

SUMMARY
Influenza B virus (IBV) infections can cause severe disease in children and the elderly. Commonly used antivirals have lower clinical effectiveness against IBV compared to influenza A viruses (IAV). Neuraminidase (NA), the second major surface protein on the influenza virus, is emerging as a target of broadly protective antibodies that recognize the NA active site of IAVs. However, similarly broadly protective antibodies against IBV NA have not been identified. Here, we isolated and characterized human monoclonal antibodies (mAbs) that target IBV NA from an IBV-infected patient. Two mAbs displayed broad and potent capacity to inhibit IBV NA enzymatic activity, neutralize the virus in vitro, and protect against lethal IBV infection in mice in prophylactic and therapeutic settings. These mAbs inserted long CDR-H3 loops into the NA active site, engaging residues highly conserved amongst IBV NAs. These mAbs provide a blueprint for the development of improved vaccines and therapeutics against IBVs.
Keywords: Influenza B virus, Neuraminidase, Monoclonal antibodies, Human, Infection, B cells, Plasmablasts
eTOC blurb
Influenza B virus (IBV) infections cause severe disease. Madsen et al. develop and characterize human monoclonal antibodies that possess broad and potent capacities to inhibit IBV neuraminidase enzymatic activity, neutralize the virus in vitro, and protect against lethal IBV infection in mice in prophylactic and therapeutic settings.
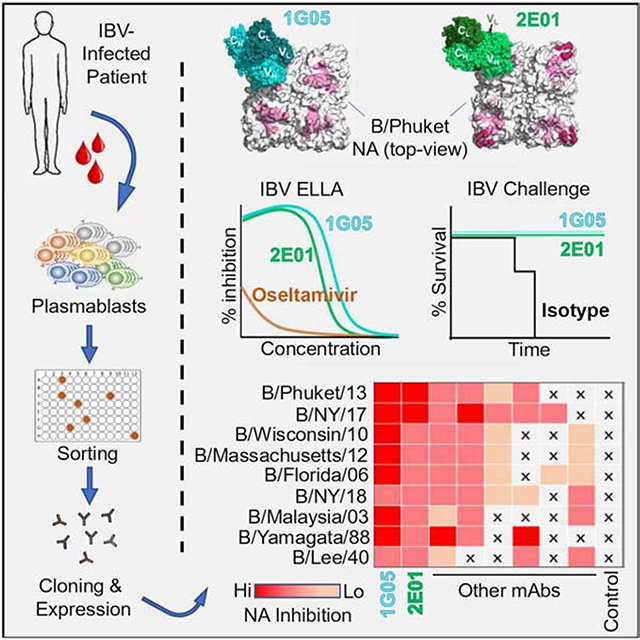
Graphical Abstract
INTRODUCTION
Seasonal influenza virus infections result in significant global morbidity and mortality. Influenza B virus (IBV) infection causes approximately 25% of all seasonal influenza virus infections (Paul Glezen et al., 2013; Tan et al., 2018). Circulating IBVs are phylogenetically divided into B/Yamagata/16/88-like (Y) and B/Victoria/2/87-like (V) lineages based on their hemagglutinin (HA) sequences (Rota et al., 1990). The B/Yamagata/16/88-like lineage divided into clades 2 and 3, and one-, two-, or three-amino acid deletion mutants of B/Victoria/2/87-like viruses have emerged, thereby expanding the antigenic diversity of IBVs (Langat et al., 2017; Virk et al., 2020). Current quadrivalent seasonal influenza virus vaccines include representative strains from both IBV lineages and the two circulating influenza A virus (IAV) strains from the H1N1 and H3N2 subtypes. These vaccines primarily elicit an antibody response directed against the major surface glycoprotein of the virus, HA (Ellebedy and Ahmed, 2012; Krammer, 2019). Vaccine-induced antibody responses can be rendered largely ineffective by the continuous antigenic drift of circulating influenza viruses, which significantly undermines overall vaccine effectiveness. Consequently, strains included in seasonal vaccines need to be reviewed on a biannual basis, creating an urgent need for new vaccines and treatment options that can provide broader and more durable protection (Ellebedy and Webby, 2009).
Neuraminidase (NA) is the second major surface protein on the influenza virus (Krammer et al., 2018). NA functions by cleaving terminal sialic acid residues from N-linked glycans, facilitating virus egress from infected cells and release of virus trapped by natural defense proteins like mucins. Antibodies targeting NA primarily block influenza virus replication by interfering with viral egress (Eichelberger et al., 2018). Anti-NA monoclonal antibodies (mAbs) and NA vaccination–induced polyclonal antibodies protect against lethal influenza virus challenge in animal models (Stadlbauer et al., 2018; Wohlbold et al., 2015). Mucosal anti-IBV NA antibodies can prevent viral transmission in guinea pigs (McMahon et al., 2019). Oseltamivir, the most widely prescribed anti-influenza antiviral drug, targets NA (Govorkova and McCullers, 2013). Oseltamivir is currently the only anti-influenza antiviral drug approved by the U.S. Food and Drug Administration (FDA) for use in children aged ≤2 years old (Burnham et al., 2013). However, oseltamivir is less effective for reducing the duration of fever and virus persistence in IBV infection compared to IAV infection, especially in pediatric populations (Kawai et al., 2006; Sato et al., 2008; Sugaya et al., 2007).
Our group isolated and characterized three clonally related mAbs derived from plasmablasts isolated from an H3N2 IAV-infected individual that show broad, heterosubtypic inhibition activity against NA in IAV groups 1 and 2 strains and a fraction of IBVs (Stadlbauer et al., 2019). These mAbs target conserved residues within the NA active site. Here, we isolated seven anti-IBV NA mAbs (BNA-mAbs) from an IBV-infected individual during the acute phase of infection. We determined that two of these BNA-mAbs neutralized virus, mediated effector functions, were broadly protective in vivo, and inhibited NA activity by blocking its enzymatic active site with long CDR-H3 loops, similar to the mechanism of NA inhibitors such as oseltamivir.
RESULTS
Isolation of Broadly Cross-Reactive Anti-NA Monoclonal Antibodies
Peripheral blood samples were obtained from a hospitalized patient with confirmed IBV infection on day 4 after onset of symptomatic illness during the 2017–2018 influenza season. IBV infection was consistent with an HA-specific plasmablast response that was exclusively directed against IBV HAs rather than HAs derived from IAV H1N1 or H3N2 influenza virus strains as measured by enzyme-linked immunospot (ELISpot) assay (Figure S1A). Plasmablasts (defined as CD19+ IgD– CD38+ CD20– CD71hi) were single-cell sorted, and the corresponding mAbs were expressed (Ellebedy et al., 2016; Wrammert et al., 2011). A total of 21 recombinant clonally distinct mAbs specific against IBV were generated (Figure 1A). Further screening revealed that ten of these mAbs recognized recombinant IBV HA, seven were IBV NA-specific, and the remaining four were directed against IBV NP and M1 proteins (Figure 1A). The seven anti-NA mAbs were derived from distinct B cell clones (Table S1). Further evaluation of the anti-NA-mAbs revealed broad cross-reactivity to recombinant NA proteins from both the B/Yamagata/16/88-like and the B/Victoria/2/87-like lineages (Figure 1B and Table S2). By contrast, the broadly neutralizing anti-IAV NA-mAb 1G01 (Stadlbauer et al., 2019) displayed binding to a very limited number of BNA, such as NA from B/Malaysia/2506/04 (Figure S1B). Competition ELISA among the BNA-mAbs indicated that 1G05, 1D05, and 2E01 recognized potentially overlapping epitopes within the B/New York/PV00094/17 NA (Figure S1C). Similarly, mAbs 3C01 and 2H09 strongly inhibited each other’s binding (>90%), indicating a potentially common epitope. The mAbs 3C01 and 2H09 share the same heavy chain variable gene (VH3–74), but not the light chain variable gene (Figure 1A and Table S1). These data indicated that IBV infection elicited a robust and cross-reactive plasmablast response to NA.
Figure 1. Broadly cross-reactive anti-neuraminidase monoclonal antibodies.

(A) Specificities of 21 clonally distinct mAbs, IGHV and IGLV gene usage, and amino acid sequence of the heavy chain CDR3 for each BNA-mAb.
(B) ELISA of binding by seven BNA-mAbs plus an IBV HA-specific mAb (2B04) to recombinant NA and HA from the indicated IBV strains. Data are representative of two experiments. See also Figure S1 and Tables S1 and S2.
Anti-BNA mAbs Exhibit Broad Enzyme Inhibition and IBV Neutralization In Vitro
The BNA-mAbs were further characterized in an enzyme-linked lectin assay (ELLA) to better assess their potential to inhibit the enzymatic activity of NA (Figures 2A and S2A–I). All mAbs showed some NA inhibition (NI) activity, and 1G05 and 2E01 demonstrated remarkable NI activities against viruses belonging to the B/Yamagata/16/88-like and B/Victoria/2/87-like lineages and the ancestral B/Lee/1940 strain, which cumulatively span more than 70 years of antigenic drift (Figure 2A). We further examined the NI capacity of 1G05 and 2E01 against two zanamivir/oseltamivir-resistant IBV strains, B/Memphis/20/1996 (Y) R152K (Gubareva et al., 1998) and B/Rochester/02/2001 (V) D198N (Ison et al., 2006) and their wild type counterparts, using oseltamivir as a control. The NI activity of oseltamivir was severely diminished against the resistant mutants, whereas the NI activities of both 1G05 and 2E01 were minimally impacted (Figure S2J–M). NA enzymatic activity can be inhibited by mAbs binding directly or proximal to the enzymatic active site through steric hindrance. ELLA uses a large substrate (fetuin), which can be blocked by steric hindrance. The NA-Star assay uses a smaller substrate, and enzymatic activity is inhibited only by mAbs that bind directly to the enzymatic active site (Chen et al., 2018; Wohlbold et al., 2017). Only BNA-mAbs 1G05 and 2E01 exhibited NI activity in the NA-Star assay (Figure 2B).
Figure 2. BNA-mAbs exhibit broadly cross-reactive virus inhibition and neutralization in vitro.

(A) NA inhibition (NI) IC50 of BNA-mAbs against the indicated IBV strains measured by ELLA. IAV NA-specific mAb 1G01 and irrelevant human IgG1 are negative controls. See also Figure S2.
(B) NI of BNA-mAbs against B/Phuket/3073/13 (Y) in an NA-Star assay. Symbols represent mean ±SD.
(C) Neutralization capacity of BNA-mAbs against B/Phuket/3073/13 (Y) and B/Brisbane/60/08 (V) measured by plaque reduction assay. See also Figure S3. Data are representative of two experiments. See also Figures S1, S2, S3.
We tested whether the BNA-mAbs inhibited virus replication in vitro using a plaque reduction neutralization assay (PRNA). All BNA-mAbs except for 1D05 exhibited, to varying degrees, neutralizing activity against B/Phuket/3073/13 (Y) and B/Brisbane/60/08 (V) viruses (Figures 2C and S3A–B), and 1G05 and 2E01 were the most potent. Anti-influenza virus antibodies can mediate protection through Fc-receptor–mediated effector functions [e.g., antibody-dependent cellular cytotoxicity (ADCC)] (DiLillo et al., 2014; Wohlbold et al., 2017). All BNA-mAbs displayed activity in an ADCC reporter assay against B/Phuket/3073/13 (Y) and B/Brisbane/60/08 (V) viruses (Figure S3C–D). These combined data indicated that the BNA-mAbs blocked virus replication in vitro by inhibiting NA activity, and suggested that mAbs directly targeting the NA enzymatic active site had potentially more potent virus neutralization capacities in vitro.
BNA-mAbs Are Broadly Protective in a Lethal Murine Model of IBV Infection
Next, we tested the protective capacities of the BNA-mAbs in vivo using a lethal murine model of IBV infection. The mAbs were tested in both prophylactic and therapeutic settings against IBVs that were isolated at the Mount Sinai Medical Center and were representative of currently circulating IBVs. All BNA-mAbs conferred robust protection (100% survival) against B/New York/PV00094/17 (Y) when 5 mg/kg was injected intraperitoneally 2 hours before intranasal virus challenge (Figure 3A–B). Remarkably, robust protection was maintained even when the mAb dose was reduced to 1 mg/kg (Figure S3E–F). Lung viral load was assessed at 3 and 6 days post-challenge. Mean lung titers trended lower in all groups treated with BNA-mAbs compared to the control-treated group by 3 days post-infection, with the 1G05-treated animals showing an almost two-log decrease in viral load (Figure 3C). By 6 days post-infection, viral replication was markedly lower or undetectable in all anti-NA treated animals compared to those injected with the negative control mAb (Figure 3D). Robust prophylactic protective capacity of the BNA-mAbs also was observed when animals were challenged with a different IBV belonging to the B/Victoria/2/87-like lineage, B/New York/PV00081/18 (V) (Figure 3E–F).
Figure 3. BNA-mAbs are broadly protective in vivo.

(A, B, E–H) Survival (A, E, G) and percent original weight (B, F, H) of mice challenged with the indicated virus 2 h after (A–F) or 72 h before (G–H) administration of the indicated mAb. Five mice were used per mAb. Symbols represent mean ±SD. *P<0.05, Mantel-Cox log rank test between each mAb and isotype.
(C, D) Lung titers of mice treated prophylactically and challenged as in (A) at 3 days (C) and 6 days (D) after infection. Error bars represent SD. Three mice were used per group. *P<0.05, Kruskal-Wallis with Dunn’s multiple comparisons test between each mAb and isotype. See also Figure S3.
We assessed the therapeutic potential of the BNA-mAbs by infecting mice with a lethal dose of B/New York/PV00094/17 (Y), and then treating with 5 mg/kg BNA-mAbs after 72 hours. All animals experienced weight loss in this setting. Only 2E01 provided 100% protection against mortality (Figure 3G–H), and 1G05 and 3C01 protected 80% of the mice against lethality (Figure 3G–H). These data indicated that the BNA-mAbs protected against lethal IBV infection in vivo, and confirmed the superiority of some (1G05 and 2E01) anti-NA mAbs that potentially targeted the NA enzymatic active site in affording protection.
Overall Structure of IBV NA-1G05 and NA-2E01 Complexes
Both 1G05 and 2E01 inhibited the enzymatic activity of NAs from a broad range of IBV strains and blocked IBV infection in vitro and in vivo. Both mAbs specifically bound to IBV NA and did not bind IVA NA (Figure S3G–I). Primary sequence alignments of 1G05 and 2E01 with their inferred germline ancestors showed that both had accumulated substantial mutations (Figure S4A–B). To determine how these mutations altered their binding to BNA, we expressed monomeric antigen-binding fragments (Fabs) of mAbs 1G05 and 2E01 and their corresponding inferred germline ancestors and performed biolayer interferometry (BLI) to measure their binding kinetics to B/Phuket/3073/2013 NA. The mAb 1G05 had higher binding affinity and longer half-life (t1/2) than 2E01 (Figure S4C–D). The inferred germline ancestor of 1G05 had substantially lower NA binding, whereas that of 2E01 displayed no detectable NA binding (Figure S4E–F). We performed single particle cryo-electron microscopy (cryo-EM) to investigate the epitopes targeted by these mAbs by solving their Fab structures in a complex with B/Phuket/3073/2013 NA, designated as NA-1G05 and NA-2E01 (Table S3). For both datasets, 2D classification showed that particle orientations on grids were well-distributed (Figure 4A–B). The 3D reconstruction of both NA-Fab complexes identified tetrameric NA decorated by one Fab per NA promoter (Figure 4C–D). The final maps were interpreted at 2.5 Å and 2.8 Å resolution for NA-1G05 and NA-2E01, respectively (Figure 4E–F).
Figure 4. Cryo-EM reconstruction of NA-1G05 and NA-2E01 particles.

(A, B) Reference model–free two dimensional classifications of NA-1G05 (A) and NA-2E01 (B).
(C, D) Cryo-EM reconstructions of Fabs 1G05 (C) and 2E01 (D) in complex with NA. One NA tetramer (gray) bound four Fabs.
(E, F) Fourier shell correlation (FSC) curves for NA-1G05 (E) and NA-2E01 (F) after post-processing with Relion-3. See also Figure S4 and Table S3.
Local resolution analyses of the final reconstructed electron density maps revealed that the resolution decreased from the NA core to the constant domains of each Fab, suggesting flexibility of the Fab elbow regions (Figures 5A–B, S5A–B, and S5E–F). The NA in NA-1G05 was highly ordered and a complete atomic model was built (Figure S5C). Four NA regions in the NA-2E01 complex were not well ordered, including residues E105-S110 and G140-Y143 in the 150-loop, G433-T437 in the 430-loop, and W456-L466 at the C-terminus (data not shown), although the atomic model for the remaining part of the NA-2E01 tetramer was built properly (Figure S5D). The variable domains of both Fabs and the complementarity-determining regions (CDRs) were very well resolved (Figure 5C–F) and provided critical information for epitope analysis. The buried surface area of the NA and 1G05 interface was ~1100 Å2, with the heavy chain (HC) accounting for ~90% of the interaction. The 1G05-HC dominated the Fab binding to NA by CDR-H3 protruding into the active pocket of NA (Figure 5C), whereas the light chain (LC) only contributed to the interface by interacting with one N-acetylglucosamine moiety attached to residue N144 on NA (Figure S6A). In NA-2E01, the buried surface area was ~960 Å2, with the LC accounting for more surface area than 1G05, representing ~20% of the total interaction (200 Å2 of the total 960 Å2 interface). The 2E01-HC displayed approximately 41.6° counterclockwise rotation compared to 1G05-HC binding to NA (Figure 6A and D). Similar to binding of 1G05, the CDR-H3 of 2E01 also played a major role in engaging the NA active site (Figure 5D). These data established that both 1G05 and 2E01 directly targeted the IBV NA active site, consistent with their NI activity.
Figure 5. Atomic models of NA-1G05 and NA-2E01 complexes.

(A, B) Local resolution of the map of one monomeric subunit of NA bound with one 1G05 (A) and one 2E01 (B) Fab.
(C, D) Ribbon diagrams of one monomeric subunit of NA bound with 1G05 (C) and 2E01 (D) in the same orientations as in (A) and (B), respectively. Gray, NA; cyan, 1G05 heavy chain; teal, light chain (C). Green, 2E01 heavy chain; dark green, light chain (D). N-linked glycan moieties are shown as yellow sticks.
(E, F) Electron density maps and atomic models of 1G05 (E) and 2E01 (F) CDR-H3 (contour level at 5.0 σ). See also Figures S5 and S6.
Figure 6. Epitope analysis of 1G05 and 2E01.

(A) The epitope of 1G05 HC is shown as a pink-colored surface. The 1G05 HC and LC are shown as spheres in cyan and teal, respectively. (B) Epitope residues making either polar or hydrophobic interactions via side chains with 1G05 are labeled in black. Crucial contacting residues on CDRs are shown as cyan sticks.
(C, F) Conservation analysis of epitopes to 1G05 (C) and 2E01 (F) with amino acid sequences from all IBV strains [upper (C) and left (F) panels], and all IBV and IAV strains [lower (C) and right (F) panels] tested. (D) The epitopes of 2E01 HC and LC are shown as pink-colored and dark pink–colored surfaces, respectively. The 2E01 HC and LC are shown as green and dark green spheres. (E) Epitope residues making polar interactions via side chains with 2E01 are labeled in black. Crucial contacting residues on CDRs are shown as green and dark green sticks, respectively. See also Figures S6 and S7 and Tables S4 and S5.
Defining NA Epitope Residues Responsible for IBV Specificity of 1G05 and 2E01
Next, we examined the structural epitopes engaged by 1G05 and 2E01 (Figure 6). CDR-H3 dominated the contact interface for 1G05, but H1 and H2 also contributed to NA binding (Figures 6B and S6B–D). We investigated the mechanism determining the strain specificities of 1G05 and 2E01 by analyzing conservation of residues within each epitope among NA sequences for multiple influenza virus strains. The 1G05 epitope residues were nearly invariant amongst IBV strains for which NA activity was inhibited (Figure 6C, upper panel). By contrast, epitope conservation analysis indicated that two key IBV NA residues engaged by 1G05 CDR-H3 (R147 and K435) were not conserved amongst IAV strains, for which no inhibition was observed (Figure 6C, lower panel). In IAV strains, the equivalent residue of IBV NA R147 is an isoleucine, whereas for K435 it is either a glutamic acid or a glutamine (Figure S7A). Thus, R147 and K435 may contribute to the IBV strain specificity of 1G05. The mAb 2E01 engaged NA primarily using CDR-H3 and all three LC CDRs (Figures 6E and S6E–G). Epitope conservation analysis indicated that all of the 2E01 CDR-H3 contacts were invariant amongst IBV strains, whereas 2E01-LC contacts exhibited considerable IBV strain sequence variation (Figure 6F, left panel), indicating a dominant role for 2E01-HC in BNA epitope contacts. Although most of the NA epitope residues engaged by 2E01 CDR-H3 were conserved in IAV strains, two variable residues (H134 and R147) may function to determine the specificity of 2E01 (Figures 6F, right panel, and S7A).
The CDR-H3 Loop from Both 1G05 and 2E01 Imitates Sialic Acid and Oseltamivir Binding to NA
In NA-1G05, the CDR-H3 had the most important role in binding by occupying the NA active site with residues D100A and R100B, which interacted with positively and negatively charged “patches” located at either end of the active site (Figure 7A). D100A engaged a three-arginine cluster formed by NA residues R116, R292 and R374, and Y409. R100B formed salt bridges with residues D149 and E226. D100A and R100B participated in extensive van der Waals contacts with additional NA epitope residues (Table S4). D100A and R100B blocked the active site in a similar manner as that observed for occupation of the active pocket by sialic acid and oseltamivir, and their carboxyl groups also were stabilized by the three-arginine cluster (Figure 7C and D). The primary amine group of oseltamivir (stabilized by D149) combined with the acetamide group (stabilized by a water molecule and E276) shared a very similar binding mode to NA as 1G05 R100B. NA residues contacting D100A and R100B from 1G05 CDR-H3 are considered important catalytic residues (Burmeister et al., 1993; Chong et al., 1992; Lentz et al., 1987; Taylor and von Itzstein, 1994), and are highly conserved in IAV and IBV NAs (Figure S7B). Residue Q100E from CDR-H3 was stabilized by NA R147, and residue E100G was stabilized by NA K435 (Figure 7A). These results explained why 1G05 served as a strong NA inhibitor.
Figure 7. Comparison of NA active site occupation by 1G05, 2E01, sialic acid, and oseltamivir.

(A) Close-up view of the interaction between NA and 1G05. H3 is shown as cartoon loops in cyan, with crucial interacting residues shown as sticks. Residues on NA are shown as gray sticks. Polar interactions are shown with dashed lines.
(B) Interactions between NA and 2E01 in the same orientation as (A). H3 is shown as cartoon loops in green with crucial interacting residues shown as sticks. Residues on NA are shown as gray sticks. Polar interactions are shown with dashed lines.
(C) Interaction of sialic acid and NA from B/Beijing/1/1987 virus (PDB ID 1NSC). Sialic acid is shown as orange sticks. Residues on NA shown as gray sticks. Polar interactions are shown as dashed lines.
(D) Interaction of oseltamivir and NA from B/Brisbane/60/2008 virus (PDB ID 4CPM). Oseltamivir shown as blue sticks. Residues on NA shown as gray sticks. Polar interactions shown with dashed lines. For sialic acid-NA and oseltamivir-NA complex structures, NA contact residue numbering was increased by 1 compared to original coordinate files to maintain consistency with NA from B/Phuket/3073/13 virus. See also Figure S7 and Tables S4 and S5.
The 1G05 CDR-H3 interacted with both basic and acidic “patches” of the NA active pocket. By contrast, the 2E01 CDR-H3 primarily made polar interactions with basic residues on NA using D100B and D100D (Figure 7B). D100B was stabilized by two conserved catalytic residues (R116 and R374), similar to D100A in 1G05 CDR-H3, and mimicked the binding mode of the sialic acid and oseltamivir carboxylate to NA (Figure 7C–D). D100D formed a salt bridge with R147 (Figure 7B). The NA R150 in NA-1G05 and NA-2E01 was not involved in ionic interactions as it was in the NA-sialic acid and NA-oseltamivir complexes, but it interacted with R100B in 1G05 and E100A in 2E01 through van der Waals contacts (Figure S6D and G, Tables S4 and S5). Similarly, in the NA-2E01 complex structure, catalytically crucial residues NA R292 and Y409 interacted with F100C and D100B in CDR-H3 of 2E01 via van der Waals contacts (Figure S6G and Table S5). In summary, both mAbs inhibited NA enzymatic activity by blocking the active pocket with long CDR-H3 loops, and bound NA using similar mechanisms as those of the NA substrate sialic acid and NA inhibitor oseltamivir. The CDR-H3 of 1G05 protruded deeper into the active pocket than 2E01 and formed a more extensive polar interaction network with NA, explaining its higher binding affinity, longer t1/2, and stronger inhibition of IBV NAs.
DISCUSSION
Studies of antibody-mediated immunity to influenza viruses traditionally focus on HA as a target (Ellebedy and Ahmed, 2012; Wilson and Andrews, 2012). However, anti-NA antibodies have an important role in providing a comprehensive immune-mediated protection against influenza virus infection (Krammer et al., 2018). In this study, we described the isolation and functional characterization of seven novel human BNA-mAbs derived from an infected patient. All mAbs protected mice in a lethal IBV challenge model using clinically isolated IBV strains that belonged to the two antigenically distinct IBV lineages. We determined the structural basis for broad NA inhibition exhibited by two mAbs that displayed the most potent reactivity in vitro and in vivo. The described BNA-mAbs are potentially valuable as therapeutics due to the limitations of currently approved antiviral drugs targeting influenza. In vitro studies show that IBVs are less susceptible than IAVs to FDA-approved NA inhibitors and cap-dependent endonuclease inhibitors, thereby complicating treatment of IBV infections (Burnham et al., 2013; Mishin et al., 2019). This is especially true in the pediatric population where oseltamivir is less effective than zanamivir, with the latter approved only for children aged ≥7 years old. Three of the isolated mAbs (2E01, 1G05, and 3C01) protected 80–100% of mice from mortality in stringent challenge models with recent IBV isolates, even when animals were treated 72 hours after virus challenge. These data clearly demonstrated the potential of the identified BNA-mAbs for use as human therapeutics.
Seasonal epidemics caused by IBVs are responsible for up to 52% of influenza-associated pediatric mortality during the last fifteen years (Burnham et al., 2013; Govorkova and McCullers, 2013). Many aspects of immunity to IBVs are understudied compared to those of IAVs, particularly antibody-mediated immunity to IBV NA. Panels of murine and human mAbs specific for IBV NA have been reported (Piepenbrink et al., 2019; Wohlbold et al., 2017), although comprehensive functional and structural analyses of how these mAbs inhibit NA or mediate protection are lacking. Anti-NA antibodies provide in vivo protection by blocking either viral transport through the mucosal layer lining the lung epithelium or viral egress from infected cells (Eichelberger et al., 2018). Here, we demonstrated that all seven of the isolated BNA-mAbs displayed NA inhibitory (NAI) activity, virus neutralization capacity, and ability to mediate secondary effector functions as evidenced by their activity in an ADCC bioreporter assay. Our detailed structural analyses indicated that the NAI activities of the most potent mAbs were mediated by a sialic acid/oseltamivir-like mode of binding to residues within the NA enzymatic active site. The in vivo protection exhibited by these mAbs was likely mediated by a combination of these mechanisms.
Structurally mapped human anti-IAV NA antibodies are predominantly subtype specific (Gilchuk et al., 2019; Zhu et al., 2019). Our group isolated 1G01, a broadly protective human anti-NA mAb that predominantly targets IAVs (Stadlbauer et al., 2019). Similar to 1G05 and 2E01, 1G01 CDR-H3 accounts for the majority of NA recognition. However, CDR-H3 loops in 1G05 and 2E01 protrude into the NA active pocket from an entirely different angle than those of 1G01. The epitope recognized by both 1G05 and 2E01 is within one protomer, whereas in NA-1G01, the side chain of Y97 from CDR-L3 is stabilized by hydrophobic interaction with W456′ from an adjacent NA protomer. The NA-1G01 catalytic arginines (R118 and R371) are engaged with a backbone carbonyl of R100C in CDR-H3, whereas the corresponding arginines (R116 and R374) in NA-1G05 and NA-2E01 are stabilized by side chains of D100A and D100B, respectively. These combined results indicate that the epitopes recognized by 1G05 and 2E01 are significantly different from those of 1G01, despite the high similarity between NA active sites in IBVs and IAVs. Similar to 1G01, 1G05 and 2E01 contain a longer than average HCDR3, which may be an important feature of mAbs that efficiently block the NA enzymatic active site. However, this feature is not sufficient to block the active site; mAb 1D05 contains the longest HCDR3 among the seven mAbs (25 aa), but did not display strong or broad NA inhibition activity.
Previously reported atomic structures of mAb/IVB NA complexes used murine mAbs targeting non-active site epitopes, which were solved at ~25 Å resolution (Wohlbold et al., 2017). A study reported a panel of broadly protective anti-BNA human mAbs from vaccinated individuals (Piepenbrink et al., 2019). Although overall mutation levels in the present study were similar with that previous report, there was no overlap among the IGHV or IGLV genes or obvious similarity in the CDR3 regions of the mAbs. Notably, 1G05 and 2E01 inhibited the NA enzymatic activity of a variety of IBVs, ranging from one of the earliest IBV isolates (B/Lee/1940) to contemporary isolates (B/New York/PVI/81/2018), spanning more than 75 years of antigenic drift. Epitope conservation analysis showed that key NA residues of the epitopes targeted by these two mAbs were highly conserved. Structure-based sequence alignment showed that these residues were conserved among the IBV strains circulating during the 2019/20 influenza season that caused the most recent IBV outbreak.
A call to action has been issued to improve the effectiveness of influenza virus vaccines (Erbelding et al., 2018; Paules et al., 2017, 2018). Ongoing efforts are predominantly focused on redesigning influenza virus immunogens to direct the antibody response toward conserved epitopes within the HA, such as those within the HA stem region (Crowe, 2017). Some of these vaccine candidates have now entered advanced stages of clinical testing. These efforts are aided by the large body of functional and structural data describing the neutralization and protection mechanisms mediated by multiple broadly cross-reactive anti-HA mAbs isolated during the last decade (Murin et al., 2019; Wu and Wilson, 2018). The results of the present study will facilitate the design of new NA-based immunogens that can elicit broad and durable antibody-mediated protection against IBVs. This is especially important in light of recent antigenic changes in the B/Victoria/2/87-like lineage (deletion mutants) and the split of the B/Yamagata/16/88-like lineage into two clades, which will further complicate HA-based vaccine development. NA-based or NA-enhanced vaccines against influenza B could provide a viable solution for this problem.
Limitations
Several gaps remain in our current understanding of human B cell response to IBV NA that this study does not address. We generated all of the monoclonal antibodies from a single patient. Therefore, we do not know how rare human B cells targeting these highly conserved IBV NA epitopes are. Along the same lines, we isolated only seven monoclonal antibodies that are directed against IBV NA, so we are unable to determine how dominant the broadly cross-reactive anti-IBV NA antibodies versus the strain-specific ones. And while we show that 1G05 and 2E01 can inhibit IBV strains that are resistant to oseltamivir, we do not know the prevalence, if any, of currently circulating IBV isolates that are resistant to these antibodies. It will be important to experimentally determine whether using a combination of 1G05 and 2E01 in prophylactic and therapeutic settings would diminish the potential of such resistant strains to emerge.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled upon reasonable request by the Lead Contact, Ali H. Ellebedy, PhD (ellebedy@wustl.edu).
Materials Availability
There are restrictions to the availability of the BNA-mAbs generated in this manuscript due to the lack of an external centralized repository for their distribution and our need to maintain the stock. We are glad to share the BNA-mAbs with reasonable compensation by requestor for their processing and shipping.
Data and Code Availability
The mAbs generated in this study were deposited with GenBank accession numbers MN888992–MN889005 and MT200637–MT200664. The NA-1G05 structure was deposited with PDB entry ID 6V4N and EMDB entry ID EMD-21042. The NA-2E01 structure was deposited with PDB entry ID 6V4O and EMDB entry ID EMD-21043.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Six- to eight-week-old female BALB/c mice were used for all animal experiments (Jackson Laboratories). All experiments were performed in accordance with protocols approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee (IACUC).
Human Materials
Human peripheral blood mononuclear cells (PBMCs) were obtained from a single subject, 1718025, enrolled into the Barnes Jewish Hospital Emergency Department Influenza (EDFLU) prospective observational cohort study in St. Louis, MO. The EDFLU study was reviewed and approved by the Washington University in Saint Louis Institutional Review Board (Approval # 2017–10-220). The patient was a 51-year-old male recruited during the 2017–2018 influenza season, and PBMCs were obtained on the fourth day of symptomatic illness following presentation for medical attention to the Barnes Jewish Hospital Emergency Department. The subject did not receive the 2017–2018 seasonal influenza virus vaccine but had received other seasonal influenza virus vaccines in previous influenza seasons. The subject was briefly admitted to the hospital and discharged 2.5 days after admission without complications.
Cell Lines
Expi293F cells were grown in Expi293™ Expression Medium (#A1435102, Gibco™). Madin Darby canine kidney (MDCK) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL). ADCC bioeffector FcγRIIIa cells (Promega) were thawed according to the manufacturer’s protocol and used directly. Sf9 cells (#12659017, Gibco™) were cultured in Sf-900™ III SFM (#12658019, Gibco™) supplemented with 0.5% penicillin-streptomycin (#15070063, Gibco™). High Five™ cells (#B85502, Gibco™) were cultured in Express Five™ SFM (#10486025, Gibco™) supplemented with 18 mM L-glutamine (#25030081, Gibco™), 10 U/ml heparin (#H3149, Sigma-Aldrich), and 0.25% penicillin-streptomycin. Insect cells were maintained in an incubator at 28°C.
METHOD DETAILS
PBMC Isolation
Blood was collected in ethylenediaminetetraacetic acid (EDTA)-anticoagulated sample tubes using standard phlebotomy techniques. PBMCs were prepared within 8 h of collection by layering blood over Ficoll and centrifuging at 400 g for 30 min. The PBMC layer at the Ficoll interface was collected, washed with 1× phosphate-buffered saline (PBS), and resuspended in Roswell Park Memorial Institute (RPMI)-1640 medium. Cell counts were obtained, and cells were cryogenically preserved in RPMI-1640 medium supplemented with 10% dimethyl sulfoxide and 40% fetal bovine serum (FBS).
Enzyme-Linked Immunospot Analysis (ELISpot)
Direct ex vivo ELISpot was used to enumerate the number of IgG-secreting, recombinant HA-specific plasmablasts present in the PBMC sample. Dilutions of washed PBMCs incubated in RPMI-1640 medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL) were incubated in 96-well ELISpot plates (Milipore) for 18 h. After washing the plates with PBS, then PBS supplemented with 0.05 % Tween, secreted antibodies were detected with anti-human IgG-biotin (Jackson ImmunoResearch) and avidin-D-horseradish peroxidase (HRP) (Vector Laboratories), and developed with 3-amino-9-ethylcarbazole (AEC) substrate (Sigma) before analysis on an ELISpot counter (Cellular Technologies, Ltd.).
Cell Sorting
Staining for sorting was performed using cryo-preserved PBMCs resuspended in PBS supplemented with 2% FBS and 2 mM EDTA. Cells were stained for 30 min at 4°C with CD71-FITC (clone CY1G4), CD19-PE (clone HIB19), CD38-BV605 (clone HIT2), CD20-APC-Fire750 (clone 2H7), and Zombie Aqua (all from BioLegend). Cells were then washed twice, and single plasmablasts (live singlet CD19+ CD38+ CD71+) were sorted with a MoFlo (Beckman-Coulter) into 96-well plates containing 10 μL of 10 mM Tris supplemented with 1 U/μL RNase inhibitor (Promega), and immediately frozen on dry ice.
Monoclonal Antibody Generation
Antibodies were cloned as described previously (Wrammert et al., 2011). Briefly, after reverse transcription of RNA from single sorted cells, VH, Vλ, and Vκ genes were amplified by polymerase chain reaction (1st PCR), and then PCR-amplified again (nested PCR) using cocktails of primers specific for IgG, Igλ, and Igκ (Smith et al., 2009), and then sequenced. To generate recombinant antibodies, heavy chain V-D-J and light chain V-J fragments were PCR-amplified from 1st round PCR products for subsequent Gibson assembly, as described previously (Ho et al., 2016). Heavy and light chain plasmids were co-transfected into Expi293F cells (Gibco) for expression, and antibody was purified with protein A agarose (Invitrogen).
Cells, Viruses, and Recombinant Proteins
Expi293F cells were grown in Expi293 Expression Medium (Gibco). Madin Darby canine kidney (MDCK) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL). ADCC bioeffector FcγRIIIa cells (Promega) were thawed according to the manufacturer’s protocol and used directly. Influenza viruses were grown in 8- to 10-day-old embryonated chicken eggs for 3 days at 37°C (IAVs) or 33°C (IBVs). Recombinant NA and HA proteins were expressed in the baculovirus expression system as described previously (Margine et al., 2013).
Enzyme-Linked Immunosorbent Assay (ELISA)
Ninety-six-well microtiter plates (Thermo Fisher) were coated with 100 μL inactivated virus diluted 1:100 in PBS or recombinant NA or HA proteins at a concentration of 1 μg/mL in PBS at 4°C overnight. Wells were blocked with 280 μL PBS supplemented with 0.05 % Tween-20 and 10 % FBS, and plates were incubated for 1.5 h at room temperature (RT). The blocking solution was removed, and 1:30 and 1:90 dilutions of mAb transfection culture supernatant or 3-fold serial dilutions of purified mAbs were added. After incubating at RT for 1 h, the plates were washed 3 times with T-PBS. HRP-conjugated anti-human IgG (Jackson ImmunoResearch) was diluted 1:2500 in blocking solution and added to all wells (100 μL/well). The plates were incubated at RT for 1 h, and washed 3 times with T-PBS and 3 times with PBS. Then, 100 μL substrate solution [phosphate-citrate buffer with 0.1% H2O2 and 0.4 mg/mL o-phenylenediamine dihydrochloride (OPD, Sigma)] was added to all wells and incubated for 5 min. The reaction was stopped with 1 M hydrochloric acid (HCl) (100 μL/well). The plates were read at a wavelength of 490 nm with a microtiter plate reader (Bio-Tek). The data were analyzed using Microsoft Excel and GraphPad Prism 7.
Passive Transfer Experiments in Mice
All animal experiments were conducted in accordance with institutional guidelines. Mouse passive transfer experiments were performed as described previously (Stadlbauer et al., 2018). For prophylactic mAb administration, 6- to 8-week-old female BALB/c mice were given 100 μL of each BNA-mAb individually at a concentration of 5 mg/kg or 1 mg/kg intraperitoneally (n=5 mice/mAb). Negative control mice received an irrelevant human IgG control mAb at the same dose. The mice were challenged intranasally with 5 × LD50 challenge virus 2 h after the mAb transfer, while being deeply anesthetized with a ketamine/xylazine mixture. Survival and weight loss were monitored daily for 14 days, and animals that lost 25% or more of their initial body weight were euthanized. For therapeutic mAb administration, mice were infected with 5 × LD50 of B/New York/PV00094/17 and given a 5 mg/kg dose of mAb 72 h post infection (n=5 mice/mAb). Survival and weight loss were monitored daily for 14 days, and animals that lost 25% or more of their initial body weight were euthanized. To determine the virus lung titers, mice received a 5 mg/kg dose of mAb. After 2 h, mice were infected with 0.1 × LD50 of B/New York/PV00094/17. Lungs were harvested at 3 (n=3 mice/mAb) and 6 (n=3 mice/mAb) days post infection. Lung virus titers were assessed by standard plaque assay (McMahon et al., 2019) and analyzed in Microsoft Excel and GraphPad Prism 7.
Enzyme-Linked Lectin Assay
Ninety-six-well flat bottom microtiter plates (Thermo Fisher) were coated with 100 μL/well fetuin (Sigma) at a concentration of 25 μg/mL in 1× coating buffer (KPL coating solution, SeraCare) at 4°C overnight. The next day, plates were washed 3 times with T-PBS. In a separate 96-well plate, mAbs were 2-fold serially diluted in sample diluent [PBS with 1% bovine serum albumin (BSA, Sigma) and 0.5% Tween-20 (Sigma)] starting at 30 μg/mL. Fifty microliters of each mAb dilution were transferred to the fetuin-coated plate in duplicate wells. Next, 50 μL of virus at a predetermined 90% maximal effective concentration (EC90) were added to the plates and incubated at 37°C for 18 h. The following day, the plates were washed 6 times, and 100 μl/well peanut agglutinin (PNA)-HRP (Sigma) was added at a concentration of 1 μg/ml in PBS with 1% BSA. After a 2-h incubation at RT, the plates were developed with 100 μL SigmaFast OPD. The reaction was stopped after 10 min by adding 50 μL 3M HCl (Thermo Fisher), and the plates were read at a wavelength of 490 nm with a microtiter plate reader (Bio-Tek). The data were analyzed using Microsoft Excel and GraphPad Prism 7, and the 50% inhibition concentration (IC50) was defined as the concentration of mAb at which 50% of the NA activity was inhibited compared to the negative control (virus with no mAb).
Neuraminidase Inhibition by NA-Star Assay
The NA-Star® Influenza Neuraminidase Inhibitor Resistance Detection Kit (Applied Biosystems) was used to quantify the inhibition of NA activity (cleavage of a small chemiluminescent substrate) in the presence of NA-mAbs. The experiments were performed according to the manufacturer’s protocol. Briefly, mAbs were diluted in NA-Star Assay Buffer to a concentration of 100 μg/mL, and subsequently serially diluted 1:3. Twenty-five microliters from each dilution were transferred to a white, flat bottom 96-well cell culture plate and mixed with 25 μL/well of B/Phuket/3073/2013 virus at a predetermined 2 × EC50 for 20 min at 37°C. NA-Star Substrate (10 μL/well) was added after the incubation, and the plates were incubated at RT for 30 min. NA-Star accelerator solution (60 μL/well) was added to the plates immediately before the readout. The chemiluminescent signal was detected by a microtiter plate reader (Bio-Tek) and analyzed using Microsoft Excel and GraphPad Prism 7.
Plaque Reduction
NA plaque reduction assays were performed as described previously (Wohlbold et al., 2017). Briefly, mAbs were serially diluted in 1× Minimum Essential Medium (MEM) [10% 10× minimal essential medium (Gibco), 2 mM L-glutamine, 0.1% of sodium bicarbonate (w/v, Gibco), 10 mM 4-HEPES (Gibco), 100 U/ml penicillin-100 μg/ml streptomycin (Gibco), and 0.2% bovine serum albumin (MP Biomedical)] starting at 100 μg/ml, and incubated with 50 μl of B/Phuket/3073 or B/Brisbane/60/08 virus at 2000 plaque forming units (PFU) per mL for 1 h at RT on a shaker. The virus and mAb mixtures were incubated on a monolayer of MDCK cells in a 12-well plate and incubated at 33°C for 3 d. After the incubation, cells were fixed with 3.7% formaldehyde for 1 h at 4°C, and blocked with 3% milk in PBS for 1 h at RT. The cells were then incubated with anti-IBV guinea pig sera diluted 1:500 in PBS with 1% milk for 1 h at RT. The plates were washed with PBS, and anti-guinea pig IgG-HRP antibody (Millipore) diluted to 1:3000 in PBS with 1% milk was added to the plates. The plates were then incubated for 1 h at RT. The plates were washed, and plaques were visualized by staining with KPL True-Blue peroxidase (Sera Care). The plaques were counted at each dilution and compared to a no-antibody control. The data were analyzed in Microsoft Excel and GraphPad Prism 7.
ADCC Reporter Assay
A commercial ADCC reporter assay kit (Promega) was used to assess the ability of the mAbs to activate ADCC pathways. Briefly, 100 μL/well MDCK cells (2 × 105 cells/mL) in RPMI 1640 media were seeded into white, flat bottom, 96-well cell culture plates (Corning) and incubated overnight at 37°C. The next day, cells were infected with B/Phuket/3073/13 or B/Brisbane/60/08 virus at a multiplicity of infection (MOI) of 3, and incubated at 33°C. After 16 h, the medium was replaced with human ADCC bioeffector FcγRIIIa cells (Promega) and 3-fold serial dilutions of mAbs in assay buffer (starting at 30 μg/mL). After a 6-h incubation at 37°C, Bio-Glo™ luciferase (Promega) was added to each well and incubated for 10 min in the dark at RT. The luminescence was measured by a microtiter plate reader (Bio-Tek), and the data were analyzed using Microsoft Excel and GraphPad Prism 7.
Competition ELISA
Ninety-six-well flat bottom microtiter plates (Thermo Fisher) were coated with 50 μL of 5 μg/mL purified B/New York/PV00094/17 (Y) virus diluted in coating solution (KLP) and incubated at 4°C overnight. The following day, the plates were washed three times with T-PBS and incubated for 1 h at RT with 200 μL/well blocking solution [PBS-T with 3% goat serum (Life Technologies, Inc.) and 0.5% milk powder]. Next, the blocking solution was discarded and unbiotinylated competing mAbs (100 μL/well) were added to the plates at a concentration of 20 μg/mL in blocking solution. Blocking solution with no mAbs was used as a negative control. The plates were incubated for 2 h at RT, and subsequently washed 3 times with T-PBS. A second set of the mAbs (target mAbs) was labeled with biotin using the EZ-Link NHS-PEG4-Biotin kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The biotinylated target mAbs were serially diluted 1:3 starting at 30 μg/mL in blocking solution, and transferred to the 96-well plate with the competing mAbs (100 μL/well). The plates were incubated for 2 h at RT, and washed 3 times with T-PBS. The plates were subsequently incubated for 1 h at RT with 50 μL/well streptavidin conjugated to HRP (Thermo Fisher Scientific) diluted 1:3000 in blocking solution. After the incubation, the plates were washed 4 times with T-PBS and developed with 100 μL SigmaFast OPD. The reaction was stopped after 10 minutes by adding 50 μL 3M HCl (Thermo Fisher), and the plates were read at a wavelength of 490 nm with a microtiter plate reader (Bio-Tek). The data were analyzed using Microsoft Excel and GraphPad Prism 7. The level of binding was measured as area under the curve. The percent competition for each mAb was calculated as the reduction in binding relative to the level of inhibition of any particular mAb against itself.
Cloning, Expression, and Purification of 1G05 and 2E01 Fabs
The VDJ region of the antibody sequences were subcloned with AgeI and SalI restriction endonucleases from pAbVec6W-hIgG1 to a modified pAbVec6W vector for Fab expression in which the encoded C-terminus of the hIgG1 constant region was replaced with a thrombin cleavage site and 6×HIS tag. Expi293F™ cells were transfected using the guidelines of the Gibco Expifectamine™ 293 Transfection Kit (A14524) and with heavy and light chain plasmid DNA in a 1:2 ratio. After 6 d of transfection, the cell culture supernatant was harvested and dialyzed against buffer (20 mM Tris-Cl, 150 mM NaCl, pH 8.0). Fabs were captured by passaging over Ni2+ affinity resin, and eluted in 500 mM imidazole. The eluate was then sized with HiLoad 16/600 Superdex 200 column (GE healthcare) in 20 mM Hepes, 150 mM NaCl, pH 7.4, with Fab fractions pooled and concentrated.
Cloning, Expression, and Purification of B/Phuket/3073/2013 NA for Structural Studies
The ectodomain of NA from B/Phuket/3073/2013 (EPI529344) was expressed using the flashBAC baculovirus expression system (Mirus) according to the manufacture’s protocol. Briefly, NA ectodomain residues W80-L466 were fused with an N-terminal gp67 signal peptide, a His-tag, and the human vasodilator-stimulated phosphoprotein tetramerization domain with a thrombin cleavage site (Xu et al., 2008). This construct was cloned into a modified pOET1 transfer vector containing green fluorescent protein as an indicator. The construct was co-transfected with flashBAC DNA into Sf9 insect cells (Gibco) to generate the corresponding baculovirus. Suspension cultured Trichoplusia ni High Five™ (Gibco) cells were infected at a density of 1.5 × 106 cells/ml with P2 virus at MOI of 1–5. The cell culture supernatant was harvested 72 h post-infection, and secreted NA protein was further purified by Ni2+ affinity chromatography and size exclusion chromatography.
Binding Affinity Measurement with Bio-Layer Interferometry
The binding affinity of NA with 1G05 and 2E01 Fabs was measured by BLI with Octet-Red96 instrument (ForteBio) as described previously (Ellebedy et al., 2020). The NA tetramer was randomly biotinylated (EZ-Link-NHS-PEG4-Biotin, Thermo Fisher), and excess biotin was removed by a desalting column (0.5 mL Zeba Spin 7K MWCO, Thermo Fisher). Briefly, for BLI monitoring, the biotinylated NA protein were loaded onto streptavidin biosensors (ForteBio), at 5 μg/mL for 2 min in HBS-EP buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% P20 surfactant) with 1% BSA. Five 3-fold serial dilutions of Fab samples were used per kinetics assay. The real-time data of BLI were recorded at 25°C and processed using Biaevaluation 3.1 (GE Healthcare). The 1:1 binding model was employed for the association and dissociation rate constants analyses and steady-state equilibrium concentration curves fitting. The t1/2 value of each Fab was then calculated using the formula t1/2=ln2/Kd, where Kd represents the dissociation rate constant.
Cryo-EM Data Acquisition and Image Processing
Purified NA at 1 mg/ml was incubated with Fabs at 2 mg/ml at a molar ratio of 1:1 in buffer (25 mM Hepes pH 7.4, 150 mM NaCl). For both samples, a 3 μL volume of the mixture was then applied to glow-discharged holey carbon-coated grids (Quantifoil), and flash frozen in liquid ethane using a FEI Vitrobot (Thermo Fisher). The grids were imaged on a Titan Krios (Thermo Fisher) microscope operating at 300 keV using Gatan K2 Summit detector with a total electron dose of 66 e-/Å2. Images were collected with 200 ms frame time over 40 frames in counting mode with a calibrated pixel size of 1.1 Å and a slit width of 20 eV at 105,000× magnification. Cryo-EM data processing was primarily conducted using RELION 3.0-beta-2 (Zivanov et al., 2018). Frame alignment was conducted using MotionCor2 (Zheng et al., 2017) with dose weighting, and contrast transfer function (CTF) estimation was performed using Gctf (Zhang, 2016). For dataset NA-1G05, a total of 385,417 particles were extracted from 1653 micrographs using crYOLO (Wagner et al., 2019). Reference-free two-dimensional (2D) classification was used to select 13 classes containing 240,330 good particles. 3D classification with good 2D-classes was then conducted using a H11N9 NA-single chain antibody complex structure (PDB code 1A14) low pass–filtered to 60 Å as a reference. The best classes containing 159,589 particles were then used in 3D consensus refinement, followed by CTF parameter refinement, Bayesian polishing, and further 3D consensus refinement. A final “gold standard” refinement produced the final map with a resolution of 2.5 Å after PostProcess masking (B factor sharpening) (Chen et al., 2013). Local resolution estimates were calculated with ResMap (Kucukelbir et al., 2014). The data processing of dataset NA-2E01 was similar to that of NA-1G05. Briefly, a total of 459,004 particles were extracted from 1660 micrographs using crYOLO. After 2D-classification, 37 classes of 301,384 particles were selected for 3D-classification. Then, the two best classes containing 150,730 particles were used for further refinement, leading to a final map with a resolution of 2.8 Å after PostProcess masking.
Atomic Model Building, Refinement, and Analysis
For NA-1G05, crystal structure B/Beijing/1/1987 NA (PDB code: 1NSC) and a Fab that binds to HA (PDB code: 6BTJ) were fitted into the Cryo-EM map using UCSF Chimera (Pettersen et al., 2004), followed by rigid-body refinement using Phenix (Adams et al., 2010). Model building was conducted manually in Coot (Emsley and Cowtan, 2004). The NA-1G05 model was refined to a final resolution of 2.5 Å using real space refinement in PHENIX, and was assesed using MolProbity (Chen et al., 2010). The final model has a real space correlation coefficient of 0.83, and contains residues W80-L466 for NA and 182 solvent molecules. For NA-2E01, the model of NA-1G05 was fit into the Cryo-EM map, and model building and refinement were performed using Coot and Phenix. The final model of NA-2E01 has a real space correlation coefficient of 0.83, and four regions from NA were unable to be built due to high flexibility (residues E105-S110, G140-Y143, G433-T437, and W456-L466). The domain movement of 2E01 heavy chain relative to 1G05 heavy chain was analyzed by DynDom (Hayward and Berendsen, 1998). Structural figures were prepared using UCSF Chimera, UCSF ChimeraX (Goddard et al., 2018), and PyMOL (Schrödinger).
BNA-mAb Sequences
Sequences were obtained from PCR reaction products and annotated using the IMGT/V-QUEST database tool (http://www.imgt.org/IMGT_vquest/input) (Brochet et al., 2008; Giudicelli et al., 2011).
NA Sequences
The NA sequences for generating the phylogenetic tree were downloaded from the Global Initiative on Sharing Avian Influenza Data (www.gisaid.org). The amino acid sequences were aligned in Clustal Omega (Sievers and Higgins, 2014), and the phylogenetic tree was generated using MEGA 6.06 (Tamura et al., 2013).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
Antibodies | ||
| anti-human IgG-biotin | Jackson ImmunoResearch | Cat#109–001-008: RRID:AB_2337530 |
| CD71-FITC (clone CY1G4) | BioLegend | Cat#334104; RRID:AB_2201482 |
| CD19-PE (clone HIB19) | BioLegend | Cat#302254: RRID:AB_302254 |
| CD38-BV605 (clone HIT2) | BioLegend | Cat#303532: RRID:AB_2562915 |
| CD20-APC-Fire750 (clone 2H7) | BioLegend | Cat#302358: RRID:AB_2572126 |
| HRP-conjugated anti-human IgG | Jackson ImmunoResearch | Cat#109–035-098: RRID:AB_2337586 |
| anti-IBV guinea pig sera | Generated in house | this manuscript |
| Donkey anti-guinea pig IgG antibody conjugated to HRP | Millipore | Cat#AP193P: RRID:AB_92662 |
| mAb 1A03 | Generated in house | This manuscript |
| mAb 1D05 | Generated in house | This manuscript |
| mAb 1G05 | Generated in house | This manuscript |
| mAb 2E01 | Generated in house | This manuscript |
| mAb 2D10 | Generated in house | This manuscript |
| mAb 2H09 | Generated in house | This manuscript |
| mAb 3C01 | Generated in house | This manuscript |
| 3% goat serum | Life Technologies, Inc. | Cat#16210–064 |
|
Oligonucleotides | ||
| oligo-dT23VN primer, random Hexamers | Integrated DNA Technologies | N/A |
| 1st PCR primers & nested PCR primers | Smith et al., 2009 | N/A |
| Gibson Cloning Primers | Ho et al., 2009 | N/A |
| NA gBlock gene fragment | Integrated DNA Technologies | N/A |
|
Experimental Models: Organisms/Strains | ||
| BALB/c mice | Jackson Laboratories | N/A |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| Penicillin-Streptomycin | Gibco™ | Cat#15070063 |
| Heparin sodium salt | Sigma-Aldrich | Cat#H3149 |
| L-Glutamine | Gibco™ | Cat#25030081 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A7906 |
| HBS-EP buffer | Teknova | Cat#H8022 |
| EZ-Link-NHS-PEG4-Biotin | Thermo Fisher Scientific | Cat#21362 |
| Ficoll-Paque PLUS | GE Healthcare | Cat#17–1440 |
| Phosphate-buffered saline | Gibco | Cat#10010 |
| Roswell Park Memorial Institute (RPMI)-1640 media | Corning | Cat#10–040-CV |
| Dimethyl Sulfoxide (DMSO) | Fisher Scientific | Cat#BP231 |
| Fetal Bovine Serum (FBS) | R&D systems | Cat#S11150H |
| Tween-20 | Sigma-Aldrich | Cat#9005–64-5 |
| Avidin-D-horseradish peroxidase (HRP) | Vector Laboratories | Cat#A-2004–5 |
| 3-amino-9-ethylcarbazole (AEC) substrate | Sigma-Aldrich | Cat#A5754 |
| EDTA | Corning | Cat#46–034Cl |
| Zombie Aqua | BioLegend | Cat#423101 |
| Tris Buffer | Invitrogen | Cat#15567027 |
| RNase inhibitor | Promega | Cat#N2111 |
| Protein A agarose | Invitrogen | Cat#15918014 |
| o-phenylenediamine dihydrochloride (OPD) | Sigma-Aldrich | Cat#P8287 |
| Hydrochloric acid (HCl) | Fisher Scientific | Cat#S25856 |
| Ketamine | KetaVed | Cat#NDC50989–161-06 |
| Xylasine | AnaSed Injection | Cat#NDC59399–110-20 |
| Fetuin | Sigma-Aldrich | Cat#F3385 |
| KPL coating solution | SeraCare | Cat#5150–0014 (50–84-00) |
| Peanut Agglutinin (PNA)-HRP | Sigma-Aldrich | Cat#L7759 |
| 10X Minimum Essential Medium (MEM) | Gibco | Cat#11430030 |
| Formaldehyde | Fisher Chemical | Cat#UN1198 |
| KPL True-Blue peroxidase | Sera Care | Cat#5510–0030 |
| streptavidin conjugated to HRP | Thermo Fisher Scientific | Cat#N100 |
| AgeI and SalI restriction endonucleases | New England Biolabs Thermo Fisher Scientific |
Cat#R3552S Cat#FD0644 |
| Nickel Agarose Beads | Goldbio | Cat# H-320–5 |
| Imidazole | Sigma-Aldrich | Cat#I5513 |
| Sf-900™ III SFM | Gibco™ | Cat#12658019 |
| Express Five™ SFM | Gibco™ | Cat#10486025 |
| Expi293™ Expression Medium | Gibco™ | Cat#A1435102 |
| Dulbecco’s modified Eagle’s medium (DMEM) | Gibco | Cat#11995–065 |
| Streptavidin Biosensors | ForteBio | Cat#18–5019 |
| NA-1G05 structure | This manuscript | PDB entry ID 6V4N |
| NA-2E01 structure | This manuscript | PDB entry ID 6V4O |
| Streptavidin Biosensors | ForteBio | Cat#18–5019 |
| Recombinant proteins used in functional assays are referenced in Table S2 | See Table S2 | |
|
Critical Commercial Assays | ||
| NA-Star® Influenza Neuraminidase Inhibitor Resistance Detection Kit | Applied Biosystems | Cat#4374422 |
| Bio-Glo™ luciferase assay system | Promega | Cat#G7940 |
| flashBAC Recombinant Baculovirus Expression Kit | Mirus | Cat#100200 |
| NEBuilder® HiFi DNA Assembly Cloning Kit | New England BioLabs | Cat#E5520S |
|
Experimental Models: Cell Lines | ||
| Sf9 insect cell line | Gibco™ | Cat#12659017 |
| High Five™ cells | Gibco™ | Cat#12659017 |
| Expi293F™ cells | Gibco™ | Cat#A14527 |
| Madin Darby Canine Kidney (MDCK) cells | IRR | Cat#FR58 |
| Human ADCC bioeffector FcγRIIIa cells | Promega | Cat#G7102 |
|
Bacterial and Virus Strains | ||
| Influenza virus strains are referenced in Table S2 | See Table S2 | |
|
Recombinant DNA | ||
| pAbVec6W based plasmids for mAb/Fab expression | Generated in house | this manuscript |
| pOET1.1_YD_NA | Generated in house | this manuscript |
| Software and Algorithms | ||
| Prism | GraphPad | N/A |
| Excel | Microsoft | N/A |
| FlowJo | FlowJo | N/A |
| Biaevaluation 3.1 | GE Healthcare | N/A |
| Clustal Omega | (Sievers and Higgins, 2013) | N/A |
| MEGA6.06 | (Tamura et al., 2013) | N/A |
| RELION 3.0-beta-2 | Zivanov et al., 2018 | N/A |
| Gctf | Zhang, 2016 | N/A |
| crYOLO | Wagner et al., 2019 | N/A |
| PostProcess | Chen et al., 2013 | N/A |
| ResMap | Kucukelbir et al., 2014 | N/A |
| UCSF Chimera | Pettersen et al., 2004 | N/A |
| UCSF ChimeraX | Goddard et al., 2018 | N/A |
| Phenix | Adams et al., 2010 | N/A |
| Coot | Emsley and Cowtan, 2004 | N/A |
| MolProbity | Chen et al., 2010 | N/A |
| DynDom | Hayward and Berendsen, 1998 | N/A |
| PyMOL | Schrödinger | N/A |
Highlights.
Generation of seven human monoclonal antibodies to influenza B virus neuraminidase
Two antibodies, 1G05 and 2E01, are broadly cross-reactive
1G05 and 2E01 are potently protective against lethal Influenza B infection in mice
1G05 and 2E01 bind conserved residues in the Influenza B neuraminidase active site
ACKNOWLEDGMENTS
We would like to thank S. House and the clinical sample collection team in the Washington University Emergency Care and Research Core. We thank J. Fitzpatrick and M. Rau of the Washington University Center for Cellular Imaging for assistance with cryo-EM data collection. The EDFLU study was reviewed and approved by the Washington University Institutional Review Board (approval no. 2017-10-220). Work in the Ellebedy laboratory was supported by NIAID R21 AI139813, U01 AI141990, and NIAID Centers of Excellence for Influenza Research and Surveillance (CEIRS, contract HHSN272201400006C). The Krammer laboratory was supported by NIAID (grant R01 AI117287), NIAID CEIRS (contract HHSN272201400008C), and NIAID Collaborative Influenza Vaccine Innovation Centers (CIVIC, contract 75N93019C00051). Work in the Fremont lab was supported by NIAID (CSGID, contract HHSN272201700060C). The Influenza Centre is funded by the Ministry of Health and Care Services, Norway, the Norwegian Research Council Globvac (284930), National Graduate School in Infection Biology and Antimicrobials (IBA, 249062), the European Union (EU IMI115672, FLUCOP), EU Nanomedicines Flunanoair (ERA-NETet EuroNanoMed2, JTC2016), and the K.G. Jebsen Centre for Influenza Vaccine Research. Human sample collection was supported by a grant from the Washington University Institute of Clinical and Translational Sciences to P.A.M., which is, in part, supported by the NIH/National Center for Advancing Translational Sciences (NCATS, CTSA grant UL1TR002345). A.M. received funding for this work from the Norway-America Association (NORAM) and the National Graduate School in Infection Biology and Antimicrobials (IBA). J.S.T. was supported by NIAID 5T32CA009547. The contents of this publication are solely the responsibility of the authors, and do not necessarily represent the official views of NIAID or NIH. The manuscript was edited by the Scientific Editing Service of the Institute of Clinical and Translational Sciences at Washington University, which is supported by an NIH Clinical and Translational Science Award (UL1 TR002345).
DECLARATION OF INTERESTS
A.H.E. is a consultant for Inbios and Fimbrion Therapeutics. D.H.F is a co-founder of Courier Therapeutics. The Ellebedy and Fremont laboratories received funding under a sponsored research agreement from Emergent BioSolutions for work that is not related to the data presented in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet X, Lefranc M-P, and Giudicelli V (2008). IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 36, W503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister WP, Henrissat B, Bosso C, Cusack S, and Ruigrok RW (1993). Influenza B virus neuraminidase can synthesize its own inhibitor. Structure 1, 19–26. [DOI] [PubMed] [Google Scholar]
- Burnham AJ, Baranovich T, and Govorkova EA (2013). Neuraminidase inhibitors for influenza B virus infection: efficacy and resistance. Antiviral Res. 100, 520–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SHW, and Henderson R (2013). High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-Q, Wohlbold TJ, Zheng N-Y, Huang M, Huang Y, Neu KE, Lee J, Wan H, Rojas KT, Kirkpatrick E, et al. (2018). Influenza Infection in Humans Induces Broadly Cross-Reactive and Protective Neuraminidase-Reactive Antibodies. Cell 173, 417–429.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong AK, Pegg MS, Taylor NR, and von Itzstein M (1992). Evidence for a sialosyl cation transition-state complex in the reaction of sialidase from influenza virus. Eur. J. Biochem 207, 335–343. [DOI] [PubMed] [Google Scholar]
- Crowe JE (2017). Principles of broad and potent antiviral human antibodies: insights for vaccine design. Cell Host Microbe 22, 193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLillo DJ, Tan GS, Palese P, and Ravetch JV (2014). Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat. Med 20, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelberger MC, Morens DM, and Taubenberger JK (2018). Neuraminidase as an influenza vaccine antigen: a low hanging fruit, ready for picking to improve vaccine effectiveness. Curr. Opin. Immunol 53, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy AH, and Ahmed R (2012). Re-engaging cross-reactive memory B cells: the influenza puzzle. Front. Immunol 3, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy AH, and Webby RJ (2009). Influenza vaccines. Vaccine 27 Suppl 4, D65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy AH, Jackson KJL, Kissick HT, Nakaya HI, Davis CW, Roskin KM, McElroy AK, Oshansky CM, Elbein R, Thomas S, et al. (2016). Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat. Immunol 17, 1226–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy AH, Nachbagauer R, Jackson KJL, Dai Y-N, Han J, Alsoussi WB, Davis CW, Stadlbauer D, Rouphael N, Chromikova V, et al. (2020). Adjuvanted H5N1 influenza vaccine enhances both cross-reactive memory B cell and strain-specific naive B cell responses in humans. Proc. Natl. Acad. Sci. USA 117, 17957–17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D, Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Erbelding EJ, Post DJ, Stemmy EJ, Roberts PC, Augustine AD, Ferguson S, Paules CI, Graham BS, and Fauci AS (2018). A universal influenza vaccine: the strategic plan for the national institute of allergy and infectious diseases. J. Infect. Dis 218, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchuk IM, Bangaru S, Gilchuk P, Irving RP, Kose N, Bombardi RG, Thornburg NJ, Creech CB, Edwards KM, Li S, et al. (2019). Influenza H7N9 Virus Neuraminidase-Specific Human Monoclonal Antibodies Inhibit Viral Egress and Protect from Lethal Influenza Infection in Mice. Cell Host Microbe 26, 715–728.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, Brochet X, and Lefranc M-P (2011). IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc 2011, 695–715. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorkova EA, and McCullers JA (2013). Therapeutics against influenza. Curr. Top. Microbiol. Immunol 370, 273–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubareva LV, Matrosovich MN, Brenner MK, Bethell RC, and Webster RG (1998). Evidence for zanamivir resistance in an immunocompromised child infected with influenza B virus. J. Infect. Dis 178, 1257–1262. [DOI] [PubMed] [Google Scholar]
- Hayward S, and Berendsen HJ (1998). Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins 30, 144–154. [PubMed] [Google Scholar]
- Ho IY, Bunker JJ, Erickson SA, Neu KE, Huang M, Cortese M, Pulendran B, and Wilson PC (2016). Refined protocol for generating monoclonal antibodies from single human and murine B cells. J. Immunol. Methods 438, 67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison MG, Gubareva LV, Atmar RL, Treanor J, and Hayden FG (2006). Recovery of drug-resistant influenza virus from immunocompromised patients: a case series. J. Infect. Dis. 193, 760–764. [DOI] [PubMed] [Google Scholar]
- Kawai N, Ikematsu H, Iwaki N, Maeda T, Satoh I, Hirotsu N, and Kashiwagi S (2006). A comparison of the effectiveness of oseltamivir for the treatment of influenza A and influenza B: a Japanese multicenter study of the 2003–2004 and 2004–2005 influenza seasons. Clin. Infect. Dis 43, 439–444. [DOI] [PubMed] [Google Scholar]
- Krammer F (2019). The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol 19, 383–397. [DOI] [PubMed] [Google Scholar]
- Krammer F, Fouchier RAM, Eichelberger MC, Webby RJ, Shaw-Saliba K, Wan H, Wilson PC, Compans RW, Skountzou I, and Monto AS (2018). NAction! How Can Neuraminidase-Based Immunity Contribute to Better Influenza Virus Vaccines? MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, and Tagare HD (2014). Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langat P, Raghwani J, Dudas G, Bowden TA, Edwards S, Gall A, Bedford T, Rambaut A, Daniels RS, Russell CA, et al. (2017). Genome-wide evolutionary dynamics of influenza B viruses on a global scale. PLoS Pathog. 13, e1006749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz MR, Webster RG, and Air GM (1987). Site-directed mutation of the active site of influenza neuraminidase and implications for the catalytic mechanism. Biochemistry 26, 5351–5358. [DOI] [PubMed] [Google Scholar]
- Margine I, Palese P, and Krammer F (2013). Expression of functional recombinant hemagglutinin and neuraminidase proteins from the novel H7N9 influenza virus using the baculovirus expression system. J. Vis. Exp e51112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Kirkpatrick E, Stadlbauer D, Strohmeier S, Bouvier NM, and Krammer F (2019). Mucosal Immunity against Neuraminidase Prevents Influenza B Virus Transmission in Guinea Pigs. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishin VP, Patel MC, Chesnokov A, De La Cruz J, Nguyen HT, Lollis L, Hodges E, Jang Y, Barnes J, Uyeki T, et al. (2019). Susceptibility of influenza A, B, C, and D viruses to baloxavir1. Emerging Infect. Dis 25, 1969–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murin CD, Wilson IA, and Ward AB (2019). Antibody responses to viral infections: a structural perspective across three different enveloped viruses. Nat. Microbiol 4, 734–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul Glezen W, Schmier JK, Kuehn CM, Ryan KJ, and Oxford J (2013). The burden of influenza B: a structured literature review. Am. J. Public Health 103, e43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules CI, Marston HD, Eisinger RW, Baltimore D, and Fauci AS (2017). The pathway to a universal influenza vaccine. Immunity 47, 599–603. [DOI] [PubMed] [Google Scholar]
- Paules CI, Sullivan SG, Subbarao K, and Fauci AS (2018). Chasing Seasonal Influenza - The Need for a Universal Influenza Vaccine. N. Engl. J. Med 378, 7–9. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Piepenbrink MS, Nogales A, Basu M, Fucile CF, Liesveld JL, Keefer MC, Rosenberg AF, Martinez-Sobrido L, and Kobie JJ (2019). Broad and Protective Influenza B Virus Neuraminidase Antibodies in Humans after Vaccination and their Clonal Persistence as Plasma Cells. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, and Nerome K (1990). Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 175, 59–68. [DOI] [PubMed] [Google Scholar]
- Sato M, Saito R, Sato I, Tanabe N, Shobugawa Y, Sasaki A, Li D, Suzuki Y, Sato M, Sakai T, et al. (2008). Effectiveness of oseltamivir treatment among children with influenza A or B virus infections during four successive winters in Niigata City, Japan. Tohoku J Exp Med 214, 113–120. [DOI] [PubMed] [Google Scholar]
- Sievers F, and Higgins DG (2014). Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol 1079, 105–116. [DOI] [PubMed] [Google Scholar]
- Smith K, Garman L, Wrammert J, Zheng N-Y, Capra JD, Ahmed R, and Wilson PC (2009). Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc 4, 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadlbauer D, Amanat F, Strohmeier S, Nachbagauer R, and Krammer F (2018). Cross-reactive mouse monoclonal antibodies raised against the hemagglutinin of A/Shanghai/1/2013 (H7N9) protect against novel H7 virus isolates in the mouse model. Emerg. Microbes Infect 7, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadlbauer D, Zhu X, McMahon M, Turner JS, Wohlbold TJ, Schmitz AJ, Strohmeier S, Yu W, Nachbagauer R, Mudd PA, et al. (2019). Broadly protective human antibodies that target the active site of influenza virus neuraminidase. Science 366, 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya N, Mitamura K, Yamazaki M, Tamura D, Ichikawa M, Kimura K, Kawakami C, Kiso M, Ito M, Hatakeyama S, et al. (2007). Lower clinical effectiveness of oseltamivir against influenza B contrasted with influenza A infection in children. Clin. Infect. Dis 44, 197–202. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Asthagiri Arunkumar G, and Krammer F (2018). Universal influenza virus vaccines and therapeutics: where do we stand with influenza B virus? Curr. Opin. Immunol 53, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NR, and von Itzstein M (1994). Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J. Med. Chem 37, 616–624. [DOI] [PubMed] [Google Scholar]
- Virk RK, Jayakumar J, Mendenhall IH, Moorthy M, Lam P, Linster M, Lim J, Lin C, Oon LLE, Lee HK, et al. (2020). Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl. Acad. Sci. USA 117, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Merino F, Stabrin M, Moriya T, Antoni C, Apelbaum A, Hagel P, Sitsel O, Raisch T, Prumbaum D, et al. (2019). SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PC, and Andrews SF (2012). Tools to therapeutically harness the human antibody response. Nat. Rev. Immunol 12, 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold TJ, Nachbagauer R, Xu H, Tan GS, Hirsh A, Brokstad KA, Cox RJ, Palese P, and Krammer F (2015). Vaccination with adjuvanted recombinant neuraminidase induces broad heterologous, but not heterosubtypic, cross-protection against influenza virus infection in mice. MBio 6, e02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold TJ, Podolsky KA, Chromikova V, Kirkpatrick E, Falconieri V, Meade P, Amanat F, Tan J, tenOever BR, Tan GS, et al. (2017). Broadly protective murine monoclonal antibodies against influenza B virus target highly conserved neuraminidase epitopes. Nat. Microbiol 2, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrammert J, Koutsonanos D, Li G-M, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, et al. (2011). Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med 208, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu NC, and Wilson IA (2018). Structural insights into the design of novel anti-influenza therapies. Nat. Struct. Mol. Biol 25, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhu X, Dwek RA, Stevens J, and Wilson IA (2008). Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol 82, 10493–10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Turner HL, Lang S, McBride R, Bangaru S, Gilchuk IM, Yu W, Paulson JC, Crowe JE, Ward AB, et al. (2019). Structural Basis of Protection against H7N9 Influenza Virus by Human Anti-N9 Neuraminidase Antibodies. Cell Host Microbe 26, 729–738.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mAbs generated in this study were deposited with GenBank accession numbers MN888992–MN889005 and MT200637–MT200664. The NA-1G05 structure was deposited with PDB entry ID 6V4N and EMDB entry ID EMD-21042. The NA-2E01 structure was deposited with PDB entry ID 6V4O and EMDB entry ID EMD-21043.