Cat#10-0006 |
| Sporozoite: P. berghei sporozoite expressing PfCSP, GFP, and luciferase | (Flores-Garcia et al., 2019) | N/A |
| Sporozoite: P. berghei sporozoite expressing PfCSP, and GFP | This Paper | N/A |
| Sporozoite: P. falciparum sporozoite, strain NF54 | (Delemarre-van de Waal and de Waal, 1981) | N/A |
| Oligonucleotides | ||
| PCR primers for amplification of antibody heavy, kappa, and lambda genes | This paper | GenBank Accession Numbers MT811859 - MT811914 |
| Recombinant DNA | ||
| Plasmodium falciparum circumsporozoite protein (clone 3D7) | PlasmoDB | PF3D7_0304600.1 |
| pVRC8400 huIgG1 | Genscript | N/A |
| pVRC8400 huIgK | Genscript | N/A |
| SBShuLambda | Genscript | N/A |
| Software and Algorithms | ||
| FlowJo 9 software | FlowJo | https://www.flowjo.com; RRID:SCR_008520 |
| The International Immunogenetics Information System | IMGT | imgt.org; RRID:SCR_006931 |
| GraphPad Prism 7 software | GraphPad Software | https://www.graphpad.com; RRID:SCR_002798 |
| RStudio | RStudio | https://rstudio.com/; RRID:SCR_000432 |
| Octet Software, version 7.0 | FortéBio | http://www.fortebio.com; |
| Living Image Software, version 4.5 | Perkin Elmer | http://perkinelmer.com; RRID:SCR_014247 |
| Imaris | Bitplane | https://imaris.oxinst.com/; RRID:SCR_007370 |
| Rosetta | Rosetta Commons | https://www.rosettacommons.org/software; |
| Geneious Prime | Geneious | http://www.geneious.com; RRID:SCR_010519 |
| lme4 | R package | https://cran.r-project.org/web/packages/lme4/index.html; RRID:SCR_015654 |
| arm | R package | https://cran.r-project.org/web/packages/arm/index.html |
| tidyverse | R package | https://cran.r-project.org/web/packages/tidyverse/index.html |
| Other | ||
| BD LSRFortessa II | BD Biosciences | N/A |
| BD FACSAria II cell sorter | BD Biosciences | N/A |
| SECTOR Image 2400 | Meso Scale Discovery | N/A |
| U-PLEX Assay Platform | Meso Scale Discovery | N/A |
| Octet HTX | FortéBio | N/A |
| VP-ITC microcalorimeter | Malvern Panalytical | N/A |
| Gallios Flow Cytometer | Beckman Coulter | N/A |
| IVIS® Spectrum In Vivo Imaging System | Perkin Elmer | N/A |
| Fluoview FVMPE-RS Multiphoton Microscope System | Olympus | N/A |
Highlights.
The subdominant NVDP minor repeats of PfCSP are neutralizing epitopes
Potent mAbs with junctional and NANP cross-reactivity bind PfCSP in two steps
Cytotoxic PfCSP mAbs kill sporozoites in the liver
PfCSP mAbs prevent sporozoites from exiting liver sinusoids to infect hepatocytes
ACKNOWLEDGMENTS
We thank B. Kim Lee Sim and Stephen L. Hoffman (Sanaria, Inc.) for providing the PfSPZ Vaccine, Youri van Waardenburg and Geert-Jan van Gemert for generating PfSPZ-infected mosquitoes and processing FRG-huHep mice samples, and David Ambrozak (Vaccine Research Center) for assistance with sorting MBCs. This work was supported by the National Institute of Allergy and Infectious Diseases, the National Cancer Institute (HHSN261200800001E) for A.S., the Dutch Research Council (NWO) talent program veni (VI.Veni.192.171) for A.S.P.Y, the Bill & Melinda Gates Foundation for N.K.H and M.P. (OPP1156262) and Y.F. and F.Z., who also thank the Bloomberg Philanthropies for continued support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
R.A.S., J.F., L.T.W., and R.V. have submitted U.S. Provisional Patent Application No. 62/842,590, filed 3 May 2019, describing mAb L9. All other authors declare no competing interests.
REFERENCES
- Alcoser SY, Kimmel DJ, Borgel SD, Carter JP, Dougherty KM, and Hollingshead MG (2011). Real-time PCR-based assay to quantify the relative amount of human and mouse tissue present in tumor xenografts. BMC Biotechnol 11, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliprandini E, Tavares J, Panatieri RH, Thiberge S, Yamamoto MM, Silvie O, Ishino T, Yuda M, Dartevelle S, Traincard F, et al. (2018). Cytotoxic anti-circumsporozoite antibodies target malaria sporozoites in the host skin. Nat Microbiol 3, 1224–1233. [DOI] [PubMed] [Google Scholar]
- Amino R, Giovannini D, Thiberge S, Gueirard P, Boisson B, Dubremetz J-F, Prévost M-C, Ishino T, Yuda M, and Ménard R (2008). Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell Host Microbe 3, 88–96. [DOI] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software 67, 1–48. [Google Scholar]
- Bejon P, White MT, Olotu A, Bojang K, Lusingu JPA, Salim N, Otsyula NN, Agnandji ST, Asante KP, Owusu-Agyei S, et al. (2013). Efficacy of RTS,S malaria vaccines: individual-participant pooled analysis of phase 2 data. Lancet Infect Dis 13, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami C, Frevert U, Sinnis P, Takacs B, Clavijo P, Santos MJ, and Nussenzweig V (1992). The basolateral domain of the hepatocyte plasma membrane bears receptors for the circumsporozoite protein of Plasmodium falciparum sporozoites. Cell 70, 1021–1033. [DOI] [PubMed] [Google Scholar]
- Chevalier A, Silva D-A, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam K-H, Yao G, et al. (2017). Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn IA, and Seder RA (2018). Malaria prevention: from immunological concepts to effective vaccines and protective antibodies. Nat. Immunol 19, 1199–1211. [DOI] [PubMed] [Google Scholar]
- Delemarre-van de Waal HA, and de Waal FC (1981). [A 2d patient with tropical malaria contracted in a natural way in the Netherlands]. Ned Tijdschr Geneeskd 125, 375–377. [PubMed] [Google Scholar]
- Dyson HJ, Satterthwait AC, Lerner RA, and Wright PE (1990). Conformational preferences of synthetic peptides derived from the immunodominant site of the circumsporozoite protein of Plasmodium falciparum by 1H NMR. Biochemistry 29, 7828–7837. [DOI] [PubMed] [Google Scholar]
- Espinosa DA, Gutierrez GM, Rojas-López M, Noe AR, Shi L, Tse S-W, Sinnis P, and Zavala F (2015). Proteolytic Cleavage of the Plasmodium falciparum Circumsporozoite Protein Is a Target of Protective Antibodies. J. Infect. Dis 212, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa DA, Christensen D, Muñoz C, Singh S, Locke E, Andersen P, and Zavala F (2017). Robust antibody and CD8+ T-cell responses induced by P. falciparum CSP adsorbed to cationic liposomal adjuvant CAF09 confer sterilizing immunity against experimental rodent malaria infection. NPJ Vaccines 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Garcia Y, Nasir G, Hopp CS, Munoz C, Balaban AE, Zavala F, and Sinnis P (2018). Antibody-Mediated Protection against Plasmodium Sporozoites Begins at the Dermal Inoculation Site. MBio 9, e02194–18, /mbio/9/6/mBio.02194-18.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Garcia Y, Herrera SM, Jhun H, Pérez-Ramos DW, King CR, Locke E, Raghunandan R, and Zavala F (2019). Optimization of an in vivo model to study immunity to Plasmodium falciparum pre-erythrocytic stages. Malar. J 18, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foquet L, Hermsen CC, van Gemert G-J, Libbrecht L, Sauerwein R, Meuleman P, and Leroux-Roels G (2013). Molecular detection and quantification of Plasmodium falciparum-infected human hepatocytes in chimeric immune-deficient mice. Malar. J 12, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foquet L, Schafer C, Minkah NK, Alanine DGW, Flannery EL, Steel RWJ, Sack BK, Camargo N, Fishbaugher M, Betz W, et al. (2018). Plasmodium falciparum Liver Stage Infection and Transition to Stable Blood Stage Infection in Liver-Humanized and Blood-Humanized FRGN KO Mice Enables Testing of Blood Stage Inhibitory Antibodies (Reticulocyte-Binding Protein Homolog 5) In Vivo. Front Immunol 9, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht F, and Matuschewski K (2017). Plasmodium Sporozoite Biology. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, Su Y-S, Yajima M, Hill J, Pittau MG, Kerman J, Zheng T, and Dorie V (2020). arm: Data Analysis Using Regression and Multilevel/Hierarchical Models. [Google Scholar]
- Ghasparian A, Moehle K, Linden A, and Robinson JA (2006). Crystal structure of an NPNA-repeat motif from the circumsporozoite protein of the malaria parasite Plasmodium falciparum. Chem. Commun. (Camb.) 174–176. [DOI] [PubMed] [Google Scholar]
- Holm S (1979). A Simple Sequentially Rejective Multiple Test Procedure. Scandinavian Journal of Statistics 6, 65–70. [Google Scholar]
- Hothorn T, Hornik K, Wiel MA van de, and Zeileis A (2008). Implementing a Class of Permutation Tests: The coin Package. Journal of Statistical Software 28, 1–23.27774042 [Google Scholar]
- Imkeller K, Scally SW, Bosch A, Martí GP, Costa G, Triller G, Murugan R, Renna V, Jumaa H, Kremsner PG, et al. (2018). Antihomotypic affinity maturation improves human B cell responses against a repetitive epitope. Science 360, 1358–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien J-P, and Wardemann H (2019). Antibodies against Plasmodium falciparum malaria at the molecular level. Nat. Rev. Immunol 19, 761–775. [DOI] [PubMed] [Google Scholar]
- Kennedy M, Fishbaugher ME, Vaughan AM, Patrapuvich R, Boonhok R, Yimamnuaychok N, Rezakhani N, Metzger P, Ponpuak M, Sattabongkot J, et al. (2012). A rapid and scalable density gradient purification method for Plasmodium sporozoites. Malar J 11, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kester KE, Cummings JF, Ofori-Anyinam O, Ockenhouse CF, Krzych U, Moris P, Schwenk R, Nielsen RA, Debebe Z, Pinelis E, et al. (2009). Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS,S/AS01B and RTS,S/AS02A in malaria-naive adults: safety, efficacy, and immunologic associates of protection. J. Infect. Dis 200, 337–346. [DOI] [PubMed] [Google Scholar]
- Kisalu NK, Idris AH, Weidle C, Flores-Garcia Y, Flynn BJ, Sack BK, Murphy S, Schön A, Freire E, Francica JR, et al. (2018). A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nat. Med 24, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmidis I, and Firth D (2009). Bias reduction in exponential family nonlinear models. Biometrika 96, 793–804. [Google Scholar]
- Krettli AU, and Miller LH (2001). Malaria: a sporozoite runs through it. Curr. Biol 11, R409–412. [DOI] [PubMed] [Google Scholar]
- Lyke KE, Ishizuka AS, Berry AA, Chakravarty S, DeZure A, Enama ME, James ER, Billingsley PF, Gunasekera A, Manoj A, et al. (2017). Attenuated PfSPZ Vaccine induces strain-transcending T cells and durable protection against heterologous controlled human malaria infection. Proc Natl Acad Sci U S A 114, 2711–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MBB, Wammes LJ, Langenberg MCC, van Gemert G-J, Walk J, Hermsen CC, Graumans W, Koelewijn R, Franetich J-F, Chishimba S, et al. (2017). Infectivity of Plasmodium falciparum sporozoites determines emerging parasitemia in infected volunteers. Sci Transl Med 9. [DOI] [PubMed] [Google Scholar]
- McNamara HA, Cai Y, Wagle MV, Sontani Y, Roots CM, Miosge LA, O’Connor JH, Sutton HJ, Ganusov VV, Heath WR, et al. (2017). Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota MM, Pradel G, Vanderberg JP, Hafalla JC, Frevert U, Nussenzweig RS, Nussenzweig V, and Rodríguez A (2001). Migration of Plasmodium sporozoites through cells before infection. Science 291, 141–144. [DOI] [PubMed] [Google Scholar]
- Murugan R, Scally SW, Costa G, Mustafa G, Thai E, Decker T, Bosch A, Prieto K, Levashina EA, Julien J-P, et al. (2020). Evolution of protective human antibodies against Plasmodium falciparum circumsporozoite protein repeat motifs. Nat. Med 26, 1135–1145. [DOI] [PubMed] [Google Scholar]
- Olotu A, Lusingu J, Leach A, Lievens M, Vekemans J, Msham S, Lang T, Gould J, Dubois M-C, Jongert E, et al. (2011). Efficacy of RTS,S/AS01E malaria vaccine and exploratory analysis on anti-circumsporozoite antibody titres and protection in children aged 5–17 months in Kenya and Tanzania: a randomised controlled trial. Lancet Infect Dis 11, 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyen D, Torres JL, Wille-Reece U, Ockenhouse CF, Emerling D, Glanville J, Volkmuth W, Flores-Garcia Y, Zavala F, Ward AB, et al. (2017). Structural basis for antibody recognition of the NANP repeats in Plasmodium falciparum circumsporozoite protein. Proc. Natl. Acad. Sci. U.S.A 114, E10438–E10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyen D, Torres JL, Cottrell CA, Richter King C, Wilson IA, and Ward AB (2018). Cryo-EM structure of P. falciparum circumsporozoite protein with a vaccine-elicited antibody is stabilized by somatically mutated inter-Fab contacts. Sci Adv 4, eaau8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyen D, Torres JL, Aoto PC, Flores-Garcia Y, Binter Š, Pholcharee T, Carroll S, Reponen S, Wash R, Liang Q, et al. (2020). Structure and mechanism of monoclonal antibody binding to the junctional epitope of Plasmodium falciparum circumsporozoite protein. PLoS Pathog. 16, e1008373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassmeyer ML, Reiter K, Shimp RL, Kotova S, Smith PD, Hurt DE, House B, Zou X, Zhang Y, Hickman M, et al. (2009). Structure of the Plasmodium falciparum circumsporozoite protein, a leading malaria vaccine candidate. J. Biol. Chem 284, 26951–26963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team R: The R Project for Statistical Computing.
- Raghunandan R, Mayer BT, Flores-Garcia Y, Gerber MW, Gottardo R, Jhun H, Herrera SM, Perez-Ramos DW, Locke E, King CR, et al. (2020). Characterization of two in vivo challenge models to measure functional activity of monoclonal antibodies to Plasmodium falciparum circumsporozoite protein. Malar. J 19, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Galán A, Salman AM, Bowyer G, Collins KA, Longley RJ, Brod F, Ulaszewska M, Ewer KJ, Janse CJ, Khan SM, et al. (2017). An in vitro assay to measure antibody-mediated inhibition of P. berghei sporozoite invasion against P. falciparum antigens. Sci Rep 7, 17011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RTS,S Clinical Trials Partnership (2015). Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet 386, 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sällberg M, Hughes J, Jones J, Phillips TR, and Milich DR (2002). A malaria vaccine candidate based on a hepatitis B virus core platform. Intervirology 45, 350–361. [DOI] [PubMed] [Google Scholar]
- Sattabongkot J, Yimamnuaychoke N, Leelaudomlipi S, Rasameesoraj M, Jenwithisuk R, Coleman RE, Udomsangpetch R, Cui L, and Brewer TG (2006). Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am. J. Trop. Med. Hyg 74, 708–715. [PubMed] [Google Scholar]
- Scally SW, and Julien J-P (2018). Peek-Peak-Pique: Repeating Motifs of Subtle Variance Are Targets for Potent Malaria Antibodies. Immunity 48, 851–854. [DOI] [PubMed] [Google Scholar]
- Scally SW, Murugan R, Bosch A, Triller G, Costa G, Mordmüller B, Kremsner PG, Sim BKL, Hoffman SL, Levashina EA, et al. (2018). Rare PfCSP C-terminal antibodies induced by live sporozoite vaccination are ineffective against malaria infection. J. Exp. Med 215, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder RA, Chang L-J, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, Holman LA, James ER, Billingsley PF, Gunasekera A, et al. (2013). Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365. [DOI] [PubMed] [Google Scholar]
- Sidjanski S, and Vanderberg JP (1997). Delayed migration of Plasmodium sporozoites from the mosquito bite site to the blood. Am. J. Trop. Med. Hyg 57, 426–429. [DOI] [PubMed] [Google Scholar]
- Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, Wellde BT, Garçon N, Krzych U, and Marchand M (1997). A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S Malaria Vaccine Evaluation Group. N. Engl. J. Med 336, 86–91. [DOI] [PubMed] [Google Scholar]
- Tan J, Sack BK, Oyen D, Zenklusen I, Piccoli L, Barbieri S, Foglierini M, Fregni CS, Marcandalli J, Jongo S, et al. (2018). A public antibody lineage that potently inhibits malaria infection through dual binding to the circumsporozoite protein. Nat. Med 24, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares J, Formaglio P, Thiberge S, Mordelet E, Van Rooijen N, Medvinsky A, Ménard R, and Amino R (2013). Role of host cell traversal by the malaria sporozoite during liver infection. J. Exp. Med 210, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R, Spaccapelo R, Bistoni F, Holder AA, and Crisanti A (2002). Function of region I and II adhesive motifs of Plasmodium falciparum circumsporozoite protein in sporozoite motility and infectivity. J. Biol. Chem 277, 47613–47618. [DOI] [PubMed] [Google Scholar]
- Triller G, Scally SW, Costa G, Pissarev M, Kreschel C, Bosch A, Marois E, Sack BK, Murugan R, Salman AM, et al. (2017). Natural Parasite Exposure Induces Protective Human Anti-Malarial Antibodies. Immunity 47, 1197–1209.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderberg JP, and Frevert U (2004). Intravital microscopy demonstrating antibody-mediated immobilisation of Plasmodium berghei sporozoites injected into skin by mosquitoes. International Journal for Parasitology 34, 991–996. [DOI] [PubMed] [Google Scholar]
- Vanderberg J, Nussenzweig R, and Most H (1969). Protective immunity produced by the injection of x-irradiated sporozoites of Plasmodium berghei. V. In vitro effects of immune serum on sporozoites. Mil Med 134, 1183–1190. [PubMed] [Google Scholar]
- Verhave JP, Leeuwenberg AD, Ponnudurai T, Meuwissen JH, and van Druten JA (1988). The biotin-streptavidin system in a two-site ELISA for the detection of plasmodial sporozoite antigen in mosquitoes. Parasite Immunol 10, 17–31. [DOI] [PubMed] [Google Scholar]
- White MT, Bejon P, Olotu A, Griffin JT, Bojang K, Lusingu J, Salim N, Abdulla S, Otsyula N, Agnandji ST, et al. (2014). A combined analysis of immunogenicity, antibody kinetics and vaccine efficacy from phase 2 trials of the RTS,S malaria vaccine. BMC Med 12, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MT, Verity R, Griffin JT, Asante KP, Owusu-Agyei S, Greenwood B, Drakeley C, Gesase S, Lusingu J, Ansong D, et al. (2015). Immunogenicity of the RTS,S/AS01 malaria vaccine and implications for duration of vaccine efficacy: secondary analysis of data from a phase 3 randomised controlled trial. Lancet Infect Dis 15, 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2015). WHO | Fact Sheet: World Malaria Report 2015.
- WHO (2018). WHO | World malaria report 2018.
- Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, et al. (2019). Welcome to the Tidyverse. Journal of Open Source Software 4, 1686. [Google Scholar]
- Wilke CO (2019). cowplot: Streamlined Plot Theme and Plot Annotations for “ggplot2.”
- Yang ASP, O’Neill MT, Jennison C, Lopaticki S, Allison CC, Armistead JS, Erickson SM, Rogers KL, Ellisdon AM, Whisstock JC, et al. (2017). Cell Traversal Activity Is Important for Plasmodium falciparum Liver Infection in Humanized Mice. Cell Rep 18, 3105–3116. [DOI] [PubMed] [Google Scholar]
- Zavala F, Cochrane AH, Nardin EH, Nussenzweig RS, and Nussenzweig V (1983). Circumsporozoite proteins of malaria parasites contain a single immunodominant region with two or more identical epitopes. J. Exp. Med 157, 1947–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The heavy and light chain gene sequences of anti-PfCSP human monoclonal antibodies isolated in this study, along with the sequences of PCR primers used to amplify the antibody genes, were deposited in GenBank (Accession Numbers MT811859–MT811914). Sequences of synthetic rPfCSP constructs created in this study were deposited in GenBank (Accession Numbers MT891160 –MT891178).