

SUMMARY
Mg2+ is the most abundant divalent cation in metazoans and an essential cofactor for ATP, nucleic acids and countless metabolic enzymes. To understand how the spatiotemporal dynamics of intracellular Mg2+ (iMg2+) are integrated into cellular signaling, we implemented a comprehensive screen to discover regulators of iMg2+ dynamics. Lactate emerged as an activator of rapid release of Mg2+ from endoplasmic reticulum (ER) stores which facilitates mitochondrial Mg2+ (mMg2+) uptake in multiple cell types. We demonstrate that this process is remarkably temperature-sensitive and mediated through intracellular but not extracellular signals. The ER-mitochondrial Mg2+ dynamics is selectively stimulated by L-lactate. Further, we show that lactate-mediated mMg2+ entry is facilitated by Mrs2, and point mutations in the intermembrane space loop limits mMg2+ uptake. Intriguingly, suppression of mMg2+ surge alleviates inflammation-induced multi-organ failure. Together, these findings reveal that lactate mobilizes iMg2+ and links the mMg2+ transport machinery with major metabolic feedback circuits and mitochondrial bioenergetics.
Graphical Abstract
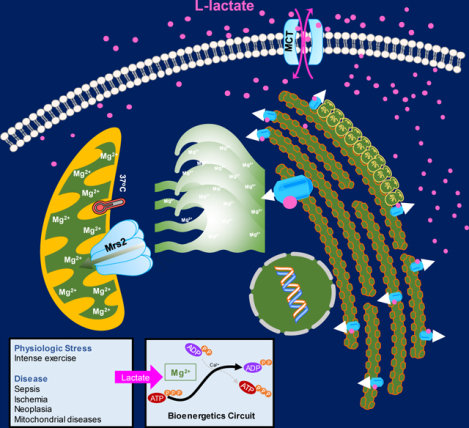
Lactate operates as a sigaling molecule to control Mg2+ handling between the ER and mitochondria
INTRODUCTION
Mg2+ is the most abundant and an essential divalent cation among eukaryotes. In metazoan cells, 95% of the intracellular Mg2+ concentration ([Mg2+]i; 10–30 mM) is bound with ATP and other molecules (de Baaij et al., 2015; Fatholahi et al., 2000; Romani, 2011). The remaining unbound free [Mg2+]i (0.5–1.2 mM) is tightly regulated, yet its effects on biomolecular functions are unknown. Remarkably, this free [Mg2+]i concentration is 100-fold below the electrochemical potential, which theoretically indicates regulated Mg2+ entry. Similar to Ca2+, Mg2+ is compartmentalized and contributes as a cofactor for enzymes in protein synthesis, ATP production, and chromatin/nucleotide stability. Regulation of [Mg2+]i occurs via numerous Mg2+ transporting proteins on the plasma membrane including the ubiquitous TRPM7, MagT1, MMGT1, SLC41A1 and tissue specific TRPM6, as well as cyclin M2 and cyclin M4 (Chaigne-Delalande et al., 2013; de Baaij et al., 2015; Li et al., 2011; Zhou and Clapham, 2009). While our understanding of cellular Mg2+ flux has improved over the years, there have been a lack of specific agonists identified for iMg2+ dynamics, preventing further assessment of Mg2+ signaling within and between specific cellular compartments, like the ER and mitochondria. Activation of receptors by a variety of agonists can rapidly generate inositol 1,4,5 trisphosphate (IP3), resulting in a steep intracellular Ca2+ increase (Berridge et al., 2003; Clapham, 2007). In contrast, metabolic and hormonal stimulation of cells leads to an alteration in [Mg2+]i in a much slower fashion (Romani and Scarpa, 1990; Rubin, 1975). While it has been suggested that metabolic stimulation promotes Mg2+ mobilization, it remains unclear whether the elevation in [Mg2+]i originates from intracellular stores or from plasma membrane entry mechanisms. Further, despite its crucial functions, how the spatiotemporal dynamics of iMg2+ control signaling systems remains a mystery.
Studies characterizing gain-of function SNPs in Parkinson disease and nephronophthisis-like phenotypes have revealed members of the solute carrier family of proteins, SLC41A1, SLC41A2, and SLC41A3, to be associated with Mg2+ transport (de Baaij et al., 2015; Kolisek et al., 2013). Further, mutations in the MagT1 Mg2+ channel are associated with immunodeficiency syndromes, suggesting an essential role for Mg2+ flux in T lymphocyte development and activation (Chaigne-Delalande et al., 2013; Li et al., 2011). While these findings have enhanced the knowledge of plasma membrane Mg2+ influx mechanisms, the iMg2+ transport systems are still not fully characterized. Originally, mitochondrial RNA splicing 2 (Mrs2) was considered to be the primary Mg2+ transporter in yeast mitochondria (Bui et al., 1999; Kolisek et al., 2003; Kuramoto et al., 2011); however, the physiologic relevance of this observation in higher order systems remains to be determined. Identification of small ligand-like activator molecules would aid in our understanding of iMg2+ dynamics and the cause-effect relationships that exist between iMg2+ flux and cellular processes.
Here, we conducted a targeted screen of cellular metabolites to identify modulators of iMg2+ dynamics. We discovered the glycolytic end-product lactate as a ligand for ER Mg2+ release and subsequent mMg2+ uptake. Lactate-mediated mMg2+ uptake is both dose-dependent and temperature-sensitive and mediated through intracellular rather than extracellular signals. We show that lactate mediated mMg2+ entry is facilitated by Mrs2 in a ΔΨm dependent fashion. These findings together reveal that lactate acts as a ligand that activates iMg2+ dynamics and establishes a link between Mrs2 with major metabolic feedback circuits and bioenergetics.
RESULTS
Lactate Is An Activator/Agonist For ER Mg2+ Release That Drives Mitochondrial Mg2+ Uptake.
The significance of Mg2+ to physiology was first appreciated ~100 years ago when this cation was identified in blood plasma by W.G. Denis, Mg2+ was demonstrated to be necessary for mouse viability by J. Leroy and a Mg2+ deficiency in humans was reported by A. Hirschfelder and V. Haury (de Baaij et al., 2015). Today, Mg2+ has been implicated and targeted in several clinical conditions, including migraines, cardiovascular diseases, diabetes, cancer, and preeclampsia (de Baaij et al., 2015), for example. Nevertheless, the regulatory mechanisms underlying iMg2+ dynamics have largely remained a mystery due to the absence of strong intercompartmental concentration gradients, lack of sensitive Mg2+ probes and, most critically, the nonidentification of ligand(s) mobilizing iMg2+ (Murphy, 2000). Since Mg2+ modulates the metabolic state of the cell through regulation of glycolytic enzymes, mitochondrial dehydrogenases and ATP synthase, yet the metabolic state of the cell regulates the concentration of cytosolic and mMg2+, we here screened several endogenous metabolites involved in cellular energy production as potential ligands that mobilize iMg2+.
To identify a ligand/activator of iMg2+ dynamics, we utilized the cell permeant [Mg2+]i indicator, Mag Green-AM, in a live cell confocal fluorescence screen. First, we tested whether GPCR agonist-induced second messengers promoted iMg2+ dynamics. To discriminate between intracellular Ca2+ and Mg2+ dynamics, HeLa cells were loaded with the cell permeant Ca2+ indicator, Fluo-4-AM or Mag Green-AM followed by stimulation with the GPCR agonist, histamine (100 μM). As expected, histamine rapidly elicited cytosolic Ca2+ (cCa2+) mobilization, while iMg2+ dynamics were absent (Figures S1A–D). These data indicate that GPCR-derived IP3 is not an activator of iMg2+ dynamics.
Suppression of oxidative phosphorylation (OXPHOS) and increased glycolysis have been linked to intense exercise, sepsis, ischemia, neoplasia, and mitochondrial diseases. To continue the search for iMg2+ modulators, we next assessed glycolytic and OXPHOS metabolites, metabolic substrates and nucleotides. Primary murine hepatocytes were loaded with Mag Green-AM and stimulated with major cellular metabolites to monitor iMg2+ dynamics. . Intriguingly, only the aerobic/anerobic glycolytic end-product lactate consistently exhibited a rapid and robust depletion of ER Mag-Green signal followed by a subsequent elevation in the mitochondria (Figures 1A and 1B). To exclude the possibility that acidification of the medium by lactate could alter membrane polarization, we normalized the pH and achieved similar results, indicating that the lactate anion is the likely ligand/activator of iMg2+ flux. Similarly, stimulation of hepatocytes with acetic acid or butyric acid did not induce ER Mg2+ release or mMg2+ uptake (Figure 1A and 1B), reinforcing a pH-independent effect. Simultaneously loading hepatocytes with the mitochondrial marker Mito-tracker Red and Mag-Green-AM confirmed that Mag-Green signals were measured from the mitochondria (Figures 1A–C). Both sodium L-lactate and L-lactic acid (5 mM) triggered a rapid depletion of ER Mg2+ with concomitant nuclear accumulation, followed by mitochondrial elevation of Mag Green signal with a lag time of ~ 25 seconds (Figures 1D). The Mg2+ changes observed in the ER and mitochondria upon stimulation with L-lactic acid were clearly reciprocal (Figures 1A, 1B, and 1D). We also observed lactate-specific robust uptake of Mg2+ into mitochondria following ER depletion in three additional cell types tested (HepG2, MEFs, and COS-7) (Figures S1E–H).
Figure 1. Lactate stimulates rapid depletion of ER magnesium stores and subsequent uptake by mitochondria.
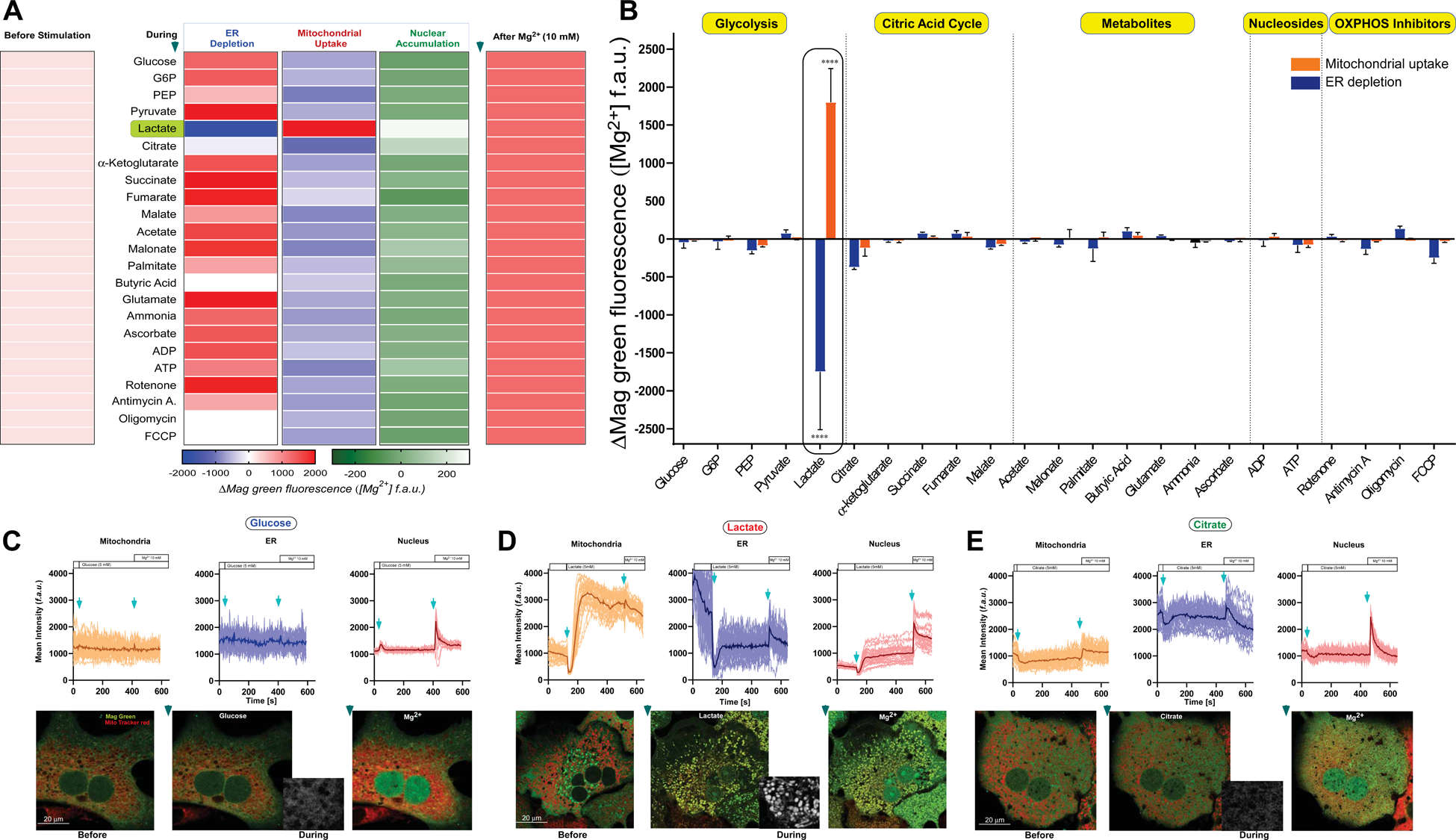
(A) Primary hepatocytes were loaded with Magnesium Green AM (Mag-Green) and MitoTracker Red (mitochondrial marker) for live confocal imaging. Relative changes in fluorescence intensity of Mag-Green in the ER, nucleus, and mitochondria were analyzed and depicted as a heatmap. n=3–10.
(B) Quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (blue) and mitochondria (orange). n=3–10. Mean ± SEM. ****p < 0.0001.
(C-E) Representative images and traces of relative changes in Mag-Green intensity of mitochondrial (orange), ER (blue) and nuclear (red) regions.
(C) Glucose (5 mM), (D) sodium L-Lactate (5 mM), or (E) Citrate (5 mM) was added after a 30 second baseline recording followed by a 10 mM Mg2+ bolus at the 125th frame. n=3–6.
Notably, pyruvate, glucose, palmitate, nucleotides, and other TCA intermediates did not induce mMg2+ uptake (Figure 1A and 1B). Low (5 mM) or high glucose (25 mM) did not induce iMg2+ dynamics (Figures 1A–C, Figures S1E–H and Table S1). Addition of citrate (5 mM), a known chelator of free Mg2+, quenched both the mitochondrial and ER Mg2+ signals (Figure 1B and 1E), as expected. Since lactate/pyruvate ratio alters NADH levels and alanine can behave similar to lactate, we tested whether alanine (5 mM) and NADH (120 μM) induce iMg2+ dynamics. These two metabolites did not stimulate iMg2+ dynamics (Figure S1I). Next, we tested whether clamping the redox state would alter lactate-induced iMg2+ dynamics. Hepatocytes were bathed in 7 mM pyruvate before L-lactate stimulation. The pyruvate supplementation did not affect lactate-induced iMg2+ dynamics, suggesting that lactate exerts an allosteric effect (Figure S1J).
We also tested whether lactate stimulates intracellular Ca2+ (iCa2+) release. Cells were loaded with the cCa2+ indicator, (Fluo-4), and stimulated with either glucose or lactate. Cells stimulated with glucose, but not lactate, elicited cCa2+ elevation, suggesting that lactate exclusively induces iMg2+ dynamics (Figure S1K). Taken together, these data indicate that lactate rapidly mobilizes ER Mg2+ stores, resulting in mMg2+ uptake in multiple cell types.
Lactate-Mediated mMg2+ Transport Is Temperature And ΔΨm Dependent
Although mammalian cells predominantly produce L-lactate, D-lactate is the major product generated by bacteria (Brooks, 2018). Remarkably, the ER depletion and mMg2+ uptake was observed only with L-lactate stimulation and not D-lactate in primary hepatocytes (Figure 2A and 2B). Additionally, we assessed other lactate analogues such as methyl L/D-lactate enantiomers and 3-phenyllactic acid using our hepatocyte system (Figure 2A). The iMg2+ mobilization was not observed with the methyl-lactate analogues, regardless of stereochemistry; however, a partial ER depletion followed by nominal mMg2+ uptake was induced by 3-phenyllactic acid (Figure 2B). Rather than a non-selective cell permeability, plasma membrane monocarboxylate transporters (MCTs) facilitate cellular entry of lactate. To assess the cellular entry of lactate and analogues, murine hepatocytes were loaded with an intracellular pH indicator (ratiometric BCECF-AM), and cytosolic pH changes were monitored. Both D-lactate and L-lactate rapidly changed the intracellular pH (Figure S2G). We also noticed that methyl L-lactate and 3-phenyllactic acid affected the intracellular pH (Figure S2G). Thus, L-lactate and the associated analogues are readily taken up by hepatocytes.
Figure 2. Mg2+ depletion from ER is L-Lactate specific, dose-dependent, while mMg2+ uptake is both dose- and temperature-dependent.
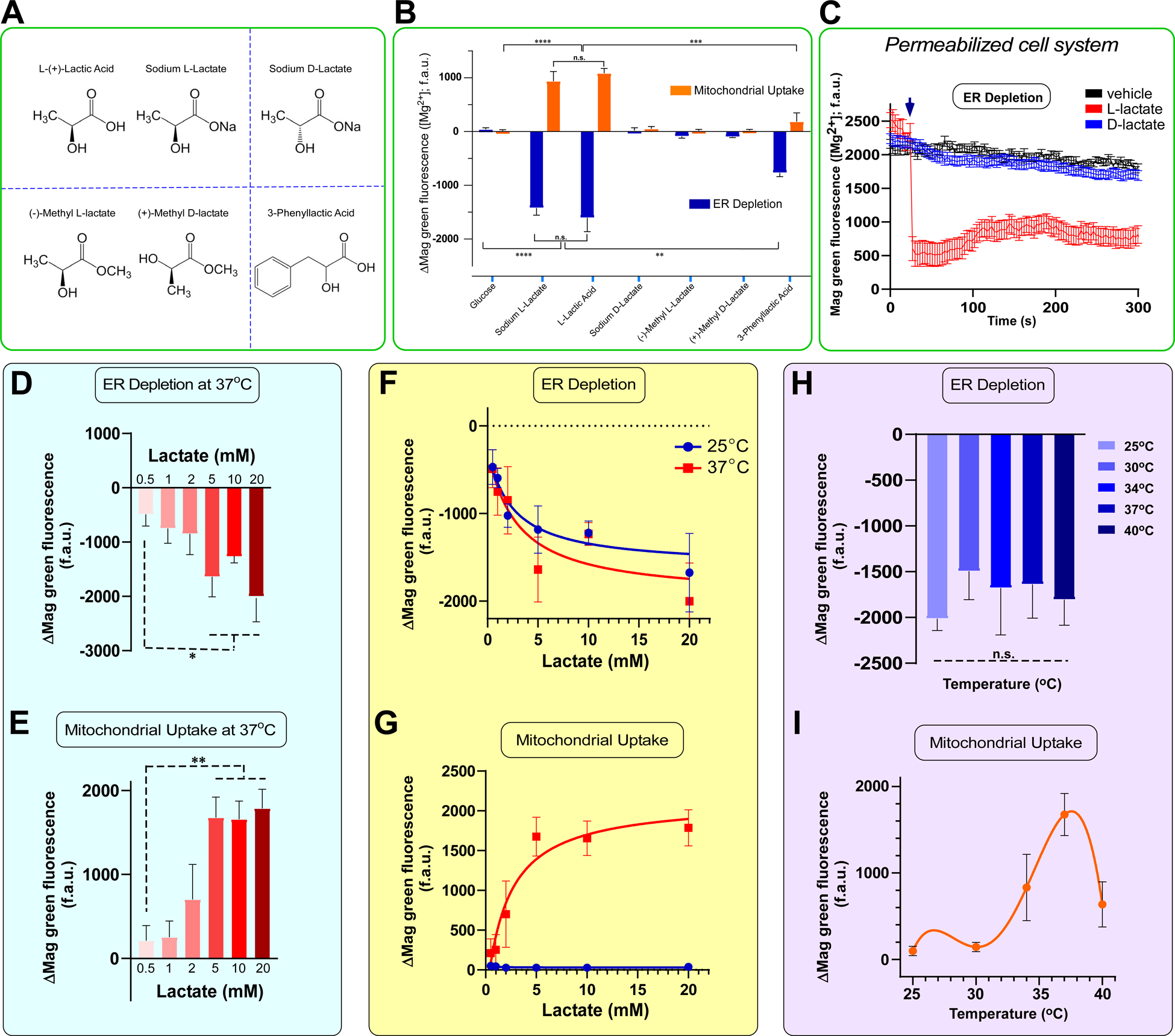
(A) Natta projections of the naturally occurring enantiomers of lactate as well as synthetic analogues that were tested in this assay.
(B) Quantification of relative changes in Mag-Green fluorescence intensity after addition of lactate and synthetic analogues (5 mM). n = 4–6 Mean ± SEM.
(C) Permeabilized hepatocytes were pulsed with either L-lactate or D-lactate (5 mM) and ER Mag-green signal was monitored using confocal imaging system. Representative mean traces show a rapid Mg2+ signal dissipation. Mean ± SEM. n=3.
(D-E) Relative changes in Mag-Green fluorescence intensity within ER or mitochondria at 37°C. n= 3–6. Mean ± SEM.
(F) Relative changes in Mag-Green fluorescence intensity within ER or (G) mitochondria at both 25°C (blue) and 37°C (red) over a range of lactate concentrations. n=3–6. Mean ± SEM.
(H-I) Relative changes in Mag-Green fluorescence intensity within ER or mitochondria after stimulation with Lactate over temperature range. n=3–6 cells for each condition. Mean ± SEM.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s. = not significant.
Since the observed iMg2+ dynamics are highly specific to L-lactate, we next assessed whether lactate functions as an upstream messenger or a ligand for ER Mg2+ release using a permeabilized cell system, which dissociates plasma membrane signaling from the ER. This permeabilized cell system also separates the ER from mitochondria. Following addition of 5 mM L-lactate but not D-lactate, the ER Mag-Green signal rapidly decreased, indicative of direct L-lactate-induced ER Mg2+ release (Figure 2C). Thus, the natural L-form of lactate found in mammals is likely a bona fide and direct activator of iMg2+ dynamics.
Given the large physiologic and pathophysiologic range of lactate accumulation in the extracellular fluid (i.e. ~0.5 – 30 mM) (Brooks, 2018; Haas et al., 2016; Pavlova and Thompson, 2016), we next evaluated the lactate concentration required to trigger ER Mg2+ depletion and uptake by the mitochondria. Lactate concentrations ≥ 2 mM induced ER depletion and partial uptake of Mg2+ into the mitochondria at 37°C (Figure 2D and 2E). However, a consistent, maximal response was observed with 5 mM lactate (Figure 2D and 2E). Serendipitously, we noticed that lactate-induced depletion of ER Mag-Green signal occurred at both 37°C and room temperature (25°C) (Figure 2F). In contrast, the mitochondrial Mag-Green signal was undetectable at 25°C, indicating that the mMg2+ entry machinery was temperature-sensitive (Figure 2G). Consequently, we repeated the experiment at a range of temperatures (i.e. 25°C – 40°C) with 5 mM lactate. Similar lactate-mediated ER Mag-Green signal depletion was seen at all temperatures (Figure 2H). Remarkably, lactate-induced mMg2+ uptake was found to occur maximally at 37°C with less uptake at 34°C and 40°C. (Figure 2I), confirming that the mMg2+ uptake machinery has temperature-sensitive properties.
We next asked whether temperature sensitivity is a distinctive property of the mMg2+ uptake machinery by measuring MCU-mediated Ca2+ uptake at 25°C and 37°C. HepG2 cells transduced with mitochondrial Ca2+ sensor (GCaMP6s-mt) were stimulated with 2.5 μM ionomycin and MCU-mediated Ca2+ uptake was measured. mCa2+ uptake was comparable at both temperatures (Figure S2A). We further demonstrated that MCU has negligible temperature-sensitivity at 25°C and 37°C using a permeabilized cell system (Figure S2B and S2C). Collectively, these findings suggest that the activity of the mMg2+ uptake machinery but not MCU is sensitive to near physiological temperature changes.
We next assessed whether mMg2+ uptake is dependent upon the highly electronegative membrane potential (ΔΨm) of the mitochondria. After a baseline recording, the mitochondrial uncoupler FCCP (10 μM) was applied to hepatocytes simultaneously loaded with Mag Green and the ΔΨm indicator TMRE. As expected, FCCP dissipated the ΔΨm as evidenced by loss of the TMRE signal, though the ER and mitochondrial Mag-Green signal intensities remained unchanged (Figure S2D and S2E). Subsequent stimulation with a 5 mM bolus of L-lactate resulted in the characteristic depletion of ER Mg2+(Figure S2F), however, loss of the ΔΨm abrogated mMg2+ elevation(Figure S2E). Thus, the machinery involved in lactate mediated mMg2+ uptake is both temperature and ΔΨm dependent.
MCT-Mediated Lactate Entry Triggers iMg2+ Dynamics
Most mammalian cells utilize monocarboxylate transporters (MCT1–4) for plasma membrane lactate entry (Brooks, 2018). To test if MCTs play a role in the iMg2+ dynamics, hepatocytes were pretreated with MCT1 (SR13800; 1 μM) or MCT1/2 (AR-C155858; 2 μM) inhibitors for 60 minutes before stimulation. Remarkably, both inhibitors suppressed lactate induced Mg2+ depletion from the ER and mMg2+ uptake (Figure 3A and 3B, left panels). The MCT1 inhibitor only partially reduced ER Mag-Green signal, but blockade of both MCT1 and 2 transporters robustly suppressed the lactate effect (Figure 3A, and 3B, left panels). Lactate stimulation in Mg2+-free buffer still elicited a decrease in the ER Mag-Green signal, but the response was slightly reduced (Figure 3A, right panel). Importantly, a significant mMg2+ uptake was observed after stimulation in Mg2+-free buffer, indicating lactate-mediated mobilization from iMg2+ stores rather than extracellular Mg2+ influx (Figure 3B, right panel).
Figure 3. Lactate-dependent Mg2+ flux into mitochondria is primarily from intracellular ER stores.
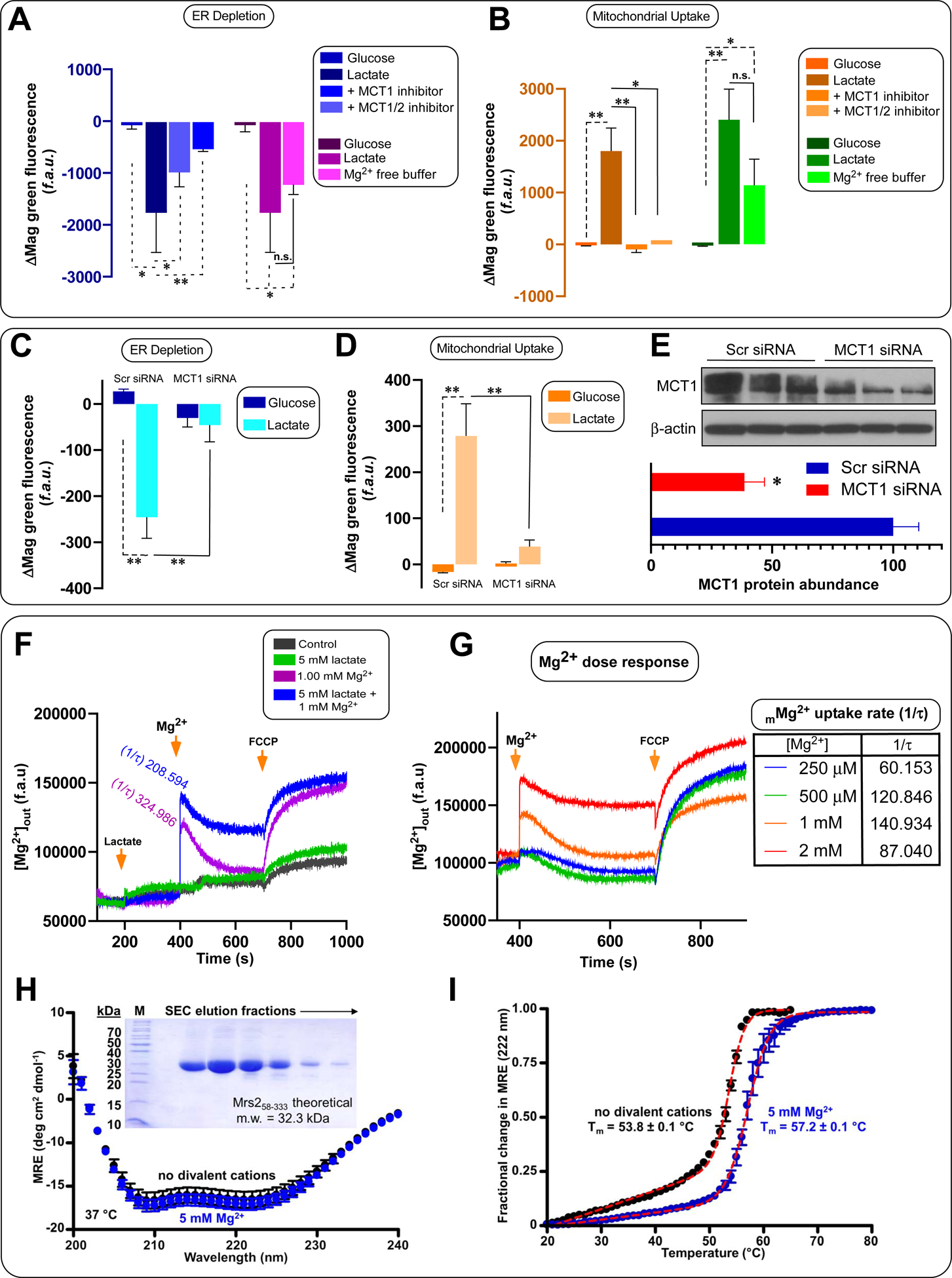
(A-B) Relative changes in Mag-Green signal within ER or (B) mitochondria after stimulation with glucose and lactate, or lactate after pretreatment with MCT1 (ARC13800; 1 μM) and MCT1/2 (AR-C155858; 2 μM) inhibitors. n=3–6. Mean ± SEM. n=3–9
(C-D) Relative changes in Mag-Green signal within ER or (D) mitochondria in MCT-1 knockdown HepG2 cells. n=3–4. Mean ± SEM.
(E) Western blot showing changes in HepG2 MCT1 protein expression following RNAi-mediated KD of MCT-1. n = 3.
(F) Permeabilized hepatocytes were pulsed with 1 mM MgCl2 or in combination with 5 mM lactate. Representative traces show bath [Mg2+] (f.a.u). Mean ± SEM. n=3.
(G) Change in bath [Mg2+] due to mMg2+ uptake in response to various bath [Mg2+] in permeabilized hepatocytes. Inset table depicts mitochondrial uptake rate. Mean ± SEM. n=3.
(H) Far-UV CD spectra of the human Mrs2 N-terminal domain (residues 58–333) in the presence (blue) and absence (black) of 5 mM Mg2+. MRE, mean residue ellipticity (×0.001). Inset shows the human Mrs2 N-terminal domain purity.
(I) Thermal stability of the human Mrs2 N-terminal domain. Thermal melts are constructed from the change in MRE at 222 nm as function of temperature. Data in panels H and I are Mean ± SEM. n=3.
*p < 0.05, **p < 0.01, n.s. = not significant.
Consistent with our pharmacological approach, siRNA-mediated knockdown of MCT1 significantly reduced both ER and mMg2+ dynamics, underscoring that lactate transport-induced depletion of ER Mg2+ is the principal source for mMg2+ uptake (Figure 3C–E). Further, we found that exogenous lactate elicited a rapid ER Mg2+ depletion and mMg2+ uptake even in the presence of the lactate dehydrogenase (LDH) inhibitor GSK2837808A (Figure S6K), reinforcing the direct effect of lactate rather any derivative.
Having established that lactate-induced ER Mg2+ store depletion is the source for mMg2+ uptake, we used permeabilized cells to probe for direct lactate effects on mMg2+ uptake. Digitonin permeabilized cells (~2.5 × 106) were challenged with 5 mM lactate, 1 mM Mg2+ or a combination of both. Cells treated with lactate alone exhibited a nominal rise of Mg2+ in the extramitochondrial medium as measured by changes in Mag-Green signal (Figure 3F). Cells exposed to 1 mM Mg2+ alone rapidly cleared the extramitochondrial Mg2+, indicating robust mMg2+ uptake in the absence of lactate. The combination of lactate and Mg2+ did not further enhance mMg2+ uptake rates (Figure 3F). We also examined the optimal extramitochondrial [Mg2+] required for mMg2+ uptake by delivering 0.25 – 2.0 mM pulses of Mg2+. We observed maximal uptake rates at 1 mM Mg2+ and reduced rates at 2 mM Mg2+, indicating a mMg2+ uptake feedback inhibition (Figure 3G).
Human Mrs2 is a homologue to yeast Mrs2p, which is a selective mMg2+ channel. To gain mechanistic insight into the basis for the feedback inhibition, we cloned, expressed in Escherichia coli and purified the human Mrs2 N-terminal matrix domain (residues 58–333), comprising ~70 % of the molecule and a key regulatory domain (Figure 3H, inset) (Pfoh et al., 2012). The far-UV circular dichroism (CD) spectrum of the domain exhibited two distinct minima at ~208 and 222 nm, indicative of high levels of α-helix and consistent with yeast Mrs2p and bacterial CorA (lower orthologue) crystal structures (Figure 3H) (Eshaghi et al., 2006; Khan et al., 2013; Lunin et al., 2006). The CD spectrum was not significantly different in the presence of 5 mM Mg2+. We also evaluated the thermal stability by monitoring the change in CD ellipticity at 222 nm as a function of temperature. The protein exhibited a high cooperativity of unfolding and midpoint of temperature denaturation (Tm) of ~53.8 °C, consistent with a folded tertiary conformation. Addition of 5 mM Mg2+ increased the Tm by ~3.4 °C, suggesting stabilizing interactions with Mg2+ (Figure 3I). Mg2+ binding to the N-terminal domain of CorA maintains a closed state by promoting inter-N-terminal domain interactions (Pfoh et al., 2012). Our data indicate that human Mrs2 also harbors a Mg2+-sensitive N-terminal domain, which could underlie feedback inhibition by binding matrix Mg2+, like CorA.
Lactate-Induced Mitochondrial Mg2+ Uptake Requires Mrs2
Having established that lactate elicits mMg2+ uptake secondary to rapid and robust ER Mg2+ depletion, we next tested whether Mrs2 is the bona fide channel responsible for mMg2+ uptake in our system. We generated a global Mrs2 knockout mouse model via CRISPR/Cas9 gene editing. Founder mice were examined for ssODN incorporation by PCR amplification and restriction enzyme digestion (aa changes introduced a BamH1 cut site) (Figure 4A). After verifying germline transmission and breeding to heterozygosity, DNA was isolated and sequenced to confirm the in-frame point mutations (Figure 4B). After 10 generations of breeding, Mrs2 deletion was confirmed by RT-qPCR (Figure 4C). We did not obtain the expected Mendelian ratios of Mrs2+/+, Mrs2+/−, and Mrs2−/− pups after splitting and fostering litters born to Mrs2+/− intercrosses (Table S2). After generation and validation of the global murine Mrs2 KO, murine WT and Mrs2 KO hepatocytes were loaded with Mag-Green and stimulated with lactate. Lactate stimulated significant depletion of ER Mg2+ in both the WT and Mrs2 KO hepatocytes; moreover, the dose-dependent effects of lactate were not different in the WT compared to Mrs2 KO (Figure S3A). Conversely, lactate-induced mMg2+ uptake was ablated in Mrs2 KO but remained intact in WT hepatocytes (Figure S3B).
Figure 4. Lactate-induced ER Mg2+ depletion and that the Mrs2 is responsible for subsequent mMg2+ uptake.
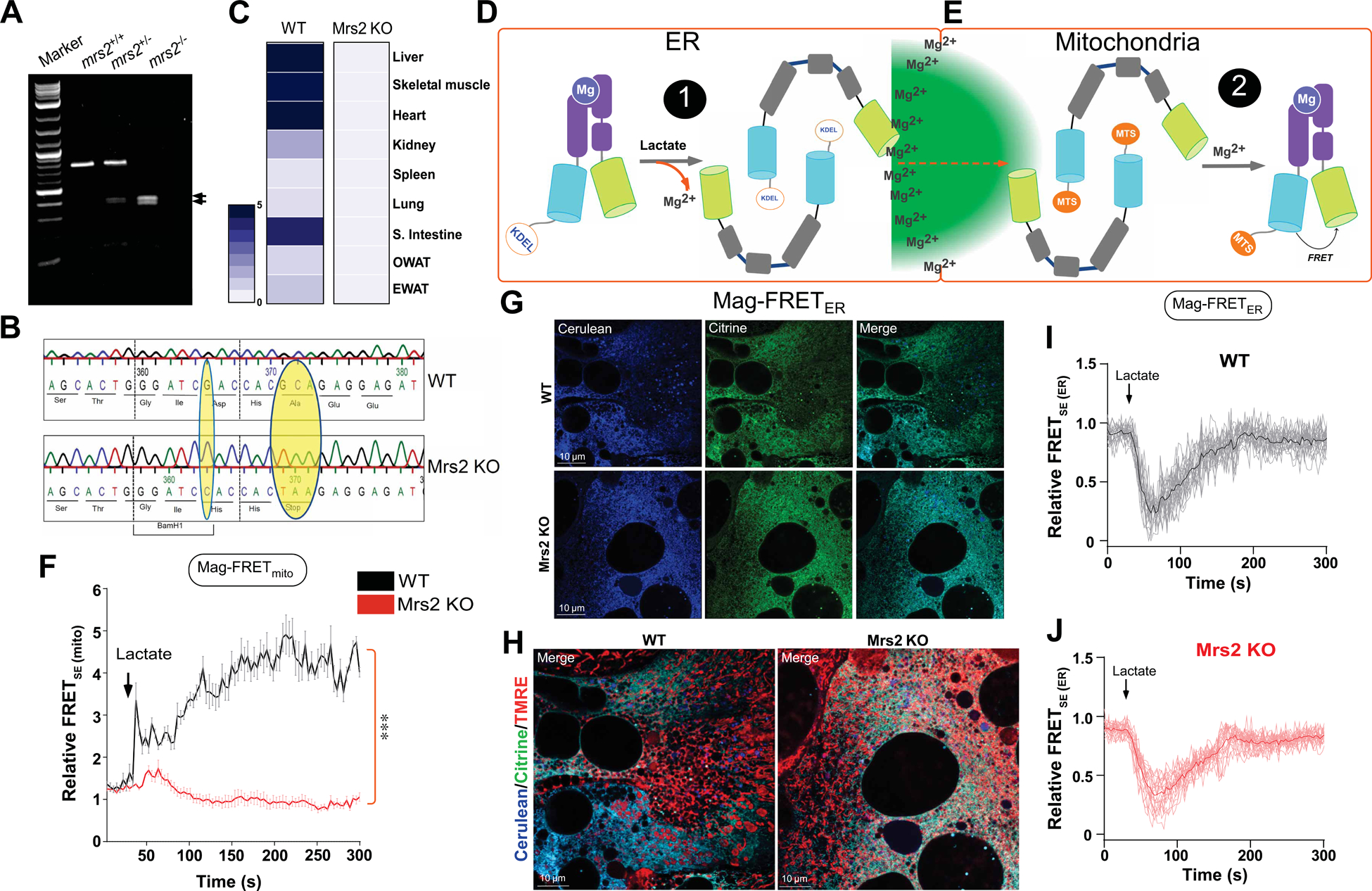
(A) Image depicts confirmation of CRISPR-Cas9-mediated global Mrs2 KO.
(B) DNA sequence analysis show the CRISPR/Cas9-mediated insertion of early stop codon that confirms Mrs2 KO.
(C) Heatmap depicts the distribution of Mrs2 mRNA in WT and Mrs2 KO tissues. n=3–5 mice per genotype.
(D-E) Schematic showing construction and operation of our modified MagFRET constructs to assess the Mg2+ dynamics in the ER and mitochondria during stimulation.
(F) Representative traces depicting MagFRET signal changes in the mitochondria of WT (black) and Mrs2 KO (red) hepatocytes after lactate stimulation. Mean ± SEM. n=4–6. ***p < 0.001.
(G) WT and Mrs2 KO hepatocytes were transduced with Mag-FRETER adenovirus. Images depict the proper localization of KDEL-tagged Mag-FRETER construct.
(H) WT and Mrs2 KO hepatocytes were transduced with Mag-FRETER adenovirus. Images depict Mag-FRETER expression and counter stained with ΔΨm indicator TMRE.
(I-J) Representative traces depicting rapid loss of MagFRET signal in the ER after lactate stimulation in both WT (I) and Mrs2 KO (J) hepatocytes. n=4–6.
Although compartmentalized Mag-Green displays robust dynamic changes in the ER and mitochondria upon lactate stimulation, Mg2+ flux was also monitored using a genetically encoded Mg2+ sensor, Mag-FRET (Lindenburg et al., 2013). We designed and generated two separate Mag-FRET constructs fused with either the ER retention sequence (KDEL) or the mitochondria targeting sequence from pyruvate dehydrogenase (PDH)-1 (Figure 4D and 4E). Primary murine hepatocytes were transduced with Adenoviral (AdV5a) versions of these Mag-FRET sensors. 36 hours post transduction, we measured the Mag-FRET signals after stimulation with 5 mM lactate (Figure 4F; Figure S3C and S3D). The mitochondrial Mag-FRET signal rapidly increased following stimulation in the WT but was almost completely absent in Mrs2 KO hepatocytes (Figure 4F). As controls, we confirmed proper mitochondrial ΔΨm and localization of the ER Mag-FRET sensor in hepatocytes (Figure 4G and 4H). Although mMg2+ uptake was absent in Mrs2 KO, the ER Mag-FRET signal was significantly depleted following stimulation with lactate in both hepatocytes (Figure 4I and 4J). These data suggest that Mrs2 is, indeed, the molecular entity responsible for the mMg2+ conductance after lactate-induced ER Mg2+ depletion.
The Conserved Mrs2 Intermembrane Space Loop Residues Control Mg2+ Uptake
Primary structure alignment of Mrs2 channels from yeast to higher eukaryotes highlights conservation of two transmembrane domains (TM1 and TM2), negatively charged amino acids in the intermembrane space loop linking TM1 and TM2 and a F/Y-G-M-N motif (Figure 5A). Single amino acid substitutions in the G-M-N motif are sufficient to abolish the selectivity of Mg2+ transport (Eshaghi et al., 2006; Lunin et al., 2006; Matthies et al., 2016; Payandeh et al., 2013; Schindl et al., 2007). Thus, we next sought to determine whether conserved residues within the putative intermembrane space loop of Mrs2 are required for lactate-mediated mMg2+ uptake. We generated Adenoviral (AdV5A) Mrs2 WT and mutant constructs [(GMNLESSLEED) to (IKLLKSSLKKK)] tagged with mRFP/Flag to analyze the protein expression, localization and impact on mMg2+ uptake (Figure 5B). Ectopic protein expression of WT and mutant Mrs2 in transduced primary murine hepatocytes was confirmed by Western blot (Figure 5C). Additionally, confocal microscopy revealed that WT and mutant Mrs2 proteins were properly localized to the mitochondria (Figure 5D). Next, we co-transduced WT and Mrs2 KO hepatocytes with the Mag-FRET sensors and WT or mutant Mrs2 constructs. WT and Mrs2 KO hepatocytes exhibited comparable ER Mag-FRETSE signal depletion upon lactate stimulation when expressing either WT or mutant Mrs2 (Figure 5E). Additionally, WT and Mrs2 KO hepatocytes expressing the WT Mrs2 construct showed significant increases in mitochondrial Mag-FRETSE signals following lactate stimulation, though the increase in signal from Mrs2 KO was lower than WT mitochondria. Strikingly, the mitochondrial Mag-FRETSE signal was largely abolished in both cell types expressing the Mrs2 mutant construct. (Figure 5F). We also confirmed the ability of Mrs2 to enhance mMg2+ uptake by overexpressing the WT Mrs2 viral construct in WT primary hepatocytes and monitoring Mag Green signals after lactate stimulation. Indeed, Mrs2 overexpression potentiated mMg2+ uptake without altering lactate-induced ER depletion (Figure S4A–C). Collectively, these results indicate that Mrs2 is the major route for mMg2+ uptake after lactate stimulation.
Figure 5. Mutations of the conserved Mrs2 transmembrane loop residues essentially inhibited mMg2+ uptake.
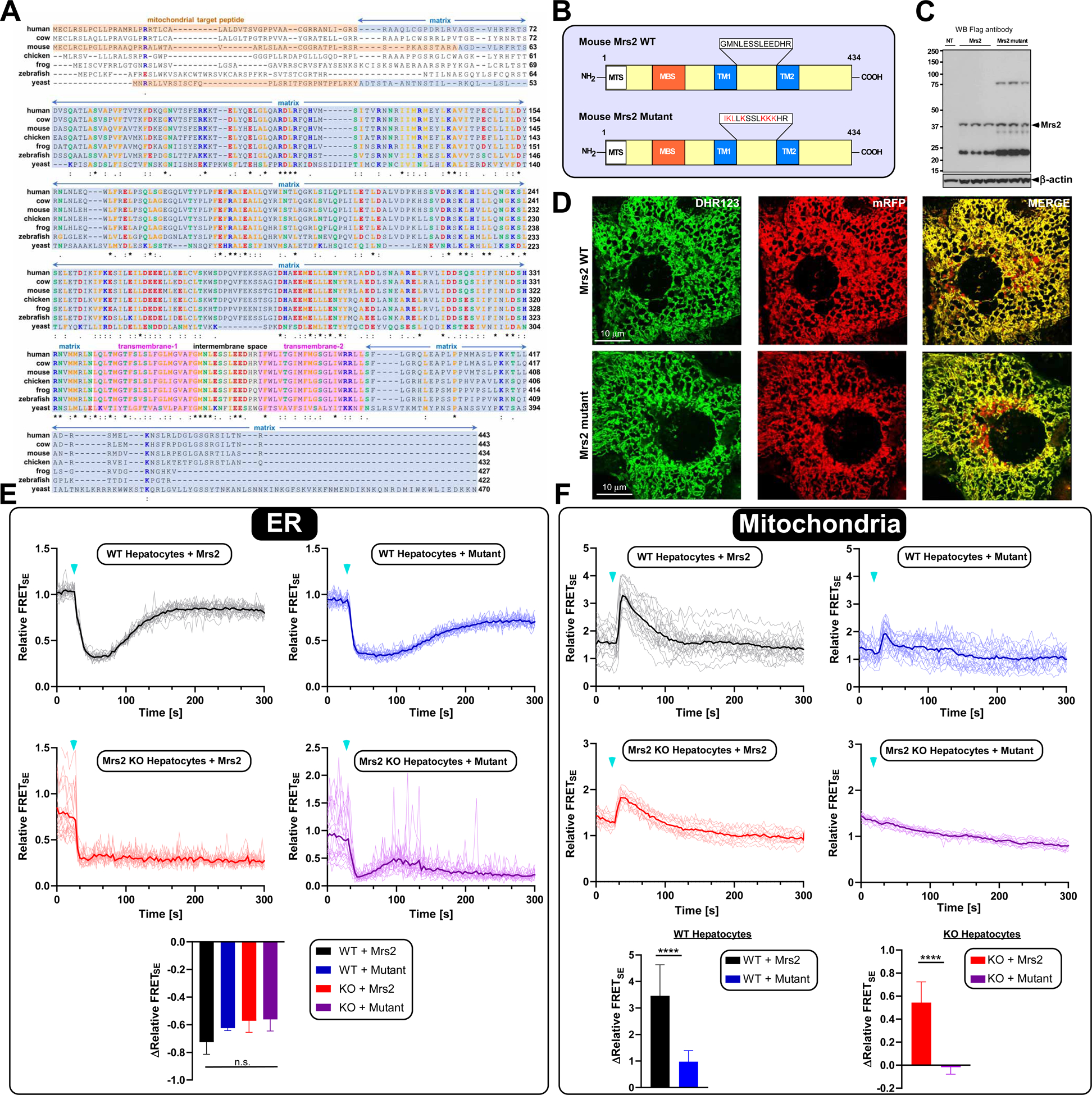
(A) Amino acid sequence alignment of the yeast Mrs2p its only mammalian homologue Mrs2.
(B) The schematic depicts the mouse Mrs2 WT (top) and Mrs2 mutant constructs (bottom).
(C) Western analysis showing the expression of Flag-tagged WT and mutant Mrs2 Adv5 constructs.
(D) Confocal micrographs of primary hepatocytes transduced with WT and mutant Mrs2-tagged with mRFP.
(E-F) Representative traces depicting MagFRET signal changes in the ER (Left) and mitochondria (right) after lactate stimulation. Bar charts show the relative FRETSE. n=4–6. Mean ± SEM. ****p < 0.0001. n.s.= not significant.
MCU-Mediated mCa2+ Uptake Is Normal In Mrs2 KO Hepatocytes
We next sought to determine if the mMg2+ uptake functional defect in Mrs2 KO cells impacts MCU. WT and Mrs2 KO hepatocytes permeabilized with digitonin were given a bolus of Ca2+ (10 μM) at 400 seconds followed by FCCP exposure at 700 seconds. The ΔΨm built similarly in WT and Mrs2 KO hepatocytes, and the mitochondrial Ca2+ uptake rates were comparable (Figure S5A). We also transduced WT and Mrs2 KO hepatocytes with GCaMP6s-mt, a mCa2+ sensor. Mrs2 deletion did not alter ionomycin or high glucose-induced mCa2+ uptake, indicating that Mrs2 is impermeable to Ca2+ (Figure S5B and S5C). Reciprocally, we also tested whether deletion of MCU could alter mMg2+ uptake. MCUfl/fl and MCU KO hepatocytes (ΔHep) were imaged for mMg2+ uptake using Mag-Green. Lactate-induced ER depletion and mMg2+ uptake were not affected by MCU deletion (Figure S5D and S5E). These results reinforce that lactate is a specific activator of Mrs2-mediated mMg2+, which functions independent of MCU activity.
Limiting Mrs2-Mediated mMg2+ Uptake Enhances Mitochondrial Bioenergetics
Ca2+ and Mg2+ ions exert major regulatory effects on cellular metabolism. For example, the activity of the PDH complex is controlled by divalent cations through ion-dependent modulation of its phosphorylation status (Glancy and Balaban, 2012; Midgley et al., 1987). In particular, the inactive form of PDH complex (phosphorylated) is enhanced by Mg2+. To assess the effects of Mrs2 deletion on cellular metabolism, we first measured basal mitochondrial matrix [Mg2+]. Permeabilized cells were exposed to FCCP, and the released mMg2+ was taken as resting matrix [Mg2+]. Strikingly, the m[Mg2+] was significantly reduced in Mrs2 KO hepatocytes, strongly corroborating Mrs2 as the major route for mMg2+ uptake (Figure 6A and 6B).
Figure 6. Genetic ablation of Mrs2 exhibits higher mitochondrial PDH activity and oxygen consumption rate.

(A) Permeabilized cells were exposed to FCCP at indicted time point to asses basal mMg2+. (B) Normalized basal mMg2+. Mean ±SEM.
(C) Representative transmission electron micrographs (TEM) of WT (left) and Mrs2 KO (right) hepatocytes.
(D and E) TEM analysis of mitochondrial length and number in WT and Mrs2 KO hepatocytes. Four images per mice. n=3 mice per group. Mean ±SEM.
(F) Western blot depicting the abundance of mitochondrial OXPHOS complex proteins in WT and Mrs2 KO hepatocytes. n = 3 mice per group.
(G) Oxygen consumption rate (OCR) of untreated WT and Mrs2 KO hepatocytes.
(H) OCR curves of WT and Mrs2 KO hepatocytes treated with 0.5 mM lactate, after basal measurements, at the indicated time point. (Bottom panels) Bar chart depicts basal, maximal, and proton leak. Mean ±SEM; n=3.
(I) OCR curves depicting hepatocytes pretreated for 24 hours with either LPS (100 μg/ml) alone or in combination with GSK2837808A (100 nM) or Oxamate (20 mM). Bar charts depict basal and maximal respiration and proton leak from the above traces. Mean ±SEM. n= 6–8.
(J) WT and Mrs2 KO hepatocytes were treated with LPS (1, 10 and 100 μg/ml) for 6 hours and immunoblotted for p-PDH E1α (S293), PDH E1α, and β-actin. Densitometric analysis shown below. n=3. Mean ±SEM.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s. = not significant.
We next assessed whether mitochondrial changes could be visualized that indicate altered metabolism. Hepatocytes were isolated from WT and Mrs2 KO mice and stained with the fluorescent lipid neutral dye BODIPY 488 as well as the ΔΨm indicator TMRE. As compared to WT, a similar number of lipid droplets were present after 24 hours for Mrs2 KO. No change was observed in ΔΨm, as assessed by TMRE staining. After 48 hours, however, Mrs2 KO cells had significantly more lipid droplets that were smaller in size, indicating that these cells may be more metabolically active than WT (Figure S6A and S6B). We also found that Mrs2 KO hepatocytes had significantly lower mitochondrial ROS signal than WT hepatocytes (Figure S6C and S6D). To gain a better understanding of the mitochondrial phenotypic differences that exist between the WT and Mrs2 KO cells, ultrastructural imaging was conducted by transmission electron microscopy (TEM). Mrs2 KO hepatocytes contained significantly longer, more filamentous mitochondria than WT (Figure 6C and 6D). Additionally, Mrs2 KO hepatocytes were observed to have significantly increased numbers of mitochondria (Figure 6C and 6E).
To further evaluate bioenergetics, we next measured the mitochondrial oxygen consumption rate (OCR) in Mrs2 KO and WT hepatocytes. Mrs2 KO hepatocytes exhibited heightened states of basal and maximal OCR compared to WT (Figure 6G). The OXPHOS complex protein abundance were relatively unchanged between WT and Mrs2 KO hepatocytes (Figure 6F), ruling out OXPHOS protein abundancy as an underlying factor. Given that increased mMg2+ flux induced by lactate might alter OCR, we next delivered lactate to both WT and Mrs2 KO hepatocytes before the sequential additions of respiratory chain inhibitors. Challenging WT hepatocytes with lactate led to a significant reduction of basal and maximal OCR (Figure S6E). In contrast, Mrs2 KO hepatocytes exhibited a marked rise in maximal OCR. These findings suggest that lactate enhances mMg2+ uptake and modulates mitochondrial respiration in WT hepatocytes (Figure 6H).
Lowering Mitochondrial Matrix Mg2+ Sustains The Dephosphorylated Form Of PDH
Given that ~95% of cellular Mg2+ is bound or sequestered by proteins and metabolites such as ATP or citrate, we asked whether depletion of cellular ATP enhances mMg2+ uptake (Corkey et al., 1986). Mag-Green and mito-Tracker Red loaded hepatocytes were challenged with a combination of oligomycin (20 μM) and 2-deoxy D-glucose (2 mM) to inhibit mitochondrial and glycolytic ATP production, respectively. The combination treatment (oligo+2DG) induced non-synchronous mito-Tracker Red oscillations, which were absent in control hepatocytes (Figure S6G and S6H top traces). Concomitantly, we found that the mMg2+ rapidly increased (Figure S6G and S6H, bottom traces and Figure S6I), indicative of ATP depletion and cytosolic Mg2+ elevation. This data indicates that the elevation of free Mg2+ dampens bioenergetics, likely through mMg2+-dependent inhibition of OXPHOS.
Intracellular elevation of lactate has been associated with several clinical conditions such as sepsis, ischemia, mitochondrial diseases, and malignancy (Brooks, 2018; Haas et al., 2016; Langley et al., 2013; Pavlova and Thompson, 2016). Given that lactate stimulation suppresses OCR, we hypothesized that modulation of mitochondrial Mg2+ by lactate would affect pyruvate oxidation. Thus, we examined mitochondrial OCR in HepG2 cells after treatment with various doses of the bacterial endotoxin lipopolysaccharide (LPS), as well as 5 mM lactate for 8 hours. Similar to the lactate effect, cells exposed to LPS had significantly suppressed OCRs (Figure 6I and Figure S6E). We next tested whether the inhibition of OCR by LPS is due to lactate-mediated ER Mg2+ release and mMg2+ uptake. Hepatocytes were pretreated with LDH inhibitors (GSK; 100 nM or Oxamate; 20 mM) prior to LPS challenge. These inhibitors significantly restored both basal as well as maximal OCR, reinforcing the lactate mediated mMg2+ uptake and suppression of OCR (Figure 6I and see Figure 7D and 7E).
Figure 7. Mrs2 Genetic Ablation Confers a Protective Effect in an Endotoxin Model of Septic Shock.
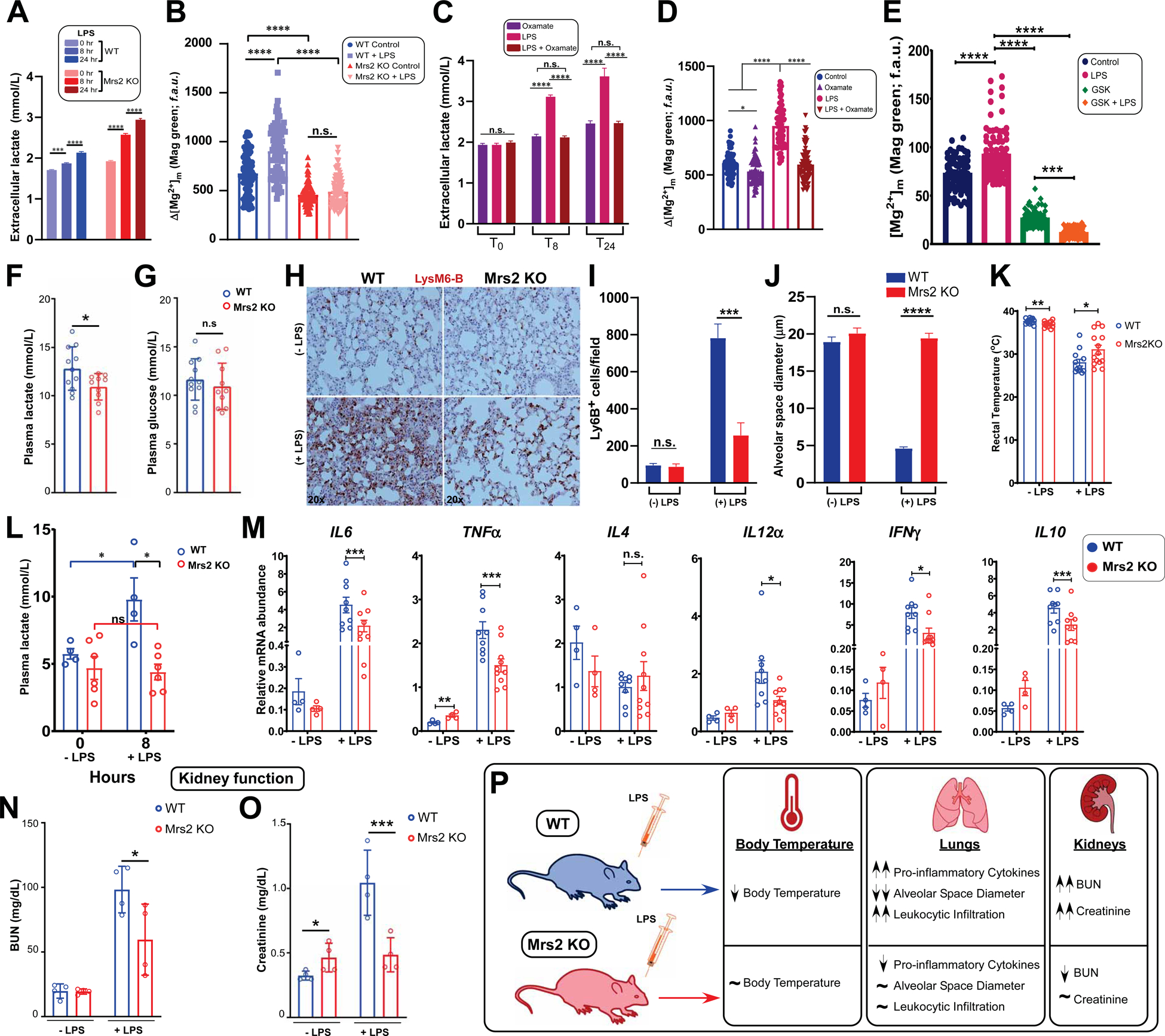
(A) Bar chart quantifying extracellular lactate accumulation in WT and Mrs2 KO hepatocytes after LPS challenge (100 μg/mL). n = 3. Mean ±SEM.
(B) Quantitative analysis comparing mMg2+ in WT and Mrs2 KO hepatocytes after 24 hours of LPS treatment. n = 3. Mean ± SEM.
(C) Extracellular lactate levels in WT hepatocytes at 0, 8, and 24 hours post LPS treatment with or without oxamate (20 mM). n = 3. Mean ±SEM.
(D) Quantification of mMg2+ in WT hepatocytes pretreated with oxamate before LPS challenge. n = 3–4.
(E) Quantification of mMg2+ in WT hepatocytes pretreated with GSK (100nM) before LPS challenge. n = 3–4.
(F-G) Measurement of plasma glucose and lactate in WT and Mrs2 KO mice. Mean ± SEM. n=10–11 mice per group.
(H-J) Lys6B immuno-staining of lung tissue from WT and Mrs2 KO mice, with and without 24-hour LPS (5mg/kg) challenge (H). (I) Quantification of Lys6B positive cells.
(J) Measurement of alveolar space diameter. n= 3 mice per group. Mean ± SEM.
(K) Measurement of body temperature in WT and Mrs2 KO mice with and without 24-hour LPS treatment. n=10–12 per group. Mean ± SEM.
(L) Plasma lactate were measured from WT and Mrs2 KO mice. Mean ± SEM. n=4–6 per group.
(M) Measurement of cytokine transcripts from WT and Mrs2 KO mice lung tissues 24-hours following LPS treatment. n=4–10 per group. Mean ± SEM.
(N-P) Serum creatinine and BUN from WT and Mrs2 KO mice 24-hours following LPS treatment. n=4 per group. Mean ± SEM.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n.s. = not significant.
To further evaluate how iMg2+ impacts OCR, hepatocytes were challenged with various concentrations of exogenous MgCl2 (1, 2, or 6 mM). Hepatocytes exposed to 2 or 6 mM MgCl2 exhibited reduced basal and maximal OCR indicating that the entry mechanism is operative at supraphysiological concentrations of extracellular Mg2+ (Figure S6F). However, exogenous lactate-mediated suppression of OCR was found to be Mrs2 dependent using Mrs2 KO cells (Figure 6G and 6H), indicating that lactate mediated ER Mg2+ release followed by mMg2+ uptake is the primary route for the marked suppression of OCR.
Given that the PDH complex is the major link between the glycolytic pathway and the TCA cycle, we also examined whether LPS-induced intracellular lactate production modulates the phosphorylation status of PDH in hepatocytes. WT and Mrs2 KO hepatocytes were treated with three different doses of LPS for 24 hours, and pPDH levels were determined by western blot analysis. When WT hepatocytes were treated with LPS at 10 and 100 μg/ml, a significant amount of phosphorylated S293PDH was detected (Figure 6J, top panel). Remarkably, the LPS-mediated phosphorylation of PDH in Mrs2 KO hepatocytes was near completely absent (Figure 6J, bottom panel). Since multiple phosphorylation sites contribute to PDH complex activity, we also assessed the status of S232 and S300. The S232 and S300 were unaffected upon LPS stimulation in WT hepatocytes, indicating that the LPS-mediated cascade targets uniquely S293 (Figure S7A, top panel and Figure 6J). In contrast, LPS-mediated phosphorylation was completely blunted, and rather hyper dephosphorylation was observed at the S232 and S300 sites of PDH in the Mrs2 KO hepatocytes, suggesting that lowering mMg2+ enhances PDH dephosphorylation upon LPS challenge (Figure S7A, bottom panel).
We next tested whether phosphorylation of PDH by LPS is contributed through an HIF1α-dependent mechanism. We found HIF1α stabilization in both WT as well as Mrs2 KO hepatocytes after LPS challenge, and the HIF1α protein abundance was higher in Mrs2 KO hepatocytes (Figure S7B). Collectively, these results reveal that lowering mitochondrial matrix [Mg2+] promotes PDH dephosphorylation, which likely enhances mitochondrial bioenergetics (i.e. OCR) (Figure 6G and 6H).
Acute Inflammation Enhances Lactate-Mediated mMg2+ Elevation That Contributes To Multi-Organ Failure
To investigate whether lactate-mediated mMg2+ flux alters organ function, we chose an LPS-induced acute inflammation model. Both WT and Mrs2 KO hepatocytes exposed to LPS showed higher lactate accumulation (Figure 7A). We next examined the mMg2+ levels under control and LPS treated conditions and found increased Mag-Green signals within the mitochondria upon LPS treatment (Figure 7B). Although LPS-induced lactate production was comparable between WT and Mrs2 KO, mMg2+ was significantly elevated in WT but not in Mrs2 KO (Figure 7B). We next asked whether targeting lactate dehydrogenase A (LDHA) would reduce mMg2+ uptake in response to LPS treatment. To test our hypothesis, we pretreated hepatocytes with oxamate (20 mM) overnight before LPS stimulation (Gandhirajan et al., 2013; Zhang et al., 2019b). Oxamate pretreatment markedly reduced lactate accumulation and mMg2+ uptake suggesting that lactate is the elicitor of iMg2+ dynamics (Figure 7C and 7D and Figure S7H). We next asked whether inhibition of lactate efflux by MCT inhibitors potentiates ER Mg2+ depletion, indeed finding that lactate accumulation promotes ER Mg2+ depletion (Figure S6J). Lactate effect on iMg2+ dynamics was further confirmed using a selective LDH inhibitor, GSK283378080A (100 nM) (Figure 7E). Conversely, LDH inhibition by GSK283378080A did not affect exogenous lactate-induced ER Mg2+ depletion (Figure S6K).
Since lactate elicits iMg2+ dynamics, we measured basal plasma glucose and lactate levels in Mrs2 KO mice and found that Mrs2 KO mice have lower lactate levels (Figure 7F and 7G). One of the major changes that occur during multi-organ-failure (MOF) in severe sepsis is mitochondrial dysfunction. To establish the role of mMg2+ flux in LPS-mediated mitochondrial reprogramming and MOF, mice were challenged with LPS (5mg/kg) for 24 hours and leukocyte infiltration was assessed. Lung sections were stained with neutrophil marker Lys6B and quantification showed that neutrophil infiltration was significantly suppressed in Mrs2 KO mice challenged with LPS as compared WT mice (Figure 7H and 7I). Reciprocally, the assessment of alveolar space diameter revealed a pulmonary edema-like phenotype in WT but was markedly reduced in Mrs2 KO mice (Figure 7J). Additionally, we found that Mrs2 KO mice challenged with LPS maintained their body temperature, as compared to WT, in which body temperature dropped (Figure 7K). As expected, WT mice challenged with LPS showed higher plasma lactate levels when compared to control (Figure 7L) while Mrs2 KO exhibited lower lactate levels (Figure 7L).
Consistent with these outcomes, proinflammatory cytokine profiles were attenuated in Mrs2 KO mice challenged with LPS (Figure 7M). Having observed a reduced cytokine expression profiles in Mrs2 KO mice following LPS challenge, we asked whether the immune response is altered under basal conditions. Splenocytes were isolated from WT and Mrs2 KO mice for CD3/CD28 co-stimulation to assess cCa2+ dynamics. We found that both cell types showed similar cCa2+ dynamics (Figure S7C–E), indicating similar T-cell receptor (TCR) crosslinking and exhibiting similar NFAT nuclear translocation (Figure S7F and S7G). Since sepsis-related acute inflammation can result in acute kidney injury (AKI), we next assessed whether lowering mMg2+ may provide protection against AKI in a LPS-induced model of septic shock. Mrs2 KO mice exhibited lower creatinine and BUN, likely linked to Mg2+-dependent control of kidney function (Figure 7N–7O). Collectively, these data suggest that lactate accumulation controls iMg2+ dynamics that coordinates mitochondrial metabolism and organ preservation during acute inflammation (Figure 7P).
DISCUSSION
In this work, we have taken on the challenge of identifying a highly specific ligand-based driver of iMg2+ dynamics that results in rapid and robust Mg2+ accumulation within mitochondria. We showed that the glycolytic end-product lactate was directly responsible for rapid depletion of ER Mg2+ stores, followed by robust mMg2+ uptake. Our findings reveal that L-lactate-induced iMg2+ dynamics is also dependent on a steep ΔΨm for mMg2+ uptake. This phenomenon demonstrated enantiomeric specificity and was not dependent upon pH. Our identification of lactate as a ligand for iMg2+ dynamics is remarkably analogous to the IP3-mediated depletion of ER Ca2+. Therefore, we conclude that the L-lactate anion acts as a signaling molecule that drives iMg2+ flux in a highly conserved phenomenon among mammalian cells.
Lactate As A Ligand For Intracellular Mg2+ Dynamics
The roles of lactic acid, lactate anions and hydrogen ions on physiology in settings of muscle fatigue, lactic acidosis, lactatemia and anaerobiosis have been debated for over a century (Brooks, 2018). For instance, it remains uncertain whether glycolysis produces lactate or lactic acid. Nevertheless, it is certain that acidosis occurs in exercise and other conditions (Brooks, 2018). Thus, we tested both L-lactic acid and sodium L-lactate in our targeted screen and found both induced mMg2+ elevation, demonstrating a powerful ligand effect on ER Mg2+ release (Figures 1, 2A and 2B). Because of the elevation of extracellular lactate noted in intermediary metabolism and exercise performance (Brooks, 2018), we questioned whether lactate could function as a ligand for plasma membrane receptors like GPR81 (Cai et al., 2008; Lee et al., 2001). Consequently, we blocked lactate influx pharmacologically as well as by RNAi-mediated silencing, revealing that lactate serves as a ligand for ER Mg2+ release rather than cell surface Mg2+ entry (Figure 3). Thus, another important fundamental feature of lactate is its activity as an intracellular ligand.
Although over 95% of iMg2+ is bound to ATP, polyphosphates, and metabolic enzymes, it was unclear whether dissociation of bound cytosolic Mg2+ in turn triggers mMg2+ uptake. Our fluorescent dye and protein FRET based assays show that ATP hydrolysis does not cause this effect, but rather mMg2+ uptake upon lactate stimulation is from ER Mg2+ release (Figures 1 and 4). Thus, the ER may contain a primary intracellular pool of free Mg2+ that is rapidly released in response to a stimulus like lactate (Figure 1 and 4). This phenomenon is analogous to IP3-induced ER Ca2+ release and subsequent MCU-mediated mCa2+ uptake (Berridge et al., 2003; Clapham, 2007). However, future studies are warranted to identify the lactate-sensitive ER Mg2+ release machinery.
Mechanistically, suppression of OXPHOS and increased glycolysis have been linked to intensive physical activity, sepsis, ischemia, neoplasia, and mitochondrial diseases (Brooks, 2018; Haas et al., 2016; Wallace, 2012). However, the effect of the dramatic elevation of cellular lactate on iMg2+ homeostasis under these conditions remains unknown. Our identification of lactate as a ligand for ER release leading to subsequent mMg2+ uptake will be an invaluable research tool for probing the role of iMg2+ in these and countless other scenarios.
Mg2+ Is A Second Messenger For Metabolic Circuits
Our experiments show a lag time of about ~25 s from lactate stimulation to mMg2+ accumulation, which is likely due to the ER depletion-dependent establishment of a concentration threshold (set-point) (Figure 1). Our in vitro calibration data show that free Mg2+ above ~2.0 mM (1.0mM pulse + 1.0mM buffer) promotes Mrs2-mediated Mg2+ uptake. Previous electrophysiological characterization of Mrs2P revealed a negative feedback mechanism when mMg2+ concentrations rose above 2 mM (Schindl et al., 2007). In support of these findings, our characterization of Mrs2 activity showed a very similar inhibition phenomenon, suggesting a highly conserved regulatory property of the Mg2+ transport machinery from yeast to mammals (Figures 3–5).
Unlike Ca2+ channels, Mg2+ channels are distributed widely across prokaryotic and eukaryotic life. In eukaryotes, plasma membrane Mg2+ flux components are controlled by multiple machineries, while mitochondrial transport is mediated primarily by Mrs2P (yeast) and mammalian Mrs2 (Bui et al., 1999; Kolisek et al., 2003). The Mrs2 channel family uniquely carry a conserved F/Y-GMN motif between TM1 and TM2, which is essential for Mg2+ selectivity. We relied on Uniprot protein annotations, which are based on multiple algorithms, to assign human and murine Mrs2 topology. Based on this curated data, a large N-terminal domain is localized in the matrix due to a 49-residue mitochondrial targeting sequence (Figure 3 and 5). Although electrophysiologic and structural studies would provide unequivocal evidence for whether Mrs2 forms a channel, our mutagenesis analysis strongly suggest that mammalian Mrs2 forms a selective pore for lactate-mediated mMg2+ uptake (Figure 5).
The transient receptor potential (TRP) superfamily of ion channels have well known thermo-sensing abilities (Julius, 2013; Moqrich et al., 2005; Voets, 2014; Xu et al., 2002). Remarkably, we showed that Mrs2-mediated mMg2+ influx is thermosensitive at near physiological temperatures, in contrast to MCU, revealing another thermosensitive ion channel. We found that the human Mrs2 N-terminal domain is thermally stabilized by Mg2+ (Figure 3H and 3I); thus, it is conceivable that temperature may regulate Mrs2 by affecting Mg2+ binding to this regulatory region. The CorA orthologue is inhibited by Mg2+-mediated N-terminal domain interactions between protomers (Pfoh et al., 2012), and this mechanism is consistent with the matrix Mg2+ dependent inhibition we observed (Figure 3F–3G). Nevertheless, future studies are needed to precisely identify the motif/location and structural basis for the temperature sensing properties of Mrs2.
It was alluded that the concentration of intracellular free Mg2+ is relatively high (~0.5–1.2 mM), and thus, Mg2+ is unlikely to act as a second messenger, like Ca2+, by rapidly changing compartmentalized concentrations. However, iMg2+ changes have profound effects on cellular metabolism, structure and bioenergetics (Romani and Scarpa, 1990). Since iCa2+ and iMg2+ antagonistically control cellular metabolism, lowering mMg2+ threshold in Mrs2 KO hepatocytes was indeed found to modulate PDH activity. However, the precise link between mMg2+ and OXPHOS pathways warrant future studies. Here, our findings reveal that iMg2+ is indeed a second messenger that controls numerous cellular metabolic circuits (Figure 6 and 7).
In conclusion, the identification of lactate as an agonist for iMg2+ dynamics represents a long-sought discovery that will rejuvenate the Mg2+ signaling field as well as prompt further studies into the downstream physiological outcomes of iMg2+ flux between cellular compartments. Furthermore, lactate production and its possible consequences has been scrutinized in cells undergoing aerobic glycolysis (Warburg effect). A common feature of cells undergoing aerobic glycolysis, such as within the tumor microenvironment or immune cell proliferation in sepsis, is the production of copious amounts of lactate. A recent report (Zhang et al., 2019a) highlights how lactate leads to epigenetic modifications that exert profound downstream transcriptional changes. Another study (Zhang et al., 2019b) identified that MAVS acts as a direct sensor of lactate controlling RLR signaling and innate immunity. Our identification of lactate as a ligand establishes a new frame of reference to ascertain how perturbation of iMg2+ ion homeostasis may contribute to the etiology of multiple disorders.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact Muniswamy Madesh (muniswamy@uthscsa.edu).
Materials Availability
This study generated new reagents. Adenoviral Mrs2 and Mag-FRET sensors and mouse lines for this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any datasets/code amenable for depositing into public repositories.
EXPERIMENTAL MODELS
Animal Experiments
Wild-type and Mrs2 KO C57BL/6 mice were housed and maintained in our animal breeding facility with prior approval and accordance with the Institutional Animal Care and Use Committee (IACUC). For LPS studies, wild-type and Mrs2 KO mice were administered LPS (E. coli O111:B4; Sigma-Aldrich; 5 mg/kg) or vehicle (sterile DPBS) via intraperitoneal injection. Body temperature was measured using a rectal thermometer (Kent Scientific Corp). After 24 hours, blood was collected for serum preparation, animals were euthanized, and tissues collected for analysis (Gandhirajan et al., 2013).
Isolation and culture of primary murine hepatocytes
Primary adult mouse hepatocytes were isolated from 8 to 12-week-old male and female animals using a two-step collagenase perfusion technique (Li et al., 2010), with slight modifications. After cannulation of the portal vein, the liver was perfused. First, with perfusion medium-I (Ca2+ and Mg2+ free HBSS; GIBCO, #14174), supplemented with 35 mM HEPES and 0.75 mM EGTA. This was followed by perfusion with perfusion medium-II (DMEM, Cat#12320, GIBCO) supplemented with collagenase D (380 μg/ml, Worthington). Liver lobes were dissected, dissociated, and the crude hepatocyte preparation passed through a 100 μM cell strainer. The crude hepatocyte preparation was centrifuged (50×g for 5 min, 4°C) to pellet parenchymal hepatocytes. The hepatocytes were washed three times with DMEM (Cat#11885, GIBCO) supplemented with 10% (v/v) FBS (Fetal bovine serum, characterized, heat-inactivated; Hyclone, Cat# SH30396.03, GE Life Sciences, UT, USA). Centrifugation during washing steps was done at 50×g for 5 min (4°C). After the third wash, cells were re-suspended in hepatocyte culture media (Williams E medium (Sigma, #W4128), supplemented with 1% (v/v) antibiotic-antimycotic solution (GIBCO, #15240), 2.0 mM L-glutamine (GIBCO, #25030), 1% (v/v) MEM non-essential amino acids (GIBCO, #11140), and 10% (v/v) FBS. Cells were plated collagen-coated plates in hepatocyte culture media. Two to four hours later, the media was replaced.
Cell Culture
All cells were cultured at 37°C and 5% CO2, unless otherwise indicated. Hepatocytes were cultured in hepatocyte culture media (see above). HepG2 (ATCC® HB-8065) cells were cultured in DMEM (Cat#11885, GIBCO) supplemented with 1% (v/v) antibiotic-antimycotic solution and 10% (v/v) FBS. HeLa (ATCC® CCL-2) and COS-7 (ATCC® CRL-1651) cells were cultured in high glucose (4.5g/L) DMEM (Hyclone, #SH30022) supplemented with 110 mg/L sodium pyruvate, 10% (v/v) FBS, and 1% (v/v) antibiotic-antimycotic solution. MEFs (ATTC ®, SCRC-1040) were cultured in high glucose (4.5g/L) DMEM (Hyclone, #SH30022) supplemented with 10% (v/v) FBS and 1% (v/v) antibiotic-antimycotic solution. Trypsin (0.25% for all cells except MEF cells, 0.05%; GIBCO) was used to detach cells from culture plates when necessary.
Generation of Mrs2 KO and liver-specific MCU KO mice
A CRISPR/Cas9 gene-editing strategy was adopted to produce Mrs2 knock out (KO) mouse embryonic fibroblasts (MEFs). Briefly, candidate sgRNAs were selected based on a CRISPR/Cas9 nickase design platform, which substantially reduces off-target effects by ~50–1500 fold. The selected paired gRNAs targeting exon 8 of Mrs2 (the Cas9 nickase mRNA and single-stranded oligodeoxynucleotide (ssODN)) for HDR-mediated gene KO were transfected into MEFs. Knockout was confirmed by genotyping by PCR followed by restriction fragment analysis with BamH1 (Figure 4B). The Mrs2 targeted allele was cleaved yielding two fragments (388 and 368 bp), whereas the wild type (WT) allele was not. After validation of sgRNAs and ssODN in vitro, gRNA1, gRNA2, ssODN containing the BamHI site along with Cas9 protein was microinjected into single-celled embryo and transferred to surrogate mother. Details of the oligo’s sequence used for the Mrs2 KO are provided in Figure 4B. The chimeras were bred with C57BL/6J (The Jackson Laboratory, USA) mice over 10 generations, and both sexes were used for in vitro and in vivo studies. Hepatocyte-specific Mcu knockouts (MCUΔhep) were generated by crossing MCUfl/fl mice (Tomar et al., 2019) with hepatocyte-specific Cre recombinase transgenic mice (B6.Cg-Tg(Alb-cre)21Mgn/J, The Jackson Laboratory, USA).
Adenoviral constructs
All adenoviral constructs were generated by Vector Biolabs (PA, USA) using Adenoviral - Human Type 5 (dE1/E3) as the backbone wherein the gene of interest is expressed under a CMV promoter. Ad-Mrs2.mRFP.FLAG was generated using the cDNA from mouse Mrs2 ORF clone (Origene; #MR217956). The Ad-Mrs2(mut).mRFP.FLAG was generated by mutating the ORF to encode the following mutant protein sequence at aa#396–406: “IKLLKSSLKKK.” Ad-ER-MagFRET-1 and Ad-Mito-MagFRET-1 were generated using pCMVMagFRET-1 (Addgene, #50742) with modifications (Lindenburg et al., 2013). For the Ad-ER-MagFRET-1 virus, the ER targeting (MLLPVLLLGLLGAAAD) and retention sequences (KDEL) were added. For the Ad-Mito-MagFRET-1 virus, the mitochondrial targeting sequence (MRKMLAAVSRVLSGASQKPASRVLVASRNFANMLHFC-SRRYRGPGIHR) was added. Cells were infected with adenoviruses at MOI’s of 20 for MagFRET constructs and 50 for Mrs2 constructs.
METHOD DETAILS
Measurement of intracellular and mitochondrial Mg2+ dynamics using a live cell confocal imaging system
Primary murine hepatocytes, HepG2, HeLa, MEFs or COS7 cells were grown on 25-mm collagen-coated glass coverslips overnight and loaded with 2.5 μM of the magnesium indicator Magnesium Green-AM (ex/em 488/510 nm) and 1.0 μM of the mitochondrial indicator MitoTracker Deep Red FM (ex/em 644/665 nm) in growth media for 30 minutes at 37°C (5% CO2). Cells were washed and imaged within a temperature-controlled environmental chamber (set at 37°C, unless otherwise indicated) using a Leica TCS SP8 live cell imaging system. After 30 seconds of baseline recording, various concentrations of metabolites were added (Table S1), and after 375 seconds, a control bolus of 10 mM MgCl2 was given. To confirm the role of lactate in mitochondrial Mg2+ dynamics, cells were treated with MCT inhibitors, SR13800 (1 μM) or AR-C155858 (2 μM), for 2 hours before measuring Mg2+ dynamics as above. Images were obtained every 3 seconds using a 100× oil objective and quantified using Leica Application Suite X.
Measurement of ER Mg2+ depletion using permeabilized cell system
Primary murine hepatocytes were grown on 25-mm collagen-coated glass coverslips overnight and loaded with 2.5 μM of the magnesium indicator Magnesium Green-AM (ex/em 488/510 nm). After dye loading cells were permeabilized with 40 μg/ml digitonin in 1.5 mL of intracellular medium (ICM) composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2 for 3 minutes at 37°C (5% CO2). Permeabilized cells were bathed in digitonin free ICM buffer. After one minute of baseline recording, Lactate derivatives (5 mM) were added and images were acquired using a Leica TCS SP8 live cell imaging system. Images were obtained every 3 seconds using a 100× oil objective and quantified using Leica Application Suite X.
Mitochondrial and cytosolic Ca2+ measurements using confocal system
Primary murine hepatocytes, HepG2, HeLa, MEFs or COS7 cells were grown on 25-mm collagen-coated glass coverslips overnight and loaded with 5 μM Fluo-4 AM (488nm/515nm) in Williams E medium supplemented with 1% (v/v) antibiotic-antimycotic solution (GIBCO), 1% (v/v) 200 mM L-glutamine, 1% (v/v) non-essential amino acids and 10% FBS for 30 minutes at 37° and cells were imaged using Leica TCS SP8 live cell imaging system. Cells were transiently transfected with GCaMP6s-mt and cells mounted on a confocal chamber and imaged at Ex:488nm/Em:515nm using a Leica TCS SP8 live cell imaging system. After 30 seconds of baseline recording, various concentrations of metabolites were added (Table S1 below), and after 375 seconds, a 10mM MgCl2 pulse was given. Images were obtained every 3 seconds using at 100x oil objective and quantified using Leica Application Suite X (Mallilankaraman et al., 2012b).
Measurement of mitochondrial Mg2+ uptake, MCU-mediated Ca2+ uptake, and basal matrix Mg2+
Primary murine hepatocytes were washed in Ca2+ and Mg2+ free HBSS, pH 7.4. An equal amount of cells (~2.5× 106 cells) were resuspended and permeabilized with 40 μg/ml digitonin in 1.5 mL of intracellular medium (ICM) composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2 and 2 μM thapsigargin to block the SERCA pump. All the measurements were performed in the presence of 1.5 μM K+/ATP or 1.5 μM Mg2+/ATP (magnesium and calcium measurements, respectively), along with 5 mM succinate. The measurement of extramitochondrial Mg2+ or Ca2+ clearance ([Mg2+]out, [Ca2+]out) was used as an indicator of [Mg2+]m or [Ca2+]m uptake; this done by loading the permeabilized cells with Mag-Green (1 μM; excitation 505 nm and emissions 535 and 595 nm), or Fura2-FF (0.5 μM; excitations 340 and 380 nm and emission 510 nm), respectively. Simultaneous measurement of mitochondrial membrane potential was measured by loading the permeabilized cells with JC-1 (800nM). Fluorescence was measured in a multi-wavelength excitation dual-wavelength emission spectrofluorometer (Delta RAM, PTI). [Ca2+]out is represented as the excitation ratio (340 nm excitation /380 nm emission) of Fura2-FF/FA fluorescence and ΔΨm as the ratio of the fluorescence of J-aggregate (ex/em 570/595 nm) and monomer (ex/em 490/535 nm) forms of JC-1. Similarly, [Mg2+]out is represented as f.a.u. (ex/em 505/595). A single Ca2+ (20 μM) or Mg2+ bolus (0.25–2.00 mM) and the mitochondrial uncoupler, FCCP (10 μM), were added at the indicated time points. All the experiments were performed at either 25°C or 37°C with constant stirring (Dong et al., 2017; Mallilankaraman et al., 2012a).
Assessment of intracellular pH
Hepatocytes were loaded with the intracellular ratiometric pH indicator (BCECF-AM; 4 μM) for 30 min. Relative changes in fluorescence intensity of BCECF was monitored using fluorescence spectroscope (Ex 440nm and Ex 490nm; and Em 538nM). L-lactate, D-lactate, methyl L-lactate, and 3-Phenyl lactate (5 mM) were added at the indicated time points. The change intracellular pH level was determined from the ratio of BCECF (Ex;490/440) (Halestrap, 2013).
Mitochondrial oxygen consumption rate
Primary murine hepatocytes or HepG2 cells were plated on 96 well Agilent Seahorse XF Cell Culture Microplates. For all primary hepatocyte experiments, the microplates were coated with collagen prior to cell addition. Primary hepatocytes were plated at a density of 4 × 105 cells per well, while HepG2 cells were plated at a density of 3 × 105 cells per well. Cells were plated in their normal growth media overnight. Media was changed to Seahorse XF Cell Mito Stress Test Kit (Agilent) assay media supplemented with glucose, glutamine, pyruvate concentrations equivalent to that of the growth media 1 hour before the experiment start time. After media change, per manufacturer instructions, cells were placed in a CO2-free incubator for 1 hour. Oxygen consumption rate (OCR) was measured at 37°C in an XF96 extracellular flux analyzer (Seahorse Bioscience, Agilent), which had been previously calibrated using Seahorse XF Calibrant solution (Seahorse Bioscience, Agilent) in a CO2-free incubator overnight. Respiratory chain inhibitors were then loaded into the XF96 flux analyzer, and during the run, added sequentially to cells at indicated time points. Primary cells received oligomycin, FCCP, and a mixture of antimycin A and rotenone at concentrations of 2 μM, 5 μM, and 1 μM, respectively, while HepG2 cells were treated with concentrations of inhibitors at 1 μM, 2 μM, and 0.5 μM, respectively. For treatment conditions with LPS (E. coli O111:B4; Sigma-Aldrich) or lactate, primary hepatocytes were treated with 0.5 mM lactate acid via syringe during the experiment at the indicated time point. HepG2 cells were pre-treated with 0.5 mM lactic acid and varying concentrations of LPS (1, 10, and 100 μg/mL) for 8 hours before the experiment start time. Data was collected using Agilent Seahorse Wave 2.6.1 Desktop software and exported to GraphPad Prism version 7 for analysis (Irrinki et al., 2011; Tomar et al., 2016).
TEM imaging and analysis
Hepatocytes isolated from WT and Mrs2 KO mice were plated on collagen-coated 6-well dishes, and samples were processed for TEM imaging as described below. Cells were washed with HBSS and fixed using 0.15 M cacodylate buffer (Ted Pella Inc., CA) pH 7.4 containing 2.5% (v/v) glutaraldehyde (Electron Microscopy Sciences, Hartfield, PA), 2% (v/v) formaldehyde (fresh from paraformaldehyde (EMS)) with 2mM CaCl2 and 20 mM KCl at 35°C for 5 minutes. Images were acquired using Zeiss LIBRA120 TEM equipped with Gatan UltraScan, 1000 2k × 2k CCD EFTEM, energy filtering. Mitochondrial length and number were quantified using ImageJ software (Nemani et al., 2018; Tomar et al., 2016).
Recombinant human Mrs2 expression and purification
Human Mrs2 (residues 58–333; NCBI accession number NP_065713.1) was cloned into a pET-28a expression vector (Novagen) using the NdeI and XhoI restrictions sites. Successful in-frame insertion was confirmed by Sanger sequencing. After transforming the pET-28a-Mrs258–333 into BL21 DE3 codon+ Escherichia coli, a single colony was grown in liquid Luria-Bertani medium containing kanamycin (60 μg mL−1) at 37 °C to an optical density (600 nm) of ~0.6–0.8. Protein expression was induced with the addition of isopropyl β-D-1-thiogalactopyranoside to a final concentration of 0.5 mM. Expression was allowed to proceed overnight (~14–16 hours) at 21 °C. After harvesting cells by centrifugation (10,000×g for 30 min) and one freeze-thaw cycle (−80 °C), the protein was natively purified according to the Ni-nitriloacetic acid (Ni-NTA) manufacturer guidelines (Qiagen), with the use of a probe sonicator for cell disruption, 0.1 % (v/v) Triton X-100, 2 mM β-mercaptoethanol and 350 mM imidazole for elution. After the hexahistidine tag was bovine thrombin cleaved (~1 U mg−1 protein) (EMD Millipore), a final size exclusion chromatography (SEC) purification step was performed through an S200 10/300 Increase GL column (GE Healthcare). The final SEC and experimental buffer was 20 mM Tris, 150 mM NaCl, 1 mM DTT, pH 8.0. Protein concentration was estimated using an extinction coefficient of 0.8084 (mg mL)−1 cm−1.
Far-UV Circular Dichroism (CD) and Thermal Stability
Protein solubilized in SEC buffer (see above) was diluted to 0.5 mg mL−1 and placed into a quartz 0.1 cm pathlength cuvette. Data was acquired between 240 – 200 nm, at a speed of 20 nm min−1, data pitch of 1 nm and response time of 8 s on an Jasco J-810 Spectrapolarimeter with a Peltier temperature control unit (Jasco). Each spectrum is an average of 3 scans acquired using these settings and is corrected for buffer contributions. Thermal melts were acquired at the same protein concentration and using the same instrument and cuvette by monitoring the change in ellipticity at 222 nm as a function of changing temperature between 20 and 90 °C. The scan rate was 1 °C min−1, and the data was acquired with an 8 s response time and 1 °C data pitch. We note that the Mg2+ spectra were acquired after “addback” to the no divalent cation samples to minimize errors associated with differences in protein concentration.
Blood and plasma analyses
Blood glucose was measure using Contour next EZ (Ascensia Diabetes Care). Serum from untreated and LPS treated mice were used for cytokine profiling (MILLIPLEX MAP Mouse Cytokine Magnetic Bead Panel, Millipore Sigma; #MCYTMAG-70K-PX32), BUN (QuantiChrom Urea Assay Kit, BioAssay Systems; #DIUR-100), Creatinine (Diazyme Creatinine Assay, Diazyme, Cat#DZ072B-K).
Leukocyte Infiltration
Lung sections were deparaffinized by 2 changes of xylene for 10 minutes each; slides were then rehydrated in 2 changes of 100% ethanol for 3 minutes, followed by 95% and 70% ethanol for 3 minutes, and rinsed in distilled water. Sections were boiled in sodium citrate buffer (10 mM sodium citrate, pH 6.0) for 20 minutes. Slides were allowed to cool and incubated in 3% hydrogen peroxide. Samples were blocked with avidin (15 min), biotin (15 min) and 10% normal goat serum (1 h). Slides were then incubated with primary alloantigen antibody anti-Ly-6B.2 (1:250 dilution; Biorad #MCA771GA) for 2 hours. Slides were washed 3 times with PBS/Tween 20, followed by incubation with diluted secondary antibodies (biotinylated goat anti-rat-antibody 1:500 dilution; Pierce, 31830) for 1 hour. Slides were incubated with ABC-HRP reagent for 30 min. and stained with DAB (Vectastain ABC-HRP kit, Vector Laboratories, PK-6101). Slides were imaged using a ×40/1.5 NA oil objective in an LSM510 META confocal imaging system. LysM6-B+ leukocyte count was quantified from histologic slide images using ImageJ software.
Quantitative RT-PCR
For quantification of mRNA, RNA was extracted from 0.2–5.0 × 106 splenocytes or ≈20mg frozen lung, kidney, brain, liver, heart, muscle, or small intestine using RNeasy Plus Universal Kits (Qiagen). cDNA was synthesized from the total RNA with the SuperScript™ VILO™ Master Mix (Thermo Fisher) with the following protocol: 25°C for 10 min, 42°C for 60 min, 85°C for 5 min. Transcript expression was measured with the appropriate primers using a QuantStudio 3 Real-Time PCR Instrument (Applied Biosystems™) using TaqMan™ OpenArray™ Real-Time PCR Master Mix as per the manufacturer’s instructions with the following protocol: 50°C for 2 min, 95°C for 2 min, 95°C for 1s and 60°C for 20s for 40 cycles. The ΔΔCt method was used for data analysis of RT-qPCR experiments.
Lactate and glucose measurements
80–200 μL of blood was collected via tail vein of C57BL/6 WT and Mrs2 KO mice and placed into K2 EDTA-coated tubes. Following collection, blood was spun at 10,000×g for 5 min at 4°C and plasma was removed from the top layer. Using a 1:3 dilution with DNAse/RNAse-free distilled water, glucose and lactate measurements were taken using a YSI 2900 Series Biochemistry Analyzer per the manufacturer’s instructions and reported as mg/dL.
To measure extracellular lactate, hepatocytes from WT and Mrs2 KO mice were plated in collagen-coated 6-well plates (BioCoat, Corning) at a density of 1.2 × 106 cells per dish in hepatocyte culture media. Cells were then treated with 100 μg/mL LPS and 20 mM sodium oxamate (Sigma-Aldrich), alone or in combination. 150 μL of media from the cells was collected at the following time points: 0 hours (2 hours post-plating), 8 hours, and 24 hours after the addition of compounds. Extracellular lactate production was then measured by subjecting collected media samples to analysis using a YSI 2900 Series Biochemistry Analyzer per the manufacturer’s instructions and results reported as mg/dL.
Lipid staining in primary hepatocytes
Primary murine hepatocytes isolated from WT and Mrs2 KO mice were grown on 25-mm collagen-coated glass coverslips overnight and loaded with lipid indicator BODIPY® (ex/em 493/503 nm, 1μg/ml) and mitochondrial membrane potential indicator tetramethylrhodamine, ethyl ester (TMRE, ex/em 556/610 nm, 100 nM) in serum-free conditions for 30 min (37°C, 5% CO2) (Tomar et al., 2019). The stained cells were washed and imaged at 24 hours and 48 hours using a Leica SP8 Confocal microscope (Manheim, Germany) coupled with a temperature-controlled environmental chamber. The acquired images were quantified using the Leica Application Suite X.
Assessment of Mrs2 localization
To visualize the ectopic expression of WT Mrs2 and mutant constructs, primary murine hepatocytes were infected with +Ad-Mrs2 WT and +Ad-Mrs2 mutant mRFP (ex/em 584/607 nm). Mitochondrial localization was determined by staining with dihydrorhodamine 123 (2.5 μM) (ex/em 505/524 nm) for 30 min (37°C, 5% CO2. Cells were washed and imaged using Leica SP8 Confocal microscope (Manheim, Germany) coupled with a temperature-controlled environmental chamber. The images were acquired using the Leica Application Suite X software and quantified.
Mitochondrial reactive oxygen species measurement
Primary murine hepatocytes isolated from WT and Mrs2 KO mice were grown on 25-mm collagen-coated glass coverslips overnight and loaded with the mitochondrial superoxide sensitive fluorophore MitoSOX red (ex/em 556/610 nm, 5 μM) for 30 min at 37°C. The stained cells were washed and imaged using a Leica SP8 Confocal microscope (Manheim, Germany) equipped with a temperature-controlled environment chamber. The images were acquired and analyzed using the Leica Application Suite X software (Mukhopadhyay et al., 2007).
Fluorescence imaging and FRET measurements
Primary murine hepatocytes isolated from WT and Mrs2 KO mice were grown on 25-mm collagen-coated glass coverslips were infected with Mito-MagFRET and ER-MagFRET adenoviruses for 48 h. The sub-cellular localization of the mitochondria and ER were visualized by targeted, genetically-encoded MagFRET-1 sensors using a Leica SP8 confocal microscope (Manheim, Germany) in FRET mode. The cells were excited using the 405 nm laser line, and the emission was collected using the hybrid detector HyD. The Cerulean channel, 460–490 nm, and Citrine channel, 510–550 nm, served to detect the emissions from the fluorescence energy transfer (Lindenburg et al., 2013). FRET sensitized emissions were acquired following donor and acceptor sequences. Selected Region of interests (ROIs) were drawn, and the acquired sequences were background corrected for acceptor cross excitation cross-talk, acceptor cross excitation, and FRET cross-talk (α=A/C; γ=B/C; δ=A/B). Time-lapse imaging was performed following evaluation in acquisition mode, and the corresponding FRET efficiencies were calculated. The mitochondrial and ER targeting of the MagFRET sensors were determined with selective region of interests and the corresponding FRET sensitized emissions (Relative FRET SE) were captured. To determine ΔΨm, cells were stained with TMRE (100 nM; ex/em 556/610 nm) for 20 minutes and imaged in a Leica SP8 Confocal microscope.
Splenocyte isolation, Ca2+ imaging, and NFATc3-GFP translocation
Whole spleen was removed from WT and Mrs2KO mice and immediately placed in 37°C RPMI media (supplemented with 10% v/v FBS). Splenocytes were isolated using a two-step lysis process. Briefly, spleens were pressed through a 100 μm cell strainer with 25 mL warm RPMI (without FBS). The cell suspension was centrifuged for five minutes at 80 × g for 5 minutes. Cell pellet was resuspended in ACK lysis buffer and incubated at 25°C temperature for 3 minutes. Lysis reaction was quenched by adding 25 mL warm RPMI supplemented with 10% v/v FBS. Suspension was centrifuged for 5 minutes at 80 × g. Cells were resuspended in RPMI (10% v/v FBS) before seeding.
The splenocytes were plated on a CELL-TAK-coated glass bottom dishes for 60 minutes and cells loaded with cytosolic Ca2+ indicator, Fluo-4-AM for 20 minutes. The splenocytes were stimulated with α-mouse-CD3/α-mouse-CD28 antibodies (2.5 μg/ml) and Ca2+ dynamics were monitored using Leica SP8 Confocal microscope (Ex at 488nm and Em at 510nm).
To assess the NFATc3-GFP translocation, Ad-NFAT-GFP was added to the splenocyte culture at 10 MOI (Gandhirajan et al., 2013). 24 hours later, cells were gently swirled and placed on glass-bottom 35 mm dishes. Cells were allowed to settle for 30 minutes before addition of nuclear marker Hoechst (5μL/mL media) and cells were stimulated with α-mouse-CD3/α-mouse-CD28 antibodies (2.5 μg/ml ) (Biolegend) or media control were added. Cells were imaged 20 minutes after treatment. NFAT3-GFP redistribution was imaged and quantified using Leica SP8 Confocal microscope (Hoechst: Ex at 355nm and Em at 460nm and NFAT-GFP Ex at 488nm and Em at 510nm)and expressed as cytosolic/nuclear intensity.
Immunoblotting
Cell extracts were prepared using RIPA buffer (50mM Tris-HCl, pH 8, 150mM NaCl, 0.5% deoxycholic acid, 1% NP-40, 10% glycerol, protease and phosphatase inhibitor cocktail (ThermoFisher Scientific)). Equal amounts of protein were separated on 4–12% Bis-Tris polyacrylamide gel (Thermo Fisher Scientific), transferred to a PVDF membrane, and probed with corresponding antibodies as specified.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as mean +/− SEM unless otherwise indicated. Statistical significance was evaluated using a Student’s unpaired t-test for one or multiple samples, and one-way ANOVA, with p < 0.5 considered to be statistically significant. All experiments were conducted at least three times unless specified otherwise. Data from cells was obtained using built-in Leica software or ImageJ software and were analyzed and plotted using GraphPad Prism version 8 or Sigma Plot 11.0 software. All other data were analyzed using Microsoft Excel 2013, GraphPad Prism version 8, or Sigma Plot 11.0 software. Charts and figures were created using GraphPad Prism version 8 software, Microsoft Office 365 Word, Microsoft Office 365 Excel, and Canvas 11 software. A p value less than 0.05 was considered statistically significant. Data are presented as the mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001
Supplementary Material
Figure S1. Lactate-induced ER Mg2+ depletion and subsequent mitochondrial Mg2+ uptake in multiple cell types, Related to Figure 1 (A) Representative trace demonstrating changes in fluorescence intensity values of Fluo-4-AM in Hela cells following stimulation with the GPCR agonist histamine.
(B) Bar chart represents basal and peak Fluo-4-AM fluorescence intensity following stimulation with histamine. n=3–4 independent experiments. Mean ± SEM. ****p< 0.0001.
(C) Representative trace demonstrating changes in fluorescence intensity values of Mag-Green in Hela cells following stimulation with the GPCR agonist histamine.
(D). Bar chart represents basal and peak Mag-Green fluorescence intensity following stimulation with histamine. n=3 independent experiments. Mean ± SEM. ****p< 0.0001.
(E-F) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (top) and mitochondria (bottom) after stimulation of HeG2 cells with lactate or glucose (5 and 25 mM) at 25°C (E) or 37°C (F). n= 3–5 independent experiments. Mean ± SEM.*P< 0.05, **p< 0.01, ****p < 0.0001. n.s. = not significant.
(G-H) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (top) and mitochondria (bottom) after stimulation of MEFs (G) or COS7 cells (H) with lactate or glucose (5 and 25 mM) at 37°C. n= 3–4 independent experiments. Mean ± SEM.*p < 0,05, **p < 0.01, ***p < 0.001.
(I) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (blue) and mitochondria (orange) after addition of Alanine (5 mM) or NADH (120 μM) to primary WT hepatocytes. n = 3–4 independent experiments; Mean ± SEM. **p < 0.01, ***p < 0.001.
(J) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (blue) and mitochondria (orange) after addition of L-lacate (5 mM) to primary WT hepatocytes bathed in 7 mM pyruvate. n = 3–4 independent experiments; Mean ± SEM. n.s.=not significant.
(K) Bar chart represents peak Fluo-4-AM fluorescence intensity following stimulation with L-lactate or glucose (5 mM). n=3–4 independent experiments. Mean ± SEM. ***p< 0.001.
Figure S2. MCU-mediated mCa2+ uptake appears Normal at varying temperatures and L-lactate-induced mMg2+ uptake is ΔΨm dependent, Related to Figure 2 (A) Quantification of mitochondrial GCaMP6s signal following ionomycin (2.5 μM) stimulation at 25°C (blue) and 37°C (red). n = 3 independent experiments per condition. Mean ± SEM. n.s. = not significant.
(B-C) Quantification of MCU-mediated mitochondrial Ca2+ levels in permeabilized HepG2 cells at both 25°C (black) and 37°C (red). (B) Representative trace shown, with a 10 μM Ca2+ bolus added to the system at indicated time point followed by FCCP addition to induce matrix Ca2+ release. (C) Analysis of mitochondrial [Ca2+] uptake rate at the two different temperatures. n = 3 independent experiments per condition. Mean ± SEM. n.s. = not significant.
(D) Average of traces of relative changes in Mag-Green intensity of mitochondrial (green) and TMRE (red) after subsequent additions of the mitochondrial uncoupler FCCP (10 μM) and lactate (5 mM). Mean ± SEM. n= 3–4 traces of each fluorescent indicator per cell condition, 20 mitochondria per cell.
(E) Quantification of mitochondrial Mag-Green fluorescence intensity in hepatocytes stimulated with lactate with or without pretreatment with FCCP. n = 4–5 independent experiments. Mean ±SEM; ****p < 0.0001.
(F) Quantification of relative ER Mag-Green fluorescence intensity changes in cells following stimulation with glucose (5 mM), lactate (5 mM), FCCP (10 μM), and FCCP (μM) followed by lactate (5 mM). n= 3–4 independent experiments per condition. Mean ± SEM.*p < 0.05, **p < 0.01, ***p < 0.001, n.s.= not significant.
(G). Hepatocytes were loaded with the intracellular ratiometric pH indicator (BCEC-FAM; 4 μM) for 30 min. Relative changes in fluorescence intensity of BCECF was monitored using fluorescence spectroscope (Ex 440nm and Ex 490nm; and Em 538nM). L-lactate, D-lactate, methyl L-lactate, and 3-Phenyl lactate (5 mM) were added at the indicated time points. n=3–4 independent experiments.
Figure S3. Mrs2 is responsible for lactate-mediated Mg2+ conductance of the mMg2+ uptake machinery, Related to Figure 4 (A) Quantification of relative Mag-Green ER signal changes in WT (red) and Mrs2 KO (blue) hepatocytes following stimulation with varying concentrations of lactate. n = 3–4 independent experiments for each concentration, 20 measurements per cell. Mean ± SEM. n.s. = not significant.
(B) Curves depict mean of relative changes in Mag-Green signal within the mitochondria of WT (red) and Mrs2 KO (black) hepatocytes following stimulation with varying concentrations of lactate. n = 3 –4 independent experiments for each concentration, 20 measurements per cell. Mean ± SEM. p** < 0.01
(C-D) Representative images depicting proper localization and response of mitochondrial Mag-FRET sensor (Mag-FRETmito) in WT (C) and Mrs2 KO (D) hepatocytes. Images demonstrate separation of both cerulean and citrine channels before and after stimulation with lactate. Below each FRET image prior to and immediately following stimulation with lactate are provided for each cell type (see Figure 4 for quantification of mito-FRET signal).
Figure S4. Ectopic expression of Mrs2 enhances mMg2+ uptake, Related to Figure 5 (A-B) WT hepatocytes were transduced with Adv5 Mrs2 WT for 48 hours. Representative traces depicting mitochondrial (A) and ER (B) Mag Green signal after 5 mM lactate stimulation. n=3 independent experiments. Mean ± SEM.
(C) Quantification of mitochondrial Mg2+ uptake and ER depletion as Mag Green signal intensity following lactate stimulation. n=3 independent experiments. Mean ± SEM. *p < 0.05, n.s.= not significant.
Figure S5. Mrs2-mediated Mg2+ uptake into the mitochondria upon lactate stimulation is distinct from MCU channel, Related to Figure 5 (A) Hepatocytes isolated from WT and Mrs2 KO mice were permeabilized and pulsed with Ca2+ (10 μM) followed by FCCP as indicated. Traces show bath [Ca2+]. The clearance of extramitochondrial Ca2+ is indicative of MCU channel activity. Bottom inset shows the similar levels of extramitochondrial Ca2+ clearance. Top left panel depicts MCU uptake rate. n=3 independent experiments. Mean ± SEM. n.s. = not significant.
(B-C) Bar graphs show relative changes in fluorescence intensity of GCaMP6-mt within the mitochondria of WT or Mrs2 KO hepatocytes after ionomycin stimulation (2.5 μM) or 25 mM glucose. n= 3–5 independent experiments. Mean ± SEM. n.s. = not significant.
(D-E) Primary hepatocytes isolated from MCUfl/fl vs MCUΔHep mice were stimulated with 5 mM and 25 mM glucose, or 5 mM lactate. Bar graphs show relative changes in Mag-Green fluorescence within ER and mitochondria. n= 3–5 cells for each condition, 20–30 measurements per cell. n=3 independent experiments. Mean ± SEM. ****p < 0.0001, n.s. = not significant.
Figure S6. Loss of Mrs2 lowers mitochondrial ROS generation and alters hepatic lipid droplet turnover. Intracellular elevation of Mg2+ by ATP depletion elicits mMg2+ uptake, Related to Figures 6 and 7 (A) Representative confocal images show lipid droplet marker BODIPY488 and ΔΨm indicator TMRE 24 and 48 hrs post hepatocyte culture. n=3 independent experiments.
(B) Quantification of lipid droplet number, size, and ΔΨm in WT and Mrs2 KO hepatocytes. Mean ±SEM; n= 3–5 cells per condition, 25 mitochondria and lipid droplets measured per cell. n=3 independent experiments. *p < 0.05, **p < 0.01, n.s. = not significant.
(C) Representative confocal images show MitoSOX Red fluorescence in WT and Mrs2 KO hepatocytes. Pseudo color images of conditions also given. n=3 independent experiments.
(D) Quantification of MitoSOX Red fluorescence intensity in WT and Mrs2 KO hepatocytes. Mean ±SEM; n = 4 cells per condition, 25 mitochondria per cell. ***p < 0.001.
(E) OCR curves depicting HepG2 cells pretreated for 8 hours with either 5 mM Lactate or varying concentrations of LPS vs. control. n= 30,000 cells per well, 4 wells per condition, 3 independent experiments. Bar charts depict maximal and basal respiration, as well as proton leak from above traces. Mean ±SEM; ***p<0.001, ****p<0.0001.
(F) OCR curves depicting HepG2 cells pretreated with various concentrations of MgCl2 for 30 minutes prior to mitochondrial inhibitors challenge. n= 30,000 cells per well, 5 wells per condition, 3 independent experiments. Bar charts depict maximal and basal respiration from above traces. Mean ±SEM; **p<0.01, ***p<0.001, ****p<0.0001.
(G-H) Simultaneous assessment of ΔΨm depolarization and mMg2+ uptake in WT hepatocytes. Hepatocytes were loaded with mitoTracker Red and Mag-Green-AM followed by oligomycin and 2-Deoxy-D-glucose treatment. Oligo+2DG were applied after 100 seconds and time lapse images were acquired every 5s. Representative traces depict MitoTracker Red (top) and Mag-Green (bottom) fluorescence changes.
(I) Quantification of mMg2+ uptake as Mag-Green fluorescence changes in the mitochondria. Mean ±SEM; n=3 independent experiments. *p < 0.05.
(J) Quantitative analysis of ER Mag Green signal depletion in WT hepatocytes pretreated with MCT-1 or MCT-1/2 (10 μM) for 2hours. Hepatocytes were loaded with Mag Green-AM and imaged to quantify ER Mg2+ (top panel). n = 3 independent experiments per group. *p < 0.05, **p < 0.01. (Bottom panel) Measurement of intracellular lactate after MCT inhibitors treatment.
(K) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (blue) and mitochondria (orange) after addition of L-lactate (5 mM) with or without GSK in WT primary hepatocytes. n = 3–4 independent experiments; Mean ± SEM. n.s. = not significant.
Figure S7. Assessment of PDH Phosphorylation Status and HIF1-α Stabilization Upon LPS Stimulation and Preservation of T-lymphocyte and NFAT Transcription Factor Activation, Related to Figures 6 and 7 WT and Mrs2 KO hepatocytes were treated with LPS (1, 10 and 100 μg/ml) for 6 hours and immunoblotted for p-PDH E1α (S232 and S300), PDH E1α, and β-actin as a loading control. Densitometric analysis shown below. n=3 independent experiments. Mean ±SEM; **p<0.01, ***p<0.001, n.s. = not significant.
(B) WT and Mrs2 KO hepatocytes were treated with LPS (1, 10 and 100 μg/ml) for 6 hours and immunoblotted for HIF1-α and β-actin as a loading control. CoCl2 treatment was used as a positive control. Densitometric analysis shown below. n=3 independent experiments. Mean ±SEM; **p<0.01, ***p<0.001.
(C) WT and Mrs2 KO splenocytes were plated on the glass bottom dishes and loaded with cytosolic Ca2+ indicator Fluo-4-AM (5μM) for 20 min. Cells were stimulated with anti-CD3/CD28 (2.5 μg/ml) and Ca2+ dynamics were monitored using confocal imaging system.
(D-E) Representative mean traces show cytosolic Ca2+ dynamics. Quantification of peak amplitude. Mean ±SEM; n.s. = not significant.
(F-G) WT and Mrs2 KO splenocytes were transduced with Ad5-NFATc3-GFP and cultured for 24 hours. Cells were stimulated with anti-CD3/CD28 (2.5 μg/ml) for 20 minutes. NFAT3-GFP redistribution was quantified and expressed as cytosolic/nuclear intensity. n=3 independent experiments. Mean ±SEM. n.s. = not significant.
(H) Measurement of intracellular lactate in WT primary hepatocytes after challenge with LPS. Hepatocytes were stimulated with LPS (100 μg/ml) for 24 hours with or without GSK or oxamate pretreatment. Mean ±SEM; *p<0.05, **p<0.01.
Highlights:
L-lactate triggers ER Mg2+ release that promotes mitochondrial Mg2+ uptake
Mg2+ is a second messenger for metabolic circuits
Limiting Mrs2-mediated Mg2+ uptake enhances mitochondrial bioenergetics
Inflammation-induced lactate contributes to organ failure via mMg2+ surge
ACKNOWLEDGEMENTS:
We thank Maarten Merkx for sharing the pCMV-MagFRET-1, Jean Ross for TEM sample preparation and imaging, and Dr. Vernon Anderson for his constant support and encouragement to the M.M. lab. This research was funded by the National Institutes of Health (R01GM109882, R01HL086699, R01HL142673, R01GM135760, and 1S10RR027327) to M.M. This work was partly supported by DOD/DHP-CDMRP PR181598P-1 and San Antonio Partnership for Precision Therapeutics (SAPPT) to M.M. and NSERC-07171 to P.B.S. B.T.E. is supported by the NIH (TL1 TR002647 and K12 GM111726).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.M. is an inventor on a patent provisionally filed by UTHSA on lactate as a ligand for Mg2+ dynamics in physiology and disease. All other authors have no financial interests to declare.
REFERENCES
- Berridge MJ, Bootman MD, and Roderick HL (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4, 517–529. [DOI] [PubMed] [Google Scholar]
- Brooks GA (2018). The Science and Translation of Lactate Shuttle Theory. Cell Metab 27, 757–785. [DOI] [PubMed] [Google Scholar]
- Bui DM, Gregan J, Jarosch E, Ragnini A, and Schweyen RJ (1999). The bacterial magnesium transporter CorA can functionally substitute for its putative homologue Mrs2p in the yeast inner mitochondrial membrane. J Biol Chem 274, 20438–20443. [DOI] [PubMed] [Google Scholar]
- Cai TQ, Ren N, Jin L, Cheng K, Kash S, Chen R, Wright SD, Taggart AK, and Waters MG (2008). Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem Biophys Res Commun 377, 987–991. [DOI] [PubMed] [Google Scholar]
- Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, Shatzer A, Biancalana M, Pittaluga S, Matthews HF, et al. (2013). Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 341, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2007). Calcium signaling. Cell 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Corkey BE, Duszynski J, Rich TL, Matschinsky B, and Williamson JR (1986). Regulation of free and bound magnesium in rat hepatocytes and isolated mitochondria. J Biol Chem 261, 2567–2574. [PubMed] [Google Scholar]
- de Baaij JH, Hoenderop JG, and Bindels RJ (2015). Magnesium in man: implications for health and disease. Physiol Rev 95, 1–46. [DOI] [PubMed] [Google Scholar]
- Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, et al. (2017). Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol Cell 65, 1014–1028 e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshaghi S, Niegowski D, Kohl A, Martinez Molina D, Lesley SA, and Nordlund P (2006). Crystal structure of a divalent metal ion transporter CorA at 2.9 angstrom resolution. Science 313, 354–357. [DOI] [PubMed] [Google Scholar]
- Fatholahi M, LaNoue K, Romani A, and Scarpa A (2000). Relationship between total and free cellular Mg(2+) during metabolic stimulation of rat cardiac myocytes and perfused hearts. Arch Biochem Biophys 374, 395–401. [DOI] [PubMed] [Google Scholar]
- Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, et al. (2013). Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest 123, 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, and Balaban RS (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, and Mauro C (2016). Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem Sci 41, 460–471. [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2013). Monocarboxylic acid transport. Compr Physiol 3, 1611–1643. [DOI] [PubMed] [Google Scholar]
- Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, et al. (2011). Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol Cell Biol 31, 3745–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D (2013). TRP channels and pain. Annu Rev Cell Dev Biol 29, 355–384. [DOI] [PubMed] [Google Scholar]
- Khan MB, Sponder G, Sjoblom B, Svidova S, Schweyen RJ, Carugo O, and Djinovic-Carugo K (2013). Structural and functional characterization of the N-terminal domain of the yeast Mg2+ channel Mrs2. Acta Crystallogr D Biol Crystallogr 69, 1653–1664. [DOI] [PubMed] [Google Scholar]
- Kolisek M, Sponder G, Mastrototaro L, Smorodchenko A, Launay P, Vormann J, and Schweigel-Rontgen M (2013). Substitution p.A350V in Na(+)/Mg(2)(+) exchanger SLC41A1, potentially associated with Parkinson’s disease, is a gain-of-function mutation. PLoS One 8, e71096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolisek M, Zsurka G, Samaj J, Weghuber J, Schweyen RJ, and Schweigel M (2003). Mrs2p is an essential component of the major electrophoretic Mg2+ influx system in mitochondria. EMBO J 22, 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto T, Kuwamura M, Tokuda S, Izawa T, Nakane Y, Kitada K, Akao M, Guenet JL, and Serikawa T (2011). A mutation in the gene encoding mitochondrial Mg(2)+ channel MRS2 results in demyelination in the rat. PLoS Genet 7, e1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, Chen B, Carin L, Suarez A, Mohney RP, et al. (2013). An integrated clinico-metabolomic model improves prediction of death in sepsis. Science translational medicine 5, 195ra195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DK, Nguyen T, Lynch KR, Cheng R, Vanti WB, Arkhitko O, Lewis T, Evans JF, George SR, and O’Dowd BF (2001). Discovery and mapping of ten novel G protein-coupled receptor genes. Gene 275, 83–91. [DOI] [PubMed] [Google Scholar]
- Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, Cohen JI, Uzel G, Su HC, and Lenardo MJ (2011). Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WC, Ralphs KL, and Tosh D (2010). Isolation and culture of adult mouse hepatocytes. Methods Mol Biol 633, 185–196. [DOI] [PubMed] [Google Scholar]
- Lindenburg LH, Vinkenborg JL, Oortwijn J, Aper SJ, and Merkx M (2013). MagFRET: the first genetically encoded fluorescent Mg2+ sensor. PLoS One 8, e82009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunin VV, Dobrovetsky E, Khutoreskaya G, Zhang R, Joachimiak A, Doyle DA, Bochkarev A, Maguire ME, Edwards AM, and Koth CM (2006). Crystal structure of the CorA Mg2+ transporter. Nature 440, 833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, et al. (2012a). MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14, 1336–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, et al. (2012b). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies D, Dalmas O, Borgnia MJ, Dominik PK, Merk A, Rao P, Reddy BG, Islam S, Bartesaghi A, Perozo E, et al. (2016). Cryo-EM Structures of the Magnesium Channel CorA Reveal Symmetry Break upon Gating. Cell 164, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley PJ, Rutter GA, Thomas AP, and Denton RM (1987). Effects of Ca2+ and Mg2+ on the activity of pyruvate dehydrogenase phosphate phosphatase within toluene-permeabilized mitochondria. Biochem J 241, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KS, Andahazy M, Story GM, and Patapoutian A (2005). Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 307, 1468–1472. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, and Pacher P (2007). Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc 2, 2295–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E (2000). Mysteries of magnesium homeostasis. Circ Res 86, 245–248. [DOI] [PubMed] [Google Scholar]
- Nemani N, Carvalho E, Tomar D, Dong Z, Ketschek A, Breves SL, Jana F, Worth AM, Heffler J, Palaniappan P, et al. (2018). MIRO-1 Determines Mitochondrial Shape Transition upon GPCR Activation and Ca(2+) Stress. Cell reports 23, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Pfoh R, and Pai EF (2013). The structure and regulation of magnesium selective ion channels. Biochim Biophys Acta 1828, 2778–2792. [DOI] [PubMed] [Google Scholar]
- Pfoh R, Li A, Chakrabarti N, Payandeh J, Pomes R, and Pai EF (2012). Structural asymmetry in the magnesium channel CorA points to sequential allosteric regulation. Proc Natl Acad Sci U S A 109, 18809–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani A, and Scarpa A (1990). Hormonal control of Mg2+ transport in the heart. Nature 346, 841–844. [DOI] [PubMed] [Google Scholar]
- Romani AM (2011). Cellular magnesium homeostasis. Arch Biochem Biophys 512, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin H (1975). Central role for magnesium in coordinate control of metabolism and growth in animal cells. Proc Natl Acad Sci U S A 72, 3551–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindl R, Weghuber J, Romanin C, and Schweyen RJ (2007). Mrs2p forms a high conductance Mg2+ selective channel in mitochondria. Biophys J 93, 3872–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, et al. (2016). MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell reports 15, 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar D, Jana F, Dong Z, Quinn WJ 3rd, Jadiya P, Breves SL, Daw CC, Srikantan S, Shanmughapriya S, Nemani N, et al. (2019). Blockade of MCU-Mediated Ca(2+) Uptake Perturbs Lipid Metabolism via PP4-Dependent AMPK Dephosphorylation. Cell reports 26, 3709–3725 e3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T (2014). TRP channels and thermosensation. Handb Exp Pharmacol 223, 729–741. [DOI] [PubMed] [Google Scholar]
- Wallace DC (2012). Mitochondria and cancer. Nat Rev Cancer 12, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, Ge P, Lilly J, Silos-Santiago I, Xie Y, et al. (2002). TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 418, 181–186. [DOI] [PubMed] [Google Scholar]
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019a). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, Chang Y, Chen Y, Lu Y, Zeng H, et al. (2019b). Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 178, 176–189 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, and Clapham DE (2009). Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci U S A 106, 15750–15755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Lactate-induced ER Mg2+ depletion and subsequent mitochondrial Mg2+ uptake in multiple cell types, Related to Figure 1 (A) Representative trace demonstrating changes in fluorescence intensity values of Fluo-4-AM in Hela cells following stimulation with the GPCR agonist histamine.
(B) Bar chart represents basal and peak Fluo-4-AM fluorescence intensity following stimulation with histamine. n=3–4 independent experiments. Mean ± SEM. ****p< 0.0001.
(C) Representative trace demonstrating changes in fluorescence intensity values of Mag-Green in Hela cells following stimulation with the GPCR agonist histamine.
(D). Bar chart represents basal and peak Mag-Green fluorescence intensity following stimulation with histamine. n=3 independent experiments. Mean ± SEM. ****p< 0.0001.
(E-F) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (top) and mitochondria (bottom) after stimulation of HeG2 cells with lactate or glucose (5 and 25 mM) at 25°C (E) or 37°C (F). n= 3–5 independent experiments. Mean ± SEM.*P< 0.05, **p< 0.01, ****p < 0.0001. n.s. = not significant.
(G-H) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (top) and mitochondria (bottom) after stimulation of MEFs (G) or COS7 cells (H) with lactate or glucose (5 and 25 mM) at 37°C. n= 3–4 independent experiments. Mean ± SEM.*p < 0,05, **p < 0.01, ***p < 0.001.
(I) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (blue) and mitochondria (orange) after addition of Alanine (5 mM) or NADH (120 μM) to primary WT hepatocytes. n = 3–4 independent experiments; Mean ± SEM. **p < 0.01, ***p < 0.001.
(J) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (blue) and mitochondria (orange) after addition of L-lacate (5 mM) to primary WT hepatocytes bathed in 7 mM pyruvate. n = 3–4 independent experiments; Mean ± SEM. n.s.=not significant.
(K) Bar chart represents peak Fluo-4-AM fluorescence intensity following stimulation with L-lactate or glucose (5 mM). n=3–4 independent experiments. Mean ± SEM. ***p< 0.001.
Figure S2. MCU-mediated mCa2+ uptake appears Normal at varying temperatures and L-lactate-induced mMg2+ uptake is ΔΨm dependent, Related to Figure 2 (A) Quantification of mitochondrial GCaMP6s signal following ionomycin (2.5 μM) stimulation at 25°C (blue) and 37°C (red). n = 3 independent experiments per condition. Mean ± SEM. n.s. = not significant.
(B-C) Quantification of MCU-mediated mitochondrial Ca2+ levels in permeabilized HepG2 cells at both 25°C (black) and 37°C (red). (B) Representative trace shown, with a 10 μM Ca2+ bolus added to the system at indicated time point followed by FCCP addition to induce matrix Ca2+ release. (C) Analysis of mitochondrial [Ca2+] uptake rate at the two different temperatures. n = 3 independent experiments per condition. Mean ± SEM. n.s. = not significant.
(D) Average of traces of relative changes in Mag-Green intensity of mitochondrial (green) and TMRE (red) after subsequent additions of the mitochondrial uncoupler FCCP (10 μM) and lactate (5 mM). Mean ± SEM. n= 3–4 traces of each fluorescent indicator per cell condition, 20 mitochondria per cell.
(E) Quantification of mitochondrial Mag-Green fluorescence intensity in hepatocytes stimulated with lactate with or without pretreatment with FCCP. n = 4–5 independent experiments. Mean ±SEM; ****p < 0.0001.
(F) Quantification of relative ER Mag-Green fluorescence intensity changes in cells following stimulation with glucose (5 mM), lactate (5 mM), FCCP (10 μM), and FCCP (μM) followed by lactate (5 mM). n= 3–4 independent experiments per condition. Mean ± SEM.*p < 0.05, **p < 0.01, ***p < 0.001, n.s.= not significant.
(G). Hepatocytes were loaded with the intracellular ratiometric pH indicator (BCEC-FAM; 4 μM) for 30 min. Relative changes in fluorescence intensity of BCECF was monitored using fluorescence spectroscope (Ex 440nm and Ex 490nm; and Em 538nM). L-lactate, D-lactate, methyl L-lactate, and 3-Phenyl lactate (5 mM) were added at the indicated time points. n=3–4 independent experiments.
Figure S3. Mrs2 is responsible for lactate-mediated Mg2+ conductance of the mMg2+ uptake machinery, Related to Figure 4 (A) Quantification of relative Mag-Green ER signal changes in WT (red) and Mrs2 KO (blue) hepatocytes following stimulation with varying concentrations of lactate. n = 3–4 independent experiments for each concentration, 20 measurements per cell. Mean ± SEM. n.s. = not significant.
(B) Curves depict mean of relative changes in Mag-Green signal within the mitochondria of WT (red) and Mrs2 KO (black) hepatocytes following stimulation with varying concentrations of lactate. n = 3 –4 independent experiments for each concentration, 20 measurements per cell. Mean ± SEM. p** < 0.01
(C-D) Representative images depicting proper localization and response of mitochondrial Mag-FRET sensor (Mag-FRETmito) in WT (C) and Mrs2 KO (D) hepatocytes. Images demonstrate separation of both cerulean and citrine channels before and after stimulation with lactate. Below each FRET image prior to and immediately following stimulation with lactate are provided for each cell type (see Figure 4 for quantification of mito-FRET signal).
Figure S4. Ectopic expression of Mrs2 enhances mMg2+ uptake, Related to Figure 5 (A-B) WT hepatocytes were transduced with Adv5 Mrs2 WT for 48 hours. Representative traces depicting mitochondrial (A) and ER (B) Mag Green signal after 5 mM lactate stimulation. n=3 independent experiments. Mean ± SEM.
(C) Quantification of mitochondrial Mg2+ uptake and ER depletion as Mag Green signal intensity following lactate stimulation. n=3 independent experiments. Mean ± SEM. *p < 0.05, n.s.= not significant.
Figure S5. Mrs2-mediated Mg2+ uptake into the mitochondria upon lactate stimulation is distinct from MCU channel, Related to Figure 5 (A) Hepatocytes isolated from WT and Mrs2 KO mice were permeabilized and pulsed with Ca2+ (10 μM) followed by FCCP as indicated. Traces show bath [Ca2+]. The clearance of extramitochondrial Ca2+ is indicative of MCU channel activity. Bottom inset shows the similar levels of extramitochondrial Ca2+ clearance. Top left panel depicts MCU uptake rate. n=3 independent experiments. Mean ± SEM. n.s. = not significant.
(B-C) Bar graphs show relative changes in fluorescence intensity of GCaMP6-mt within the mitochondria of WT or Mrs2 KO hepatocytes after ionomycin stimulation (2.5 μM) or 25 mM glucose. n= 3–5 independent experiments. Mean ± SEM. n.s. = not significant.
(D-E) Primary hepatocytes isolated from MCUfl/fl vs MCUΔHep mice were stimulated with 5 mM and 25 mM glucose, or 5 mM lactate. Bar graphs show relative changes in Mag-Green fluorescence within ER and mitochondria. n= 3–5 cells for each condition, 20–30 measurements per cell. n=3 independent experiments. Mean ± SEM. ****p < 0.0001, n.s. = not significant.
Figure S6. Loss of Mrs2 lowers mitochondrial ROS generation and alters hepatic lipid droplet turnover. Intracellular elevation of Mg2+ by ATP depletion elicits mMg2+ uptake, Related to Figures 6 and 7 (A) Representative confocal images show lipid droplet marker BODIPY488 and ΔΨm indicator TMRE 24 and 48 hrs post hepatocyte culture. n=3 independent experiments.
(B) Quantification of lipid droplet number, size, and ΔΨm in WT and Mrs2 KO hepatocytes. Mean ±SEM; n= 3–5 cells per condition, 25 mitochondria and lipid droplets measured per cell. n=3 independent experiments. *p < 0.05, **p < 0.01, n.s. = not significant.
(C) Representative confocal images show MitoSOX Red fluorescence in WT and Mrs2 KO hepatocytes. Pseudo color images of conditions also given. n=3 independent experiments.
(D) Quantification of MitoSOX Red fluorescence intensity in WT and Mrs2 KO hepatocytes. Mean ±SEM; n = 4 cells per condition, 25 mitochondria per cell. ***p < 0.001.
(E) OCR curves depicting HepG2 cells pretreated for 8 hours with either 5 mM Lactate or varying concentrations of LPS vs. control. n= 30,000 cells per well, 4 wells per condition, 3 independent experiments. Bar charts depict maximal and basal respiration, as well as proton leak from above traces. Mean ±SEM; ***p<0.001, ****p<0.0001.
(F) OCR curves depicting HepG2 cells pretreated with various concentrations of MgCl2 for 30 minutes prior to mitochondrial inhibitors challenge. n= 30,000 cells per well, 5 wells per condition, 3 independent experiments. Bar charts depict maximal and basal respiration from above traces. Mean ±SEM; **p<0.01, ***p<0.001, ****p<0.0001.
(G-H) Simultaneous assessment of ΔΨm depolarization and mMg2+ uptake in WT hepatocytes. Hepatocytes were loaded with mitoTracker Red and Mag-Green-AM followed by oligomycin and 2-Deoxy-D-glucose treatment. Oligo+2DG were applied after 100 seconds and time lapse images were acquired every 5s. Representative traces depict MitoTracker Red (top) and Mag-Green (bottom) fluorescence changes.
(I) Quantification of mMg2+ uptake as Mag-Green fluorescence changes in the mitochondria. Mean ±SEM; n=3 independent experiments. *p < 0.05.
(J) Quantitative analysis of ER Mag Green signal depletion in WT hepatocytes pretreated with MCT-1 or MCT-1/2 (10 μM) for 2hours. Hepatocytes were loaded with Mag Green-AM and imaged to quantify ER Mg2+ (top panel). n = 3 independent experiments per group. *p < 0.05, **p < 0.01. (Bottom panel) Measurement of intracellular lactate after MCT inhibitors treatment.
(K) Bar chart showing quantitative analysis of relative changes in Mag-Green fluorescence intensity in ER (blue) and mitochondria (orange) after addition of L-lactate (5 mM) with or without GSK in WT primary hepatocytes. n = 3–4 independent experiments; Mean ± SEM. n.s. = not significant.
Figure S7. Assessment of PDH Phosphorylation Status and HIF1-α Stabilization Upon LPS Stimulation and Preservation of T-lymphocyte and NFAT Transcription Factor Activation, Related to Figures 6 and 7 WT and Mrs2 KO hepatocytes were treated with LPS (1, 10 and 100 μg/ml) for 6 hours and immunoblotted for p-PDH E1α (S232 and S300), PDH E1α, and β-actin as a loading control. Densitometric analysis shown below. n=3 independent experiments. Mean ±SEM; **p<0.01, ***p<0.001, n.s. = not significant.
(B) WT and Mrs2 KO hepatocytes were treated with LPS (1, 10 and 100 μg/ml) for 6 hours and immunoblotted for HIF1-α and β-actin as a loading control. CoCl2 treatment was used as a positive control. Densitometric analysis shown below. n=3 independent experiments. Mean ±SEM; **p<0.01, ***p<0.001.
(C) WT and Mrs2 KO splenocytes were plated on the glass bottom dishes and loaded with cytosolic Ca2+ indicator Fluo-4-AM (5μM) for 20 min. Cells were stimulated with anti-CD3/CD28 (2.5 μg/ml) and Ca2+ dynamics were monitored using confocal imaging system.
(D-E) Representative mean traces show cytosolic Ca2+ dynamics. Quantification of peak amplitude. Mean ±SEM; n.s. = not significant.
(F-G) WT and Mrs2 KO splenocytes were transduced with Ad5-NFATc3-GFP and cultured for 24 hours. Cells were stimulated with anti-CD3/CD28 (2.5 μg/ml) for 20 minutes. NFAT3-GFP redistribution was quantified and expressed as cytosolic/nuclear intensity. n=3 independent experiments. Mean ±SEM. n.s. = not significant.
(H) Measurement of intracellular lactate in WT primary hepatocytes after challenge with LPS. Hepatocytes were stimulated with LPS (100 μg/ml) for 24 hours with or without GSK or oxamate pretreatment. Mean ±SEM; *p<0.05, **p<0.01.
Data Availability Statement
This study did not generate any datasets/code amenable for depositing into public repositories.