

SUMMARY
The non-receptor protein tyrosine phosphatase (PTP) SHP2, encoded by PTPN11, plays an essential role in RAS-MAPK signaling during normal development. It has been perplexing as why both enzymatically activating and inactivating mutations in PTPN11 result in human developmental disorders with overlapping clinical manifestations. Here, we uncover a common liquid-liquid phase separation (LLPS) behavior shared by these disease-associated SHP2 mutants. SHP2 LLPS is mediated by the conserved well-folded PTP domain through multivalent electrostatic interactions and regulated by an intrinsic autoinhibitory mechanism through conformational changes. SHP2 allosteric inhibitors can attenuate LLPS of SHP2 mutants, which boosts SHP2 PTP activity. Moreover, disease-associated SHP2 mutants can recruit and activate WT SHP2 in LLPS to promote MAPK activation. These results not only suggest that LLPS serves as a gain-of-function mechanism involved in the pathogenesis of SHP2-associated human diseases, but also provide evidences that PTP may be regulated by LLPS that can be therapeutically targeted.
In Brief
Disease-associated mutants of a critical phosphatase in the RAS-MAPK pathway undergo phase separation through a dominant gain-of-function mechanism, explaining how both enzymatically activating and -inactivating mutations dysregulate the pathway and can be therapeutically targeted.
Graphical Abstract
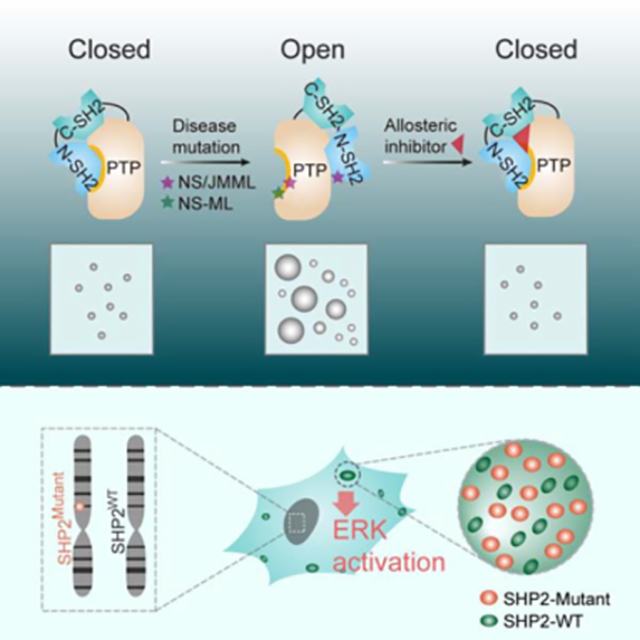
INTRODUCTION
Protein tyrosine phosphorylation is crucial for regulating numerous cellular signaling transduction. Disruption of the normal homeostasis of protein tyrosine phosphorylation has been implicated in the etiology of a variety of major human diseases (Böhmer et al., 2013; He et al., 2014; Tonks, 2006). PTPs represent the largest family of phosphatases regulating the homeostasis of protein tyrosine phosphorylation. PTPs have long been erroneously assumed to be constitutively active and housekeeping enzymes, while it has been recognized that cells have to tightly control PTP activity to fine-tune and limit the extent and duration of signaling transduction (Tonks, 2006). However, the underlying regulatory mechanism is not well elucidated, which is of great importance to develop new therapeutic strategy in the untapped resource for many human diseases.
SHP2, a ubiquitous non-receptor PTP encoded by PTPN11, is required for RAS-MAPK signaling pathway activation and normal development (Tajan et al., 2015). Mutations in PTPN11 are linked to numerous human diseases (Bowen et al., 2011; Digilio et al., 2002; Tartaglia and Gelb, 2005). Germline heterozygous mutations of PTPN11 are associated with 50% Noonan syndrome (NS) (Tartaglia and Gelb, 2005) and 90% Noonan syndrome with multiple lentigines (NS-ML, formally called Leopard syndrome) (Digilio et al., 2002). Somatic SHP2 mutations are found in 35% of juvenile myelomonocytic leukemias (JMML) and other related malignancies (Tartaglia et al., 2004; Miyamoto et al., 2008). In marked contrast to the activating mutations associated within NS and cancer, which are viewed as gain-of-function (GOF), all NS-ML mutations cause a decrease in SHP2 phosphatase activity and therefore are regarded as loss-of-function (LOF) (Kontaridis et al., 2006). However, NS-ML clinically resembles NS, raising a long debated question why both GOF and LOF mutations of SHP2 result in similar clinical manifestations (Edouard et al., 2007; Zheng et al., 2009). Remarkably, both NS and NS-ML are predisposed to elevated risk of malignancy (Cheng et al., 2013; Colmant et al., 2018; Laux et al., 2008; Liu et al., 2016; Tartaglia et al., 2004). These observations suggest that LOF mutations of SHP2 may, similarly as GOF mutations, also acquire capability to activate RAS-MAPK signaling pathway.
SHP2 is an allosteric enzyme composed of two tandem SH2 domains at its NH2-terminus (N-SH2 and C-SH2), a central PTP catalytic domain, and a C-terminal tail (Hof et al., 1998). The phosphatase activity of SHP2 is regulated by an elegant “molecular switch” mechanism in which SHP2 is autoinhibited by an intramolecular interaction between its N-SH2 domain and the PTP domain in the basal state (Hof et al., 1998). An intermolecular engagement of the N-SH2 domain with unique phosphor-tyrosine (pTyr) sequences from upstream growth factor receptors and/or scaffold proteins diminishes its intramolecular autoinhibitory interaction with the PTP domain, leading to an open and activated SHP2 state. GOF mutations of SHP2 in NS and JMML found in the interface of the N-SH2 and PTP domains abolish the autoinhibitory interaction between the two domains, whereas LOF mutations in NS-ML are located in the PTP domain causing impaired PTP activity (Yu et al., 2013; Yu et al., 2014; Qiu et al., 2014; LaRochelle et al., 2018). Interestingly, previous studies have suggested that both NS/JMML SHP2 mutants and NS-ML SHP2 mutants have an increased tendency for adapting the open conformation (Yu et al., 2013; Yu et al., 2014; Qiu et al., 2014; LaRochelle et al., 2018), while the link of the conformation changes in SHP2 mutants to the molecular mechanism underlying SHP2-associated human diseases has not been fully understood.
Phase separation has emerged as a fundamental mechanism that regulates various biological processes. Biomolecular condensates mediated by phase separation form coherent structures that can compartmentalize and concentrate biochemical reactions within cells (Alberti et al., 2019; Banani et al., 2017). Previous studies reveal that LLPS is triggered by multivalent interactions mediated by intrinsically disordered regions (IDRs) in proteins or by protein-interacting domains (Li et al., 2012; Shin and Brangwynne, 2017; Uversky, 2017). Research in the past years has provided mounting evidence that aberrant phase separation and liquid-to-solid phase transitions are involved in the process of neurodegenerative diseases (Alberti and Dormann, 2019; Hofweber et al., 2018; Murakami et al., 2015). However, very little is known about whether dysregulation of LLPS takes part in other human diseases, such as developmental disorders and cancers.
Here, we demonstrate that both NS/NS-ML and cancer-associated SHP2 mutants gain acquired capability of LLPS, which promotes its PTP enzymatic activity. The LLPS of SHP2 is driven by the conserved well-folded PTP domain through intermolecular electrostatic interactions and subjected to the intramolecular conformation regulation. The disease-associated SHP2 mutants can recruit WT SHP2 in phase-separated condensates and promote RAS-MAPK signaling. Moreover, SHP2 allosteric inhibitors can attenuate LLPS of SHP2 mutants. Our study reveals that LLPS provides a gain-of-function mechanism in the pathogenesis of SHP2-associated human diseases and possibility to therapeutically target LLPS for the treatment of human diseases.
RESULTS
Disease-associated SHP2 mutants form discrete puncta in cells
To understand how disease-associated mutations affect SHP2 cellular function, we stably expressed six mEGFP-labelled SHP2 variants (WT, 3 NS/JMML and 2 NS-ML mutants) in KYSE520 cells, which is a SHP2 dependent esophagus cancer cell line (Chen et al., 2016) (Figure S1A). The expression levels of SHP2-mEGFP were comparable to that of endogenous SHP2 (Figure S1B). Remarkably, live cell fluorescence microscopy revealed that all of the SHP2 variants bearing disease-associated mutations formed discrete puncta, whereas SHP2WT was apparently dispersed throughout the cell (Figure 1A). Disease-associated SHP2 mutants exceeded SHP2WT not only in the number but also in the average size of puncta as revealed by high-content image analysis (Figures 1B, S1C and S1D). The number of the puncta correlates with the average puncta size among the SHP2 variants (Figure S1E). Further, similar mutation-specific puncta formation was also observed in experiments using either mEGFP- or mScarlet-labelled SHP2 in several other cell lines, including HEK293T, A549 and HUVEC cells (Figures S1F–S1K). To rule out the possibility that the observed puncta formation was due to increased intracellular protein concentration by exogenous SHP2 expression, we further stably expressed mEGFP-labelled SHP2 variants and WT in SHP2 knockout HEK293T cells. The SHP2-mEGFP proteins were expressed at the same level as that of the endogenous SHP2 in parental HEK293T cells (Figure S1L). Again, we observed the puncta formation by mutant SHP2, but not by SHP2WT (Figure S1M).
Fig. 1. Disease-associated SHP2 mutants form discrete puncta in cells.

(A) Live imaging of SHP2WT and indicated SHP2mut-mEGFP in KYSE520 cells. Scale bar, 10μm. See Figure S1A for schematic representation for NS and NS-ML mutations. See Figure S1B for expression levels of SHP2-mEGFP. (B) Quantification of high content image data (means ± SEM) for SHP2WT and SHP2mut-mEGFP puncta. ***p<0.001. See Figure S1C, S1D and S1E for representative images of SHP2-mEGFP and related analysis result. (C) Immunofluorescence imaging of SHP2 in MEF cells derived from Ptpn11D61G/+ and control mice. Scale bar, 10 μm. (D) Quantification result (means ± SEM, N = 104 cells) of (C) was shown. ***p<0.001. See Figure S1O for other representative images. (E) Immunofluorescence imaging of SHP2 in mesenchymal stem cells (MSCs) derived from Ptpn11E76K/+ and control mice. Scale bar, 10 μm. (F) Quantification result (means ± SEM, N = 149 cells) of (E) was shown. ***p<0.001. See Figure S1P for other representative images. (G) Immunofluorescence imaging of SHP2 in oral mucosal epithelial cells collected form two Noonan syndrome patients and two healthy volunteers. Scale bar, 10 μm. (H) Quantification result (means ± SEM, N = 34 cells) of (G) was shown. ***p<0.001.
Notably, two cancer cell lines H661 and CCF-STTG1, which harbor an endogenous NS-associated SHP2N58S and NS-ML-associated SHP2R498W mutation respectively, also exhibited SHP2 puncta, whereas the KYSE520 cells harboring two WT SHP2 alleles only had occasional SHP2 puncta revealed by immunofluorescence (Figure S1N). We next examined the presence of SHP2 puncta in more physiological settings. We found that both the MEF cells derived from Ptpn11D61G/+ mice (Araki et al., 2004) and the mesenchymal stem cells (MSCs) derived from Ptpn11E76K/+ mice (Dong et al., 2016; Xu et al., 2011) exhibited highly statistically significant numbers of SHP2 puncta, while very few puncta were found in WT MEFs or MSCs (Figures 1C, 1D, 1E,1F,S1O and S1P). Moreover, a massive amount of SHP2 puncta was found in the oral mucosa cells from two NS patients harboring SHP2A72S and SHP2Q79R mutation respectively, but not in those of the two healthy donors (Figures 1G and 1H). These results clearly demonstrate that disease-associated mutant SHP2 exhibits a higher tendency to form puncta in cells.
The puncta of disease-associated SHP2 mutants exhibited liquid-like features in cells
LLPS has emerged as a fundamental regulatory mechanism in various biological processes (Alberti and Dormann, 2019; Alberti et al., 2019; Banani et al., 2017; Shin and Brangwynne, 2017). We assessed whether the mutant SHP2 puncta displayed any liquid-like features. Indeed, we observed that the puncta formed by both NS/JMML and NS-ML mutant SHP2 could mobilize and spontaneously fuse together upon encountering (Figures 2A, S2A, S2B, Movie S1 and S2). Fluorescence recovery after photobleaching (FRAP) experiments showed that upon photobleaching, the fluorescence of mutant SHP2-mEGFP puncta recovered within minutes (Figures 2B, 2C, S2C–S2F and Movies S3–5). To determine whether endogenous mutant SHP2 forms puncta displaying liquid-like features in live cells, we used CRISPR-Cpf1 to tag endogenous SHP2N58S mutant allele with mEGFP in H661 cells followed by single clone expansion and confirmation by sequencing (Figure S2G). SHP2N58S-mEGFP in the engineered H661 cells showed the same obvious condensate formation as the endogenous SHP2N58S did in the parental H661 cells (Figures S1N and S2H). Moreover, the puncta of endogenously tagged SHP2N58S-mEGFP in the engineered H661 cells also exhibited condensate fluidity assessed by FRAP experiments (Figure 2D). These results indicate that both NS/JMML and NS-ML mutations of SHP2 promote the formation of puncta that exhibit dynamic liquidlike behavior.
Fig. 2. The puncta of disease-associated SHP2 mutants exhibited liquid-like features in cells.

(A) Fusion of two SHP2E76K-mEGFP puncta. Scale bar, 5 μm. See Figure S2A and S2B for other SHP2 mutants. (B) Representative images of the FRAP experiment with SHP2E76K -mEGFP in KYSE520 cells. Scale bar, 5 μm. (C) Quantification of FRAP data (means ± SEM, N = 3 experiments) for SHP2E76K –mEGFP puncta. K (exponential constant) = 0.005497±0.000888s−1 and R (normalized plateau after fluorescence recovery) = 107.5% ± 7.45%. See Figure S2C, S2D, S2E and S2F for other SHP2 mutants. (D) Representative images of the FRAP experiment with endogenous tagged SHP2-mEGFP in H661(SHP2N58S) cells. Scale bar,10 μm.
Disease-associated mutations of SHP2 promote SHP2 LLPS in vitro
We next sought to examine if SHP2 possesses phase separation potential in vitro. We prepared recombinant WT and various disease-associated mutant SHP2 proteins (Figures S3A and S3B). We found that both NS/JMML and NS-ML mutants exhibited stronger capability for droplet formation than SHP2WT (Figures 3A and 3B). Notably, the in vitro droplet formation potential of SHP2 mutants nicely correlated with their puncta formation capability inside the cells (Figure 3C). Droplets of mutant SHP2 tended to coalesce together over time (Figure 3D and S3C). Moreover, FRAP experiments showed that when bleaching was performed, the fluorescence of mutant SHP2-mEGFP in the droplets was efficiently recovered (Figure 3E, S3D, and S3E), which further confirmed that SHP2 variants underwent typical LLPS in vitro.
Fig. 3. Disease-associated mutations of SHP2 promote SHP2 LLPS in vitro.

(A) Representative images of 8 μM recombinant SHP2WT and indicated SHP2mut-mEGFP protein droplet formation in the presence of 10%(w/v) PEG3350. Scale bar, 5 μm. (B) Quantification of SHP2WT and indicated SHP2mut-mEGFP droplet formation by solution turbidity OD600 (means ± SEM, N = 5 experiments). ***p<0.001. (C) The correlation between cellular and in vitro LLPS capability of SHP2WT and SHP2mut. (D) Fusion event of SHP2E76K protein was shown. Scale bar, 5 μm. See Figure S3C for other SHP2 mutants. (E) Quantification of FRAP data (means ± SEM, N = 5 experiments) for SHP2E76K –mEGFP droplet. See Figure S3D and S3E for other SHP2 mutants. (F) Phase diagrams of SHP2WT, SHP2E76K and SHP2R498L protein with the concentration ranging from 0.125–8 μM in 20mM Tris pH8.0, 10%(w/v) PEG3350, sodium chloride (ranging from 50–500 mM). Blue dots: no phase separation; red dots: phase separation. The LLPS ability of SHP2 under different conditions was color-coded on the basis of droplet turbidity measured at OD600 when the proteins were incubated with phase separation buffer at 37°C for 120 min.
Furthermore, we established the phase diagrams of SHP2WT and two disease-associated mutants SHP2E76K and SHP2R498L by performing the droplet formation assay with varying concentrations of SHP2 variants and sodium chloride. We observed that in the presence of 125 mM NaCl, which is close to the physiological salt concentration in cells (Tadashi et al., 2010), both SHP2E76K and SHP2R498L proteins were able to undergo LLPS starting from 0.25–0.5 μM, while LLPS of SHP2WT appears from the concentration of 2–4 μM (Figure 3F). We then measured the protein concentrations of endogenous SHP2 in various cell lines (Du, et al. 2018) and found that the concentration of SHP2 protein in cells is approximately 0.3 to 0.4 μM (0.3 μM in A549, 0.4 μM in HEK293T, and 0.4 μM in KYSE520) (Figure S3F). Assuming that SHP2 mutation occurs in one allele of PTPN11, the concentration of endogenous SHP2 mutant (0.15–0.2 μM) is approximately close to the critical concentration of SHP2 mutants required for LLPS in vitro (Figure 3F), while the protein concentration of SHP2WT in cells is much lower than the critical concentration of SHP2WT for LLPS in vitro. This may help to explain why SHP2 mutants tend to form puncta in cells while SHP2WT does not (Figure 1A). Together, these results demonstrate the differential LLPS behavior of disease-associated SHP2 mutants and SHP2WT in cells and in vitro.
Transition from closed to open conformation promotes SHP2 LLPS
Previous structural and biophysical analyses of WT, NS/JMML, and NS-ML mutations of SHP2 revealed that the degree of their propensities for the open conformation nicely correlated with the extent of increased dynamic and solvent exposure at the N-SH2/PTP domain interface and decreased N-SH2/PTP inter-domain interaction as a result of the mutations (LaRochelle et al., 2018; Qiu et al., 2014; Yu et al., 2014; Yu et al., 2013) (Figures 4A and 4B). Interestingly, by quantifying the openness status of SHP2 mutants based on the results of hydrogen/deuterium exchange mass spectrometry (H/DX-MS) (Yu et al., 2014), we found that the LLPS capabilities of droplet formation of SHP2 mutants perfectly correlated with their propensities for the open conformation regardless of NS/JMML or NS-ML mutations (Figures 4C and 4D). This observation suggests that the ability to transition from the closed to the open conformation may determine the potential of SHP2 LLPS. To test this hypothesis, we used the SHP2 allosteric inhibitor SHP099 that could stabilize SHP2 in its auto-inhibited/closed conformation (Chen et al., 2016; LaRochelle et al., 2018) (Figure 4E). Indeed, the single-molecule fluorescence resonance energy transfer (smFRET) experiments revealed that SHP2E76A was predominantly present in the open state; however, the addition of SHP099 dramatically reshaped the conformational landscape of SHP2E76A towards the closed conformation (Figure 4F and Figure S4A). Interestingly, the addition of SHP099 significantly attenuated the droplet formation of mutant SHP2 in a dose-dependent manner (Figures 4G and 4H). Consistently, the treatment with SHP099 also reduced the puncta formation of SHP2 mutants in cells (Figures 4I and 4J).
Fig. 4. Open conformation promotes SHP2 LLPS.

(A) Representative disease-associated SHP2 mutants. NS/JMML mutations (blue) and NS-ML (green). (B) A scheme representing the intrinsic propensity for open conformation in SHP2WT and indicated SHP2mut. (C) The correlation between in vitro LLPS capability and openness of SHP2 mutants. (D) Representative images of recombinant indicated SHP2mut-mEGFP protein droplet formation in the presence of 10%(w/v) PEG3350. Scale bar, 5 μm. (E) A scheme illustrating allosteric inhibitor SHP099 locks SHP2 in the closed conformation. (F) Single molecule FRET results show the conformation change of SHP2E76A-87/266 induced by SHP099. See Figure S4A for representative FRET time-trajectories. (G, H) Indicated SHP2mut droplets were treated with SHP099 and ET070. Time course of droplet turbidity OD600 (means ± SEM, N = 3 experiments) **p<0.01; ***p<0.001. (G) and representative images (H). Scale bar, 5 μm. (I, J) KYSE520 cells stably expressing indicated SHP2mut–mEGFP treated with SHP099 or ET070. Quantification of puncta/nuclei (means ± SEM) **p<0.01; ***p<0.001. N=46 cells. (I) and representative images of indicated SHP2mut–mEGFP (J). Scale bar, 10 μm. (K) The cartoon for binding of the bis-P peptide to SHP2. (L) SHP2Y279C proteins were stimulated w/o 1 μM bis-P peptide and the droplet turbidity (means ± SEM, N=2 experiments) was assessed. ***p<0.001. (M) Images of SHP2Y279C droplets treated with DMSO and 1 μM bis-P peptide. Scale bar, 5 μm.
Furthermore, the degree of puncta reduction was correlated with the extent of SHP2 inhibition as ET070, a more potent SHP2 allosteric inhibitor than SHP099 that we recently identified (Figures S4B–E), exhibited much stronger inhibition on the LLPS of SHP2 mutants in vitro (Figures 4G and 4H) and in cells than that of SHP099 (Figures 4I, 4J and Movie S6). Moreover, puncta formation of both SHP2D61G in Ptpn11D61G/+ MEFs and SHP2E76K in Ptpn11E76K/+ MSCs was strongly abolished by the treatment of ET070 (Figure S4F and S4G). Conversely, treatment with the stimulating peptide 2P-IRS-1, which contains bi-pTyr that can bind to both N- and C-SH2 domains and unlock the intramolecular interaction of N-SH2 domain with the PTP domain (Chen et al., 2016) (Figure 4K), promoted the droplet formation of SHP2Y279C which had relatively weak LLPS capability (Figures 4L and 4M).
We next investigated whether LLPS is an intrinsic property of SHP2 and if the open conformation could drive SHP2WT to undergo LLPS. We found that 2P-IRS-1 also promoted SHP2WT condensate formation in vitro (Figure S4H). Remarkably, treatment of growth factors such as bFGF and EGF induced SHP2WT-mEGFP puncta formation in KYSE520 cells (Figures S4I and Movie S7), suggesting that SHP2WT is able to undergo LLPS when the closed conformation is unlocked.
PTP domain drives SHP2 LLPS mediated by electrostatic interactions
We next sought to reveal the molecular basis underlying SHP2 phase separation. Unlike most well-documented examples of proteins that can undergo LLPS, SHP2 does not contain low-complexity intrinsic disordered region (IDRs) or repetitive multivalent modular domain (Alberti et al., 2019; Banani et al., 2017; Franzmann et al., 2018; Li et al., 2012; Nott et al., 2015; Shin and Brangwynne, 2017). To identify the domain responsible for SHP2 LLPS, we prepared recombinant full-length WT SHP2 (FL-SHP2) and three truncated SHP2 (SHP2ΔC, N/C-SH2, SHP2-PTP) (Figures 5A and S5A). Notably, the catalytic PTP domain showed much stronger LLPS capability than that of FL-SHP2 and other variants (Figures 5B and 5C). The droplets of SHP2-PTP underwent fusion revealed by fusion assay (Figure S5B). Intriguingly, the allosteric inhibitor SHP099 which efficiently attenuates full-length SHP2 mutants LLPS does not influence the droplet formation of SHP2-PTP (Figure S5C).
Fig. 5. PTP domain drives SHP2 LLPS mediated by electrostatic interactions.

(A) Schematic of full-length SHP2 (FL-SHP2) and truncated SHP2 (SHP2ΔC, N/C-SH2, SHP2-PTP). (B) Representative images showing 8 μM FL-SHP2, SHP2ΔC, SHP2-PTP and N/C-SH2 protein droplet formation in the presence of 10%(w/v) PEG3350. Scale bar, 5 μm. (C) Quantification of droplet turbidity (means ± SEM, N = 5 experiments) for 8 μM FL-SHP2, SHP2ΔC, SHP2-PTP and N/C-SH2 in LLPS buffer (10%(w/v) PEG3350). ***p<0.001. (D) Representative images of liquid droplets formed by SHP2-PTP in the presence of indicated concentration of NaCl. Scale bar, 5 μm. (E) Schematic illustrating that R362E/K364E mutation converts the positively charged patch (in yellow circle) to the negatively charged patch on the surface of SHP2-PTP (PDB:4DGP). (F) Microscopy images of PTP and PTPR362E/K364E droplets. Scale bar, 5 μm. (G) Microscopy images of the indicated mutant SHP2-mEGFP in KYSE520 cells. Scale bar, 10 μm.
Because the surface of PTP domain contains several negative and positive patches (Böhmer et al., 2013; Tonks, 2006), we next examined whether electrostatic interactions might be involved in mediating SHP2 LLPS. Indeed, the droplet formation of SHP2-PTP was highly sensitive to the increased concentration of NaCl (Figure 5D). To further pinpoint the essential charged patches mediating SHP2-PTP LLPS, we generated totally 17 SHP2 mutants including 16 double mutations and 1 single mutation distributed on the different charged patches of the SHP2-PTP surface (Figures S6A and S6B). We found that 6 mutants (R362E/K364E, E523K/D437K, E447K/E481K, E441K/D437K, E447K/D451K and R265E) significantly impaired the droplet formation of SHP2-PTP (Figure S6C). Among them, the E523/D437, E447/E481, E441/D437 and E447/D451 mutations are distributed on the two negatively charged patches (NCPs) while R362/K364 and R265 are located on the two positively charged patches (PCPs) (Figure S6D). These results suggest that multiple negatively and positively charged patches on the surface of SHP2-PTP proteins mediate intermolecular electrostatic interactions for SHP2-PTP LLPS. Among the different charged patches, the R362/K364-containing patch may play a dominant role, since the R362E/K364E mutation nearly abolished the LLPS capability of SHP2-PTP (Figures 5E and 5F) without perturbing its overall structure or PTP activity (Figures S6B, S6E and S6F). Strikingly, when introduced in disease-associated SHP2 mutants, the R362E/K364E mutation totally eliminated the puncta formation by different SHP2 mutants in cells (Figure 5G).
LLPS stimulates SHP2 PTP activity and is indispensable for ERK1/2 hyperactivation induced by SHP2 mutants
SHP2 is a key positive mediator for RAS-MAPK signaling. NS and NS-ML syndromes, caused by PTPN11 mutations (Tajan et al., 2015), belong to a newly classified family of autosomal dominant syndromes termed “RASopathies” (Tajan et al., 2018; Tomoki et al., 2009). We found that both NS and NS-ML mutant SHP2, but not SHP2WT, enhanced both MEK1/2 and ERK1/2 phosphorylation levels when stably expressed in cells (Figure 6A). Remarkably, although the LLPS-defective R362E/K364E mutation had no impact on SHP2 PTP activity (Figures S6E and S6F), they attenuated ERK1/2 activation induced by these disease-associated SHP2 mutants (Figures 6B and 6C), suggesting that LLPS of disease-associated SHP2 mutants is essential for ERK1/2 hyperactivation.
Fig. 6. LLPS stimulates SHP2 PTP activity and is indispensable for ERK hyperactivation induced by SHP2 mutants.

(A) Immunoblot of the indicated proteins in HEK293T cells stably expressing SHP2WT and SHP2mut. (B) Immunoblots of the indicated proteins in HEK293T cells stably expressing indicated SHP2 variants. (C) The densitometry analysis of pERK/ERK levels in (B). Data are plotted as means ± SEM, (n=3 experiments). *p<0.05; **p<0.01. RK (R362E/K364E). (D) Kinetic phosphatase activity (n ≥ 2 experiments) of SHP2WT and SHP2mut under conditions of LLPS and solution using pNPP as substrate. (E) Images of SHP2E76A droplets using DiFMUP substrate. Scale bar, 5 μm. DIC, differential and interference contrast.
Given that LLPS can locally concentrate molecules in condensates to accelerate reactions (Alberti et al., 2019; Banani et al., 2017; Huang et al., 2019; Shin and Brangwynne, 2017; Su et al., 2016), we next examined if LLPS could boost the PTP activity of SHP2. Indeed, enzymatic assays revealed that droplets of SHP2 variants displayed more robust PTP activity under LLPS condition than the corresponding mutants in solution condition (Figure 6D). The dephosphorylation product (DiFMU) of DiFMUP, an in vitro artificial substrate of SHP2, was concentrated in SHP2E76A droplets (Figure 6E), suggesting that SHP2 PTP activity was enhanced in the phase-separated condensates.
LLPS of NS-ML SHP2 mutants recruit and activate SHP2WT to promote ERK1/2 activation
As shown in Figure 6D, the enhanced PTP activity of NS-ML mutants by LLPS was still weaker than that of SHP2WT, which does not support the observation that MAPK signaling is elevated by NS-ML SHP2 mutations (Figure 6A and 6B). Given that NS-ML patients are genetically heterozygous mutation carriers, we next asked if the WT allele of SHP2 could contribute to ERK1/2 hyperactivation in cells expressing NS-ML mutant SHP2. We transfected HEK293T cells with 1:1 mixture of SHP2WT and SHP2Y279C plasmids and observed a clear elevation of pERK1/2 levels compared to that the equal amounts of SHP2WT or SHP2Y279C expression alone (Figure 7A). Interestingly, the ratio of WT and NS-ML SHP2 was critical because ERK1/2 was maximally activated when the levels of SHP2WT and SHP2Y279C were approximately equal when transfected in either parental HEK293T or SHP2 KO HEK293T cells (Figures 7B, 7C and S7A). Moreover, we generated a Tet-inducible SHP2Y279C-mEGFP stable KYSE520 cell line and titrated the expression of SHP2Y279C-mEGFP with different concentrations of doxycycline. The level of pERK1/2 peaked when SHP2Y279C-mEGFP was expressed at the same level as the endogenous SHP2 (Figure S7B). These results suggest that interaction of WT and NS-ML mutant SHP2 might provide a mechanistic basis for ERK1/2 hyperactivation as reported by independent lines of research in knock-in animal models and NS-ML patient-specific iPSCs that NS-ML SHP2 mutations result in hyperactivated RAS-MAPK ( Carvajal-Vergara et al., 2011; Oishi et al., 2009).
Fig. 7. LLPS of SHP2 mutants recruit and activate SHP2WT to promote ERK1/2 activation.

(A) Immunoblots of the indicated proteins in HEK293T cells transiently transfected with indicated expression plasmids. (B, C) HEK293T cells were transiently transfected with the indicated amount of SHP2WT and SHP2Y297C plasmids. The immunoblots (B) and the densitometry analysis (C) of pERK/ERK levels(means ± SEM, N = 3 experiments) was shown. See Figure S7A for SHP2 knock-out cells. (D) Representative images showing SHP2WT-mEGFP was readily distributed into SHP2R498L-mScarlet droplets. Scale bar, 2.5 μm. See also Figure S7C, S7D and S7E.(E) Enzymatic activity (means ± SEM, N = 3 experiments) of indicated proteins under the conditions of LLPS and solution using pNPP as substrate. ***p<0.001. (F) Living images of KYSE520 cells co-expressed with SHP2WT-mEGFP and SHP2R498L-mScarlet. Scale bar, 10 μm. See also Figure S7G. (G) Cartoon illustrating that SHP2mut induces strong LLPS to recruit SHP2WT and promote ERK activation.
We then questioned if NS-ML mutant SHP2 could form condensates to recruit SHP2WT and promote ERK1/2 activation. Indeed, we observed that SHP2WT was readily distributed into NS-ML mutant SHP2 droplets under which conditions SHP2WT itself could not efficiently form droplets (Figure 7D). Although the overall droplet formation capability of WT/mutant SHP2 mixture was compromised compared to mutant SHP2 alone (Figure S7C), a significant portion of SHP2WT were enriched in mutant SHP2 droplets (Figure S7D and S7E). Moreover, SHP2WT exhibited rapid recovery upon photobleaching (Figure S7F). Remarkably, droplets composed of the mixture of WT and NS-ML mutant SHP2 displayed stronger PTP activity than the droplets of each protein (Figure 7E). We next examined if SHP2WT in SHP2 mutant cells could be recruited by the phase-separated puncta of NS-ML mutant SHP2. SHP2WT-mEGFP did not form puncta when transfected in KYSE520 cells (Figure 1A); however, when coexpressed with SHP2R498L, SHP2WT-mEGFP exhibited puncta that colocalized with SHP2R498L puncta (Figure 7F). Similar results were obtained when SHP2WT was coexpressed with SHP2Y279C (Figure S7G). These results suggested that NS-ML mutant SHP2 could form condensates to recruit SHP2WT and activate MAPK signaling (Figure 7G).
DISCUSSION
Genetic mutations of SHP2 involved in human developmental disorders and cancers promote a gain-of-function LLPS
LLPS has been extensively studied as a regulatory mechanism of normal proteins in membraneless cellular compartments. In this study, we provided strong evidence demonstrating that disease-causing SHP2 mutant proteins present at endogenous levels display a dramatically higher propensity than that of WT SHP2 to form LLPS in several engineered cell lines, MEF and MSC cells derived from Ptpn11D61G/+ and Ptpn11E76K/+ mice, and oral mucosa cells from NS patients. Given the broad spectrum of SHP2 mutations in human diseases, it will be interesting to expand this work and further examine whether the acquired LLPS capability is a general property for all disease-associated SHP2 mutants in NS, NS-ML, as well as JMML and solid tumors. More importantly from this study, we describe genetic mutations in SHP2 directly endow phase separation capability as a gain-of-function mechanism to activate MAPK signaling, which is critically important in RASopathy and tumorigenesis. Our discovery provides insights into a previously unappreciated function of LLPS in mediating pathogenesis of human diseases. Our results suggest that catalytically impaired SHP2 mutants may exert gain-of-function activity by facilitating LLPS to promote the activity of SHP2WT protein and MAPK hyperactivation in NS-ML patients, explaining the clinical similarities between NS and NS-ML. This finding also explains a long-standing puzzle as why NS-ML patients carrying SHP2 inactivating mutations have an increased risk of developing hematological malignancies ( Uçar et al., 2006; Laux et al., 2008; Merks et al., 2005; Seishima et al., 2010), similar to NS patients who harbor SHP2 activating mutations. Moreover, our results reveal that somatic mutations of SHP2 in cancer cells also promote SHP2 LLPS (Figure S1N), suggesting that LLPS of SHP2 mutants may provide a cancer-driving mechanism that directly links to tumorigenesis by promoting ERK1/2 activation.
Regulation of WT SHP2 PTP activity by LLPS
Given that the substrate dephosphorylation by PTP is biochemically passive and spontaneous, it is vital for PTP activity to be tightly fine-tuned during signaling transduction in cells. Here we showed that SHP2WT also formed condensates when the cells were stimulated with various factors such as EGF and FGF, suggesting that LLPS may serve as a regulatory mechanism to control WT SHP2 PTP activity under physiological conditions. We found that the protein concentration of endogenous SHP2 is about 0.3–0.4 μM, close to the critical concentration of disease-associated SHP2 mutants for LLPS in vitro, but largely below the critical concentration of SHP2WT for LLPS (Figures 3F and S3F). This may explain why SHP2 mutants but not SHP2WT are prone to form phase separated condensates in cells. In addition, other factors may also modulate SHP2 LLPS in cells. For instance, phosphorylated tyrosine residues in upstream growth factor receptor could bind to the two SH2 domains of SHP2 and unlock its intramolecular autoinhibitory interaction with the PTP domain, which may lower the critical concentration of both WT and mutant SHP2 for LLPS in cells. In line with this, when cells are stimulated with growth factors, we do observe SHP2WT puncta formation on the cell membrane (Figures S4H and Movie S7). SHP2 may adopt LLPS as a reaction crucible in stimulating its enzymatic activity to promote dephosphorylation of its substrates and MAPK signaling. Notably, SHP2 can also function as adaptor proteins to recruit signal proteins (Dance et al., 2008), suggesting that LLPS of SHP2 may also serve as a node to amplify signaling magnitude.
SHP2 protein does not contain low-complexity IDRs that are typical for mediating proteins LLPS (Franzmann et al., 2018; Nott et al., 2015). Our study reveals that the well-folded conserved PTP domain can drive SHP2 LLPS mediated by the multivalent intermolecular electrostatic interactions. Given that PTP domains in other protein tyrosine phosphatases commonly contain multiple positively and negatively charged patches on the protein surface areas (Böhmer et al., 2013), our finding may bring up an interesting hypothesis that LLPS may function as a general regulatory mechanism of protein tyrosine phosphatases.
NS-ML mutant SHP2 recruit SHP2WT to promote ERK1/2 activation
Our study demonstrates that NS-ML SHP2 mutants induce a robust phase transition to liquid-like droplets in cells, which recruit and activate SHP2WT to promote ERK1/2 activation. In our study, we found that although overexpression of NS-ML SHP2 alone may sustain or mildly increase ERK1/2 phosphorylation (Figure S7A) as observed previously (Yu et al., 2013; Yu et al., 2014), SHP2WT plays a critical role in ERK1/2 hyperactivation and the maximal pERK1/2 level was reached when the levels of WT and NS-ML SHP2 were approximately equal. This result may reflect the NS-ML disease condition, where both WT and mutant allele of PTPN11 are expressed at comparable levels. This finding may also address the perplexing observation reported in previous studies on ERK1/2 activation induced by NS-ML SHP2 mutants (Tajan et al., 2015). The different expression levels of mutant SHP2 compared to SHP2WT may result in the different outcome in term of ERK1/2 activation. Notably, it is reported that both pERK1/2 and pMEK1/2 levels are elevated in NS-ML patient-specific iPSC (Carvajal-Vergara et al., 2011), providing a strong evidence that NS-ML SHP2 mutations indeed are GOF mutations to promote MAPK signaling pathway via acquired capability of LLPS.
Regulation of SHP2 LLPS through conformational change
Our results demonstrate that the R362/K364-containing positively charged patch (362/364PCP) of SHP2-PTP is important in mediating LLPS of SHP2 both in vitro and in cells. Intriguingly, despite that the 362/364PCP is exposed to solvent in the isolated PTP domain for mediating LLPS, it is partially masked by the N-SH2 in the full-length SHP2 when the protein is in the close conformation (Figure S6G). However, since the disease-associated mutations induce conformational transition of SHP2 from the close to the open state, the N-SH2 is detached from PTP which may allow 362/364PCP to be fully accessible for driving SHP2 LLPS (Figure S6G). This finding further supports that the open conformation promotes SHP2 LLPS.
An interesting hypothesis is that SHP2WT may also adopt an open conformation when recruited in NS-ML mutant SHP2 droplets as the 362/364PCP has to be exposed to enable phase separation. In the droplets, 362/364PCP, which is partially masked by the N-SH2 domain in the closed conformation, may gain new intermolecular interactions with other negative patches. These new intermolecular interactions may thus compete with the intramolecular interaction of the N-SH2 and induce the conformational transition towards the open conformation. This may explain why WT/mutant SHP2 droplets exhibited more potent PTP activity than WT or mutant SHP2 droplets alone (Figure 7E). However, further study is needed to directly probing the conformations of SHP2 variants in the droplet. At last, the acquired LLPS capability of mutant SHP2 can be inhibited by SHP2 allosteric inhibitors which lock SHP2 in the closed formation, providing a therapeutic strategy to treat SHP2-associated developmental disorders.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jidong Zhu (zhujd@sioc.ac.cn).
Materials Availability
Plasmids, compounds, and cell lines generated in this study will be made available upon request. We may require a payment and/or a completed Materials Transfer Agreement in case there is potential for commercial application.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study. No unique code was generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Oral mucosal epithelial cells were collected from two NS patients harboring SHP2A72S (male) and SHP2Q79R mutation (female) and 2 healthy donors (1 male and 1 female), ages between 4 and 8 years old. This study was approved by the Ethics Committee of Xin Hua Hospital affiliated to Shanghai Jiao Tong University School of Medicine. We have also certified that the study was strictly in accordance with the Declaration of Helsinki and International Ethical Guidelines for Health-related Research Involving Humans. Oral mucosal epithelial cells were obtained by gently scraping palate and gum using cotton swab, right after oral hygiene through brushing of teeth.
Cell line
Cells were maintained accordingly to the guidance from American Type Culture Collection. Human HEK293T (female) and HEK293FT (female) cells were cultured in DMEM supplemented with 10% (v/v) FBS, 100 units/mL penicillin and 100 mg/mL streptomycin. Human H661(male), CCF-STTG1(female), KYSE520(female), NCI-H1299(male), A549(male), SF268(female) and MV4;11(male) cells were cultured in RPMI1640 supplemented with 10% (v/v) FBS, 100 units/mL penicillin and 100 mg/mL streptomycin. All cells were cultured at 37 °C in a humidified atmosphere of 95% air and 5% CO2.
All cell line identifiers are listed in the Key Resources Table. H661, H1299 and A549 cells obtained from Cell bank of Shanghai Institute of Biochemistry and Cell Biology (SIBCB) and KYSE520, CCF-STTG1 and MV4;11 cells from Cobioer company have been authenticated by STR analysis and mycoplasma detection. HEK293T, HEK293FT and SF268 cells have been verified through periodic morphology checks and mycoplasma detection.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SH-PTP2(B-1) | Santa Cruz | Cat# sc-7834; RRID:AB_628252 |
| Phospho-p44/p42 MAPK(Erk1/2)(Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370S; RRID:AB_2315112 |
| p44/p42 MAPK(Erk1/2) | Cell Signaling Technology | Cat# 9102S; RRID:AB_330744 |
| Phospho-MEK1/2(Ser217/221)(41G9) | Cell Signaling Technology | Cat# 9154; RRID:AB_2138017 |
| MEK1/2 | Cell Signaling Technology | Cat# 9122; RRID:AB_823567 |
| SHP2 | Cell Signaling Technology | Cat# 3397T; RRID:AB_2174959 |
| HA | Cell Signaling Technology | Cat# 2367S; RRID:AB_10691311 |
| FLAG | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| GAPDH | Cell Signaling Technology | Cat# 2118S; RRID:AB_561053 |
| α-tubulin | Abcam | Cat# ab52866; RRID:AB_869989 |
| Bacterial and Virus Strains | ||
| Escherichia coli BL21(DE3) | TIANGEN | Cat# CB105–01 |
| Escherichia coli Stbl3 | Exinbio | Cat# CC104–01 |
| Escherichia coli DH5α | TIANGEN | Cat# CB101–01 |
| Biological Samples | ||
| Human oral mucosal epithelial cells | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2P-IRS-1 peptide | QiangYao | N/A |
| 6,8-difluoro-4-methylumbelliferyl phosphate(DiFMUP) | Invitrogen | Cat# D22065 |
| p-Nitrophenyl Phosphate(PNPP) | NEB | Cat# P0757S |
| Doxycycline hyclate | Yeasen | Cat# 60204ES08 |
| G418 Sulfate | Yeasen | Cat# 60220ES03 |
| Puromycin Dihydrochloride | ThermoFisher Scientific | Cat# A1113802 |
| SHP099 | This study | N/A |
| ET070 | This study | N/A |
| Recombinant SHP2(WT) protein | This study | N/A |
| Recombinant SHP2(D61G) protein | This study | N/A |
| Recombinant SHP2(E76A) protein | This study | N/A |
| Recombinant SHP2(E76K) protein | This study | N/A |
| Recombinant SHP2(Y279C) protein | This study | N/A |
| Recombinant SHP2(R498L) protein | This study | N/A |
| Recombinant SHP2(Q506P) protein | This study | N/A |
| Recombinant SHP2(G464A) protein | This study | N/A |
| Recombinant SHP2(PTP) protein | This study | N/A |
| Recombinant SHP2(1–528) protein | This study | N/A |
| Recombinant SHP2(C+N-SH2) protein | This study | N/A |
| Recombinant SHP1 protein | This study | N/A |
| Recombinant PTP1B protein | This study | N/A |
| Recombinant FAP1 protein | This study | N/A |
| Recombinant CD45 protein | This study | N/A |
| Recombinant VHR protein | This study | N/A |
| Recombinant LMWPTP protein | This study | N/A |
| Recombinant MKP3 protein | This study | N/A |
| Recombinant SHP2(WT) –mEGFP protein | This study | N/A |
| Recombinant SHP2(E76A) –mEGFP protein | This study | N/A |
| Recombinant SHP2(E76K) –mEGFP protein | This study | N/A |
| Recombinant SHP2(Y279C) –mEGFP protein | This study | N/A |
| Recombinant SHP2(R498L) –mEGFP protein | This study | N/A |
| Recombinant SHP2(G464A) –mEGFP protein | This study | N/A |
| Recombinant SHP2(Q506P) –mEGFP protein | This study | N/A |
| Recombinant SHP2(T468M) –mEGFP protein | This study | N/A |
| Recombinant SHP2(E76A) –mScarlet protein | This study | N/A |
| Recombinant SHP2(E76K) –mScarlet protein | This study | N/A |
| Recombinant SHP2(Y279C) –mScarlet protein | This study | N/A |
| Recombinant SHP2(R498L) –mScarlet protein | This study | N/A |
| Recombinant PTPE523K/D437K protein | This study | N/A |
| Recombinant PTPR278E/K280E protein | This study | N/A |
| Recombinant PTPR265E protein | This study | N/A |
| Recombinant PTPE249K/E250K protein | This study | N/A |
| Recombinant PTPR362E/K364E protein | This study | N/A |
| Recombinant PTPE447K/E481K protein | This study | N/A |
| Recombinant PTPE441K/D437K protein | This study | N/A |
| Recombinant PTPE447K/D451K protein | This study | N/A |
| Recombinant PTPD431K/D395K protein | This study | N/A |
| Recombinant PTPD373KE390K protein | This study | N/A |
| Recombinant PTPD294K/E299K protein | This study | N/A |
| Recombinant PTPD296K/E299K protein | This study | N/A |
| Recombinant PTPD373A/E374A protein | This study | N/A |
| Recombinant PTPD303K/D296K protein | This study | N/A |
| Recombinant PTPK358A/R421A protein | This study | N/A |
| Recombinant PTPK266E/R270E protein | This study | N/A |
| Recombinant PTPK274E/K276E protein | This study | N/A |
| Recombinant PTPE523K/D437K –mEGFP protein | This study | N/A |
| Recombinant PTPR278E/K280E –mEGFP protein | This study | N/A |
| Recombinant PTPR265E –mEGFP protein | This study | N/A |
| Recombinant PTPE249K/E250K –mEGFP protein | This study | N/A |
| Recombinant PTPR362E/K364E –mEGFP protein | This study | N/A |
| Critical Commercial Assays | ||
| CellTiter-Glo reagent | Promega | Cat# G7570 |
| Deposited Data | ||
| Crystal structure of SHP2 | Yu et al., 2013 | PDB: 4DGP |
| Crystal structure of catalytic domain of human SHP2 | Grosskopf et al. 2015 | PDB: 3ZM0 |
| Experimental Models: Cell Lines | ||
| Human: KYSE520 | Cobioer, Nanjing | Cat# CBP60658 |
| Human: CCF-STTG1 | Cobioer, Nanjing | Cat# CBP60580 |
| Human: H661 | Cell Bank of SIBCB | Cat# TCHu121 |
| Human: NCI-H1299 | Cell Bank of SIBCB | Cat# SCSP-589 |
| Human: A549 | Cell Bank of SIBCB | Cat# SCSP-503 |
| Human: MV4;11 | Cobioer, Nanjing | Cat# CBP60522 |
| Human: SF268 | Zhang Wei-Min lab | N/A |
| Human: HEK293FT | Wang Wenyuan lab | N/A |
| Human: HEK293T | Hu Junhao lab | N/A |
| Human: HUVEC | Promocell | C-12203 |
| Mouse: MSC (bone marrow) | Shi Yufang lab | N/A |
| Mouse: E76K/+ MSC (bone marrow) | Wang Siying lab | N/A |
| Mouse: MEF | Wang Siying lab | N/A |
| Mouse: D61G/+ MEF | Wang Siying lab | N/A |
| Oligonucleotides | ||
| SHP2 gRNA for Knock out 5’-GTTACTGACCTTTCAGAGGT-3’ | This study | N/A |
| SHP2 gRNA for Knock in 5’- AGATGAGAAAACCTGCCAAAACT-3’ | This study | N/A |
| Primer for PCR amplification of Knock in fragment: Forward: 5’ -TGACATCGCGGAGATGGTTT- 3’ | This study | N/A |
| Primer for PCR amplification of Knock in fragment: Reverse: 5’-GCTGAACTTGTGGCCGTTTA- 3’ | This study | N/A |
| Forward primer for Sanger sequencing: 5’-GAGAGAAGACAGTGCTAGAGTCTATG-3’ | This study | N/A |
| Reverse primer for Sanger sequencing: 5’-CATCTCCATTCTTCTCTTTTAATTGC-3’ | This study | N/A |
| Recombinant DNA | ||
| pET28a-SHP2(WT) | This study | N/A |
| pET28a-SHP2(PTP) | This study | N/A |
| pET28a-SHP2(1–528) | This study | N/A |
| pET28a-SHP2(C+N-SH2) | This study | N/A |
| pET28a-SHP2(E76A) | This study | N/A |
| pET28a-SHP2(E76K) | This study | N/A |
| pET28a-SHP2(Y279C) | This study | N/A |
| pET28a-SHP2(R498L) | This study | N/A |
| pET28a-SHP2(G464A) | This study | N/A |
| pET28a-SHP2(Q506P) | This study | N/A |
| pET28a-SHP2(T468M) | This study | N/A |
| pET28a-SHP1(243–541) | This study | N/A |
| pET28a-PTP1B(1–298) | This study | N/A |
| pET28a-FAP1(2163–2477) | This study | N/A |
| pET28a-CD45(624–1233) | This study | N/A |
| pET28a-VHR(2–185) | This study | N/A |
| pET28a-LMWPTP(2–158) | This study | N/A |
| pET28a-MKP3(206–381) | This study | N/A |
| pET28a-SHP2(WT) -mEGFP | This study | N/A |
| pET28a-SHP2(E76A) -mEGFP | This study | N/A |
| pET28a-SHP2(E76K) -mEGFP | This study | N/A |
| pET28a-SHP2(Y279C) -mEGFP | This study | N/A |
| pET28a-SHP2(R498L) -mEGFP | This study | N/A |
| pET28a-SHP2(G464A) -mEGFP | This study | N/A |
| pET28a-SHP2(Q506P) -mEGFP | This study | N/A |
| pET28a-SHP2(T468M) -mEGFP | This study | N/A |
| pET28a-SHP2(E76A) -mScarlet | This study | N/A |
| pET28a-SHP2(E76K) -mScarlet | This study | N/A |
| pET28a-SHP2(Y279C) -mScarlet | This study | N/A |
| pET28a-SHP2(R498L) -mScarlet | This study | N/A |
| pET28a- PTPE523K/D437K | This study | N/A |
| pET28a- PTPR278E/K280E | This study | N/A |
| pET28a- PTPR265E | This study | N/A |
| pET28a- PTPE249K/E250K | This study | N/A |
| pET28a- PTPR362E/K364E | This study | N/A |
| pET28a- PTPE447K/E481K | This study | N/A |
| pET28a- PTPE441K/D437K | This study | N/A |
| pET28a- PTPE447K/D451K | This study | N/A |
| pET28a- PTPD431K/D395K | This study | N/A |
| pET28a- PTPD373KE390K | This study | N/A |
| pET28a- PTPD294K/E299K | This study | N/A |
| pET28a- PTPD296K/E299K | This study | N/A |
| pET28a- PTPD373A/E374A | This study | N/A |
| pET28a- PTPD303K/D296K | This study | N/A |
| pET28a- PTPK358A/R421A | This study | N/A |
| pET28a- PTPK266E/R270E | This study | N/A |
| pET28a- PTPK274E/K276E | This study | N/A |
| pET28a- PTPE523K/D437K -mEGFP | This study | N/A |
| pET28a- PTPR278E/K280E -mEGFP | This study | N/A |
| pET28a- PTPR265E -mEGFP | This study | N/A |
| pET28a- PTPE249K/E250K -mEGFP | This study | N/A |
| pET28a- PTPR362E/K364E -mEGFP | This study | N/A |
| pMSCV-HA-SHP2(WT) | This study | N/A |
| pMSCV-FLAG-SHP2(WT) | This study | N/A |
| pMSCV-HA-SHP2(E76A) | This study | N/A |
| pMSCV-HA-SHP2(E76K) | This study | N/A |
| pMSCV-HA-SHP2(Y279C) | This study | N/A |
| pMSCV-HA-SHP2(R498L) | This study | N/A |
| pMSCV-HA-SHP2(D61G) | This study | N/A |
| pMSCV-HA-SHP2(WT)-mEGFP | This study | N/A |
| pMSCV-HA-SHP2(D61G)-mEGFP | This study | N/A |
| pMSCV-HA-SHP2(E76A)-mEGFP | This study | N/A |
| pMSCV-HA-SHP2(E76K)-mEGFP | This study | N/A |
| pMSCV-HA-SHP2(Y279C)-mEGFP | This study | N/A |
| pMSCV-HA-SHP2(R498L)-mEGFP | This study | N/A |
| pMSCV-HA-SHP2(WT)-mScarlet | This study | N/A |
| pMSCV-HA-SHP2(D61G)-mScarlet | This study | N/A |
| pMSCV-HA-SHP2(E76A)-mScarlet | This study | N/A |
| pMSCV-HA-SHP2(E76K)-mScarlet | This study | N/A |
| pMSCV-HA-SHP2(Y279C)-mScarlet | This study | N/A |
| pMSCV-HA-SHP2(R498L)-mScarlet | This study | N/A |
| pMSCV-HA-SHP2E76A/R362E/K364E-mEGFP | This study | N/A |
| pMSCV-HA-SHP2E76K/R362E/K364E-mEGFP | This study | N/A |
| pMSCV-HA-SHP2Y279C/R362E/K364E-mEGFP | This study | N/A |
| pMSCV-HA-SHP2R498L/R362E/K364E-mEGFP | This study | N/A |
| pInducer20-HA-SHP2Y279C-mEGFP | This study | N/A |
| pcDNA3.1-hAsCpf1 | Zetsche et al.,2015 | Addgene plasmid # 69982 |
| pUC57-U6-gRNA | This study | N/A |
| pUC57-PTPN11-HR-donor | This study | N/A |
| Software and Algorithms | ||
| LAS X | Leica | https://www.leicamicrosystems.com/products/microscopesoftware/details/product/leica-las-x-ls/ |
| PyMOL | PyMOL | https://pymol.sourceforge.net/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| ImageLab software | Bio-Rad | https://www.biorad.com |
| Fiji | NIH | https://imagej.nih.gov/ij/ |
| Graphpad Prism version 6.0 | GraphPad Software | https://www.graphpad.com |
Primary cell culture
HUVEC cells isolated from the vein of the umbilical cord were purchased from Promocell(C-12203) and cultured in Endothelial Cell Growth Medium(C-22010, Promocell). Mouse MSC isolation has been described previously (Dong et al., 2016). Briefly, mice were sacrificed and rinsed with 75% ethanol for 3 minutes. Then, the compact bones and bone marrow were collected after muscle dissection. Bone marrow cells were seeded in a culture dish and then moved into the cell incubator. Compact bones were then crushed and digested with collagenase II for 1.5 h at 37°C and finally cultured in an incubator. MSCs were observed after 3 days of incubation without any disturbance. Both bone marrow and compact bone MSCs were purified by controlling the digesting time (no more than 3 minutes at room temperature), and then they were growth together. MSC markers were used to determine the purity of MSCs.
Escherichia coli strains
Escherichia coli BL21(DE3) cells were used in this study for the production of recombinant proteins. Cells were cultured in lysogeny broth (LB) medium at 37 °C, 220rpm.
METHOD DETAILS
Plasmids Transfection and viral infection
Transfection of plasmids into HEK293T/293FT was performed using Polyjet (SignaGen, SL100688) according to the manufacturer’s instructions. To generate stable cells, retroviral and lentiviral infections were used. Briefly, HEK293FT cells were co-transfected with viral plasmids and packaging plasmids. Forty-eight hours after transfection, culture medium was filtered through a 0.45 μm filter (Millipore), and used to infect cells of interest.
Live cell imaging
Cells were grown on 24-well glass bottom plate (Cellvis, P24–1.5H-N) and images were taken with the Leica TCS SP8 confocal microscopy system using a 100x oil objective (NA=1.4). Cells were imaged on a heated stage (37 °C) and supplemented with warmed (37 °C) humidified air.
For experiments that SHP2mut-mEGFP cells were treated with SHP099 or ET070, cells were imaged every 15 min after 10 μM SHP099 or ET070 was added.
Fluorescent images were processed and assembled into figures using LAS X (Leica) and Fiji.
High content image
KYSE520 cells expressing SHP2WT and SHP2mut (SHP2D61G, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L)–mEGFP were seeded in 24-well glass bottom plate (Cellvis P24–1.5H-N) and images were obtained with the Operetta CLS™ high-content cell imaging analysis system (PerkinElmer Inc.,Waltham,MA,USA). The 63x objective lens was applied in each condition. Data was analyzed by images collected from 10 representative fields in each group. Single cells were identified based on Hoechst 33342 as reference, while the spots quantification was performed based on the area and intensity of the spots through mEGFP channel. GraphPad Prism is used to plot and analyze the high content image results.
Fluorescence Recovery After Photobleaching (FRAP)
FRAP assay was conducted using the FRAP module of the Leica SP8 confocal microscopy system. The SHP2mut-mEGFP was bleached using a 488-nm laser beam. Bleaching was focused on a circular region of interest (ROI) using 100% laser power and time-lapse images were collected. Fluorescence intensity was measured using Fiji. Background intensity was subtracted and values are reported relative to pre-bleaching time points. GraphPad Prism is used to plot and analyze the FRAP results.
Protein expression and purification
The full-length SHP2 (WT) gene was PCR amplified from a human cDNA library (reference sequence NCBI:NP_002825.3, 593 aa). Various mutants of SHP2 were generated using standard PCR-based methods and confirmed by DNA sequencing. All constructs of human SHP2 were inserted into a pET28a vector. A coding sequence for a 6× histidine tag was added on the N terminus to the constructs. These constructs were transformed into Escherichia coli BL21(DE3) cells and grown in lysogeny broth (LB) medium at 37 °C to an optical density at OD600 of 0.8. The expression of recombinant proteins was induced by 0.8 mM IPTG at 16 °C for 18h. Briefly, cells were harvested by centrifugation at 4000 rpm for 30 min at 4°C. After centrifugation, cells were lysed in buffer containing 25 mM Tris-HCl pH 8.0, 500 mM NaCl and 1mM PMSF, followed by supercentrifugation at 16000 rpm for 40 min at 4 °C. The supernatant was loaded onto a HisTrap HP chelating column (GE healthcare) in 50 mM Tris-HCl pH 8.0, 500mM NaCl, 2 mM DTT and proteins were eluted with the addition of 500 mM imidazole. Fractions containing SHP2 were concentrated and loaded onto a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) using buffer containing 25 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM DTT. Fractions were collected according to the results of SDS-PAGE, and then these proteins were concentrated to 10 mg/mL or more, and stored at −80°C.
In vitro liquid-liquid phase separation (LLPS) assay
For the LLPS assay, the purified SHP2WT or SHP2mut protein at 8 μM was mixed with a LLPS buffer containing 20 mM Tris pH 8.0, 12%(w/v) PEG3350 (Sigma) and incubated for 5 min at room temperature. Finally, 2 μL of each sample was pipetted onto a glass dish and imaged using a Leica microscope. Under the same conditions, 30 μL of each sample was added into 384-well white polystyrene plate with clear flat bottom, and the value of OD600 was measured by using a TECAN Infinite F PLEX Pro microplate reader (37 °C, 10 hrs).
For LLPS assays treated with allosteric inhibitor, 20/50μM SHP099 or ET070 was incubated with 8 μM SHP2mut phase-separated droplets at 37 °C for 15 min, and further transferred to 384-well white polystyrene plate for OD600 kinetics study. The microplate reader reads every 5 minutes and shakes 10 seconds before each reading (37 °C, 10 hrs).
For SHP2Y279C in the presence of 2P-IRS-1 peptide, 4 μM SHP2Y279C protein was incubated with 1 μM peptide or DMSO at 37 °C for 15 min and further mixed with the LLPS buffer. Each sample was then subjected to OD600 kinetics study at 37 °C for 10 hrs.
Phase diagrams were generated by mixing SHP2 protein (varying from 0.125–8 μM, final) in phase separation buffer 20 mM Tris pH 8.0, 12%(w/v) PEG3350 (Sigma), sodium chloride (varying from 50–500 mM). Droplet turbidity OD600 was measured by microplate reader as described above.
Endogenously-tagged cell line generation
CRISPR-Cpf1 was used (Li et al., 2018, Zetsche et al.,2015) to generate endogenously-mEGFP-tagged SHP2 in SHP2N58S H661 and SHP2WT HEK293T cells. Oligos coding for guide RNAs targeting the C-terminus of SHP2 was 5’-AGATGAGAAAACCTGCCAAAACT-3’. Repair templates were cloned into a pUC57 vector containing mEGFP, a GGS linker and 800 bp homology arms flanking the insert. H661 and HEK293T cells were co-transfected with 0.6 μg pcDNA3.1-hAsCpf1 vector, 0.6 μg pUC57-U6-gRNA and 0.8 μg pUC57-PTPN11-HR-donor repair templates using lipofectamine 3000. Cells were sorted 3 days after transfection for mEGFP and colonies were picked 14 days after seeding into 96 well plates. Cells were further passaged into 2 plates. One was used for genotyping by Sanger sequencing and the other was for imaging.
Primers for PCR amplification of Knock in fragment:
F: 5’ -TGACATCGCGGAGATGGTTT- 3’
R: 5’-GCTGAACTTGTGGCCGTTTA- 3’
And following primers were used for Sanger sequencing:
F: 5’-GAGAGAAGACAGTGCTAGAGTCTATG-3’
R: 5’-CATCTCCATTCTTCTCTTTTAATTGC-3’
CRISPR/Cas9 knock-out in 293T cells and rescue experiments
Oligos coding for guide RNAs targeting the N-terminus of SHP2 were cloned into lentiCRISPRv2 backbone. The sequence targeted for SHP2 is 5’- GTTACTGACCTTTCAGAGGT-3’. Cloned plasmids were co-transfected with psPax2 and pMD2.G viral packaging plasmids using polyjet in HEK293FT cells and viral supernatants were harvested after 48h. 293T cells were infected with lentiviral supernatants and selected by 1 μg/mL puromycin. After two days selection, media was replaced and selected pool cells were expanded in normal media for rescue experiments. HEK293T knock-out pool cells were infected with SHP2WT-mEGFP, SHP2E76K-mEGFP and SHP2R498L-mEGFP virus supernatants and sorted for further imaging. For re-expressing different ratios of WT/Y279C, HEK293T knock-out pool cells plated on 12-well plate were transiently transfected with 0.8 μg pMSCV-SHP2(Flag-WT+HA-Y279C) plasmids per well by polyjet.
Generation of tet-inducible stable cell line expressing SHP2Y279C-mEGFP
SHP2Y279C-mEGFP was cloned into pInducer20 vector and packaged into lentivirus with psPax2 and pMD2.G as described above. KYSE520 cells were infected with lentiviral supernatants and selected by 400 μg/ml G418. Cells were treated with different concentrations (100, 50, 10, 5, 2.5, 1, 0.5 and 0.25 ng/mL) of doxycycline for 3 days to induce the expression of SHP2Y279C-mEGFP and western blot was performed to valid the expression level.
Estimation of endogenous SHP2 protein concentrations
The concentration of endogenous SHP2 protein was measured following the previously reported protocol (Du, et al., 2018). Briefly, the quantification was based on the western blot densitometry analysis performed on cell lysates and purified SHP2WT protein. A549, HEK293T and KYSE520 cells were lysed in WB/IP lysis buffer with protease inhibitors and subjected to western blot with 3–0.063 μM purified SHP2WT protein. After densitometry analysis of western blot result by Fiji, we plotted band density against purified SHP2WT concentration. The measured volume of collected cells was calibrated with the maximal spherical stacking coefficient (0.74).
Single Molecule Fluorescence Resonance Energy Transfer (Sm-FRET)
Through an engineered tRNA/aminoacyl-tRNA synthetase system, SHP2E76A with two Azido-p-Phe incorporated at Q87/K266 was expressed and purified. The fluorophores (Cy3 as FRET donor and Cy5 as FRET acceptor) were further conjugated to Azido- SHP2E76A −87/266 via click reaction. Imaging was collected w/o SHP099 in imaging buffer and incubated for 10 min before image acquisition was started. Single-molecule imaging was performed on a prism-type total internal reflection fluorescence (TIRF) microscope equipped with a dual-laser excitation system (532 and 640 nm Crystal Laser) to excite Cy3 or Cy5. Fluorescence signals were collected by a water immersion objective lens (60X, NA: 1.2) and then passed through a notch filter to block out excitation beams. The emission signals from Cy3 and Cy5 were separated by a dichroic mirror and detected by the electron-multiplying charge-coupled device (EMCCD) camera (iXon 897; Andor Technology). Data were recorded with a time resolution of 200 ms as a stream of imaging frames and analyzed with scripts written in interactive data language to give fluorescence intensity time trajectories of individual molecules. An oxygen scavenging system consisting of 0.8 mg/ml glucose oxidase, 0.625% glucose, 3 mM Trolox and 0.03 mg/ml catalase was added in the imaging buffer to avoid photobleaching.
Preprocessing and analyses of single-channel current recordings were performed offline with smCamera software written in C++ (Microsoft) which was generously provided by Taekjip Ha (Johns Hopkins University). FRET efficiency, E, was calculated as EFRET = IA/(IA+ID), in which IA and ID are the donor and acceptor intensity after background subtraction. Cy3 and Cy5 channel were mapped using tetraSpeck fluorescent microsphere beads (Invitrogen, 0.1 μM) by mapping tool in smCamera. Traces extracted from the movies were selected based on the following acceptance criteria (Wang et al. 2016, Tuboyama et al. 2018): (1) no more than one bleaching step for both donor and acceptor fluorophores; (2) traces showing a clear anticorrelated pattern; (3) lifetimes of both donor and acceptor fluorophore longer than 8 s. All smFRET trajectories that met these criteria were used to calculate the apparent FRET efficiency and to generate population FRET histograms. Error bars in the histograms represent the s.e.m. from at least three independent movies. The bin size of all histograms was set as 0.02. To make sure real FRET transitions, all transitions were manually inspected based on anti-correlation of the donor and acceptor intensity signals. All the experiments were repeated at least three times and no significant difference was found between these replicates.
Circular Dichroism Measurements
The secondary structure of SHP2 and variants was measured by a Chirascan CD spectrometer (Applied Photophysics, UK) at the room temperature. PTP or PTPmut proteins were purified with His-tag and diluted into 4 μM in a buffer containing 25 mM Tris pH 8.0, 150 mM NaCl and 2 mM DTT. Spectra were recorded at 200–260 nm with a step size of 1 nm and a cell path length of 1mm. Each sample was scanned three times. Pro-Data Viewer is used to analyze the data. Secondary structural content of each protein was determined by analysis of the CD spectrum using BeStSel.
In vitro centrifugation-based phase separation assay
The purified SHP2WT-mEGFP and mutants (SHP2E76A, SHP2E76K, SHP2Y279C, SHP2R498L) were pre-cleared via high-speed centrifugation. Briefly, the two proteins of SHP2WT-mEGFP and each SHP2 mutant were mixed at a molar ratio of 1:1 at final concentration 8 μM and incubated at 37 °C overnight. Then, samples were centrifuged at 15000 rpm for 30 min at room temperature. Supernatant and pellet were separated into two tubes immediately after centrifugation. The pellet fraction was washed with the LLPS buffer and re-suspended with the same buffer to the equal volume as supernatant. Proteins from supernatant and pellet fraction were separated by 8% SDS-PAGE and the gel was stained with Coomassie blue.
In vitro SHP2 phosphatase assay
The surrogate substrate 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) (Invitrogen) was used to monitor the catalytic activity of SHP2 in a fluorescence assay format. The phosphatase reactions were performed at room temperature in 96-well black polystyrene plate, flat bottom (Costar) with a final volume of 100 μL. The assay buffer contains 60 mM HEPES pH 7.2, 75 mM NaCl, 75 mM KCl, 1 mM EDTA. The full-length wild type various mutations of SHP2 enzymes (5 ng/μL) were diluted with reaction buffer, then the substrate DiFMUP was added, and immediately Ex/Em at 358/455 was measured with a kinetic process for 20 min. The SHP2WT (5 nM) was co-incubated with of bisphosphorylated IRS-1 peptide (2.5 μM, sequence: LN(p-Y)IDLDLV(PEG8)LST(p-Y)ASINFQK-NH2) and the inhibitory compounds ( SHP099 or ET070, 0.001μM −10μM ) for 30 min at room temperature. The substrate DiFMUP was added and incubated for 30min at room temperature, and Ex/Em at 358/455 was measured. The SHP2E76A or SHP2E76K enzyme (5 μg/μL) was co-incubated with the compounds (SHP099 or ET070, 0.001μM −10μM) for 20min, then the substrate DiFMUP was added to the reaction, incubated for another 20 min, and Ex/Em at 358/455 was measured by a microplate reader (Envision, Perkin-Elmer). The IC50 data was derived from the binding curve, which was fitted by the responsed-variable slop equation by using the GraphPad Prism5 software.
For phosphatase assay conducted in droplet condition, the purified wild type, mutants or truncations of SHP2 at 8 μM was added to a LLPS buffer or solution buffer and incubated for 10 min at 37 °C. The enzymatic reaction was initiated by the addition of 20 mM pNPP(NEB) substrate and the OD405 of product was acquired every 10s (shakes 10 seconds before each reading) by the microplate reader (Envision, Perkin-Elmer). Images to examine the phosphatase activity were collected using substrate DiFMUP. After the formation of phase-separated droplets, 2 μL of each sample was pipetted onto a coverslip. Images were collected once the substrate was added.
Immunofluorescence cell staining
Cells were seeded in 24-well plate with a coverslip in each well and grown overnight. After 4% PFA fixation for 15minutes at room temperature, cells were treated with 0.4% Triton X-100 and blocked with 5% BSA. Diluted SHP2 antibody (Santa Cruz, sc-7384) was applied to the coverslip and incubated overnight at 4°C. After PBS washing, corresponding fluorochrome-labeled secondary antibody was applied at room temperature for 1h and further PBS washed. After mounting with DAPI, examined the slides under the fluorescence microscope.
Western blot
Whole cell lysates were prepared in RIPA lysis buffer (P0013B, Beyotime) or 2×loading buffer, then separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and subsequently transferred onto a PVDF membrane (Millipore, Bedford, MA, USA). Membrane was blocked with 5% (m/v) BSA for 1 hr at room temperature and then incubated with primary antibodies at 4 °C overnight and second antibodies at room temperature. The following antibodies were used: pERK (CST#4370S), ERK (CST#9102S), SHP2 (CST#3397T), HA (CST#2367S), FLAG(Sigma# F1804) GAPDH (CST #2118S) and α-tubulin (abcam# ab52866), pMEK1/2(S217/221) (CST#9154), MEK1/2(CST#9122).
Cell survival assay
KYSE520 and MV4;11 cells (3000 cells per well) were seeded in 96-well plate culture overnight and treated with SHP099 or ET070 at concentrations varying from 10 μM to 0.0005 μM for 96 h. 50 μL CellTiter-Glo reagent (Promega) was added and the luminescent signal was determined according to the supplier’s instruction. The percentage of inhibition was normalized by the DMSO vehicle control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Images were analyzed with Fiji. All data are presented as the mean ± standard error of mean (s.e.m.) or standard deviation (s.d.) from independent determinations, and statistical analyses were done using the software Graphpad Prism (GraphPad Software, Inc,; La Jolla, CA, USA).Differences of means were tested for statistical significance with unpaired two-tailed Student’s t test.*p<0.05; **p<0.01; ***p<0.001.
Supplementary Material
Fig. S1. Disease-associated mutations of SHP2 form discrete puncta in cells. Related to Figure. 1. (A) Human disease-associated mutations in SHP2. NS/JMML mutations were shown in blue, and NS-ML mutations in green. (B) The immunoblot analysis of the indicated proteins in KYSE520 cells stably expressing SHP2-mEGFP (SHP2WT, SHP2D61G, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L). (C) Quantification of puncta size for SHP2WT and SHP2mut-mEGFP expressing KYSE520 cells. N=150 puncta. ***p<0.001. (D) Representative images of SHP2WT and SHP2mut (SHP2D61G, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L) -mEGFP in KYSE520 cells collected from high content image experiments. Scale bar, 20 μm. (E) The correlation between puncta size and number of SHP2WT and SHP2mut-mEGFP expressing KYSE520 cells. (F) Live imaging of SHP2WT and SHP2mut (SHP2D61G, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L) -mEGFP in HEK293T cells. Scale bar, 5 μm. (G) Live imaging of SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L)-mEGFP in A549 cells. Scale bar, 10 μm. (H) Live imaging of SHP2E76A-mEGFP in NCI-H1299, SF268 and HUVEC cells. Scale bar,10 μm. (I) The immunoblot analysis of the indicated proteins in A549 cells stably expressing SHP2-mEGFP (SHP2WT, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L). (J) The immunoblot analysis of the indicated proteins in KYSE520 cells stably expressing SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L)-mScarlet (K) Live imaging of SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L)-mScarlet in KYSE520 cells. Scale bar, 10 μm. (L) The immunoblot analysis of the indicated proteins in HEK293T knock-out cells stably re-expressing SHP2-mEGFP (SHP2WT, SHP2E76K and SHP2R498L). (M) Live cell images of HEK293T knock-out cells stably re-expressing SHP2-mEGFP (SHP2WT, SHP2E76K and SHP2R498L). Scale bar, 10 μm. (N) Immunofluorescence (IF) imaging of SHP2 in KYSE520, H661 and CCF-STTG1 cells. Scale bar, 10 μm. (O) Immunofluorescence imaging of SHP2 in MEF cells derived from Ptpn11D61G/+ and control mice. Scale bar, 10 μm. (P)Immunofluorescence imaging of SHP2 in mesenchymal stem cells (MSCs) derived from Ptpn11E76K/+ and control mice. Scale bar, 10 μm.
Fig. S2. The puncta of SHP2mut exhibited liquid-like features in cells. Related to Figure. 2. (A) Fusion of two SHP2E76A-mEGFP puncta was shown. Scale bar, 10 μm. (B) Fusion of two SHP2Y279C-mEGFP puncta was shown. Scale bar, 10 μm. (C) Representative images of the FRAP experiment with SHP2E76A-mEGFP in KYSE520 cells. Scale bar, 2 μm. (D) Quantification of FRAP data for SHP2E76A –mEGFP puncta. Data are plotted as means ± SEM, (n=3 experiments). (E) Representative images of the FRAP experiment with SHP2Y279C-mEGFP in KYSE520 cells. Scale bar, 2 μm. (F) Quantification of FRAP data for SHP2Y279C–mEGFP puncta. Data are plotted as means ± SEM, (n=3 experiments). (G) Schematic of SHP2-mEGFP knock-in strategy. (H) Live cell imaging of endogenous tagged SHP2-mEGFP in H661 cells. Scale bar,10 μm.
Fig. S3. Disease-associated mutations of SHP2 promote SHP2 LLPS in vitro. Related to Figure. 3. (A) SDS-PAGE results of purified recombinant SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2D61G, SHP2Y279C, SHP2R498L, SHP2G464A, SHP2T468M and SHP2Q506P) protein. (B) SDS-PAGE results of purified recombinant SHP2WT, SHP2mut-mEGFP (SHP2E76A, SHP2E76K, SHP2D61G, SHP2Y279C, SHP2R498L, SHP2G464A, SHP2T468M, SHP2Q506P-mEGFP) and SHP2mut-mScarlet (SHPE76A, SHP2E76K, SHP2Y279C, SHP2R498L-mScarlet) protein. (C) Fusion event of SHP2R498L and SHP2E76A–mEGFP droplet were shown. Scale bar, 2.5 μm. (D) Quantification of FRAP data for SHP2R498L –mEGFP droplet. Data are plotted as means ± SEM, (n=3 experiments). (E) Quantification of FRAP data for SHP2E76A–mEGFP droplet. Data are plotted as means ± SEM, (n=3 experiments). (F) Quantification result of endogenous SHP2 protein concentration based on immunoblot densitometry analysis performed on cell lysates and purified SHP2(WT) protein.
Fig. S4. Open conformation promotes SHP2 LLPS. Related to Figure. 4. (A) Representative smFRET trajectories for E76A87/266 molecules (left) and E76A87/266 with SHP099(right). Real-time donor (green) and acceptor (red) intensity traces and FRET trace (blue) obtained from individual E76A-87/266 molecules. F, fluorescence; a.u., arbitrary units. (B) Structure of ET070. (C) ET070 more potently inhibited SHP2WT (stimulated with p-IRS-1 peptide), SHP2E76A and SHP2E76K than SHP099. Data presents as means ± SEM, (n=2 experiments). (D) Selectivity of ET070 against a panel of phosphatases. (E) Effects of SHP099 and ET070 on the cell viability of SHP2-dependent cancer cell (KYSE520 and MV4;11 cells) Data presents as means ± SEM, (n=2 experiments). (F) Immunofluorescence imaging of SHP2 in MEF cells derived from Ptpn11D61G/+ mice treated w/o 10μM ET070. Scale bar, 10 μm. Quantification result (means ± SEM, N = 104 cells) was shown. ***p<0.001. (G) Immunofluorescence imaging of SHP2 in mesenchymal stem cells (MSCs) derived from Ptpn11E76K/+ mice treated w/o 10μM ET070. Scale bar, 10 μm. Quantification result (means ± SEM, N = 149 cells) was shown.***p<0.001. (H) 6μM SHP2WT were stimulated w/o 10μM bis-P peptide (pY1172-PEG8-pY1222) and the droplet turbidity was assessed after droplet formation. Data are plotted as mean±s.e.m. (n=3 experiments) **p<0.01 (I) Live cell images of SHP2WT-mEGFP in KYSE520 cells stimulated with bFGF and EGF. Scale bar, 10μm.
Fig. S5. PTP domain drives SHP2 LLPS. Related to Figure. 5. (A) SDS-PAGE results of purified recombinant full-length SHP2 (FL-SHP2) and truncated SHP2 (SHP2ΔC, SHP2-PTP, N/C-SH2). (B) Fusion event of SHP2-PTP droplet was shown. Scale bar, 5 μm. (C) SHP2WT, SHP2E76A, SHP2R498L and SHP2-PTP droplets were treated with different concentrations of SHP099. Quantification results of droplet turbidity OD600 were shown. Data are plotted as means ± SEM, (n = 5 experiments). **p<0.01; ***p<0.001.Representative images of SHP2-PTP droplets w/o SHP099 (right panel). Scale bar, 5 μm.
Fig. S6. LLPS of SHP2-PTP is mediated by electrostatic interactions. Related to Figure. 5. (A) SDS-PAGE results of purified recombinant PTP and 17 charge-mutant PTP proteins. (B) Circular dichroism measurements result of PTP and 17 PTPmut proteins. (C) Droplet turbidity OD600 of purified recombinant PTP and 17 PTPmut proteins. Data are plotted as means ± SEM, (n=3 experiments). *p<0.05; **p<0.01; ***p<0.001.(D) Schematic representation of the two negatively charged patches and two positively charged patches on the surface of SHP2-PTP. (E) Enzymatic activity of purified recombinant PTP and PTPR362E/K364E proteins. Phosphatase assays were conducted using pNPP as substrate. Data are plotted as means ± SEM, (n=3 experiments). (F) Enzymatic activity of purified recombinant full-length SHP2E76A, SHP2E76A/R362E/K364E, SHP2E76K, SHP2E76K/R362E/K364E proteins. Phosphatase assays were conducted using DiFMUP as substrate. Data are plotted as means ± SEM (n=2 experiments). (G) Conformation transition of SHP2 protein (PDB:4DGP). In basal state, SHP2 adopts a closed auto-inhibited conformation in which the R362/K364-containing positively charged surface (362/364PCS) of PTP is partially masked by N-SH2. However, once SHP2 is activated by either activator or mutation, the N-SH2 is proposed to be detached from PTP which may cause 362/364PCS fully accessible for driving SHP2 LLPS.
Figure S7. LLPS of NS-ML SHP2 mutants recruit and activate SHP2WT to promote ERK1/2 activation. Related to Figure. 7. (A) HEK293T SHP2 knock out cells were transiently transfected with the indicated amount of SHP2WT and SHP2Y297C plasmids. The immunoblots (left) and the densitometry analysis (right) of pERK levels. (means ± SEM, N = 4 experiments) (B) Immunoblot of the indicated proteins in tet-inducible SHP2Y279C-mEGFP KYSE520 cells treated with various concentrations of doxycycline (100, 50, 10, 5, 2.5, 1, 0.5, 0.25 ng/mL). (C) Droplet turbidity of SHP2WT, SHP2R498L, SHP2WT/SHP2R498L condensates. (means ± SEM, N = 3 experiments). (D, E) The distribution of SHP2WT and SHP2R498L in solution and condensed pellets of R498L/WT mixed condensates(D). Quantification is performed by analyzing the SDS-PAGE result of in vitro centrifugation based phase separation assay for SHP2WT-mEGFP and NS-ML mutant SHP2 mixtures (E). (F) Quantification results of FRAP data for SHP2WT-mEGFP distributed in SHP2R498L-mScarlet droplets. (means ± SEM, N = 3 experiments). (G) Living images of KYSE520 cells co-expressed with SHP2WT-mEGFP and SHP2Y279C-mScarlet. SHP2WT-mEGFP formed puncta co-localized with SHP2Y279C-mScarlet puncta. Scale bar, 10 μm.
Movie S1. Related to Figure.2.
Fusion of two SHP2R498L-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S2. Related to Figure.2.
Fusion of two SHP2E76A-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S3. Related to Figure.2.
Time-lapse imaging video of FRAP experiments with SHP2R498L-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S4. Related to Figure.2.
Time-lapse imaging video of FRAP experiments with SHP2Y279C-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S5. Related to Figure.2.
Time-lapse imaging video of FRAP experiments with SHP2E76K-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S6. Related to Figure.4.
Time-lapse imaging video of SHP2R498L-mEGFP puncta in KYSE520 cells treated with ET070. Scale bar, 10μm.
Movie S7. Related to Figure.4.
Time-lapse imaging video of SHP2WT-mEGFP KYSE520 cells stimulated with FGF. Scale bar, 10μm.
Highlights.
Disease-associated mutations endow SHP2 liquid-liquid phase separation capability.
SHP2 LLPS is driven by electrostatic interactions mediated by PTP domain.
SHP2 allosteric inhibitors block SHP2 LLPS by locking SHP2 in closed conformation.
Mutant SHP2 can recruit and activate WT SHP2 in LLPS to promote MAPK activation.
ACKNOWLEDGMENTS
We thank J. Yuan for critical reading of the manuscript and P. Li for helpful discussion. We thank J. Hu and members for help with fluorescent protein constructs, imaging and sorting, C. Zhang and W. Yang for tet-on plasmids. We thank Z. Zhang for the protein purification platform, Y. Geng for assistance in microscopy imaging, G. Chen, K. Liu, Q. Li and Y. Wang for providing mesenchymal stem cells.
Funding: This work was supported by the National Key R&D Program of China (2016YFA0501900 to J. Zhu and C. Liu), the National Natural Science Foundation of China (21532002, 21877123 to J. Zhu, 81803560 to L. Sun and 31872716, 91853113 to C. Liu,), the Shanghai Municipal Science and Technology Major Project (Grant No. 2019SHZDZX02) to J. Zhu and C. Liu, the Science and Technology Commission of Shanghai Municipality (18JC1420500) to C. Liu, and the joint postdoc program with Roche Innovation Center. Z.Y.Z. is supported by NIH RO1 CA207288.
Footnotes
Competing interests: J.Z. is a co-founder of Etern Biopharma Co. Ltd. and a member of its scientific advisory board. Q.Z. is a consultant to Etern Biopharma Co. Ltd. L.D. is an employee of Etern Biopharma Co. Ltd. H.C.S. is an employee of Roche. Etern Biopharma Co. Ltd. holds the patent WO-2020094018 which includes ET070 as one exemplified compound. The other authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND NOTES
- Alberti S, and Dormann D (2019). Liquid–Liquid Phase Separation in Disease. Annu. Rev. Genet 53,3.1–3.24 [DOI] [PubMed] [Google Scholar]
- Alberti S, Gladfelter A, and Mittag T (2019). Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA and Neel BG(2004). Mouse model of Noonan syndrome reveals cell type- and gene dosage- dependent effects of Ptpn11 mutation. Nat. Med 10, 849–857. [DOI] [PubMed] [Google Scholar]
- Böhmer F, Szedlacsek S, Tabernero L, Ostman A, and Den HJ (2013). Protein tyrosine phosphatase structure: function relationships in regulation and pathogenesis. Febs J. 280, 413–431. [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafé L, Superti-Furga A, Ikegawa S, Cormier-Daire V, Bovée JV, and Pansuriya TC (2011). Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet. 7, e1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Vergara X, Sevilla A, DʹSouza SL, Ang Y-S, Schaniel C, Lee D-F, Yang L, Kaplan AD, Adler ED, Rozov R, Ge YC, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, and Lemischka IR (2011). Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465, 808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH-T, Chen Z, and Cooke VG (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148. [DOI] [PubMed] [Google Scholar]
- Cheng YP, Chiu HY, Hsiao TL, Hsiao CH, Lin CC, and Liao YH (2013). Scalp melanoma in a woman with LEOPARD syndrome: Possible implication of PTPN11 signaling in melanoma pathogenesis. J. Am. Acad. Dermatol 69, e186–e187. [DOI] [PubMed] [Google Scholar]
- Colmant C, Franck D, Marot L, Matthijs G, Sznajer Y, Blomme S, and Tromme I (2018). Patient with confirmed LEOPARD syndrome developing multiple melanoma. Dermat. Pract. Concept 8, 59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance M, Montagner A, Salles JP, Yart A, and Raynal P (2008). The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal 20, 453–459. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, and Dallapiccola B (2002). Grouping of Multiple-Lentigines/LEOPARD and Noonan Syndromes on the PTPN11 Gene. Am. J. Hum. Genet 71, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, Huang G, Zhou MX, Broxmeyer HE, Scadden DT and Qu C-K (2016) Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 539: 304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361,704–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edouard T, Montagner A, Dance M, Conte F, Yart A, Parfait B, Tauber M, Salles J, and Raynal P (2007). How do Shp2 mutations that oppositely influence its biochemical activity result in syndromes with overlapping symptoms? Cell. Mol. Life Sci 64, 1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann TM, Jahnel M, Pozniakovsky A, Mahamid J, Holehouse AS, Nüske E, Richter D, Baumeister W, Grill SW, and Pappu RV (2018). Phase separation of a yeast prion protein promotes cellular fitness. Science 359, eaao5654. [DOI] [PubMed] [Google Scholar]
- Grosskopf S, Eckert C, Arkona C, Radetzki S, Böhm K, Heinemann U, Wolber G, Kries J-P, Birchmeier W, and Rademann J (2015). Selective Inhibitors of the Protein Tyrosine Phosphatase SHP2 Block Cellular Motility and Growth of Cancer Cells in vitro and in vivo. ChemMedChem 10, 815–826. [DOI] [PubMed] [Google Scholar]
- He R-J, Yu Z-H, Zhang R-Y, and Zhang Z-Y (2014). Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmaco. Sin 35, 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, and Shoelson SE (1998). Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450. [DOI] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, and Madl T (2018). Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 173, 706–719.e713. [DOI] [PubMed] [Google Scholar]
- Huang WY, Alvarez S, Kondo Y, Lee YK, Chung JK, Lam HYM, Biswas KH, Kuriyan J, and Groves JT (2019). A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 363, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontaridis MI, Swanson KD, David FS, Barford D, and Neel BG (2006). PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem 281, 6785–6792. [DOI] [PubMed] [Google Scholar]
- LaRochelle JR, Fodor M, Vemulapalli V, Mohseni M, Wang P, Stams T, LaMarche MJ, Chopra R, Acker MG, and Blacklow SC (2018). Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun 9, 4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux D, Kratz C, and Sauerbrey A (2008). Common acute lymphoblastic leukemia in a girl with genetically confirmed LEOPARD syndrome. J. Pediatr. Hematol. Oncol 30, 602–604. [DOI] [PubMed] [Google Scholar]
- Li B, Zeng CX, and Dong YZ (2018). Design and assessment of engineered CRISPR–Cpf1and its use for genome editing. Nat. Protoc 13, 899–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng H-C, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, and Banani SF (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zheng H, Li X, Wang S, Meyerson HJ, Yang W, Neel BG, and Qu C-K (2016). Gain-of-function mutations of Ptpn11 (Shp2) cause aberrant mitosis and increase susceptibility to DNA damage-induced malignancies. Proc. Natl. Acad. Sci. USA 113, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merks JHM, Caron HN, and Hennekam RCM (2005). High incidence of malformation syndromes in a series of 1,073 children with cancer. Am. J. Med. Genet. A 134A, 132–143. [DOI] [PubMed] [Google Scholar]
- Miyamoto D, Miyamoto M, Takahashi A, Yomogita Y, Higashi H, Kondo S, and Hatakeyama M (2008). Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene 27, 3508. [DOI] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GK, Rees E, Miyashita A, Costa A, Dodd R, Chan FS, and Michel C (2015). ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 88, 678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, and Forman-Kay JD (2015). Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 57, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi K, Zhang H, Gault WJ, Wang CJ, Tan CC, In-Kyong K, Huiwen Y, Tabassum R, Natalie P, and Marco T (2009). Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum. Mol. Genet 18, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Wang X, Romanov V, Hutchinson A, Lin A, Ruzanov M, Battaile KP, Pai EF, Neel BG, and Chirgadze NY (2014). Structural insights into Noonan/LEOPARD syndrome-related mutants of protein-tyrosine phosphatase SHP2 (PTPN11). BMC Struct. Biol 14, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seishima M, Shibuya YM, Arakawa C, Yoshida R, and Ogata T (2010). Malignant melanoma in a woman with LEOPARD syndrome: identification of a germline PTPN11 mutation and a somatic BRAF mutation. Brit. J. Dermat 157, 1297–1299. [DOI] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382. [DOI] [PubMed] [Google Scholar]
- Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, and Vale RD (2016). Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadashi A and Jeffrey S (2010). Crowding and hydrodynamic interactions likely dominate in vivo macromo lecular motion. Proc. Natl. Acad. Sci. USA 107, 18457–18462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajan M, Paccoud R, Branka S, Edouard T, and Yart A (2018). The RASopathy family: consequences of germline activation of the Ras/MAPK pathway. Endocr. Rev 39, 676–700. [DOI] [PubMed] [Google Scholar]
- Tajan M, Serra ADR, Valet P, Edouard T, and Yart A (2015). SHP2 sails from physiology to pathology. Eur. J. Med. Genet 58, 509–525. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Lavarone L, Spinelli M, Palmi C, Carta C, Pession A, Aricò M, Masera G, Basso G, Sorcini M, Gelb BD, and Biondi A (2004). Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood 104, 307–313. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, and Gelb BD (2005). Noonan syndrome and related disorders: genetics and pathogenesis. Annu. Rev. Genom. Hum. Genet 6, 45–68. [DOI] [PubMed] [Google Scholar]
- Tomoki N, James G, Ronald P, and Jeffrey R (2009). Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc. Natl. Acad. Sci. USA 106, 15436–15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK (2006). Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol 7, 833–846. [DOI] [PubMed] [Google Scholar]
- Tsuboyama K, Tadakuma H, and Tomari Y(2018). Conformational activation of argonaute by distinct yet coordinated actions of the Hsp70 and Hsp90 chaperone systems. Mol. cell, 70, 722–729. [DOI] [PubMed] [Google Scholar]
- Uçar C, Çalýþkan Ü, Martini S, and Heinritz W (2006). Acute myelomonocytic leukemia in a boy with LEOPARD syndrome (PTPN11 gene mutation positive). J. Pediatr. Hematol. oncol 28, 123–125. [DOI] [PubMed] [Google Scholar]
- Uversky VN (2017). Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struc. Biol 44, 18–30. [DOI] [PubMed] [Google Scholar]
- Wang SW, Vafabakhsh R, Borschel WF, Ha T, and Nichols CG(2016). Structural dynamics of potassium-channel gating revealed by single-molecule FRET. Nat. Struct. Mol. Biol, 23, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL and Qu CK(2011) Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J. Exp. Med. 208: 1977–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z-H, Zhang R-Y, Walls CD, Chen L, Zhang S, Wu L, Liu S, and Zhang Z-Y (2014). Molecular basis of gain-of-function LEOPARD syndrome-associated SHP2 mutations. Biochemistry 53, 4136–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZH, Xu J, Walls CD, Chen L, Zhang S, Zhang R, Wu L, Wang L, Liu S, and Zhang ZY (2013). Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J. Biol. Chem 288, 10472–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, Oost J, Regev A, Koonin EV and Zhang F(2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Alter S, and Qu C-K (2009). SHP-2 tyrosine phosphatase in human diseases. Int. J. Clin. Exp. Med 2, 17–25. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Disease-associated mutations of SHP2 form discrete puncta in cells. Related to Figure. 1. (A) Human disease-associated mutations in SHP2. NS/JMML mutations were shown in blue, and NS-ML mutations in green. (B) The immunoblot analysis of the indicated proteins in KYSE520 cells stably expressing SHP2-mEGFP (SHP2WT, SHP2D61G, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L). (C) Quantification of puncta size for SHP2WT and SHP2mut-mEGFP expressing KYSE520 cells. N=150 puncta. ***p<0.001. (D) Representative images of SHP2WT and SHP2mut (SHP2D61G, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L) -mEGFP in KYSE520 cells collected from high content image experiments. Scale bar, 20 μm. (E) The correlation between puncta size and number of SHP2WT and SHP2mut-mEGFP expressing KYSE520 cells. (F) Live imaging of SHP2WT and SHP2mut (SHP2D61G, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L) -mEGFP in HEK293T cells. Scale bar, 5 μm. (G) Live imaging of SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L)-mEGFP in A549 cells. Scale bar, 10 μm. (H) Live imaging of SHP2E76A-mEGFP in NCI-H1299, SF268 and HUVEC cells. Scale bar,10 μm. (I) The immunoblot analysis of the indicated proteins in A549 cells stably expressing SHP2-mEGFP (SHP2WT, SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L). (J) The immunoblot analysis of the indicated proteins in KYSE520 cells stably expressing SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L)-mScarlet (K) Live imaging of SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2Y279C and SHP2R498L)-mScarlet in KYSE520 cells. Scale bar, 10 μm. (L) The immunoblot analysis of the indicated proteins in HEK293T knock-out cells stably re-expressing SHP2-mEGFP (SHP2WT, SHP2E76K and SHP2R498L). (M) Live cell images of HEK293T knock-out cells stably re-expressing SHP2-mEGFP (SHP2WT, SHP2E76K and SHP2R498L). Scale bar, 10 μm. (N) Immunofluorescence (IF) imaging of SHP2 in KYSE520, H661 and CCF-STTG1 cells. Scale bar, 10 μm. (O) Immunofluorescence imaging of SHP2 in MEF cells derived from Ptpn11D61G/+ and control mice. Scale bar, 10 μm. (P)Immunofluorescence imaging of SHP2 in mesenchymal stem cells (MSCs) derived from Ptpn11E76K/+ and control mice. Scale bar, 10 μm.
Fig. S2. The puncta of SHP2mut exhibited liquid-like features in cells. Related to Figure. 2. (A) Fusion of two SHP2E76A-mEGFP puncta was shown. Scale bar, 10 μm. (B) Fusion of two SHP2Y279C-mEGFP puncta was shown. Scale bar, 10 μm. (C) Representative images of the FRAP experiment with SHP2E76A-mEGFP in KYSE520 cells. Scale bar, 2 μm. (D) Quantification of FRAP data for SHP2E76A –mEGFP puncta. Data are plotted as means ± SEM, (n=3 experiments). (E) Representative images of the FRAP experiment with SHP2Y279C-mEGFP in KYSE520 cells. Scale bar, 2 μm. (F) Quantification of FRAP data for SHP2Y279C–mEGFP puncta. Data are plotted as means ± SEM, (n=3 experiments). (G) Schematic of SHP2-mEGFP knock-in strategy. (H) Live cell imaging of endogenous tagged SHP2-mEGFP in H661 cells. Scale bar,10 μm.
Fig. S3. Disease-associated mutations of SHP2 promote SHP2 LLPS in vitro. Related to Figure. 3. (A) SDS-PAGE results of purified recombinant SHP2WT and SHP2mut (SHP2E76A, SHP2E76K, SHP2D61G, SHP2Y279C, SHP2R498L, SHP2G464A, SHP2T468M and SHP2Q506P) protein. (B) SDS-PAGE results of purified recombinant SHP2WT, SHP2mut-mEGFP (SHP2E76A, SHP2E76K, SHP2D61G, SHP2Y279C, SHP2R498L, SHP2G464A, SHP2T468M, SHP2Q506P-mEGFP) and SHP2mut-mScarlet (SHPE76A, SHP2E76K, SHP2Y279C, SHP2R498L-mScarlet) protein. (C) Fusion event of SHP2R498L and SHP2E76A–mEGFP droplet were shown. Scale bar, 2.5 μm. (D) Quantification of FRAP data for SHP2R498L –mEGFP droplet. Data are plotted as means ± SEM, (n=3 experiments). (E) Quantification of FRAP data for SHP2E76A–mEGFP droplet. Data are plotted as means ± SEM, (n=3 experiments). (F) Quantification result of endogenous SHP2 protein concentration based on immunoblot densitometry analysis performed on cell lysates and purified SHP2(WT) protein.
Fig. S4. Open conformation promotes SHP2 LLPS. Related to Figure. 4. (A) Representative smFRET trajectories for E76A87/266 molecules (left) and E76A87/266 with SHP099(right). Real-time donor (green) and acceptor (red) intensity traces and FRET trace (blue) obtained from individual E76A-87/266 molecules. F, fluorescence; a.u., arbitrary units. (B) Structure of ET070. (C) ET070 more potently inhibited SHP2WT (stimulated with p-IRS-1 peptide), SHP2E76A and SHP2E76K than SHP099. Data presents as means ± SEM, (n=2 experiments). (D) Selectivity of ET070 against a panel of phosphatases. (E) Effects of SHP099 and ET070 on the cell viability of SHP2-dependent cancer cell (KYSE520 and MV4;11 cells) Data presents as means ± SEM, (n=2 experiments). (F) Immunofluorescence imaging of SHP2 in MEF cells derived from Ptpn11D61G/+ mice treated w/o 10μM ET070. Scale bar, 10 μm. Quantification result (means ± SEM, N = 104 cells) was shown. ***p<0.001. (G) Immunofluorescence imaging of SHP2 in mesenchymal stem cells (MSCs) derived from Ptpn11E76K/+ mice treated w/o 10μM ET070. Scale bar, 10 μm. Quantification result (means ± SEM, N = 149 cells) was shown.***p<0.001. (H) 6μM SHP2WT were stimulated w/o 10μM bis-P peptide (pY1172-PEG8-pY1222) and the droplet turbidity was assessed after droplet formation. Data are plotted as mean±s.e.m. (n=3 experiments) **p<0.01 (I) Live cell images of SHP2WT-mEGFP in KYSE520 cells stimulated with bFGF and EGF. Scale bar, 10μm.
Fig. S5. PTP domain drives SHP2 LLPS. Related to Figure. 5. (A) SDS-PAGE results of purified recombinant full-length SHP2 (FL-SHP2) and truncated SHP2 (SHP2ΔC, SHP2-PTP, N/C-SH2). (B) Fusion event of SHP2-PTP droplet was shown. Scale bar, 5 μm. (C) SHP2WT, SHP2E76A, SHP2R498L and SHP2-PTP droplets were treated with different concentrations of SHP099. Quantification results of droplet turbidity OD600 were shown. Data are plotted as means ± SEM, (n = 5 experiments). **p<0.01; ***p<0.001.Representative images of SHP2-PTP droplets w/o SHP099 (right panel). Scale bar, 5 μm.
Fig. S6. LLPS of SHP2-PTP is mediated by electrostatic interactions. Related to Figure. 5. (A) SDS-PAGE results of purified recombinant PTP and 17 charge-mutant PTP proteins. (B) Circular dichroism measurements result of PTP and 17 PTPmut proteins. (C) Droplet turbidity OD600 of purified recombinant PTP and 17 PTPmut proteins. Data are plotted as means ± SEM, (n=3 experiments). *p<0.05; **p<0.01; ***p<0.001.(D) Schematic representation of the two negatively charged patches and two positively charged patches on the surface of SHP2-PTP. (E) Enzymatic activity of purified recombinant PTP and PTPR362E/K364E proteins. Phosphatase assays were conducted using pNPP as substrate. Data are plotted as means ± SEM, (n=3 experiments). (F) Enzymatic activity of purified recombinant full-length SHP2E76A, SHP2E76A/R362E/K364E, SHP2E76K, SHP2E76K/R362E/K364E proteins. Phosphatase assays were conducted using DiFMUP as substrate. Data are plotted as means ± SEM (n=2 experiments). (G) Conformation transition of SHP2 protein (PDB:4DGP). In basal state, SHP2 adopts a closed auto-inhibited conformation in which the R362/K364-containing positively charged surface (362/364PCS) of PTP is partially masked by N-SH2. However, once SHP2 is activated by either activator or mutation, the N-SH2 is proposed to be detached from PTP which may cause 362/364PCS fully accessible for driving SHP2 LLPS.
Figure S7. LLPS of NS-ML SHP2 mutants recruit and activate SHP2WT to promote ERK1/2 activation. Related to Figure. 7. (A) HEK293T SHP2 knock out cells were transiently transfected with the indicated amount of SHP2WT and SHP2Y297C plasmids. The immunoblots (left) and the densitometry analysis (right) of pERK levels. (means ± SEM, N = 4 experiments) (B) Immunoblot of the indicated proteins in tet-inducible SHP2Y279C-mEGFP KYSE520 cells treated with various concentrations of doxycycline (100, 50, 10, 5, 2.5, 1, 0.5, 0.25 ng/mL). (C) Droplet turbidity of SHP2WT, SHP2R498L, SHP2WT/SHP2R498L condensates. (means ± SEM, N = 3 experiments). (D, E) The distribution of SHP2WT and SHP2R498L in solution and condensed pellets of R498L/WT mixed condensates(D). Quantification is performed by analyzing the SDS-PAGE result of in vitro centrifugation based phase separation assay for SHP2WT-mEGFP and NS-ML mutant SHP2 mixtures (E). (F) Quantification results of FRAP data for SHP2WT-mEGFP distributed in SHP2R498L-mScarlet droplets. (means ± SEM, N = 3 experiments). (G) Living images of KYSE520 cells co-expressed with SHP2WT-mEGFP and SHP2Y279C-mScarlet. SHP2WT-mEGFP formed puncta co-localized with SHP2Y279C-mScarlet puncta. Scale bar, 10 μm.
Movie S1. Related to Figure.2.
Fusion of two SHP2R498L-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S2. Related to Figure.2.
Fusion of two SHP2E76A-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S3. Related to Figure.2.
Time-lapse imaging video of FRAP experiments with SHP2R498L-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S4. Related to Figure.2.
Time-lapse imaging video of FRAP experiments with SHP2Y279C-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S5. Related to Figure.2.
Time-lapse imaging video of FRAP experiments with SHP2E76K-mEGFP puncta in KYSE520 cells. Scale bar, 10μm.
Movie S6. Related to Figure.4.
Time-lapse imaging video of SHP2R498L-mEGFP puncta in KYSE520 cells treated with ET070. Scale bar, 10μm.
Movie S7. Related to Figure.4.
Time-lapse imaging video of SHP2WT-mEGFP KYSE520 cells stimulated with FGF. Scale bar, 10μm.
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. No unique code was generated in this study.