

Abstract
Background and aims:
Coronary artery disease (CAD) arises from the interaction of genetic and environmental factors. Although genome-wide association studies (GWAS) have identified multiple risk loci and single nucleotide polymorphisms (SNPs) associated with risk of CAD, they are predominantly located in non-coding or intergenic regions and their mechanisms of effect are largely unknown. Accordingly, our objective was to develop a data-driven informatics pipeline to understand complex CAD risk loci, and to apply this to a poorly understood cluster of SNPs in the vicinity of ZEB2.
Methods:
We developed a unique informatics pipeline leveraging a multi-tissue CAD genetics-of-gene-expression dataset, GWAS datasets, and other resources. The pipeline first dissected SNP locations and their linkage disequilibrium relationships, and progressed through analyses of tissue-specific expression quantitative trait loci, and then gene-gene, gene-phenotype, SNP-phenotype relationships. The pipeline concluded by exploring CAD-relevant gene regulatory networks (GRNs).
Results:
We identified three independent CAD risk SNPs in close proximity to the ZEB2 coding region (rs6740731, rs17678683 and rs2252641/rs1830321). Our pipeline determined that these SNPs likely act in concert via the atherosclerotic arterial wall and adipose tissues, by governing metabolic and lipid functions. In addition, ZEB2 is the top key driver of a liver-specific GRN that is related to lipid levels, metabolic and anthropometric measures, and CAD severity.
Conclusions:
Using a novel informatics pipeline, we disclosed the multi-faceted mechanisms of action of the ZEB2-associated CAD risk SNPs. This pipeline can serve as a roadmap to dissect complex SNP-gene-tissue-phenotype relationships and to reveal targets for tissue- and gene-specific therapeutic interventions.
Graphical Abstract
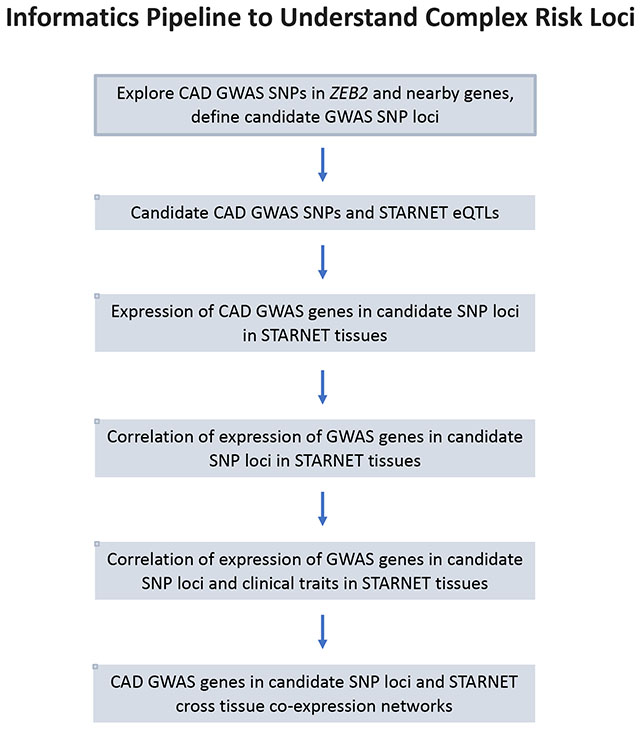
INTRODUCTION
The pathology of coronary artery disease (CAD) is driven by an interaction between genetic and environmental factors (1, 2). As a disease with complex traits, it is understood that multiple loci, each imparting a small-modest effect, contribute to the polygenic nature of CAD, and accordingly multiple risk loci for the disease have been identified by genome wide association studies (GWAS) (3, 4). However, the CAD susceptibility loci identified by GWAS are predominantly located in non-coding or intergenic regions and their effects on genes or CAD are largely unknown (3–5).
As an important challenge, it is increasingly clear that there is substantial and under-appreciated complexity underlying the causality of CAD (3). For example, our group recently showed that a previously unrecognized mechanism accounts for a significant proportion of the heritability of CAD, namely, the heritability carried in gene regulatory networks (GRNs) (2). As another dimension adding to the complexity of CAD, at each genetic locus that is associated with CAD, there are often multiple single nucleotide polymorphisms (SNPs) that are of potential importance (6). In some instances, this may be because there is a single causal SNP and other SNPs in that vicinity have merely been identified due to their being in linkage disequilibrium (LD), while in reality these additional SNPs are not causal. However, other instances exist where there appear to be multiple, independent SNPs arising in close vicinity of each other. One specific example of this latter phenomenon exists in a cluster of SNPs in close proximity to the coding region for Zinc Finger E-Box Binding Homeobox 2 (ZEB2), with various GWAS repeatedly identifying and replicating differing SNPs in this region that are related to the risk of CAD. It is also noteworthy that the first description of a SNP in the vicinity of ZEB2 that is related to the risk of CAD appeared in 2013 (7), however, very few insights have arisen regarding the potential mechanism(s) of association of these SNPs or ZEB2 with CAD since that time.
To address these issues leveraged the recently curated Stockholm-Tartu Atherosclerosis Reverse Networks Engineering Task (STARNET) study datasets (8), and other informatics resources, to develop a data-driven pipeline to gain mechanistic insights on complex CAD genetic loci, which we applied to a cluster of SNPs in the vicinity of the coding region for ZEB2 that are associated with risk of CAD. This revealed significant complexity and interaction among these SNPs, with evidence for independent disease-promoting effects of ZEB2 in at least two separate CAD-relevant tissues, as well as a key driver role whereby ZEB2 governs an important GRN that is related to CAD.
MATERIALS AND METHODS
Previously identified SNPs associated with CAD in the vicinity of ZEB2
Known GWAS SNPs associated with CAD in the ZEB2 gene and its vicinity were obtained from published reports (6, 7, 9–13) and public databases, including the GWAS catalog (14) and PhenoScanner (15). These GWAS SNPs were plotted and visualized in LocusZoom (Figure 1A) (16). Stepwise regression was calculated in R (3.6.0) to identify independent SNP signals (17) and a q-q plot of these results is presented Figure 1C.
Figure 1.
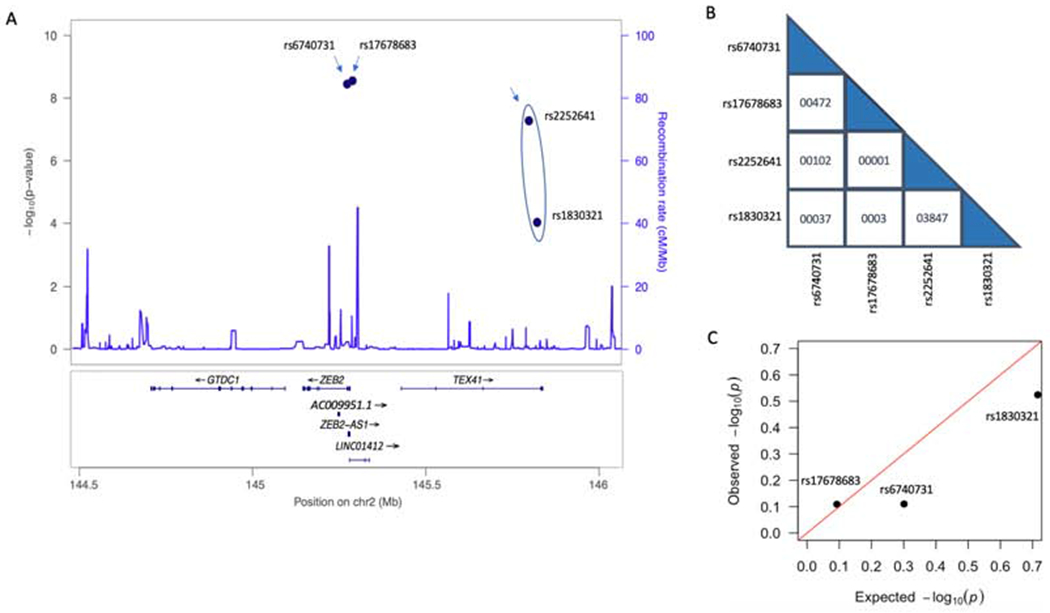
Known ZEB2-associated GWAS SNPs related to risk of CAD.
(A) Locus zoom plot of the four known GWAS SNPs related to the risk of CAD in the vicinity of ZEB2: rs6470731, rs17678683, rs2252641 and rs1830321. Independent SNPs are indicated by arrows, with the circle around rs2252641 and rs1830321 signifying that they are in low LD. (B) Linkage disequilibrium (LD) score between each of the four SNPs with respect to the other SNPs. (C) Stepwise regression of ZEB2 expression in AOR and the SNP genotypes for rs17678683, rs6740731 and rs1830321 in STARNET showing that these SNPs are independent.
Study description, tissue collection
Subject recruitment and tissue collection in STARNET were performed as previously described (8). Briefly, patients with CAD who were eligible for open-thorax surgery at the Department of Cardiac Surgery, Tartu University Hospital in Estonia as well as control subjects without CAD were enrolled into this approved protocol and after informed consent. From each STARNET subject, venous blood (BLOOD) as well as biopsies from atherosclerotic aortic wall (AOR), non-atherosclerotic mammary artery (MAM), liver (LIV), skeletal muscle (SKLM), subcutaneous fat (SF) and visceral fat (VAF) were obtained and RNA was extracted as described (8). BLOOD was also used to obtain DNA which was isolated for genotyping using the Illumina Infinium assay. Additional details are available in the original manuscript (8).
RNA sequencing
RNA sequencing for STARNET cases in different tissues was performed as described (8). The Ribo-Zero library preparation method was used for all AOR and MAM samples, and for some LIV, SKLM, SF and VAF samples due to lower total amounts of RNA, while all BLOOD and most of LIV, SF, VAF and SKLM samples were sequenced by poly(A)+ selection. Samples were sequenced on an Illumina HiSeq with single-end read lengths of 50 or 100 base pairs. In the case-control matched study cohorts, cases and controls with matched age, gender and BMI were selected from AOR, LIV, SKLM, SF and VAF tissues for RNAseq. Samples were sequenced with poly(A)+ selection on Illumina HiSeq with single-end at read lengths of 100 base pairs. STARNET RNAseq datasets have been previously described, including the number of samples passing quality control (8).
Quality control and differential gene expression
Quality control was performed using FASTQC (18) that checks raw sequence data for per-base quality, per-sequence quality, number of duplicate reads, number of reads with an adaptor, sequence length distribution, per-base GC content, per-sequence GC content and Kmer content. GENCODE v.19 was used as reference annotation to quantify gene and isoform expression. Sequencing reads (fastq files) were mapped with STAR (19) onto the human genome. Raw reads were summarized by feature counts (20). Samples with less than 1,000,000 uniquely mapped reads were discarded. Low counts were removed by keeping genes where the count per million (cpm) is greater than 1 in at least two samples. For differential gene expression analysis, cases and controls were differentiated by Duke CAD index and Syntax scores. The number of subjects for each tissue was: AOR 102 versus 79; LIV 154 versus 100; SF 131 versus 91, SKLM 168 versus 115; VAF 135 versus 102 (CAD cases versus controls, respectively). Covariates (age, gender, BMI and batch) were not added to the linear module for adjustment since all 1177 samples were randomized before sequencing to remove batch effects, while cases and controls in each tissue were selected with matched age, gender and BMI. Differential gene expression between cases and controls was analyzed using R package limma (21). Statistical significance was defined as adjusted p < 0.05.
Co-expression module, network and key driver analysis
R package Weighted Gene Co-expression Network Analysis (WGCNA) (22) was used to identify correlation patterns among genes across RNA seq data. Cross-tissue co-expression networks were constructed based upon 30,716 genes from seven tissues. Unsigned, weighted correlation network construction and module detection were performed using default parameters. Genes with similar expression patterns were assigned into the same module. The correlations of differing modules with clinical traits were assessed using Pearson’s correlation. Regulatory networks were reconstructed by GENIE3, an algorithm inferring gene regulatory network from expression data based on feature selection with tree-based ensemble methods, by using transcription factor and eQTL regulated genes as regulators (23). Key driver analyses were performed using Mergeomics (24). Protein-protein interaction networks for genes in co-expression modules were built using String software (25). An edge was defined by seven types of interaction evidence, including gene fusions, gene co-occurrence, gene neighborhood, co-expression, protein homology, experimentally determined, text mining and curated database. The minimum required interaction score was 0.5.
Array-based genotyping, imputation and expression quantitative trait loci analysis
DNA genotyping was performed using the Illumina Infinium assay with the human OmniExpressExome-8v1 bead chip. Data was analyzed using GenomeStudio 2011.1 (Illumina) which produced 951,117 genomic markers (genome build 37) (8). Quality control was performed using PLINK v.1.07 and IMPUTE2 v.2.3.0 was used for genotype imputation to increase the power of analysis (13). Of the four SNPs in the vicinity of ZEB2 studied here in detail (Figure 1), three of the four being rs2252641, rs1830321 and rs6740731, were directly genotyped by the human OmniExpressExome-8v1 bead chip. Genotyping for rs17678683 was obtained by imputation. Genotyping for all four SNPs passed quality control. In each tissue, cis and trans- regulated expression quantitative trait loci (eQTLs) were identified with the R package Matrix eQTL v.2.1.1 (8).
Association of GWAS SNPs and gene expression levels with clinical phenotypes
STARNET phenotypic and demographic data were used to explore relationships with our SNPs and genes of interest. These data include indices of CAD severity: Duke CAD index (26), Syntax score (27), number of coronary lesions per patient (Lesions) and number of diseased coronary vessels per patient (Diseased vessels). Other data included body mass index (BMI) and waist to hip ratio (Waist:Hip); and the following parameters from peripheral blood: hemoglobin A1C concentration (HbA1c), c-reactive protein concentration (CRP), white blood cell concentration (WBC), hemoglobin concentration (HbG), platelet concentration (PLT), aspartate aminotransferase concentration (AST), alanine aminotransferase concentration (ALT), cholesterol concentration (Chol), low-density lipoprotein cholesterol concentration (LDL), high-density lipoprotein cholesterol concentration (HDL), triglyceride concentration (TG).
Pearson correlation coefficient in R (3.6.0) was used to measure the strength of linear correlation between expression of the same gene in different tissues, of different genes in the same tissue, as well as the correlation between gene expression and clinical phenotypes. The association of the expression of different alleles in GWAS SNPs with clinical phenotypes were analyzed using STARNET data and computed by one-way ANOVA in R (3.6.0). P value was adjusted by Bonferroni correction for multiple comparisons and an adjusted p < 0.05 was considered statistically significant.
RESULTS
We applied a data-driven approach to study a cluster of SNPs in close proximity to the coding region for ZEB2 that are related to risk of CAD. We first sought to understand the linkage relationships of these SNPs, and then used gene expression data to identify expression quantitative trait loci (eQTLs) at these SNPs to further determine their relative independence and the likely tissues in which their CAD-causal effects are operative. We also undertook various functional analyses of gene expression data, including exploring GRNs where ZEB2 is operative and appears causal for CAD (Graphical Abstract).
Four different SNPs in the region of ZEB2 are linked to risk of CAD
There are four SNPs in close proximity to ZEB2 on chromosome 2 (2q22.3) that have been identified by GWAS and which are related to risk of CAD (Figure 1A, Table 1).
Table 1.
Known ZEB2-associated GWAS SNPs that are related to the risk of CAD.
| SNP ID | Location on chromosome 2 | Reference allele | CAD risk allele | Allele frequency | Gene name | Function | Nearby gene(s) | Beta of risk allele | GWAS p value* | References |
|---|---|---|---|---|---|---|---|---|---|---|
| rs2252641 | 145043894 | T | C | T=0.307; C=0.693 | TEX41 | intron | LINC01966, AC096666.1 | −0.0368 | 5.00× 10−13 | (6, 7, 14, 15) |
| rs1830321 | 145067988 | C | T | C=0.503; T=0.497 | TEX41 | intron | LINC01966, AC096666.1 | 0.033 | 3.19×10−8 | (6, 9) |
| rs6740731 | 144513025 | G | A/C/T | G=0.679; A=0.321 | ZEB2 | intron or 3’UTR | ZEB2-AS1 | 0.0457 | 2.77×10−9 | (6, 14, 15) |
| rs17678683 | 144528992 | T | G | T=0.938; G=0.062 | LINC01412 | intron | ZEB2, ZEB2-AS1 | −0.0988 | 3.00×10−9 | (6, 7, 12, 14, 15) |
The lowest p value is provided from across all the cited CAD GWAS studies, respectively. See also Figure 1.
Rs2252641 was identified by the CARDIoGRAMplusC4D Consortium (total 67,394 cases and 142,664 controls from European and South Asian decedents) with p = 5.3×10−8 (7). This SNP was replicated in the UK biobank (p = 1.94×10−5) and in a meta-analysis of the combined CARDIoGRAMplusC4D and UK biobank datasets (p = 7.34×10−13) (13). This SNP is located within an intron of a long non-protein coding RNA (lncRNA) gene called testis expressed 41 TEX41).
The second SNP rs1830321 was identified in a UK Biobank GWAS analysis (34,541 CAD cases and 261,984 controls) with p = 3.5×10−6, and in a second stage meta-analysis using 122,733 cases and 42,4528 controls it reached genome-wide significance with p = 3.19×10−8 (6). This SNP was also found to be associated aortic stenosis in an Icelandic clinical trait association analysis with p = 1.8×10−13, where the association with CAD was also confirmed (CAD association p = 9.3×10−5) (9).
The third SNP rs6740731 is associated with risk of CAD and was identified using GWAS data from the UK biobank (34,541 cases and 261,984 controls, p = 4.94×10−5) and CARDIoGRAMplusC4D (p = 1.87×10−5) as discovery cohorts, and a meta-analysis of a combination of the two datasets (including a total 122,733 cases and 424,528 controls) with p = 3.86×10−9 (13). Depending on the specific isoform of ZEB2 (47 isoforms are currently recognized), this SNP is located either in an intron (for 42 of the ZEB2 isoforms) or 5’ UTR (for 5 of the ZEB2 isoforms) in the ZEB2 gene on the reverse (minus) strand of the double stranded DNA.
The fourth CAD-associated SNP in the vicinity of ZEB2, rs17678683, was identified by CARDIoGRAMplusC4D (p = 3×10−9) (7, 13). The SNP was not replicated by UK biobank data, but was reported to have a p = 3.44×10−7 in a meta-analysis combining these two datasets (13). This SNP is located in lncRNA LINC01412, which is a long intergenic non-protein coding RNA. rs17678683 also lies very close to the gene encoding ZEB2-AS1. ZEB2-AS1 produces a spliced lncRNA which is a natural antisense transcript corresponding to the 5’ UTR of ZEB2, which is thought to be involved in regulation of ZEB2 expression (28).
We next explored potential LD relationships between these four CAD-associated SNPs in the region of ZEB2. As shown in Figure 1B, other than rs2252641 and rs1830321 which are in low LD (LD score = 0.3847), there were no other significant LD relationships. In particular, despite their relatively close proximity, rs6740731 and rs17678683 were not in LD (LD score = 0.0472; Figure 1B). Therefore, assuming the LD relationship between rs2252641/rs1830321 is true, there are potentially three SNPs in the vicinity of ZEB2 that are each independently associated with the likelihood of CAD (rs6740731, rs17678683 and rs2252641/rs1830321) (Figure 1). Whether the three SNPs rs1830321, rs6740731 and rs17678683 are in LD was further tested by a stepwise regression analysis of SNP genotypes and ZEB2 expression in AOR in STARNET. The results confirmed that these three SNPs are independent (Figure 1C).
ZEB2-associated CAD SNPs modulate gene expression in the aortic wall and adipose tissue
We sought to determine potential gene-regulatory effects for each of these three CAD-associated SNPs in the vicinity of ZEB2 (rs6740731, rs17678683, rs2252641/rs1830321) by exploring their effect on transcript expression (i.e. eQTLs) in STARNET tissues.
As shown in Table 2, while there were no cis eQTLs identified for rs2252641, the other 3 SNPs exhibited corresponding eQTLs that were operative in the atherosclerotic aortic wall and in adipose tissue. Of most relevance, rs1830321 was found to be an eQTL for ZEB2 expression in AOR. We were able to further confirm that rs1830321 is an eQTL for ZEB2 expression in AOR using GTEx study data (Table 2). In addition, we found that rs17678683 is an eQTL for ZEB2 expression in both VAF and SF. We also detected cis eQTLs for TEX41 (in SF at rs1830321), GTDC1 (in AOR at rs17678683) and for the non-coding and uncharacterized gene AC009951.1 (in MAM at rs6740731) (Table 2). Importantly, the risk alleles of these SNPs were consistently associated with reduced ZEB2 expression levels at rs1830321 in AOR, and rs17678683 in VAF and SF (Figures 2A, E and F; CAD risk alleles are shown in red). While reduced TEX41 (in SF at rs1830321) and AC009951.1 (in MAM at rs6740731) were also associated with risk of CAD, increased levels of GTDC1 (in AOR at rs17678683) were associated with risk of CAD (Figures 2B, C, and D, respectively).
Table 2.
cis eQTLs in GWAS CAD SNPs in the vicinity of ZEB2 in STARNET (p < 0.05 is considered statistically significant).
| SNP id | SNP chromosome location | Reference allele | CAD risk allele | Tissue | Target gene name | Beta | p value | FDR | GTEx p value | GTEx eQTL tissue |
|---|---|---|---|---|---|---|---|---|---|---|
| rs1830321 | chr2:145067988 | C | T | AOR | ZEB2 | 0.1727 | 0.00697 | 0.8931 | 0.0001 | Aorta |
| rs1830321 | chr2:145067988 | C | T | SF | TEX41 | −0.1916 | 0.00199 | 0.4918 | - | - |
| rs6740731 | chr2:144513025 | G | A/C/T | MAM | AC009951.1 | 0.2425 | 0.00445 | 0.7921 | 0.000011 | Skelet. mu |
| rs17678683 | chr2:144528992 | T | G | AOR | GTDC1 | −0.2844 | 0.00879 | 0.9435 | 0.0000012 | Skelet. mu |
| rs17678683 | chr2:144528992 | T | G | VAF | ZEB2 | 0.2920 | 0.00994 | 0.9530 | - | |
| rs17678683 | chr2:144528992 | T | G | SF | ZEB2 | 0.3380 | 0.00180 | 0.4978 | - |
There were no cis eQTLs identified for rs2252641. These results are also presented in Figure 2. The final 2 columns are data derived from the GTEx study for the same SNPs/eQTLs. Skelet. Mu, skeletal muscle.
Figure 2.
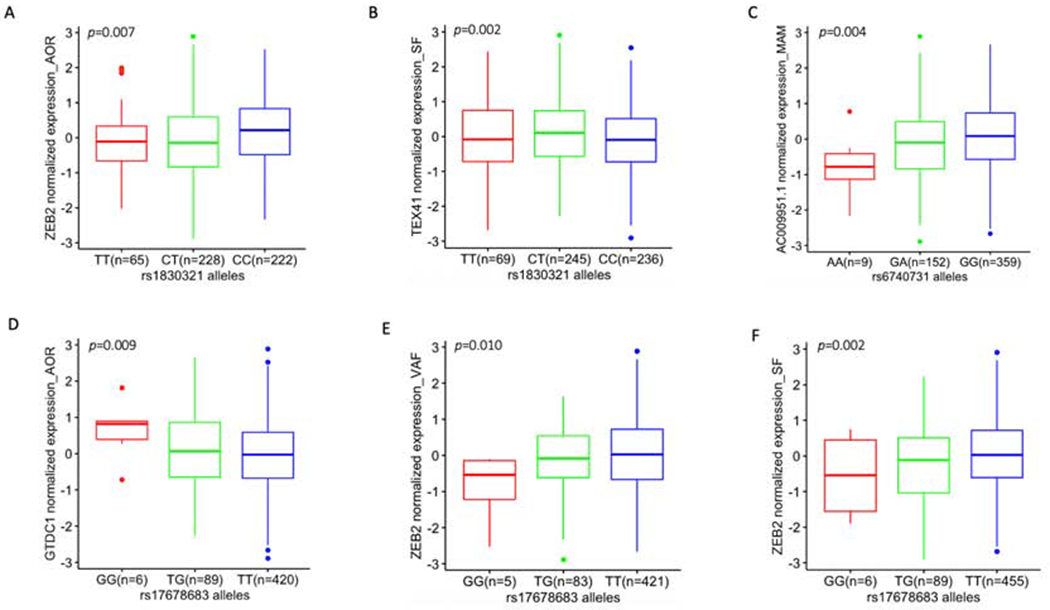
eQTL box plots of significant associations between genotypes of CAD GWAS SNPs and gene expression in STARNET tissues (see also Table 2).
(A) eQTL at rs1830321 for ZEB2 expression in AOR, (B) eQTL at rs1830321 for TEX41 in SF, (C) eQTL at rs6740731 for AC009951.1 in MAM, (D - F) eQTLs at rs17678683 for GTDC1 in AOR, for ZEB2 in VAF and for ZEB2 in SF, respectively. Homozygous CAD risk alleles are colored red and are on the left of each panel, heterozygous alleles are in green and homozygous reference (CAD protective) alleles are in blue. The number of subjects with 3 different genotypes at rs17678683 vary slightly between tissues because the availability of expression data across these tissues also differs slightly (due to reasons such as isolated samples failing to pass quality checks, or insufficient sample obtained from isolated tissues – see original STARNET manuscript for full details (8)). Although rs6740731 and rs17678683 are in close proximity (Figure 1A), allele frequency of rs6740731 and rs17678683 are different since they are located in different genes and they are not in linkage disequilibrium (Figure 1B).
Expression levels of genes in the vicinity of ZEB2
We next explored the expression levels of genes in close proximity to the CAD SNPs in this region (Figure 1A), being ZEB2, GTDC1, ZEB2-AS1, TEX41 and LINC01412. Expression levels were examined and compared according to RPKM (Reads Per Kilobase Million) mapped reads (Supplementary Figures 1 and 2). Overall, the mean levels of ZEB2 were ~10-fold higher (2.5-22.5 RPKM) than any of the other ZEB2-associated genes in this region. Thus, expression levels of GTDC1 were intermediate (1-5 RPKM), while as expected the levels of the non-coding genes TEX41, ZEB2-AS1 or LINC01412 were much lower (generally < 1 RPKM). For ZEB2 specifically, higher transcript levels were observed in MAM, SF, VAF and AOR – a pattern that was closely mirrored by transcript levels of GTDC1 (Supplementary Figures 1 and 2).
We also explored the levels of these transcripts in the Genotype-Tissue Expression (GTEx) project. As shown in Supplementary Figure 3, the levels of these transcripts were broadly similar between STARNET and GTEx. Due to its very low expression levels (particularly in STARNET tissues), expression of LINC01412 was not further considered in this study.
We explored the potential differential gene expression between matched cases and controls in STARNET for ZEB2, GTDC1, ZEB2-AS1 and TEX41 (Supplementary Table 1 and Supplementary Figure 4). While CAD cases in STARNET were patients undergoing coronary artery bypass graft surgery, matched controls underwent open chest surgery for other reasons (e.g. mitral valve replacement) and CAD was excluded on pre-operative angiograms. ZEB2 expression was significantly increased in SF and SKLM in CAD cases versus controls. Furthermore, ZEB2-AS1 expression was decreased in VAF in CAD cases versus controls; TEX41 was increased in AOR and SKLM in CAD cases; and GTDC1 was decreased in AOR in CAD cases, but was increased in CAD cases in LIV, SKLM, SF and VAF. These differences affirm that altered regulation and expression of these transcripts may play a role in the development of CAD.
Gene expression correlations between ZEB2, GTDC1, ZEB2-AS1 and TEX41
We sought to define any relationships among the respective expression levels of each of these individual genes in different tissues. As shown in Supplementary Figure 5 and Supplementary Table 2, positive correlations were found between ZEB2 expression levels in MAM and LIV, while a negative association was present between ZEB2 expression levels in BLOOD and AOR. GTDC1 expression levels were also positively correlated between several tissues, including: MAM and LIV, AOR and SF, SKLM and SF, SKLM and VAF. There were no correlations among ZEB2-AS1 expression levels in different tissues, nor TEX41. Therefore, with possible exception of GTDC1, this result confirmed the tissue-specificity of the eQTLs at these SNPs. In other words, as indicated in Table 2, the effects of the ZEB2-associated CAD risk SNPs (and their corresponding eQTLs) on gene expression are largely unique for each tissue presented in this study.
Additional regulatory control of ZEB2, GTDC1, ZEB2-AS1 and TEX41 expression
To understand any additional regulatory influences on the expression levels of ZEB2, GTDC1, ZEB2-AS1 and TEX41, we sought additional eQTLs that govern expression levels of these genes. We identified a single additional cis eQTL that governed ZEB2 expression in AOR (Supplementary Table 3). There were a further 46 cis eQTLs identified which governed either TEX41 or GTDC1 expression, with 45/46 of these eQTLs being in SF (Supplementary Table 3). Throughout the entire genome, there were no other cis or trans-regulated eQTLs that governed expression levels of ZEB2, GTDC1, ZEB2-AS1 or TEX41.
We also sought eQTLs residing within ZEB2, GTDC1, ZEB2-AS1 and TEX41 which govern the expression levels of other transcripts (besides ZEB2, GTDC1, ZEB2-AS1 and TEX41), but there were no such cis or trans eQTLs identified.
In summary, with the exception of TEX41 and GTDC1 in SF, these data affirm that there appears to be minimal additional genomic influence on the expression of ZEB2, GTDC1, ZEB2-AS1 and TEX41 beyond that exerted by the CAD-associated GWAS SNPs (Figure 1A).
Correlation of gene expression and GWAS SNPs with clinical phenotypes
We compared the clinical phenotypes of STARNET subjects with the transcript expression levels of ZEB2, GTDC1, ZEB2-AS1 and TEX41. There were no gene expression level correlations with HbA1c, Chol, TG, Diseased vessels, Syntax score or Duke CAD index (Figure 3A and Supplementary Table 4). AOR showed the greatest number of phenotypic associations of the seven tissues. Specifically, ZEB2 levels in AOR were positively correlated with WBC, ALT, AST and CRP; ZEB2-AS1 levels in AOR were positively correlated with ALT and LDL; while TEX41 levels in AOR were positively correlated with WBC and LDL but negatively correlated with HbG and HDL. In terms of phenotypes, the strongest associations were with LDL, HDL and liver enzymes (ALT and AST). The strongest of all relationships was a positive correlation between ZEB2-AS1 expression and HDL in BLOOD (correlation coefficient 0.24, adjustedp = 6.72×10−7). Also of note, levels of ZEB2 in LIV were negatively correlated with HDL cholesterol levels (p = 0.0504).
Figure 3.
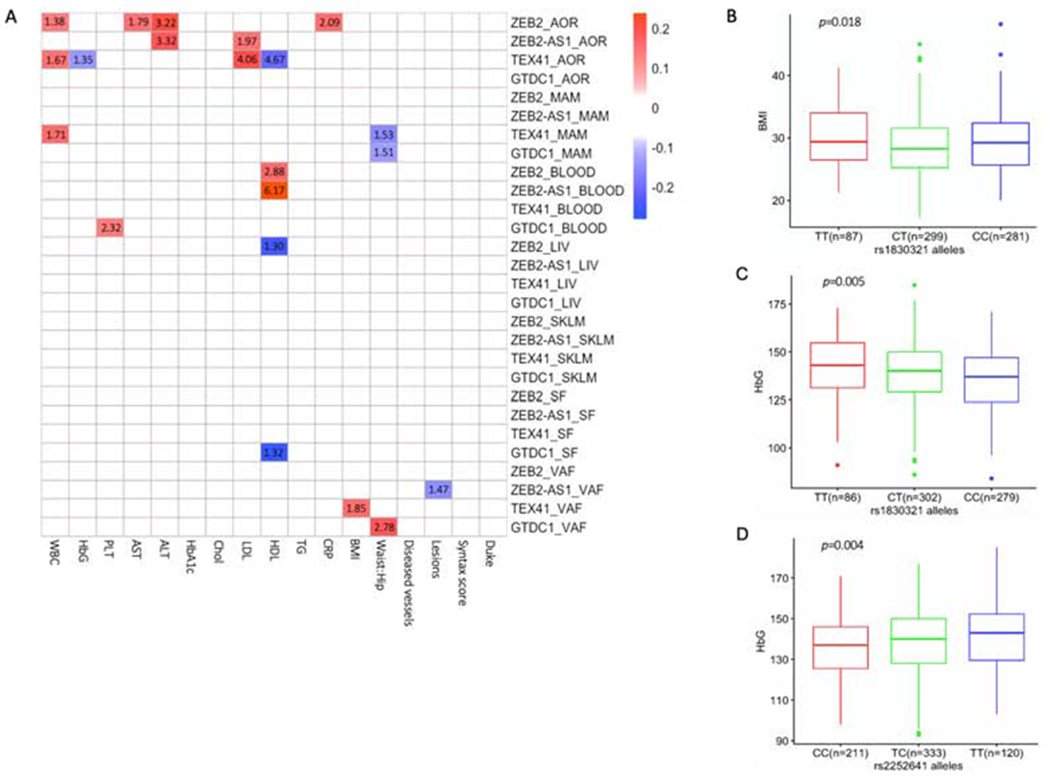
Correlations of ZEB2, GTDC1, ZEB2-AS1 and TEX41 gene expression levels, and CAD GWAS SNPs, with clinical phenotypes.
(A) Heatmap showing Pearson correlation coefficients for the significant correlations of ZEB2, GTDC1, ZEB2-AS1 and TEX41 expression levels with clinical phenotypes in STARNET tissues. Blue and red correspond to significant negative and positive correlations (respectively); non-significant correlations were not color coded. The number within each box represents the -log10 of the adjusted p value for that association. The association between ZEB2 in LIV and HDL was borderline significant after correction for multiple testing (p = 0.0504). Full results are presented in Supplementary Table 4. (B, C) Box plots showing associations of rs1830321 alleles with BMI and HbG. (D) Box plot indicating associations of rs2252641 alleles with HbG. The homozygous CAD risk allele is on the left (red color), the heterozygous allele is in the middle (green) and the homozygous reference (CAD protective) allele is on the right (blue). P values represent nominal significance thresholds. After adjustment for multiple comparisons, only the associations of rs2252641 and rs1830321 with HbG remained significant (Supplementary Table 5).
We also evaluated the relationship between the four reported CAD GWAS SNPs in the vicinity of ZEB2 (Figure 1A) and their effects on clinical traits in STARNET CAD subjects. At nominal significance thresholds, SNPs at rs2252641 and rs1830321 were correlated with HbG, while rs1830321 was also associated with BMI (Figures 3B–D). None of these SNPs were associated with any other clinical traits (Supplementary Table 5). After adjustment for multiple comparisons, only the associations of rs2252641 and rs1830321 with HbG remained significant. Interestingly, both the T allele in rs2252641 and also the T allele in rs1830321 were associated with increased HbG. However, while T is the CAD risk allele in rs1830321, it is not the CAD risk allele in rs2252641.
Co-expression modules, cross tissue network and key drivers
Cross-tissue co-expression modules were generated from AOR, MAM, BLOOD, LIV, SKLM, SF and VAF tissues. A total of 224 modules were generated from 30,716 transcripts, with the majority of co-expression modules involving genes from multiple tissues. Co-expression modules containing ZEB2 were found in all seven tissues (Figure 4A). ZEB2 was identified as hierarchically being the top key driver for module 20 (p =8.16×10−135; Figures 4A and 4B, Supplementary Table 6). The module contained 103 genes, with all of these being expressed in liver. The module was significantly associated with lipids level in blood, including LDL, HDL, Chol and TG (Figure 4C). The module was also associated with certain metabolic indices including Waist:Hip ratio, BMI, HbA1c. Importantly, module 20 was also associated with the severity of CAD as assessed by various parameters including Syntax score, number of coronary lesions and Duke CAD index (Figure 4C). At the protein level, there were 8 connected protein networks for the 103 genes in module 20 (Figure 4D). ZEB2 was directly connected with CTBP2 identified by evidence of co-expression, text mining and experimental determination. ZEB2 was also connected with ARHGAP31 identified by co-expression and text mining. Other proteins in the network that contained ZEB2 were ZFPM2, MECOM, RHOJ, ARHGAP15 and ARHGEF6. In terms of its patho-biological roles, pathway analysis indicated that module 20 is involved in multiple cardiovascular functions (Supplementary Figure 6A).
Figure 4.
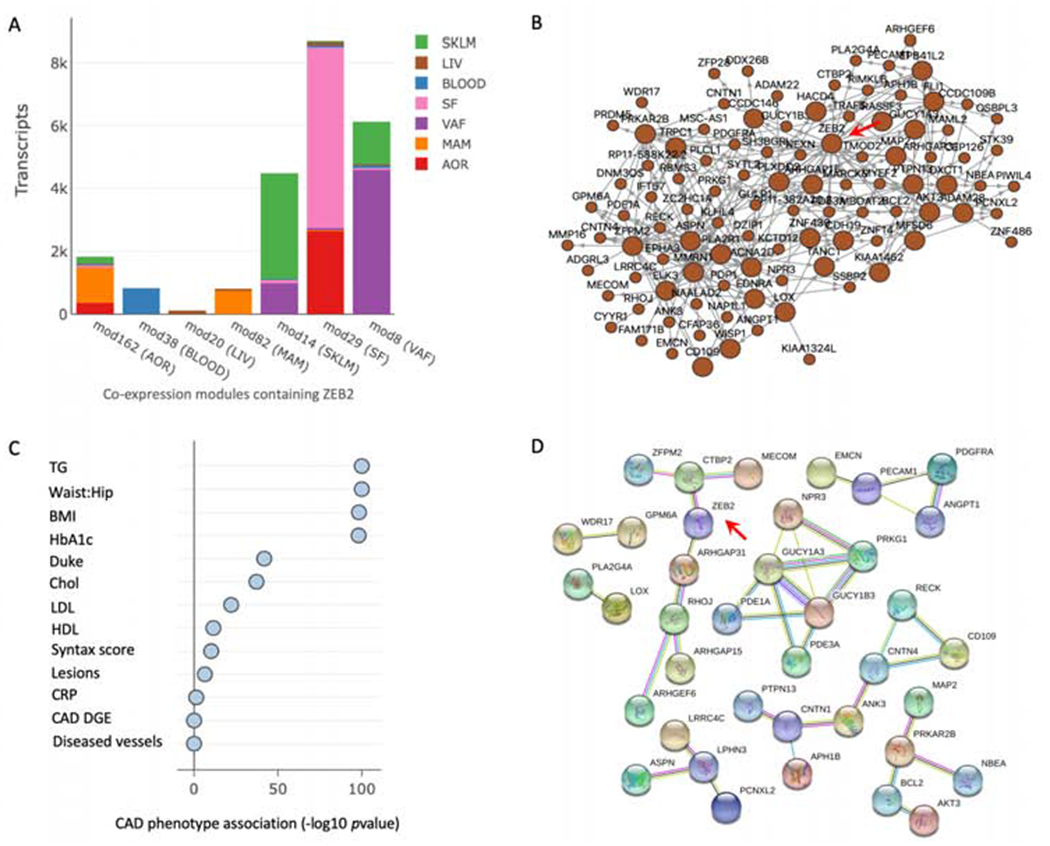
ZEB2 co-expression modules and their associations in STARNET tissues.
(A) ZEB2 co-expression modules. As indicated, the color codes represent differing tissues acquired in the STARNET study, while the tissue named on the horizontal axis (i.e. mod 162 (AOR)) indicates the tissue containing ZEB2 in that particular module. The vertical axis (‘Transcripts’) represents the total number of transcripts in each module. (B) Visualization of module 20, with ZEB2 identified as hierarchically the top key driver as indicated by red arrow (p = 8.16×10−135; Supplementary Table 6). (C) Association of module 20 with clinical traits assessed in the STARNET study. CAD DGE represents the enrichment of differential gene expression in the module between cases and controls. (D) Protein-protein interaction network of genes in module 20. Only connected nodes in the network are shown. A red line indicates evidence of gene fusion; green line indicates gene neighborhood evidence; blue line indicates co-occurrence evidence; purple line indicates experimental evidence; yellow line indicates text mining evidence; light blue line indicates curated database evidence; black line indicates co-expression evidence.
Apart from module 20, ZEB2 was not a key driver in any other co-expression modules. However, of the other modules that contained ZEB2, co-expression modules 162 and 38 were associated with various clinical traits, with both being highly correlated with LDL and HDL (Supplementary Figures 6B and C).
In terms of other genes in the vicinity of ZEB2, co-expression modules containing GTDC1 were found in all seven STARNET tissues, while co-expression modules for ZEB2-AS1 and TEX41 were each found in six tissues (Supplementary Figure 7). However, ZEB2-AS1, GTDC1 and TEX41 were not key drivers in any co-expression modules.
DISCUSSION
Using an array of datasets and bioinformatics tools, our study has shed light on the potential mechanisms of CAD causality that arises from SNPs in the vicinity of ZEB2. This includes: (1) Of the four SNPs in the region of ZEB2 that are associated with CAD, only two are in potential LD, and therefore there are potentially three SNPs in this region that are each independently associated with the likelihood of CAD (rs6740731, rs17678683 and rs2252641/rs1830321) (Figure 1); (2) eQTL findings at rs1830321 in AOR, and rs17678683 in VAF and SF indicate that these tissues may be of particular importance for governing the effect of ZEB2 in CAD (Figure 2); (3) Cross-tissue comparisons of gene expression levels indicated that, with the possible exception of GTDC1, levels of ZEB2, ZEB2-AS1 and TEX41 were tissue specific (Supplementary Figure 5); (4) rs2252641 and rs1830321 are related to HbG levels; (5) Gene expression levels of ZEB2, GTDC1, ZEB2-AS1 and TEX were related to various clinical phenotypes, with many of these relationships being detected in AOR, and the strongest of these associations being to HDL, LDL and liver enzyme levels (ALT and AST). ZEB2 gene expression levels in liver were also negatively associated with HDL cholesterol levels (p = 0.0504); (6) While ZEB2, GTDC1, ZEB2-AS1 and TEX41 were in multiple GRNs related to CAD, most notable was that ZEB2 is the top key driver of a liver-specific GRN that is associated with HDL and other lipid levels, metabolic and anthropometric measures, and CAD severity (Figure 4); (7) Collectively, these findings can be summarized as indicating that the ZEB2-associated CAD SNPs appear to act in concert via the atherosclerotic aortic arterial wall and adipose tissues, likely by governing metabolic and lipid functions. Furthermore, ZEB2 also exhibits a key driver role whereby it governs a CAD-associated GRN in liver.
To date, the scientific community has collectively deciphered the mechanism of action of only a small proportion of GWAS SNPs for complex diseases. Notable examples of SNPs and their associated genes for which we have a strong understanding of their causal mechanisms for CAD include PCSK9 and the LDL receptor (29). Accordingly, one can speculate that GWAS SNPs that are ‘understood’ may be biased toward those with simpler or more obvious mechanisms of action that could be deciphered using contemporary research tools. For example, it is logical that variants in the LDL receptor would promote CAD by influencing LDL cholesterol levels. Conversely, SNPs or genetic regions with more complex mechanisms of action are perhaps more likely to be those that we do not yet understand. In support of this concept, as well as the findings of the present study there are many examples of this potential bias toward only understanding the more straightforward causal SNP-phenotype relationships for complex diseases. For example, the first genetic risk variant to be identified for CAD resides on the small arm p of chromosome 9 and is referred to as 9p21. Although first described in 2007 (30, 31), after over a decade of intense efforts the precise mechanisms of action of this risk variant remain under investigation, but certainly, these mechanisms appear to of similar complexity as described here for the ZEB2-associated CAD SNPs (29). As another example, at rs9349379, which is located in an exon of PHACTR1, either an adenine (A) or guanine (G) may be present. Remarkably, a disease association exists regardless of which nucleotides are present (AA, AG, or GG). Thus, an (A) at rs9349379 is associated with increased risk of cervical artery dissection (32), migraine headache (33), fibromuscular dysplasia (34) and spontaneous coronary artery dissection (35). Conversely, the (G) allele at rs9349379 is associated with CAD (12), coronary artery calcification (36), and myocardial infarction (12, 36). While many theories exist, at present we have few insights on this complex reciprocal risk of having these diverse vascular diseases (37). The implications of this potential bias toward understanding the more straightforward causal pathways for complex diseases, are that it may require a substantial amount of time, effort, and resources before we begin to fully understand complex genetic diseases. We suggest that the pipeline of analyses undertaken in this study (Graphical Abstract) may be a useful and important first step toward unravelling genetic regions or related SNPs that are linked to complex disease traits.
While many of the results to emerge from this study are novel, our finding that rs2252641 and rs1830321 are associated with HbG concentration is not without precedent (Figure 3). ZEB2 is known to be involved in controlling hematopoiesis and hematopoietic stem cell differentiation (38), and a GWAS study in the Japanese population found that other SNPs in the ZEB2 region are associated with erythrocyte count and various erythrocyte indices (e.g. mean red cell corpuscular volume) (39). However, in our study both the T allele in rs2252641 and also in rs1830321 were associated with increased HbG, but while T is the CAD risk allele in rs1830321 it is not the CAD risk allele in rs2252641. Interestingly, both low (< 15 g/dL) and high HbG levels (≥ 17 g/dL) have been found to be independently associated with increased risk for cardiac events (40). Therefore, at the present time the precise link between HbG levels, rs2252641 and rs1830321, ZEB2 and causality for CAD remains to be defined. Nevertheless, based on the totality of our other findings, the regulation of HbG levels does not appear to be the predominant mechanism of action of these ZEB2-associated SNPs for causing CAD.
As an unexpected finding to emerge from this study, we identified that ZEB2 is the top key driver of a liver-specific GRN (Figure 4). This GRN appears to be involved in various vascular and cardiac disease pathways (Supplementary Figure 6A), and showed strong associations with multiple CAD-relevant sub-phenotypes such as lipid levels, Waist:Hip ratio and BMI, HbA1c levels and CAD severity (Figure 4C). The importance of this finding is underscored by the fact that we have previously shown that GRNs capture a major portion of genetic variance and contribute to heritability, playing pivotal roles as mediators of gene-environmental interactions in CAD (2). The fact that eQTLs in the vicinity of ZEB2 did not control ZEB2 expression in the liver (Table 2) is consistent with this key driver role, because prior studies of gene networks have indicated that hub nodes tend to be essential and evolutionarily conserved, and that ‘disease genes’ do not encode hubs (41). Therefore, the contribution of ZEB2 as the top key driver of this GRN likely represents an additional aspect of its role in CAD causality, in addition to the causality driven via the ZEB2-associated CAD SNPs.
There are certain limitations of this study that should be acknowledged. This is the first study to examine the effect(s) of multiple GWAS SNPs located near a gene of interest in CAD using this bioinformatics pipeline. More studies using this pipeline are needed to further prove its applicability. RNA expression data used in this study were obtained from bulk tissues and it is challenging to infer detailed causal mechanisms or gene interactions from different cell types involved in the disease. Indeed, we hypothesize that ZEB2 might also govern endothelial cell fate in atherosclerosis and CAD (42), however, in the absence of endothelial-specific RNA sequence data is was not possible to reliably prove or refute this hypothesis. Furthermore, while this study offers evidence to support causal roles for ZEB2 in CAD, our results do not prove causality. These potential roles and proof of causality require further validation in direct functional studies. Such functional studies of the role of ZEB2 in atherosclerosis are currently underway in our laboratory.
In conclusion, we applied a pipeline of investigations to a cluster of ZEB2- associated SNPs that begins with a careful dissection of SNP locations and LD relationships, and which moves through a series of eQTL, gene-gene, gene-phenotype, SNP-phenotype and GRN analyses. While control of HbG levels may also play a role, our major finding was that the ZEB2-associated CAD SNPs appear to act in concert via the atherosclerotic arterial wall and adipose tissues, likely by governing metabolic and lipid functions. Moreover, ZEB2 is the top key driver of a GRN in liver that is related to lipid levels, metabolic and anthropometric measures, and CAD severity. These findings indicate several possibilities for manipulating ZEB2 as a clinical therapeutic target, such as liver-specific ZEB2 inhibition aiming to decrease the activity of module 20. Furthermore, this analytic pipeline can serve as a roadmap for future studies aiming to dissect other complex SNP and gene relationships with complex clinical phenotypes.
Supplementary Material
HIGHLIGHTS.
Hundreds of genes and single nucleotide polymorphisms (SNPs) are associated with coronary artery disease (CAD)
However the mechanisms of effect of these genes and SNPs are largely unknown
To address these concerns we developed a unique informatics pipeline
This pipeline disclosed several potential mechanisms underlying a cluster of ZEB2-associated CAD risk SNPs
This pipeline can serve as a roadmap to understand other complex CAD risk loci, and complex SNP-gene-tissue-phenotype relationships
Acknowledgments
FINANCIAL SUPPORT
Jason Kovacic acknowledges research support from the US National Institutes of Health (NIH) (R01HL130423, R01HL135093, R01HL148167). Nirupama Chandel was supported by US National Institutes of Health grant T32HL007824. Johan Björkegren acknowledges research support from the NIH R01HL125863, Swedish Research Council (2018-02529) and Heart Lung Foundation (20170265), Foundation Leducq (PlaqueOmics: Novel Roles of Smooth Muscle and Other Matrix Producing Cells in Atherosclerotic Plaque Stability and Rupture, 18CVD02; and CADgenomics: Understanding CAD Genes, 12CVD02]) and Astra-Zeneca.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, et al. Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med. 2016;375(24):2349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeng L, Talukdar HA, Koplev S, Giannarelli C, Ivert T, Gan LM, et al. Contribution of Gene Regulatory Networks to Heritability of Coronary Artery Disease. J Am Coll Cardiol. 2019;73(23):2946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Björkegren JLM, Kovacic JC, Dudley JT, Schadt EE. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol. 2015;65(8):830–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braenne I, Civelek M, Vilne B, Di Narzo A, Johnson AD, Zhao Y, et al. Prediction of Causal Candidate Genes in Coronary Artery Disease Loci. Arterioscler Thromb Vasc Biol. 2015;35(10):2207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Harst P, Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res. 2018;122(3):433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consortium CAD, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franzen O, Ermel R, Cohain A, Akers NK, Di Narzo A, Talukdar HA, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016;353(6301):827–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helgadottir A, Thorleifsson G, Gretarsdottir S, Stefansson OA, Tragante V, Thorolfsdottir RB, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat Commun. 2018;9(1):987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McPherson R, Tybjaerg-Hansen A. Genetics of Coronary Artery Disease. Circ Res. 2016; 118(4) :564–78. [DOI] [PubMed] [Google Scholar]
- 11.Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49(9):1385–91. [DOI] [PubMed] [Google Scholar]
- 12.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Laan SW, Siemelink MA, Haitjema S, Foroughi Asl H, Perisic L, Mokry M, et al. Genetic Susceptibility Loci for Cardiovascular Disease and Their Impact on Atherosclerotic Plaques. Circ Genom Precis Med. 2018;11(9):e002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016;32(20):3207–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26( 18):2336–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8(4):e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrews S FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- 19.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- 21.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arneson D, Bhattacharya A, Shu L, Makinen VP, Yang X. Mergeomics: a web server for identifying pathological pathways, networks, and key regulators via multidimensional data integration. BMC Genomics. 2016;17(1):722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mark DB, Nelson CL, Califf RM, Harrell FE Jr, Lee KL, Jones RH, et al. Continuing evolution of therapy for coronary artery disease. Initial results from the era of coronary angioplasty. Circulation. 1994;89(5):2015–25. [DOI] [PubMed] [Google Scholar]
- 27.Kovacic JC, Limaye AM, Sartori S, Lee P, Patel R, Chandela S, et al. Comparison of six risk scores in patients with triple vessel coronary artery disease undergoing PCI: competing factors influence mortality, myocardial infarction, and target lesion revascularization. Catheter Cardiovasc Interv. 2013;82(6):855–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, Pena R, et al. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snaill-induced epithelial-mesenchymal transition. Genes Dev. 2008;22(6):756–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts R Genetics of coronary artery disease. Circ Res. 2014;114(12):1890–903. [DOI] [PubMed] [Google Scholar]
- 30.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491–3. [DOI] [PubMed] [Google Scholar]
- 32.Debette S, Kamatani Y, Metso TM, Kloss M, Chauhan G, Engelter ST, et al. Common variation in PHACTR1 is associated with susceptibility to cervical artery dissection. Nat Genet. 2015;47(1):78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anttila V, Winsvold BS, Gormley P, Kurth T, Bettella F, McMahon G, et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet. 2013;45(8):912–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiando SR, Tucker NR, Castro-Vega LJ, Katz A, D’Escamard V, Treard C, et al. PHACTR1 Is a Genetic Susceptibility Locus for Fibromuscular Dysplasia Supporting Its Complex Genetic Pattern of Inheritance. PLoS Genet. 2016;12(10):e1006367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adlam D, Olson TM, Combaret N, Kovacic JC, Iismaa SE, Al-Hussaini A, et al. Association of the PHACTR1/EDN1 Genetic Locus With Spontaneous Coronary Artery Dissection. J Am Coll Cardiol. 2019;73(1):58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Donnell CJ, Kavousi M, Smith AV, Kardia SL, Feitosa MF, Hwang SJ, et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation. 2011;124(25):2855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovacic JC. Unraveling the Complex Genetics of Coronary Artery Disease. J Am Coll Cardiol. 2017;69(7):837–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goossens S, Janzen V, Bartunkova S, Yokomizo T, Drogat B, Crisan M, et al. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood. 2011;117(21):5620–30. [DOI] [PubMed] [Google Scholar]
- 39.Kanai M, Akiyama M, Takahashi A, Matoba N, Momozawa Y, Ikeda M, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. 2018;50(3):390–400. [DOI] [PubMed] [Google Scholar]
- 40.Chonchol M, Nielson C. Hemoglobin levels and coronary artery disease. Am Heart J. 2008;155(3):494–8. [DOI] [PubMed] [Google Scholar]
- 41.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, et al. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(2):190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.