

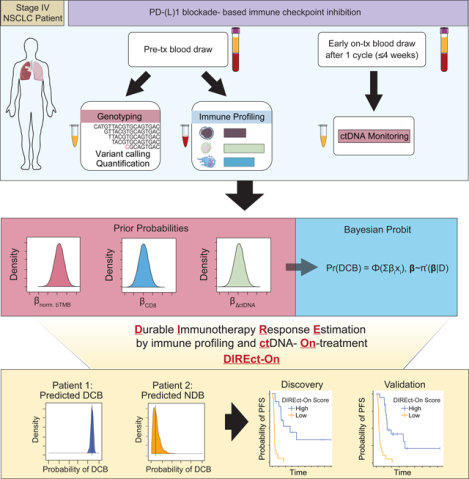
Summary
Although treatment of non-small cell lung cancer (NSCLC) with immune checkpoint inhibitors (ICI) can produce remarkably durable responses, most patients develop early disease progression. Furthermore, initial response assessment by conventional imaging is often unable to identify which patients will achieve durable clinical benefit (DCB). Here, we demonstrate that pre-treatment circulating tumor DNA (ctDNA) and peripheral CD8 T cell levels are independently associated with DCB. We further show that ctDNA dynamics after a single infusion can aid in identification of patients who will achieve DCB. Integrating these determinants, we developed and validated an entirely noninvasive multiparameter assay (DIREct-On, Durable Immunotherapy Response Estimation by immune profiling and ctDNA- On-treatment) that robustly predicts which patients will achieve DCB with higher accuracy than any individual feature. Taken together, these results demonstrate that integrated ctDNA and circulating immune cell profiling can provide accurate, noninvasive, and early forecasting of ultimate outcomes for NSCLC patients receiving ICI.
Keywords: Circulating tumor DNA, immunotherapy, response classification, non-small cell lung cancer
Graphical Abstract
In-brief
Multiparameter noninvasive models that integrate pre-treatment ctDNA and peripheral CD8+ T cell features, together with early on-treatment ctDNA dynamics, show promise in predicting durable clinical response to immune checkpoint blockade treatment in patients with non-small cell lung cancer.
Introduction
Antibody-based blockade of programmed death 1 and programmed death-ligand 1 (PD-[L]1) signaling has shown remarkable promise for treatment of advanced non-small lung cancer (NSCLC) (Reck et al., 2016; Socinski et al., 2018). Clinical trials combining PD-(L)1 blockade with cytotoxic therapy or with other immune checkpoint inhibition (ICI) strategies have shown higher response rates at the risk of higher toxicity (Gandhi et al., 2018; Hellmann et al., 2018). However, only a minority of patients achieve durable benefit from ICI and reliable biomarkers that can accurately identify these patients before or early during treatment have thus far remained elusive (Camidge et al., 2019). The most heavily studied biomarkers for predicting response to PD-(L)1 blockade-based ICI prior to therapy include assessment of tumor PD-L1 expression and tumor mutational burden (TMB) (Cristescu et al., 2018; Reck et al., 2016). Moreover, PD-L1 has several major shortcomings as a predictive biomarker of durable benefit while TMB is continuing to be evaluated clinically (Camidge et al., 2019; Cristescu et al., 2018; Rizvi et al., 2018).
Current practice in the US for front-line therapy of advanced, driver mutation negative NSCLC is to treat patients with PD-L1 ≤ 49% (percentage of PD-L1+ tumor cells by immunohistochemistry) with concurrent chemotherapy plus pembrolizumab, and those with PD-L1 ≥ 50% with pembrolizumab alone or with concurrent chemotherapy (Hanna et al., 2020). However, a significant subset of patients whose tumors have ≤ 49% PD-L1 staining can respond to immunotherapy alone. For example, the KEYNOTE-042 trial which compared pembrolizumab versus chemotherapy in advanced non-squamous NSCLC demonstrated that pembrolizumab alone had an objective response rate of 16% in patients with PD-L1 1–49%, comprising of 32% of all objective responses observed (Mok et al., 2019). Thus, PD-L1 staining alone is not sufficiently accurate to identify all potential responders to PD-(L)1 blockade-based ICI in NSCLC.
In the search for better biomarkers to predict which patients will respond to immunotherapy, several groups have recently demonstrated that multivariable models based on molecular analyses of tumor biopsy tissue collected prior to treatment can predict response to ICI (Anagnostou et al., 2020; Auslander et al., 2018; Cristescu et al., 2018; Jiang et al., 2018). Each of these methods achieves modest improvement for predicting benefit (area under the curve, [AUC]= 0.7–0.8) over PD-L1 expression (AUC = 0.6–0.7) (Mok et al., 2019) or TMB alone (AUC = 0.6–0.7) (Rizvi et al., 2018). Moreover, few studies have applied this type of approach to NSCLC. Additionally, the requirement for tumor tissue can be problematic in advanced NSCLC, where obtaining sufficient tissue for molecular analysis is not possible in a substantial subset of patients (Green et al., 2014; Lim et al., 2015). Reliance on single biopsy specimens also risks confounding of biomarker results due to intra-tumoral heterogeneity or low tumor content, leading to lower than desired inter-observer concordance (Camidge et al., 2019).
A separate approach for predicting ultimate clinical benefit of ICI would be early response assessment. Clinically, response is currently evaluated by conventional imaging, usually performed six to twelve weeks after treatment start. To assess how accurately the first on-treatment imaging study identifies which patients ultimately achieve long-lasting clinical benefit from ICI, we analyzed response data from 273 NSCLC patients receiving PD-(L)1-based ICI (αPD-(L)1, 75%; αPD-1+chemotherapy, 13%; αPD-(L)1+αCTLA-4, 12%). Strikingly, we found that among patients achieving stable disease (SD, n = 112) at the first scan, about half (51%; n=57) did not ultimately achieve durable clinical benefit (DCB), defined as progression-free survival (PFS) of at least 6 months (Fig. 1A) (Rizvi et al., 2015). We observed similar ultimate outcomes after initial stable scans irrespective of PD-(L)1 blockade-based therapy regimen (Fig. S1A). Further, 39% of patients (35/90) who achieved SD as best overall response ultimately experienced DCB, suggesting objective responses assessed by imaging are not fully capturing patients that are benefitting. Therefore, there is considerable unmet need for early response assessment methods that more accurately identify who will achieve DCB or NDB.
Figure 1: Association of radiologic response and durable clinical benefit in NSCLC.
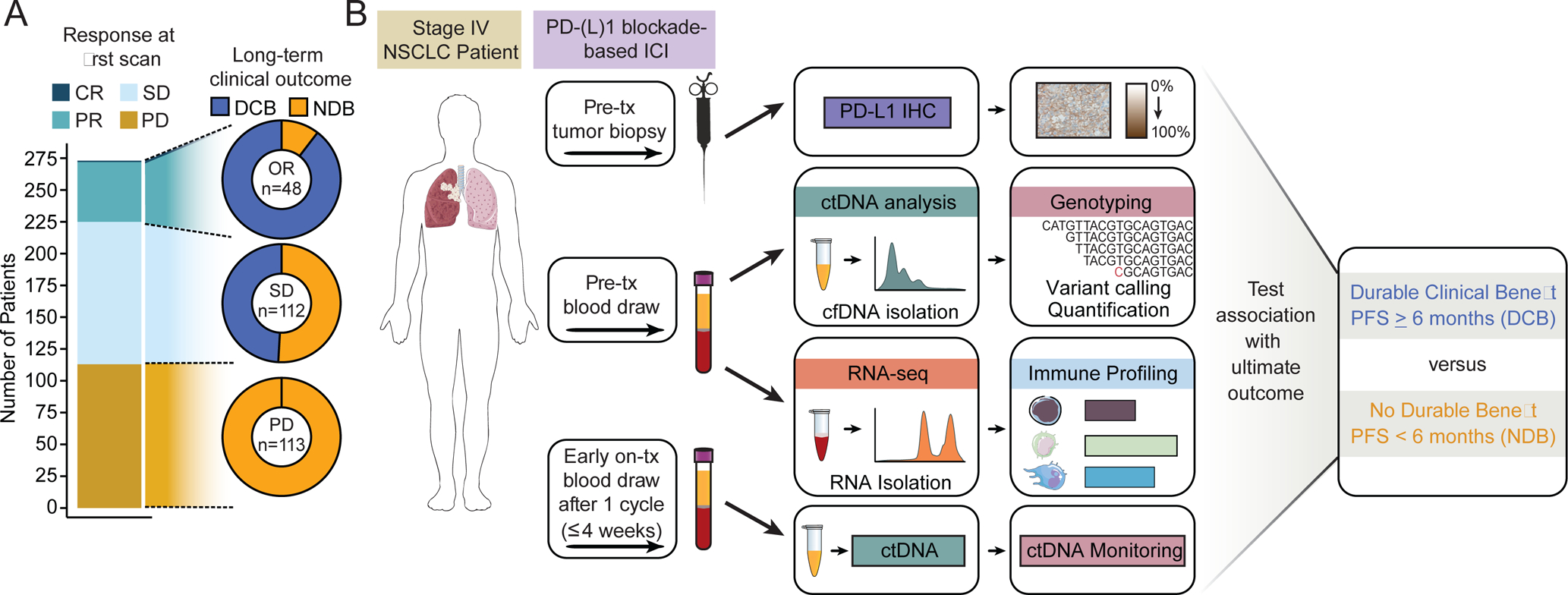
A) Rate of DCB or NDB in advanced NSCLC patients receiving PD-(L)1 blockade-based ICI achieving partial response (PR), stable disease (SD), or progressive disease (PD) at the first scan by RECIST v1.1 criteria.
B) Study schematic. Pre-treatment blood from NSCLC patients receiving PD-(L)1 blockade-based ICI was collected and fractionated for ctDNA analysis and RNA-seq for immune profiling. Pre-treatment tumor biopsies were used to assess PD-L1 expression. Early on-treatment blood was collected within 4 weeks for ctDNA monitoring. Tumor and immune features were tested for their association with ultimate outcomes. See also Figure S1.
One such potential approach, is comparison of circulating tumor DNA (ctDNA) levels before and after the start of treatment to assess treatment response. Indeed, several recent publications have demonstrated that ctDNA changes 4–8 weeks after therapy initiation can classify response to ICI in NSCLC with modest accuracy (AUC = 0.65–0.75) (Anagnostou et al., 2019; Goldberg et al., 2018; Raja et al., 2018). Each of these studies used a different ctDNA change threshold and timepoint to assess molecular responses, leaving questions about the optimal approach. Furthermore, even earlier classification of ultimate clinical benefit is desirable to improve personalized medicine approaches.
We therefore set out to develop a noninvasive approach for early identification of which advanced NSCLC patients will achieve DCB upon treatment with PD-(L)1 blockade-based ICI. We hypothesized that both tumor and immune factors can contribute to PD-(L)1 blockade-based ICI outcome prediction and can be measured noninvasively. Specifically, we integrated ctDNA profiling, which measures tumor properties, and analysis of circulating immune cells, which reflect the immune milieu, using pre- and early on-treatment blood samples. To combine these factors into a single biomarker, we applied a Bayesian framework for integrating diverse risk predictors that we recently demonstrated has significant advantages compare to Cox models for time-dependent biomarker development (Kurtz et al., 2019). Using a discovery and validation approach we demonstrate that integration of tumor-intrinsic and -extrinsic features assessable from noninvasive blood draws can robustly identify which NSCLC patients will achieve durable clinical benefit from ICI.
Results
Assembly of patient cohorts
To develop a multiparameter biomarker for predicting clinical benefit from ICIs in NSCLC, we assembled a cohort of 99 advanced NSCLC patients that received PD-(L)1 blockade-based ICI. Tumors were characterized for PD-L1 expression and we analyzed ctDNA in pre-treatment plasma specimens using CAPP-Seq (Fig. 1B, Tables S1–2) (Chabon et al., 2020; Newman et al., 2014, 2016). We performed CAPP-Seq on both cell-free DNA from plasma and cellular DNA from leukocytes to enable censoring of clonal hematopoiesis variants (see STAR Methods). To investigate the ability of circulating leukocyte profiles to predict ICI response, we also profiled available pre-treatment blood specimens for leukocyte immune composition using CIBERSORT (Newman et al., 2015, 2019), flow cytometry, or both. Lastly, we performed CAPP-Seq ctDNA analysis on early on-treatment plasma samples to allow incorporation of ctDNA responses. Of the 99 patients, 94 had detectable pre-treatment ctDNA by CAPP-Seq (95%, Tables S3–4). These 94 patients were split into 3 groups for downstream analyses: 1) 22 patients who did not have early on-treatment plasma samples or material for immune profiling available and were therefore only informative as part of feature-discovery analyses, 2) 34 patients that had all necessary samples (pre-treatment plasma, pre-treatment leukocytes, and early on-treatment plasma) and were used to train the multiparameter model (“DIREct Discovery Cohort”), and 3) 38 patients that had all necessary samples and constituted the DIREct Validation Cohort. The latter were held out from all analyses until utilized for validation of findings from (1) and (2) (Fig. S1B).
Pre-treatment ctDNA features are associated with durable clinical benefit from ICI.
Responses induced by ICI and their durability are known to be influenced both by the burden of mutations as neoantigenic substrates for immune recognition as captured by TMB (Mandal et al., 2019; Rizvi et al., 2015; Snyder et al., 2014) and by the total anatomic disease burden (Ito et al., 2019; Kaira et al., 2017). Prior studies have demonstrated that tissue-based TMB can be noninvasively estimated from ctDNA using sequencing panels of various sizes, as might be useful for predicting response to ICI (Chaudhuri et al., 2017; Gandara et al., 2018; Wang et al., 2019). To explore this further, we began by analyzing data from a recently published study of 853 advanced NSCLC patients treated with either the PD-L1 inhibitor atezolizumab (POPLAR/OAK ICI Cohort, n = 429) or chemotherapy (POPLAR/OAK Chemo Cohort, n = 424), and examined whether ctDNA metrics and PD-L1 expression could accurately classify DCB versus NDB (Fig. S1C) (Gandara et al., 2018). While high bTMB (≥14 mutations per megabase) was significantly associated with durable benefit from ICI, we found the strength of this association to be modest (Fig. 2A, 2D). Separately, we reasoned that pre-treatment ctDNA concentration may be associated with treatment benefit since it is a surrogate for total body disease burden (Chaudhuri et al., 2017; Ito et al., 2019). Indeed, patients ultimately achieving DCB had significantly lower ctDNA levels prior to ICI therapy (Fig. 2B, 2D). Furthermore, since these two blood-based measurements had reciprocal associations with DCB, we used their ratio to integrate them as ctDNA-normalized bTMB (normalized bTMB). Patients achieving DCB from ICI had significantly higher normalized bTMB (Fig. 2C–D). Accordingly, ICI-treated patients with higher normalized bTMB had significantly better ultimate clinical outcomes (Fig. 2E, P = 0.001, PFS HR = 1.46). Moreover, when comparing hazard ratios of normalized bTMB to either bTMB or ctDNA alone, we find that normalized bTMB is superior to both individual metrics in ICI-treated patients (Fig. S2C).
Figure 2: Pre-treatment ctDNA-normalized tumor mutation burden predicts response to ICI.
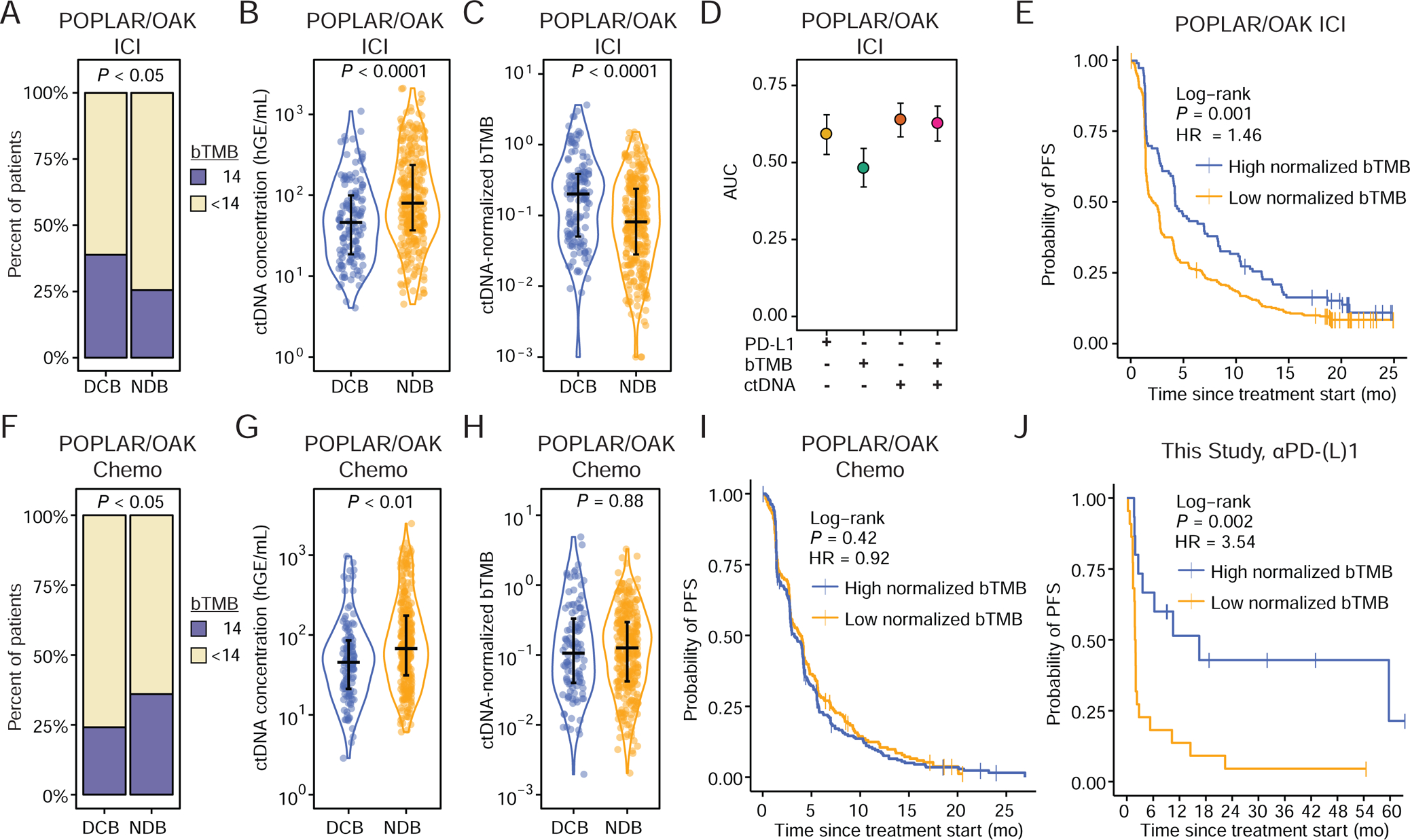
A) Outcomes of PD-L1 blockade treated patients from the POPLAR/OAK Cohort (Gandara et al., 2018) (POPLAR/OAK ICI Cohort) stratified by high bTMB/MB (≥14) and low bTMB/MB (<14). P-value calculated by two-sided Fisher’s Exact Test (DCB n = 139; NDB n = 290).
B) Pre-treatment ctDNA concentration (haploid genome equivalents per mL of plasma, hGE/mL) and C) ctDNA-normalized bTMB (norm. bTMB) in POPLAR/OAK ICI Cohort. P-values were calculating using a Wilcoxon test.
D) Area under the curve (AUC) for individual parameters in immunotherapy patients generated by leave-one-out cross-validation (LOOCV) ROC analysis.
E) Probability of PFS for high norm. bTMB (median = 4.14 mo.) and low norm. bTMB (median = 2.16 mo.) PD-L1 blockade patients stratified by the LOOCV-identified optimal cut-point in the POPLAR OAK ICI Cohort (n = 429).
F) Outcomes of chemotherapy treated patients from the POPLAR/OAK Cohort (Gandara et al., 2018) (POPLAR/OAK Chemo Cohort) stratified by high bTMB/MB (≥14) and low bTMB/MB (<14). P-value calculated by two-sided Fisher’s Exact Test (DCB n=118; NDB n = 306).
G) Pre-treatment ctDNA concentration and H) norm. bTMB (POPLAR/OAK Chemo Cohort) in chemotherapy patients. P-values were calculated using a Wilcoxon test.
I) Probability of PFS for high norm. bTMB (median = 3.25 mo.) and low norm. bTMB (median = 4.00 mo.) chemotherapy patients stratified by the LOOCV-identified optimal cut-point in the POPLAR/OAK Chemo Cohort (n = 424).
J) Probability of PFS for high norm. bTMB (median = 16.49 mo.) and low norm. bTMB (median = 1.92 mo.) patients who received single-agent PD-(L)1 blockade in this study, exclusive of the DIREct Validation Cohort (n = 37) stratified by the cut-point of norm. bTMB identified in the POPLAR/OAK ICI Cohort. See also Figure S2.
To assess if normalized bTMB was predictive of durable benefit specifically for ICI, we separately analyzed its relationship to outcomes in patients who received chemotherapy alone (Fig. 2F–I). Interestingly, in chemotherapy treated patients, lower bTMB and lower ctDNA burden were individually associated with higher likelihood of DCB, such that normalized bTMB was not associated with outcome in the absence of ICI (P = 0.88, Fig. 2F–I). As expected, tumor PD-L1 expression was also associated with DCB only in the setting of ICI (P < 0.01 for ICI; P = 0.41 for chemotherapy). Therefore, while ctDNA and bTMB were associated with outcomes in patients treated with either ICI or chemotherapy, ctDNA normalized bTMB was only predictive in the context of ICI.
Independent validation of normalized bTMB as a predictor of ICI response
Next we tested the validity of the association of ctDNA concentration and normalized bTMB with ultimate outcomes in response to single-agent PD-L1 blockade using an independent cohort of patients newly profiled in this study. We first determined the relationship between TMB measured in cell-free DNA by CAPP-Seq and tumor TMB measured by whole-exome sequencing using matched tissue samples (Fig. S2A–B). In patients treated with single-agent PD-(L)1 blockade, the associations between clinical benefit and ctDNA concentration or normalized bTMB in our cases mirrored those observed in the POPLAR/OAK ICI Cohort (Fig. S2D–G). Utilizing the normalized bTMB threshold defined in the POPLAR/OAK ICI Cohort to stratify patients in the validation cohort, patients with high normalized bTMB had significantly better outcomes (Fig 2J, P = 0.002, PFS HR = 3.54). Collectively, these data suggest that pre-treatment normalized bTMB can noninvasively identify patients most likely to experience durable benefit in response to ICI. However, nearly one third of patients were misclassified using normalized bTMB alone: ~25% of patients predicted to have NDB nevertheless achieved PFS ≥6 months, and conversely, ~40% of patients predicted to have DCB had PFS < 6 months. This suggests that further improvement in predictive accuracy would be desirable.
Circulating immune profiles predict outcomes to PD-(L)1 blockade-based ICI.
We therefore explored other biomarkers as potential determinants of durable benefit from ICI. Specifically, we reasoned that the frequency and representation of circulating leukocyte subsets might vary between patients achieving or failing to achieve DCB from ICI. We therefore profiled leukocyte transcriptomes in patients with available specimens (excluding the DIREct Validation Cohort, Fig. S1B) using RNA-seq and applied CIBERSORTx (Newman et al., 2015, 2019) to quantify the relative proportions of major leukocyte subpopulations (Tables S5–6). We confirmed highly correlated composition estimates of leukocyte composition, whether using RNA-seq deconvolution or conventional flow cytometric immunophenotyping (Fig. S3A–B). Interestingly, we observed fewer circulating CD8 T cells prior to ICI therapy in patients ultimately achieving DCB—while no other cell type was significantly associated with clinical benefit (Fig. 3A–B, Fig. S3C). Indeed, CD8 T cell levels alone had comparable classification accuracy for predicting durable benefit (Accuracy = 70%), compared to other pre-treatment features including PD-L1 and normalized bTMB (Fig. 3C). Furthermore, there were no significant differences in pre-treatment circulating CD8 T cell fractions based on prior receipt of chemotherapy, radiation, or corticosteroids (data not shown). Of note, none of the three pre-treatment biomarkers were correlated with each other, suggesting that they are independent and might therefore be useful in the context of a multiparameter predictor of ICI benefit (Fig. S3D).
Figure 3: Pre-treatment circulating immune profiles and early on-treatment ctDNA dynamics predict outcomes to PD-(L)1 blockade-based ICI.

A) Comparison of pre-treatment circulating immune cell populations by CIBERSORTx restricted to those with >1% median frequency in patients achieving DCB (n = 20) vs. NBD (n = 17). Shown are AUCs and 95% confidence intervals (CIs) generated by bootstrapping for classifying DCB versus NDB by each cell population. Shown on the top is the median relative abundance of each cell type in these patients.
B) Pre-treatment relative CD8 T cell fraction in circulation of ICI DCB (n = 20) and NDB (n = 17) with available CIBERSORTx immune profiling. P-value was calculating using a Wilcoxon test.
C) Bootstrapping-generated accuracy of outcome classification using the indicated pre-treatment tumor, ctDNA, and/or immune parameters in cases with all data types available (n = 27).
D) Early on-treatment (≤4 weeks) ctDNA concentration normalized to pre-treatment ctDNA concentration (median = 2.4 weeks, n = 46, DCB = 27, NDB = 19). Colors indicate the ultimate clinical outcome. The dashed line indicates 50% of pre-treatment ctDNA concentration (ctDNA molecular response).
E) Early on-treatment ctDNA concentration at the first timepoint after the first cycle of ICI normalized to pre-treatment ctDNA concentration, stratified by ultimate clinical outcome. Colors indicate if the sample was collected ≤2 or 2–3.3 weeks after treatment initiation. The dashed line indicates ctDNA molecular response.
F) Probability of PFS in patients with at least a 0.5-fold ctDNA drop from baseline (median = 22.4 mo.) and those without at least a 0.5-fold ctDNA drop (median = 2.30 mo.) within 4 weeks of treatment start. See also Figure S3.
Early on-treatment ctDNA dynamics classify durable benefit from PD-(L)1 blockade-based ICI
Recent studies have reported that ctDNA responses within 4–8 weeks after starting ICI correlate with best radiographic responses and modestly with DCB (Anagnostou et al., 2019; Goldberg et al., 2018; Raja et al., 2018). We therefore explored whether ctDNA kinetics within 4 weeks of starting treatment could help determine likelihood of durable benefit from ICI. We measured post-treatment ctDNA responses early after ICI therapy initiation in 46 patients with available samples (excluding the DIREct Validation Cohort, Fig. S1B) with detectable ctDNA at baseline (Tables S3–4). The median time of blood collection from treatment start was 2.4 weeks and 98% (n=45) of patients had the sample collected after only a single cycle of ICI. Based on a prior study of intra-day ctDNA variation in untreated advanced NSCLC, we defined a threshold of 50% decrease in ctDNA concentration from pre-treatment as a “ctDNA molecular response” (Wang et al., 2017). Even at these early time points, ctDNA levels showed significant changes compared to baseline in most patients (Fig. 3D). ctDNA burden dropped in 59% of patients achieving DCB while it did not meet the 0.5-fold threshold in 95% of NDB patients (P < 0.0001; Fig. 3D–3E). Strikingly, ctDNA responses after a single cycle of ICI therapy distinguished the majority of DCB from NDB patients (Fig. 3E). Furthermore, early ctDNA dynamics outperformed all individual pre-treatment factors (Fig. S3E, P < 0.05, Accuracy = 73%) and patients with ctDNA molecular responses had significantly better clinical outcomes (Fig. 3F, P = 0.013, PFS HR = 2.28). Thus, early ctDNA molecular response after just one cycle of ICI appears to be a promising approach for early response assessment. However, given that over 25% of patients are incorrectly classified, further improvements in predicting ultimate clinical outcomes would be desirable.
Multiparameter models for predicting DCB after PD-(L)1 blockade-based ICI.
Having identified tumor-cell intrinsic and extrinsic determinants of ICI therapeutic benefit, we next combined these into integrated multi-parametric models. We developed two related Bayesian biomarker models, one relying on pre-treatment factors alone, and another incorporating early on-treatment ctDNA dynamics (Fig. 4A). A Bayesian approach was chosen because we have previously demonstrated that it is a robust method for building risk-based models that include dynamic ctDNA measurements (Kurtz et al., 2019). Four key features of the Bayesian approach that make it desirable are that it: 1) enables incorporation of prior knowledge from established biomarkers to guard against overfitting to our own discovery cohort, 2) allows for missing parameters in cases where a specific feature may be unavailable (e.g. assay failure), 3) is well suited to binary classification problems often faced in the clinic such as DCB versus NDB, and 4) requires fewer training samples before predictions stabilize (Kurtz et al., 2019).
Figure 4: Multiparameter Bayesian frameworks for noninvasive outcome classification.

A) Schematic depicting the discovery and validation approach for generating and testing the DIREct models.
B) Patient characteristics. Each column represents an individual patient. Tumor histology, smoking status, best overall response, tumor PD-L1 expression, and PD-(L)1 blockade-based ICI therapy type (PD-(L)1 blockade alone; PD-(L)1 blockade with either CTLA-4 or chemotherapy) are indicated. PFS is shown in months, where asterisks signify ongoing responses. TMB is presented as the number of nonsynonymous mutations per megabase of the coding exome captured, measured in the blood (See Figure S2A–B). Mutations in the most recurrently mutated genes in TCGA NSCLC cases that also overlapped the NSCLC-focused CAPP-Seq selector in our cohort are shown at the bottom. See also Figure S4.
We trained the models in a discovery cohort consisting of patients with available pre-treatment and early on-treatment (≤4 weeks from start) blood samples (Fig. S1B, DIREct Discovery Cohort, n = 34 patients) and then validated these in an independent DIREct Validation Cohort (n=38 patients; Fig. S1B, Fig. 4A). The patients in the two cohorts were all treated PD-(L)1 blockade-based ICI (PD-1, n = 45; PD-L1, n = 2; PD-1+CTLA-4, n = 13; PD-1+Chemotherapy, n = 12) and had an expected distribution of histology, smoking status, tumor PD-L1 expression, and mutations for advanced NSCLC patients receiving ICI (Fig 4B).
Hyperparameters for the Bayesian models were inferred in two ways. For tumor PD-L1 expression and normalized bTMB the hyperparameters were empirically inferred from the independent POPLAR/OAK ICI Cohort. For parameters unique to this study (circulating CD8 T cell fraction and early ctDNA dynamics) hyperparameters were inferred in a leave-one-out cross-validation framework (LOOCV) using the samples from the DIREct Discovery Cohort. The models were then trained in a LOOCV framework within the DIREct Discovery Cohort. For each patient, the models generate a distribution of the probability of achieving DCB and the median of this distribution is used for predicting ultimate outcomes. Receiver operating characteristic curve analysis was used to determine the optimum threshold for classification of DCB versus NDB. The models and thresholds were then locked and we tested the performance of each of the parameters, models, and thresholds in the independent DIREct Validation Cohort of 38 advanced NSCLC patients receiving PD-(L)1 blockade-based ICI (DIREct Validation Cohort, Table S7).
DIREct-Pre forecasts ultimate outcomes using only pre-treatment parameters.
Using the outlined approach, a model based on pre-treatment tumor PD-L1 expression, normalized bTMB, and circulating CD8 T cells (DIREct-Pre: Durable Immunotherapy Response Estimation by immune profiling and ctDNA- Pre-treatment) was trained in the DIREct Discovery Cohort. Cross-validation analyses revealed that the DIREct-Pre model achieved 88% sensitivity (classifying DCB), at 56% specificity (Accuracy = 71%, AUC = 0.68, Precision = 64%, Recall = 88%, Fig. 5A, S4B). Patients with higher DIREct-Pre scores achieved significantly longer PFS (Fig. 5B, PFS HR = 2.66, P = 0.012, Table S7). DIREct-Pre had similar classification performance in the validation cohort as in the discovery cohort (Accuracy = 74%, AUC = 0.74, Precision = 79%, Recall = 71%, Table S7). Moreover, patients with higher DIREct-Pre scores had significantly longer PFS than those with low DIREct-Pre scores (Fig. 5C, P = 0.03, PFS HR = 2.18).
Figure 5: DIREct-On enables fully noninvasive outcome classification.

A) Proportion of patients expected to achieve DCB (blue) or NDB (orange) by the DIREct-Pre model stratified by clinical outcome determined by RECIST in the DIREct Discovery Cohort (n = 34).
B) Probability of PFS for high DIREct-Pre score (median = 8.2 mo.) and low DIREct-Pre score (median = 2.0 mo.) patients in the DIREct Discovery Cohort, using the optimal cut-point identified by LOOCV analysis (n = 34).
C) Probability of PFS for high DIREct-Pre score (median = 8.4 mo.) and low DIREct-Pre score (median = 2.6 mo.) patients in the DIREct Validation Cohort using the cut-point defined in the DIREct Discovery Cohort (n = 38).
D) Hazard ratio (top) or accuracy (bottom) for low scores (below LOOCV-generated cut-point) of each model with the indicated parameters only considering patients with all parameters available in the DIREct Discovery Cohort (n = 26). Error bars represent the 95% CIs generated by bootstrapping. NS = not significant, ** = P < 0.01.
E) Proportion of patients expected to achieve DCB (blue) or NDB (orange) by DIREct-On stratified by clinical outcome determined by RECIST in the DIREct Discovery Cohort (n = 34).
F) Probability of PFS for high DIREct-On score (median = 16.5 mo.) and low DIREct-On score (median = 1.9 mo.) patients in the DIREct Discovery Cohort, using the optimal cut-point identified by LOOCV analysis (n = 34).
G) Probability of PFS for high DIREct-On score (median = 8.5 mo.) and low DIREct-On score (median = 2.1 mo.) patients in the DIREct Validation Cohort using the cut-point defined in the DIREct Discovery Cohort (n = 38).
H) Accuracy for each individual parameter and DIREct-On in the combined DIREct Discovery and Validation Cohorts for those cases with all data types available, using the cut-points identified the DIREct Discovery Cohort (n = 58). Error bars represent 95% CIs generated by bootstrapping. ** = P < 0.01, *** = P < 0.001.
I) Net reclassification improvement of DIREct-On compared to each individual feature (top) and DIREct-On compared to Bayesian models with each feature that comprises DIREct-On removed (bottom). Errors bars represent 95% CIs generated by bootstrapping. * = P < 0.01, ** = P < 0.01. See also Figure S5.
Addition of ctDNA dynamics enable noninvasive classification of ultimate outcomes.
While promising, we reasoned that incorporation of early ctDNA dynamics would further improve response classification performance. Indeed, the addition of early ctDNA response to DIREct-Pre significantly improved classification accuracy (Fig. 5D). Furthermore, addition of early on-treatment ctDNA dynamics rendered tumor PD-L1 expression dispensable for outcome classification and therefore we removed it from our final model (DIREct-On, Durable Immunotherapy Response Estimation by immune profiling and ctDNA- On-treatment, Fig. 5D, Fig. S4A–B). Cross-validation analyses revealed the DIREct-On model to achieve 94% sensitivity (classifying DCB) and 89% specificity in the DIREct Discovery Cohort (Fig. 5E, S4B). Patients with higher DIREct-On scores had substantially longer PFS than those with lower scores (Fig. 5F, P < 0.0001, HR = 8.93). Overall, DIREct-On achieved excellent classification performance across various performance metrics in the discovery cohort (Accuracy = 92%, AUC = 0.93, Precision = 88%, Recall = 94%, Table S7). Importantly, application of the locked DIREct-On model and same threshold to the validation cohort demonstrated similarly excellent performance (Accuracy = 92%, AUC = 0.93, Precision = 95%, Recall = 90%, Table S7). As expected, patients in the validation cohort with high DIREct-On scores also had significantly longer PFS than those with low DIREct-On scores, with a median PFS of 8.1 months versus 2.1 months, respectively (Fig. 5G, P < 0.0001, PFS HR = 7.11). Thus, DIREct-On is a fully non-invasive multiparameter biomarker that robustly classifies ultimate outcomes to PD-(L)1 blockade-based ICI.
DIREct-On outperforms ctDNA dynamics alone
To assess performance of DIREct-On compared to each individual feature, we compared accuracy of DCB versus NDB classification and hazard ratios of DIREct-On with that of individual features in the combined discovery and validation cohorts. DIREct-On had significantly better classification accuracy than each individual metric or tumor PD-L1 expression (Fig. 5H). Moreover, comparison of hazard ratios also demonstrated that patients with high DIREct-On scores had a significantly lower risk of progression than patients with ctDNA molecular responses, low circulating CD8 T cells, high normalized bTMB, or high tumor PD-L1 expression (P < 0.0001). (Fig. S5A). Similarly, we also assessed the superiority of DIREct-On to each of the individual features that comprise the model using Net Reclassification Improvement (NRI), which quantifies classification performance differences between models. DIREct-On was significantly superior to normalized bTMB (NRI = −1.48, P < 0.05), CD8 T cell fraction (NRI = −1.64, P < 0.01), and ctDNA dynamics (NRI = −1.13, P < 0.01) (Fig. 5I, top). Thus, the multiparameter DIREct-On model significantly outperforms each individual feature.
DIREct-On requires each feature for optimal classification efficacy
To explore the importance of all three features of the DIREct-On model we examined the ability of the other two features to correctly predict outcome when the third feature incorrectly predicts outcome. In each reclassification scenario, each feature was able to rescue classification in ≥45% of the cases (Fig. S5B–D). Notably, for the pre-treatment features, normalized bTMB and CD8 T cell fraction were able to correctly classify at least 62% of their respective misclassifications (Fig. S5B–C).
To further assess if each of the features is required for optimal DIREct-On outcome classification, we constructed DIREct models that had each one of the parameters removed and compared the performance of these models to that of the full DIREct-On model. This enabled us to measure the impact of each feature on the overall performance of DIREct-On. We found that each feature that comprises DIREct-On is required for optimal performance (Fig. 5I, bottom). As expected, removal of ctDNA dynamics had the most detrimental effect (NRI = −1.60, P < 0.001), but removal of either normalized bTMB or CD8 T cell fractions also significantly decreased performance (NRI = −1.25, P < 0.001; NRI = −1.13, P < 0.001, respectively). Comparison of HRs for the full DIREct-On model and the versions with each parameter removed showed similar results (Fig. S5E). Thus, each feature is required for optimal response classification.
DIREct-On forecasts response to PD-(L)1 blockade-based ICI
We next tested calibration of DIREct-Pre and DIREct-On in the validation cohort to evaluate how accurately they forecast ultimate clinical outcomes (Steyerberg et al., 2010) and found that both models were well-calibrated (slope = 0.64, 0.98, respectively; Fig. S5F–G). Similarly, the DIREct-Pre and DIREct-On scores forecasted PFS as continuous variables in the validation cohort (Cox likelihood ratio test: PFS HR = 6.97, P = 0.007; PFS HR = 11.53, P < 0.0001, respectively).
Overall, in the cases that DIREct-On classified correctly, the second DIREct-On blood draw was performed on average 5.52 weeks prior to the first scan (Fig. 6). Moreover, DIREct-On correctly stratified patient outcomes on average 6.7 weeks prior to developing radiologically-defined progression in NDB cases (n = 35) and on average 24.5 weeks prior to the best overall radiographic response in DCB cases (n = 37), indicating significant lead times (Fig. 6). Moreover, DIREct-On stratified patients equally well whether the early on-treatment blood draw was collected prior to 3 weeks or between 3 and 4 weeks (data not shown). Additionally, we assessed performance of age, ECOG performance status, line of therapy, and DIREct-On in a multivariable Cox proportional hazards model to test if DIREct-On was independent from clinical factors known to be associated with outcome. DIREct-On was the only feature that was independently associated with PFS in this analysis (Fig. S6A, HR = 0.06, P < 0.0001).
Figure 6: Clinical course for patients in the DIREct Discovery and Validation Cohorts.
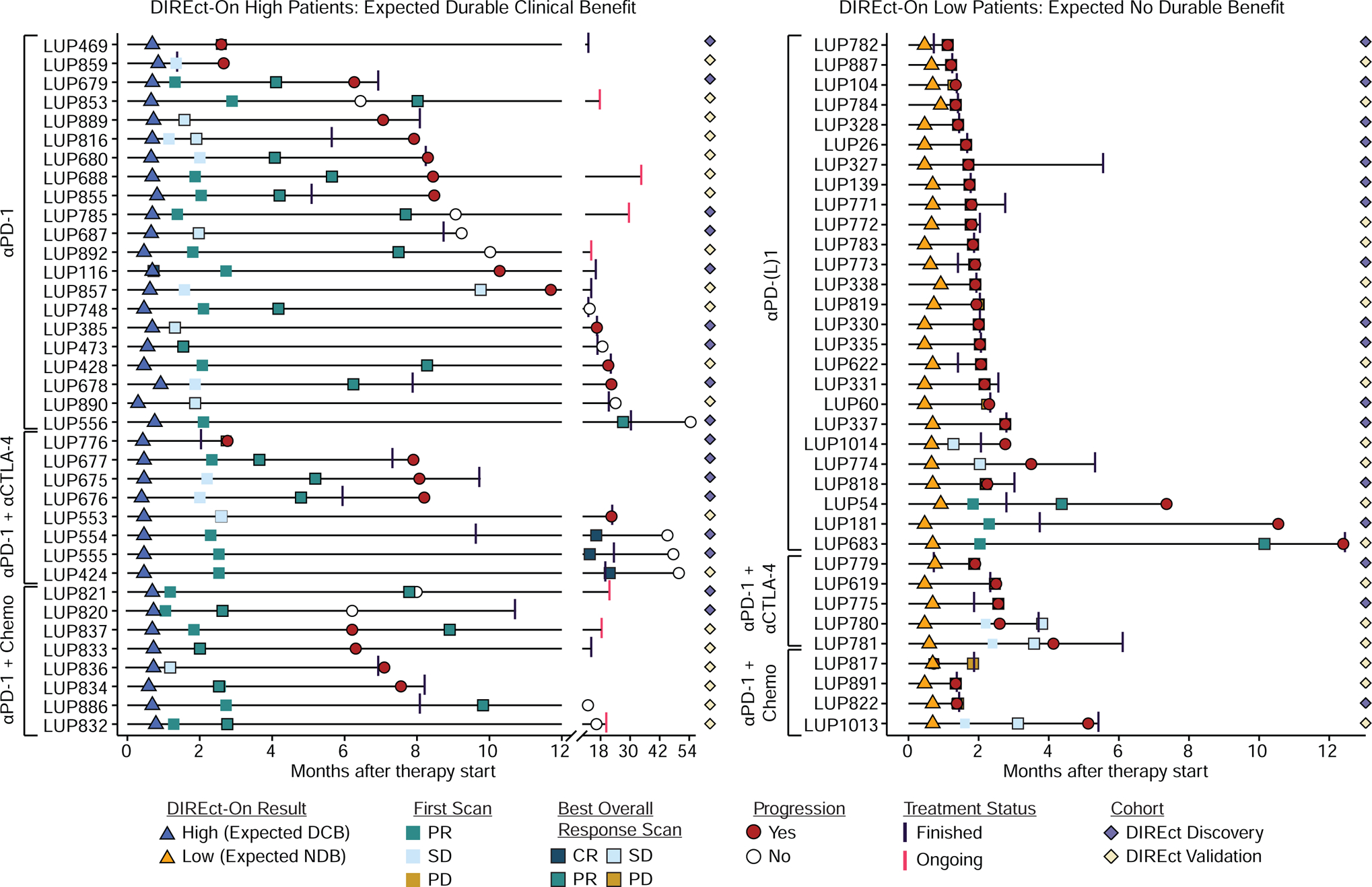
Swimmers chart for patients with high (left) or low (right) DIREct-on scores. Chart depicts timing of on-treatment blood draw for DIREct-On results (triangles), RECIST v1.1 status at the first scan (squares without outline) and the scan demonstrating the best overall response (squares with outline). In cases where the first scan was also the best overall response scan only the first scan is shown. Progression events or time of censoring is shown (red circle = progression, open circle = no progression at last follow-up). Last infusion date is depicted as a vertical line (black = treatment finished, pink = treatment ongoing). Patients belonging to the DIREct Discovery (purple) or Validation Cohort (tan) are indicated by diamonds. See also Figure S6.
We further assessed the ability of DIREct-On to classify responses to PD-(L)1 blockade-based ICI in our cohort and found high accuracy for prediction of patients achieving objective responses (26/29 [90%] of patients achieving a complete response [CR] or partial response [PR] had high DIREct-On scores) or progressive disease (PD, 27/29 [93%] of patients who had PD as best overall response had DIREct-On low scores) (Fig. S6B–C). Thus, although DIREct-On was trained to distinguish between durable clinical benefit versus no durable benefit, it is also appears useful for predicting the likelihood of objective response. Furthermore, we found no significant difference in PFS between patients who achieved DCB or NDB versus those expected to do so by DIREct-On (Fig. 7A).
Figure 7: DIREct-On enables early forecasting of ultimate outcomes.
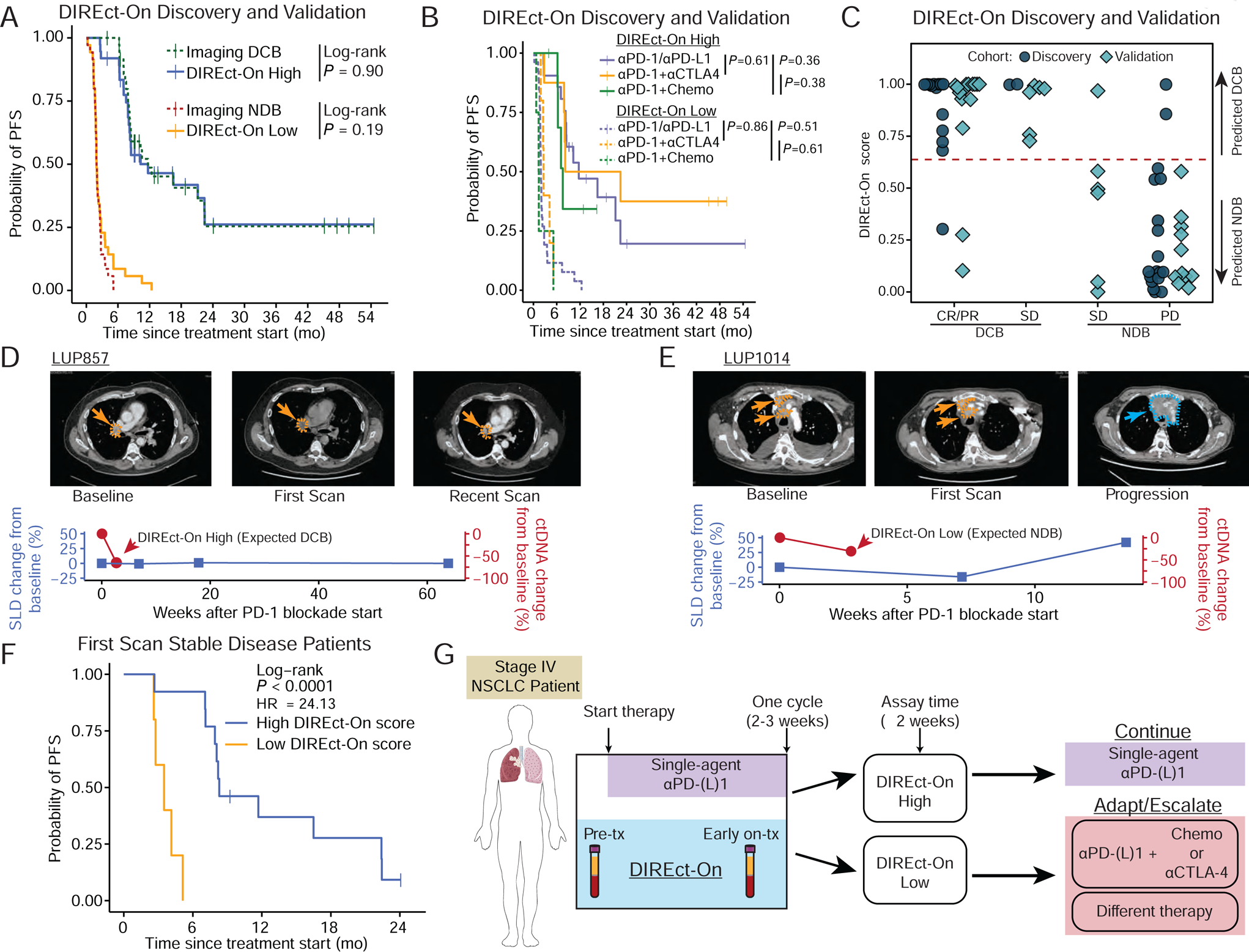
A) Probability of PFS for high DIREct-On score (median = 11.69 mo.) patients, actual DCB patients measured by RECIST (median = 11.69 mo.), low DIREct-On score (median = 1.94 mo.) patients, and actual NDB patients measured by RECIST (median = 1.94 mo.) in the combined DIREct Discovery and Validation cohorts (n = 72).
B) Probability of PFS from start of therapy stratified by DIREct-On score (solid line = expected DCB, dashed lines = expected NDB) in patients in the DIREct Discovery and Validation Cohorts treated with PD-1/PD-L1 single-agent blockade (purple), PD-1 and CTLA-4 combination therapy (orange), or the combination of PD-1 and chemotherapy (green).
C) DIREct-On score in the combined DIREct Discovery and Validation Cohorts (indicated by shape and color) stratified by response measured by RECIST and DCB versus NDB. The horizontal line indicates the threshold identified in the discovery cohort to best classify DCB versus NDB.
D) Vignette for patient with high DIREct-On score and stable disease at the first scan.
E) Vignette for patient with low DIREct-On score and stable disease at the first scan.
F) Probability of PFS for high DIREct-On score (median = 10.26 mo.) and low DIREct-On score (median = 3.62 mo.), in those patients with RECIST stable disease at the first available scan in the combined DIREct Discovery and Validation Cohorts (n = 18).
G) Potential application of DIREct-On to personalize immunotherapy in front-line treatment of advanced NSCLC. Patients could begin by receiving single agent PD-(L)1 blockade for one cycle and could then either remain on PD-(L)1 blockade if DIREct-On forecasts durable response or undergo treatment adaptation or escalation if DIREct-On forecasts lack of durable benefit. See also Figure S7.
Additionally, we performed subset analyses to assess the performance of DIREct-On in various clinical contexts in our cohort. Although DIREct-On was primarily trained and validated on patients treated with single-agent PD-1 blockade, it performed equally well in all three types of treatment regimens included in our study (Table S7). Moreover, patients with high DIREct-On scores had similarly prolonged PFS regardless of treatment type, while patients with low DIREct-On scores had equally abbreviated PFS (Fig. 7B, Fig S7A–C). Similarly, DIREct-On displayed similar classification efficacy in non-squamous or squamous tumors (Fig S7D–E, non-squamous, n = 7; squamous, n = 65). Therefore, DIREct-On appears to accurately stratify patient outcomes in PD-(L)1 blockade-based ICI strategies in NSCLC.
DIREct-On classifies patients with ambiguous imaging responses
Next, we explored the potential utility of DIREct-On for reconciling response status in patients with advanced lung cancer with ambiguous imaging results. First, we examined patients with a best overall response of stable disease (SD), meaning a change in the sum of the largest diameters (SLD) no greater than 20% increase or 30% decrease. In both the discovery and validation cohorts, DIREct-On accurately classified the durability of ICI response for 93% of patients (13/14) who achieved SD as their best radiographic response (Fig. 7C). We also tested if the presence of some shrinkage or growth that does not meet criteria for RECIST PR or PD could predict if a patient will achieve DCB or NDB. Interestingly – and highlighting the need for improved tools to adjudicate radiologically-defined stable disease – whether the tumors grew or shrunk within the bounds of +20% to −30% did not reliably classify whether these patients achieved DCB or NDB (Fig. S7F, Fisher’s Exact Test P = 1.00) and did not stratify PFS (Fig. S7G).
Two vignettes from our cohort highlight the ability of DIREct-On to adjudicate stable disease (SD) evaluations at the first on-treatment scan. LUP857 had a high DIREct-On score after three weeks and stable disease at the first scan seven weeks after therapy initiation. As predicted by DIREct-On, this patient derived durable benefit from PD-1 blockade and maintained radiographic SD for over one year (Fig. 7D). Another exemplar patient, LUP1014 also had stable disease at the first scan seven weeks after therapy start, yet had a low DIREct-On score at three weeks. As predicted by DIREct-On, this patient progressed at the second scan at 14 weeks after therapy initiation. (Fig. 7E). Overall, in patients whose first scan after treatment initiation was classified as stable, DIREct-On correctly identified 94% of patients (17/18) ultimately achieving DCB (Fig. 7F). Therefore, DIREct-On allows early identification of patients with initially stable disease who are most likely to achieve durable clinical benefit from PD-(L)1 blockade-based ICI.
Discussion
Here, we describe parameters associated with durable clinical benefit from ICI in patients with advanced NSCLC that integrate both tumor-intrinsic and -extrinsic features and that can be measured noninvasively using blood. We found lower baseline circulating CD8 T cell levels in patients with lung cancer ultimately achieving durable benefit from ICI, a result consistent with observations in melanoma responders to immunotherapy (Krieg et al., 2018). We also demonstrate that a pre-treatment composite model (DIREct-Pre) combining tumor PD-L1 expression with pre-treatment ctDNA and circulating immune cell profiling accurately predicts outcomes.
Moreover, we developed DIREct-On, which is a fully noninvasive response classifier that incorporates pre-treatment ctDNA and immune profiling with early on-treatment ctDNA response assessment to most accurately classify the likelihood of durable benefit after one cycle of immunotherapy. We demonstrate that DIREct-On outperforms each of the individual features alone and that each of the features are required for optimal classification efficacy. Our results also highlight and contrast the predictive power of measurements taken before and after therapy. Additionally, we believe there is significant biological rationale for each of the components of DIREct-Pre and DIREct-On: 1) pre-treatment ctDNA: reflects total body tumor burden and patients with higher burden are known to respond less well to ICIs (Ito et al., 2019; Kaira et al., 2017); 2) tumor mutation burden: likely correlates with the number of tumor neoantigens and therefore higher values increase immunogenicity; 3) circulating CD8 T cell fraction: fewer CD8 T cells in circulation are associated with response and we speculate that this could reflect greater homing of CD8 T cells to tumor deposits in patients with immunogenic tumors; and 4) early ctDNA dynamics: reflects change in disease burden early during therapy and is a direct reflection of tumor response or resistance. We envision that either model could be further improved by exploring additional biomarkers of clinical benefit, such as deeper immunophenotyping.
DIREct-On allows for classification of ultimate clinical outcomes significantly earlier than previously reported ctDNA-based approaches (Anagnostou et al., 2019; Goldberg et al., 2018; Raja et al., 2018). For on-treatment biomarkers, being able to assess response as early as possible is important since it would facilitate earlier changes in management. Moreover, we demonstrate that DIREct-On has significantly better performance than ctDNA dynamics alone. Therefore, the integration of pre-treatment parameters that can be measured noninvasively with on-treatment ctDNA dynamics allows for optimal classification of response. Furthermore, while cross-study comparisons of different techniques are fraught with caveats due to potential differences in patient cohorts, DIREct-On appears to outperform previously described integrative models relying on tumor RNA and/or DNA-seq (Anagnostou et al., 2020; Auslander et al., 2018; Cristescu et al., 2018; Jiang et al., 2018). The prior approaches were largely focused on melanoma rather than NSCLC and have substantially inferior performance compared to DIREct-On (prior studies: HR = 0.30–0.37, AUC = 0.70–0.83; DIREct-On: HR = 0.04–0.11, AUC = 0.89–0.93).
The endpoints of DCB and NDB were utilized to enable a binary classification approach. Unlike standard RECIST-based overall response classification criteria, the definition of DCB is inclusive of patients with long-term stable disease and we have previously demonstrated that it robustly identifies patients with prolonged overall survival to PD-(L)1 blockade-based therapy (Rizvi et al., 2018). Overall, DIREct-On demonstrated robust, early, and noninvasive classification of DCB and progression-free survival in a discovery and validation approach. Moreover, DIREct-On had excellent performance in various subsets of patients such as those receiving PD-(L)1 blockade with other ICI or chemotherapy, squamous versus non-squamous histology, and crucially, those without objective responses or progression at the first scan. Of note, we do not envision that DIREct-On would replace radiologic surveillance of NSCLC patients treated with PD-(L)1 blockade since in patients predicted to develop early progression the anatomic distribution of progressing lesions could inform subsequent therapy (e.g. systemic therapy for progression in many sites versus local therapy such as radiotherapy for progression in a limited number of sites).
To test the clinical utility of DIREct-On, prospective clinical trials will be required. One potential treatment strategy for patients with driver mutation negative Stage IV NSCLC that could be tested in clinical trials is to treat with single-agent PD-(L)1 blockade for one cycle (irrespective of PD-L1 expression) and to personalize subsequent cycles based on DIREct-On measured at 2–3 weeks after treatment start. Patients with high DIREct-On scores (expected durable benefit) could remain on single-agent PD-(L)1 blockade whereas patients with low DIREct-On scores (expected lack of benefit) could be treated with several options including 1) adding chemotherapy to PD-(L)1 blockade, 2) adding an additional ICI such as CTLA-4 blockade for potential synergy, or 3) stopping the current ICI and changing to another agent/regimen (Fig. 7G). If successful, such a trial could optimize the number of patients receiving immunotherapy alone and reserve combination therapy for patients who are destined not to respond to single-agent immunotherapy.
Limitations of our study include that it was retrospective and that patients were treated with somewhat heterogeneous PD-(L)1 regimens. Nevertheless, we found that DIREct-On performed similarly well regardless of type of PD-(L)1 regimen. Furthermore, since our training and validation cohorts were derived from patients receiving immunotherapy as part of standard-of-care management, they were different than the cohorts from the randomized POPLAR/OAK studies used for training some of the model parameters. Separately, while we validated the DIREct models in a held out patient cohort, this cohort was relatively small, resulting in relatively broad confidence intervals. While further validation in larger cohorts and prospective clinical trials will therefore be necessary, our data suggest that DIREct-On has strong prognostic power in real-world clinical settings. An additional limitation is that our cohort included few patients with targetable driver alterations (n=9, 13%) including EGFR (L858R n=4, Del19 n=2) and BRAF (V600E n=3), since these patients did not routinely receive immunotherapy during the time period of sample collection. Our cohort also did not include patients with active central nervous system disease. Therefore, it remains unclear how DIREct-On would perform in these important subsets of patients and further exploration is warranted.
Finally, we anticipate that our models could also have utility for predicting response to ICI in other tumor types since similar tumor-intrinsic and –extrinsic features have been identified to associate with response to ICI in other cancers (Cristescu et al., 2018; Krieg et al., 2018). Given the difficultly to date of developing robust predictive biomarkers for immunotherapy, approaches based on early response assessment could help fill the unmet need of improving personalization of therapy for patients treated with ICIs.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Maximilian Diehn (diehn@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The code to apply DIREct-On, model coefficients, and patient-level parameters for the discovery and validation cohorts are available for download at direct.stanford.edu. Clinical and demographic data for the cohorts generated as part of this study, sample timing information, ctDNA analysis metrics, transcripts per million quantified RNA sequencing data, immune profiling results, as well as parameter and model performance metrics are provided in the Supplemental Tables. Due to restrictions related to dissemination of germline sequence information included in the informed consent forms used to enroll study subjects, we are unable to provide access to the raw cfDNA, germline DNA, or RNA sequencing data because germline sequence information could be gleaned from these. Requests for additional data such as additional patient-level clinical or demographic data will be reviewed by the Lead Contact to determine whether they can be fulfilled in accordance with these privacy restrictions.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PD-(L)1 blockade-based Immune checkpoint inhibitor treated patients
All patients had stage IV non-small cell lung cancer (NSCLC) and were treated with PD-(L)1 blockade-based ICI (PD-(L)1 blockade alone or in combination with CTLA-4 blockade or chemotherapy) at Stanford University Cancer Center or Memorial Sloan Kettering Cancer Center (Tables S1–2). All patients consented to Institutional Review Board-approved protocols permitting specimen collection and genetic sequencing for analysis of tumor biopsies, leukocytes, and cfDNA.
Clinical efficacy analysis
Response was quantified in a blinded fashion by thoracic radiologists using RECIST v1.1 (Eisenhauer et al., 2009). Stable disease was defined by RECIST 1.1 criteria, meaning not significant shrinkage of the target lesions to be classified as partial response (≤30% decrease) or significant growth to be classified as progressive disease (≥20%). Durable clinical benefit (DCB) was defined as confirmed absence of progressive disease for at least 6 months after ICI; whereas, no durable benefit (NDB) was defined as patients experiencing progression or death within 6 months (Rizvi et al., 2015). Progression-free survival was determined from the start of PD-(L)1 blockade, with outcomes determined or censored as of the 01/22/2019 database lock.
Tumor samples
Thirty of 82 patients had tumor tissue available for whole-exome sequencing. All tumor tissue was obtained prior to treatment with immunotherapy. Tumor PD-L1 expression was assessed by clinical immunohistochemistry and the percentage of PD-L1 positive tumor cells was used as input into the relevant Bayesian models.
Plasma and leukocyte samples
Plasma was processed from whole blood samples collected in EDTA tubes. Tubes were centrifuged at 2500 × g at room temperature for 10 minutes. The plasma supernatant was collected and the remaining plasma-depleted whole blood was collected for DNA and/or RNA purification. In indicated cases, density gradient centrifugation was used to collect PBMC for flow cytometry, DNA, and/or RNA isolation. Early on-treatment blood samples were collected using the first available on-treatment samples. In the majority of patients (96%) this specimen was collected at the time of the second immunotherapy infusion. The earliest collection timepoint for any patient as 12 days after initiation of therapy (median = 18.5; range = 12–28 days).
METHOD DETAILS
cfDNA extraction
Cell-free DNA was extracted from three to six mL of plasma utilizing the QiaAmp Circulating Nucleic Acid Kit per manufacturer’s instructions. DNA was quantified using the Qubit dsDNA High Sensitivity Kit and quality and size was assessed by the Agilent 5400 Fragment Analyzer. For samples collected in CPT tubes, cfDNA was treated with Heparinase II (Sigma) for 2 hours at 37°C and re-purified by 1.8X Ampure XP bead selection prior to quantitation and size analysis.
Germline DNA extraction
Germline DNA was extracted from 100μL of PDWB or ~30,000 PBMCs with the QiaAmp DNA Micro Kit per manufacturer’s instructions. 100–1000ng of genomic DNA was then sheared using the Covaris S2 Focused-ultrasonicator using the following settings: 10% duty cycle, intensity level 5, 200 cycles per burst, and 2 minute duration. After sonication, sheared DNA was re-purified using the QiaQuick PCR Purification Kit per manufacturer’s instructions.
CAPP-Seq
CAPP-Seq was performed as previously described (Newman et al., 2014, 2016). Briefly, a 20–55ng of cfDNA or sheared genomic DNA was utilized for library preparation with the KAPA HyperPrep Kit with some modifications to the manufacturer’s instructions, as described (Chabon et al., 2016; Chaudhuri et al., 2017). After library preparation, custom-designed biotinylated DNA oligonucleotides were utilized for hybridization and subsequent enrichment with a capture panel covering 355 kb and targeting 270 genes could. Following hybridization capture, samples were sequenced on an Illumina HiSeq4000 as 2 × 150bp lanes of 8–12 samples multiplexed per lane yielding ~40 million paired-end reads with a median deduped depth of 3037X per case (Table S4). Data were then processed using a custom bioinformatics pipeline (Newman et al., 2014, 2016).
To abrogate the need for invasive biopsies, the CAPP-Seq analyses performed in this study were completed in a tumor-naïve manner. For tumor naïve calling we: 1) limited variants to coding positions, 2) removed any variants with greater than 1 reads in the matched germline samples, 3) removed variants with more than 2 reads in 5% of healthy control plasma samples (n = 54), 4) removed variants present in ≥0.05% of samples in the Genome Aggregation Database (Lek et al., 2016). For monitoring of these variants in early on-treatment samples we used a previously described Monte Carlo-based ctDNA detection index with a significance cut-point of P ≤ 0.05 (Newman et al., 2014). ctDNA fold change was calculated by dividing the ctDNA concentration at an on-treatment timepoint by the pre-treatment ctDNA concentration. In on-treatment cases where ctDNA was undetectable, the limit of detection for that sample was used as the ctDNA concentration, as described previously (Moding et al., 2020). To define the threshold for a ctDNA molecular response we analyzed previously published data from 23 treatment-naïve advanced NSCLC patients who had three plasma collections over the course of 6 hours. The maximum deviation from the average ctDNA concentration amount these patients was 1.87-fold (Wang et al., 2017). Therefore, we considered a ctDNA molecular response as a ≤0.5-fold decrease from the pre-treatment time point.
Tumor mutation burden estimation by CAPP-Seq
Tumor mutation burden was estimated by CAPP-Seq by identifying the relationship between the number of nonsynonymous mutations identified in pre-treatment tumors by whole-exome sequencing per megabase of coding exome captured to the number of coding mutations (nonsynonymous and synonymous) identified in the pre-treatment cfDNA by CAPP-Seq per megabase of coding exome captured in a discovery cohort of 24 patients (Figure S2B). We derived a linear regression model in a leave-one-out cross validation (LOOCV) framework in the discovery cohort 24 patients and found that TMB measured in cell-free DNA was significantly correlated with tissue TMB (Figure S2B, R = 0.86, P < 0.0001). This resulted in the following relationship:
We then validated this model in the independent group of patients and again observed strong correlation (Figure S2C, R = 0.99, P < 0.001, n=6).
RNA extraction
We did not have access to viably preserved peripheral blood mononuclear cells (PBMCs) amenable to flow cytometry for most patients but did have frozen plasma depleted whole blood (PDWB) containing leukocyte RNA for the majority of our cohort. Leukocyte RNA was extracted from either PDWB or PBMCs. For PDWB, RNA was extracted from 200μL of PDWB by mixing with 600μL TRIzol LS Reagent (ThermoFisher), 200μL of chloroform was used for phase separation. For PBMCs, pellets were lysed with 600μL of TRIzol Reagent and 150μL of chloroform was used for phase separation. For both starting materials, the aqueous phase from the initial phase separation was then mixed with equal volume of 100% ethanol, then loaded onto columns for the RNeasy Micro Kit (Qiagen) and the protocol was followed per manufacturer’s instruction, including on-column DNase treatment. The GLOBINclear Kit (ThermoFisher) was utilized to deplete globin mRNA per the manufacturer’s instructions in the PDWB samples. Before proceeding to RNA-seq, RNA was quantified using the Quant-iT RiboGreen RNA Assay Kit (ThermoFisher) and quality was confirmed by DV200 and RIN after RNA Pico Bioanalyzer Analysis. All RNA samples sequenced had DV200 ≥ 80%.
RNA-seq
The Illumina TruSeq RNA Exome kit was used for RNA-seq library preparation with 20ng of input PBMC RNA or globin-depleted PDWB RNA per manufacturer’s instructions. In brief, RNA was fragmented and stranded cDNA libraries were created per the manufacturer’s protocol. The RNA libraries were then enriched for the coding transcriptome by exon capture. Hybridization captures were then pooled and samples were sequenced on an Illumina HiSeq4000 as 2 × 150bp lanes of 16–20 samples per lane, yielding ~20 million paired-end reads per case. After demultiplexing the data were aligned and expression levels summarized using Salmon to Gencode version 27 transcript models (Patro et al., 2017). Expression levels per gene were summarized as transcripts per million (TPM), and then used as input for CIBERSORTx for deconvolution with the LM22 signature matrix with B-mode batch correction (Newman et al., 2019).
Tumor whole-exome sequencing
Whole-exome libraries were prepared using the Agilent SureSelect Human All Exon v2.0, v4.0, or the Illumina Rapid Capture Exome Kit, as described previously (Hellmann et al., 2020). Captured exome libraries for tumor and germline DNA were sequenced to equivalent depths on HiSeq2000, HiSeq2500, or HiSeq4000 platforms as 2×75bp or 2×150bp reads and aligned to the hg19 human genome build using the Burrows-Wheeler Aligner (Li and Durbin, 2009). Further indel realignment, base-quality score recalibration, and duplicate-read removal were performed utilizing the Genome Analysis Toolkit (DePristo et al., 2011). Single nucleotide variants were identified using Mutect with default parameters (Cibulskis et al., 2013). Tumor mutational burden per megabase for bTMB estimation was calculated by dividing the total number of nonsynonymous mutations by the coding region of each respective capture kit (Agilent SureSelect Human All Exon v2.0 = 29.9MB, Agilent SureSelect Human All Exon v4.0 = 34.2 MB, Illumina Rapid Capture Exome Kit = 42.7MB).
Flow cytometry
Flow cytometry was used to confirm the fidelity of CIBERSORTx estimates for the major lymphoid and myeloid lineages among the cellular subsets captured by the LM22 signature matrix, whether viable or nonviable leukocytes were used as input RNA for sequencing and subsequent transcriptome deconvolution. To do so, single cell suspensions were prepared from viably frozen PBMC aliquots. Live/dead cell discrimination was performed using 7-AAD Viability Staining Solution (BioLegend). Human Fc receptors were blocked using Human TruStain FcX (BioLegend). Cell surface staining was performed for 30 min at 4°C with the following antibodies: CD14 (Clone: M5E2, Color: BV421, BD Biosciences), CD3 (Clone: UCHT1, Color: PE, BioLegend), CD4 (Clone: RPA-T4, Color: PerCP-Cy5.5, BD Biosciences), CD8 (Clone: RPA-T8, Color: FITC, BD Biosciences), CD19 (Clone: SJ25C1, Color: BV711, BD Biosciences), and CD56 (Clone: HCD56, Color: PE-Cy7,BioLegend). All data acquisition was done using a FACS Aria Fusion (BD Biosciences) or a CytoFLEX Flow Cytometer (Beckman Coulter) and analyses were performed using FlowJo v10. CD4 T cells were gated by CD14−, CD19−, CD3+, CD8−, CD4+; CD8 T cells were gated by CD14−, CD19−, CD3+, CD8+, CD4−; monocytes were gated by CD56−, CD3−, CD19−, CD14+; B cells were gated by CD3−, CD19+; and NK cells were gated by CD14−, CD3−, CD19−, CD56+.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
The Wilcoxon test was used to compare groups where indicated. Fisher’s exact test was used to compare bTMB high versus bTMB low in the normalized bTMB Discovery and Validation cohort. Receiver operating characteristic curves were used to generate area under the curve (AUC), and 95% confidence intervals for AUCs were generated by bootstrapping (Robin et al., 2011). Significance analysis of performance evaluation metrics (AUC, NRI, hazard ratio, and accuracy) were also computed by bootstrapping, where the difference in the metrics curves for each bootstrap replicate is computed and compared across conditions to generate an empiric P-value (Kurtz et al., 2019).
For progression-free survival analysis, the log-rank test was used to compare Kaplan-Meier survival curves. Cox proportional hazards regression models were used to generate hazard ratios as stratified relative to cut-points defined in each figure, as captured in corresponding legends. Stratification thresholds were determined using optimal cut-point selection by ROC analysis only within the discovery cohorts. For ctDNA dynamics, the cut-point was not optimized and 0.5 was used as the cut-point in both discovery and validation cohorts. For PFS analysis of the validation cohorts, the cut-points from the discovery cohorts were applied to the respective validation sets to stratify response. DIREct-Pre and DIREct-On were also tested as continuous variables in the validation cohort by Cox likelihood ratio test. When comparing performance of individual biomarkers to each other or to Bayesian probit models analyses were restricted to patients with all data types available, as described in the corresponding legends. All analyses were conducted using R v.3.5.1 using the pROC, survminer, MCMCpack, survIDINRI, and survival packages.
Bayesian probit models
To classify DCB versus NDB we utilized a Bayesian probit approach (Figure 4A). In brief, patients were split into the DIREct Discovery (n = 34) and DIREct Validation Cohort (n = 38). Features that associated with ultimate outcomes were identified in the DIREct Discovery Cohort. For the Bayesian probit models, hyperparameters without prior knowledge in publicly available data were inferred using a LOOCV framework in the DIREct Discovery Cohort samples (CD8 T cell fraction and early ctDNA dynamics). For features with publicly available data available (tumor PD-L1 expression and normalized bTMB), hyperparameters were empirically inferred from the POPLAR/OAK ICI Cohort (n = 424). The models were then trained in the DIREct Discovery Cohort and DCB versus NDB was stratified based on the best threshold (Youden’s J). Based on the performance of DIREct-On in the discovery cohort, a validation cohort of >20 provides >99% power to detect a similar difference between the two groups (one-sided two-arm binomial with alpha = 0.05). The models and thresholds were then applied to the DIREct Validation Cohort.
Specifically, likelihood of DCB for each sample is modeled as probabilityDCB=Φ(xTβ), where Φ(.) denotes the cumulative distribution function of a standard normal variable, x denotes the feature vector (i.e., normalized bTMB, PD-L1, and CD8 fraction for DIREct-Pre and normalized bTMB, CD8 fraction, and ΔctDNA for DIREct-On), and β denotes the model parameters from prior knowledge derived from the literature or from our discovery cohort (as described below). The model parameters were assumed to be governed by a Gaussian distribution, i.e. , where β0 and denote the mean and covariance matrix of the distribution, respectively. This approach allows for uncertainty in the underlying distributions (as might be expected for example between different ctDNA profiling techniques or patient populations) and therefore expected to be more generalizable for response prediction.
Construction of Bayes priors from public data
To identify the parameters of the Gaussian distribution governing the probit coefficients, we denoted the features from the POPLAR/OAK ICI Cohort (Gandara et al., 2018), by Xprior and the corresponding labels (DCB/NDB) by Yprior. Here, we subsampled of n=100 cases for m = 2000 times. For each iteration and for each set of covariates (e.g., PD-L1 and normalized bTMB), we solved a probit regression model to generate a matrix of associated coefficients (β). We used this matrix as a random realization of this multi-variable normal distribution, and then estimated the Bayes prior probability parameters (i.e., hyper-parameters).
Prior construction within LOOCV and Markov-Chain Monte Carlo (MCMC)
We used a LOOCV framework to fully exploit the available data, and for the features not estimated by Xprior (e.g., CD8 T cell fraction). Specifically, we used n − 1 samples to find the hyperparameters as described above. We then derived the final full prior probability distributions by concatenating two components: (1) the prior knowledge-based priors, and (2) the priors derived from our DIREct Discovery Cohort, such that where β0,prior and β0,ourcohort are the mean vectors of the variables, estimated from the prior set and our cohort, respectively. Similarly and denote the covariance matrices. We then used the same n − 1 samples with all available covariates in a MCMC step to calculate the posterior probabilities (i.e., updated prior probabilities). In cases where tumor PD-L1 expression was unavailable (DIREct Discovery Cohort, n = 8; DIREct Validation Cohort, n = 4), this parameter was left out.
Final prediction score for both DIREct models
To generate a single score representing the probability of DCB, we used the MCMC posterior samples, and generated 10,000 pseudo-samples with 1000 initial burn-in. These realizations of the β vectors (i.e. { β1, β2,…,β10000 } along with the sample feature vector, x) were then used to calculate 10,000 realizations of the DCB score, such that Φ(xTβi) for i ∈ {1,…,10000}. We then used the median of these realizations as the final prediction score.
Threshold selection for DIREct models
To select the optimal thresholds in the discovery cohort for the DIREct-Pre and DIREct-On scores, we used the optimal ROC corner point approach (Youden’s J) for classification of DCB, where the optimality was defined as the point with maximal unweighted accuracy, i.e. specificity plus sensitivity (Youden, 1950). These cut-points were then applied to the validation set for calculation of precision and survival analyses.
Calibration testing of DIREct models
We evaluated DIREct predictions for quantitative accuracy via model calibration regression (Steyerberg et al., 2010). To test calibration of the DIREct-Pre and DIREct-On models we performed a bootstrap resampling with n=2000 and reported the “true event rate”, estimated via the Kaplan-Meier maximum likelihood estimates of observed events versus expected events from our models’ predictions. A perfect calibration leads to a slope of 1, i.e. the predicted probability of the model is in fact representative of the true event rate in the population.
ADDITIONAL RESOURCES
We have created a web resource for the DIREct-On model described in this study. Here, readers can input parameters to generated DIREct-On scores and predictions for individual and batched samples and download the code used to generate the model. URL: https://direct.stanford.edu
Supplementary Material
Figure S1: Data related to Figure 1. A) Percentage of patients who achieved DCB or NDB in those who had first scan of stable disease. Shown are the subsets of patients who received single-agent PD-(L)1 blockade (total n = 204) , PD-(L)1 with CTLA-4 blockade (total n = 34), or PD-1 blockade with chemotherapy (total n = 36). Within each pie chart are the number of first scan SD patients in each treatment regimen. P-values were calculated with a two-sided Fisher’s Exact Test.
B) Cohorts from this study. In the 94 patients with detectable pre-treatment ctDNA 22 patients (A1) did not have all noninvasive parameters available (n=11 did not have early on-treatment blood draws, n = 11 did not have viable or non-viable leukocytes for immune profiling) and were used only for discovery of features that are associated with DCB vs. NDB. (A2) Samples from 34 patients were used to both discover features as well as train models to forecast ultimate outcomes. Samples from A1 and A2 were utilized to independently validate the thresholds for normalized bTMB defined in the POPLAR/OAK ICI cohort. (A3) Samples from 38 patients were held out from all analyses except to validate the DIREct models and thresholds as well as individual model features.
C) Cohorts from publicly available data (Gandara et al., 2018). B1 was utilized in this study to discover the relationship between bTMB and ctDNA burden and empirically infer the hyperparameters for PD-L1 status and normalized bTMB for the DIREct models. B2 was used to assess if pre-treatment ctDNA parameters were predictive or response to ICI or prognostic.
Figure S2: Data related to Figure 2. A) Tumor mutation burden per megabase (TMB/MB) from tumor whole-exome sequencing (WES) as compared with cfDNA mutational burden per MB (n = 24). R = Pearson’s correlation coefficient.
B) Validation of TMB/MB from tumor WES versus estimated TMB/MB from cfDNA using the linear regression relationship identified in B (n = 6).
C) Hazard ratios and 95% confidence intervals for each of the indicated variables in the POPLAR/OAK ICI Cohort (n=429) treating each as a standardized continuous variable. Normalized bTMB has a significantly greater hazard ratio compared to ctDNA or bTMB alone.
D) Percent of patients stratified by high bTMB/MB (≥14) and low bTMB/MB (<14) in ICI patients that achieve DCB (n = 15) versus NDB (n = 22) in our independent cohort, exclusive of the DIREct Validation Cohort. P-value calculated by two-sided Fisher’s Exact Test.
E) Pre-treatment ctDNA concentration and F) the ctDNA-normalized bTMB for patients receiving single-agent PD-(L)1 blockade in our independent cohort, exclusive of the DIREct Validation Cohort (n = 37). P-values were calculated using a Wilcoxon test.
G) AUC for individual parameters in the normalized in our independent cohort, exclusive of the DIREct Validation Cohort generated by LOOCV ROC analysis.
Figure S3: Data related to Figure 3. A) Comparison of cell proportions from flow cytometry of peripheral blood mononuclear cells (PBMCs) versus RNA-seq followed by CIBERSORTx in PBMCs (left) or plasma-depleted whole blood (PDWB, right) in matched samples from healthy donors (n = 3). R = Pearson’s correlation coefficient.
B) Comparison of cell proportions from flow cytometry of PBMCs versus RNA-seq followed by CIBERSORTx in PBMCs (left) or PDWB (right) in matched samples from NSCLC patients (n = 10). R = Pearson’s correlation coefficient.
C) Pre-treatment immune composition in circulation of ICI DCB (n = 20) and NDB (n = 17) with available CIBERSORTx immune profiling. Shown are major circulating immune cell populations, excluding CD8 T cells (shown in Fig. 3B). P-values were calculating using a Wilcoxon test.
D) Spearman correlation of pre-treatment biomarkers (CD8 T cell fraction, normalized bTMB, and tumor PD-L1 expression). Shown are standardized values for each parameter.
E) ROC curve for classification of DCB and NDB by ctDNA dynamics within 4 weeks. Shown is sensitivity and specificity at the threshold of 50% pre-treatment ctDNA concentration (ctDNA molecular response).
Figure S4: Data related to Figure 4. A) Response prediction score by incorporating pre-treatment bTMB, ctDNA burden, CD8 T cell fraction, and the ctDNA change within 4 weeks with (x-axis) or without (y-axis) including tumor PD-L1 expression cases with all data types available (n = 26).
B) ROC curve for classification of DCB and NDB by DIREct-Pre and DIREct-On.
Figure S5: Data related to Figure 5 A) Hazard ratio for each individual parameter and DIREct-On in the combined DIREct Discovery and Validation Cohorts for those cases with all data types available, using the cut-points identified the DIREct Discovery Cohort (n = 58). Error bars are 95% confidence intervals generated by bootstrapping. ** = P < 0.01, *** = P < 0.001.
B) Prediction by CD8 T cell fraction and ctDNA dynamics in cases in which normalized bTMB incorrectly classified outcomes (n = 26).
C) Prediction by normalized bTMB and ctDNA dynamics in cases in which CD8 T cell fraction incorrectly classified outcomes (n = 30).
D) Prediction by normalized bTMB and CD8 T cell fraction in cases in which ctDNA dynamics incorrectly classified outcomes (n = 11).
E) Hazard ratios and 95% confidence intervals for each of models with the indicated feature removed in the combined DIREct Discovery and Validation Cohorts. DIREct-On high scores results in a significantly lower hazard ratio compared to each model with one feature removed. Errors bars are 95% confidence intervals generated by bootstrapping.
F) Predicted probability of progression within 6 months versus observed probability of progression within 6 months in the DIREct Validation Cohort for DIREct-Pre (n = 38).
G) Predicted probability of progression within 6 months versus observed probability of progression within 6 months in the DIREct Validation Cohort for DIREct-On (n = 38).
Figure S6: Data related to Figure 6 A) Hazard ratios of each indicated parameter and DIREct-On in a multivariable Cox proportional hazards model utilizing the patients in the combined DIREct Discovery and Validation Cohorts (n = 72).
B) Patients in the DIREct Discovery and Validation cohorts with best overall response by RECIST v1.1 indicated, excluding patients with stable disease (left). DIREct-On classification of patients with objective responses (middle) or progressive disease (right) by RECIST.
C) Waterfall depiction of sum of the lesion change from baseline at the time of best overall response by RECIST v1.1 for patients in the DIREct Discovery and Validation Cohorts. Colors indicate the DIREct-On status for each patient. Patients with progression of non-target lesions are indicated with asterisks and arbitrarily assigned to 21%.
Figure S7: Data related to Figure 7 Probability of PFS from start of therapy stratified by DIREct-On score in patients in the DIREct Discovery and Validation Cohorts treated with A) PD-1/PD-L1 single-agent blockade, B) PD-1 and CTLA-4 combination therapy, C) or the combination of PD-1 and chemotherapy.
Probability of PFS from start of therapy stratified by DIREct-On score in patients with tumors of D) non-squamous, or E) squamous histology.
F) Percent change in the sum of largest diameters (SLD) at the first scan for the 18 patients that had stable disease at the first scan in the DIREct Discovery and Validation Cohorts.
G) Probability of PFS from the time of treatment start for patients with increasing or decreasing SLD at the first scan within the stable disease bounds of +20% to −30%.
Table S3: Noninvasively detected mutations in genes recurrently mutated in NSCLC. Related to Figures 2–7.
Table S4: ctDNA analysis metrics in patients with available pre- or on-treatment plasma. Related to Figures 2–7.
Table S6: Pre-treatment circulating immune composition of major leukocyte populations by CIBERSORTx. Related to Figures 3–7.
Table S7: Parameter and model performance evaluation in the DIREct Discovery and Validation Cohorts. Related to Figures 5–7.
Highlights.
Pre-treatment ctDNA features are associated with checkpoint blockade response
Pre-treatment peripheral T cell levels are associated with checkpoint blockade response
Early on-treatment ctDNA dynamics are associated with checkpoint blockade response
Multiparameter noninvasive models can predict checkpoint blockade response in NSCLC
Acknowledgments
We thank the patients and families who participated in this study. This work was supported by the National Cancer Institute (B.Y.N. and M.S.E., R25CA180993; M.S.E, T32CA009302; M.Diehn and A.A.A., R01CA188298), the US National Institutes of Health Director’s New Innovator Award Program (M.Diehn; 1-DP2-CA186569), the Virginia and D.K. Ludwig Fund for Cancer Research (M.Diehn and A.A.A.), the Bakewell Foundation (to M.D. and A.A.A.) and the SDW/DT and Shanahan Family Foundations (to A.A.A.), and the CRK Faculty Scholar Fund (M.Diehn). A.A.A. is a scholar of the Leukemia and Lymphoma Society. M.D.H. and this research supported, in part, by the Damon Runyon Cancer Research Foundation (Grant No. CI-98-18), the Memorial Sloan Kettering Cancer Center Support Grant/Core Grant No. P30 CA008748, and a Stand Up to Cancer (SU2C)-American Cancer Society Lung Cancer Dream Team Translational research grant (SU2C-AACR-DT17-15). SU2C is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. M.D.H. is a member of the Parker Institute for Cancer Immunotherapy. B.Y.N. is supported by the Postdoctoral Research Fellowship (134031-PF-19-164-01-TBG) from the American Cancer Society. The schematic in Figure 1B was produced using Servier Medical Art (https://smart.servier.com) licensed under CC BY 3.0.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
J.J.C. reports paid consultancy from Lexent Bio Inc. A.A.C. reports speaker honoraria and travel support from Roche Sequencing Solutions (RSS), Varian, and Foundation Medicine; a research grant from RSS, and has served as a paid consultant for Oscar Health. T.M. is a co-founder of Imvaq. J.W.N. reports research support from Genentech (GNE)/Roche, Merck, Novartis, Boehringer Ingelheim, Exelixis, Takeda Pharmaceuticals, Nektar Therapeutics, Adaptimmune, and GSK, and has served in a consulting or advisory role for AstraZeneca (AZ), GNE/Roche, Exelixis Inc, Jounce Therapeutics, Takeda Pharmaceuticals, and Eli Lilly. H.A.W. has received honoraria from Novartis and AZ and has participated on the advisory boards of Xcovery, Janssen, and Mirati. S.K.P. reports grant support from EpicentRx, Forty Seven, Bayer, and Boehringer Ingelheim and serves in a consulting or advisory role for AZ, AbbVie, G1 Therapeutics, and Pfizer. M.Das reports grant support from Abbvie, United Therapeutics, Varian, and Celgene and serves in a consulting role for AZ and Bristol-Myers Squibb (BMS). A.M.N. has patent filings related to expression deconvolution and cancer biomarkers and has served as a consultant for Roche, Merck and CiberMed. M.D.H. reports paid consultancy from BMS, Merck, GNE, AZ/MedImmune, Nektar, Syndax, Janssen, Mirati Therapeutics, Shattuck Labs, and Blueprint Medicines; travel/honoraria from BMS and AZ; research funding from BMS; and a patent has been filed by MSK related to the use of tumor mutation burden to predict response to immunotherapy (PCT/US2015/062208), which has received licensing fees from Personal Genome Diagnostics. A.A.A. reports ownership interest in CiberMed and FortySeven, patent filings related to cancer biomarkers, and paid consultancy from GNE, Roche, Chugai, Gilead, and Celgene. M.Diehn reports research funding from Varian, ownership interest in CiberMed, patent filings related to cancer biomarkers, paid consultancy from Roche, AZ, and BioNTech, and travel/honoraria from Reflexion. M.Diehn, A.A.A., B.Y.N., and M.S.E., are co-inventors on a provisional patent application filed by Stanford University relating to this manuscript. The remaining authors declare no competing interests.
References
- Anagnostou V, Forde PM, White JR, Niknafs N, Hruban C, Naidoo J, Marrone K, Sivakumar IKA, Bruhm DC, Rosner S, et al. (2019). Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Res. 79, 1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostou V, Niknafs N, Marrone K, Bruhm DC, White JR, Naidoo J, Hummelink K, Monkhorst K, Lalezari F, Lanis M, et al. (2020). Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat. Cancer 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, Tian T, Wei Z, Madan S, Sullivan RJ, et al. (2018). Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med 24, 1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camidge DR, Doebele RC, and Kerr KM (2019). Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol 16, 341–355. [DOI] [PubMed] [Google Scholar]
- Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, et al. (2016). Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun 7, 11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, Schroers-Martin J, Nabet BY, Chen B, Chaudhuri AA, et al. (2020). Integrating genomic features for non-invasive early lung cancer detection. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, Khodadoust MS, Esfahani MS, Liu CL, Zhou L, et al. (2017). Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 7, 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science (80-.) 362, eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. (2009). New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247. [DOI] [PubMed] [Google Scholar]
- Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, Rittmeyer A, Fehrenbacher L, Otto G, Malboeuf C, et al. (2018). Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med 24, 1441–1448. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. (2018). Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med 378, 2078–2092. [DOI] [PubMed] [Google Scholar]
- Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, Lee A, Nemati R, Nath SK, Mane SM, et al. (2018). Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res 24, 1872–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Willey J, Buettner A, Lankford M, Neely DB, and Ramalingam SS (2014). Molecular testing prior to first-line therapy in patients with stage IV nonsquamous non-small cell lung cancer (NSCLC): A survey of U.S. medical oncologists. J. Clin. Oncol 32, 8097–8097. [Google Scholar]
- Hanna NH, Schneider BJ, Temin S, Baker S, Brahmer J, Ellis PM, Gaspar LE, Haddad RY, Hesketh PJ, Jain D, et al. (2020). Therapy for Stage IV Non–Small-Cell Lung Cancer Without Driver Alterations: ASCO and OH (CCO) Joint Guideline Update. J. Clin. Oncol. JCO 1903022. [DOI] [PubMed] [Google Scholar]
- Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, Minenza E, Linardou H, Burgers S, Salman P, et al. (2018). Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med 378, 2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann MD, Nabet BY, Rizvi H, Chaudhuri AA, Wells DK, Dunphy MP, Chabon JJ, Liu CL, Hui AB, Arbour KC, et al. (2020). Circulating Tumor DNA Analysis to Assess Risk of Progression after Long-term Response to PD-(L)1 Blockade in NSCLC. Clin. Cancer Res 26, 2849–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Schöder H, Teng R, Humm JL, Ni A, Wolchok JD, and Weber WA (2019). Prognostic value of baseline metabolic tumor volume measured on 18F-fluorodeoxyglucose positron emission tomography/computed tomography in melanoma patients treated with ipilimumab therapy. Eur. J. Nucl. Med. Mol. Imaging 46, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaira K, Higuchi T, Naruse I, Arisaka Y, Tokue A, Altan B, Suda S, Mogi A, Shimizu K, Sunaga N, et al. (2017). Metabolic activity by 18F–FDG-PET/CT is predictive of early response after nivolumab in previously treated NSCLC. Eur. J. Nucl. Med. Mol. Imaging 56–66. [DOI] [PubMed] [Google Scholar]
- Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, and Becher B (2018). High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med 24, 144–153. [DOI] [PubMed] [Google Scholar]
- Kurtz DM, Esfahani MS, Scherer F, Soo J, Jin MC, Liu CL, Newman AM, Dührsen U, Hüttmann A, Casasnovas O, et al. (2019). Dynamic Risk Profiling Using Serial Tumor Biomarkers for Personalized Outcome Prediction. Cell 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C, Tsao MS, Le LW, Shepherd FA, Feld R, Burkes RL, Liu G, Kamel-Reid S, Hwang D, Tanguay J, et al. (2015). Biomarker testing and time to treatment decision in patients with advanced nonsmall-cell lung cancer. Ann. Oncol 26, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, et al. (2019). Genetic diversity of tumors with mismatch repair deficiency influences anti–PD-1 immunotherapy response. Science (80-.) 364, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moding EJ, Liu Y, Nabet BY, Chabon JJ, Chaudhuri AA, Hui AB, Bonilla RF, Ko RB, Yoo CH, Gojenola L, et al. (2020). Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nat. Cancer 1, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, Castro G, Srimuninnimit V, Laktionov KK, Bondarenko I, et al. (2019). Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 393, 1819–1830. [DOI] [PubMed] [Google Scholar]
- Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, et al. (2014). An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med 20, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C, et al. (2016). Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol 34, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol 37, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, and Ranade K (2018). Early Reduction in ctDNA Predicts Survival in Patients with Lung and Bladder Cancer Treated with Durvalumab. Clin. Cancer Res 24, 6212–6222. [DOI] [PubMed] [Google Scholar]
- Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. (2016). Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med 375, 1823–1833. [DOI] [PubMed] [Google Scholar]
- Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, Plodkowski A, Long N, Sauter JL, Rekhtman N, et al. (2018). Molecular Determinants of Response to Anti–Programmed Cell Death (PD)-1 and Anti–Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non–Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol 36, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (80-.) 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, and Müller M (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D, Thomas CA, Barlesi F, et al. (2018). Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med 378, 2288–2301. [DOI] [PubMed] [Google Scholar]
- Steyerberg EW, Vickers AJ, Cook NR, Gerds T, Gonen M, Obuchowski N, Pencina MJ, and Kattan MW (2010). Assessing the Performance of Prediction Models. Epidemiology 21, 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Bai H, Hong C, Wang J, and Mei TH (2017). Analyzing epidermal growth factor receptor mutation status changes in advanced non-small-cell lung cancer at different sampling time-points of blood within one day. Thorac. Cancer 8, 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Duan J, Cai S, Han M, Dong H, Zhao J, Zhu B, Wang S, Zhuo M, Sun J, et al. (2019). Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients With Non–Small Cell Lung Cancer With Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youden WJ (1950). Index for rating diagnostic tests. Cancer 3, 32–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Data related to Figure 1. A) Percentage of patients who achieved DCB or NDB in those who had first scan of stable disease. Shown are the subsets of patients who received single-agent PD-(L)1 blockade (total n = 204) , PD-(L)1 with CTLA-4 blockade (total n = 34), or PD-1 blockade with chemotherapy (total n = 36). Within each pie chart are the number of first scan SD patients in each treatment regimen. P-values were calculated with a two-sided Fisher’s Exact Test.
B) Cohorts from this study. In the 94 patients with detectable pre-treatment ctDNA 22 patients (A1) did not have all noninvasive parameters available (n=11 did not have early on-treatment blood draws, n = 11 did not have viable or non-viable leukocytes for immune profiling) and were used only for discovery of features that are associated with DCB vs. NDB. (A2) Samples from 34 patients were used to both discover features as well as train models to forecast ultimate outcomes. Samples from A1 and A2 were utilized to independently validate the thresholds for normalized bTMB defined in the POPLAR/OAK ICI cohort. (A3) Samples from 38 patients were held out from all analyses except to validate the DIREct models and thresholds as well as individual model features.
C) Cohorts from publicly available data (Gandara et al., 2018). B1 was utilized in this study to discover the relationship between bTMB and ctDNA burden and empirically infer the hyperparameters for PD-L1 status and normalized bTMB for the DIREct models. B2 was used to assess if pre-treatment ctDNA parameters were predictive or response to ICI or prognostic.
Figure S2: Data related to Figure 2. A) Tumor mutation burden per megabase (TMB/MB) from tumor whole-exome sequencing (WES) as compared with cfDNA mutational burden per MB (n = 24). R = Pearson’s correlation coefficient.
B) Validation of TMB/MB from tumor WES versus estimated TMB/MB from cfDNA using the linear regression relationship identified in B (n = 6).
C) Hazard ratios and 95% confidence intervals for each of the indicated variables in the POPLAR/OAK ICI Cohort (n=429) treating each as a standardized continuous variable. Normalized bTMB has a significantly greater hazard ratio compared to ctDNA or bTMB alone.
D) Percent of patients stratified by high bTMB/MB (≥14) and low bTMB/MB (<14) in ICI patients that achieve DCB (n = 15) versus NDB (n = 22) in our independent cohort, exclusive of the DIREct Validation Cohort. P-value calculated by two-sided Fisher’s Exact Test.
E) Pre-treatment ctDNA concentration and F) the ctDNA-normalized bTMB for patients receiving single-agent PD-(L)1 blockade in our independent cohort, exclusive of the DIREct Validation Cohort (n = 37). P-values were calculated using a Wilcoxon test.
G) AUC for individual parameters in the normalized in our independent cohort, exclusive of the DIREct Validation Cohort generated by LOOCV ROC analysis.
Figure S3: Data related to Figure 3. A) Comparison of cell proportions from flow cytometry of peripheral blood mononuclear cells (PBMCs) versus RNA-seq followed by CIBERSORTx in PBMCs (left) or plasma-depleted whole blood (PDWB, right) in matched samples from healthy donors (n = 3). R = Pearson’s correlation coefficient.
B) Comparison of cell proportions from flow cytometry of PBMCs versus RNA-seq followed by CIBERSORTx in PBMCs (left) or PDWB (right) in matched samples from NSCLC patients (n = 10). R = Pearson’s correlation coefficient.
C) Pre-treatment immune composition in circulation of ICI DCB (n = 20) and NDB (n = 17) with available CIBERSORTx immune profiling. Shown are major circulating immune cell populations, excluding CD8 T cells (shown in Fig. 3B). P-values were calculating using a Wilcoxon test.
D) Spearman correlation of pre-treatment biomarkers (CD8 T cell fraction, normalized bTMB, and tumor PD-L1 expression). Shown are standardized values for each parameter.
E) ROC curve for classification of DCB and NDB by ctDNA dynamics within 4 weeks. Shown is sensitivity and specificity at the threshold of 50% pre-treatment ctDNA concentration (ctDNA molecular response).
Figure S4: Data related to Figure 4. A) Response prediction score by incorporating pre-treatment bTMB, ctDNA burden, CD8 T cell fraction, and the ctDNA change within 4 weeks with (x-axis) or without (y-axis) including tumor PD-L1 expression cases with all data types available (n = 26).
B) ROC curve for classification of DCB and NDB by DIREct-Pre and DIREct-On.
Figure S5: Data related to Figure 5 A) Hazard ratio for each individual parameter and DIREct-On in the combined DIREct Discovery and Validation Cohorts for those cases with all data types available, using the cut-points identified the DIREct Discovery Cohort (n = 58). Error bars are 95% confidence intervals generated by bootstrapping. ** = P < 0.01, *** = P < 0.001.
B) Prediction by CD8 T cell fraction and ctDNA dynamics in cases in which normalized bTMB incorrectly classified outcomes (n = 26).
C) Prediction by normalized bTMB and ctDNA dynamics in cases in which CD8 T cell fraction incorrectly classified outcomes (n = 30).
D) Prediction by normalized bTMB and CD8 T cell fraction in cases in which ctDNA dynamics incorrectly classified outcomes (n = 11).
E) Hazard ratios and 95% confidence intervals for each of models with the indicated feature removed in the combined DIREct Discovery and Validation Cohorts. DIREct-On high scores results in a significantly lower hazard ratio compared to each model with one feature removed. Errors bars are 95% confidence intervals generated by bootstrapping.
F) Predicted probability of progression within 6 months versus observed probability of progression within 6 months in the DIREct Validation Cohort for DIREct-Pre (n = 38).
G) Predicted probability of progression within 6 months versus observed probability of progression within 6 months in the DIREct Validation Cohort for DIREct-On (n = 38).
Figure S6: Data related to Figure 6 A) Hazard ratios of each indicated parameter and DIREct-On in a multivariable Cox proportional hazards model utilizing the patients in the combined DIREct Discovery and Validation Cohorts (n = 72).
B) Patients in the DIREct Discovery and Validation cohorts with best overall response by RECIST v1.1 indicated, excluding patients with stable disease (left). DIREct-On classification of patients with objective responses (middle) or progressive disease (right) by RECIST.
C) Waterfall depiction of sum of the lesion change from baseline at the time of best overall response by RECIST v1.1 for patients in the DIREct Discovery and Validation Cohorts. Colors indicate the DIREct-On status for each patient. Patients with progression of non-target lesions are indicated with asterisks and arbitrarily assigned to 21%.
Figure S7: Data related to Figure 7 Probability of PFS from start of therapy stratified by DIREct-On score in patients in the DIREct Discovery and Validation Cohorts treated with A) PD-1/PD-L1 single-agent blockade, B) PD-1 and CTLA-4 combination therapy, C) or the combination of PD-1 and chemotherapy.
Probability of PFS from start of therapy stratified by DIREct-On score in patients with tumors of D) non-squamous, or E) squamous histology.
F) Percent change in the sum of largest diameters (SLD) at the first scan for the 18 patients that had stable disease at the first scan in the DIREct Discovery and Validation Cohorts.
G) Probability of PFS from the time of treatment start for patients with increasing or decreasing SLD at the first scan within the stable disease bounds of +20% to −30%.
Table S3: Noninvasively detected mutations in genes recurrently mutated in NSCLC. Related to Figures 2–7.
Table S4: ctDNA analysis metrics in patients with available pre- or on-treatment plasma. Related to Figures 2–7.
Table S6: Pre-treatment circulating immune composition of major leukocyte populations by CIBERSORTx. Related to Figures 3–7.
Table S7: Parameter and model performance evaluation in the DIREct Discovery and Validation Cohorts. Related to Figures 5–7.
Data Availability Statement
The code to apply DIREct-On, model coefficients, and patient-level parameters for the discovery and validation cohorts are available for download at direct.stanford.edu. Clinical and demographic data for the cohorts generated as part of this study, sample timing information, ctDNA analysis metrics, transcripts per million quantified RNA sequencing data, immune profiling results, as well as parameter and model performance metrics are provided in the Supplemental Tables. Due to restrictions related to dissemination of germline sequence information included in the informed consent forms used to enroll study subjects, we are unable to provide access to the raw cfDNA, germline DNA, or RNA sequencing data because germline sequence information could be gleaned from these. Requests for additional data such as additional patient-level clinical or demographic data will be reviewed by the Lead Contact to determine whether they can be fulfilled in accordance with these privacy restrictions.