

Abstract
Recently, mutations in speckle-type pox virus and zinc finger protein (SPOP) gene (mutant SPOP [mtSPOP]) have been associated with improved outcomes to abiraterone in the castration-resistant setting. We hypothesized that mtSPOP would be associated with improved outcomes to systemic therapy in men with de novo metastatic castration-sensitive prostate cancer (d-mCSPC). Retrospective data of newly diagnosed d-mCSPC patients were collected from four institutions. Eligibility criteria included standard androgen deprivation therapy without intensification, and SPOP mutational status (mtSPOP or wild-type SPOP [wtSPOP]) determination by targeted next-generation sequencing from tumor biopsies. A total of 121 men (25 mtSPOP [21%] and 96 wtSPOP [79%]) were included. After adjusting for covariates, mtSPOP was significantly associated with better median progression-free survival (35 vs 13 mo; adjusted hazard ratio [HR] 0.47; p = 0.016) and overall survival (97 vs 69 mo; adjusted HR 0.32; p = 0.027), with similar HR and p value on the univariate analysis. These findings, upon external validation, may assist with counseling and prognostication in the clinic, and inform the design of future clinical trials in this setting.
Keywords: SPOP, Androgen deprivation therapy, Progression-free survival, Overall survival, Metastatic hormone-sensitive prostate cancer
Patient summary:
Presence of tumor mutation in speckle-type pox virus and zinc finger protein (SPOP) gene was associated with improved survival outcomes in men with de novo metastatic castration-sensitive prostate cancer receiving standard androgen deprivation therapy.
Genomic markers associated with outcomes to systemic therapy are not currently used in the clinic in men with metastatic prostate cancer. These markers have the potential to assist with prognostication and shared decision making with patient, and inform the design of future clinical trials in this setting.
Recurrent missense mutations in the gene encoding speckle-type pox virus and zinc finger (POZ) protein (SPOP) are the most common point mutations in primary prostate cancer, with an estimated incidence of 10% in clinically localized prostate cancer and a slightly lower prevalence in metastatic castration-resistant prostate cancer (mCRPC) [2,3]. Recently, Boysen et al [4] reported, in a single-center study involving 89 men diagnosed with mCRPC, that men harboring mutant SPOP (mtSPOP) treated with abiraterone (n = 17) had an improved prostate-specific antigen (PSA) 50% response rate (odds ratio 14.5, p = 0.001) and a longer time on abiraterone (hazard ratio [HR] 0.37; p = 0.002) as compared with men with wild-type SPOP (wtSPOP).
Based on these data, we hypothesized that mtSPOP would be associated with improved outcomes to systemic therapy in men with de novo metastatic castration-sensitive prostate cancer (d-mCSPC). Herein, we report the results of a retrospective multicenter investigation on the impact of mtSPOP on outcomes in men diagnosed with d-mCSPC.
Patient data were retrospectively collected from four academic institutions. Eligibility criteria included the receipt of standard androgen deprivation therapy (sADT) without treatment intensification, diagnosis of d-mCSPC (defined as metastatic prostate cancer with no prior history of treatment for prostate cancer), and established SPOP mutational analysis (mtSPOP or wtSPOP) as determined by targeted next-generation sequencing (NGS) of a tumor tissue biopsy by a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory (Foundation Medicine, Caris Life Sciences, Myriad Genetics, and previously published platforms [5]; Supplementary Table 1). The primary outcomes of interest included progression-free survival (PFS) and overall survival (OS). The study was performed with the approval of the Institutional Review Boards of each respective institution.
The primary analysis compared the hazard of having mtSPOP on PFS and OS (vs wtSPOP), as estimated using the Mantel-Haenszel test. PFS was determined as PSA or radiographic or clinical progression as per the modified Prostate Cancer Working Group 2 (PCWG2) criteria [6], from the time of starting sADT. OS was defined as the time from starting sADT to death from any cause or loss to follow-up (the last date of evaluation by the treating physician).
Baseline patient characteristics were compared between men with mtSPOP and wtSPOP (Table 1). Differences in age and baseline PSA were evaluated by Kruskal-Wallis test, while race and Gleason score were compared using chi-square test. A Cox proportional hazards model was fit to estimate the covariate-adjusted hazard. The adjusted model accounted for age, baseline PSA, and Gleason score. Missing data were imputed from 100 chained equations and conditioning on mtSPOP exposure and the adjusting covariates. Wald test was used to determine statistical significance of the HR comparing mtSPOP versus wtSPOP with respect to PFS and OS. Interactions between mtSPOP and each covariate were tested to assess effect modifications.
Table 1 –
Multivariable Cox proportional hazards model
| Variable | Progression-free survival | Overall survival | ||||
|---|---|---|---|---|---|---|
| Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | |
| Mutant SPOP | 0.47 | 0.25–0.87 | 0.016 | 0.32 | 0.12–0.88 | 0.027 |
| Age | 0.93 | 0.68–1.28 | 0.7 | 1.00 | 0.62–1.62 | >0.9 |
| Baseline PSA | 1.04 | 1.00–1.10 | 0.044 | 0.97 | 0.85–1.11 | 0.7 |
| Gleason Sum | 1.40 | 1.01–1.93 | 0.040 | 1.36 | 0.84–2.2C | 0.21 |
CI = confidence interval; PSA = prostate-specific antigen; SPOP = Speckle-type pox virus and zinc finger protein.
Of 937 patients with metastatic prostate cancer, who received treatment between May 21, 2007 to May 28, 2019 and had targeted NGS testing, 121 men met all the eligibility criteria and were included in the analysis (flow diagram in Supplementary Fig. 1). In this cohort, 82 patients did not experience death and were followed for a median of 33.9 mo, and 36 patients did not progress on disease and were followed for a median of 13.3 mo. Twenty-five men (21%) harbored an mtSPOP and 96 (79%) had wtSPOP. Baseline characteristics including age, Gleason score, and PSA were similar between the mtSPOP and wtSPOP cohorts (Supplementary Table 2). The patterns of metastasis at initial presentation are presented in Supplementary Table 3.
Men harboring mtSPOP had significantly improved median PFS compared with those with wtSPOP after adjusting for clinical covariates (35 vs 13 mo, adjusted HR [aHR] 0.47, 95% confidence interval [CI] 0.25–0.87; p = 0.016; Table 1 and Fig. 1A). Similarly, the median OS was significantly higher in men with mCSPC with mtSPOP than in those with wtSPOP (97 vs 69 mo, aHR 0.32, 95% CI 0.12–0.88; p = 0.027; Fig. 1B). The HR and p value for PFS and OS on univariate analysis were similar to aHR.
Fig. 1 –
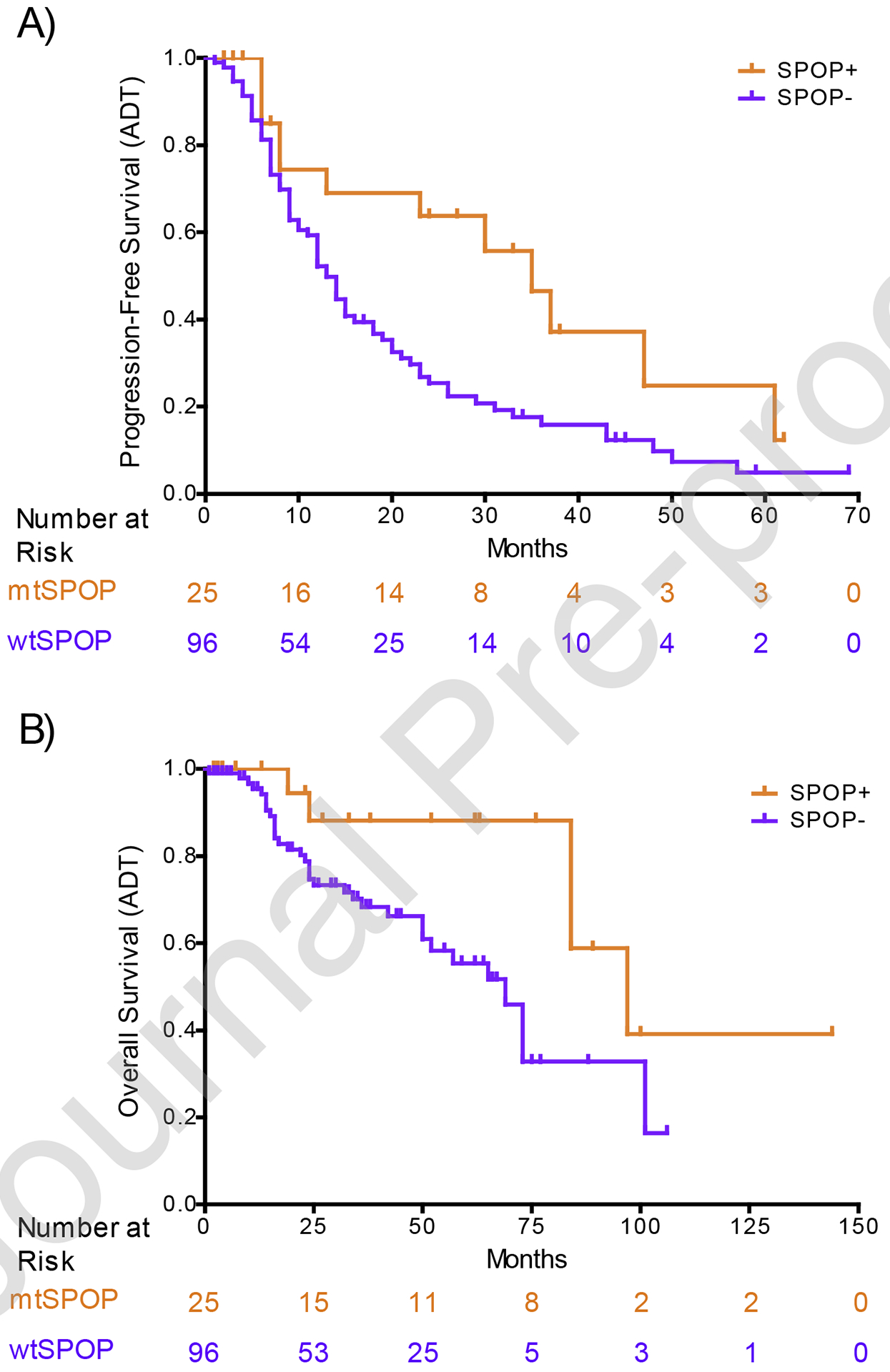
Kaplan-Meier (A) progression-free survival and (B) overall survival curves of men with de novo metastatic castration-sensitive prostate cancer with mutant SPOP (mtSPOP) and wild-type SPOP (wtSPOP). ADT = androgen deprivation therapy; SPOP = speckle-type pox virus and zinc finger protein.
There was no observed effect modification of baseline age or total Gleason score; however, lower baseline PSA was associated with a stronger mtSPOP protective effect. An increase in PSA of 50 ng/ml further increased the PFS HR by 1.10 (95% CI 1.01–1.20; p = 0.033) and the OS HR by 1.14 (95% CI 1.02–1.27; p = 0.019) in patients with mtSPOP. Supplementary Figure 2 demonstrates PFS and OS of individual patients (in the same chronological order as Supplementary Table 1). In a post hoc multivariable analysis, after adjusting for BRCA2, PTEN, RB1, and TMPRSS2-ERG mutations, mtSPOP was still significant for PFS (aHR 0.48, 95% CI 0.24–0.95; p = 0.034), and although we saw some evidence of improved OS, it did not meet conventional levels of statistical significance (aHR 0.32, 95% CI 0.12–1.02; p = 0.054).
We show significantly improved survival outcomes in men with d-mCSPC harboring mtSPOP and undergoing sADT. Recently, longer time on treatment with first-line abiraterone and/or enzalutamide has been reported in men with mCRPC [4,7]. SPOP mutations often occur in MATH domain and are determined to be an early event in prostate cancer tumorigenesis (Supplemental Fig. 3) [8]. SPOP protein acts as a substrate adaptor for the CUL3-based E3 ubiquitin-protein ligase complex for ubiquitinating target proteins, which thereafter undergo proteasomal degradation [9,10]. Androgen receptor (AR) is a target for ubiquitination in prostate cancer. Hence, SPOP mutations may lead to increased tumor AR protein levels [9,11–13]. Additionally, evidence suggests that SPOP functions as a tumor suppressor gene by enhancing degradation of multiple oncogenic substrates, such as AR, SRC3, ERG, TRIM24, c-Myc, DEK, SENP7, EglN2, ATF2, Cdc20, BRD4, PD-L1, and cyclin E1, and mtSPOP leads to tumor genomic instability [8,14,15]. Furthermore, results from The Cancer Genome Atlas shows that mtSPOP (along with FOXA1) has the highest AR transcriptional activity of all genotypically distinct prostate cancer subsets [16]. Based on these data, we hypothesized that mtSPOP prostate cancer may primarily be driven by AR signaling and thus in turn will be more responsive to androgen/AR targeted therapy.
The strength of our study is its multi-institutional cohort design and homogeneity in the study population. We included men with d-mCSPC only, as prior ADT (with or without other systemic therapies) in the localized prostate cancer setting can attenuate the response to ADT in the subsequent metastatic setting. Moreover, inclusion of patients who received ADT with intensified treatment (docetaxel or novel androgen-axis inhibitors) would have resulted in treatment and outcome heterogeneity. Many of these biases, confounders, and effect modifiers were reduced by the inclusion of only de novo mCSPC patients treated with ADT alone. A limitation of our study is the inability to analyze the impact of CHD1 loss due to limitations of testing platforms. CHD1 deletions have been reported to coexist with SPOP mutations [4]. It is possible that sensitivity to hormonal therapies is driven by CHD1 loss and not by SPOP mutation. Finally, our study still remains prone to the most common limitations associated with a retrospective design.
Men with d-mCSPC and mtSPOP have improved PFS and OS on standard first-line ADT compared with men with d-mCSPC and wtSPOP. These findings warrant prospective validation in larger datasets. If confirmed, these novel results could aid in the design of future trials involving men with d-mCSPC.
Supplementary Material
Acknowledgments:
We thank the patients and their families for ongoing support.
Financial disclosures: Neeraj Agarwal certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Neeraj Agarwal has consulted for Pfizer, Exelixis, Merck, Argos, EMD Serono, Eisai, Bayer, Novartis, Genentech, BMS, Astra Zeneca, Medivation, Clovis, Foundation One, Astellas, Eli Lilly, Nektar, Active Biotech, Bavarian Nordic, Calithera, Celldex, GlaxoSmithKline, Immunomedics, Janssen, Merck, New Link Genetics, Prometheus, Rexahn, Sanofi, Takeda, and Tracon. Emmanuel Antonarakis is a paid consultant/advisor to Janssen, Astellas, Sanofi, Dendreon, Medivation, ESSA, AstraZeneca, Clovis, and Merck; he has received research funding to his institution from Janssen, Johnson & Johnson, Sanofi, Dendreon, Genentech, Novartis, Tokai, Bristol Myers-Squibb, AstraZeneca, Clovis, and Merck; and he is the coinventor of a biomarker technology that has been licensed to Qiagen. Benjamin Maughan has consulted for Janssen Oncology, Exelixis, Tempus, Peloton Therapeutics, and Astellas. Roberto Nussenzveig has consulted for Tempus. Remaining authors have nothing to declare.
Funding/Support and role of the sponsor: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA042014 to Huntsman Cancer Institute at the University of Utah, P30CA006973 to Sidney Kimmel Comprehensive Cancer Center at John Hopkins University, and 3P30CA042014-25S2 to N. Agarwal. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].National Comprehensive Cancer Network. Prostate cancer (version 4.2019).
- [2].Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012;44:685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rubin MA, Demichelis F. The genomics of prostate cancer: emerging understanding with technologic advances. Mod Pathol 2018;31:S1–11. [DOI] [PubMed] [Google Scholar]
- [4].Boysen G, Rodrigues DN, Rescigno P, et al. SPOP-mutated/CHD1-deleted lethal prostate cancer and abiraterone sensitivity. Clin Cancer Res 2018;24:5585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26:1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Abida W, Cyrta J, Heller G, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 2019;116:11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Song Y, Xu Y, Pan C, Yan L, Wang ZW, Zhu X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol Cancer 2020;19:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Geng C, He B, Xu L, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A 2013;110:6997–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kwon JE, La M, Oh KH, et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem 2006;281:12664–72. [DOI] [PubMed] [Google Scholar]
- [11].An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep 2014;6:657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Geng C, Rajapakshe K, Shah SS, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 2014;74:5631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wei X, Fried J, Li Y, et al. Functional roles of Speckle-Type Poz (SPOP) protein in genomic stability. J Cancer 2018;9:3257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Armenia J, Wankowicz SAM, Liu D, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet 2018;50:645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Boysen G, Barbieri CE, Prandi D, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife 2015;4:e09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.