

SUMMARY
The mammalian genome has hundreds of nuclear-encoded tRNAs, but the contribution of individual tRNA genes to cellular and organismal function remains unknown. Here, we demonstrate that mutations in a neuronally enriched arginine tRNA, n-Tr20, increased seizure threshold and altered synaptic transmission. n-Tr20 expression also modulated seizures caused by an epilepsy-linked mutation in Gabrg2, a gene encoding a GABAA receptor subunit. Loss of n-Tr20 altered translation initiation by activating the integrated stress response and suppressing mTOR signaling, the latter of which may contribute to altered neurotransmission in mutant mice. Deletion of a highly expressed isoleucine tRNA similarly altered these signaling pathways in the brain, suggesting that regulation of translation initiation is a conserved response to tRNA loss. Our data indicate that loss of a single member of a tRNA family results in multiple cellular phenotypes, highlighting the disease-causing potential of tRNA mutations.
INTRODUCTION
Eukaryotes have hundreds of nuclear-encoded tRNA genes, and in general, the number of these genes increases with organismal complexity. These molecules are classified into multigene families based on their anticodon; for instance, mice have ~400 tRNA genes that are grouped into 46 different families (Chan and Lowe, 2016). The multicopy nature of nuclear-encoded, cytoplasmic tRNAs has been proposed to buffer the effect of deleterious mutations in these molecules, and indeed, deletion of individual genes from multicopy tRNA families in yeast largely resulted in no appreciable phenotypes under normal growth conditions (Bloom-Acker-mann et al.,2014). However, deletion of individual tRNA genes from the same anticodon family was found in some cases to differentially impact cellular fitness, suggesting imperfect compensation by the remaining family members. In addition, evidence from multicellular organisms indicates that members of a tRNA family may be differentially expressed during development and between tissues, suggesting that the existence of multiple copies may have a function beyond providing protective redundancy (Ishimura et al., 2014; Kutter et al., 2011; Sagi et al., 2016; Schmitt et al., 2014).
A recent study suggests that the transcription-associated mutagenesis rate at nuclear-encoded tRNA genes can be 7 to 10 times greater than the genome-wide average and reflective of their high expression (Thornlow et al., 2018). Indeed, genome sequencing has revealed many cytoplasmic tRNA variants in the human population, some of which are predicted to be deleterious to tRNA function (Iben and Maraia, 2014; Lant et al., 2019; Parisien et al., 2013). However, whether mutations in individual tRNA genes can alter cellular and organismal fitness is poorly understood.
We previously discovered a mutation in a member of the nuclear-encoded tRNAArgUCU family, n-Tr20 (tRNA-Arg-TCT-4–1), in the widely used mouse strain C57BL/6J (B6J) (Ishimura et al., 2014). tRNAs are transcribed by RNA polymerase III (Pol III) and undergo a series of processing steps in order to be functional, including removal of leader and trailer sequences, post-transcriptional modification of bases, addition of the non-templated 3′-CCA end, and finally, aminoacylation with their cognate amino acid (Phizicky and Hopper, 2010). The B6J mutation (C50T) in the T-stem loop of n-Tr20 impairs its processing, leading to accumulation of a precursor form and a significant reduction in the production of mature, translational-competent n-Tr20 (Ishimura et al., 2014). Although mice have four other tRNAArg UCU genes that can decode cognate AGA codons, the mutation in n-Tr20 causes a 60% reduction in the total tRNAArgUCU pool in the brain, demonstrating that it is a prominently expressed member of this gene family. Indeed, ribosome profiling of cerebella from B6J mice revealed increased pausing on AGA codons. Of note, although the internal A and B box promoter sequences used by Pol III are identical among the five members of the tRNAArgUCU family, n-Tr20 expression is limited to the nervous system, while the other tRNAArgUCU genes are widely expressed. Little is known about how the expression of individual tRNA genes is regulated, and to date, n-Tr20 is the only known tissue-specific mammalian tRNA.
Here, we demonstrate that loss of n-Tr20 alters signaling pathways that control mRNA translation, leading to changes in synaptic transmission. Accompanying these neurophysiological changes, we observed that expression of this tRNA altered the seizure threshold of mice and was sufficient to modulate the effect of an epilepsy-associated mutation. Our data demonstrate that mutations in individual nuclear-encoded tRNAs, even those that are members of multicopy gene families, can perturb normal cellular function by altering gene expression and physiological processes. Importantly, these findings provide an impetus for future investigations of tRNA mutations in disease.
RESULTS
n-Tr20 Levels Alter Seizure Susceptibility
B6J mice are seizure resistant compared with other inbred strains, with a high threshold for both electrically and pharmacologically induced seizures. The genetic influences underlying seizure threshold differences between B6J mice and more seizure-susceptible strains have been partially dissected by quantitative trait loci (QTL) mapping (Ferraro et al., 1997, 1998, 1999; Frankel et al., 2001; Winawer et al., 2011). Intriguingly, a 6-Mb region of distal chromosome 1 containing ~140 genes, including n-Tr20, has been identified as a major determinant of seizure threshold (Ferraro et al., 1997, 1999, 2001, 2004). The unique, brain-specific expression of n-Tr20 led us to consider whether its expression could impact neurological function.
The large degree of genetic variation between inbred strains makes it difficult to refine a QTL to the causative gene variant(s). To investigate the role of n-Tr20 in the regulation of seizure susceptibility, we first analyzed F2 progeny from an intercross of B6J and the closely related substrain C57BL6/NJ (B6N), which, like all other mouse strains, lacks the mutation in n-Tr20 (Ishimura et al., 2014). As the B6J and B6N strains diverged relatively recently, there is significantly less genetic variation between them in contrast to other, unrelated mouse strains (Bailey, 1978; Keane et al., 2011), allowing us to hone in on the putative contribution of n-Tr20 to seizure susceptibility. The electroconvulsive seizure threshold (ECT) in B6N mice was significantly lower than in B6J mice (Figures 1A and S1A). Variation in seizure threshold segregated with the n-Tr20 genotype in F2 mice; i.e., a higher seizure threshold was observed in mice homozygous for the mutant B6J allele of n-Tr20 (n-Tr20J/J), and a lower threshold was observed in mice homozygous for the wild-type B6N (n-Tr20N/N) allele.
Figure 1. n-Tr20 Expression Modulates Seizure Susceptibility.

(A–C) Electroconvulsive seizure threshold (ECT) in male mice. Boxplots show the median, quartiles (boxes), and range (whiskers).
(D) Pentylenetetrazol (PTZ)-induced seizure incidence. The number of mice tested is indicated.
(E and F) Northern blot of hippocampal tRNAArgUCU levels in transgenic mice overexpressing either nTr21 or n-Tr22, using pooled probes. Bands were normalized to 5S rRNA and quantified relative to B6J. Data are reported as mean + SEM.
(G) PTZ-induced seizure incidence. Number of mice tested are indicated in hatched (seizure) and solid (no seizure) bars.
(H and I) Average spike-wave discharge (SWD) event (H) frequency and length (I).
****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05. See also Figures S1 and S2.
To determine whether the n-Tr20 mutation indeed underlies the high seizure threshold of B6J mice, we measured the ECT of transgenic B6J mice expressing the wild-type n-Tr20 gene at levels comparable to those observed in B6N mice (B6JTg(n-Tr20N/N), hereafter referred to as B6J-Tgwt) (Ishimura et al., 2014) (Figures 1B, S1B, and S1C). Expression of wild-type n-Tr20 significantly reduced seizure threshold. Moreover, the threshold of B6J-nTr20−/− mice was similar to that of B6J mice, indicating that the accumulation of the mutant precursor n-Tr20 in B6J mice does not contribute to the seizure phenotype and that the B6J mutation in n-Tr20 is a loss-of-function mutation. We next deleted n-Tr20 on the B6N background. Loss of n-Tr20 significantly increased ECT of B6N mice. As expected, overexpression of mutant or B6J allele of n-Tr20 in B6N mice (B6N-Tgmut) did not alter seizure threshold (Figures 1C, S1B, and S1D).
B6J mice are also resistant to seizures induced by treatment with pentylenetetrazol (PTZ), a noncompetitive GABAA receptor antagonist (Ferraro et al., 1999). PTZ administration resulted in generalized tonic-clonic seizures in only 3.6% of B6J mice, whereas 89.5% of B6N mice seized (Figure 1D). Transgenic expression of wild-type n-Tr20 increased the sensitivity of B6J mice to PTZ treatment, while deletion of n-Tr20 on the B6N background decreased sensitivity. Together, these data demonstrate that the expression of n-Tr20 is sufficient to alter seizure susceptibility in mice in response to different stimuli.
To determine if changes in seizure threshold were due to a specific translational role of n-Tr20 or a non-canonical role not shared by other tRNAArgUCU family members, we generated B6J transgenic lines for n-Tr21 (tRNA-Arg-TCT-3–1) and n-Tr22 (tRNA-Arg-TCT-1–1), both of which are widely expressed. Levels of these tRNAs in transgenic mice were 5- to 10-fold higher than in B6J mice (Figures S2A and S2B). Overexpression of either nTr21 or n-Tr22 increased the total tRNAArgUCU pool and rescued neurodegeneration induced by loss of n-Tr20 and the ribosome rescue factor Gtpbp2 (Ishimura et al., 2014) (Figures 1E, 1F, S2C, and S2D). We measured PTZ-induced seizures in four independent transgenic lines–two overexpressing n-Tr21 and two overexpressing n-Tr22. Seizure susceptibility was significantly increased in all lines, and the extent of rescue correlated with the level of tRNAArgUCU in the brain (Figures 1E–1G and S2C). These data demonstrate that n-Tr21 and n-Tr22 can indeed functionally substitute for n-Tr20, suggesting that depletion of the total tRNAArgUCU pool underlies the phenotypes caused by n-Tr20 mutation.
Epilepsies can be phenotypically variable, even when caused by the same mutation (Baulac et al., 2001; Frankel, 2009; Wallace et al., 2001). Mutation of the GABAA receptor γ2 subunit (Gabrg2) is linked to childhood absence epilepsy, and interestingly, spike-wave discharge (SWD) events in mice heterozygous for a patient mutation (R43Q) are strongly influenced by genetic background (Tan et al., 2007; Tyler et al., 2014). We investigated whether SWD events in R43Q mice can be modulated by n-Tr20 expression (Figures 1H and 1I). True SWD events are rarely observed in B6J-Tgwt mice, as has been previously reported for B6N and B6J mice (Letts et al., 2014). Consistent with prior studies (Tan et al., 2007), the GABAARγ2 (R43Q) mutation only modestly increased the incidence of SWD events on the B6J background. However, transgenic expression of expression of wild-type n-Tr20 exacerbated the effect of the GABAARγ2 (R43Q) mutation, causing a significant increase in SWD incidence, but not duration, indicating that n-Tr20 genetically interacts with the GABAARγ2 mutation in a nonadditive fashion.
n-Tr20 Mutations Result in Dysfunctional Synaptic Transmission
Expression of n-Tr20 in the mouse brain dramatically increased between embryonic day 12.5 (E12.5) and E15.5, a period of active neurogenesis (Figure 2A). In contrast, expression of other tRNAArgUCU genes was high between E9.5 and E12.5 but decreased at later embryonic stages. Indeed, n-Tr20 was highly expressed in cultured primary neurons, including cortical and hippocampal neurons and cerebellar granule cells, but barely detectable in immunopurified astrocytes or microglia. Expression was also detected in immature oligodendrocytes but decreased upon their differentiation. In situ hybridization demonstrated that n-Tr20 was expressed throughout the hippocampus, cortex, and cerebellum (Figures 2B–2E). n-Tr20 expression was detected in many excitatory neuronal populations, including CA1 pyramidal neurons and cerebellar granule cells, as well as in inhibitory neurons such as Purkinje cells in the cerebellum and in GABAergic interneurons in the cortex and hippocampus (Figures 2B, 2D, and 2E). As expected, expression was not observed in astrocytes (Figure 2C).
Figure 2. n-Tr20 Is Primarily Expressed in Neurons.
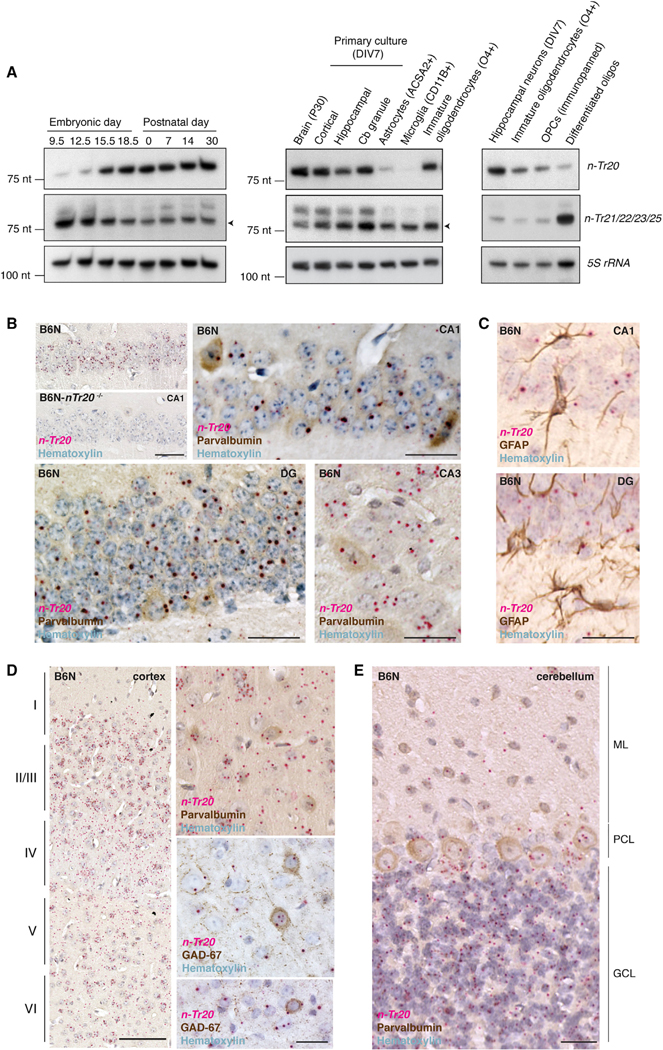
(A) Northern blot of the expression of n-Tr20 and other tRNAArgUCU genes. 5S rRNA was used as a loading control.
(B–E) In situ hybridization of n-Tr20 (red) and immunohistochemistry for cell-type markers (brown) in the hippocampus (B and C), cortex (D), and cerebellum (E). Cortical layers are indicated by Roman numerals. DG, dentate gyrus, ML, molecular layer; PCL, Purkinje cell layer; GCL, granule cell layer. Scale bars represent 100 μm (D), 50 μm (B), and 25 μm (B and D [higher magnification], C, and E).
The increased seizure threshold observed in n-Tr20 mutant mice suggests that the balance of excitation and inhibition may be altered, as seen in models of epilepsy and other neurological disorders (Lee et al., 2017). Multiple brain regions, including the hippocampal formation, have been implicated in seizure activity (Barton et al., 2001; Chawla et al., 2018). Due to its well-defined circuitry and the widespread expression of n-Tr20 therein, we examined the effect of n-Tr20 loss on synaptic transmission in the CA1 region of the hippocampus. As B6J mice have a SNP in n-Tr20 that impairs its processing, they produce significantly less mature n-Tr20 and thus, despite being the parental strain, are partial loss-of-function mutants for n-Tr20. To investigate the role of n-Tr20 in synaptic transmission on the B6J background, B6J and B6J-nTr20−/− mice were always compared to transgenic mice expressing wild-type n-Tr20 (B6J-Tgwt).
We first examined spontaneous excitatory neurotransmission. We observed a small but significant increase in the frequency of miniature excitatory postsynaptic currents (mEPSCs) in B6J and B6J-nTr20−/− CA1 neurons relative to B6J-Tgwt neurons; however, loss of n-Tr20 on the B6N background did not alter mEPSC frequency (Figures 3A and 3B). No significant difference in mEPSC amplitude or kinetics was observed on either genetic background (Figure 3C; data not shown).
Figure 3. Loss of n-Tr20 Alters Synaptic Neurotransmission.

(A) Representative mEPSC traces.
(B and C) Mean mEPSC frequency and amplitude.
(D) Representative mIPSC traces.
(E and F) Mean mIPSC frequency and amplitude.
(G and H) Paired-pulse response (PPR) of excitatory postsynaptic currents (EPSCs). Representative traces of responses evoked by pairs of stimuli (arrowheads) delivered 50, 75, or 100 ms apart. (G) The responses evoked from B6J-Tgwt and B6J-nTr20−/− neurons are overlaid. B6J, n = 7 cells; Tgwt and −/−, n = 8 cells). (H) The responses evoked from B6N and B6N-nTr20−/− neurons are overlaid. B6N, n = 8 cells; −/−, n = 10 cells.
(I and J) Paired-pulse response (PPR) of inhibitory postsynaptic currents (IPSCs). Representative traces of responses evoked by pairs of stimuli (arrowheads) delivered 50, 75, or 100 ms apart. (I) The responses evoked from B6J-Tgwt and B6J-nTr20−/− neurons are overlaid. B6J, n = 10 cells; Tgwt, n = 11 cells; −/−, n = 12 cells. (J) The responses evoked from B6N and B6N-nTr20−/− neurons are overlaid. B6N, n = 11 cells; −/−, n = 10 cells.
(K) Excitatory/inhibitory (E/I) ratio in CA1 neurons.
Error bars represent SEM. ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. See also Figures S3 and S4.
Loss of n-Tr20 expression also increased the frequency of miniature inhibitory postsynaptic currents (mIPSCs) relative to control neurons expressing wild-type n-Tr20 on both the B6J and B6N background, with little change in kinetics (Figures 3D and 3E; data not shown). mIPSC amplitude was also increased in B6N-nTr20−/− neurons relative to B6N neurons but was not significantly altered between B6J-nTr20−/− and B6J-Tgwt neurons (Figure 3F). Together, these data indicate that loss of n-Tr20 alters spontaneous inhibitory transmission on both the B6J and B6N genetic backgrounds, while excitatory transmission is only affected by n-Tr20 levels on the B6J genetic background.
Our observation that n-Tr20 expression altered mEPSC frequency on the B6J background, without any change in amplitude or kinetics, suggested possible changes in synapse number or in presynaptic properties. However, deletion of n-Tr20 did not affect the number of excitatory synapses relative to B6J-Tgwt mice (Figure S3A). Excitatory synapses in CA1 neurons usually undergo facilitation, showing enhanced neurotransmitter release in response to the second of two closely placed stimuli (Thomson, 2000). Strikingly, the paired-pulse ratio (PPR) was significantly decreased in B6J-n-Tr20−/− neurons relative to B6JTgwt neurons, indicating that n-Tr20 expression alters release probability of presynaptic vesicles at excitatory synapses (Figures 3G and S3B). Despite the increased frequency of mEPSCs in B6J mice relative to B6J-Tgwt mice, we did not observe a significant difference in PPR between B6J and B6J-Tgwt neurons, suggesting that spontaneous vesicle release may be more sensitive to reduced levels of n-Tr20 than evoked release. In contrast to the effect of n-Tr20 expression on PPR on the B6J background, we observed no difference in PPR between B6N and B6N-nTr20−/− neurons (Figure 3H).
Similarly, the increased mIPSC frequency upon n-Tr20 mutation indicated changes in GABAergic synapse number or presynaptic release. Mutation or deletion of n-Tr20 did not change GABAergic synapse number relative to mice expressing wild-type n-Tr20 on either genetic background (Figures S3C and S3D). Unlike excitatory synapses, inhibitory CA1 synapses generally express paired-pulse depression (PPD) in response to closely spaced stimuli (Wu and Leung, 1997). Strikingly, in contrast to B6N and B6J-Tgwt neurons, which exhibited the expected depression, PPD was abolished at short interstimulus intervals in n-Tr20 knockout neurons, demonstrating that deletion of n-Tr20 alters presynaptic properties at GABAergic synapses (Figures 3I and 3J). PPD was also reduced in B6J neurons relative to B6J-Tgwt neurons, though not to the same extent as it was reduced in B6J-nTr20−/− neurons, possibly due to the presence of the residual levels of n-Tr20 in these mice (Figures 3I and S3E). Together, our data indicate that deletion of n-Tr20 alters inhibitory neurotransmission at the CA1 synapse on both genetic backgrounds. Finally, we investigated whether the loss of nTr20 and the resultant changes in synaptic transmission caused an excitatory/inhibitory (E/I) imbalance. We found that deletion of n-Tr20 on the B6J background significantly reduced the E/I ratio compared to control B6J-Tgwt CA1 neurons, indicating that loss of n-Tr20 increases inhibitory drive to these cells (Figure 3K).
We also assessed whether n-Tr20 expression modulated the intrinsic excitability of CA1 neurons. While excitability was not altered by n-Tr20 expression on the B6J background, B6N-nTr20−/− neurons fired fewer action potentials (APs) than B6N neurons in response to depolarizing current (Figures S4A–S4D). This reduced excitability was not due to changes in the input resistance (Figure S4E). Rather, loss of n-Tr20 elevated the AP threshold, and while in B6J-nTr20−/− neurons this increase is countered by a more depolarized resting membrane potential relative to B6J-Tgwt neurons, B6N-nTr20−/− neurons required a larger increase in their membrane potential to trigger an AP relative to B6N neurons (Figures S4F–S4H). Taken together, these data indicate that the n-Tr20 loss impacts intrinsic neuronal excitability, particularly in B6N mice.
Loss of n-Tr20 Leads to Widespread Transcriptional Changes
The region of distal chromosome 1 containing n-Tr20 has been identified as a locus that controls the variation in expression (expression QTL [eQTL]) of almost 1,000 genes expressed in the nervous system, including multiple genes associated with mRNA translation, such as aminoacyl tRNA synthetases (Table S1) (Mozhui et al., 2008). This led us to investigate whether nTr20 levels could modulate gene expression. RNA-sequencing (RNA-seq) analysis of the forebrain found that deletion of n-Tr20 had no influence on the expression of genes located within a 4-Mb region surrounding n-Tr20 relative to mice expressing wild-type n-Tr20 (Table S2). However, we observed differential expression of hundreds of unlinked genes (q-value ≤ 0.05; Tables S3A and S3B). Many of these changes were shared between the B6J and B6N backgrounds, though the genetic background did influence the effect of n-Tr20 deletion (Figure S5A). Transcriptome changes in the hypomorphic B6J mice showed a similar directional trend to that observed in B6J-nTr20−/− mice; however, fewer of these differences were significant (Table S3C).
To dissect the effect of n-Tr20 loss versus the influence of genetic background on gene expression, we performed differential expression analysis on our RNA-seq datasets from B6J-Tgwt, B6N, B6J, B6J-n-Tr20−/−, and B6N-n-Tr20−/− mice. 136 genes were differentially expressed between the B6N and B6J strains independent of n-Tr20 genotype and intriguingly, they were enriched for synaptic localization (Figure 4A; Table S3D and S3E). SNPs and indels have been found between B6J and B6N mice in some of these genes (Table S3F), including an intronic mutation in Gabra2 in B6J mice that disrupts its splicing, resulting in reduced expression (Mulligan et al., 2019). These data suggest that differential expression of synaptic genes between the B6J and B6N strains leads to different neurophysiological setpoints and likely underlies the different effects of n-Tr20 deletion on synaptic physiology on the two genetic backgrounds.
Figure 4. n-Tr20 Mutation Leads to Widespread Transcriptional Changes and Activates the Integrated Stress Response.

(A) Differentially expressed genes between the B6N and B6J genetic backgrounds (independent of the effect of n-Tr20 genotype). Synaptic genes are highlighted in red.
(B) Differentially expressed genes due to n-Tr20 deletion (independent of genetic background). (Inset) Heatmap of differentially expressed neocortical and hippocampal genes regulated by distal chromosome 1. Effect of homozygosity for B6J versus DBA/J alleles at distal chromosome 1 (eQTL) (left) and loss of nTr20 versus wild type (right). Red indicates increased expression and blue indicates decreased expression associated with the B6J allele or n-Tr20 deletion.
(A and B) q-value = 0.05; dashed line. The b value is analogous to the natural log fold change of the transcript.
(C and D) B6J-Tgwt mice or B6N mice (C) or congenic B6J mice in which the wild-type n-Tr20 gene was transferred from B6N (J.N; D). Data are represented as mean + SEM.
***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. See also Figure S5.
We next examined genes that were differentially expressed due to deletion of n-Tr20, independent of genetic background (Figure 4B; Table S3G). Deletion of n-Tr20 upregulated 174 genes and downregulated 62 genes relative to mice expressing wild-type n-Tr20. In addition to altering the expression of many translation-associated genes, n-Tr20 also regulated genes associated with transcriptional control and RNA metabolism, including several small ribonucleoproteins involved in mRNA splicing. Indeed, significant differences in the splicing of 377 genes were observed upon loss of n-Tr20 (Tables S4A–S4E). Few of the differentially spliced genes (7/377) were also differentially expressed upon n-Tr20 deletion, indicating that loss of n-Tr20 can impact distinct sets of genes at different stages of mRNA metabolism. Pathway analysis of the differentially spliced genes revealed enrichment for calcium signaling and the endocannabinoid neuronal synapse pathway, suggesting that these changes in mRNA splicing could impact synaptic function (Table S4F).
Interestingly, loss of n-Tr20 modulated the expression of multiple genes associated with synaptic transmission including Gad1 (glutamate decarboxylase), Rims1 (regulating synaptic membrane exocytosis), and numerous potassium channels, and also led to the reduced expression of immediate early genes including Fos and Npas4 (Figure 4B). The expression of many of these genes has been previously reported to be regulated by distal chromosome 1 (Mozhui et al., 2008), and strikingly, we found that n-Tr20 expression explained almost a quarter of the distal chromosome 1 eQTL in the hippocampus and neocortex (Table S1). The direction of differential expression upon n-Tr20 deletion was largely shared with the eQTL; that is, genes for which the B6J allele was associated with higher expression were upregulated, whereas those for which it was associated with lower expression were downregulated upon deletion of nTr20 (Figure 4B, inset).
Pathway analysis revealed significant enrichment for the eIF2α (eukaryotic initiation factor 2, alpha subunit) signaling pathway and its upstream regulators (Figure S5B). Phosphorylation of eIF2α by stress-activated kinases activates the integrated stress response (ISR), characterized by global repression of translation initiation and enhanced translation of transcripts with upstream open reading frames, including the transcription factor ATF4 (Kapur et al., 2017). Indeed, ATF4-target genes were significantly upregulated in the forebrain upon loss of n-Tr20 (Figure S5C). Levels of phosphorylated eIF2α (p-eIF2α) were also increased in the hippocampus and cortex upon n-Tr20 loss (Figures 4C and S5D). Deletion of the eIF2α kinase Gcn2 (general control non-depressible protein 2) abolished the increase in p-eIF2α, consistent with our previous finding that this kinase is responsible for eIF2α phosphorylation in the cerebellum of mice with mutations in both n-Tr20 and Gtpbp2 (Figures 4D and S5E) (Ishimura et al., 2016).
ATF4 is known to induce expression of amino acid transporters and enzymes involved in the amino acid metabolism (Han et al., 2013; Harding et al., 2003), a number of which showed increased expression upon deletion of n-Tr20. We observed a significant increase in the levels of serine and glycine in the n-Tr20 knockout hippocampus relative to mice expressing wild-type n-Tr20 (Figures S5F and S5G), consistent with the observed upregulation of Phgdh (D-3-phosphoglycerate dehydrogenase), the gene encoding the enzyme that catalyzes the first step of serine biosynthesis, in n-Tr20 knockout mice and the established role of ATF4 as a vital regulator of serine-glycine metabolism (Zhao et al., 2016). Thus, our data suggest that the loss of n-Tr20 alters amino acid homeostasis in the brain.
Translational Reprogramming Caused by n-Tr20 Deletion
We next investigated whether n-Tr20 loss, and the accompanying activation of the ISR altered the global translational landscape. Ribosome profiling of B6N and B6N-nTr20−/− hippocampi revealed that loss of n-Tr20 increased the number of strong pauses detected at AGA codons, and genome-wide ribosome occupancy at AGA codons increased by 20% (Figure 5A; Tables S5A and S5B). Pathway analysis on genes that have strong AGA pauses revealed no significant enrichment, suggesting that AGA stalling is likely stochastic.
Figure 5. Loss of n-Tr20 Alters mRNA Translation and Suppresses mTOR Signaling.

(A) Ribosome profiling of hippocampal translation upon deletion of n-Tr20. Fraction of genome-wide pauses (z ≥ 10) at AGA codons (top). Average occupancy at AGA codons (bottom).
(B) Analysis of differential translation efficiency upon deletion of n-Tr20. p-adj = 0.05; dashed line.
(C) Western blot of S6 ribosomal protein phosphorylation in the hippocampus upon mutation of n-Tr20. The level of p-S6 is shown relative to either B6J-Tgwt mice or B6N mice. Data are represented as mean + SEM.
(D) p-S6 (S240/244) and parvalbumin immunofluorescence in the CA1 region of the hippocampus. Scale bars represent 100 μm (20 μm, higher magnification).
(E–H) Synaptic properties of mice treated with either vehicle (Veh) or the mTOR inhibitor rapamycin (Rapa). Mean frequency and amplitude of mEPSCs (E) and mIPSCs (F). Paired-pulse response of evoked EPSCs (G) (rapamycin, n = 9 cells; vehicle, n = 10 cells) and IPSCs (H) (n = 11 cells for both). Data are represented as mean ± SEM.
***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. See also Figures S6–S8.
To identify genes regulated at the translational level, we determined the translational efficiency (TE) of each gene by normalizing the footprint abundance to the level of its corresponding transcript (Table S5C). Exclusion of footprints with the AGA codon in the A-site or adjacent sites prior to measurement of TE did not significantly alter our results, suggesting that ribosome stalling at AGA codons does not noticeably bias TE analysis (Figures S6A and S6B.
Loss of n-Tr20 significantly decreased the TE of 330 genes and increased the TE of 104 genes (Figure 5B; Table S5C). Very few of the differentially translated genes (<1.5%) were also differentially expressed between B6N-nTr20−/− and B6N hippocampi (Figure S6C). Despite the lack of overlap of genes with changes in transcription and translation, pathway analysis revealed shared enriched pathways between differentially expressed and translated genes, including eIF2 signaling and oxidative phosphorylation (Table S5D). Although histone mRNAs were excluded from TE analysis because they lack poly(A) tails, we observed a significant reduction in the abundance of ribosome footprint reads on these mRNAs, suggesting suppression of their translation and that changes in histone occupancy may contribute to the altered transcriptome (Figures S6D–S6F; Table S5E).
Analysis of genes with decreased TE upon n-Tr20 deletion revealed several translation initiation factors, consistent with dysregulation of this process. Strikingly, we found that n-Tr20 loss decreased the TE of numerous ribosomal protein genes, many of which are known to be regulated by mTOR complex 1 (mechanistic target of rapamycin complex 1; mTORC1) via their 5′ terminal oligopyrimidine (TOP) motifs (Figure 5B). While inhibition of mTORC1 suppresses global translation, the translation of mRNAs that contain 5′ TOP motifs is particularly sensitive to mTORC1 activity (Saxton and Sabatini, 2017; Thoreen, 2017; Thoreen et al., 2012). Indeed, upon deletion of n-Tr20, the TE of mRNAs with 5′ TOP motifs was significantly reduced compared to mRNAs lacking this motif (Figure S6G). Additionally, the TE was decreased for several other genes associated with mTOR signaling, including the kinase Akt2 and the inhibitory regulator of protein phosphatase-1, Ppp1r2, both members of the PI3K (phosphatidylinositol 3-kinase)/Akt pathway and Prkab2, a regulatory member of the AMPK signaling pathway. These data suggest that loss of n-Tr20 leads to changes in mTOR signaling in the brain.
To more rigorously examine possible changes in mTOR activity, we assessed the phosphorylation status of several mTORC1 substrates. The ribosomal S6 kinase (p70 S6K) is phosphorylated by mTORC1, leading to its activation and downstream phosphorylation of the S6 ribosomal protein (Saxton and Sabatini, 2017). Phosphorylation of p70 S6K in the hippocampus of B6J-nTr20−/− mice was significantly reduced relative to mice expressing the wild-type n-Tr20 transgene (B6J-Tgwt), and a similar trend (p = 0.13) was observed upon n-Tr20 deletion in the B6N background (Figure S7A). The level of phosphorylated S6 (pS6) was also reduced upon loss of n-Tr20 expression on both genetic backgrounds relative to mice expressing wild-type n-Tr20 (Figures 5C, S7B, and S7C). Reduced p-S6 was apparent in both CA1 neurons and inhibitory neurons in the hippocampus upon loss of n-Tr20 expression (Figures 5D, S7C, and S7D). nTr20 deletion also decreased the levels of phosphorylated 4EBP, another direct target of mTORC1, relative to mice expressing wild-type n-Tr20 (Figure S7E). Lastly, we observed a significant increase in the levels of the phagophore-promoting protein ATG16L and the ATG5~ATG12 conjugate upon the loss of nTr20 expression, consistent with the role of mTOR in the regulation of autophagy (Figures S7F–S7H). Together, these data demonstrate that n-Tr20 loss leads to reduced mTORC1 signaling.
GCN2 and mTORC1 signaling can impinge on common pathways; however, the regulatory interactions between them are still poorly understood. Although recent studies in fibroblasts have found that activation of GCN2 is necessary for the suppression of mTORC1 signaling during amino acid deprivation (Averous et al., 2016; Ye et al., 2015), we found that deletion of Gcn2 did not significantly increase p-S6 levels in the B6J hippocampus, indicating that the mTORC1 inhibition observed upon loss of n-Tr20 is at least partially independent of the ISR (Figure S8A).
Both the ISR and mTOR signaling pathways have been implicated in the regulation of synaptic function. The ISR, including the eIF2α kinases and Atf4, have been shown to be essential for hippocampal synaptic plasticity, and several studies suggest that it is also involved in the maintenance of basal synaptic transmission (Buffington et al., 2014; Costa-Mattioli et al., 2005, Costa-Mattioli et al., 2007; Pasini et al., 2015; Zhu et al., 2011). Thus, we investigated neurotransmission in B6J-Gcn2−/− mice, which do not mount the ISR in response to the loss of n-Tr20. mEPSC frequency was significantly reduced in B6J-Gcn2−/− mice relative to B6J mice. However, loss of Gcn2 did not alter spontaneous GABAergic transmission (Figures S8B and S8C). These data suggest that while the ISR may mediate the increased spontaneous excitatory transmission observed in B6J or B6J-nTr20−/− CA1 neurons relative to B6J-Tgwt neurons, it does not underlie the changes in inhibitory neurotransmission. mTORC1 signaling modulates neuronal function by regulating intrinsic excitability, synaptic transmission, and long-term plasticity, and disruption of mTORC1 signaling has been associated with numerous neurological disorders, including epilepsies (Bateup et al., 2011, 2013; Buffington et al., 2014). Both the cellular context and the genetic manipulation can dramatically influence the physiological phenotype caused by dysregulated mTORC1 signaling (Benthall et al., 2018). We investigated the possible contribution of altered mTOR signaling to the changes in synaptic transmission observed upon n-Tr20 loss. To test whether mTOR inhibition was sufficient to induce changes in synaptic transmission similar to those observed upon n-Tr20 deletion, we treated B6N mice with the mTOR inhibitor rapamycin for 7 days, which significantly reduced p-S6 levels in the forebrain (Figure S8D). Rapamycin treatment increased mEPSC frequency, without any change in amplitude (Figure 5E). However, unlike Gcn2 loss, rapamycin treatment also affected inhibitory neurotransmission, increasing both the frequency and amplitude of mIPSCs (Figure 5F). While rapamycin did not alter PPR at excitatory synapses, PPD at inhibitory synapses was significantly reduced upon treatment, suggesting that acute mTORC1 inhibition can alter presynaptic release properties at GABAergic synapses (Figures 5G and 5H). Together, these data indicate that pharmacological inhibition of mTOR signaling in the context of wild-type n-Tr20 mimics many of the changes in neurotransmission observed upon deletion of the tRNA, suggesting that reduced mTOR signaling may mediate the changes in inhibitory transmission observed in n-Tr20 mutants.
Finally, we investigated whether rapamycin treatment could increase the ECT of B6N mice. While we did not observe a significant difference in the threshold for male B6N mice after a 7-day treatment with rapamycin, a modest but significant increase in seizure threshold was observed in rapamycin-treated female B6N mice, indicating that mTOR inhibition may contribute to the increased seizure threshold observed upon loss of n-Tr20 (Figures S8E and S8F).
Translation Initiation Signaling Pathways Are Sensitive to the Loss of Other Nuclear-Encoded tRNAs
To investigate whether translation initiation signaling pathways are also sensitive to mutations in other tRNA gene families, we targeted the tRNAIleUAU family for mutagenesis. This family consists of four high-confidence tRNA genes, and the mature sequence of three of these genes is identical (Figure 6A). Using CRISPR-Cas9-mediated mutagenesis, we deleted a 15-nt region, overlapping with the A-box, from the 5′ exon of n-Ti17 (Ile-TAT-2–3). Deletion of n-Ti17 reduced the tRNAIleUAU pool in the forebrain to ~50% of that in wild-type mice (Figure 6B).
Figure 6. Deletion of other tRNAs Regulates the Integrated Stress Response and mTORC1 Signaling.

(A) Sequences of mouse tRNAIleUAU genes. The anticodon is highlighted in blue, introns are in lowercase, and the SNP in n-Ti16 is highlighted in red. The nucleotides deleted from n-Ti17 by CRISPR-Cas9-induced mutagenesis are shown in green, and the northern probes used are depicted.
(B) Northern blot of tRNAIleUAU expression in the forebrain of control (+/+) and n-Ti17 knockout mice. The expression of tRNAIleUAU genes was assessed using two different probes: one that recognized n-Ti14, n-Ti15, and n-Ti17, but not n-Ti16; and a second that recognized all four members of the tRNAIleUAU family. 5S rRNA was used as a loading control.
(C) Western blot of eIF2α phosphorylation in the forebrain of control (+/+) and n-Ti17 knockout mice.
(D) Western blot analysis of ribosomal protein S6 phosphorylation in the forebrain of control (+/+) and n-Ti17 knockout mice.
All data are shown as mean + SEM. ****p ≤ 0.0001, **p ≤ 0.01, and *p ≤ 0.05.
n-Ti17 deletion increased p-eIF2α levels in the forebrain, indicative of activation of the ISR (Figure 6C). In addition, mTORC1 signaling was suppressed upon loss of n-Ti17, as demonstrated by reduced levels of p-S6 (Figure 6D). This suggests that activation of the ISR and inhibition of mTOR signaling in response to tRNA mutations is not unique to mutations in neuron-enriched n-Tr20 but rather represents a conserved response to tRNA deficiency in the brain.
DISCUSSION
One of the major challenges of genetics is identifying the sequence differences that underlie phenotypic diversity. This is particularly problematic for complex behaviors that are regulated by many variants, and in general, the attribution of causality is based on prior knowledge of gene function (Ferraro et al., 2007). Here, we show that the loss of a single member of a multicopy nuclear-encoded tRNA family is sufficient to cause transcriptional and translational reprogramming and alter neuronal function and mouse behavior. We demonstrate that expression of n-Tr20 contributed significantly to seizure threshold and eQTL and also modulated the phenotype of an epilepsy-linked mutation in the GABAA receptor γ2 subunit. These surprising results fundamentally alter our understanding of tRNA biology.
Recent studies have found that the cytoplasmic tRNA repertoire can change in response to stress, during proliferation, and in cancer. It has been proposed that these changes in the tRNA pool coordinate or match the codon usage of certain mRNAs, enhancing their stability and translation (Gingold et al., 2014; Goodarzi et al., 2016; Pavon-Eternod et al., 2009; Torrent et al., 2018). In contrast, we find that loss of n-Tr20 and the concomitant reduction in the tRNAArgUCU pool in the brain leads to ISR activation via the eIF2α kinase GCN2 and suppression of mTORC1 signaling, both of which inhibit translation initiation. Depletion of the tRNAIleUAU pool in the brain also modulates these signaling pathways, indicating that translation repression likely represents a conserved, codon-independent response to tRNA mutation.
Classically, GCN2 was thought to be activated by deacylated tRNA that accumulated as a result of amino acid starvation (Dong et al., 2000). However, we have found that GCN2 can be activated when a single tRNA gene is deleted, in the absence of any amino acid depletion or accumulation of uncharged tRNA. Recent studies suggest that the ribosome can directly activate GCN2. The P-stalk, a uL10/P12/P22 pentameric protein complex that is part of the ribosomal GTPase-associated center, has been shown to activate GCN2 in vitro, leading to eIF2α phosphorylation (Inglis et al., 2019). Furthermore, mutations of P-stalk proteins in mammalian cells abolish GCN2-mediated activation of the ISR caused by amino acid starvation (Harding et al., 2019). During translation elongation, the interaction between the ribosomal P-stalk proteins and GCN2 is believed to be blocked by steric hinderance with elongation factors. Ribosome profiling revealed that deletion of n-Tr20 leads to stalling at the cognate AGA codon, and we propose that these ribosomes are stalled in a conformation that exposes the P-stalk, allowing for GCN2 activation. Our study highlights the sensitivity of GCN2 to the detection of even small disruptions in translational elongation.
The observed inhibition of mTORC1 signaling downstream of the mutation in n-Tr20 was not dependent on ISR activation or accompanied by reduced levels of any amino acid, suggesting that tRNA mutations may regulate this pathway, either directly or indirectly, through an as-yet-unknown mechanism. Previous studies have shown that mTOR regulates the transcription of tRNA genes via phosphorylation of the repressor Maf1 (Kantidakis et al., 2010; Michels et al., 2010). Here, in turn, we find that the disruption of a single tRNA gene can alter mTOR activity in the mammalian nervous system.
Loss of n-Tr20 altered synaptic transmission in the hippocampus, resulting in an apparent increase in inhibitory drive. Although n-Tr20 is widely expressed in the hippocampus, its loss predominantly altered presynaptic function, leading to increased spontaneous GABAergic release and altered depression at CA1 synapses on both genetic backgrounds. Strikingly, these changes were recapitulated in B6N mice treated with the mTORC1 inhibitor rapamycin. The role of mTOR signaling in presynaptic function, particularly in GABAergic neurons, remains relatively poorly understood; however, recent studies have suggested that presynaptic autophagy and protein synthesis, key processes regulated by mTORC1 signaling, can impact neurotransmitter release (Henry et al., 2012; Hernandez et al., 2012; McCabe et al., 2020; Scarnati et al., 2018). Although rapamycin acutely inhibits mTORC1, mTORC2 has been reported to be inhibited by chronic administration of rapamycin (Sarbassov et al., 2006), and we cannot exclude the possibility that mTORC2 inhibition may also contribute the changes observed upon rapamycin treatment of B6N mice.
Seizures are circuit-level events that involve complex interactions across several brain regions (Bertram, 2013; Goldberg and Coulter, 2013). Thus, it is difficult to directly link physiological changes in a specific neuronal population to seizure probability. The relatively modest effect of rapamycin on seizure threshold may reflect a developmental contribution to the altered seizure threshold in n-Tr20 mutants that cannot be recapitulated by briefly treating adult mice with an mTOR inhibitor. In addition, systemic rapamycin treatment has been reported to have differential effects depending on the cell type; for instance, it does not appear to inhibit mTOR in dopaminergic neurons (Kosillo et al., 2019). Finally, additional molecular pathways may also be involved in modulating seizure threshold.
There are ~10,000 SNPs and 20,000 indels between the B6J and B6N substrains, including the SNP in n-Tr20 (Keane et al., 2011). Indeed, although the seizure threshold difference between B6J and B6N mice is entirely accounted for in an F2 cross, expression of n-Tr20 alone only explains 60% of the differential seizure sensitivity of these two strains, indicating that additional genetic factors likely contribute to the seizure susceptibility differences between the parental strains. Genetic background can also modulate the effect of n-Tr20 levels on synaptic physiology and gene expression. For instance, changes in n-Tr20 expression altered mEPSC frequency and PPR in CA1 neurons on the B6J background but did not change excitatory neurotransmission in B6N mice. These differences may be caused by genetic interactions between the n-Tr20 mutation and other variants or expression differences between the two strains. A direct comparison of the synaptic physiology of the parental B6J and B6N mice examines the cumulative effect of thousands of genetic differences between the strains. Indeed, although loss of n-Tr20 on both the B6J and B6N background increased mIPSC frequency and decreased PPD relative to that of mice expressing the wild-type tRNA, mIPSC frequency did not significantly differ between the parental strains. Furthermore, B6N mice actually had slightly reduced PPD (two-way ANOVA, p = 0.003) at CA1-inhibitory synapses compared to B6J mice, indicating that the two strains have different baselines upon which the n-Tr20 mutation exerts its effect.
Finally, our data indicate that the brain is sensitive to the total pool of tRNA available to decode a particular codon rather than the levels of any individual tRNA gene. Transgenic overexpression of other members of the tRNAArgUCU family rescued phenotypes caused by n-Tr20 loss, including changes in seizure threshold and neurodegeneration induced by mutation of the translational GTPase Gtpbp2. In addition, the extent of rescue of seizure threshold appeared to correlate with the total pool of tRNAArgUCU. Bolstering this hypothesis, many of the neurophysiological and molecular phenotypes of B6J mice, which have low levels of n-Tr20, were intermediate between those observed in nTr20 knockout mice and transgenic mice expressing the wild-type tRNA. However, the sensitivity of different phenotypes to the level of n-Tr20 varied; for instance, while the ECT of B6J mice was similar to that of B6J-nTr20−/− mice, the PPR at CA1 excitatory synapses appeared similar to transgenic mice expressing wild-type n-Tr20 (B6J-Tgwt), suggesting a possible threshold effect of this phenotype to n-Tr20 level.
We speculate that the inhibition of translation initiation induced by tRNA mutations represents a conserved response in the brain and that mutation of any tRNA gene that significantly depletes the tRNA pool for a particular codon could alter molecular and cellular physiology. In addition to being disease-causing themselves, an intriguing possibility is that mutations in tRNA genes could alter phenotypes caused by mutations in other genes. Given the critical role of signaling pathways regulating translation initiation in practically every aspect of cellular function, we anticipate that tRNA genes will show epistasis with many genes involved in other cellular pathways. We suggest that mutations in nuclear-encoded tRNAs may influence the penetrance and expressivity of multiple types of diseases and that the extent of the effect of such mutations will depend on the wild-type level and spatial and temporal expression pattern of each tRNA.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Susan Ackerman (sackerman@health.ucsd.edu).
Materials Availability
Mouse lines generated in this study are available on request to the Lead Contact.
Dataset and Code Availability
The datasets generated during this study are available at GEO: GSE151742.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse husbandry
All mouse studies were performed under the guidance of the University of California, San Diego and The Jackson Laboratory Animal Care and Use Committee in accordance with institutional and regulatory guidelines. Mice were group-housed in a 12-hour light/dark cycle (light between 7:00 and 19:00 hours) with free access to food and water. The ages used are indicated in the methods. Naive mice from both sexes were used for all experiments.
Mouse strains
Generation of the B6J-Tg(n-Tr20N/N) / B6J-Tgwt and B6N-Tg(n-Tr20J/J) / B6N-Tgmut transgenic lines, B6J-nTr20−/− knockout line, and B6J-Gtpbp2−/− line has been previously described (Ishimura et al., 2014, 2016). B6N-nTr20−/− mice were generated using the same targeting strategy as previously used to create B6J-nTr20−/− mice, except that the recombineered BAC was electroporated into C57BL6/NJ (B6N) ES cells and chimeras were mated to B6N mice for germline transmission. B6J.Gcn2/ mice (Eif2ak4tm1.2Dron) and B6.129(Cg)-Gabrg2tm1Spet/Frk mice (carrying the R43Q patient mutation in the GABAAγ2) were obtained from The Jackson Laboratory. To generate the n-Tr21 and n-Tr22 transgenic mice, we amplified a 1.2 kb region containing the respective tRNAs from B6J genomic DNA. This PCR fragment was then injected into the pronuclei of B6J zygotes. Founders were identified by genotyping across the head to tail tandem repeat insertion, and expression levels of n-Tr21 and n-Tr22 respectively were assessed using Northern blot analysis. B6J-nTi17−/− mice were generated at The Jackson Laboratory using CRISPR-Cas9 via pronuclear injection of B6J fertilized oocytes with Cas9 mRNA and a single-guide RNAs (sgRNA) targeting the 5′ region of the tRNA. The injected zygotes were transferred into pseudo-pregnant female mice to obtain live pups. The offspring were screened for deletions within n-Ti17, and a founder with a 15 bp deletion within the 5′ exon of n-Ti17 was selected and crossed to congenic B6J mice in which the wild-type n-Tr20 allele has been transferred from B6N mice (B6J.B6NnTr20 or J.N mice, previously described in Ishimura et al., 2014 (Ishimura et al., 2014)). Mice from both sexes were used for all experiments.
Primary neuronal and glial cultures
Cortical and hippocampal neuronal cultures were prepared from B6N mice at embryonic day 15, and cerebellar granule cultures were prepared from P4 mice. In brief, the region of interest was dissected out and dissociated in 0.25% trypsin (Invitrogen), and plated on poly-L-lysine coated dishes. The cells were maintained in media supplemented with the anti-mitotic agent cytosine β-D-arabinofuranoside hydrochloride (Sigma, 5 μM) for 7 days before RNA isolation.
Differentiated oligodendrocytes and oligodendrocyte precursor cells (OPCs) were purified as previously described (Dugas and Emery, 2013). Briefly, OPCs were purified by immunopanning with an antibody against O4 (R&D Systems, MAB1326) from a single cell suspension of cortices derived from P7 rats. The immunopanned cells were trypsinized and pelleted by spinning for 10 minutes at maximum speed at 4C. To obtain differentiated oligodendrocytes, OPCs were plated into a 10 cm dish in serum-free media and differentiated the next day by removing PDGF and adding 59.4nM T3. Cells were differentiated for 5 days before RNA isolation.
METHOD DETAILS
Electroconvulsive threshold (ECT)
ECT test were performed as previously published (Frankel et al., 2001). Briefly, tests were performed on experimentally naive mice€ beginning at 8–9 weeks of age. A drop of anesthetic containing 0.5% tetracaine and 0.9% NaCl was placed onto each eye and a fixed electrical current was applied via silver transcorneal electrodes using a customized electroconvulsive stimulator (Ugo Basile model 57800-D01, Stoelting Co., Wood Dale, IL). The stimulator was set to produce rectangular wave pulses with the following parameters: 299 Hz, 0.2 s duration, 1.6 ms pulse width.
To find the generalized tonic-clonic seizure threshold, the seizure response was determined for a particular current, beginning with a current approximately 1 mA below the known average threshold for that genetic background and sex. Seizures were scored using a modified Racine scale (seizure grades: 0, no observable symptoms; 3, rearing, forelimb and jaw clonus; 5, tonic hindlimb extension). The current was increased in +0.3 mA increments daily until the mouse had a generalized seizure (Racine score = 3). As male and female mice have different thresholds, the sexes were analyzed separately. For analysis, the mean integrated root mean square (iRMS) current is reported for each genotype-sex group.
Pentylenetetrazole (PTZ) seizure test
To determine susceptibility to chemically induced tonic-clonic seizures, näive mice between 8- and 9-weeks old were injected subcutaneously under the dorsal skin with a 50 mg/kg bodyweight dose of PTZ. After injection, mice were observed for generalized tonic-clonic seizures (at least a 3 on the modified Racine scale) for at least 30 minutes. The incidence and latency to seizure endpoints were recorded.
EEG recording and analysis
Mice aged between 6 and 8 weeks were anesthetized with tribromoethanol (400 mg/kg i.p.). Small burr holes were drilled (1 mm anterior to the bregma and 2 mm posterior to the bregma) on both sides of the skull 2 mm lateral to the midline. Four teflon-coated silver wires were soldered onto the pins of a microconnector (Mouser Electronics, Texas). The wires were placed between the dura and the brain and a dental cap was then applied. The mice were given a post-operative analgesic of carprofen (5 mg/kg subcutaneous) and allowed at least 48 hours of recovery before recordings. Differential amplification recordings were recorded between all four electrode-pairs, providing 6 channels for each subject. Mice were connected to the EEG Stellate Lamont Pro-36 programmable amplifier (Lamont Medical Instruments, Madison, WI) for a 2-hour period on 2 separate days, between 9 AM and 4 PM during the lights-on period. EEG data were recorded with Stellate Harmonie software (Stellate Systems, Inc., Montreal, Canada) into a database. SWD consist of adjacent, connected spike-wave (or wave-spike) complexes. Recordings were reviewed using low/hi bandpass filters at 0.3 Hz and 35 Hz respectively, and SWD episodes were scored blinded to genotype using the following criteria: at least 2 connected spike-wave complexes (typically spanning at least 0.5 s) with amplitudes at least two fold higher than background and observed concurrently in the majority of the 6 recording channels per mouse.
Northern blot analysis
RNA extraction and Northern blot analysis was performed as previously described (Ishimura et al., 2014). Briefly, total RNA was extracted from tissue (8-week old mice) or primary cells using TRIzol reagent (Invitrogen), resolved on denaturing 15% urea-acrylamide gels, and blotted onto Hybond-N+ membranes (GE Healthcare Life Sciences). The membranes were hybridized with 5′ 32P-labeled oligonucleotide probes at 55°C in hybridization buffer containing 6x SSC, 0.01M sodium phosphate, 1mM EDTA, 0.25% SDS and 100 mg/ml salmon sperm DNA. When multiple oligonucleotide probes were pooled, or increased stringency was required, hybridization was performed at 60C in hybridization buffer containing 3M TMACl rather than 6x SSC.
Histology and immunofluorescence
Anesthetized mice were transcardically perfused with Bouin’s fixative (for histology) or 10% neutral buffered formalin (for immunofluorescence and BaseScope hybridization). Brains were post-fixed overnight and embedded in paraffin. For histological analysis, sections were deparaffinated, rehydrated and were stained with hematoxylin and eosin according to standard protocols. For immunofluorescence, antigen retrieval was performed on deparaffinated sections in 0.01M sodium citrate buffer (pH 6.0). The buffer was brought to a boil, and then slides were immersed and incubated for 15 minutes at sub-boiling temperatures. Sections were stained with rabbit anti-phosphorylated S6 ribosomal protein (p-S6 S240/244) (Cell Signaling Technologies, #5364) and mouse anti-parvalbumin (Sigma, P3088) or mouse anti-GAD67 (MBL, M018–3). Signal was visualized with Alexa Fluor 488-congugated or 555-conjugated goat secondary antibodies (Invitrogen). Sections were counterstained with DAPI and autofluorescence was quenched using Sudan Black. A minimum of three mice per genotype were used for histology, and two mice per genotype were used for immunofluorescence.
BaseScope in situ hybridization
In situ hybridization for n-Tr20 was performed using the BaseScope Assay (ACDBio). In situ hybridization for tRNAs can be problematic because of their short length, and the existence of many highly similar genes. Multiple custom BaseScope probes with a single Z pair targeting the region of n-Tr20 between nucleotide 15 and nucleotide 55 were designed and screened for signal strength and specificity on B6N brain sections, using the B6N-nTr20−/− sections as negative controls. BaseScope assays were performed according to manufacturer’s instructions (BaseScope Detection Reagent Kit–RED), with some modifications for compatibility with immunohistochemistry. Briefly, mice were transcardially perfused with 10% neutral buffered formalin. Brains were postfixed for no more than 16 hours before processing and embedding. 7 mm sections were cut on a microtome, and air-dried overnight. Sections were then baked for 1 hour at 60°C in a dry oven, and deparaffinated in accordance with the BaseScope protocol. Deparaffinated slides were then treated with RNAscope hydrogen peroxide solution for 10 minutes at room temperature, followed by washes in distilled water. Target retrieval was performed by boiling the RNAscope target retrieval reagents on a hot plate. Slides were submerged in a beaker containing mildly boiling (95–98°C) target retrieval reagent on the hot plate for 5 minutes, the beaker was then removed from the hot plate for 1 minute, and finally returned for an additional 4 minutes. Slides were immediately washed in distilled water, followed by 100% ethanol, and allowed to dry at room temperature. A hydrophobic barrier was drawn around the brain section, which was then treated with RNAscope Protease III for 20 minutes. Incubations were performed at 40°C in a humidified tray. Slides were washed with distilled water, and then incubated with the BaseScope probe (n-Tr20, start position nucleotide 15, end position nucleotide 49) for 2 hours, followed by washes in RNAscope 1x wash buffer. The slides were sequentially incubated with BaseScope AMP1–4 in accordance with the product protocol. Next, the slides were incubated at room temperature with BaseScope AMP 5 for 45 minutes, followed a 15-minute incubation in AMP 6. Slides were washed in 1x wash buffer between each incubation. The signal was developed using BaseScope Fast RED for approximately 7 minutes and the development of the signal was monitored under a microscope. Slides were rinsed in tap water and immediately blocked in 5% goat serum/0.3% Triton X-100/PBS for 1 hour at room temperature. Slides were then incubated in primary antibody, diluted in blocking buffer for 2 days at 4C. Primary antibodies used were: mouse anti-parvalbumin (Sigma, P3088, 1:100), mouse anti-GFAP (2E1, Santa Cruz, sc33673, 1:50), and mouse anti-GAD67 (MBL, M018–3, 1:100). Slides were washed in PBS/0.3% Triton X-100 (PBS-T) and incubated with goat anti-mouse IgG Biotin conjugate (Sigma, B7264, 1:100) diluted in blocking buffer for 30 minutes at room temperature. After washing with PBS-T, slides were incubated with ExtraAvidin-peroxidase (Sigma, E2886, 1:50) diluted in blocking buffer for 30 minutes at room temperature. Slides were washed with PBS and then the signal was developed using SIGMAFAST 3,3′-Diaminobenzidine tablets (Sigma, D4293) according to manufacturer’s instructions. Slides were washed in PBS and then counterstained with hematoxylin, before mounting with Vectamount permanent mounting media (Vector Labs, H-5000).
Glial cell isolation
Enriched glial populations were prepared from B6N mice at postnatal day (P) 3–5. Briefly, brains from 4–6 mice were pooled and then enzymatically dissociated using the Neural Tissue Dissociation Kit-P (Miltenyi Biotec). Microglia, astrocytes and immature oligodendrocytes were isolated using magnetic Cd11b microbeads, ACSA-2 microbeads, or O4 microbeads, respectively (Miltenyi Biotec) according to the manufacturer’s protocol. Enrichment for the desired population was confirmed by semiquantitative PCR for markers including GFAP, MBP, CD11b, and NeuN.
Rapamycin treatment
Rapamycin (LC laboratories) was prepared in ethanol at a concentration of 50mg/ml. The stock dilution was diluted fresh on the day of injection in vehicle consisting of equal volumes of a 10% PEG-400/8% ethanol solution and a 10% Tween-80 solution. B6N (7 weeks old) mice were injected intraperitoneally with 5mg/kg rapamycin daily for 6 days and on the morning of electrophysiology analysis or ECT determination. Control B6N mice were injected daily for 6 days and on the morning of analysis with vehicle. The experimenter was blinded to the treatment conditions during the electrophysiology experiments and analysis. Three mice were tested per group. For ECT determination, treatment with rapamycin or vehicle continued until minimal seizure threshold was reached (usually 2 additional days).
Evaluation of synapse density
Acute hippocampal sections (300 mm) were prepared described for current clamp recordings. Immunostaining of synapses was performed as described by McLeod et al. (McLeod et al., 2017). Briefly, sections for fixed for 40 minutes in 4% PFA/4% sucrose/PBS at room temperature with gentle agitation, washed with PBS, and then permeabilized for 6 hours at room temperature in 10% goat serum/0.5% Triton X-100/PBS. Sections were incubated with primary antibodies diluted in the permeabilization buffer overnight at 4C with shaking.
Antibodies used: Glutamatergic synapses (guinea pig x vGlut1,1:2000, Millipore Sigma, #5905; mouse x PSD95, 1:200, MA1–046) and GABAergic synapses (rabbit x vGAT, 1:1000, Synaptic Systems #131 002; mouse x gephyrin, 1:200, Santa Cruz #25311). One section per mouse was stained for MAP2 (mouse x MAP2, 1:1000, Sigma #9942) to assess section quality and integrity. Sections were then washed in PBS and incubated with the appropriate secondary antibodies for 4 hours at RT, washed again, and mounted with Fluoromount G aqueous mounting medium. Synapses were imaged on a Zeiss Observer Z.1 microscope using a 100x oil immersion lens. 9 adjacent image stacks of 15 (330 nm) planes were taken along the CA1 stratum radiatum, from three slices per mouse, and three mice per genotype. The image stacks were deconvolved (fast iterative) using Zen 2.3 Pro software, and the number of presynaptic, postsynaptic and colocalized puncta were quantified on 5 (1 μm) maximum projections for each image stack using the Synapse Counter ImageJ plug-in (Dzyubenko et al., 2016). All data is represented as mean ± SEM.
Western blot analysis
Proteins were extracted from relevant tissues of 8-week-old mice as previously described (Carnevalli et al., 2004). Briefly, tissue was homogenized in 5 volumes of cold homogenization buffer (50 mM HEPES/ KOH, pH 7.5, 140 mM potassium acetate, 4 mM magnesium acetate, 2.5 mM dithiothreitol, 0.32 M sucrose, 1 mM EDTA, 2 mM EGTA), supplemented with phosphatase and protease inhibitors (PhosStop and cOmplete Mini, EDTA-free Protease inhibitor Cocktail, Roche). Proteins were resolved on SDS-Page gels prior to transfer to PVDF membranes. Western blotting was performed according to standard protocols. Unless otherwise specified, all antibodies used for western blotting were from Cell Signaling Technology. Primary antibodies used: rabbit anti-phospho-EIF2α (Ser51, CST# 9721), rabbit anti- EIF2α (CST#9722), rabbit anti-phospho-p70S6 Kinase (T389, CST#9234), rabbit anti-p70S6 Kinase (CST#2708), rabbit anti-phospho S6 ribosomal protein (S240/244, CST#5364), mouse anti-S6 ribosomal protein (Santa Cruz Biotechnology, sc-74459), rabbit anti-phospho 4EBP1 (T37/46, CST#2855), rabbit anti-4EBP1 (CST#9644), rabbit anti-ATG16L (MBL, PM040), rabbit anti-ATG5 (MBL, PM050), Rabbit anti-GAPDH (CST#2118). Signals were detected with SuperSignal West Pico PLUS Chemiluminescent substrate (ThermoScientific) and band intensities were quantified using ImageJ. A minimum of three mice were tested for each genotype. All data is represented as mean + SEM.
Amino acid metabolomics
Amino acids were extracted from samples as previously described (Yuan et al., 2012). Briefly, hippocampi were isolated from 8week-old mice, and immediately flash frozen in liquid nitrogen. Metabolites were extracted from tissue samples with 500 mL of 80% methanol (ice cold) containing heavy internal standards (Metabolomics Amino Acids Standard, MSK-A2, Cambridge Isotope Laboratories). Samples were sonicated, vortexed and incubated for 4h at −80°C. Samples were centrifuged (14,000 × g, 4°C, 10 minutes), supernatants were transferred to new tubes and dried in SpeedVac. Samples were reconstituted in 45 μL water prior to injection.
Amino acids were analyzed on a Dionex Ultimate 3000 LC system (ThermoFisher) coupled to a TSQ Quantiva mass spectrometer (ThermoFisher) fitted with a XBridge Amide HILIC column (3.5 μm, 100 × 4.6 mm i.d., Waters). The following LC solvents were used: solution A, 95:5 water:acetonitrile, 20 mM ammonium hydroxide, 20 mM ammonium acetate, solution B, 100% acetonitrile. The following gradient was used (flow rate: 0.4ml/minute): 85%–60% solution B in 2 minutes, 60%–50% solution B in 8 minutes, 50%–2% solution B in 2 minutes, 2% solution B for 4 minutes, up to 85% solution B in 1 minute and re-equilibration at 85% solution B for 7 minutes, for a total run time of 24 minutes. The injection volume was 10 μL, the column oven temperature was set to 25°C and the autosampler kept at 4°C. MS analyses were performed using electrospray ionization in positive ion mode, with spay voltages of 3.5 kV, ion transfer tube temperature of 325°C, and vaporizer temperature of 275°C. Multiple reaction monitoring (MRM) was performed by using mass transitions between specific parent ions into corresponding fragment ions for each analyte (Table S6). Skyline was used to measure peak areas and absolute quantitation was obtained by using heavy internal standards (MacLean et al., 2010). Three mice were tested per genotype.
Electrophysiology
Chemicals and inhibitors
All chemicals, inhibitors and drugs were purchased from Sigma-Aldrich unless otherwise mentioned. DNQX, picrotoxin and kynurenic acid were purchased from Tocris and tetrodotoxin was purchased from Abcam.
Electrophysiology in acute hippocampal slices
All experiments were performed in mice 6–9 weeks of age. A minimum of three mice per genotype or treatment were analyzed. Mice were anesthetized with isoflurane followed by rapid decapitation. The brain was isolated in ice cold NMDG aCSF containing: 93 mM NMDG, 30 mM NaHCO3, 1.2 mM NaH2PO4, 2.5 mM KCl, 20 mM HEPES, 25 mM glucose, 5 mM sodium ascorbate, 2 mM thiourea, 3 mM sodium pyruvate, 10 mM MgSO4.7H2O and 0.5 mM CaCl2.2H2O (pH-7.35–7.38) in less than 60 s. The brain was cut before gluing on to the sectioning stage by the ‘magic-cut technique’ (Bischofberger et al., 2006). Transverse hippocampal slices (300 mm) were cut using a Leica VT1200S vibratome in ice cold NMDG aCSF under constant oxygenation with 95% O2 and 5% CO2. For all current clamp recordings, the slices were recovered in NMDG aCSF for 6 minutes at 30°C followed by recovery in external aCSF containing: 124 mM NaCl, 24 mM NaHCO3, 1.2 mM NaH2PO4, 2.5 mM KCl, 5 mM HEPES, 12.5 mM glucose, 2 mM MgSO4.7H2O and 2 mM CaCl2.2H2O (pH-7.35–7.38) for at least 45 minutes at room temperature under constant oxygenation. For all other recordings, the slices were recovered in external aCSF for 8 minutes at 30°C, followed by recovery in external aCSF for at least 45minutes at room temperature under constant oxygenation. The osmolarity of all internal solutions used in this study was adjusted to 289–291mOsMol/kg whereas that of the external aCSF was adjusted to 298–302 mOsMol/kg. For all recordings, thin-walled borosilicate patch pipettes (G150TF-4, Warner Instruments) were pulled using a Narishige PC-10 pipette puller to attain a pipette resistance of 2.8–4.2 MΩ. Slices were visualized using an upright Olympus BX51WI microscope. CA1 pyramidal hippocampal neurons were identified and the patch pipette was guided on to the neuron to be patched under an Olympus 40x water-immersion lens (N.A 0.80) aided by a 700nm IR filter. All recordings were performed at 32°C with constant perfusion of external aCSF at a flow rate of 1 ml/minute. Data was acquired using the Axon Instruments CV-7B head stage in combination with the Multiclamp 700B amplifier and an Axon Digidata 1550B digitizer. All data was acquired using the Axon pClamp10.7 and Multiclamp 700B Commander software packages (Axon Instruments).
Voltage clamp recordings
i). mEPSC and mIPSC recordings:
Miniature EPSC (mEPSC) events were recorded in voltage clamp mode by holding the patched neuron at 70mV. All mEPSC recordings were performed in external aCSF (as used for slice recovery) containing 1μM tetrodotoxin and 100 μM picrotoxin (to block GABAergic transmission) using a cesium-based internal solution containing: 120 mM cesium methanesulfonate, 15 mM CsCl, 10 mM TEA-Cl, 8 mM NaCl, 10 mM HEPES, 2 mM EGTA, 5 mM QX-314, 4 mM ATP-Mg2, 0.3 mM GTP-Na2, and 10 mM phosphocreatine-Na2 (pH-7.28–7.3) in the recording pipette.
Miniature IPSC (mIPSC) events were also recorded in voltage clamp mode by holding the patched neuron at 70mV. mIPSCs were recorded in external aCSF (as used for slice recovery) containing 1μM tetrodotoxin, 10 μM DNQX and 2mM kynurenic acid (to block glutamatergic transmission) in combination with a cesium chloride internal solution containing: 120 mM CsCl, 20 mM TEA-Cl, 10 mM HEPES, 0.5 mM EGTA, 4 mM QX-314, 4 mM ATP-Mg2, and 0.3 mM GTP-Na2 (pH-7.28–7.3) in the recording pipette.
For both mEPSC and mIPSC recordings, each neuron was recorded for 5 minutes starting at least 5 minutes after break-in. Series resistance compensation was not performed for any of the recorded neurons. Access/series resistance was constantly monitored during the recording and any cell with access/series resistance larger than 20 MΩ was rejected for analysis. Any instances where the hold-current changed by more than 50pA or where the access/series resistance changed by more than 20% during the 5 minutes of recording was rejected for analysis. All data was acquired at a gain of 20 and digitized with the Bessel filter set to 4 kHz. All miniature synaptic current events were analyzed using the built-in functions in Mini Analysis program (Synaptosoft, Decatur, CA).
ii). Paired-pulse ratio (excitatory and Inhibitory) recordings:
Paired-pulse ratio (PPR) at monosynaptic excitatory synapses was measured by holding the patched hippocampal neuron at −70mV in external aCSF (as used for slice recovery) without inhibitors. The same cesium-based internal solution as used for recording mEPSCs was included in the recording pipette. A concentric bipolar electrode (CBARC75, FHC Inc., Bowdoin ME) with an internal diameter of 25 microns was placed ~400–500 microns from the neuron being recorded to stimulate the Schaffer collateral axon tracts. Two consecutive voltage pulses spaced 25, 50, 75, 100, 150, 200, 250 and 300 ms apart were delivered at 0.1 Hz frequency to stimulate the axon tracts. Voltage pulses were delivered using a Digitimer DS2A (Digitimer Ltd., UK) constant voltage injector and the data was digitized at 10 kHz (Bessel filter) with the gain set to 2. The PPR was measured as the ratio of the peak amplitude of the second EPSC to that of the first EPSC.
Paired-pulse ratio (PPR) at the inhibitory monosynaptic synapses was measured by holding the patched hippocampal neuron at 0 mV in external aCSF (as used for slice recovery) in the presence of 10 mM DNQX, 10 μM CPP and 5 mM CGP55845 to block all disynaptic inhibitory input. The same cesium-based internal solution as used for recording mEPSCs was included in the recording pipette. The bipolar electrode was placed ~200–300 microns from the patched neuron in the middle of the stratum radiatum to directly stimulate interneurons and evoke IPSCs. Two consecutive voltage pulses spaced 25, 50, 75, 100, 150, 200, 250 and 300 ms apart were delivered at 0.2 Hz frequency to stimulate the interneurons. Voltage pulses were delivered using a Digitimer DS2A (Digitimer Ltd., UK) constant voltage injector and the data was digitized at 10 kHz (Bessel filter) with the gain set to 2. Access/series resistance was constantly monitored during the recording and any cell with access/series resistance larger than 20 MU or where the access resistance changed by more than 20% during recording was rejected for analysis. The inhibitory monosynaptic PPR was measured as the ratio of the peak amplitude of the second IPSC to that of the first IPSC.
iii). Excitation to Inhibition ratio recordings:
To measure E/I ratio, voltage clamp recordings from CA1 pyramidal neurons were performed in external aCSF (as used for slice recovery) with no inhibitors. The same cesium-based internal solution as used for recording mEPSCs was included in the recording pipette. Schaffer collaterals axons were stimulated at 0.4 Hz using the concentric bipolar electrode placed ~400–500 microns from the patched neuron to evoke both monosynaptic EPSPs and compound di-synaptic/ monosynaptic inhibitory post-synaptic potentials (IPSPs). EPSCs were recorded at 56mV based on the reversal potential of chloride in the internal solution, whereas the IPSCs were recorded at 0 mV. A single pulse of voltage was delivered to evoke IPSCs and EPSCs, where the strength of the pulse was chosen in order to not saturate the response from the patched neuron. The amplitude of the excitatory to inhibitory voltage pulse was maintained at a ratio of 3:2 for all genotypes. Access/series resistance was constantly monitored during the recording and any cell with access/series resistance larger than 20 MU or where the access resistance changed by more than 20% during recording was rejected for analysis. E/I ratio was calculated from baseline subtracted traces as the maximum depolarization amplitude (in mV) divided by the maximum hyperpolarization amplitude 500 ms after the stimulus.
Current clamp recordings
All recordings were performed in external aCSF (as used for slice recovery) with the addition of 10 μM CPP, 10 μM DNQX and 100 μM picrotoxin. A potassium-based internal solution containing: 145 mM K-Gluconate, 10 mM HEPES, 1 mM EGTA, 2 mM MgCl2.6H2O, 2 mM Na2-ATP and 0.3 mM Na2-GTP (pH-7.28–7.3) was added to the recording pipette. After break-in, the resting membrane potential of the patched neuron was monitored in I = 0 mode for 2–3 minutes until it remained relatively unchanged before proceeding with any recordings. Bridge balance was fully compensated on all patched neurons and any neuron with a resting membrane potential more depolarized than −55mV was rejected for recording. To determine the action potential threshold 10 pA steps of current was injected incrementally from −20 to 200pA for 50 ms. The input resistance of the neuron was determined by injecting 10pA steps of current for 1 s from −80pA to +40pA. To plot the action potential frequency upon current injection, 20pA steps of current was sequentially injected into the patched neuron from −100 to 300pA for 500 ms. All data was acquired was digitized with Bessel filter set at 10kHz and analyzed by using the AxoGraph 1.7.2 (AxoGraph Scientific) and Clampfit 10.7 (Molecular Devices) software packages.
RNA-Seq library preparation
Library preparation for total gene expression analysis of B6J, B6J-Tgwt, B6J-nTr20−/−, B6N, and B6N-nTr20−/− mice was performed using the Illumina TruSeq methodology. Briefly, forebrains (the cortex, hippocampus, and striatum together) were isolated from 8week-old mice (n = 4 mice for each genotype) and immediately stored in RNAlater solution (Invitrogen, AM7020) at 20°C. Total RNA was extracted using Trizol reagent and the quality of the isolated RNA was assessed using an Agilent 2100 Bioanalyzer instrument and RNA 6000 Nano LabChip assay. Following ribosomal RNA depletion, the remaining RNA was purified, fragmented, and libraries were prepared using TruSeq Stranded Total RNA with RiboZero-Mouse (Illumina). The quality of libraries was assessed on Agilent 2100 Bioanalyzer. Paired-end reads (2×100bp) were obtained using the NextSeq sequencer (Illumina).
mRNA libraries for the assessment of translational efficiency in the B6N and B6N-nTr20−/− hippocampus were prepared using the TruSeq v2 mRNA kit (Illumina). Briefly, total hippocampal RNA was isolated from 8-week-old mice (n = 3 mice for each genotype), and the purified RNA was assessed on Agilent Tapestation. mRNA was purified using biotin-tagged polydT oligonucleotides and streptavidin-coated magnetic beads. After fragmentation of mRNA, libraries were prepared according to manufacturer’s instructions. Paired end reads (2×100bp) were obtained using the HiSeq 4000 (Illumina).
Ribosome profiling library construction
Ribosome profiling libraries were generated from B6N and B6N-nTr20−/− hippocampi (8-week old mice). Libraries were constructed as previously described with certain modifications as described below (Ingolia et al., 2012; Ishimura et al., 2014). Briefly, mice were euthanized by cervical dislocation, and hippocampi were immediate dissected out and frozen in liquid nitrogen. Two mice were pooled for each biological replicate, and three biological replicates were prepared for each genotype. Tissue homogenization was performed with a mixer mill (Retsch MM400) in 350 μL lysis buffer (20mM Tris-Cl, pH 8.0, 150 mM NaCl, 5mM MgCl2, 1mM DTT, 100 mg/ml CHX, 1% (v/v) Triton X-100, 25units/ml Turbo DNaseI). RNase I treated lysates were overlaid on top of a sucrose cushion in 5ml Beckman Ultraclear tubes and centrifuged in a SW55Ti rotor for 4 hours at 4°C at 46,700 rpm. Pellets were resuspended and RNA was extracted using the miRNeasy kit (QIAGEN) according to manufacturer’s instructions. 26–34 nucleotide RNA fragments were purified by electrophoresis on a denaturing 15% gel. Linker addition, cDNA generation, circularization, rRNA depletion, and amplification of cDNAs with indexing primers was performed as previously described (Ingolia et al., 2012). Library quality and concentration was assessed using high sensitivity D1000 screen tape on the Agilent tape station, Qubit 2.0 Fluorometer, and qPCR. Six libraries (three biological replicates from each genotype) were pooled and run on a single lane on HiSeq4000 (SR75).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Seq data analysis
Reads were quantified using kallisto version 0.42.4 (Bray et al., 2016) pseudo-aligning to a Gencode M20 transcript reference. Differential expression was performed using sleuth version 0.30.0 (Pimentel et al., 2017) accounting for strain, batch, or tRNA genotype covariates when applicable. For the splicing analysis, reads were trimmed to 75 bases using trim_galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) (Martin, 2011) and mapped to mm10 mouse reference using a Gencode M20 annotation using hisat2 version 2.1.0 (Kim et al., 2019). Splicing events were then quantified using rMATS version 3.2.5 (Shen et al., 2014).
Analysis of the trans eQTL
Trans eQTLs for hundreds of genes have been reported to map to distal chromosome 1 in BXD and other crosses (Mozhui et al., 2008). In this study, eQTLs mapping between 172–176.2 Mb on Chromosome 1 (mm10), with an LRS score R 15 were retrieved from GeneNetwork (http://gn2.genenetwork.org). Information about the datasets queried is in Table S1. Genes located between 170–180 Mb on Chromosome 1 were eliminated to ensure that our analysis was limited to genes with bona fide trans eQTL. The gene lists acquired from GeneNetwork included expressed sequence tags, Agilent probes, and Riken clones, which were eliminated for the purposes of our analyses. Genes with reported trans eQTLs on Chromosome 1 whose expression was not detected in the forebrain RNA-Seq datasets were also eliminated. For the heatmap (Figure 3B, inset), the additive effect of the eQTL with the highest LRS score was used, and log2(foldchange) values, that is, twice the additive effect, is shown.
Gene (GO) ontology and pathway analysis
RNA-Seq data were analyzed through the use of IPA (QIAGEN Inc., https://digitalinsights.qiagen.com/products/ingenuity-pathwayanalysis) (Kramer et al., 2014). Gene Ontology (GO) and Kegg pathway analysis was performed as previously described using the DAVID bioinformatics web server (https://david.ncifcrf.gov/) (Huang et al., 2009) by uploading the gene lists from our RNA-Seq analysis (differentially regulated genes, q-value % 0.05) and ribosome profiling analysis (AGA stalling transcripts, z ≤ 10, stalls detected in at least 2 biological replicates). The functional annotation chart and clustering analysis modules were employed for gene-term enrichment analysis. GO terms and Kegg pathway terms with a Benjamini-Hochberg adjusted p value % 0.05 were considered enriched.
Ribosome profiling data analysis
Reads were clipped to remove adaptor sequences (CTGTAGGCACCATCAAT) using fastx_clipper and trimmed so that reads start on the second base using fastx_trimmer (http://hannonlab.cshl.edu/fastx_toolkit/). Reads containing ribosomal RNA were then filtered out by mapping to a ribosomal RNA reference using bowtie2 version 2.2.3 using parameters -L 13 (Langmead and Salzberg, 2012). Remaining reads were mapped to a mm10 mouse reference using a Gencode M20 annotation, or a Gencode M20 transcript reference using hisat2 version 2.1.0 (Kim et al., 2019). Ribosomal A-sites were identified using RiboWaltz version 1.0.1(Lauria et al., 2018) and pauses were identified using previous methodology (Ishimura et al., 2014) using a 0.5 reads/codon threshold. Reads counts for translational efficiency (TE) analysis were quantified using featureCounts (Liao et al., 2014) with footprints overlapping CDS features and RNA read pairs overlapping gene exon features. Histone mRNAs were removed from the analysis and TE was quantified and tested for using riborex version 2.3.4 using the DESeq2 engine (Li et al., 2017). TE analysis of AGA A-site filtered datasets was performed by identifying AGA codons in the transcriptome and transferring the coordinates to the genome using ensembldb v.2.6.8 (Rainer et al., 2019). Then reads with AGAs in the A-site (±1 codon based on riboWaltz A-site offset for each read length) were removed and the above TE analysis was performed. Sets of mouse transcripts with known TOP motifs were identified (Yamashita et al., 2008) and histone genes were identified using histonedb2.0 or the prefix “Hist” in the gene name (Draizen et al., 2016). Differential footprint analysis was performed using DESeq2 version 1.22.2 (Love et al., 2014).
Statistics
Graphs were generated and statistical analysis was performed in GraphPad Prism 7, unless otherwise specified. Statistical parameters including the value of n and statistical significance (p value) are reported in the figures and figure legends. For seizures tests, Northern blot analyses, and western blot analyses, n represents individual mice, and the individual datapoints are shown in the figure. For electrophysiology experiments, n represents individual cells recorded from at least three mice, and the individual datapoints are shown in the figure. In the case of paired-pulse recordings, the number of cells is reported in the figure legend. No statistical methods were used to predetermine sample size. Statistical tests were selected based on the desired comparison. Mann-Whitney test (2 genotypes) or Kruskal-Wallis test followed by Dunn’s multiple comparison (≥3 genotypes) were used to assess significance of ECT, spike-wave discharge data, and mEPSC/mIPSC frequency and amplitude. Fisher’s exact test was used to analyze PTZ seizure data. Unpaired two-tailed Student’s t test with Welch’s correction (2 genotypes) or one-way ANOVA followed by Tukey’s multiple comparison test (≥3 genotypes) was used for statistical analysis of northern and western blot data. Two-way ANOVA followed by either Tukey (2 genotypes) or Sidak (≥3 genotypes) was used for analysis of paired-pulse ratio data. The Mann-Whitney test (performed in R project) was used to compare distributions of mRNA/ribosome footprint/translational efficiency fold changes between two gene sets.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-phospho-EIF2α | Cell Signaling Technology | CST #9721; RRID:AB_330951 |
| Rabbit anti- EIF2α | Cell Signaling Technology | CST #9722; RRID:AB_2230924 |
| Rabbit anti-phospho-p70S6 Kinase, T389 | Cell Signaling Technology | CST #9234; RRID:AB_2269803 |
| Rabbit anti- p70S6 Kinase | Cell Signaling Technology | CST #2708; RRID:AB_390722 |
| Rabbit anti-phospho S6 ribosomal protein, S240/244 | Cell Signaling Technology | CST #5364; RRID:AB_10694233 |
| Mouse anti-S6 ribosomal protein | Santa Cruz Biotechnology | sc-74459; RRID:AB_1129205 |
| Rabbit anti-phospho 4EBP1, T37/46 | Cell Signaling Technology | CST #2855; RRID:AB_560835 |
| Rabbit anti-4EBP1 | Cell Signaling Technology | CST #9644; RRID:AB_2097841 |
| Rabbit anti-ATG16L | MBL International | PM040; RRID:AB_1278757 |
| Rabbit anti-ATG5 | MBL International | PM050; RRID:AB_1520800 |
| Rabbit anti-GAPDH | Cell Signaling Technology | CST #2118; RRID:AB_561053 |
| Guinea pig anti-vGlut1 | Millipore Sigma | AB5905; RRID:AB_2301751 |
| Mouse anti-PSD95 | Thermo Fisher Scientific | MA1–046; RRID:AB_2092361 |
| Rabbit anti-vGAT | Synaptic Systems | #131 002; RRID:AB_887871 |
| Mouse anti-gephyrin | Santa Cruz Biotechnology | sc-25311; RRID:AB_627670 |
| Mouse anti-parvalbumin | Sigma Aldrich | P3088; RRID:AB_477329 |
| Mouse anti-GFAP | Santa Cruz Biotechnology | sc-33673; RRID:AB_627673 |
| Mouse anti-GAD67 | MBL International | M018–3; RRID:AB_591728 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pentylenetetrazol | Sigma-Aldrich | P6500 |
| Rapamycin | LC Laboratories | R5000 |
| DNQX | Tocris | # 0189/10 |
| Picrotoxin | Tocris | # 1128/1G |
| Kynurenic acid | Tocris | # 0223/1G |
| Tetrodotoxin | Abcam | ab120054 |
| Critical Commercial Assays | ||
| BaseScope Reagent Kit--RED | Advanced Cell Diagnostics | # 322971 |
| Neural Tissue Dissociation Kit-P | Miltenyi Biotec | # 130–092-628 |
| CD11b (Microglia) MicroBeads, human and mouse | Miltenyi Biotec | # 130–093-634 |
| Anti-ASCA2 MicroBead Kit | Miltenyi Biotec | # 130–097-678 |
| Anti-O4 MicroBeads, human, mouse, rat | Miltenyi Biotec | # 130–096-670 |
| Deposited Data | ||
| RNA-seq and ribosome profiling data | This study | GEO: GSE151742 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| B6J.Gcn2−/− mice (Eif2ak4tm1.2Dron) | The Jackson Laboratory | RRID:IMSR_JAX:008240 |
| B6.129(Cg)-Gabrg2tm1Spet/Frk | The Jackson Laboratory | RRID:IMSR_JAX:013209 |
| B6J-Gtpbp2−/− (C57BL/6J-Gtpbp2nmf205/J) | Ishimura et al., 2014 | RRID:IMSR_JAX:004823 |
| B6J-Tg(n-Tr20N/N) / B6J-Tgwt | Ishimura et al., 2014 | N/A |
| B6J.B6NnTr20 | Ishimura et al., 2014 | N/A |
| B6N-Tg(n-Tr20J/J) / B6N-Tgmut | Ishimura et al., 2016 | N/A |
| B6J-nTr20 −/− | Ishimura et al., 2016 | N/A |
| B6N-nTr20−/− | This study | N/A |
| B6J-Tg(n-Tr21)516 | This study | N/A |
| B6J-Tg(n-Tr21)310 | This study | N/A |
| B6J-Tg(n-Tr22)557 | This study | N/A |
| B6J-Tg(n-Tr22)529 | This study | N/A |
| B6J.N-nTi17−/− | This study | N/A |
| Oligonucleotides | ||
| n-Tr20 (B6N allele): GGGACTCGAACCCGGAACCTCTGGATT | Ishimura et al., 2014 | N/A |
| n-Tr20 (B6J allele): GGGACTCGAACCCAGAACCTCTGGATT | Ishimura et al., 2014 | N/A |
| n-Tr21: GGGACTCGAACCCACAACCTTTGAATT | Ishimura et al., 2014 | N/A |
| n-Tr22: GGGACTCGAACCCGGAACCTTTGAATT | Ishimura et al., 2014 | N/A |
| n-Tr23: GGGACTCGAACCCGCAACCTTTGAATT | Ishimura et al., 2014 | N/A |
| n-Tr25: GGGATTCGAACCCACAACCTTTGAATT | Ishimura et al., 2014 | N/A |
| n-Tr20 (both B6J and B6N alleles): CTGGATTAGAAGTCCAGCGCGCTCGTCC | Ishimura et al., 2014 | N/A |
| n-Tr21/22/23/25: TAGAAGTCCAATGCGCTATCCATTGCG | Ishimura et al., 2014 | N/A |
| n-Ti14/15/16/17: TCGAACTCACAACCTCGGCAT | This paper | N/A |
| nTi14/15/17: TGAGGCTCGAACTCACAACCTCGGC | This paper | N/A |
| 5S rRNA: CATCCAAGTACTAACCAGGCCCGAC | Ishimura et al., 2014 | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Axon pClamp 10.7 | Molecular Devices | N/A |
| Multiclamp 700B Commander software package | Molecular Devices | N/A |
| Mini Analysis program | Synaptosoft | N/A |
| Synapse Counter Image J plug-in | Dzyubenko et al., 2016 | N/A |
| Image J | NIH | https://imagej.nih.gov/ij |
| GraphPad Prism 7 | GraphPad software | N/A |
| kallisto v0.42.4 | Bray et al., 2016 | RRID:SCR_016582; https://pachterlab.github.io/kallisto/about |
| sleuth v0.30.0 | Pimentel et al., 2017 | RRID:SCR_016883; https://pachterlab.github.io/sleuth/about |
| featureCounts | Liao et al., 2014 | RRID:SCR_012919; http://bioinf.wehi.edu.au/featureCounts |
| Trim Galore | Martin, 2011 | RRID:SCR_011847; http://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| hisat2 v2.1.0 | Kim et al., 2019 | RRID:SCR_015530; https://daehwankimlab.github.io/hisat2/ |
| rMATS v3.2.5 | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net |
| fastx_clipper | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| fastx_trimmer | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| bowtie2 v 2.2.3 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| RiboWaltz v1.0.1 | Lauria et al., 2018 | RRID:SCR_016948; https://github.com/LabTranslationalArchitectomics/RiboWaltz |
| DESeq2 v1.22.2 | Love et al., 2014 | RRID:SCR_015687; https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| riborex v2.3.4 | Li et al., 2017 | https://github.com/smithlabcode/riborex |
| ensembldb v2.6.8 | Rainer et al., 2019 | https://www.bioconductor.org/packages/release/bioc/html/ensembldb.html |
| Pause site identification algorithm | Ishimura et al., 2014 | N/A |
| Ingenuity Pathway Analysis (IPA) | QIAGEN Inc | RRID:SCR_008653; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis |
| DAVID bioinformatics web server | Huang et al., 2009 | RRID:SCR_001881; http://david.abcc.ncifcrf.gov |
| Skyline | Maclean et al., 2010 | RRID:SCR_014080 |
| Other | ||
ACKNOWLEDGMENTS
We thank A. Pinto, A. Saghatelian, and the Mass Spectrometry Core of the Salk Institute for technical support for amino acid metabolomics; L.C. Foo for oligodendrocyte RNA; M. Terrey for embryonic brain RNA; A. Kano for mouse husbandry assistance; and Z.-W. Zhang for feedback on the manuscript. Libraries were sequenced at the IGM Genomics Center at University of California, San Diego (UCSD). This work was supported in part by NIH grants R01 NS094637 (S.L.A.) and R37 NS031349 (W.N.F.). S.L.A. is an investigator of the Howard Hughes Medical Institute. Amino acid metabolomics was supported by Mass Spectrometry Core of the Salk Institute with funding from NIH-NCI CCSG P30 014195 and the Helmsley Center for Genomic Medicine.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2020.07.023.
REFERENCES
- Averous J, Lambert-Langlais S, Mesclon F, Carraro V, Parry L, Jousse C, Bruhat A, Maurin AC, Pierre P, Proud CG, and Fafournoux P. (2016). GCN2 contributes to mTORC1 inhibition by leucine deprivation through an ATF4 independent mechanism. Sci. Rep 6, 27698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DW (1978). Sources of subline divergence and their relative importance for sublines of six major inbred strains of mice In Origins of Inbred Mice, Morse HC, ed. (Academic Press; ), pp. 197–215. [Google Scholar]
- Barton ME, Klein BD, Wolf HH, and White HS (2001). Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 47, 217–227. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, and Sabatini BL (2011). Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J. Neurosci 31, 8862–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, and Sabatini BL (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, Baulac M, Brice A, Bruzzone R, and LeGuern E. (2001). First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the γ2-subunit gene. Nat. Genet 28, 46–48. [DOI] [PubMed] [Google Scholar]
- Benthall KN, Ong SL, and Bateup HS (2018). Corticostriatal transmission is selectively enhanced in striatonigral neurons with postnatal loss of Tsc1. Cell Rep. 23, 3197–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram EH (2013). Neuronal circuits in epilepsy: do they matter? Exp. Neurol 244, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JRP, and Jonas P. (2006). Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat. Protoc 1, 2075–2081. [DOI] [PubMed] [Google Scholar]
- Bloom-Ackermann Z, Navon S, Gingold H, Towers R, Pilpel Y, and Dahan O. (2014). A comprehensive tRNA deletion library unravels the genetic architecture of the tRNA pool. PLoS Genet. 10, e1004084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Buffington SA, Huang W, and Costa-Mattioli M. (2014). Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci 37, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevalli LS, Pereira CM, Longo BM, Jaqueta CB, Avedissian M, Mello LEAM, and Castilho BA (2004). Phosphorylation of translation initiation factor eIF2α in the brain during pilocarpine-induced status epilepticus in mice. Neurosci. Lett 357, 191–194. [DOI] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2016). GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44 (D1), D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla MK, Gray DT, Nguyen C, Dhaliwal H, Zempare M, Okuno H, Huentelman MJ, and Barnes CA (2018). Seizure-induced arc mRNA expression thresholds in rat hippocampus and perirhinal cortex. Front. Syst. Neurosci 12, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, et al. (2005). Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436, 1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, et al. (2007). eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Qiu H, Garcia-Barrio M, Anderson J, and Hinnebusch AG (2000). Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279. [DOI] [PubMed] [Google Scholar]
- Draizen EJ, Shaytan AK, Mariño-Ramírez L, Talbert PB, Landsman D, and Panchenko AR (2016). HistoneDB 2.0: a histone database with variants–an integrated resource to explore histones and their variants. Database (Oxford) 2016, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, and Emery B. (2013). Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harb. Protoc. 2013, 745–758. [DOI] [PubMed] [Google Scholar]
- Dzyubenko E, Rozenberg A, Hermann DM, and Faissner A. (2016). Colocalization of synapse marker proteins evaluated by STED-microscopy reveals patterns of neuronal synapse distribution in vitro. J. Neurosci. Methods 273, 149–159. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, Schork NJ, St Jean P, Ballas C, Choi H, and Berrettini WH (1997). Mapping murine loci for seizure response to kainic acid. Mamm. Genome 8, 200–208. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Snyder R, Laibinis M, Smith GG, Buono RJ, and Berrettini WH (1998). Genetic influences on electrical seizure threshold. Brain Res. 813, 207–210. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, St Jean P, Schork NJ, Mulholland N, Ballas C, Schill J, Buono RJ, and Berrettini WH (1999). Mapping loci for pentylenetetrazol-induced seizure susceptibility in mice. J. Neurosci 19, 6733–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, Longman RL, Snyder RL, DeMuth D, Szpilzak I, Mulholland N, Eng E, Lohoff FW, et al. (2001). Quantitative genetic study of maximal electroshock seizure threshold in mice: evidence for a major seizure susceptibility locus on distal chromosome 1. Genomics 75, 35–42. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, Martin JF, Lohoff FW, Gieringer TA, Zamboni D, Schwebel CL, Press DM, Kratzer SO, et al. (2004). Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm. Genome 15, 239–251. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Dahl JP, Smith GG, Schwebel CL, MacDonald R, Lohoff FW, Berrettini WH, and Buono RJ (2007). Analysis of a quantitative trait locus for seizure susceptibility in mice using bacterial artificial chromosome-mediated gene transfer. Epilepsia 48, 1667–1677. [DOI] [PubMed] [Google Scholar]
- Frankel WN (2009). Genetics of complex neurological disease: challenges and opportunities for modeling epilepsy in mice and rats. Trends Genet. 25, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel WN, Taylor L, Beyer B, Tempel BL, and White HS (2001). Electroconvulsive thresholds of inbred mouse strains. Genomics 74, 306–312. [DOI] [PubMed] [Google Scholar]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD, et al. (2014). A dual program for translation regulation in cellular proliferation and differentiation. Cell 158, 1281–1292. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, and Coulter DA (2013). Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat. Rev. Neurosci 14, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, and Tavazoie SF (2016). Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165, 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Harding HP, Ordonez A, Allen F, Parts L, Inglis AJ, Williams RL, and Ron D. (2019). The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. eLife 8, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry FE, McCartney AJ, Neely R, Perez AS, Carruthers CJL, Stuenkel EL, Inoki K, and Sutton MA (2012). Retrograde changes in presynaptic function driven by dendritic mTORC1. J. Neurosci 32, 17128–17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez D, Torres CA, Setlik W, Cebrián C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE, et al. (2012). Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Iben JR, and Maraia RJ (2014). tRNA gene copy number variation in humans. Gene 536, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis AJ, Masson GR, Shao S, Perisic O, McLaughlin SH, Hegde RS, and Williams RL (2019). Activation of GCN2 by the ribosomal P-stalk. Proc. Natl. Acad. Sci. USA 116, 4946–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang X-L, Schimmel P, Senju S, Nishimura Y, Chuang JH, and Ackerman SL (2014). RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Chuang JH, and Ackerman SL (2016). Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. eLife 5, e14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, and White RJ (2010). mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc. Natl. Acad. Sci. USA 107, 11823–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur M, Monaghan CE, and Ackerman SL (2017). Regulation of mRNA translation in neurons—a matter of life and death. Neuron 96, 616–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosillo P, Doig NM, Ahmed KM, Agopyan-Miu AHCW, Wong CD, Conyers L, Threlfell S, Magill PJ, and Bateup HS (2019). Tsc1mTORC1 signaling controls striatal dopamine release and cognitive flexibility. Nat. Commun 10, 5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr., and Tugendreich S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutter C, Brown GD, Gonçalves A, Wilson MD, Watt S, Brazma A, White RJ, and Odom DT (2011). Pol III binding in six mammals shows conservation among amino acid isotypes despite divergence among tRNA genes. Nat. Genet 43, 948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lant JT, Berg MD, Heinemann IU, Brandl CJ, and O’Donoghue P. (2019). Pathways to disease from natural variations in human cytoplasmic tRNAs. J. Biol. Chem 294, 5294–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria F, Tebaldi T, Bernabò P, Groen EJN, Gillingwater TH, and Viero G. (2018). riboWaltz: Optimization of ribosome P-site positioning in ribosome profiling data. PLoS Comput. Biol. 14, e1006169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Lee J, and Kim E. (2017). Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81, 838–847. [DOI] [PubMed] [Google Scholar]
- Letts VA, Beyer BJ, and Frankel WN (2014). Hidden in plain sight: spike-wave discharges in mouse inbred strains. Genes Brain Behav. 13, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang W, Uren PJ, Penalva LOF, and Smith AD (2017). Riborex: fast and flexible identification of differential translation from Ribo-seq data. Bioinformatics 33, 1735–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2014). Feature Counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-through put sequencing reads. EMBnet. J 17, 10. [Google Scholar]
- McCabe MP, Cullen ER, Barrows CM, Shore AN, Tooke KI, Laprade KA, Stafford JM, and Weston MC (2020). Genetic inactivation of mTORC1 or mTORC2 in neurons reveals distinct functions in glutamatergic synaptic transmission. eLife 9, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod F, Marzo A, Podpolny M, Galli S, and Salinas P. (2017). Evaluation of synapse density in hippocampal rodent brain slices. J. Vis. Exp 128, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, and Hernandez N. (2010). mTORC1 directly phosphorylates and regulates human MAF1. Mol. Cell. Biol 30, 3749–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozhui K, Ciobanu DC, Schikorski T, Wang X, Lu L, and Williams RW (2008). Dissection of a QTL hotspot on mouse distal chromosome 1 that modulates neurobehavioral phenotypes and gene expression. PLoS Genet. 4, e1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Abreo T, Neuner SM, Parks C, Watkins CE, Houseal MT, Shapaker TM, Hook M, Tan H, Wang X, et al. (2019). Identification of a functional non-coding variant in the GABAA receptor α2 subunit of the C57BL/6J mouse reference genome: major implications for neuroscience research. Front. Genet 10, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Wang X, and Pan T. (2013). Diversity of human tRNA genes from the 1000-genomes project. RNA Biol. 10, 1853–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini S, Corona C, Liu J, Greene LA, and Shelanski ML (2015). Specific downregulation of hippocampal ATF4 reveals a necessary role in synaptic plasticity and memory. Cell Rep. 11, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, and Pan T. (2009). tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 37, 7268–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, and Hopper AK (2010). tRNA biology charges to the front. Genes Dev. 24, 1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L. (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Rainer J, Gatto L, and Weichen berger CX (2019). ensembldb: an R package to create and use Ensembl-based annotation resources. Bioinformatics 35, 3151–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagi D, Rak R, Gingold H, Adir I, Maayan G, Dahan O, Broday L, Pilpel Y, and Rechavi O. (2016). Tissue- and Time-Specific Expression of Otherwise Identical tRNA Genes. PLoS Genet. 12, e1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, and Sabatini DM (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarnati MS, Kataria R, Biswas M, and Paradiso KG (2018). Active presynaptic ribosomes in the mammalian brain, and altered transmitter release after protein synthesis inhibition. eLife 7, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt BM, Rudolph KLM, Karagianni P, Fonseca NA, White RJ, Talianidis I, Odom DT, Marioni JC, and Kutter C. (2014). High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA-tRNA interface. Genome Res. 24, 1797–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, et al. (2007). Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc. Natl. Acad. Sci. USA 104, 17536–17541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM (2000). Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 23, 305–312. [DOI] [PubMed] [Google Scholar]
- Thoreen CC (2017). The molecular basis of mTORC1-regulated translation. Biochem. Soc. Trans 45, 213–221. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornlow BP, Hough J, Roger JM, Gong H, Lowe TM, and Corbett-Detig RB (2018). Transfer RNA genes experience exceptionally elevated mutation rates. Proc. Natl. Acad. Sci. USA 115, 8996–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent M, Chalancon G, de Groot NS, Wuster A, and Madan Babu M. (2018). Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler AL, McGarr TC, Beyer BJ, Frankel WN, and Carter GW (2014). A genetic interaction network model of a complex neurological disease. Genes Brain Behav. 13, 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, and Berkovic SF (2001). Mutant GABA(A) receptor g2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet 28, 49–52. [DOI] [PubMed] [Google Scholar]
- Winawer MR, Gildersleeve SS, Phillips AG, Rabinowitz D, and Palmer AA (2011). Mapping a mouse limbic seizure susceptibility locus on chromosome 10. Epilepsia 52, 2076–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, and Leung LS (1997). Partial hippocampal kindling decreases efficacy of presynaptic GABAB autoreceptors in CA1. J. Neurosci 17, 9261–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R, Suzuki Y, Takeuchi N, Wakaguri H, Ueda T, Sugano S, and Nakai K. (2008). Comprehensive detection of human terminal oligo-pyrimidine (TOP) genes and analysis of their characteristics. Nucleic Acids Res. 36, 3707–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Palm W, Peng M, King B, Lindsten T, Li MO, Koumenis C, and Thompson CB (2015). GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 29, 2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, and Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc 7, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E, Ding J, Xia Y, Liu M, Ye B, Choi JH, Yan C, Dong Z, Huang S, Zha Y, et al. (2016). KDM4C and ATF4 cooperate in transcriptional control of amino acid metabolism. Cell Rep. 14, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, Zhou H, Bell JC, Friedlander MJ, Krnjevic K, et al. (2011). Suppression of PKR promotes network excitability and enhanced cognition by interferon-g-mediated disinhibition. Cell 147, 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.