

Summary.
Oncogenic transformation alters lipid metabolism to sustain tumor growth. We define a mechanism by which cholesterol metabolism controls the development and differentiation of pancreatic ductal adenocarcinoma (PDAC). Disruption of distal cholesterol biosynthesis by conditional inactivation of the rate limiting enzyme Nsdhl or treatment with cholesterol-lowering statins switches glandular pancreatic carcinomas to a basal (mesenchymal) phenotype in mouse models driven by KrasG12D expression and homozygous Trp53 loss. Consistently, PDACs in patients receiving statins show enhanced mesenchymal features. Mechanistically, statins and NSDHL loss induce SREBP1 activation, which promotes the expression of Tgfb1 enabling epithelial-mesenchymal transition. Evidence from patient samples in this study suggests activation of TGFβ signaling and epithelial-mesenchymal transition by cholesterol-lowering statins may promote basal type of PDAC conferring poor outcomes in patients.
Graphical Abstract
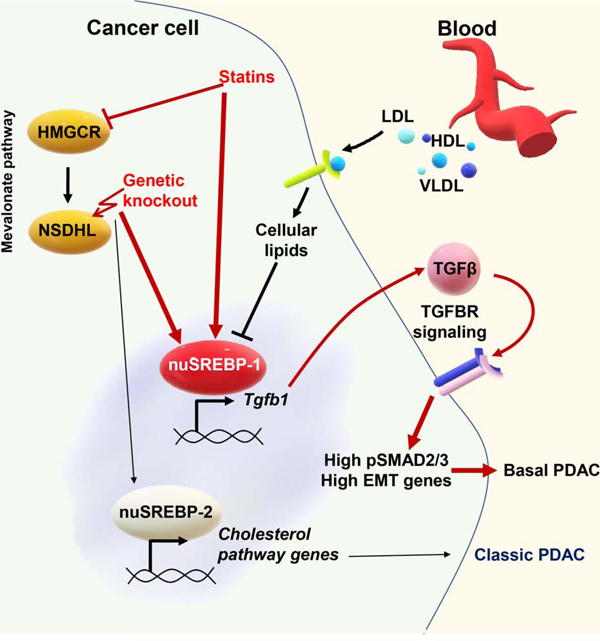
In brief.
Gabitova-Cornell et al. show that disruption of cholesterol biosynthesis by Nsdhl knockout or treatment with statins switches glandular pancreatic carcinomas to a basal subtype via activation of SREBP1, which induces Tgfb1 expression and autocrine TGFβ-SMAD2/3 signaling, inducing epithelial-mesenchymal transition.
Introduction.
Pancreatic ductal adenocarcinoma (PDAC) is poised to become the second leading cause of cancer deaths in the USA by 2030 (Chari et al., 2015; Rahib et al., 2014). The similar numbers of PDAC diagnoses and deaths reflect the high metastatic propensity and treatment resistance of this cancer. Epithelial-to-mesenchymal transition (EMT) has been proposed as a key PDAC mechanism linking drug resistance (Sabnis and Bivona, 2019; Zheng et al., 2015), invasive growth, and metastatic dissemination (Aiello et al., 2017). Gene expression analyses established at least two molecular subtypes of PDAC, the classic (or glandular), and the basal (or mesenchymal) (Aung et al., 2018; Moffitt et al., 2015), each of which is associated with distinct prognoses and sensitivity to chemotherapy. The median survival of basal PDAC is much lower than that in the classic subtype (respectively, 6.3 versus 10.4 months in a recent study (Aung et al., 2018)). Activation of the transforming growth factor beta (TGFβ) pathway a defining feature of basal PDAC, leading to increased expression of genes associated with a mesenchymal phenotype, including transcription factors ZEB1, ZEB2, TWIST, SNAI1, and SNAI2 (Scheel et al., 2011), as well as reduced expression of epithelial adhesion molecule E-cadherin (CDH1) and epithelial lineage transcription factors GATA6, SOX17, and HNF4A (Bailey et al., 2016; Collisson et al., 2011; Moffitt et al., 2015; TCGA., 2017). Although TGFβ signaling plays a central role in EMT, it encompasses a tumor suppressive role in untransformed cells; as reflected by frequent inactivation of components of the TGFβ pathway (TGFBR1, TGFBR2, SMAD4) in about a quarter of PDAC cases (David et al., 2016; Stankic et al., 2013).
The possible mechanistic linkage between PDAC incidence and cholesterol metabolism has been raised by epidemiological studies, nominating elevated serum cholesterol (dyslipidemia) and obesity as risk factors (Genkinger et al., 2015). Accelerated uptake and endogenous biosynthesis of cholesterol and phospholipids is a feature of oncogene-transformed cells (Pitroda et al., 2009; Silvente-Poirot and Poirot, 2014). Increased expression of genes of the cholesterol pathway is allied to classic PDAC (Karasinska et al., 2019). Yet, the mechanism for this linkage remains elusive. In mouse models of PDAC, driver mutations in the oncogene Kras coupled with loss of the tumor suppressor Trp53 reprogram cellular metabolism in numerous ways affecting utilization of energy sources, accelerating cholesterol biosynthesis and uptake (Freed-Pastor et al., 2012; Ying et al., 2012). Although increased cholesterol biosynthesis driven by activity of sterol response element binding protein (SREBP2) is linked to common loss-of-function mutations in TP53 (Moon et al., 2019), the clinical activity of cholesterol-lowering medications such as statins has been relatively modest in reducing PDAC risk (Bang et al., 2018) or PDAC mortality (Huang et al., 2017).
An alternative possibility is that cholesterol, its precursors, and/or metabolites modulate tumor cell oncogenic functions to modify the disease course at early PDAC stages. To this end, cholesterol, its metabolites, and other components of the cholesterol biosynthetic pathway are known to impact progression in some cancer types (Gabitova et al., 2015; Nelson et al., 2013). For example, liver X-receptor (LXR) activation, induced by accumulation of 27-hydroxycholesterol, fosters the development of the highly aggressive basal breast carcinoma characterized by its mesenchymal features (Nelson et al., 2013). These findings suggest that cholesterol homeostasis regulating genes and metabolites could affect cancer differentiation. In this study, we dissect the causal relationship between endogenous cholesterol metabolism and PDAC development and differentiation. Our results indicate that a metabolically-determined dichotomy of PDAC differentiation is mediated by cholesterol-sensitive SREBP1-dependent regulation of TGFβ expression, TGFβ receptor activation, and induction of a canonical SMAD2/3 effector cascade.
Results.
Distinct cholesterol metabolic programs define transcriptional subsets of human pancreatic cancer.
To investigate the relationship between cholesterol biosynthesis and epigenetic programs in human PDAC, we compared gene signatures between classic and basal subsets of PDAC using data from 76 high purity (estimated >30% of transcripts originating from cancer cells) samples profiled by The Cancer Genome Atlas Research Network (TCGA., 2017). Basal PDAC showed significantly increased expression of TGFβ and EMT pathway genes, among other hallmark tumor aggressiveness pathways like PI3K-mTOR, mitosis, and hypoxia (Fig. 1A, Tables S1-2). In contrast, the fatty acid and cholesterol metabolism genes (Molecular Signature Database, (Liberzon et al., 2015)) were significantly higher in classic PDAC compared to basal (Fig. 1B, C). These patterns were confirmed in a curated set of samples from the International Cancer Genome Consortium (ICGC, Fig. S1A, B). We further confirmed that the Hallmark Cholesterol Homeostasis gene signature independently impacted the overall survival of patients in both datasets (Fig. 1D and S1C). Overall, PDAC cases with gene expression enrichment for cholesterol homeostasis had a similar longer survival (17.5 months versus not reached, p=0.4, logrank test) for the Moffitt classic and basal subtypes, respectively. Further, low expression of the same set of genes (Fig. 1D) was associated with a shorter 2-year survival; 18.4% in the basal subclass of PDAC compared to 60% in the classic subtype (hazard ratio 0.23, 95% confidence interval, 0.06–0.87, p=0.02, logrank test). This difference was not evident in patients with PDAC characterized by high cholesterol homeostasis signature: classic (n=33, 2 year survival 47%) and basal (n=14, 2 year survival 64.3%; p=0.4, logrank test), suggesting a specific relationship between reduced activity of the cholesterol biosynthesis and aggressiveness of basal PDAC.
Figure 1. A cholesterol homeostasis gene signature differentiates classic versus basal human PDAC.
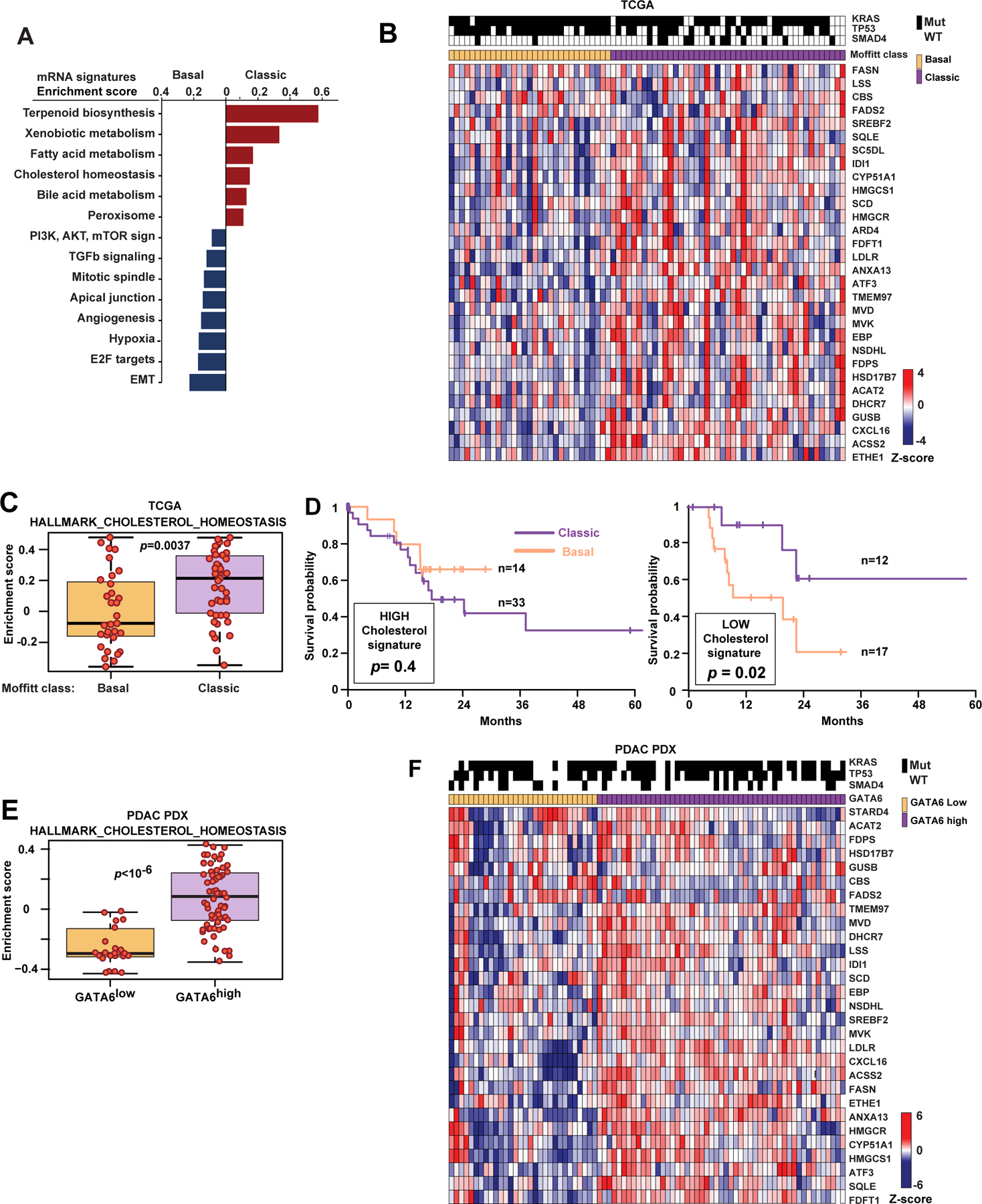
(A) Comparison of Hallmark mRNA transcriptional signatures (p-values <0.01; Molecular Signature Database (Liberzon et al., 2015)) between classic and basal PDAC among 76 TCGA cases with an estimated tumor cell fraction greater than 30%. (B) Heat map of normalized expression of representative genes in the “hallmark cholesterol homeostasis” signature for TCGA cases. (C) Comparison of “hallmark cholesterol homeostasis” gene signature in 76 PDAC cases from TCGA. (D) Kaplan-Meier survival of classic and basal PDAC in the TCGA cohorts stratified by high (left) or low (right) enrichment for “hallmark cholesterol homeostasis” mRNA signature. (E) Comparison of “hallmark cholesterol homeostasis” signature, and (F) a heat map of normalized expression of representative genes in the “hallmark cholesterol homeostasis” signature in 85 patient-derived xenografts stratified by expression of human GATA6 mRNA (shown as Z-score above or below zero). In C and E, y-axes illustrate positive and negative enrichment scores comparing basal and classic subtypes of PDAC. Boxplots represent median (black bar) and range (colored panel, 25–75th percentile) of enrichment scores for individual cases shown as red dots for each sample in that subtype. In B and F, Z-scores calculated for each gene are plotted on a red (higher expression) and blue (low expression) scale. Top color bar, subtype of PDAC; mutations, shown as black lines if present. See also Figure S1 and Tables S1–2.
To more clearly distinguish transcripts derived from human epithelial cells versus murine stroma, we analyzed transcriptomes of 85 human PDAC patient-derived xenografts (PDXs). Stratification by high or low GATA6 mRNA expression in the human cancer cell component to the classic or basal PDAC subsets, respectively (Aung et al., 2018), demonstrated significantly higher expression of cholesterol homeostasis genes in classic (i.e., GATA6high samples; Fig. 1E). The cholesterol homeostasis genes, underrepresented in basal PDAC (Fig. 1B, F), included 16 canonical cholesterol biosynthesis genes, including NSDHL.
Disruption of epithelial cholesterol biosynthesis conditions PDAC development.
To test the dependency of pancreatic carcinogenesis on tumor-intrinsic cholesterol biosynthesis, we used mice in which conditional inactivation of Nsdhl (NAD(P)-dependent steroid dehydrogenase-like catalyzing irreversible oxidative decarboxylation of methyl moieties at the C4 position of a cholesterol precursors (Cunningham et al., 2015; Gabitova et al., 2015)) via a Pdx1-Cre transgene takes place in the developing pancreatic bud at embryonic day 8.5 (Offield et al., 1996). Nsdhl inactivation in the pancreatic epithelium was efficiently achieved (Fig. S2A-C). Nsdhl∆Panc mice produced normal-sized litters at term with an approximate 1:1 male to female ratio; given the location of Nsdhl on the X chromosome, this implied no embryonic lethality. Comparison between Nsdhl∆Panc and wild type mice, at 6–8 weeks, included normal pancreatic size (Fig. S2D), ductal and acinar anatomy, and proliferation rates (Fig. S2E). To probe the regenerative capacity of Nsdhl-deficient pancreas, we treated animals with a two-day course of caerulein, an known acute pancreatitis inducer (Renner et al., 1983). No significant morphological or proliferation differences were evident (Fig. S2F).
The effects of Pdx1-Cre conditional Nsdhl knockout were evaluated in a pancreatic model driven by oncogenic KrasG12D and inactivation of both alleles of Trp53 (KPPC mice, Pdx1-Cre;LSL-KrasG12D;Trp53f/f), known to induce rapid PDAC development (Bardeesy et al., 2006; Hingorani et al., 2005; Jackson et al., 2001; Tuveson et al., 2004). Presence of pre-malignant lesions, acinar-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasia (PanIN), was significantly delayed in KPPCN (N denoting Nsdhlf/f alleles) compared to KPPC controls (Fig. 2A). At 4 weeks, PDAC was detectable in 4/7 KPPC, while no evidence of tumors was apparent in the age-matched KPPCN animals. Despite this marked delay in the absence of Nsdhl, the median survival of KPPCN mice was only increased by 12 days, compared to KPPC (median survival, 76 vs. 64 days, respectively, Fig. 2B).
Figure 2. Effects of NSDHL deficiency on pancreatic adenocarcinoma development.
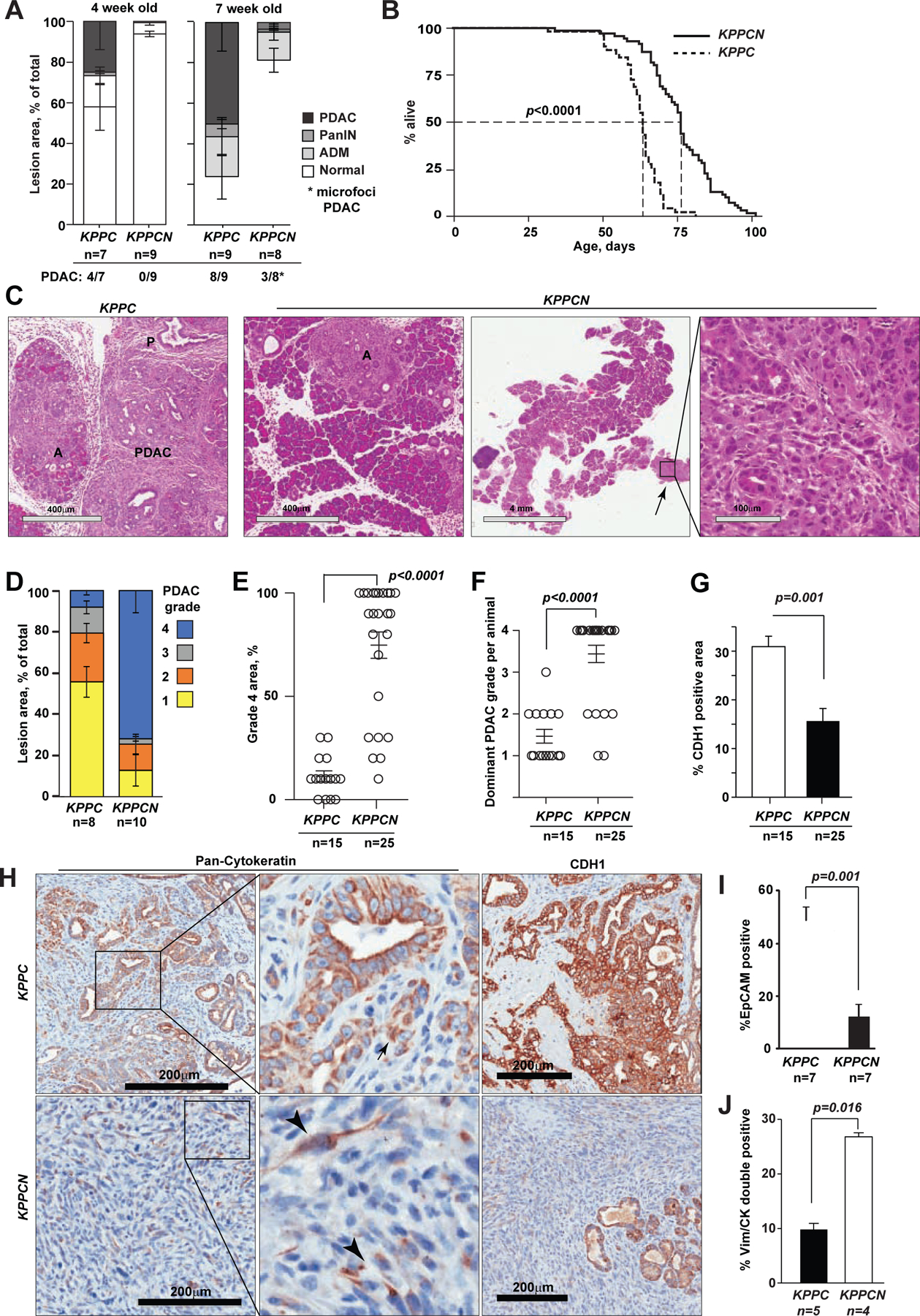
(A) Histological grading of pancreatic epithelial lesion in KPPC and KPPCN mice at 4 and 7 weeks of age; p<0.02 for comparisons of PDAC and normal areas. (B) Kaplan-Meier survival of KPPC (n=64) and KPPCN (n=76) mice; p<0.0001, Logrank test. (C) Hematoxylin and eosin stained pancreas sections of KPPC and KPPCN mice at 7 weeks of age. Small foci of grade 4 PDAC (arrow) are seen on the background of nearly normal KPPCN pancreatic tissue with ADM (A) and early PanIN (P) lesions. Scale bars, as shown. (D) Histological grading of pancreatic adenocarcinoma; p<0.01 for grades 1, 3 and 4. (E) Comparison of per cent of grade 4 PDAC areas; p<0.0001. (F) Histological grades of predominant PDAC per animal; p=0.0009. (G) Quantification of CDH1 expression as percent of positive areas per section; p=0.001. (H) Representative pan-cytokeratin (CK) and E-cadherin (CDH1) staining of pancreatic tumor tissues. Arrows, CK-positive PDAC cells. Scale bars, 200 μm. (I) Percentage of EPCAM-positive epithelial cells assessed by FACS in primary KPPC and KPPCN tumors; p=0.0012. (J) Quantification of CK+/VIM+ double positive cells by simultaneous multi-channel immunofluorescence (see images in Figure S2); p=0.016. In all graphs, data are represented as mean±SEM, p-values by Wilcoxon test. See also Figure S2 and Table S3.
To investigate the course of PDAC development in KPPCN, we examined pancreatic tissues at 7 weeks, when survival curves separation was evident (Fig. 2A, B). At this time, 8/9 KPPC mice had developed multifocal large tumors comprising more than 50% of the pancreas, while 3/8 KPPCN mice developed microscopic PDAC foci covering <5% (Fig. 2A, C). Results obtained with micro-magnetic resonance imaging (mMRI), at 7-weeks, also showed larger mases for KPPC compared to KPPCN (Fig. S2G). Differences were also apparent in pancreas weight (Fig. S2H). Advanced KPPC PDAC were mostly well-differentiated (grade 1–2), while tumors in KPPCN mice were nearly uniform grade 4 (Fig. 2C–F, and Table S3).
In depth analyses of KPPC, revealed cytokeratin-positive glandular structures with strong expression of E-cadherin (CDH1 in Fig. 2G, H), while the majority of KPPCN lesions comprised of single cells or small clusters of spindle-shaped tumor cells with weak expression of cytokeratin and no CDH1 (Fig. 2G, H). Since loss of CDH1 is a feature of EMT (Scheel et al., 2011), we examined the differentiation markers of KPPCN and KPPC carcinomas. We performed direct labeling of cells isolated from tumor tissues with EPCAM antibody to distinguish populations of differentiated epithelial carcinoma cells from cancer-associated fibroblasts (CAFs) and mesenchymal carcinoma cells, which are typically positive for PDGFRα (Fig. S2I). Fluorescence-activated cell sorting (FACS) of spontaneous pancreatic tumors showed significant reduction in EPCAM-positive cells and increase in EPCAM-/PDGFRα− and PDGFRα+ cells in KPPCN compared to KPPC (10% vs. 50%, respectively; Fig. 2I, S2I).
To discern between the epithelial and stromal compartments, we used multiplex immunofluorescence; identifying carcinoma cells with pan-cytokeratin (CK) and mesenchymal cells with vimentin (VIM) antibodies. About 10% of cells in KPPC tumors were VIM+/CK+ (Fig. S2J). In contrast, in KPPCN tumors, VIM+/CK+ cells were more prevalent (Fig. 2J, S2J). The prevalence of VIM+/CK+ cells together with the loss of CDH1 expression in KPPCN tumors, is akin to mesenchymal differentiation, and consistent with the reported PDAC basal subtype (Aung et al., 2018; Moffitt et al., 2015).
Activation of EMT transcriptional program in NSDHL-deficient PDAC.
To resolve the differentiation heterogeneity due to NSDHL deficiency, we analyzed the transcriptomes of single cells isolated from advanced pancreatic tumors (Fig. 3). After quality control and pooled analysis batch corrections (see Methods; Table S4), we obtained transcriptomes of 16,832 cells from advanced KPPC (n=9,501) and KPPCN (n=7,331) tumors. After dimensionality reduction, we performed graph-based clustering using Uniform Manifold Approximation and Projection (UMAP, Fig. 3A, B and Table S4). This identified 18 cells clusters based on established lineage markers (Biffi et al., 2019; Dominguez et al., 2020). Cells in each cluster were denoted in all replicates.
Figure 3. Conditional knockout of Nsdhl promotes epithelial-to-mesenchymal transition (EMT) switch in mouse pancreatic adenocarcinoma.
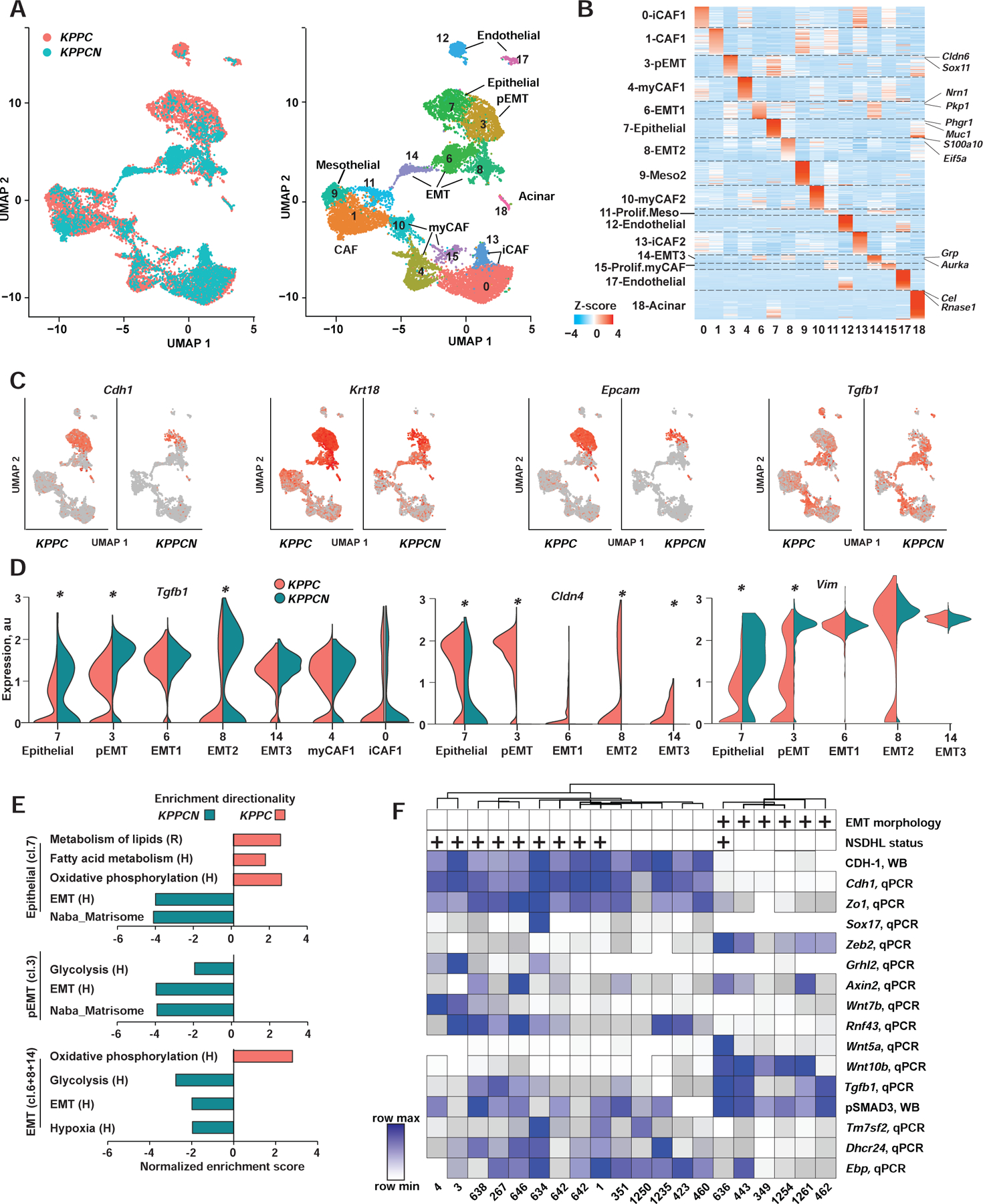
(A) UMAP-embedding of transcriptomes of 16,832 single cells isolated from 3 KPPC and 2 KPPCN advanced tumors. Eighteen cell types were identified by graph-based clustering are indicated by color (T-, B- and myeloid cells excluded). (B) Heat map of differentially expressed genes. Z-score normalized expression of the enriched genes for each cluster is shown as a log2-fold change in cells within a cluster relative to all other cells in the dataset. Representative genes are highlighted for each cluster. iCAF, inflammatory cancer associated fibroblasts; myCAF, myofibroblasts; pEMT, partial EMT; Meso, mesothelial. (C) UMAP-embedding with color proportionate to the Log2-normalized expression of indicated gene transcripts. (D) Violin plot of normalized expression of Tgfb1, Cldn4 and Vim in indicated clusters of carcinoma cells; * false discovery rate-adjusted p<10−10 are indicated for significant differences. Y-axis, normalized expression; violin width, cell density in each population. (E) Gene Set Enrichment Analysis (GSEA) of differentially expressed gene signatures in carcinoma cell from clusters 7, 3 and combined EMT (clusters 6,8, and 14). Sources of signatures: H (Hallmark, (Liberzon et al., 2015)); R, www.Reactome.org. Shown are selected signatures with family-wise error rate, FWER<1%. (F) Unsupervised hierarchical clustering of quantified expression of indicated genes. qPCR, quantitative reverse transcription PCR; WB, Western blot. Positive mesenchymal morphology in vitro (EMT) and NSDHL status is indicated above the heatmap by (+). See also Figure S3 and Table S4.
Keratin expressing cancer cells, representative of epithelial carcinomas (Fig. 3C), equaled 35% and 41% of cells from KPPC and KPPCN tumors, respectively. Among these differentiated PDAC cells (i.e., retaining high Epcam, Cdh1, and Cldn4), 67% were gauged in KPPC while only 21% were detected in KPPCN tumors. Contrastingly, a markedly expanded population of cancer cells expressed the mesenchymal markers Vim, Zeb2, and Nes in KPPCN tumors (80%), while KPPC tumors included 33% of these cells (Fig. 3C, S3A and Table S4). Detailed analysis of cells that are well-differentiated (cluster 7), versus those with partial EMT (pEMT; cluster 3) features, showed increased expression of Tgfb1, Vim, Nes and Zeb2 in KPPCN compared to KPPC. Reciprocally, KPPCN cells had lower expression of the biomarkers of a differentiated epithelial lineage (exemplified by Cldn4, Cdh1, and Epcam, Fig. 3C,D and S3A) in cluster 7, and further depletion of these transcripts in the partial EMT (cluster 3). Together, these data suggested that an epithelial cell-intrinsic mechanism was activated by Nsdhl loss leading to EMT via increased transcription of Tgfb1 (Scheel et al., 2011).
Using gene set expression analyses (GSEA), we compared the extended expression profiles of epithelial cluster 7, pEMT cluster 3, and pooled EMT cells from clusters 6, 8 and 14, in KPPC versus KPPCN cells (Fig. 3E). This demonstrated enrichment for EMT, glycolysis and matrisome gene signatures (Liberzon et al., 2015) in all KPPCN clusters indicating progressive acquisition of mesenchymal features concomitant with Tgfb1 upregulation in KPPCN PDAC cells. The transcriptional metabolic reprograming associated with basal PDAC seems to occur early in well-differentiated KPPCN epithelial (cluster 7) cells, which also showed elevated Tgfb1 (Fig. 3D), reduced oxidative phosphorylation, fatty acid, and lipid metabolism signatures as reported in suppression of cholesterogenic program of TGFβ-activated basal PDAC (Fig. 1, (Karasinska et al., 2020)).
The single-cell RNA sequencing generated patterns were validated by qRT-PCR in 11 KPPC and 10 KPPCN primary tumors (Fig. 3G, Fig. S3B and Table S5). Cell lines with both mesenchymal and epithelial features were obtained; for all models, epithelial origin was validated by positivity for CK, and Kras gene rearrangement (Fig. S3C-D). The epithelial vs. mesenchymal features of cells were stable over multiple passages and cloning in vitro. Syngeneic orthotopic implantation of mesenchymal PDAC cells led to development of undifferentiated carcinomas, whereas phenotypically epithelial cells led to emergence of classically differentiated lesions (e.g. classic KPC3 and mesenchymal KPCN349, Fig. S3E).
Five epithelial KPPCN cell lines segregated with epithelial KPPC lines, based on shared expression of CDH1. Mesenchymal cells including 5/10 KPPCN and one KPPC included high expression of Tgfb1, EMT transcription factor Zeb2, and reduced expression of Cdh1, Zo1, Sox17, and Grhl2 (Fig. 3F, S3F). Genes of canonical and non-canonical WNT signaling were upregulated in mesenchymal PDAC cells including Wnt5a, Wnt10b, while a negative regulator of WNT signaling, Rnf43 (Wu et al., 2011), was nearly undetectable (Fig. 3G). In addition to mRNA expression differences, we confirmed elevated secreted TGFβ1 protein in culture supernatants of KPPCN mesenchymal cell lines (Fig. S3G). The expression of CDH1 inversely correlated with phosphorylated SMAD3, a canonical TGFβ pathway effector (Kretzschmar et al., 1999), and with elevated mRNA for Zeb2, Wnt5a, and Wnt10a. In contrast, increased expression of epithelial biomarkers Zo1, Sox17, components of the cholesterol pathway Tm7sf2, Dhcr24 and Ebp, and WNT signaling negative regulator Rnf43, positively correlated with CDH1 (Fig. S3F,H).
NSDHL loss limits PDAC development in the presence of intact p53.
We tested the biological effect of Nsdhl loss in a more indolent PDAC model ((Morton et al., 2010); oncogenic KrasG12D mutation coupled with hemizygous rather than homozygous loss of Trp53 (KPC and KPCN)) and saw that the effect of Nsdhl loss was magnified. Histological evaluation of pancreata from age-matched mice, at 5–6 months, revealed low numbers of high grade PanINs and PDAC lesions in KPCN mice vs. KPC (Fig. 4A), and the PDAC-free survival of KPCN mice was extended (Fig. 4B), while control KPC animals developed PDAC at 2 months with a medium overall survival of ~5 months. Delayed PDAC was observed only in 3 out of 37 KPCN mice (Fig. 4B). PDAC-free survival, at 6 months, in KPCN was 92% compared to 30% in KPC (log rank test p<0.0001). The majority of PDAC in KPC had glandular differentiation (i.e., grade 1/2; Table S3). In contrast, 2 KPCN PDAC showed basal/sarcomatoid histology (grade 4; Fig. S4A).
Figure 4. NSDHL deficiency protects from pancreatic adenocarcinoma development in Trp53+/− heterozygotes.
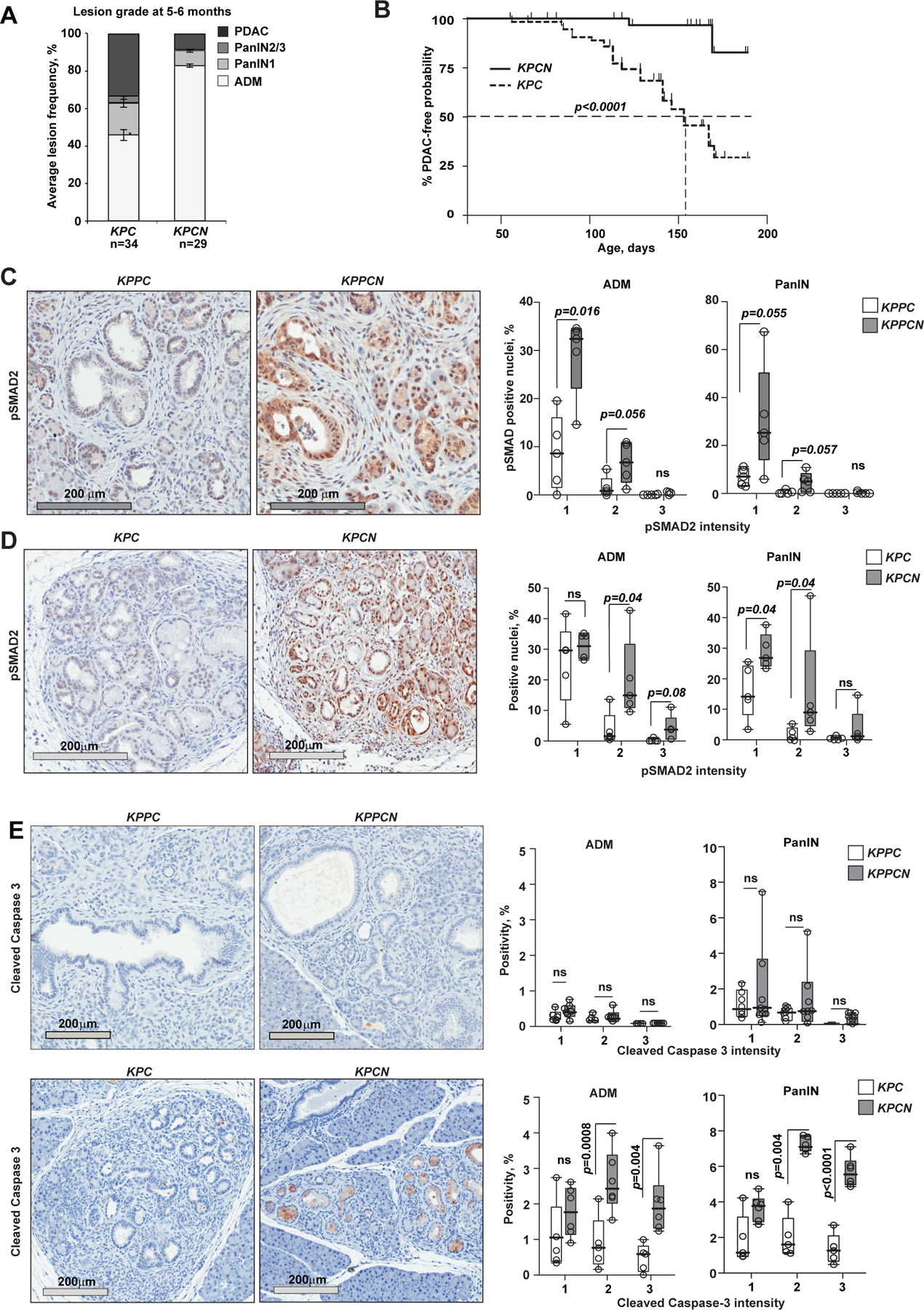
(A) Enumeration of pancreatic epithelial lesion by grade per section in KPC and KPCN mice aged 5–6 months; p=0.035 for PDAC (Fisher’s exact test); p=0.02 for PanIN2/3; p=0.0006 for ADM, Wilcoxon rank-sum test; error bars, SEM. (B) Kaplan-Meier representation of PDAC-free survival of KPC (n=34) and KPCN (n=37) mice. p<0.0001, logrank test. (C, D) Activated TGFβ pathway signaling in NSDHL-deficient pancreatic lesions with homozygous (C) and heterozygous (D) Trp53 knockout as assessed by phosphorylated SMAD2 immunohistochemistry. Top, 5–6 months old KPC and KPCN mice; bottom, 5 week old KPPC and KPPCN pancreatic lesions. Right panel, quantification of pSMAD2-positive nuclei in acinar-to-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasm (PanIN) lesions. (E) Cleaved caspase 3 in pancreatic ADM and PanIN lesions. In C-E, staining intensity was quantified separately in ADM and PanIN lesions; p-values are calculated by Wilcoxon test; ns, not significant; data are represented as boxplots: median (black bar), box (25% to 75% confidence interval), whiskers (full range of measurements). See also Figure S4.
We found that activated SMAD2 (pSMAD2), indicative of EMT related canonical TGFβ activation (David et al., 2016), was elevated in NSDHL-deficient ADM and PanIN lesions in both KPPCN and KPCN, compared to KPPC and KPC lesions (Fig. 4C,D). The tumor-suppressive effect of TGFβ has been linked to caspase-induced apoptosis in KRAS-transformed cells (David et al., 2016); we found cleaved caspase 3 expression exclusively in transformed premalignant lesions of KPCN mice, but neither in normally appearing acini, nor in pancreatic tissues of Nsdhl-wild type KPC mice, or in pancreatic tissues with homozygous deletion of Trp53 (i.e. Trp53−/− mice with KPPC and KPPCN genotypes, Fig. 4E). In contrast, Ki67 showed no difference between KPC and KPCN (Fig. S4B).
To determine if Nsdhl could affect TGFβ signaling, we blocked Nsdhl expression in KPPC cells (KPC3), using CRISPR inhibition of transcription (CRISPRi; (Gilbert et al., 2013)). Loss of NSDHL, confirmed by Western blot, resulted in increased cellular pSMAD2 expression, and TGFB1 secretion (Fig. 5A,B). Increased mesenchymal features of Nsdhl knockdown were stable in vivo, as orthotopic syngeneic implantation of NsdhlCRISPRi cells produced predominantly undifferentiated tumors. In contrast, similarly implanted control KPC3 cells mostly formed well-differentiated tumors (Fig. 5C).
Figure 5. Cholesterol depletion activates TGFβ pathway signaling in PDAC cells.
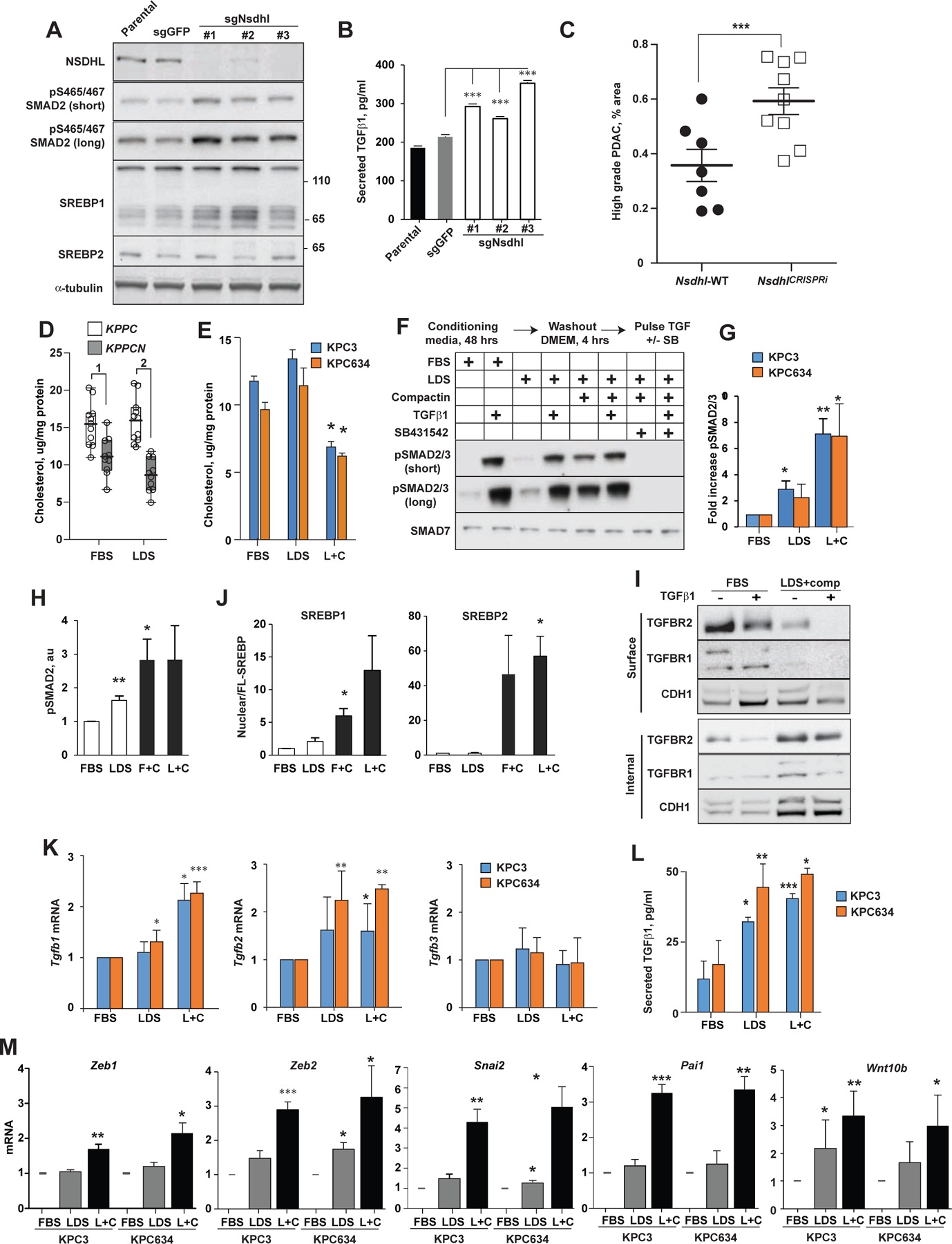
(A) NSDHL inactivation in well-differentiated KPC3 PDAC cells by CRISPRi is confirmed by absence of NSDHL band on Western blot of total cellular lysates; KPC3 parental cells and GFP-targeted gRNA used as controls. (B) Secreted TGFB1 as determined by ELISA using 48 hour supernatants. Shown, averaged results from 3 independent repeats; error bars, SEM. (C) Histological grading of glandular (grades 1–2) versus solid (grades 3–4) tumor areas in tumors generated via orthotopic implantation of KPC3wt or KPC3 NsdhlCRISPRi cells; p=0.007, two-way Student t-test. Symbols represent individual tumors; black bars, mean±SEM. (D) Cholesterol level in KPPC (n=10) and KPPCN (n=10) clones grown for 48 hours in FBS or LDS media; (1) p=0.004; (2) p=0.0001, Wilcoxon test. Boxplots represent median (black bar) and full range of measurements. (E) Cholesterol levels in PDAC cells conditioned for 48 hours as indicated; L+C, 5%LDS with 1 μM compactin. (F) Representative Western blot of phosphorylated pSMAD2 (Ser465/467) and pSmad3 (Ser423/425) in KPC3 cells cultured for 48 hours in fetal bovine serum (FBS), lipid depleted serum (LDS), or LDS with 1 μM of compactin (L+C) followed by incubation in serum-free DMEM for 4 hours. Indicated samples were treated with TGFβ1 at 10 ng/ml for 30 minutes, and/or SB431542 at 25 μM for 1 hour. (G) Summary results of levels of phosphorylated pSMAD2(Ser465/467) and pSmad3 (Ser423/425) in cholesterol depleted PDAC cells. Results from 3 independent experiments normalized to α-tubulin are shown. (H) Phosphorylated pSMAD2(Ser465/467) in human Capan-2 carcinoma cells conditioned for 48 hours in FBS, LDS with or without 1 μM compactin. Summary results from 3 independent experiments normalized to α-tubulin are shown. (J) Increased nuclear SREBP1 and SREBP2 in Capan-2 cells as in H. (I) Surface versus internalized pools of TGFBR1 and TGFBR2 in KPC3 cells conditioned for 48 hours in 5% FBS or in 5% LDS with 1 μM compactin. Biotinylated (surface) and non-biotinylated (internalized) proteins were affinity separated using streptavidin-agarose beads. (K) Levels of Tgfb1, Tgfb2 and Tgfb3 mRNA as assessed by qRT-PCR, in cells grown in indicated media for 48 hrs. (L) ELISA measurement of secreted TGFβ1 in supernatants of KPC3 cells conditioned in indicated media for 48 hrs; (M) Expression of Zeb2, Tgfb1 and Wnt10b mRNA as assessed by qRT-PCR in KPC3 and KPC634 PDAC cells cultured for 48 hours in media supplemented with FBS, LDS or LDS+compactin (1 μM). In graphs B, E, G-J and K-M, statistical p-values by two-way Student t-test are indicated as: *, <0.05, **, <0.01, ***, <0.001. See also Figure S5.
Inhibition of the cholesterol pathway regulates autocrine TGFβ signaling via SREBP1.
To investigate how loss of Nsdhl activates TGFβ signaling, we modulated lipid pools in PDAC cells. Baseline values for total cellular cholesterol were ~25% lower in KPPCN vs. KPPC cells cultured in lipid containing medium (with fetal bovine serum, FBS, Fig. 5D). This suggested that exogenous lipids cellular uptake does not compensate for the reduced endogenous cholesterol biosynthesis. These differences were increased (40% lower in KPPCN; Fig. 5D) with lipid-depleted serum (LDS). Expression of NSDHL substrate 4α-carboxy-4-methyl-cholest-8(9)-en-3β-ol (Gabitova et al., 2015) was selectively elevated in KPPCN cells during a short-term incubation in serum-free media, confirming a stage specific block in cholesterol biosynthesis (Fig. S5A). Conditioning KPPCN PDAC and NsdhlCRISPRi KPC3, but not parental KPC3 cells, in LDS resulted in growth arrest (Fig. S5B).
We also cultured KPPC cells (KPC3 and KPC634) for 48 hours in LDS medium, with or without non-toxic concentrations (1 μM) of compactin (also known as mevastatin), a 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor (Brown et al., 1978; Radhakrishnan et al., 2008), which resulted in 40% decrease in cellular cholesterol composition and in activation of SREBP targets (Fig. 5E, S5C,D). LDS+compactin activated the TGFβ pathway, reaching comparable levels to the ones following a 30-minute pulse with TGFβ1 in cholesterol-rich media containing FBS (Fig. 5F, G). Increased pSMAD2/3, induced by LDS+compactin, was eliminated with SB431542, an inhibitor of type I TGFβ receptor 1 (TGFBR1; Fig. 5F). Silencing of TGFBR1, but not of homologous activing receptors ACVR1B and ACVR1C, with siRNA abrogated induction of pSMAD2/3 in cholesterol-depleted KPC3, indicating activation was predominantly mediated via TGFβ receptors (Fig. S5E). Similar results were obtained in human PDAC Capan-2 cells (Fig. 5H,J), which showed activation of SREBP1 and SREBP2 (Fig. 5J).
We next evaluated whether cholesterol deprivation altered cellular distribution of TGFBR1 and TGFBR2. Indeed, surface biotin labeling of KPC3 PDAC cells cultured for 48 hours in LDS medium+1 μM compactin showed marked reduction in surface TGFBR1, TGFBR2, whereas the unlabeled intracellular fractions increased (Fig. 5I), suggesting internalization of ligand-bound receptor. Addition of 10 ng/ml recombinant TGFβ1 further exacerbated this phenotype. Use of LDS+compactin in KPC3 and KPC634 cells induced Tgfb1 and Tgfb2, but not Tgfb3 transcripts (Fig. 5K), and increased secretion of TGFβ1 (Fig. 5L). We determined that cholesterol starvation of epithelial PDAC cells increased the expression of Zeb1, Zeb2, Snai2, Pai1 and Wnt10b (Fig. 5M), known to be induced by canonical TGFβ signaling (Scheel et al., 2011)(Fig. 3G).
We next tested if TGFβ-induced EMT is regulated by SREBPs in lipid-starved pancreatic cancer cells. Whereas cholesterol supplementation repressed SREBP2 cleavage (Adams et al., 2004; Radhakrishnan et al., 2008), SREBP1, in contrast, is negatively regulated by non-sterol lipids, such as unsaturated fatty acids and phosphatidylcholine, repress SREBP1 (Ou et al., 2001; Walker et al., 2011). Accordingly, supplementation of compactin-treated KPC3 cells with cholesterol did not suppress nuclear SREBP1 and had no effect on pSMAD2 expression. Adding low-density lipoproteins (LDL) to compactin-treated KPC3 cells robustly reduced nuclear SREBP1 and pSMAD2, implying a reduction of TGFβ signaling (Fig. 6A, B). Examination of the Tgfb1 promoter, using TFBIND algorithm (Tsunoda and Takagi, 1999), revealed multiple candidate SREBP1 binding motifs (Fig. S6A); no candidate sites were found in the Tgfb3 promoter (not shown). These patterns were consistent with the changes in the mRNA levels for these genes upon cholesterol starvation (Fig. 5K). To assess SREBP responsiveness of Tgfb1 transcription, we used fatostatin, which binds to the SREBP cleavage-activating protein SCAP and blocks proteolytic formation of the active nuclear fragments of SREBPs (Fig. S5C) (Kamisuki et al., 2009). Addition of fatostatin to KPC3 cells in LDS abrogated secreted TGFβ1 and cellular pSMAD2 (Fig. 6C, D).
Figure 6. Cholesterol-sensitive transcription factor SREBP1 regulates Tgfb1 expression.
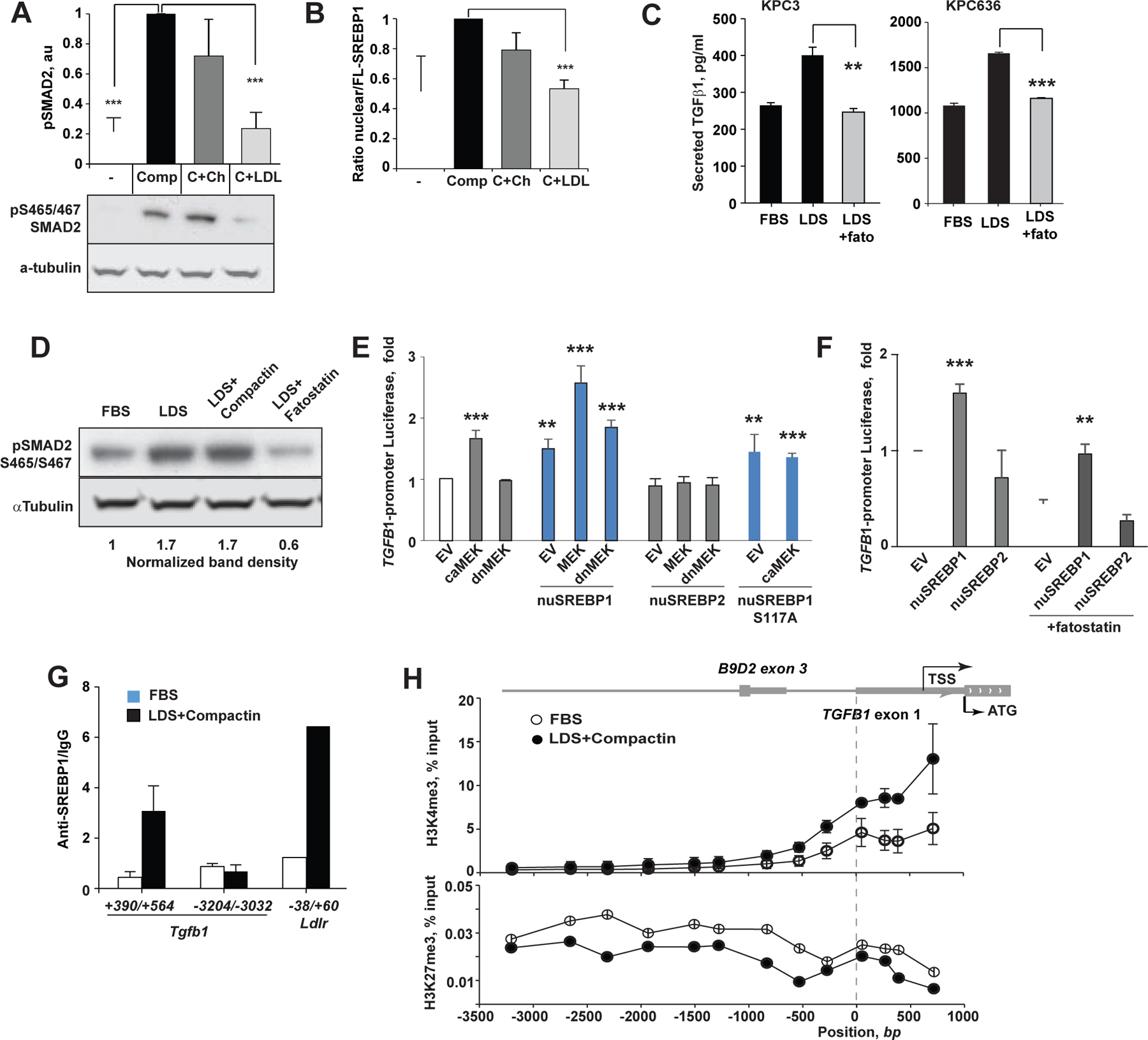
(A) Supplementation of serum-free KPC3 cultures with LDL (100 μg/ml), but not with 50 μM ethanol-solubilized cholesterol, reverses pSMAD2 induction by compactin. Shown are results of 3 independent repeats and a representative panel below. (B) Compactin-induced activation of SREBP1 is reversed by addition of LDL quantified as ratio of nuclear and full length (FL) protein. (C) Secreted TGFβ1 suppression by SREBP inhibitor. Fatostatin (20 μM) was added to KPC3 cells grown in the indicated media for 48 hours. Averaged results of 3 independent ELISA assays are shown. (D) Representative Western blot of pSMAD2 of cellular lysates corresponding to (C). (E) Human TGFB1 promoter-dependent luciferase reporter activity following co-transfection into HEK293T cells with plasmids expressing nuclear fragments of SREBP1 (aa 1–480), SREBP2 (aa 1–473), constitutively active MEK1 (S218D/S222D), or dominant negative MEK1 mutant (S218A/S222A). Empty vectors (EV) were used as negative controls. (F) TGFB1-luciferase reporter activity in human PDAC cells MiaPaCa2 co-transfected with nuclear SREBP1 or SREBP2. Fatostatin at 10 μM was used to block the endogenous SREBP activation. (G) Chromatin immunoprecipitation and quantitative PCR (ChIP-qPCR) determination of genomic Tgfb1 DNA binding (amplicon +390 bp to +564 bp) by the endogenous SREBP1 in KPC3 cells conditioned for 48 hours in FBS, or LDS+ 1 μM compactin. Amplicon −3204 bp to −3032 bp distant to TSS served as negative control, whereas a canonical SREBP1 binding site in Ldlr promoter (−38 bp to +60 bp) served as a positive control for SREBP1 activity. (H) Increased association of open chromatin (H3K4me3) and reduced association of repressed chromatin marks (H3K27me3) with the proximal Tgfb1 promoter of cholesterol-depleted KPC3 cells as determined by ChIP-qPCR. The map of genomic Tgfb1 locus is drawn to scale. Data were pooled from two independent experiments. In all figures, data are represented as mean±SEM, p-values determined by independent two-sample Student t-test: *, p<0.05; **, p<0.01, ***, p<0.001. See also Figure S6.
We then co-transfected a 843 bp fragment of the human TGFB1 promoter containing 5 putative SREBP1 binding motifs linked to a luciferase reporter (Yeh et al., 2018) (Fig. S6A) with or without a plasmid expressing the activated nuclear fragment of SREBP1, into HEK293 and MiaPaCa-2 cells. Two-fold induction of luciferase expression was observed, compared to luciferase reporter alone, while nuclear SREBP2 fragment did not induce the TGFB1 promoter (Fig. 6E, F). Activation of an LDLR reporter by both SREBP constructs served as controls (Fig.S6B-D). Activated ERK1/2 (downstream to KRAS) phosphorylates serine 117 of SREBP1 (Roth et al., 2000). We introduced a constitutively active form of MEK1 (caMEK, mutations S218D/S222D (Murakami et al., 1999)), into HEK293, to activate mitogen-activated protein kinase (MAPK) pathway. Transfection of nuclear SREBP1 in combination with caMEK1 induced Tgfb1 luciferase reporter to a greater level than with either construct alone, while overexpression of the inactive form of MEK1 (dnMEK, mutations S218A/S222A (Zheng and Guan, 1994)), or Ser117 to Ala mutant of nuclear SREBP1 (S117A, Fig. 6E) did not.
We validated SREBP1 binding to the endogenous Tgfb1 non-coding 5’-locus downstream of the transcription start site (TSS) in KPC3 PDAC cells conditioned in LDS+compactin. In contrast, binding to this site was at background level in cells cultured in FBS (Fig. 6G). Treatment of KPC3 cells with LDS+compactin resulted in increased association of active chromatin marks (tri-methylated lysine 4 of histone H3 (H3K4me3) with sequences proximal to transcription start site (TSS) of Tgfb1, and reciprocal decrease in repressed chromatin marks (HeK27me3) as compared to cholesterol replete conditions (Fig. 6H). We conclude that cholesterol depletion promotes the EMT in PDAC by inducing sterol-responsive SREBP1 transcription to activate autocrine TGFβ signaling.
Statins condition PDAC basal differentiation.
To investigate the effect of pharmacological inhibition of cholesterol biosynthesis on PDAC differentiation, we treated cohorts of KPPC mice with atorvastatin (10 mg/kg/day) or vehicle starting at age 4 weeks (Fig. S7A) until PDAC development required euthanasia. Most animals were exposed to atorvastatin for only 3–5 weeks, due to rapid PDAC development in this model (Fig. S7A). Treatment with atorvastatin resulted in shorter survival of the animals (p=0.008, log rank test, Fig. S7B). In the 3 mice with the largest grade 4 areas, PDAC tumors showed poorly differentiated (grade 4) histology and loss of CDH1 expression (Fig. S7C, D). Use of multiplex immunofluorescence (Franco-Barraza et al., 2017) (Fig. 7A) demonstrated that atorvastatin-treated mice presented with a preponderance of EMT-PDAC cells; estimated as percent of tumor area covered with CK+/VIM+ cells (Fig. 7B), which corresponded to areas occupied by grade 4 carcinoma (two-tailed Mann-Whitney test, p=0.001, Fig. 7C).
Figure 7. Inhibition of cholesterol biosynthesis with statins promotes basal PDAC development.
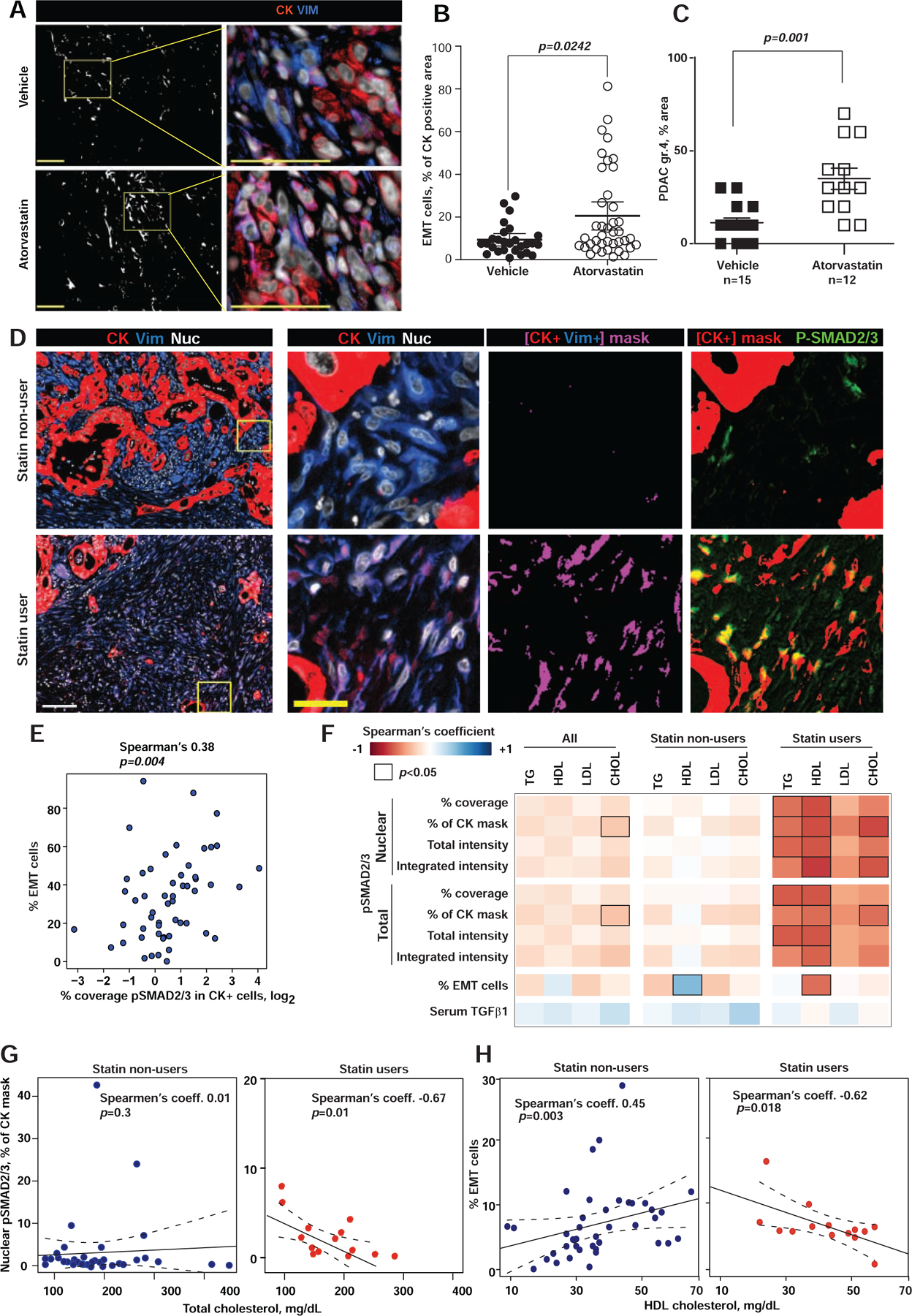
(A) Epithelial (cytokeratin, CK, red) and mesenchymal (vimentin, VIM, blue) compartments in murine pancreatic KPPC tumors. Digital mask in white corresponds to CK+/Vim+ areas, used to quantify EMT cells (magenta). Nuclei are in gray. Scale bars = 100 μm. (B) Graph shows EMT areas (dot represent single images). (C) Percent area with grade 4 PDAC in KPPC mice treated with atorvastatin (open symbols) or vehicle (closed symbols). (D) From left to right, first column shows Epithelial (CK, red), mesenchymal (VIM, blue) and nuclei (grey) compartments in human PDAC tissue. The following three columns correspond to magnifications of the yellow boxed regions. Third column depicts EMT areas (i.e., CK+/Vim+ in magenta masks). Last column includes CK+ masks in red, which were overplayed with immunofluorescent pSMAD2/3 (Green) to highlight co-localization areas (yellow). Scale bars: white=100 μm, yellow=25 μm. (E) Spearman’s correlations of pSMAD2/3 and the percent of CK+/VIM+ cells (EMT-PDAC cells). (F) Heat map of Spearman’s correlation coefficient for nuclear and total pSMAD2/3 and serum lipids, correlations with p<0.05 are outlined. (G, H) Correlation of total serum cholesterol with nuclear pSMAD2/3 expression in PDAC cells (G) and percent of EMT-PDAC cells (H) in statin users. In figures B and C, data are represented as mean±SEM, p-values determined by Mann-Whitney test with indicated p-values. See also Figure S7.
We next determined if systemic blood levels of cholesterol and long-term exposure to statins stratify with PDAC differentiation states. Archival surgical PDAC tissues from 55 untreated patients were matched with blood samples collected prior to pancreatectomy. Statin use information in these patients was available. Multiplex immunofluorescence of the tissues showed higher CK+/VIM+ (i.e., EMT) and pSMAD2/3 (Spearman’s coefficient 0.38, p=0.004) in statins treated patients (Fig. 7D, E).
We compared serum lipids with PDAC EMT numbers and pSMAD2/3 levels and cellular distributions. Serum levels of total cholesterol, triglycerides, cholesterol in high density lipoproteins (HDL) and low-density lipoproteins (LDL) were comparable between patients taking (n=15) and not taking statins (Fig. S7C). Data showed and inverse correlation between total and nuclear pSMAD2/3 (Fig. 7F,G) plus EMT percentages vs. serum triglycerides and cholesterol levels, in statin users (p=0.018, Fig. 7H). Suggesting that restricting cholesterol content in PDAC cells with statins may support basal PDAC differentiation.
Discussion.
Given the known linkages between diet, obesity, and the risk and prognosis of aggressive cancers (Golemis et al., 2018), there has long been interest in the relationship between cholesterol and PDAC pathogenesis (Huang et al., 2017). The complex signaling mechanism revealed in our study offers one explanation as to why such relationships have been difficult to establish, and for the poor clinical outcomes in trials of statins for PDAC (Hong et al., 2014). Data collected in this study provide evidence for metabolically-determined plasticity in PDAC differentiation mediated by cholesterol-sensitive, SREBP1-dependent regulation of TGFβ expression, which causes TGFβ receptor activation and induction of a canonical SMAD2/3 effector cascade (Graphical Abstract). Our study proposes the cholesterol pathway as a metabolic trigger for the EMT, whereas other factors, such as recently reported GLI1 and GLI2 (Adams et al., 2019), may regulate lineage commitment and viability in established EMT cells.
Genetic ablation of Nsdhl provides a useful model for dissection of cholesterol signaling (Gabitova et al., 2015). While high expression of cholesterol biosynthesis genes is a feature of classic PDAC, we report that low mRNA expression of NSDHL and other cholesterol biosynthesis genes, is prevalent in human basal PDAC (Fig. 1E). Our model integrating the data from genetic and pharmacological studies demonstrates that lowering cholesterol in non-EMT epithelial cancer cells triggers activation of both SREBP1 and SREBP2. We parsed out the transcriptional activity of SREBP1, from SREBP2, and saw it mediates increased Tgfb1 transcription (Fig. 6). Supplementation of serum-free cultures containing compactin with cholesterol failed to suppress SREBP1, whereas addition of LDL suppressed SREBP1 nuclear fragment generation and lowered secreted TGFB1 (Fig. 6A). Mouse single-cell RNA sequencing data, focusing on carcinoma cells, showed that the Nsdhl conditional knockout and pharmacologic treatment of KPPC tumors with atorvastatin render similar results. In both cases, the arrest of endogenous cholesterol biosynthesis induces autocrine TGFβ abundance and increases the preponderance of partial and/or full EMT cell clusters (Fig. 3, 7).
There are some limitations of this study. Modeling EMT in vitro is notoriously difficult because epithelial cells do not fully acquire mesenchymal characteristics (Katsuno et al., 2019; Scheel et al., 2011; Zhang et al., 2014). This is thought to reflect the absence of pro-mesenchymal stimuli provided by the in vivo tumor microenvironment. Our in vitro experiments do not fully recapitulate the complexity of EMT, based on comparison of transcriptional profiles of in vitro versus in vivo transcriptomes (Ligorio et al., 2019). In spite of these differences, the correlation between suppression of the cholesterol biosynthetic pathway, and activation of SREBP1 and Tgfb1 is markedly consistent across assay systems.
Our findings also provide a mechanistic framework for cholesterol suppression as means of cancer prevention (Bang et al., 2018). Inactivation of Nsdhl in the context of a heterozygous Trp53+/− dramatically delayed malignant progression of the precursor lesions and nearly abrogated PDAC development, in contrast to the accelerated growth of late-arising PDACs in the Trp53−/− genetic background (Fig. 2A, 4B). We hypothesize that blockade of cholesterol biosynthesis, which triggers activation of TGFβ signaling, could initiate apoptosis (David et al., 2016) and prevent PDAC selectively in a functional p53 dependent manner (Cordenonsi et al., 2003).
It is possible that basal PDAC are selected to bypass strong pro-apoptotic effects of TGFβ induced in tumors arising in individuals on statins. If statins act differently in patients treated with these drugs, prior versus post acquisition of TP53 mutations, this would considerably complicate identification of a clear relationship between statin use, PDAC incidence, and survival. More speculatively, a precipitous drop in blood lipids could contribute to PDAC aggressiveness, associated with the pre diagnosis (Chen et al., 2019) cachexia and malnutrition (Judge et al., 2018). Hence, our findings suggest an explanation for the reported strong favorable effect of high expression of the cholesterol homeostasis mRNA signature on PDAC life expectancy (Karasinska et al., 2019). Further, this study reveals that reduced expression of cholesterol homeostasis signature in a subset of basal of PDAC tumors confers low survival (Fig. 1D).
STAR Methods.
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Igor Astsaturov (igor.astsaturov@fccc.edu).
Materials Availability.
Mouse lines generated in this study are maintained at Fox Chase Cancer Center and will be available upon request.
Data and Code Availability.
The datasets generated during this study are available at Sequence Read Archive (SRA), https://www.ncbi.nlm.nih.gov/sra, deposition PRJNA530747, and GSE156210.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models.
Mice carrying a conditional knockout allele of Nsdhl (Cunningham et al., 2015) were kindly provided by Dr. Gail Herman (The Research Institute at Nationwide Children’s Hospital, Columbus, OH). These mice (official name: Nsdhltm1.1Hrm, MGI:5581334, designated as Nsdhlf/f here) are congenic on a C57BL/6J background. KPC (LSL-KrasG12D;Trp53f/f; Pdx1-Cre) and KC (LSL-KrasG12D; Pdx1-Cre) mice were provided by Dr. Kerry Campbell (Fox Chase Cancer Center, Philadelphia, PA) and are congenic on a C57BL/6J background. Mice were bred to obtain the desired genotype with Nsdhl depletion in normal pancreas (Nsdhlf/f; Pdx1-Cre, designated as Nsdhl∆Panc here) and Nsdhl depletion in pancreas of KPPC/KPC mice with loss two or one floxed p53 alleles (KPC mice carrying Trp53f/f or Trp53f/+) or wt p53 (KC mice). All mice were bred and maintained under defined-flora pathogen-free conditions at the AAALAC-approved Animal Facility of the Fox Chase Cancer Center, Philadelphia, PA. Mice of both genders, equally distributed, were used for experiments. Tumor-bearing mice were observed twice weekly until signs of sickness appeared or animals showed distress or weight loss of more than 10%, per the local Institutional Animal Care and Use Committee (IACUC) guidelines.
Cell Lines.
Mouse pancreatic cancer cell lines (KPPC and KPPCN) were derived from mouse pancreatic tumors by tumor dissociation and subsequent fluorescence-activated cell sorting (FACS). Sorted cells were propagated for first two passages in enriched media (RPMI-1640 supplemented with 15% v/v FBS, 2mM L-glutamine, 100μg/ml Penicillin/Streptomycin, 20ng/ml EGF, 25μg/ml Insulin, NEAA 1x, 1mM Na-Pyruvate and 2μg/ml Hydrocortizone) and for subsequent passages in DMEM supplemented with 10% v/v FBS and 2mM L-glutamine with 100μg/ml Penicillin/Streptomycin. Cells were used at passages 3–5. HEK293T, Capan-2, and MIA PaCa-2 cells were obtained from ATCC.
Human pancreatic adenocarcinoma tissue and blood samples.
Human tissues were collected under exemption-approval of the Center’s Institutional Review Board. Patients signed a written informed consent agreeing to donate samples to be used solely for research purposes. In strict observance of patients’ identities, samples were coded and distributed by the Institutional Biosample Repository Facility to the researchers. Tissue microarrays were assembled, by a certified pathologist, as previously described (Franco-Barraza et al., 2017). Briefly, two cores representative of individual pancreatic adenocarcinoma cases were obtained from de-identified surgical samples and assembled in tissue microarrays at the Institutional Biosample Repository Facility. Blood sera samples matched to the PDAC tissues were obtained 2–3 weeks before surgery and stored at −80°C until used for cholesterol measurements (Fox Chase Cancer Center Clinical Laboratory) and TGFβ1 ELISA.
METHOD DETAILS
Isolation of murine pancreatic carcinoma cells.
Tumor dissociation was performed using a Gentle MACS Tumor Dissociation Kit (Miltenyi Biotec, Order No. 130–096-730) according to the manufacturer’s instructions. Briefly, each tumor was isolated from the animal in a sterile environment and washed in PBS. A 1 mm3 piece was taken for genotyping and a larger piece was taken for histopathology analysis. The rest of the tumor tissue was placed in the dissociation enzyme mix and minced quickly to get pieces ~2mm3 in size. Then the enzyme-tissue mixture was transferred into the gentleMACS C tube and incubated at 37°C with constant rotation for 40 minutes. After that the tissue was further mechanically processed by the gentleMACS Dissociator. A single-cell suspension was obtained by passing the tissue mixture through a 70μm cell strainer. Dead cells were subsequently removed by the Dead Cell Removal Kit (Miltenyi Biotec, Order No. 130–090-101). In order to separate cells of different lineage we further stained the cell suspension with antibodies against CD45 (Biolegend #103107; 1:200), FAP (Abcam #ab28244; 1:50), EPCAM (Biolegend #118212; 1:200) and PDGFRα (CD140a) (Biolegend #135905; 1:80). To prevent antibodies from binding to Fc-receptors, cell suspensions were treated with Fc-block (Biolegend #101301; 1:50) prior to using other antibodies. Live cells were selected based on propidium iodide staining (Biolegend #421301). Fluorescence detection and sorting were performed with a BD FACS Aria II flow cytometer.
Induction of acute pancreatitis in mice with caerulein.
Acute pancreatitis was induced by caerulein (#C9026, Sigma-Aldrich) treatment as previously described (Morris et al., 2010). Mice were injected with 50 μg/kg of caerulein i/p every hour for 6 hours (6 injections total) daily for two days. Control mice were injected with saline. In 2, 5, and 7 days, pancreatic tissues were collected and immediately fixed in formalin. Fixed tissues were embedded in paraffin and stained with H&E and Ki-67 for further histological analysis.
Atorvastatin treatment in vivo.
To analyze the effect of statin treatment on differentiation and progression of pancreatic tumors, KPC mice were treated with Atorvastatin (PHR1422–1G, Sigma) prepared as aqueous suspension at 10 mg/kg of mouse body weight by oral gavage. Animals were treated beginning from their weaning date (approximately 4 weeks old) daily, five days a week. Mice were kept under defined-flora pathogen-free conditions at the AAALAC-approved Animal Facility of the Fox Chase Cancer Center, Philadelphia, PA. Mice of both genders, equally distributed, were used for the experiment. Tumor-bearing mice were observed daily five days a week at the time of oral gavage. Treatment was performed until signs of sickness appeared or animals showed distress or weight loss of more than 10%, per the local Institutional Animal Care and Use Committee (IACUC) guidelines. Tissue was collected, fixed and stained as described above.
Micro-magnetic resonance imaging (mMRI).
Animals were imaged in a 7 Tesla vertical wide-bore magnet, using a Bruker DRX 300 spectrometer (Billerica, MA) with Magnevist contrast (#50419–188-82, Bayer Healthcare Pharmaceuticals Inc.). A two-dimensional spin echo pulse sequence was employed with echo time 15 msec, repetition time 630 msec, field of view=2.56 cm, acquisition matrix=256x256, slice thickness=0.75 mm, 2 averages, and scan time=5 minutes. Fat suppression (standard on Bruker DRX systems) was used for all scans.
Orthotopic implantations.
Orthotopic implantations of murine PDAC tumor cells to mouse pancreas were performed as described by Kim et al. (Kim et al., 2009): 106 cells per 50 μl of a 30% Matrigel:70% PBS mixture were injected into the pancreatic tails of syngeneic C57BL/6J mice. Anesthetics and analgesics were used according to the local IACUC guidelines. Tumor-bearing mice were observed twice weekly until signs of sickness appeared, or animals showed distress or weight loss of more than 10%, per the local IACUC guidelines.
Western blot analyses of protein expression.
For Western blot analysis, dispersed tissue or cultured cells were homogenized in RIPA buffer (#24928, Santa Cruz) with phosphatase and protease inhibitors (#1862495, #1861278, Thermo Fisher Scientific) on ice and cleared then by centrifugation. The protein concentration was measured with a Pierce BCA Protein Assay Kit (#23225, Thermo Fisher Scientific). Proteins were separated on 4–12% Bis-Tris Protein gels (Invitrogen) and then horizontally transferred to the Immobilon-FL PVDF membrane (#IPFL00010, Millipore). Primary and secondary antibodies were used at the concentrations indicated in the Key Resources Table according to manufacturer’s instructions. The density of obtained bands was quantified with Image Studio software (LI-COR).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Pan-cytokeratin (for IHC 1:100) | Abcam | Cat#ab9377; RRID:AB_307222 |
| Mouse monoclonal anti-EpCAM/TROP1 (clone MOC-31) (for SMI 1:200) | Novus Biologicals | Cat# NBP2-48287 RRID:AB_2857921 |
| Mouse monoclonal anti-CD70 (clone 113-16) (for SMI 1:50) | Biolegend | Cat# 355102 RRID:AB_2561429 |
| Mouse monoclonal anti-insulin (clone 2D11-H5) (for SMI 1:30000 | Santa Cruz | Cat#sc-8033; RRID:AB_627285 |
| Mouse monoclonal anti-amylase clone G-10 (for SMI 1:200) | Santa Cruz | Cat# sc-46657; RRID:AB_626668 |
| Rabbit monoclonal anti-pSMAD3 (pS423+S425) (clone EP823Y), (for WB 1:1000) | Abcam | Cat#ab52903; RRID:AB_882596 |
| Rabbit monoclonal anti-pSMAD2 (Ser465+467)/Smad3 (Ser423+425) (clone D27F4) (for SMI 1:200) | Cell Signaling | Cat# 8828; RRID:AB_2631089 |
| Rabbit polyclonal anti-FAP (for FC 1:50) | Abcam | Cat#ab28244; RRID:AB_732312 |
| Rabbit monoclonal anti-vimentin (clone EPR3776) (for IF 1:200) | Abcam | Cat#ab92547; RRID:AB_10562134 |
| Rabbit polyclonal anti-Ki-67 (for IF 1:50) | Abcam | Cat#ab15580; RRID:AB_443209 |
| Rabbit anti-SREBP2 polyclonal antibody | Abcam | Cat#ab30682; RRID:AB_779079 |
| Goat polyclonal anti-rabbit BV421secondary antibody (for FC 1:50) | BD Biosciences | Cat#565014; RRID:AB_2716308 |
| Rat monoclonal anti-CD45-FITC (clone 30-F11) (for FC 1:200) | Biolegend | Cat#103107; RRID:AB_312972 |
| Rat monoclonal anti-Ep-CAM-Alexa Fluor 647 (clone G8.8) (for FC 1:200) | Biolegend | Cat#118212; RRID:AB_1134101 |
| Rat monoclonal anti-PDGFRa-PE (clone APA5) (for FC 1:80) | Biolegend | Cat#135905; RRID:AB_1953268 |
| Rat monoclonal anti-CD16/32 (clone 93) (Fc-block) (for FC 1:50) | Biolegend | Cat#101301; RRID:AB_312800 |
| Rabbit monoclonal anti-Tri-Methyl-Histone H3 (Lys27) (clone C36B11) (for ChIP 10µl/10µg of chromatin) | Cell Signaling | Cat#9733S; RRID:AB_2616029 |
| Rabbit monoclonal anti-Tri-Methyl-Histone H3 (Lys4) (clone C42D8) (for ChIP 10µl/10µg of chromatin) | Cell Signaling | Cat#9751S; RRID:AB_2616028 |
| Rabbit polyclonal anti-Caspase-3 (for IHC 1:200) | Cell Signaling | Cat#9662; RRID:AB_331439 |
| Rabbit monoclonal anti-E-Cadherin (clone 24E10) (for IHC 1:100, for WB 1:1000) | Cell Signaling | Cat#3195; RRID:AB_2291471 |
| Mouse monoclonal anti-alpha-Tubulin (clone DM1A) (for WB 1:1000) | Cell Signaling | Cat#3873; RRID:AB_1904178 |
| Rabbit monoclonal anti-pSMAD2 (Ser465+467)/Smad3 (Ser423+425) (clone D27F4) (for IF on tissues 1:200) | Cell Signaling | Cat# 8828; RRID:AB_2631089 |
| Goat anti-rabbit IgG HRP-linked (for WB 1:3000) | Cell Signaling | Cat#7074; RRID:AB_2099233 |
| Horse anti-mouse IgG HRP-linked (for WB 1:3000) | Cell Signaling | Cat#7076; RRID:AB_330924 |
| Rabbit polyclonal anti-TGFBR2 (for WB 1:1000) | Cell Signaling | Cat#79424; RRID:AB_2799933 |
| Rabbit monoclonal anti-beta-actin (clone 13E5) (for WB 1:1000) | Cell Signaling | Cat#4970; RRID:AB_2223172 |
| Mouse monoclonal anti-HA-tag (clone 6E2) (for WB 1:1000) | Cell Signaling | Cat#2367S; RRID:AB_10691311 |
| Rabbit polyclonal anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (for WB 1:1000) | Cell Signaling | Cat#9101; RRID:AB_331646 |
| Mouse monoclonal anti-p44/42 MAPK (Erk1/2) (clone 3A7) (for WB 1:1000) | Cell Signaling | Cat#9107; RRID:AB_10695739 |
| Rabbit monoclonal anti-c-Jun (clone 60A8) (for WB 1:1000) | Cell Signaling | Cat#9165; RRID:AB_2130165 |
| Normal rabbit IgG (for ChIP 10µl/10µg of chromatin) | Cell Signaling | Cat#2729S; RRID:AB_1031062 |
| Rabbit monoclonal anti-phospho-Smad2 (Ser465/467)/Smad3 (Ser423/425) (clone D27F4) (for WB 1:1000; for IF 1:200) | Cell signalling | Cat#8828; RRID:AB_2631089 |
| EnVision+ System- HRP Labelled Polymer Anti-Rabbit | Dako | Cat#K4003; RRID:AB_2630375 |
| EnVision+ System- HRP Labelled Polyme Anti-Mouse | Dako | Cat#K4001 |
| Mouse monoclonal anti-cytokeratin (clone AE1/AE3) (for IF 1:40) | DAKO | Cat#M3515; RRID:AB_2132885 |
| Rat monoclonal anti-E-Cadherin, Biotin (clone DECMA-1) (for FC 1:200) | eBioscience | Cat#13-3249-82; RRID:AB_1659688 |
| Rabbit polyclonal anti-phospho-SMAD2 (Ser465, Ser467) (for IHC 1:1000; for WB 1:1000) | Invitrogen | Cat#44-244G; RRID:AB_2533614 |
| Polyclonal Cy™3 AffiniPure Donkey Anti-Rabbit IgG (H+L) (for IF 1:100) | Jackson ImmunoResearch | Cat#711-165-152; RRID:AB_2307443 |
| Polyclonal Cy™2 AffiniPure Donkey Anti-Mouse IgG (H+L) (for IF 1:100) | Jackson ImmunoResearch | Cat#715-225-151; RRID:AB_2340827 |
| IRDye 800CW Goat polyclonal anti-Rabbit IgG (for WB 1:20000) | LI-COR | Cat#925-32211; RRID:AB_2651127 |
| IRDye 680RD Goat polyclonal anti-Mouse IgG (for WB 1:20000) | LI-COR | Cat#925-68070; RRID:AB_2651128 |
| Rabbit polyclonal anti-NSDHL (for WB 1:500) | Proteintech Group Inc. | Cat#15111-1-AP; RRID:AB_2155681 |
| Mouse monoclonal anti-SMAD7 (clone #293039) (for WB 1:1000) | R&D Systems | Cat#MAB2029-SP; RRID:AB_2193479 |
| Rat monoclonal anti-TGFBR1 (clone #141231) (for WB 1:1000) | R&D Systems | Cat#MAB5871; RRID:AB_2202335 |
| Mouse monoclonal anti-SREBP-1 (clone 2A4) (for WB 1:250, for ChIP 4µg/10µg of chromatin) | Santa Cruz | Cat#sc-13551; RRID:AB_628282 |
| Normal mouse IgG (for ChIP 4µg/10µg of chromatin) | Santa Cruz | Cat#sc-2025; RRID:AB_737182 |
| Rabbit monoclonal anti-Ki-67 (for IHC 1:100) | Vector Laboratories | Cat#VP-RM04; RRID:AB_2336545 |
| Bacterial and Virus Strains | ||
| pLEX-HA-MYC empty vector | Thermo Scientific Open biosystems | Cat#OHS4492 |
| pLEX-HA-nuSREBP1a, nuclear fragment aa1-480 of mouse Srebp1a | Cloned in the lab from Addgene #32017 | N/A |
| pLEX-HA-nuSREBP2, nuclear fragment aa1-473 of mouse Srebp2 | Cloned in the lab from Addgene #32018 | N/A |
| pLEX-HA-nuSREBP1a (mutation S117A) | Cloned in the lab | N/A |
| pBabe-Puro | Addgene | Cat#1764; RRID: Addgene_1764 |
| pBabe-Puro-MEK-DD (mutations S218D/S222D) | Addgene | Cat#15268 RRID: Addgene_15268 |
| pBabe-Puro-MEK-DN (mutations S218A/S222A) | Cloned in the lab | N/A |
| pGL3-TGFb1 | Addgene | Cat#101762 RRID: Addgene_101762 |
| pRL-SV40P | Addgene | Cat#27163; RRID: Addgene_27163 |
| psPAX2 | Addgene | Cat#12260 RRID: Addgene_12260 |
| pMD2.G (VSV-G envelope) | Addgene | Cat#12259 RRID: Addgene_12259 |
| pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro modified in the lab to include a “staffer” at the gRNA cloning site | Addgene | Cat#27163; RRID: Addgene_71236 |
| Biological Samples | ||
| Patient-derived xenografts (PDX) | Champions Oncology Inc. | https://championsoncology.com/ |
| Pancreatic adenocarcinoma tissue microarrays | Biosample Repository, Fox Chase Cancer Center, Philadelphia, PA) | https://studies.fccc.edu/bsrreport/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SiteClick™ Qdot™ 605 Antibody Labeling Kit | ThermoFisher Scientific | S10469 |
| SiteClick™ Qdot™ 565 Antibody Labeling Kit | ThermoFisher Scientific | S10450 |
| Compactin | Santa Cruz | Cat#sc-200853 |
| Lipid depleted serum (LDS) | Prepared as described (Cunningham et al., 2005) | N/A |
| Recombinant TGFβ1 | Sigma | Cat#T7039 |
| SB431542 | Sigma | Cat#616464 |
| Fatostatin | Cayman Chemical | Cat#13562 |
| Matrigel Matrix | Corning | Cat#356234 |
| Terra PCR Direct Red Dye Premix | TaKaRa | Cat#639286 |
| GoTaq Green Master Mix | Promega | Cat#M7122 |
| Caerulein | Sigma-Aldrich | Cat#C9026 |
| Magnevist | Bayer Healthcare Pharmaceuticals Inc. | Cat#50419-188-82 |
| DRAQ5 | Biostatus | Cat#DR05500 |
| Puromycin | Sigma | Cat#P8833 |
| Streptavidin-BV421 (for FC 1:300) | Biolegend | Cat#405226 |
| siTgfbr1_6 | Qiagen | Cat#SI02735194 |
| siTgfbr1_4 | Qiagen | Cat#SI01447040 |
| siTgfbr1_3 | Qiagen | Cat#SI01447033 |
| siTgfbr1_1 | Qiagen | Cat#SI01447019 |
| siAcvr1b_4 | Qiagen | Cat#SI00888916 |
| siAcvr1b_3 | Qiagen | Cat#SI00888909 |
| siAcvr1b_2 | Qiagen | Cat#SI00888902 |
| siAcvr1b_1 | Qiagen | Cat#SI00888895 |
| siAcvr1c_4 | Qiagen | Cat#SI00888944 |
| siAcvr1c_3 | Qiagen | Cat#SI00888937 |
| siAcvr1c_2 | Qiagen | Cat#SI00888930 |
| siAcvr1c_1 | Qiagen | Cat#SI00888923 |
| siGl2 | Qiagen | Cat#SI03650353 |
| X-tremeGene9 Transfection Reagent | Sigma-Aldrich | Cat#6365787001 |
| Polybrene | Santa-Cruz | Cat#sc-134220 |
| HiPerfect Transfection Reagent | Qiagen | Cat#301704 |
| Triton X-100 | Fisher Scientific | Cat#BP151-100 |
| Atorvastatin Calcium | Sigma | Cat#PHR1422-1G |
| Turbofect transfection reagent | Thermo Scientific | Cat#R0533 |
| NHS-Biotin | Sigma | Cat#203118 |
| Pierce NeutrAvidin Agarose beads | ThermoFisher Scientific | Cat#29200 |
| Pierce Lane Marker Reducing Sample Buffer | ThermoFisher Scientific | Cat#39000 |
| Protein A/G beads | ThermoFisher Scientific | Cat#20421 |
| Propidium iodide | Biolegend | Cat#421301 |
| RIPA buffer | Santa Cruz | Cat#24928 |
| Phosphatase inhibitor | ThermoFisher Scientific | Cat#1862495 |
| Protease inhibitor | ThermoFisher Scientific | Cat##1861278 |
| TRIzol | Life Technologies | Cat#15596-026 |
| Critical Commercial Assays | ||
| Amplex Red kit | Life Technologies | Cat#A12216 |
| Mouse TGF-beta 1 DuoSet ELISA | R&D Systems | Cat#DY1679-05 |
| Sample Activation Kit 1 | R&D Systems | Cat#DY010 |
| DuoSet ELISA Ancillary Reagent Kit 1 | R&D Systems | Cat#DY007 |
| CellTiter-Blue Cell Viability Assay | Promega | Cat#G8081 |
| Gentle MACS Mouse Tumor Dissociation Kit | Miltenyi Biotec. | Cat#130-096-730 |
| Dead Cell Removal Kit | Miltenyi Biotec. | Cat#130-090-101 |
| CD45 MicroBeads Mouse | Miltenyi Biotec | Cat#130-052-30 |
| QIAamp® DNA Micro Kit | Qiagen | Cat#56304 |
| Accucore C30 2.1-mm i.d × 150 µm column | ThermoFisher Scientific | Cat#27826-152130 |
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 |
| EpiTect ChIP kit | Qiagen | Cat#334471 |
| QIAquick PCR Purification Kit | Qiagen | Cat#28104 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Strategene | Cat#200521 |
| Chromium Single Cell 3′ Library, Gel Bead & Multiplex Kit and Chip Kit, V3 | 10X Genomics | Cat#PN-1000092 |
| Bioanalyzer High Sensitivity DNA Kit | Agilent | Cat#5067-4626 |
| Pico Input SMARTer Stranded Total RNA-Seq Kit | Takara | Cat#634411 |
| Qubit dsDNA HS Assay Kit | Thermal Fisher | Cat#Q32851 |
| Deposited Data | ||
| RNA sequencing data of murine pancreatic carcinoma cells and cell lines | Sequence Read Archive (SRA), https://www.ncbi.nlm.nih.gov/sra | PRJNA530747 |
| Single-cell RNA sequencing of murine pancreatic tumors | Sequence Read Archive (SRA), https://www.ncbi.nlm.nih.gov/sra | GSE156210 |
| Experimental Models: Cell Lines | ||
| Mouse: KPC (pancreatic tumor cell lines) | This paper | N/A |
| Mouse: KPCN (pancreatic tumor cell lines) | This paper | N/A |
| HEK293T | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| Capan-2 | ATCC | Cat#HTB-80 RRID:CVCL_0026 |
| MIA PaCa-2 | ATCC | Cat#CRM-CRL-1420 RRID:CVCL_0428 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Nsdhltm1.1Hrm, to be designated as Nsdhlf/f here (The Research Institute at Nationwide Children’s Hospital, Columbus, OH) | Laboratory of Dr. Gail Herman(Cunningham et al., 2015) | MGI:5581334 |
| Mice were obtained from JAX.org and crossed in-house to generate KC (LSL-KrasG12D; Pdx1-Cre) and KPC (LSL-KrasG12D;Trp53f/f; Pdx1-Cre) | Jackson Laboratory, Bar Harbor, ME | JAX 014647; 019104; 008462 |
| Oligonucleotides | ||
| See Supplementary Table S6 for genotyping primers | ||
| See Supplementary Table S6 for qPCR primers | ||
| See Supplementary Table S6 for primers for the SREBP-1 site-directed mutagenesis | ||
| Software and Algorithms | ||
| ImageScope software | Leica Biosystems Imaging, Inc. | http://www2.leicabiosystems.com/l/48532/2014-11-18/35cqc; RRID:SCR_014311 |
| SMIA-CUKIE software (SCR_014795) | Laboratory of Dr. Edna Cukierman (Fox Chase Cancer Center, Philadelphia, PA) (Franco-Barraza et al., 2017) | https://github.com/cukie/SMIA; RRID:SCR_014795 |
| Morpheus on-line software | Morpheus | https://software.broadinstitute.org/morpheus; RRID:SCR_014975 |
| ImageJ software | Rasband et al., 1997-2016 | http://rsbweb.nih.gov/ij/download.html; RRID:SCR_003070 |
| FiJi software | (Schindelin et al., 2012) | https://imagej.net/Fiji/Downloads; RRID:SCR_002285 |
| Image Studio software | LI-COR | https://www.licor.com/bio/products/software/image_studio/index.html; RRID:SCR_015795 |
| FlowJo | FlowJo LLC | https://www.flowjo.com/solutions/flowjo; RRID:SCR_008520 |
| ParaVision | Bruker | http://www.bruker.com/service/support-upgrades/software-downloads/mri.html; RRID:SCR_001964 |
| CellRanger 2.1.0 | 10X Genomics | https://support.10xgenomics.com/ |
| Seurat version 2.3.4 | New York Genome Center | https://satijalab.org/seurat/install.html |
Quantification of TGFBR1 and TGFBR2 levels on the cell surface.
KPC3 cells conditioned for 48 hours in 5% FBS or 5% LDS with 1 μM compactin were collected by trypsinization and counted. Equal numbers of cells were washed twice with cold PBS and incubated with 1 mg/ml of NHS-Biotin (sulfosuccinimidobiotin, Cat#203118, Sigma) for 2 hrs at +4°C on a rotating platform. Biotinylation was quenched in TBS (pH=7.5) followed by two washes in PBS, and lysed with RIPA buffer (#24928, Santa Cruz). Pierce NeutrAvidin Agarose beads (#29200, ThermoFisher Scientific) for the pulldown were prepared by being washed in RIPA buffer once. Then beads were added to the cell lysates and incubated overnight at +4°C on rotating platform. The next day beads were spun down and unbound lysates were removed. Proteins captured on beads were washed twice with RIPA lysis buffer. Then beads were heated to +94°C for 5 minutes with Pierce Lane Marker Reducing Sample Buffer (#39000, ThermoFisher Scientific) and RIPA lysis buffer and vigorously shaken for two hours. Subsequently beads were spun down, and the obtained supernatants, in parallel with the unbound lysates and aliquots of the total lysates, were loaded on the polyacrylamide gel. Western blotting was performed as described above, using anti-TGFBR1 (#MAB5871, Millipore), anti-TGFBR2 (#79424S, Cell Signaling), and anti-CDH1 (#3195, Cell Signaling) antibodies. The concentrations of antibodies are indicated in Key Resource Table.
TGFβ ELISA.
The amount of TGFβ secreted by mouse pancreatic tumor cells was quantified using the Mouse TGFβ1 DuoSet ELISA (#DY1679–05, R&D systems), complemented with Sample Activation Kit 1 (#DY010, R&D systems) and DuoSet ELISA Ancillary Reagent Kit 1 (#DY007, R&D systems) according to the manufacturer’s instructions. Conditioned media for TGFβ secretion quantification was collected from KPC and KPCN tumor cells growing in DMEM supplemented with 1% v/v FBS and 2mM L-glutamine with 100μg/ml Penicillin/Streptomycin for 48 hours. Protein concentration of lysates, prepared from remaining cell pellets, was used for data normalization. Human TGFβ1 in serum samples from patients was measured by human DuoSet ELISA kit (#DY240, R&D systems).
Cholesterol measurement.
Cholesterol level in KPC and KPCN cells as well as in FBS and LDS containing media was measured by Amplex Red Kit (#A12216, Life Technologies), according to the manufacturer’s instructions. Measurements of lipids in patients’ serum samples was performed at the clinical laboratory of Fox Chase Cancer Center.
Lipid extraction, and gas chromatography-mass spectrometry analysis of sterols.
Sterol analysis was performed using ion-ratio GC/MS on an Agilent 6390N/5973 GC/MS system as previously described (Kelley, 1995) with modifications to the GC/MS method to include ions for additional intermediates in the cholesterol biosynthetic pathway between lanosterol and cholesterol.
TGFB1 luciferase reporter assay.
To evaluate the level of transcriptional activation of the human TGFB1 promoter, a dual luciferase reporter assay was performed (#E1910, Promega). HEK293T (#CRL-3216, ATCC) cells were transiently transfected with the following plasmids: pGL3-TGFb1 containing human TGFB1 gene promoter and Firefly luciferase reporter (Addgene#101762 (Yeh et al., 2018)), nuclear fragments of mouse Srebp1a (aa1–490) and Srebp2 (aa1–490), constitutively active MAP2K1 containing mutations S218D/S222D (pBabe-Puro-MEK-DD, Addgene#15268), or its dominant-negative form S218A/S222A (Mansour et al., 1994). All plasmids are listed in Key Resource Table. Cells were transfected with Turbofect transfection reagent (#R0533, Thermo Scientific) according to the manufacturer’s instructions. In 72 hours after transfection the culture medium was removed, cells were gently washed with cold PBS and processed with Dual-Luciferase Reporter System according to the manufacturer’s instructions (#E1910, Promega), and luminescence measured with a Perkin-Elmer plate reader. Ratios of Firefly and Renilla luciferase were calculated and normalized by control sample and data was presented as relative luminescent units.
PCR genotyping.
Small (1–2 mm3) pieces from mouse tails or mouse pancreata were used for genotyping. For cell line genotyping, we isolated genomic DNA with QIAamp® DNA Micro Kit (#56304, Qiagen). Primers for genotyping are listed in Supplementary Table S6. The PCR reaction was performed using Terra PCR Direct Red Dye Premix (#639286, TaKaRa) for LSL-KRas construct detection, and GoTaq Green Master Mix (#M7122, Promega) for all other constructs.
Quantitative RT-PCR.
For evaluation of target gene expression, total RNA was extracted using the RNeasy Mini Kit (#74104, Qiagen). RNA was reverse-transcribed (RT) using Moloney murine leukemia virus (MMLV) reverse transcriptase (#28025013, Ambion) and a mixture of anchored oligo-dT and random decamers (IDT). Two reverse-transcription reactions were performed for each sample using either 100 or 25ng of input RNA in a final volume of 50μl. Taqman or SYBR Green assays were used (see Supplementary Table S6) in combination with Life Technologies Universal Master mixes and run on a 7900 HT sequence detection system (Life Technologies). Cycling conditions were 95°C, 15 minutes, followed by 40 (two-step) cycles (95°C, 15s; 60°C, 60s). Ct (cycle threshold) values were converted to quantities (in arbitrary units) using a standard curve (five points, four fold dilutions) established with a calibrator sample.
Chromatin immunoprecipitation and quantitative PCR.
A ChIP assay for various nuclear proteins was performed using EpiTect ChIP kit (#334471, Qiagen). In brief, KPC3 murine pancreatic cancer cells were conditioned for 48 hours in 5% FBS or in 5% LDS with 1 μM compactin. Cells were briefly washed with warm PBS, fixed with 1% formaldehyde at 37°C temperature for 10 min, neutralized with glycine buffer, and suspended in lysis buffers provided in the kit. After DNA was sheared to 200–400 bp fragments using sonication, samples were incubated with 10 μl of antibodies, or isotype control rabbit or mouse IgG, per 10 μg of chromatin overnight at 4°C. After the addition of protein A/G beads (#20421, Thermo Scientific), the beads were sequentially washed as per the kit manual. The DNA-protein complex was eluted by heating at 45°C with vigorous shaking for 30 minutes in the presence of proteinase K. DNA was recovered with the QIAquick PCR Purification Kit (#28104, Qiagen) and then subjected to real-time PCR analysis. The primer sets are described in Table S6.
SREBP1 site-directed mutagenesis.
Serine-117 to alanine mutant in nuclear fragment of Srebp1a was produced by site-directed mutagenesis of TCG (Ser) to GCG (Ala) using the QuikChange II XL Site-Directed Mutagenesis Kit (#200521, Strategene) according to the manufacturer’s instructions. Primers used for the mutagenesis are listed in Supplementary Table S6.
siRNA transfection.
siRNAs targeting the Tgfbr1, Acvr1b, Acvr1c genes and control (Gl2) were obtained from Qiagen (#SI02735194, #SI01447040, #SI01447033, #SI01447019, #SI00888916, #SI00888909, #SI00888902, #SI00888895, #SI00888944, #SI00888937, #SI00888930, #SI00888923, #SI03650353, Qiagen). For control (Gl2) one siRNA was used and for targets of our interest (Tgfbr1, Acvr1b, Acvr1c) mix of four siRNAs per one gene was used. Cells were transfected with siRNA at 30 nM (total for four siRNAs) concentrations mixed with HiPerfect Transfection Reagent (#301704, Qiagen) according to the manufacturer’s reverse transfection protocol. In 24 hours after plating, the media was changed to 5%FBS, 5%LDS, or 5%LDS with 1μM compactin (#sc-200853, Santa Cruz). RNA and protein lysates were collected at 48 hours after treatment.
CRISPR interference (CRISPRi).
An all-in-one lentiviral CRISPRi system, with a plasmid containing both nuclease-dead Cas9 (dCas9) fused to the transcriptional repressor domain KRAB and gene specific gRNAs (Gilbert et al., 2013), was employed to achieve knockdown of target genes. Briefly, the top 3 most efficient predicted gRNAs were chosen from the published study (Horlbeck et al., 2016) and individually cloned into the all-in-one lentiviral vector, CRISPRi-Puro (modified from the addgene plasmid #71236, a gift from Charles Gersbach (Thakore et al., 2015)), to contain a “stuffer” at the gRNA cloning site.) Next, lentiviruses were generated by transfecting the CRISPRi-Puro plasmid, along with the packaging plasmids psPAX2 (a gift from Didier Trono; Addgene plasmid #12260) and pMD2.G (VSV-G envelope, a gift from Didier Trono; Addgene plasmid #12259) with X-tremeGene9 transfection reagent (#6365787001, Sigma-Aldrich) into 293T cells in serum free/antibiotic free media overnight. The following day, media was replaced with complete media containing FBS and media containing lentiviruses were collected at days 2 and 4 post-transfection. The lentiviral media was then filtered through a 0.45 uM filter (#HAWP14250, Millipore) and used to transduce target cells, by culturing cells in lentiviral media plus 10 ug/mL Polybrene (#sc-134220, Santa Cruz Biotechnology). After 24 hours, lentiviral media was replaced with complete media and cells were selected 48 hours later with 10 ug/mL puromycin for 14 days. Surviving cells were subsequently expanded and knockdown of target genes was confirmed at the protein level by western blotting.
Lesion and tumor grading criteria.
Normal tissue, acinar-to-ductal metaplasia (ADM), pancreatic intraepithelial neoplasia (PanINs) and pancreatic ductal adenocarcinoma (PDAC) were determined according to standard classification guidelines (Hruban et al., 2006). For histopathological scoring, tumors were classified by a Pathologist (K.Q.C.) using the standard pathological grading scheme into either well differentiated (grade 1), moderately differentiated (grade 2), poorly differentiated (grade 3) or undifferentiated (which includes sarcomatoid) (grade 4).
Analyses of protein expression in formalin-fixed paraffin-embedded pancreatic cancer tissues.
For immunohistochemistry, formalin-fixed pancreatic tissue was embedded in paraffin and stained with indicated antibodies diluted per the manufacturer’s instructions (listed in Key Resources Table). Antibody binding was visualized via HRP reaction together with using the Liquid DAB+ Substrate Chromogen System (Dako). Samples were counterstained for 1 minute with hematoxylin. Slides were scanned by an Aperio ScanScope CS scanner (Aperio) and selected regions of interest were outlined manually. The surface area different categories of lesion, and the expression levels of cleaved Caspase-3, Ki-67, CDH1 and pSMAD2 positive area were measured using the ImageScope software (Leica Biosystems Imaging, Inc.) with the help of a certified pathologist.
For detection of VIM+/CK+ EMT cells and to query pSMAD2/3 expression in epithelial compartment of human PDAC, we used simultaneous multiplex immunofluorescent (SMI) on formalin-fixed paraffin-embedded (FFPE) tissues as described (Franco-Barraza et al., 2017). Briefly, primary rabbit monoclonal anti-pSMAD2/SMAD3 (clone D27F4, Cell Signaling) antibody was conjugated with the SiteClick™ Qdot labeling kit (ThermoFisher Scientific). Slides were de-paraffinized and stained with Q-dot pre-labeled antibodies overnight at 4°C in the dark, followed by detection of epithelial and mesenchymal compartments for 2 hr at room temperature. To detect epithelial components, human tissue slides were stained with a cocktail of primary antibodies containing mouse monoclonal anti-pan-cytokeratin (clones AE1/AE3, DAKO), mixed with anti-EpCam/TROP1 (clone MOC-31, Novus Biologicals) and anti-CD-70 (clobe 113–16, Biolegend); for murine tissue the cocktail was complemented with anti-Insulin (clone 2D11-H5, Santa Cruz) and anti-amylase (clone G-10, Santa Cruz) antibodies. For the mesenchymal compartment, we used rabbit monoclonal anti-vimentin (clone EPR3776, Abcam). These primary antibodies were detected with secondary donkey anti-mouse/Cy3 and donkey anti-rabbit/Cy2 antibodies, respectively. Nuclei were stained with DRAQ5 dye (1:105, #DR50050, Biostatus). Images were collected using Vectra multispectral imaging system (Perkin Elmer, Waltham, MA) or Nuance-FX multispectral imaging system (Caliper LifeSciences, PerkinElmer), and analyzed using the SMIA-CUKIE (SCR_014795) 2.1.0 software (https://github.com/cukie/SMIA), as described (Franco-Barraza et al., 2017).
Generation of single-cell sequencing libraries.
For single-cell sequencing, KPPC and KPPCN mice with advanced tumors, 3 of each genotype, were sacrificed 7–8 weeks of age. Single-cell suspensions were isolated from minced tumors using Miltenyi Biotec GentleMacs dissociator in gentleMACS C Tubes (#130–093-23) and mouse tumor tissue dissociation kit (#130–096-730) as per the manufacturer’s instructions. Dead cells were removed by use of Dead Cell removal microbeads (#130–090-101), hematopoeitic cells were depleted by CD45 MicroBeads (#130–052-30). The Chromium controller was used to make single-cell droplet with GEM bead. Single-cell suspensions were converted to barcoded scRNA-seq libraries by using the Chromium Single Cell 3′ Library, Gel Bead & Multiplex Kit and Chip Kit V3 (10X Genomics, #PN-1000092), loading an estimated 6,000 cells per library and following the manufacturer’s instructions. Samples were processed using kits pertaining to the V3 barcoding chemistry of 10x Genomics. For each replicate, all tumor samples were processed in parallel in the same thermal cycler. The final libraries were profiled using the Bioanalyzer High Sensitivity DNA Kit (Agilent Technologies) and quantified using the Qubit 2.0 (Thermal Fisher, #Q32851). Each single-cell RNA-seq library was sequenced twice in two lanes of HiSeq 4000 (Illumina) to obtain single-end, 98 bp, approximately 500 million reads per library.
Bioinformatics of mouse single-cell RNA sequencing.
Single-cell RNA-seq data for each replicate were processed using CellRanger 2.1.0 (10x Genomics) to align and quantify sequencing reads using a mouse reference genome (GRCm38). Individual count tables were merged using CellRanger aggr function. Subsequent data analysis was carried out in R 3.5.1 and the Seurat package (v 3.0.2). We applied the following filters to exclude beads without cells and dead cells by imposing a threshold of at least 500 transcripts measured per cell, and less than 5% mitochondrial reads were set as a threshold to exclude dead cells. Overall, 9501 KPPC cells and 7331 KPPCN cells were available for further analyses. Subsequently, data was normalized to log (CPM/100+1) and scaled regressing out the number of distinct UMIs and the fraction of mitochondrial, ribosomal reads during scaling. The top 2000 most variable genes were identified (Seurat, FindVariableGenes using the mean of log-transformed values and the variance to mean ratio in non-logspace) for principal component analysis (PCA). We then integrated the normalized expression data from KPPC and KPPCN using IntegrateData function of Seurat. This unified dataset was further processed to regress on the genes induced due to dissociation stress of single cells (van den Brink et al., 2017), ribosomal and percentage of mitochondrial genes. To score the stress and ribosomal genes we used AddModuleScore function in Seurat. We applied principal component analysis (PCA) to cells in this gene space followed by dimensionality reduction using UMAP in the Seurat package (Stuart et al., 2019).
We used 20 principal components for dimensionality reduction via UMAP with default parameters. Clusters of cells were identified on the basis of a shared-nearest neighbor graph between cells and the smart moving (SLM) algorithm (resolution = 0.1). Markers for each cluster were identified by reducing the number of candidate genes to those genes which were (i) at least log (0.25)-fold higher expressed in the cluster under consideration compared to all other clusters and (ii) expressed in at least 10% of cells in the cluster under consideration. For genes passing those criteria, significance between cells in the cluster versus all other cells was calculated using model-based analysis of single-cell transcriptomics (MAST) (Finak et al., 2015) and adjusted with the Benjamini–Hochberg method. Differentially expressed genes were used as input for gene set expression analysis.
RNA sequencing of mouse pancreatic cancer cells.
Murine PDAC cells were freshly isolated from dissociated pancreatic tumor tissues followed by FACS sorting (see Cell Lines). Total RNA was isolated using TRIzol (#15596–026, Life Technologies) according to the manufacturer’s protocol. RNA from established cell lines was extracted with the use of an RNeasy Mini Kit (#74104, Qiagen). For RNAseq analysis, total RNA libraries were prepared by using Pico Input SMARTer Stranded Total RNA-Seq Kit (#634411, Takara). Briefly, 250 pg-10 ng total RNA from each sample was reverse-transcribed via random priming and reverse transcriptase. Full-length cDNA was obtained with SMART (Switching Mechanism At 5’ end of RNA Template) technology. The template-switching reaction maintained the strand orientation of the RNA. The ribosomal cDNA was hybridized to mammalian-specific R-Probes and then cleaved by ZapR. Libraries containing Illumina adapters with TruSeq HT indexes were subsequently pooled and loaded to the Hiseq 2500. Single end reads at 75 bp were generated for gene expression analyses. Sequencing reads were analyzed for quality using FastQC (S.Andrews, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were aligned to mouse genome (mm10) using TopHat2 (Kim et al., 2013) and absolute gene counts were quantified using HTSeq (Anders et al., 2015). The resulting gene counts were used as input for differential expression analysis between KPC and KPCN clones and primary cells using DESeq2 (Love et al., 2014). Genes that are differentially expressed were selected for subsequent downstream analysis for identification of biological pathways and ontologies using Gene Set Enrichment Analysis (GSEA) (p-value < 0.001) (Subramanian et al., 2005) using MsigDB datasets (Liberzon et al., 2015). To graphically represent the significantly enriched datasets (False Discovery Rate < 25%) Enrichment Map was used (Merico et al., 2010).
Clustering analysis.
Hierarchical clustering analysis of KPPC and KPPCN cell lines was performed based on data obtained from qPCR and WB results. The expression level of each gene (for qPCR) or the level of each protein in total cell lysates (for WB) for all analyzed cell lines was first normalized by converting them to a percentage, wherein the highest level of expression for each gene was set to 100%. The obtained results were further clustered using Morpheus online software (Morpheus, https://software.broadinstitute.org/morpheus) with using average linkage method and a one minus Pearson correlation metric.
Transcriptome analyses of human PDAC.
Data for mRNA expression and patient outcomes for human pancreatic ductal adenocarcinoma were extracted from publicly available data portals. RNA-Seq data for the TCGA PDAC tumor tissues were downloaded from Broad GDAC Firehose portal (https://gdac.broadinstitute.org/) Subsequent analyses were limited to 76 cases with the PDAC fraction inferred to be >30% by the ESTIMATE method (Yoshihara et al., 2013) as described in (TCGA., 2017). The mRNA gene expression array dataset from 103 primary pancreatic ductal adenocarcinoma samples was obtained from GSE50827 and matched to survival information from the International Cancer Genome Consortium (ICGC) data portal (http://dcc/icgc.org). One case was excluded from the ICGC series due to early death. The PDAC cases in both the TCGA and ICGC series were classified as classic or basal according to a weighted gene expression algorithm (Moffitt et al., 2015). The RNA sequencing data from 85 human PDAC patient-derived xenografts was obtained from Champions Oncology, Inc., Hackensack, NJ, via the TumorGraft® database (https://database.championsoncology.com/). The raw gene expression data used for analyzing subtype-specific pathway alterations in PDAC were normalized using Robust Multi-array Average (RMA) procedure (Irizarry et al., 2003). Gene set variation analysis (GSVA, (Hanzelmann et al., 2013)) was applied to the RMA normalized expression data to identify pathways that are enriched in a single sample (method arguments: function=‘gsva’; mx. Diff=TRUE; verbose=FALSE). Pathways that differed significantly between basal and classical cases were identified using Wilcoxon-Rank sum test using enrichment scores resulting from GSVA. Analyses to compare the overall survival among the basal and classical subtypes were performed by comparing survival curves with log-rank tests. These were calculated using the R ‘survival’ package (Therneau and Grambsch, 2000).
QUANTIFICATION AND STATISTICAL ANALYSIS.
For analysis of continuous data, we used Wilcoxon tests, Mann-Whitney and Student t-test as indicated, and binary outcomes were compared using Fisher’s exact test. Repeated measures (i.e. multiple measures within a single mouse) were analyzed using generalized linear regression models with Generalized Estimating Equations (Liang and Zeger, 1986). Growth curves were modeled using linear regression with interactions between treatment and time, again using GEE to account for within-sample correlation. Survival time outcomes were assessed using Kaplan-Meier curves with log-rank tests. The statistical details of experiments can be found in the figure legends, figures and text of the Results.
Supplementary Material
Table S3. Histological characterization of mouse adenocarcinoma. Related to Figure 2.
Table S6. Primers for genotyping, CRISPRi and qPCR assays. Related to STAR Methods.
Highlights.
Knockout of Nsdhl switches pancreatic carcinoma from glandular to a basal
Statins or Nsdhl knockout activate SREBP1-dependent Tgfb1 expression and EMT
PDACs in patients receiving statins have enhanced mesenchymal features
LDL cholesterol in vitro or in patients antagonizes SREBP1 and autocrine TGFβ
Acknowledgments.
We are grateful to Catherine Reiner for technical assistance with histological analyses and immunohistochemistry experiments; to Dr. Harvey Hensley for assistance with micro-MRI, to Dr. Lisa E. Kratz for assistance with GC-MS determination of sterols, to Ms. Asal Radjabova, Molecular Diagnostics Laboratory, Hamilton, NJ for assistance with histological sections preparation, and to Ms. Krista Saufler, Invivotek, Hamilton, NJ for animal experiments during the COVID-19 pandemic.
Grant Support: This work was supported by NIH core grant CA-06927, by the Pew Charitable Fund, and by a generous gift from Mrs. Concetta Greenberg to the M&C Greenberg Pancreatic Cancer Institute at Fox Chase Cancer Center. Some of the authors were supported by NIH R01 CA188430, K22 CA160725, R21 CA164205, R21 CA231252 (I.A., E.C.), a career development award from Genentech; by Tobacco Settlement funding from the State of Pennsylvania (I.A., E.C.), and by a grant from the Bucks County Board of Associates (L.G.C., I.A.) a grant by the Worldwide Cancer Research foundation (E.C), the 5th AHEPA Cancer Research Foundation, Inc., grants by DOD WX81XWH-15–1-0170 (E.C., R.F., J.F.-B.) and NIH R01 DK108195 (E.A.G.); R01 CA113451 (E.C.); R01 CA232256 (E.C., J.F.-B.), T32 CA009035 (R.F., L.G.C.), R01 HD065800 (A.M.O.), the Program of Competitive Growth of Kazan Federal University (L.G.C.) and NCI core grant CA006927.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
IA served as a consultant for Caris Life Sciences Inc.
References
- Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, and Goldstein JL (2004). Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. The Journal of biological chemistry 279, 52772–52780. [DOI] [PubMed] [Google Scholar]
- Adams CR, Htwe HH, Marsh T, Wang AL, Montoya ML, Subbaraj L, Tward AD, Bardeesy N, and Perera RM (2019). Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello NM, Brabletz T, Kang Y, Nieto MA, Weinberg RA, and Stanger BZ (2017). Upholding a role for EMT in pancreatic cancer metastasis. Nature 547, E7–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, Southwood B, Liang SB, Chadwick D, Zhang A, et al. (2018). Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Bang UC, Watanabe T, and Bendtsen F (2018). The relationship between the use of statins and mortality, severity, and pancreatic cancer in Danish patients with chronic pancreatitis. Eur J Gastroenterol Hepatol 30, 346–351. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. (2006). Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A 103, 5947–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, and Tuveson DA (2019). IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer discovery 9, 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Faust JR, Goldstein JL, Kaneko I, and Endo A (1978). Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. The Journal of biological chemistry 253, 1121–1128. [PubMed] [Google Scholar]
- Chari ST, Kelly K, Hollingsworth MA, Thayer SP, Ahlquist DA, Andersen DK, Batra SK, Brentnall TA, Canto M, Cleeter DF, et al. (2015). Early detection of sporadic pancreatic cancer: summative review. Pancreas 44, 693–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WC, Boursi B, Mamtani R, and Yang YX (2019). Total Serum Cholesterol and Pancreatic Cancer: A Nested Case-Control Study. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 28, 363–369. [DOI] [PubMed] [Google Scholar]
- Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. (2011). Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 17, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, and Piccolo S (2003). Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell 113, 301–314. [DOI] [PubMed] [Google Scholar]
- Cunningham D, DeBarber AE, Bir N, Binkley L, Merkens LS, Steiner RD, and Herman GE (2015). Analysis of hedgehog signaling in cerebellar granule cell precursors in a conditional Nsdhl allele demonstrates an essential role for cholesterol in postnatal CNS development. Human molecular genetics 24, 2808–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, and Massague J (2016). TGF-beta Tumor Suppression through a Lethal EMT. Cell 164, 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. (2020). Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer discovery 10, 232–253. [DOI] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome biology 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Barraza J, Francescone R, Luong T, Shah N, Madhani R, Cukierman G, Dulaimi E, Devarajan K, Egleston BL, Nicolas E, et al. (2017). Matrix-regulated integrin alphavbeta5 maintains alpha5beta1-dependent desmoplastic traits prognostic of neoplastic recurrence. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. (2012). Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabitova L, Restifo D, Gorin A, Manocha K, Handorf E, Yang DH, Cai KQ, Klein-Szanto AJ, Cunningham D, Kratz LE, et al. (2015). Endogenous Sterol Metabolites Regulate Growth of EGFR/KRAS-Dependent Tumors via LXR. Cell reports 12, 1927–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genkinger JM, Kitahara CM, Bernstein L, Berrington de Gonzalez A, Brotzman M, Elena JW, Giles GG, Hartge P, Singh PN, Stolzenberg-Solomon RZ, et al. (2015). Central adiposity, obesity during early adulthood, and pancreatic cancer mortality in a pooled analysis of cohort studies. Ann Oncol 26, 2257–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golemis EA, Scheet P, Beck TN, Scolnick EM, Hunter DJ, Hawk E, and Hopkins N (2018). Molecular mechanisms of the preventable causes of cancer in the United States. Genes & development 32, 868–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hong JY, Nam EM, Lee J, Park JO, Lee SC, Song SY, Choi SH, Heo JS, Park SH, Lim HY, et al. (2014). Randomized double-blinded, placebo-controlled phase II trial of simvastatin and gemcitabine in advanced pancreatic cancer patients. Cancer chemotherapy and pharmacology 73, 125–130. [DOI] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, and Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, Furth EE, Furukawa T, Klein A, Klimstra DS, et al. (2006). Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer research 66, 95–106. [DOI] [PubMed] [Google Scholar]
- Huang BZ, Chang JI, Li E, Xiang AH, and Wu BU (2017). Influence of Statins and Cholesterol on Mortality Among Patients With Pancreatic Cancer. Journal of the National Cancer Institute 109. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, and Speed TP (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development 15, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge SM, Nosacka RL, Delitto D, Gerber MH, Cameron ME, Trevino JG, and Judge AR (2018). Skeletal Muscle Fibrosis in Pancreatic Cancer Patients with Respect to Survival. JNCI cancer spectrum 2, pky043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, et al. (2009). A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chemistry & biology 16, 882–892. [DOI] [PubMed] [Google Scholar]
- Karasinska JM, Topham JT, Kalloger S, Jang GH, Denroche RE, Culibrk L, Williamson LM, Wong HL, Lee MK, O’Kane GM, et al. (2019). Altered gene expression along the glycolysis-cholesterol synthesis axis is associated with outcome in pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. [DOI] [PubMed]
- Karasinska JM, Topham JT, Kalloger SE, Jang GH, Denroche RE, Culibrk L, Williamson LM, Wong HL, Lee MKC, O’Kane GM, et al. (2020). Altered Gene Expression along the Glycolysis-Cholesterol Synthesis Axis Is Associated with Outcome in Pancreatic Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 135–146. [DOI] [PubMed] [Google Scholar]
- Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, and Derynck R (2019). Chronic TGF-beta exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Science signaling 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RI (1995). Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin Chim Acta 236, 45–58. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, and Gallick GE (2009). Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 4, 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, and Massague J (1999). A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes & development 13, 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KY, and Zeger SL (1986). Longitudinal Data-Analysis Using Generalized Linear-Models. Biometrika 73, 13–22. [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, Ebright RY, Karabacak MN, Kulkarni AS, Liu A, et al. (2019). Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 178, 160–175.e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, and Ahn NG (1994). Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966–970. [DOI] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. (2015). Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47, 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris J. P. t., Tschaharganeh DF, Kastenhuber ER, Barsotti AM, Culp-Hill R, et al. (2019). p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 176, 564–580 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J. P. t., Cano DA, Sekine S, Wang SC, and Hebrok M (2010). Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 120, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. (2010). Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A 107, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Ui M, and Iba H (1999). Fra-2-positive autoregulatory loop triggered by mitogen-activated protein kinase (MAPK) and Fra-2 phosphorylation sites by MAPK. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 10, 333–342. [PubMed] [Google Scholar]
- Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, et al. (2013). 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342, 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, and Wright CV (1996). PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122, 983–995. [DOI] [PubMed] [Google Scholar]
- Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, and Brown MS (2001). Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A 98, 6027–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitroda SP, Khodarev NN, Beckett MA, Kufe DW, and Weichselbaum RR (2009). MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc Natl Acad Sci U S A 106, 5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, and Brown MS (2008). Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell metabolism 8, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM (2014). Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- Renner IG, Wisner JR Jr., and Rinderknecht H (1983). Protective effects of exogenous secretin on ceruletide-induced acute pancreatitis in the rat. J Clin Invest 72, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth G, Kotzka J, Kremer L, Lehr S, Lohaus C, Meyer HE, Krone W, and Muller-Wieland D (2000). MAP kinases Erk1/2 phosphorylate sterol regulatory element-binding protein (SREBP)-1a at serine 117 in vitro. The Journal of biological chemistry 275, 33302–33307. [DOI] [PubMed] [Google Scholar]
- Sabnis AJ, and Bivona TG (2019). Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol Med 25, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, and Weinberg RA (2011). Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvente-Poirot S, and Poirot M (2014). Cancer. Cholesterol and cancer, in the balance. Science 343, 1445–1446. [DOI] [PubMed] [Google Scholar]
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massague J, and Benezra R (2013). TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell reports 5, 1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA. (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, and Gersbach CA (2015). Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nature methods 12, 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Modeling survival data : extending the Cox model, (New York: Springer; ). [Google Scholar]
- Tsunoda T, and Takagi T (1999). Estimating transcription factor bindability on DNA. Bioinformatics 15, 622–630. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et al. (2004). Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer cell 5, 375–387. [DOI] [PubMed] [Google Scholar]
- van den Brink SC, Sage F, Vertesy A, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature methods 14, 935–936. [DOI] [PubMed] [Google Scholar]
- Walker AK, Jacobs RL, Watts JL, Rottiers V, Jiang K, Finnegan DM, Shioda T, Hansen M, Yang F, Niebergall LJ, et al. (2011). A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 147, 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, Eshleman JR, Goggins MG, Wolfgang CL, Canto MI, et al. (2011). Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A 108, 21188–21193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Vought BW, Satterlee JS, Walker AK, Jim Sun ZY, Watts JL, DeBeaumont R, Saito RM, Hyberts SG, Yang S, et al. (2006). An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442, 700–704. [DOI] [PubMed] [Google Scholar]
- Yeh HW, Hsu EC, Lee SS, Lang YD, Lin YC, Chang CY, Lee SY, Gu DL, Shih JH, Ho CM, et al. (2018). PSPC1 mediates TGF-beta1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol 20, 479–491. [DOI] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nature communications 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F, Elankumaran S, and Xing J (2014). TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Science signaling 7, ra91. [DOI] [PubMed] [Google Scholar]
- Zheng CF, and Guan KL (1994). Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. The EMBO journal 13, 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, and Kalluri R (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Histological characterization of mouse adenocarcinoma. Related to Figure 2.
Table S6. Primers for genotyping, CRISPRi and qPCR assays. Related to STAR Methods.
Data Availability Statement
The datasets generated during this study are available at Sequence Read Archive (SRA), https://www.ncbi.nlm.nih.gov/sra, deposition PRJNA530747, and GSE156210.