

Abstract
The dimeric diketopiperazine (DKPs) alkaloids are a diverse family of natural products (NPs) whose unique structural architectures and biological activities have inspired the development of new synthetic methodology to access these molecules. However, catalyst-controlled methods that enable the selective formation of constitutional and stereoisomeric dimers from a single monomer are lacking. To resolve this long-standing synthetic challenge, we sought to characterize the biosynthetic enzymes that assemble these NPs for application in biocatalytic syntheses. Genome mining enabled identification of the cytochrome P450, NzeB (derived from Streptomyces sp. NRRL F-5053), which catalyzes both intermolecular carbon-carbon (C–C) and carbon-nitrogen (C–N) bond formation, generating all currently known DKP dimer scaffolds isolated from bacterial sources. To identify the molecular basis for the flexible site-, stereo-, and chemoselectivity of NzeB, we obtained high-resolution crystal structures (1.5Å) of the protein in complex with native and non-native substrates. This, to our knowledge, represents the first crystal structure of an oxidase catalyzing direct, intermolecular C–H amination. Site-directed mutagenesis was employed to assess the role individual active site residues play in guiding selective DKP dimerization. Finally, computational approaches were employed to evaluate plausible mechanisms regarding NzeB function and its ability to catalyze both C–C and C–N bond formation. These results provide a structural and computational rationale for the catalytic versatility of NzeB, as well as new insights into variables that control selectivity of CYP450 diketopiperazine dimerases.
Keywords: cytochrome P450, diketopiperazine, diketopiperazine dimers, natural product biosynthesis, C–H functionalization
Introduction
The tryptophan-linked dimeric diketopiperazine (DKP) alkaloids are a family of over 50 natural products isolated from bacteria, fungi, and plants.1–4 Their dimerization modes encompass an array of stereo- and regiochemical connectivities, and potent biological activity of these natural products elicited the development and application of effective methods for their total synthesis.3–5 In particular, biomimetic strategies enabled access to C2-symmetrical dimers, such as (−)-ditryptophenaline (3, Figure 1), wherein two identical monomers are coupled at a late-stage resulting in highly convergent and efficient syntheses.6, 7 However, the chemical synthesis of structurally challenging non-symmetrical DKP dimers such as (+)-naseseazines A (1) and B (2),8, 9 (−)-naseseazine C (4),10, 11 and the related alkaloid (−)-aspergilazine A (5),10, 12 continues to inspire the development of new synthetic routes.4, 9, 13–16 The synthesis of structurally complex non-symmetrical dimeric DKPs requires independent synthesis of appropriately substituted precursors to allow selective formation of a key C–C or C–N bond that adjoin the two subunits.9, 17, 18 Seeking to develop an efficient approach towards DKP dimer assembly, we envisioned a process wherein the site- and stereochemical outcome of dimerization would be dictated by the biocatalyst, and a single DKP monomer could be dimerized selectively to a variety of site- and stereo-isomeric products.
Figure 1.
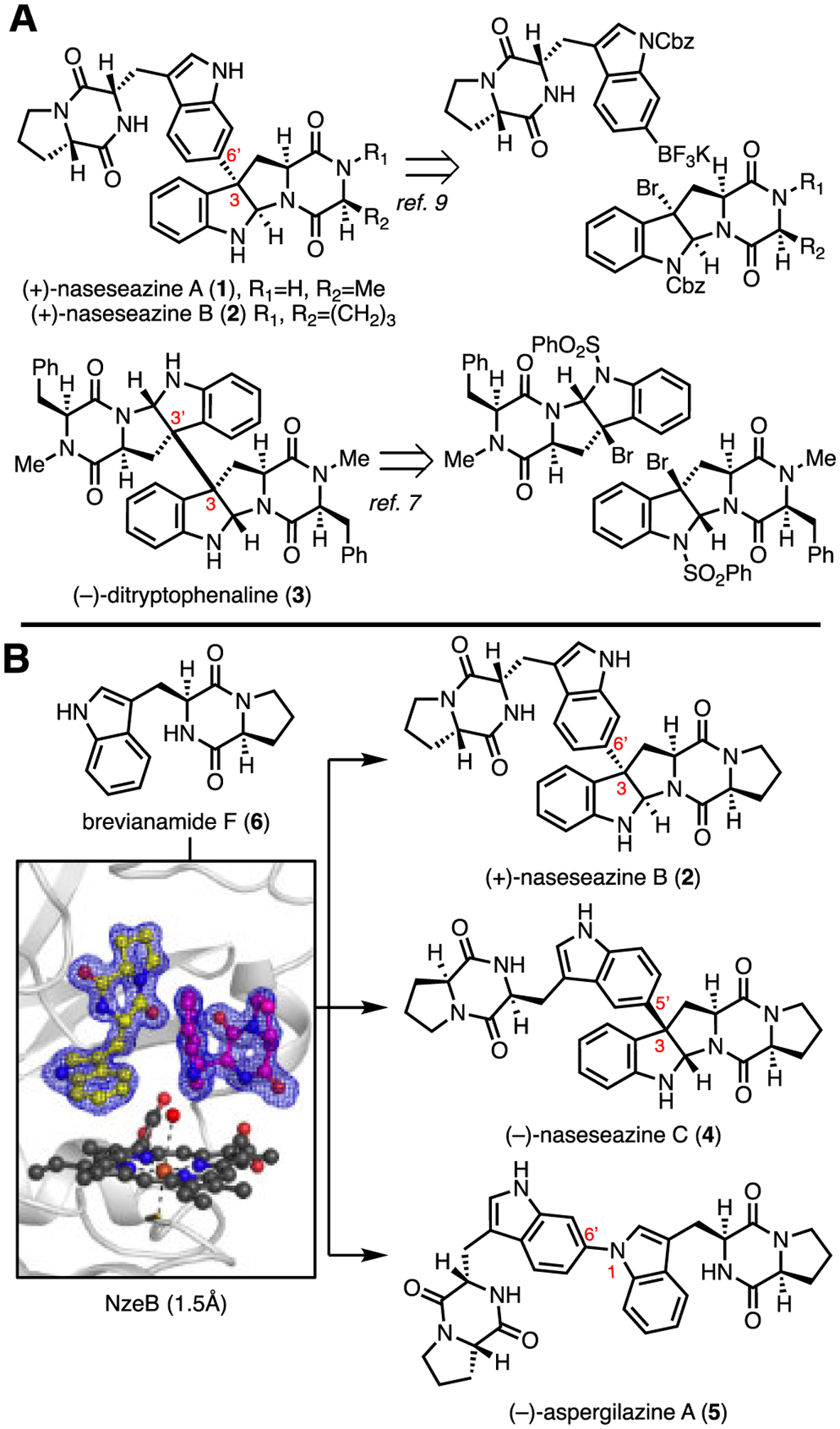
A: Representative examples of substrate controlled, biomimetic total syntheses of dimeric diketopiperazine natural products. B: Current work describing the molecular basis for catalyst-controlled NzeB mediated assembly of isomeric dimers.
While dozens of fungal dimeric diketopiperazine natural products have been reported since the isolation of 2,1–4,6 fungal biosynthetic gene clusters (BGCs) responsible for dimerization of DKPs have only recently been identified, and have been limited to biosynthesis of C2 symmetrical dimers.19 Characterization of these BGCs identified cytochromes P450 that mediate the oxidative convergence of two DKP monomers to generate C2 symmetrical dimers, validating long-standing hypotheses proposed in isolation, characterization, and total syntheses of these molecules.19 While the majority of DKP dimers have been isolated from fungi, marine actinomycetes have recently emerged as a new source for non-symmetrical dimeric DKP natural products. Notably, bacterial DKP dimers display coupling patterns distinct from their fungal counterparts, with the exception of 4, a rare example of a secondary metabolite isolated from both fungal and bacterial sources.8, 10, 11 Contemporaneous efforts to identify DKP dimer biosynthetic gene clusters from bacteria have led to the recent identification of gene clusters for dimers 4 (nasc)20 (Streptomyces sp. CMB-MQ030), and the (+)-naseseazines A (1) and B (2) (nas) and (−)-aspergilazine A (5) gene clusters (Streptomyces sp. NRRL-S1868).21 While functional characterization of these BGCs have provided insight into the biogenesis of these natural products, the molecular basis for the selective formation of non-symmetrical dimers by these P450s has remained elusive.
Herein, we report the identification of NzeB, a versatile P450 that catalyzes both C–C and C–N bond formation through bioinformatics and sequence similarity network (SSN) analysis. Structural and functional characterization of NzeB provides the first insights into the molecular basis of selectivity displayed by the DKP dimerizing class of P450s. Quantum mechanics (QM) calculations were performed to elucidate plausible mechanisms for NzeB catalyzed C–C and C–N dimerization.
Identification of the nzn gene cluster.
Previous studies by Tian et al. identified a gene cluster (nasc) from Streptomyces sp. CMB-MQ030 composed of a cyclodipeptide synthase (CDPS), nascA, and P450, nascB, which mediates assembly of cryptic diketopiperazine dimer (−)-naseseazine C (4).20 Our independent sequencing and annotation of the Streptomyces sp. CMB-MQ030 genome (Supplementary Information, Table S1) identified an additional operon, nzn, also composed of a CDPS and P450, each with high sequence homology to nasc counterparts (NznA/NascB = 61%, NznB/NascB = 68%). Given the high sequence homology to the nasc gene cluster, we hypothesized P450 NznB catalyzes formation of the site- and stereoisomeric (+)-naseseazines A (1) and B (2), the original dimeric DKP natural products isolated from this strain. To assess NznB as the (+)-naseseazines A and B synthase as well as evaluate relative catalytic activities, both recombinant proteins (NznB and NascB) were subjected to dimerization conditions with predicted native substrate brevianamide F (6). Homodimerization reactions demonstrated NznB catalyzes formation of 6 into 2, albeit in modest total turnover number (TTN), 145 TTN, relative to the conversion of 6 to 4 by NascB, >500 TTN (Figure 2).22
Figure 2.
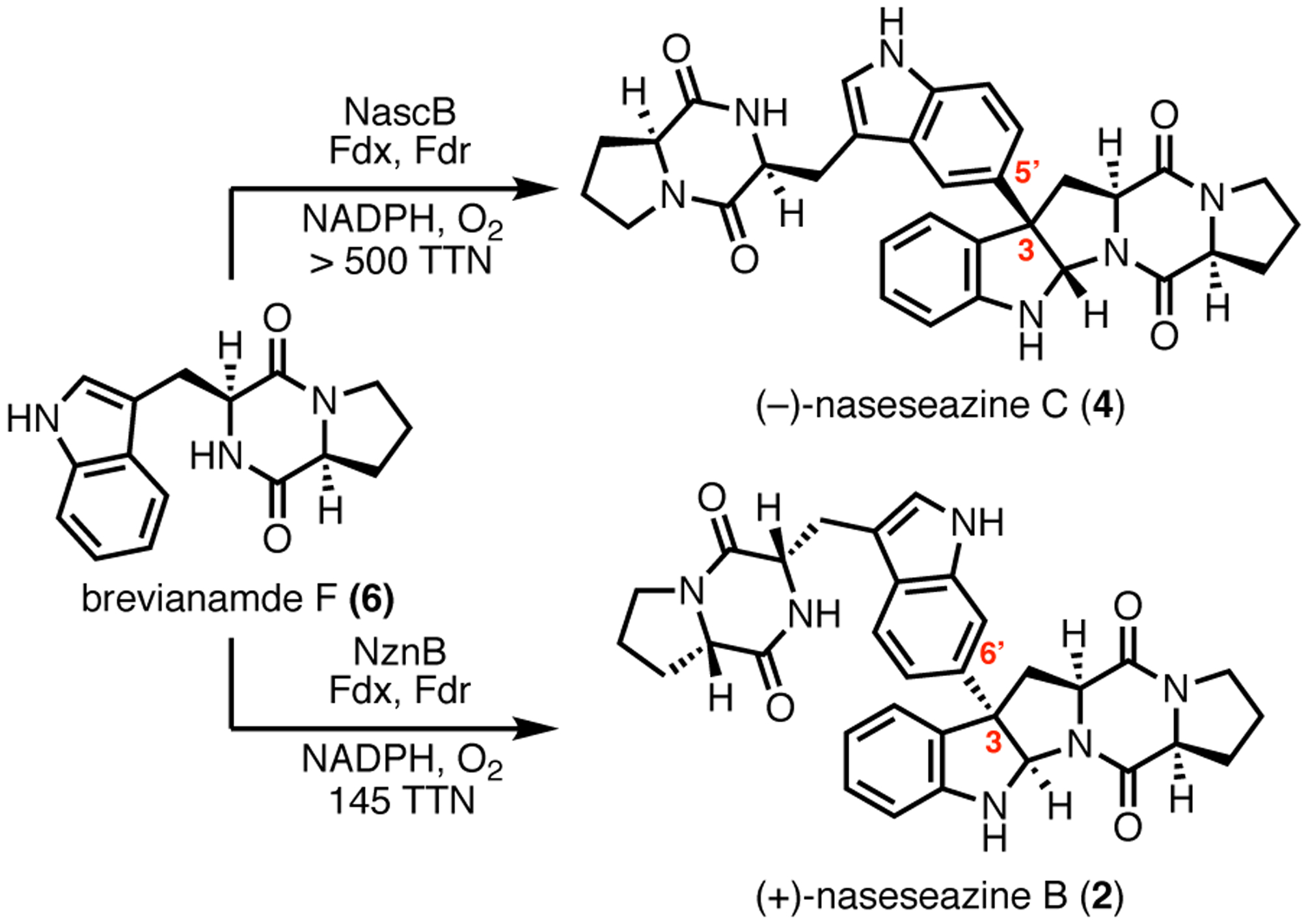
Products (−)-naseseazine C (4) and (+)-naseseazine B (2) formed from homodimerizations of 6 catalyzed by NascB and NznB, respectively.
To confirm the structure of (−)-naseseazine C (4), as well as resolve a discrepancy in the literature regarding its connectivity,10, 11 the first total synthesis of (−)-naseseazine C (4) was performed, using the undirected Friedel-Crafts methodology described in the first generation total synthesis of (+)-naseseazines A and B (Scheme 1).9 This route was provided dimers with connectivities at both C5 and C6, encompassing the constitutional isomers proposed in both isolation reports. Detailed analysis of key HMBC correlations confirmed both the stereochemical configuration of the dimerization axis and connectivity of (−)-naseseazine C (4) at C5, as well as its C6 constitutional isomer, which we name (−)-isonaseseazine C (11) based on previous precedent.9
Scheme 1. Total synthesis of (−)-naseseazine C (4) and (−)-iso-naseseazine C (11).
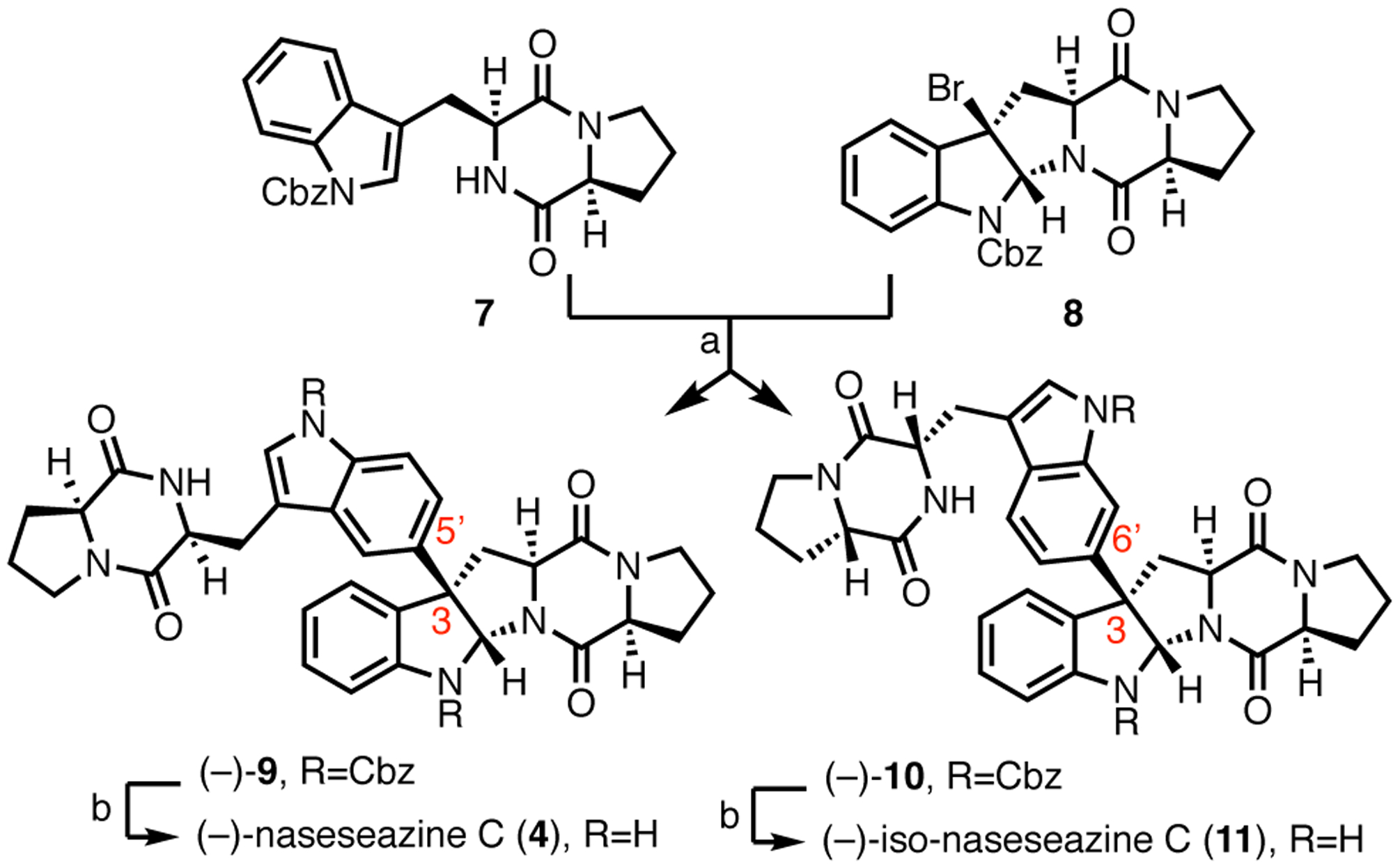
aReagents and conditions: (a), AgSbF6, EtNO2, (−)-9:(−)-10 = 1:1.4, 40%. (b) H2/Pd/C, EtOH, (80% for (−)-4 and 84% for (−)-11.
Discovery and functional characterization of NzeB.
Given the divergent site- and stereoselectivities displayed by NascB and NznB, we reasoned that comparative sequence analysis may uncover regions conferring selectivity to the dimerization cascade. Therefore, we sought to identify additional DKP dimerases through sequence similarity network (SSN) analysis.23 Using NascB and NznB as in silico probes, along with known DKP functionalizing P450s from the thaxtomin (12),24 pulcherrimic acid (13),25 guanitrypmycin (14),26–29 bicyclomycin (15),30, 31 and mycocyclosin (16)32, 33 BGCs, we mined the NCBI database to identify candidate DKP dimerases. The resulting sequences were assembled into a SSN, which revealed a series of nodes clustered with NascB and NznB. From these data we identified a BGC, nze, consisting of a CDPS (NzeA) and P450 (NzeB) pair with high sequence homology to the nasc and nzn operons (Figure 3).
Figure 3.
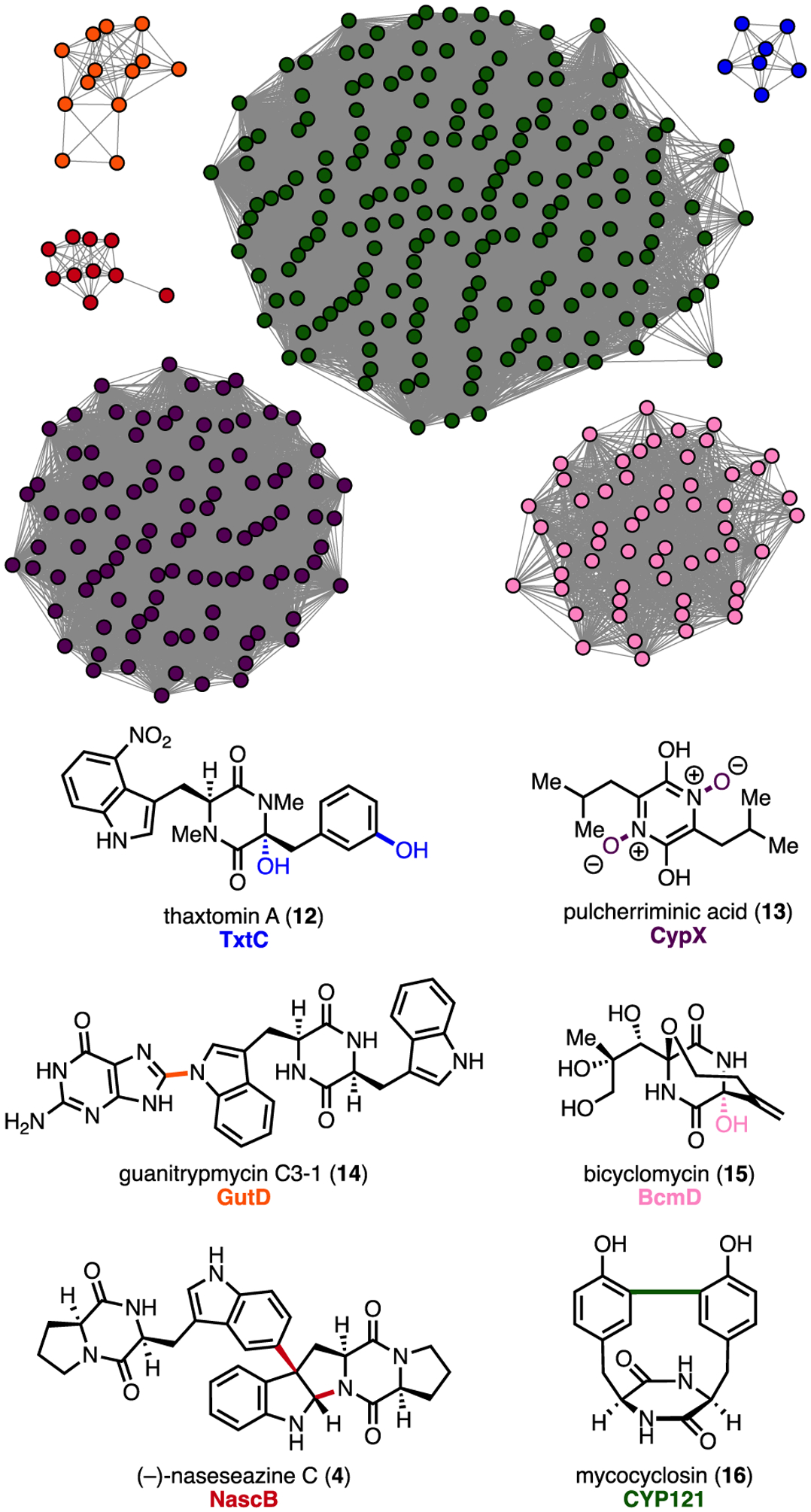
Sequence similarity network with clusters representing characterized DKP functionalizing P450s (clusters of unknown function and orphan nodes omitted for clarity).
Heterologous expression of CDPS, NzeA, and analysis of its product profile identified 6 as the sole DKP metabolite (Supporting Information, Supplementary Figure 8). Subsequent NzeB-catalyzed homodimerization of 6 resulted in formation of three dimeric products; (−)-naseseazine C (4) as the major product, and two minor products, (+)-naseseazine B (2) and unknown dimer 5 (Figure 4). Scale up and NMR characterization revealed 5 to be C–N linked dimer (−)-aspergilazine A. This product distribution exemplifies the versatility of NzeB, as it is able to form site- and stereoisomeric Csp3–Csp2 linked dimeric products 2, and 4, as well as display variant chemoselectivity in the formation of Csp2–Nsp2 linked 5 (Figure 1b).10, 12, 14, 15 The multifunctional nature of NzeB enables assembly of all currently known bacterial-derived DKP dimer scaffolds from a single biocatalyst.
Figure 4.
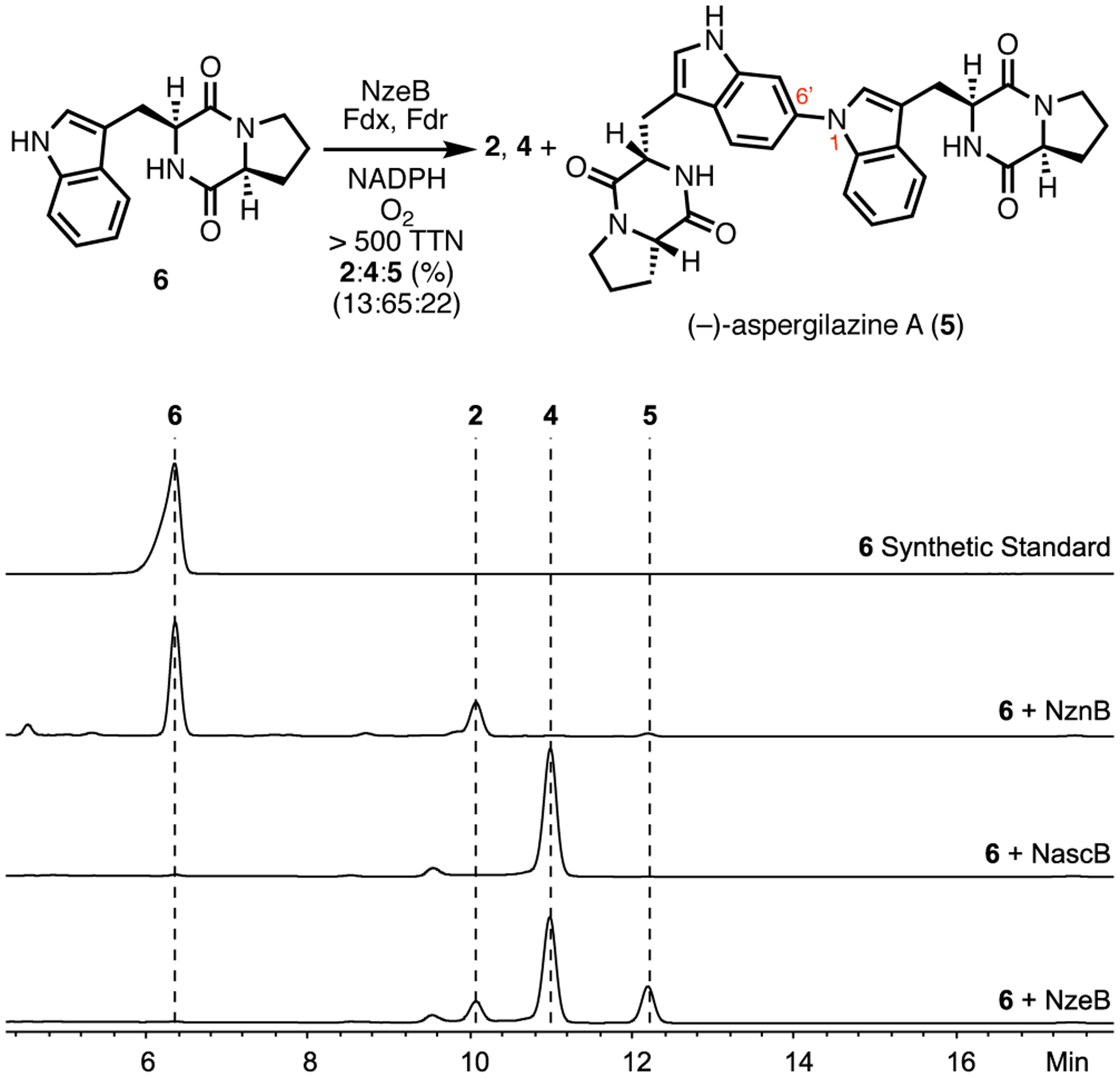
Products formed and HPLC traces (absorbance measured at 280 nm) from homodimerization of 6 by NzeB.
Crystal structure of NzeB.
Given the catalytic flexibility displayed by NzeB, we sought to identify the molecular basis for this versatility using X-ray crystallography. We obtained high-resolution (1.5 Å) crystal structures of NzeB both in the presence and absence of native substrate 6 (Figure 5, Supporting Information Supplementary Figure 21). NzeB shares highest structural homology with DKP functionalizing cytochrome P450, CYP121, which catalyzes oxidative intramolecular C–C bond formation to generate mycocyclosin (30% Sequence ID, 2.4 Å RMSD, 45.8 Z-score).34 NzeB adopts the prototypical triangular prism P450 fold, with only minor structural rearrangement between the ligand-bound and ligand-free forms (0.45 Å RMSD). Notably, both ligand-free and substrate bound structures of NzeB display electron density for the conserved axial water 3Å from the heme-iron.
Figure 5.
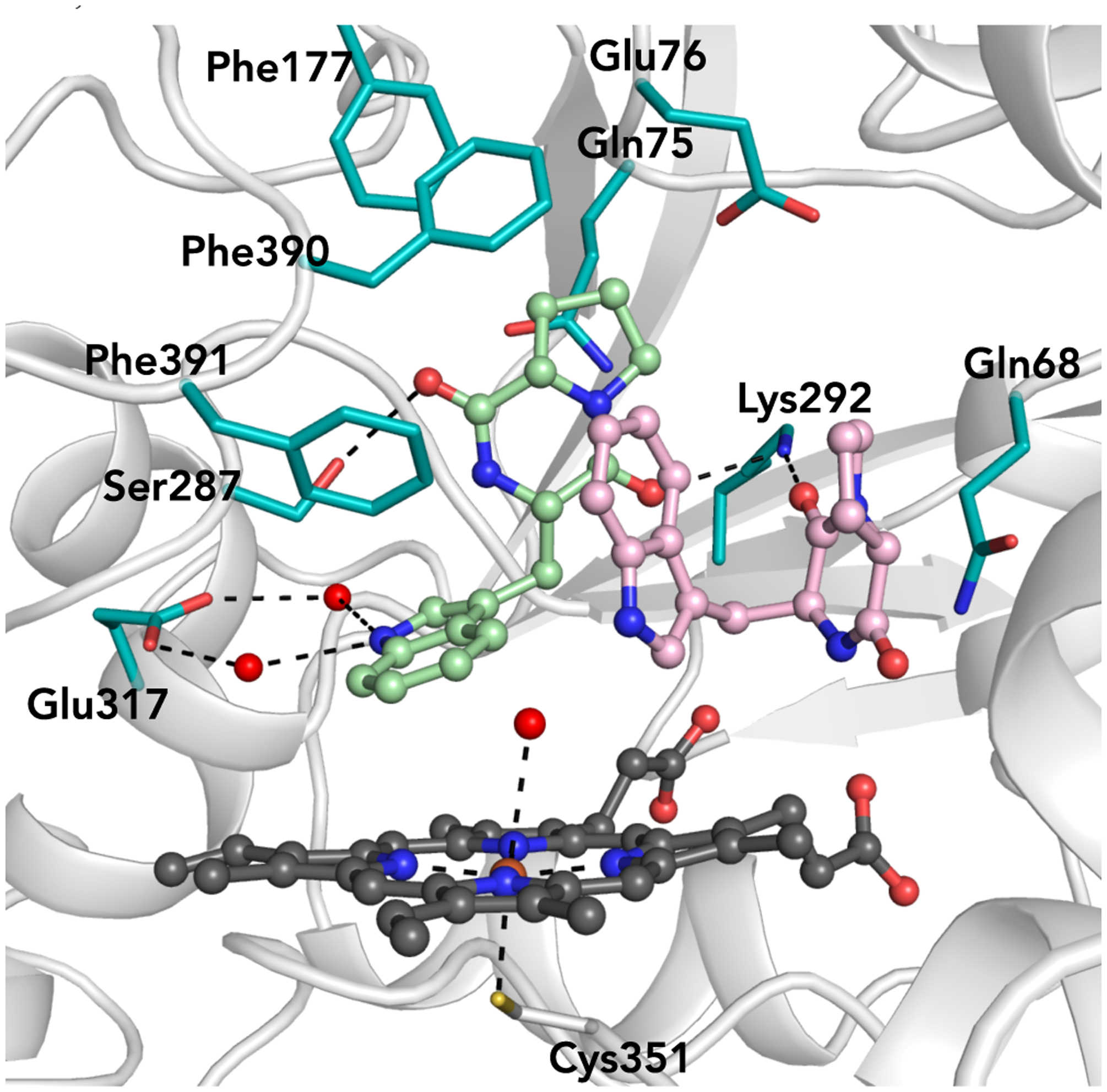
Active site of NzeB with concave substrate (light pink) bound in the “cyclization site” and extended substrate (mint green) bound in the “dimerization site.”
The active site of the ligand bound crystal structure of NzeB contains two units of 6, with each substrate occupying a discrete pocket in the active site. The DKP core and tryptophan side chain of the first substrate (Figure 5, light pink) are “sandwiched” by Gln68 and Phe391, forcing the DKP to adopt a concave architecture. The concave pose of this substrate is suggestive of the pyrroloindoline scaffold in the C–C linked dimeric products 2 and 4 and as such we named this site the “cyclization site.” The proline side chain of the second substrate (Figure 5, mint green) is encapsulated by predominately hydrophobic residues (Gln75, Glu76, Phe177, Phe390, Phe391) and binds in an extended pose. This extended conformation positions the indole atoms that undergo C–H functionalization to form the dimeric axis (C5 for 4 and C6 for 2 and 5) proximal to the iron center of the heme cofactor; therefore, we named this the “dimerization site.”
Inspection of this complex revealed close packing of the substrate proline side chain in the dimerization site compared to the relatively open cavity surrounding the proline side chain in the cyclization site (Supporting Information, Supplementary Figure 22).
We reasoned that the more open cyclization site could selectively accommodate non-native substrates with bulkier side chains. We hypothesized that reactions involving heterodimerization of the larger substrate and the native DKP 6 would show pyrroloindoline ring formation in the larger substrate due to selective binding in the putative cyclization site.
To confirm the roles of the cyclization and dimerization sites, non-native DKP substrates with bulky side chains in place of proline were synthesized and screened for their heterodimerization capacity with native substrate 6 (Supporting Information, Supplementary Figure 12). Although a variety of these substrates were capable of forming heterodimers with 6, only heterodimerizations with cyclo(L-Trp-L-Trp) (17) resulted in the exclusive formation of a single heterodimeric product (Figure 6). Suppression of homo-dimeric product formation in the presence of 11 and formation of a single heterodimer suggested that each substrate bound selectively and preferentially to a single DKP binding site.
Figure 6.
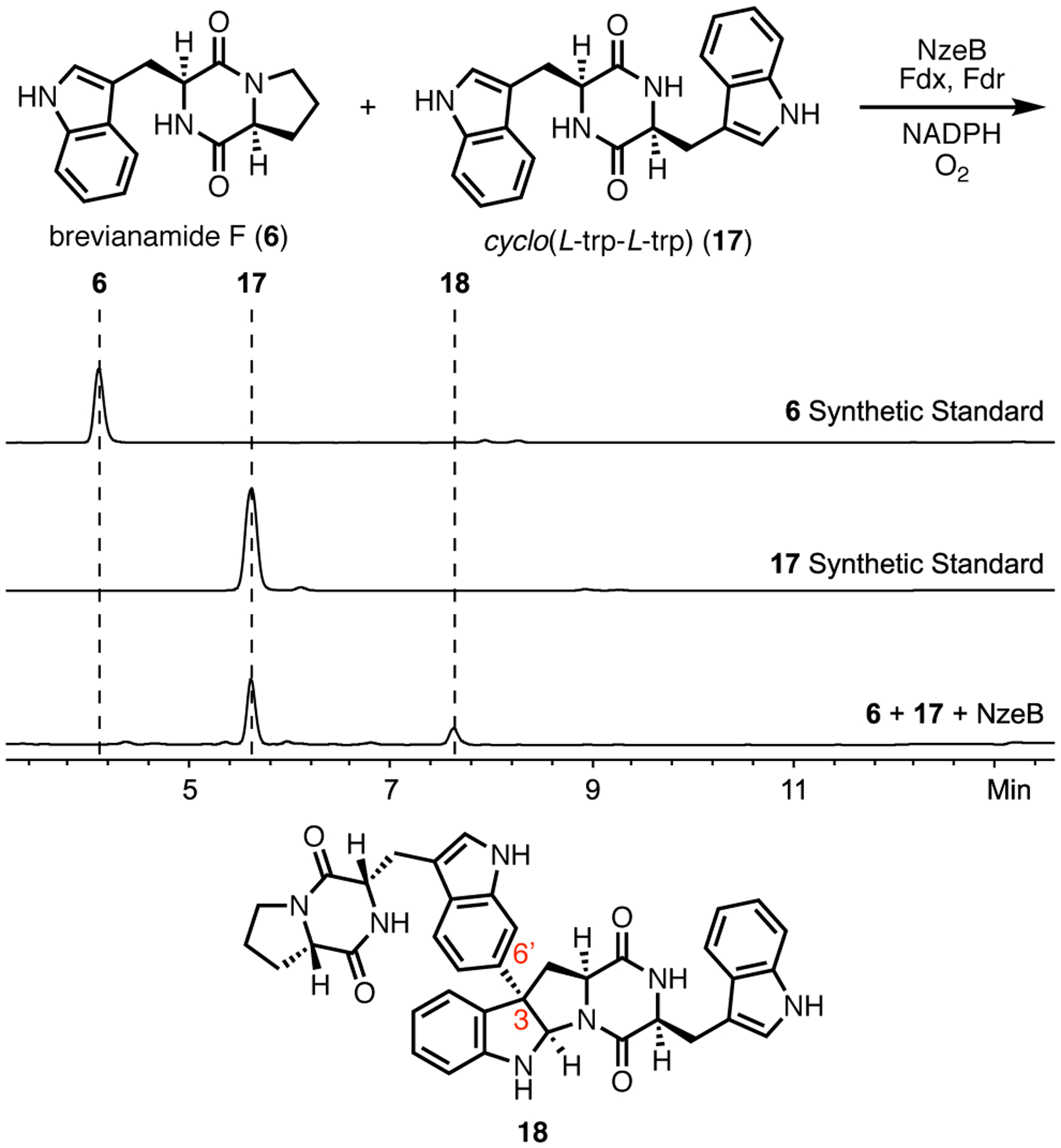
Heterodimerization reaction of NzeB with substrates 6 and 17, HPLC traces (absorbance measured at 280 nm) from heterodimerization reactions and structure of heterodimer 18.
This biocatalytic union of substrates 6 and 17 was performed on preparative scale and the structure of product 18 was determined. This result suggests that binding of 17 is limited to the cyclization site. We obtained a crystal structure of a mixed-ligand complex of NzeB with 17 binding in the cyclization site and 6 in the dimerization site, confirming our assignment of the functional roles of each of these active site pockets (Figure 7).
Figure 7.
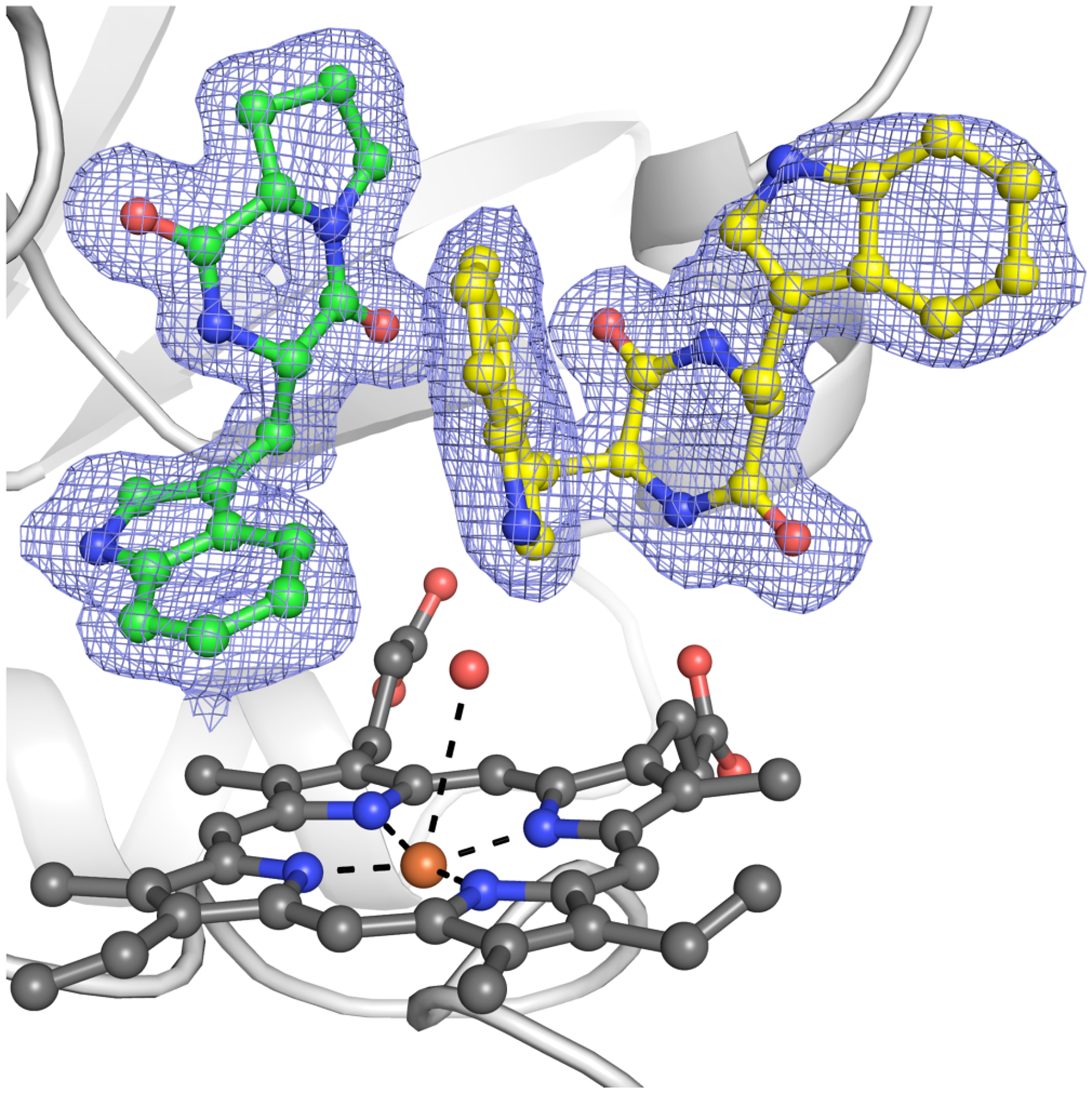
Crystal structure of NzeB heterocomplex with diketopiperazine 17 (yellow) bound in the cyclization site and diketopiperazine 6 (green) bound in the dimerization site, with omit map (F0-Fc) displayed as blue mesh.
Site-directed mutagenesis of NzeB.
To identify key residues that control chemoselectivity in the NzeB catalyzed dimerization cascade, we examined the conservation of amino acid residues in both the cyclization and dimerization sites between NzeB, NascB and NznB. As (−)-naseseazine C (4) is the major product of the NzeB and the exclusive product of NascB, we began our analysis by comparing the active sites of these two homologs. A single residue variation exists in the active sites of NascB and NzeB, (NascB: Ala 287, NzeB: Ser 287). In the ligand-bound structures of NzeB, Ser287 forms a hydrogen bond with the proline carbonyl of the substrate in the dimerization site (Figure 5). NzeB variant NzeBS287A was generated and homodimerization reactions of 6 with this mutant protein resulted in exclusive formation of (−)-naseseazine C (4) (Figure 9). Functionally, this mutation completely reconstituted the specificity of NascB and pinpointed Ser287 as a key determinant of dimerase selectivity.
Figure 9.
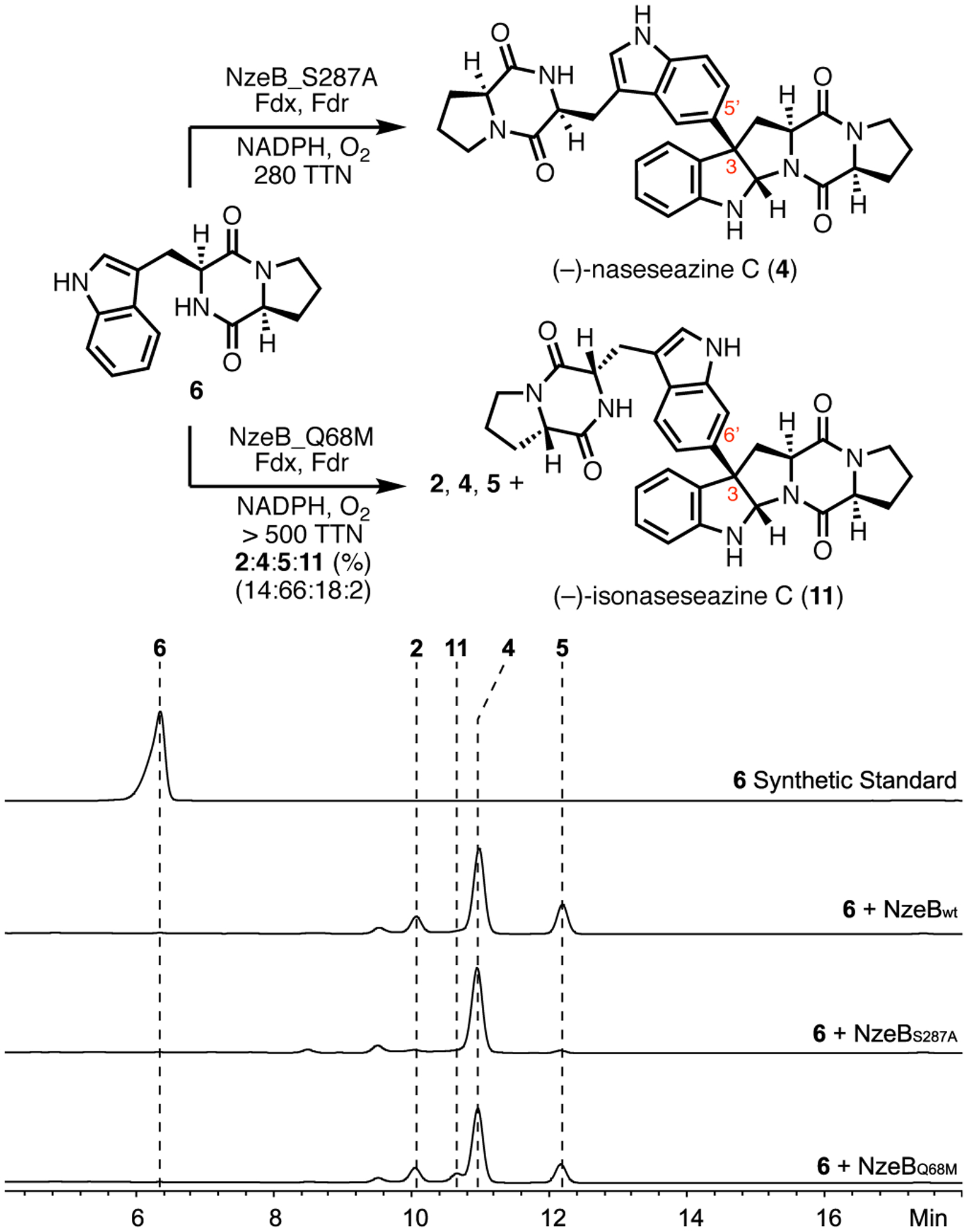
Homodimerization reactions of NzeB variants with 6 and corresponding HPLC traces. Minor peaks (around 9.5 min) are unrelated to either DKP substrate or dimeric products of the reaction.
To examine the generality of remodeling active sites to recapitulate selectivity, we compared the active site residues of (+)-naseseazine B (2) synthase, NznB, to NzeB. This revealed a single active site variation (NznB: Met68, NzeB: Gln68), located in the cyclization site. To assess the consequence of this variation, NzeBQ68M was constructed and subjected to homodimerization reactions with 6. Rather than exclusive formation of 2, homodimerization reactions of 6 with NzeBQ68M generated dimers 2, 4 and 5, as well as a new metabolite, 11 (Figure 9). Scale up and characterization of this product revealed its identity as the C6 constitutional isomer of 2, (−)-isonaseseazine C (11) based on comparison to synthetic standard (Scheme 1).9
To further profile the role of active site residues in controlling selectivity in the C–C/C–N bond-forming cascade, we performed alanine scanning mutagenesis on residues within 5 Å of either substrate. The resulting variants (S287A, Q75A, E76A, Q75A/E76A, F177A, E317A, F390A, F391A and F390A/F391A) were profiled for catalytic activity and selectivity against native substrate 6 (Supporting Information, Supplementary Figure 13). All functional variants from this experiment displayed the same product profile as NzeBQ68M, formation of 2, 4, 5 and 11, and differed only in the relative distributions of dimers. Given the increased formation of 13 across a variety of active site variants at various positions within the NzeB active site, we hypothesize that 13 results from loss of selectivity during the dimerization cascade.
NzeB dimerization mechanism and quantum mechanics (QM) calculations.
The first biomimetic total synthesis of the (+)-naseseazines proposed two alternative biosynthetic mechanisms for DKP dimer formation.9 Both mechanisms involved oxidative formation of a pyrroloindoline radical, but diverged on formation of the intermolecular C–C bond. The first mechanism proposed directed pyrroloindoline radical coupling to a second DKP followed by oxidative re-aromatization to afford the non-symmetrical DKP scaffold. The second involved oxidation of the pyrroloindoline radical to a tertiary, benzylic cation followed by electrophilic aromatic substitution (EAS) to form the C–C bond. The reaction was completed by re-aromatization via deprotonation of the resulting Wheland intermediate to forge the dimeric axis. To shed light on the viability of these proposed mechanisms for intermolecular C–C bond formation, as well as the intermolecular C–N bond formation to give 5 (catalyzed by NzeB), we turned to quantum mechanical (QM) calculations.
Given the proximity and orientation of the substrate in the cyclization site relative to the heme iron, we propose that the dimerization cascade is initiated by the oxidation of the substrate occupying the cyclization site. The mechanistic pathways (Figure 8) compare two possible modes for oxidative formation of the cyclized pyrroloindoline radical: hydrogen abstraction of 1) the proximal indole N–H bond or 2) the diketopiperazine N–H bond starting from separated brevianamide F (6) and P450 iron-oxo (Compound I). While indole N–H abstraction has a lower free energy barrier (path 1, 11.5 kcal/mol) than DKP N–H abstraction (path 2, 20.3 kcal/mol), both pathways are energetically plausible and subsequent steps have equal or higher free energy barriers. Furthermore, the diketopiperazyl radical (R-II) can cyclize directly to the pyrroloindoline radical (R-III) with a free energy barrier of 20.7 kcal/mol, while cyclization of the indolyl radical (R-I) would need to be accompanied by the net movement of a hydrogen atom from the DKP nitrogen to the indole nitrogen. A unique feature of the active site of NzeB is the hydrogen bonding of the DKP N–H to a heme propionate. We considered the heme propionate as a possible hydrogen relay to enable cyclization of indolyl radical (R-I), however calculations using acetate as a surrogate for the heme propionate gave a large free energy barrier (30.8 kcal/mol). Furthermore, there is an overall absence of participant residues (both side chain and backbone atoms) in the active site to catalyze this proton transfer. Single electron oxidation of indole to give a radical carbocation was also considered (Supporting Information, Supplementary Figure 23), however given the high energy radical cation intermediate (41.2 kcal/mol) we did not to pursue this pathway further. In both depicted N–H abstraction pathways, a significant contribution to the overall free energy barrier is the formation of a pre-complex (PC) between the heme iron-oxo species and the substrate N–H bond to be abstracted (Figure 8, PC-I and PC-II). Compared to separated starting materials (diketopiperazine 6 and the heme iron-oxo), the free energy for the formation of the PC for indole N–H abstraction (PC-I) and the PC for DKP N–H abstraction (PC-2) are 5.3 kcal/mol and 9.2 kcal/mol, respectively. However, as evidenced by the crystal structure of NzeB, both DKP substrates are oriented to facilitate PC formation, substantially lowering the free energy barrier for the entire transformation.
Figure 8.
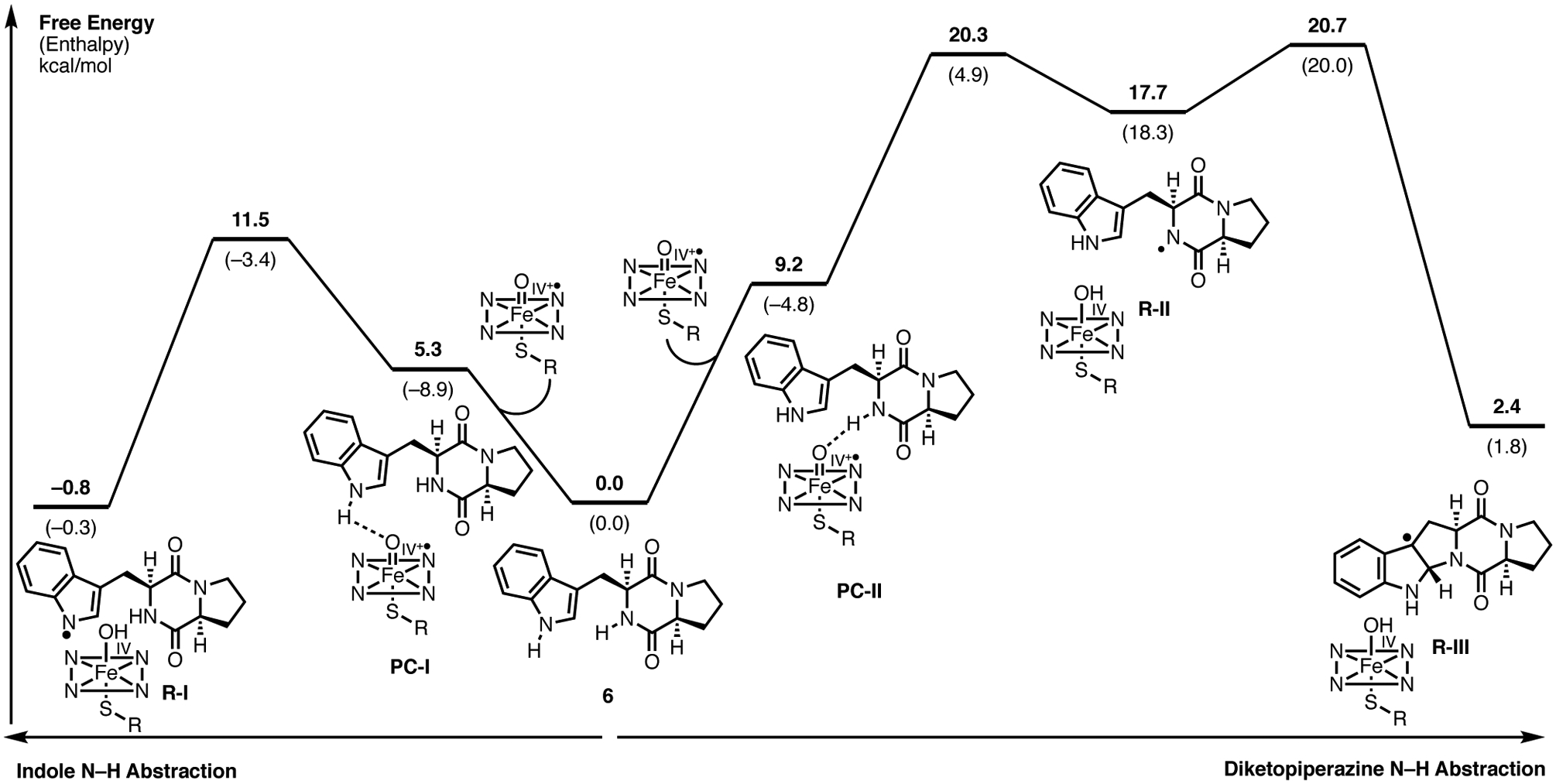
Computed mechanistic pathways for the formation of radical cyclized pyrroloindoline intermediate (R-III) via indole N–H abstraction (left) and DKP N–H abstraction (right).
Following cyclization to the pyrroloindoline radical (Figure 10, R-III), formation of the C–C bond with a second unit of 6 (modeled as 3-methylindole) may occur through either a radical-mediated, or a cationic Friedel-Crafts mechanism. The depicted energetic landscape of both dimerization mechanisms lead to formation of (−)-naseseazine C (4) (Figure 10), however, analogous computations of the pathway leading to the formation of (+)-naseseazine B (2) (Supporting Information), were qualitatively similar and led to the same conclusions. In the radical-mediated mechanism, C–C bond formation is the rate-determining step with a free energy barrier of 24.3 kcal/mol, while in the cationic mechanism the C–C bond formation barrier of 20.3 kcal/mol falls slightly below the preceding cyclization. Both mechanisms remain viable given that these computations are performed on a model system in the absence of active site residues, and that the computed barriers are relatively close. However, NzeB lacks any side chains, backbone amides, or water molecules at an appropriate location in the active site to act as a general base for deprotonation of Wheland intermediate (C-IV) generated in the cationic Friedel-Crafts pathway. Furthermore, alanine mutants of distal acidic/basic residues in the active site (Q78, Q75, E76, E317), which could act to re-aromatize the Wheland intermediate (C-IV) are still catalytically competent for dimerization (Supporting Information, Supplementary Figure S23). However, re-aromatization of intermediate (R-IV) via H-atom abstraction by the iron(IV)-hydroxo (Compound II) is likely to be facile given the proximity of C5 and C6 to the heme co-factor. Alternatively, localization of C5 and C6 to the heme may also allow a single electron oxidation of intermediate R-IV to intermediate C-IV by Compound II followed by deprotonation and re-aromatization of C-IV by the resulting iron(III)-hydroxo species.
Figure 10.
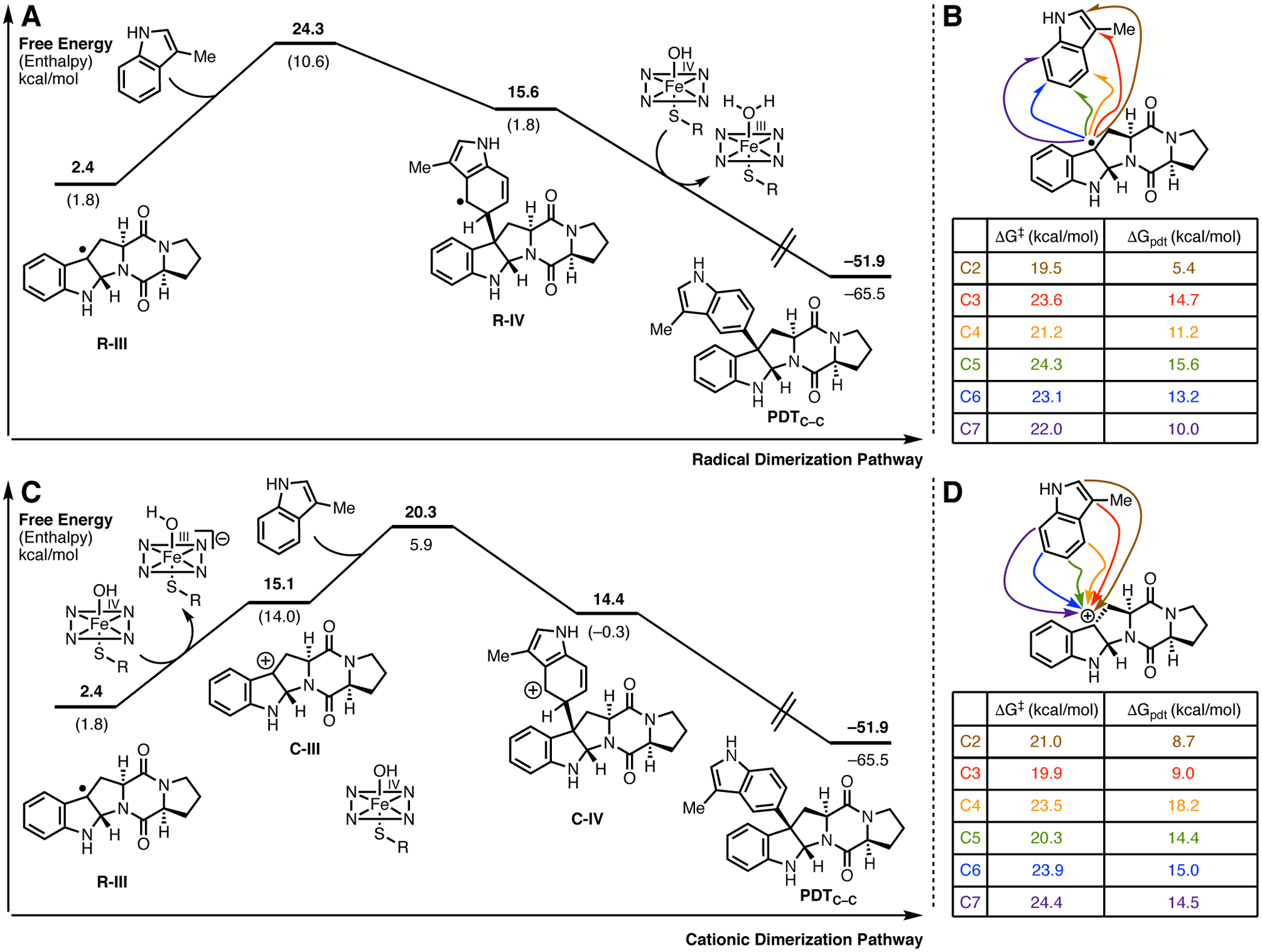
A. Computed radical dimerization pathways to generate (−)-naseseazine C model PDTC–C and B: Transition state and product free energies for both radical C–C bond formation at indole positions C2-C7 for PDTC–C. C: Computed cationic dimerization pathways to generate PDTC–C and D: Transition state and product free energies for cationic C–C bond formation at indole positions C2-C7 for PDTC–C.
In addition to catalyzing intermolecular C–C bond formation, NzeB also catalyzes the intermolecular C–N bond formation to generate (−)-aspergilazine A (5). Given the viability of both a radical and cationic dimerization mechanism for NzeB catalyzed C–C bond formation, we also compared the energetic landscapes of radical and cationic pathways for C–N bond formation starting from the uncyclized indolyl radical (Figure 11). In the radical-mediated pathway, the rate-limiting C–N bond formation has a thermodynamically plausible free energy barrier of 21.7 kcal/mol. In contrast, a potential cationic pathway would require oxidizing the indolyl radical (R-V) to a cation (C-V), which is thermodynamically demanding (45.5 kcal/mol). Given that the oxidation needed to access a cationic pathway is so highly unfavorable, quantum mechanical computations clearly support a radical-mediated pathway in the case of C–N bond formation.
Figure 11.
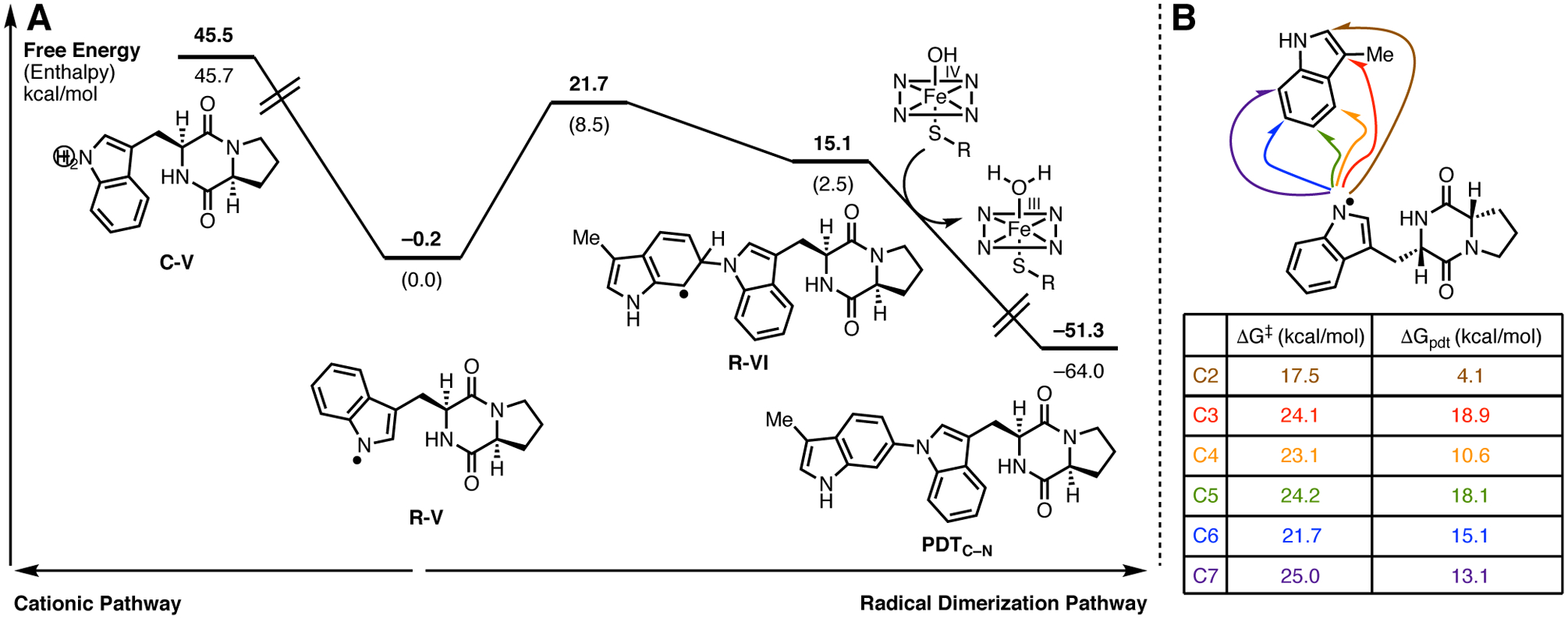
A. Computed dimerization pathways for the formation of (−)-aspergilazine A (5): radical dimerization pathway and demonstration that a cationic pathway is thermodynamically unfavorable. B. Transition state and product free energies for radical C–N bond formation at indole positions C2-C7 for (−)-aspergilazine A (5) model PDTC–N.
The C–C bond forming cascade catalyzed by NzeB results in the formation of two C–C linked dimers 2 and 4, as well as the C–N bond linked dimer 5. Computations were performed to examine the innate selectivity for cation and radical mechanisms for intermolecular C–C bond formation (Figure 10B & 10D), and analogous computations were performed for radical C–N bond formation (Figure 11B). For all computations relating to C–C and C–N bond formation, regardless of radical or cation mechanism, the observed dimerization site selectivity displayed by NzeB does not coincide with either the transition states or products that are lowest in free energy. Thus, these computations suggest that DKP-dimerization is catalyst-controlled rather than driven by the innate reactivity of the DKP substrates, with NzeB guiding the selective intermolecular C–C and intramolecular C–N bond formation.
Discussion
Biosynthesis of the tryptophan-linked dimeric DKP alkaloids reveal Nature’s ability to build molecular complexity from simple and abundant building blocks captured from primary metabolism. The production of DKP dimers from both fungal and bacterial origin indicates the biological utility of this natural product scaffold. Characterized fungal systems utilize nonribosomal peptide synthetases (NRPS) to generate DKP monomers, while bacterial pathways generate the same monomers via CDPSs. Characterization of CDPS-associated gene clusters has decrypted the biosynthetic pathways for known natural products such as bicyclomycin,30, 31 albonoursin,35 and the drimentines.36 Furthermore, these investigations have also identified new classes of natural products as well as unique DKP tailoring reactions including dehydrogenation,35 C–H hydroxylation,30, 31 C–, O–, and N–methylation,37 prenylation and cyclization of terpenes,36 and oxidative coupling with purine nucleobases.26–29 P450-mediated diketopiperazine dimerization further expands the repertoire of tailoring events found in CDPS-associated pathways.
The isolation of isomeric DKP dimers from a single organism is relatively common, which suggested two biosynthetic postulates; 1) a single dimerase with low selectivity enabled the assembly of isomeric products, or 2) a discrete dimerase was required for each coupling mode. Our results demonstrate there is no general axiom for bacterial biosynthesis of DKP dimers. P450s NascB and NznB from Streptomyces sp. CMB-MQ030 each catalyze a site- and stereoselective dimerization of 6 resulting in (−)-naseseazine C (4) and (+)-naseseazine B (2), respectively. By contrast, NzeB catalyzes both known bacterial C–C bond forming dimerization modes generating 2 and 4, while also displaying chemodivergent reactivity in the formation of C–N bond linked dimer, (−)-aspergilazine A (5). While “multifunctional” bacterial P450s are well known,38 this typically refers to the ability for a P450 to perform multiple monooxygenase reactions, such as hydroxylations at multiple sites,39 hydroxylation and epoxidation activity,40 hydroxylation with continued reaction to higher oxidation states, or a combination of these processes. NzeB differs from these enzymes in its ability to mediate formation of site- and stereoisomeric C–C linked products 1 and 2, and also catalyzes direct Csp2–H amination in the formation of 5. Recently, multiple reports have disclosed engineered enzymes capable of performing inter- and intramolecular C–H amination.41, 42 However this transformation is relatively rare in nature with few examples in the literature,43, 44 and to our knowledge, NzeB represents the first structurally characterized, wild-type P450 to catalyze intermolecular Csp2–H amination.
We were able to obtain multiple high-resolution crystal structures, including NzeB ligand free, and in complex with select native and non-native substrates. Our biosynthetic hypotheses for the C–C bonded dimers 2 and 4 were supported by direct observation of the NzeB-DKP complex, with the enzyme possessing two distinct binding sites to pre-organize substrates for diastereoselective cyclization as well as site-selective C–C bond formation. Synthesis and screening of non-native substrates for their selective heterodimerization enabled validation of the cyclization and dimerization sites. Moreover, we were able to capture a complex of NzeB with sterically differentiated ligands, brevianamide F (6) and cyclo(L-Trp-L-Trp) (17), which unambiguously confirmed our assignment of roles to each DKP binding site. The connectivity and configuration of the dimerization axis of heterodimeric product 18 is identical to that of the NzeB minor product (+)-naseseazine B (2), and is consistent with the binding mode of the two monomeric precursors (Figure 7). We hypothesize this is due to the markedly larger side chain of 17 which occupies the additional channel in the cyclization site wherein the native substrates may occupy during catalysis in order for both substrate and enzyme to undergo the conformational changes necessary to access either the (−)-naseseazine C or (−)-aspergilazine A scaffolds. Notably, all other structurally characterized DKP functionalizing CYP450s (CYP121,33 CypX,25 and TxtC24) bind DKPs in the analogous position of the active site, which corresponds to the cyclization site of NzeB. The dimerization site of NzeB is a previously unobserved pocket for DKPs in P450s and accounts for an approximate doubling of the active site volume of NzeB (832 Å3) relative to other DKP binding CYP450s.45 Previous efforts in the characterization of NascB demonstrated that in heterodimerization reactions the bulky substrates undergo cyclization to form pyrroloindoline functionality.20 Our structural investigations of NzeB and assignment of the role of the dimerization site provides a structural basis for this phenomenon. Furthermore, identifying the sidechains in the dimerization site that encapsulate the proline moiety of its substrate paves the way to reshaping this binding pocket. Engineering DKP dimerases to enable coupling of DKPs with larger side chains may enable formation of products similar to those isolated from fungi.46–48
Mutagenesis of the active site of NzeB provided further insight into how NzeB guides the site- and stereoselective formation of C–C and C–N bonds. Comparison of the active site residues of NzeB to NascB and NznB, identified that each possessed a single, active site variation (NzeB: Ser287, NascB: Ala287). To examine the role of this active site variation in selectivity the NzeBS287A variant was generated and homodimerization reactions with native substrate 6, NzeBS287A exclusively generates (−)-naseseazine C (4) completely reconstituting the selectivity of NascB with a single substitution. Given this result, we reasoned that through homologous substitution of the active site variation between NznB and NzeB, located in the cyclization site (NzeB: Gln68 vs NznB: Met68) would provide access to a surrogate of NznB. The prospect of engineering the dimerization selectivity of NzeB to recapitulate that of NznB is particularly attractive, as wildtype NznB has low TTN relative to NascB and NzeB (Figure 2). However, counter to the results from NzeBS287A, dimerization reactions, NzeBQ68M did not recapitulate the observed NznB selectivity, rather led to the formation of previously unobserved dimer (−)-isonaseseazine C (11).
These results demonstrate that the selectivity of a given DKP dimerase cannot be reconstituted simply by interchanging active site residues, and therefore the selectivity of NzeB and its homologues must also be determined by residues outside the active site motif. Thus, we propose that the observed crystal structure represents one of many catalytically relevant conformations, and that the active site must undergo conformational remodeling during catalysis, with the selectivity of the dimerization reaction being influenced by a combination of residues both inside and outside of the active site. Notably, the overall sequence conservation between NzeB and NznB (69% identity) is decreased compared to that of NzeB and NascB (87% identical), and conformational changes during catalysis could affect these regions of low conservation present in NznB, but absent in NascB. This could result in a shift into or near the active site that modulates the selectivity of the dimerization reaction. Support for active site remodeling is best represented by the discrepancy between the conformation of substrates in the ligand bound crystal structure of NzeB and the observed product distribution from the dimerization reaction. In the ligand-bound structure of NzeB, both substrates are apparently pre-organized toward formation of (+)-naseseazine B (2), despite this dimer being a minor product. To access conformations that would lead to the major product, diketopiperazine dimer 4, the orientation of substrate in the cyclization site would require inversion from a concave to convex posture. Furthermore, in spin-shift binding assays of NzeB,49 substrate 6 was able to displace the water molecules axial to the heme to induce a low- to high-spin transition of the heme iron indicative of direct binding to the active site (Supporting Information, Supplementary Figure 10). However, this axial water is present in both the ligand-bound and ligand-free crystal structures at high-occupancy, suggesting that solution-phase dynamics provide access to currently unobserved conformations. In addition, it is well-precedented that binding of electron transport proteins alters the architecture of cytochromes P450 during catalysis.50 Given these factors we propose residues and regions distal to the active site are influencing selectivity and catalysis, and efforts to identify them in bacterial DKP dimerases are currently underway.
Finally, quantum mechanical computations provided new insights into NzeB’s ability to catalyze formation of C–C and C–N bonds. Relative to C–O bond forming P450s, far fewer C–C bond forming P450s have been structurally characterized; examples include both intramolecular such as StaP (PDB: 2Z3U)51 and CYP121 (PDB: 3G5H),33 as well as intermolecular such as CYP158A1/CYP158A2 (PDB: 1T93, 2D09),52, 53 and recently the intramolecular C–N bond forming indolactam cytochrome P450 (PDB: 6J82).54 A common feature among the characterized intra-and intermolecular C–C/C–N bond forming P450s is that one of the reactive atoms participating in C–C/C–N coupling is proximal to the heme iron while the second reactive atom is distal, and oxidation of this distal atom is facilitated by a basic amino acid residue or water via proton-coupled electron transfer (PCET).55, 56 However, unique to the structure of NzeB, reactive atoms from both ligands are positioned within 4 Å of the axial water ligand, indicating the possibility for direct oxidation of both ligands catalyzed by the heme without intervention of neighboring residues or solvent (Figure 5). Regardless of mechanism, they indicate that a major role of NzeB is to orient substrates for reaction with the heme iron oxo species. By facilitating the formation of a pre-complex, NzeB can reduce the free energy barrier for the overall transformation by up to 5 to 10 kcal/mol. Previous investigations into the mechanism for C–C bond formation excluded a cationic mechanism due to reactivity of electron poor substrates.20 However, the utilization of electronically differentiated substrates for mechanistic studies such as Hammett plots is well represented in the literature, including enzymatic electrophilic aromatic substitution (EAS) type reactions.57 Our computations demonstrate that both radical-mediated and cationic pathways are thermodynamically plausible for the formation of C–C bonds, with rate-determining transition states of comparable free energies. While there are residues present in the active site that could be positioned for deprotonation of Wheland intermediates, their distance appears too great from the appropriate carbon atoms, and mutation of these residues to alanine does not prevent dimerization. While either N–H abstraction from the indole or diketopiperazine could initiate the dimerization cascade, abstraction of the indole N–H would require multiple proton transfer steps in order to allow for cyclization of the pyrroloindoline. The active site of NzeB has a general lack of basic residues as well as active site waters that could act as proton shuttles. Furthermore, distal residues that could be implicated in proton transfer were mutagenized to alanine, and the resulting variants were still catalytically competent repudiating their side chains participation in proton transfer during catalysis. Taken together we propose that in the case of a C–C linked dimer, the dimerization cascade is likely initiated by abstraction of the DKP N–H. Given this lack of residues or active site water molecules, which would also be necessary to deprotonate Wheland intermediates generated from a cationic pathway dimerization, and the localization of reactive atoms from both substrates proximal to the heme iron, we propose that C–C bond forming dimerization modes likely also proceed via a radical-mediated mechanism. However, re-aromatization of the C–C bond formed radical intermediate may occur through either concerted HAT or stepwise PCET by Compound II. In contrast, computations relating to formation of the intermolecular C–N bond in (−)-aspergilazine A (5) demonstrate that the cationic pathway is thermodynamically prohibitive and the radical-mediated mechanism is strongly favored. To examine the site selectivity of NzeB we also explored computationally the innate selectivity for C–C and C–N bond forming dimerizations. These computations demonstrate that regardless of cationic or radical mechanism, NzeB overrides the innate selectivity for C–C and C–N bond formation and the connectivity of the product dimers 2, 4, and 5 are the result of a catalyst-controlled dimerization, rather than the reactivity of the diketopiperazine substrates themselves. Further analysis is required to unambiguously exclude other mechanisms for C–C and C–N bond formation and is currently underway.
Conclusion
In conclusion, we identified and characterized the chemically versatile cytochrome P450, NzeB, capable of generating both (+)- and (−)-naseseazine frameworks as well as the non-cyclized Csp2–Nsp2 linked (−)-aspergilazine dimeric scaffold. To gain insight into the molecular basis for its versatility, we obtained the first high-resolution crystal structure of a DKP dimerizing P450 in complex with select native and non-native substrates. This structural information revealed a previously unrecognized DKP binding site compared to other reported crystal structures of DKP functionalizing P450s.24, 25, 33 NzeB is distinguished from the flaviolin dimerase, in its relatively close binding of DKP substrates proximal (within 4Å) to the heme cofactor. Site directed mutagenesis of NzeB demonstrated that active site residues are not the sole drivers of site-, stereo- and chemoselectivity. Tandem structural, biochemical, and computational approaches explored the viability of cationic and radical mechanisms in intermolecular C–C bond formation, and provided evidence for a likely radical mechanism for intermolecular C–N bond formation. The structural, biochemical, and computational characterization of NzeB provides the first insight into the molecular basis for DKP dimerization. This new information opens the door to engineering NzeB and its functional homologs for improved total turnover number, broadened substrate scope, and new non-natural dimerization modes furthering our efforts towards a catalyst controlled method for DKP dimerization.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the NSF CCI Center for Selective C−H Functionalization (CHE-1700982), NIH grant R35 GM118101 and the Hans W. Vahlteich Professorship (to D.H.S.). M.M. also acknowledges financial support by NIH grant GM089732. We are grateful to Dr. Robert J. Capon for providing Streptomyces sp. CMB MQ-030 under a materials transfer agreement. J.N.S. acknowledges support from the National Institutes of Health for award F32 GM122218. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and by the San Diego Supercomputing Center (SDSC) through XSEDE (ACI-1548562).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Full experimental and computational details, NMR spectra, tables, and figures. (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Hino T; Nakagawa M, Chemistry and Reactions of Cyclic Tautomers of Tryptamines and Tryptophans The Alkaloids: Chemistry and Pharmacology, Brossi A, Ed. Academic Press: New York, 1989; Vol. 34, pp 1–75. [Google Scholar]
- 2.Anthoni U; Christophersen C; Nielsen PH, Naturally Occurring Cyclotryptophans and Cyclotryptamines Alkaloids: Chemical and Biological Perspectives, Pelletier SW, Ed. Pergamon: London, 1999; Vol. 13, pp 163–236. [Google Scholar]
- 3.Borthwick AD, 2,5-Diketopiperazines: synthesis, reactions, medicinal chemistry, and bioactive natural products. Chem. Rev 2012, 112 (7), 3641–716. [DOI] [PubMed] [Google Scholar]
- 4.Tadano S; Ishikawa H, Synthesis of Tryptophan-Based Dimeric Diketopiperazine Alkaloids Using Bioinspired Reactions. Synlett 2014, 25 (2), 157–162. [Google Scholar]
- 5.Kim J; Movassaghi M, Biogenetically-inspired total synthesis of epidithiodiketopiperazines and related alkaloids. Acc. Chem. Res 2015, 48 (4), 1159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Springer JP; Bűchi G; Kobbe B; Demain AL; Clardy J, The structure of ditryptophenaline - a new metabolite of. Tet. Lett 1977, 18 (28), 2403–2406. [Google Scholar]
- 7.Movassaghi M; Schmidt MA; Ashenhurst JA, Concise total synthesis of (+)-WIN 64821 and (−)-ditryptophenaline. Angew. Chem. Int. Ed. Engl 2008, 47 (8), 1485–7. [DOI] [PubMed] [Google Scholar]
- 8.Raju R; Piggott AM; Conte M; Aalbersberg WG; Feussner K; Capon RJ, Naseseazines A and B: a new dimeric diketopiperazine framework from a marine-derived actinomycete, Streptomyces sp. Org. Lett 2009, 11 (17), 3862–5. [DOI] [PubMed] [Google Scholar]
- 9.Kim J; Movassaghi M, Concise total synthesis and stereochemical revision of (+)-naseseazines A and B: regioselective arylative dimerization of diketopiperazine alkaloids. J. Am. Chem. Soc 2011, 133 (38), 14940–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong ZQ; Liu QX; Pan ZL; Zhao N; Feng ZX; Wang Y, Diversity and bioprospecting of culturable actinomycetes from marine sediment of the Yellow Sea, China. Arch. Microbiol 2015, 197 (2), 299–309. [DOI] [PubMed] [Google Scholar]
- 11.Buedenbender L; Grkovic T; Duffy S; Kurtboke DI; Avery VM; Carroll AR, Naseseazine C, a new anti-plasmodial dimeric diketopiperazine from a marine sediment derived Streptomyces sp. Tet. Lett 2016, 57 (52), 5893–5895. [Google Scholar]
- 12.Cai SX; Kong XL; Wang W; Zhou HN; Zhu TJ; Li DH; Gu QQ, Aspergilazine A, a diketopiperazine dimer with a rare N-1 to C-6 linkage, from a marine-derived fungus Aspergillus taichungensis. Tet. Lett 2012, 53 (21), 2615–2617. [Google Scholar]
- 13.Kieffer ME; Chuang KV; Reisman SE, Copper-catalyzed diastereoselective arylation of tryptophan derivatives: total synthesis of (+)-naseseazines A and B. J. Am. Chem. Soc 2013, 135 (15), 5557–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyd EM; Sperry J, Total synthesis of (-)-aspergilazine A. Org. Lett 2014, 16 (19), 5056–9. [DOI] [PubMed] [Google Scholar]
- 15.Chuang KV; Kieffer ME; Reisman SE, A Mild and General Larock Indolization Protocol for the Preparation of Unnatural Tryptophans. Org. Lett 2016, 18 (18), 4750–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alonso I; Alvarez R; de Lera ÁR, Indole-Indole Ullmann Cross-Coupling for CAr-N Bond Formation: Total Synthesis of (−)-Aspergilazine A. Eur. J. Org. Chem 2017, 2017 (33), 4948–4954. [Google Scholar]
- 17.Loach RP; Fenton OS; Movassaghi M, Concise Total Synthesis of (+)-Asperazine, (+)-Pestalazine A, and (+)-iso-Pestalazine A. Structure Revision of (+)-Pestalazine A. J. Am. Chem. Soc 2016, 138 (3), 1057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson BM; Loach RP; Schiesser S; Movassaghi M, Concise total synthesis of (+)-asperazine A and (+)-pestalazine B. Org. Biomol. Chem 2018, 16 (2), 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saruwatari T; Yagishita F; Mino T; Noguchi H; Hotta K; Watanabe K, Cytochrome P450 as dimerization catalyst in diketopiperazine alkaloid biosynthesis. Chembiochem 2014, 15 (5), 656–9. [DOI] [PubMed] [Google Scholar]
- 20.Tian W; Sun C; Zheng M; Harmer JR; Yu M; Zhang Y; Peng H; Zhu D; Deng Z; Chen SL; Mobli M; Jia X; Qu X, Efficient biosynthesis of heterodimeric C(3)-aryl pyrroloindoline alkaloids. Nat. Commun 2018, 9 (1), 4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H; Li SM, Two Cytochrome P450 Enzymes from Streptomyces sp. NRRL S-1868 Catalyze Distinct Dimerization of Tryptophan-Containing Cyclodipeptides. Org. Lett 2019, 21 (17), 7094–7098. [DOI] [PubMed] [Google Scholar]
- 22. Due to the limited availability of NznB protein, we were only able to perform scale up reactions for characterization of (+)-naseseazine B. Analytical reactions were performed to detect formation of (+)-naseseazine A and its structure was assigned by analogy and via comparison of retention time to an analytical standard (Supplementary Figure 11).
- 23.Gerlt JA; Bouvier JT; Davidson DB; Imker HJ; Sadkhin B; Slater DR; Whalen KL, Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 2015, 1854 (8), 1019–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alkhalaf LM; Barry SM; Rea D; Gallo A; Griffiths D; Lewandowski JR; Fulop V; Challis GL, Binding of Distinct Substrate Conformations Enables Hydroxylation of Remote Sites in Thaxtomin D by Cytochrome P450 TxtC. J. Am. Chem. Soc 2019, 141 (1), 216–222. [DOI] [PubMed] [Google Scholar]
- 25.Cryle MJ; Bell SG; Schlichting I, Structural and biochemical characterization of the cytochrome P450 CypX (CYP134A1) from Bacillus subtilis: a cyclo-L-leucyl-L-leucyl dipeptide oxidase. Biochem 2010, 49 (34), 7282–96. [DOI] [PubMed] [Google Scholar]
- 26.Yu H; Xie X; Li SM, Coupling of Guanine with cyclo-l-Trp-l-Trp Mediated by a Cytochrome P450 Homologue from Streptomyces purpureus. Org. Lett 2018, 20 (16), 4921–4925. [DOI] [PubMed] [Google Scholar]
- 27.Shi J; Xu X; Zhao EJ; Zhang B; Li W; Zhao Y; Jiao RH; Tan RX; Ge HM, Genome Mining and Enzymatic Total Biosynthesis of Purincyclamide. Org. Lett 2019, 21 (17), 6825–6829. [DOI] [PubMed] [Google Scholar]
- 28.Yu H; Xie X; Li SM, Coupling of cyclo-l-Trp-l-Trp with Hypoxanthine Increases the Structure Diversity of Guanitrypmycins. Org. Lett 2019, 21 (22), 9104–9108. [DOI] [PubMed] [Google Scholar]
- 29.Liu J; Xie X; Li SM, Guanitrypmycin Biosynthetic Pathways Imply Cytochrome P450 Mediated Regio- and Stereospecific Guaninyl-Transfer Reactions. Angew. Chem. Int. Ed. Engl 2019, 58 (33), 11534–11540. [DOI] [PubMed] [Google Scholar]
- 30.Patteson JB; Cai W; Johnson RA; Santa Maria KC; Li B, Identification of the Biosynthetic Pathway for the Antibiotic Bicyclomycin. Biochem 2018, 57 (1), 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng S; Han W; Zhao J; Jian XH; Pan HX; Tang GL, A Six-Oxidase Cascade for Tandem C-H Bond Activation Revealed by Reconstitution of Bicyclomycin Biosynthesis. Angew. Chem. Int. Ed. Engl 2018, 57 (3), 719–723. [DOI] [PubMed] [Google Scholar]
- 32.Vetting MW; Hegde SS; Blanchard JS, The structure and mechanism of the Mycobacterium tuberculosis cyclodityrosine synthetase. Nat. Chem. Biol 2010, 6 (11), 797–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belin P; Le Du MH; Fielding A; Lequin O; Jacquet M; Charbonnier JB; Lecoq A; Thai R; Courcon M; Masson C; Dugave C; Genet R; Pernodet JL; Gondry M, Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106 (18), 7426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holm L, Benchmarking fold detection by DaliLite v.5. Bioinf 2019, 35 (24), 5326–5327. [DOI] [PubMed] [Google Scholar]
- 35.Lautru S; Gondry M; Genet R; Pernodet JL, The albonoursin gene Cluster of S noursei biosynthesis of diketopiperazine metabolites independent of nonribosomal peptide synthetases. Chem. Biol 2002, 9 (12), 1355–64. [DOI] [PubMed] [Google Scholar]
- 36.Yao T; Liu J; Liu Z; Li T; Li H; Che Q; Zhu T; Li D; Gu Q; Li W, Genome mining of cyclodipeptide synthases unravels unusual tRNA-dependent diketopiperazine-terpene biosynthetic machinery. Nat. Commun 2018, 9 (1), 4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giessen TW; von Tesmar AM; Marahiel MA, A tRNA-dependent two-enzyme pathway for the generation of singly and doubly methylated ditryptophan 2,5-diketopiperazines. Biochem 2013, 52 (24), 4274–83. [DOI] [PubMed] [Google Scholar]
- 38.Podust LM; Sherman DH, Diversity of P450 enzymes in the biosynthesis of natural products. Nat. Prod. Rep 2012, 29 (10), 1251–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sherman DH; Li S; Yermalitskaya LV; Kim Y; Smith JA; Waterman MR; Podust LM, The structural basis for substrate anchoring, active site selectivity, and product formation by P450 PikC from Streptomyces venezuelae. J. Biol. Chem 2006, 281 (36), 26289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlson JC; Li S; Gunatilleke SS; Anzai Y; Burr DA; Podust LM; Sherman DH, Tirandamycin biosynthesis is mediated by co-dependent oxidative enzymes. Nat. Chem 2011, 3 (8), 628–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McIntosh JA; Coelho PS; Farwell CC; Wang ZJ; Lewis JC; Brown TR; Arnold FH, Enantioselective intramolecular C–H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew. Chem. Int. Ed. Engl 2013, 52 (35), 9309–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh R; Bordeaux M; Fasan R, P450-catalyzed intramolecular sp(3) C-H amination with arylsulfonyl azide substrates. ACS Catal 2014, 4 (2), 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baunach M; Ding L; Bruhn T; Bringmann G; Hertweck C, Regiodivergent N–C and N–N aryl coupling reactions of indoloterpenes and cycloether formation mediated by a single bacterial flavoenzyme. Angew. Chem. Int. Ed. Engl 2013, 52 (34), 9040–3. [DOI] [PubMed] [Google Scholar]
- 44.Tsutsumi H; Katsuyama Y; Izumikawa M; Takagi M; Fujie M; Satoh N; Shin-Ya K; Ohnishi Y, Unprecedented Cyclization Catalyzed by a Cytochrome P450 in Benzastatin Biosynthesis. J. Am. Chem. Soc 2018, 140 (21), 6631–6639. [DOI] [PubMed] [Google Scholar]
- 45.Tian W; Chen C; Lei X; Zhao J; Liang J, CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 2018, 46 (W1), W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li XB; Li YL; Zhou JC; Yuan HQ; Wang XN; Lou HX, A new diketopiperazine heterodimer from an endophytic fungus Aspergillus niger. J. Asian Nat. Prod. Res 2015, 17 (2), 182–7. [DOI] [PubMed] [Google Scholar]
- 47.Ding G; Jiang L; Guo L; Chen X; Zhang H; Che Y, Pestalazines and pestalamides, bioactive metabolites from the plant pathogenic fungus Pestalotiopsis theae. J. Nat. Prod 2008, 71 (11), 1861–5. [DOI] [PubMed] [Google Scholar]
- 48.Varoglu M; Corbett TH; Valeriote FA; Crews P, Asperazine, a Selective Cytotoxic Alkaloid from a Sponge-Derived Culture of Aspergillus niger. J. Org. Chem 1997, 62 (21), 7078–7079. [DOI] [PubMed] [Google Scholar]
- 49.Noble MA; Miles CS; Chapman SK; Lysek DA; MacKay AC; Reid GA; Hanzlik RP; Munro AW, Roles of key active-site residues in flavocytochrome P450 BM3. Biochem. J 1999, 339 (Pt 2), 371–9. [PMC free article] [PubMed] [Google Scholar]
- 50.Tripathi S; Li H; Poulos TL, Structural basis for effector control and redox partner recognition in cytochrome P450. Science 2013, 340 (6137), 1227–30. [DOI] [PubMed] [Google Scholar]
- 51.Makino M; Sugimoto H; Shiro Y; Asamizu S; Onaka H; Nagano S, Crystal structures and catalytic mechanism of cytochrome P450 StaP that produces the indolocarbazole skeleton. Proc. Natl. Acad. Sci. USA 2007, 104 (28), 11591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao B; Guengerich FP; Bellamine A; Lamb DC; Izumikawa M; Lei L; Podust LM; Sundaramoorthy M; Kalaitzis JA; Reddy LM; Kelly SL; Moore BS; Stec D; Voehler M; Falck JR; Shimada T; Waterman MR, Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J. Biol. Chem 2005, 280 (12), 11599–607. [DOI] [PubMed] [Google Scholar]
- 53.Zhao B; Guengerich FP; Voehler M; Waterman MR, Role of active site water molecules and substrate hydroxyl groups in oxygen activation by cytochrome P450 158A2: a new mechanism of proton transfer. J. Biol. Chem 2005, 280 (51), 42188–97. [DOI] [PubMed] [Google Scholar]
- 54.He F; Mori T; Morita I; Nakamura H; Alblova M; Hoshino S; Awakawa T; Abe I, Molecular basis for the P450-catalyzed C-N bond formation in indolactam biosynthesis. Nat. Chem. Biol 2019, 15 (12), 1206–1213. [DOI] [PubMed] [Google Scholar]
- 55.Dumas VG; Defelipe LA; Petruk AA; Turjanski AG; Marti MA, QM/MM study of the C-C coupling reaction mechanism of CYP121, an essential cytochrome p450 of Mycobacterium tuberculosis. Proteins 2014, 82 (6), 1004–21. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y; Chen H; Makino M; Shiro Y; Nagano S; Asamizu S; Onaka H; Shaik S, Theoretical and experimental studies of the conversion of chromopyrrolic acid to an antitumor derivative by cytochrome P450 StaP: the catalytic role of water molecules. J. Am. Chem. Soc 2009, 131 (19), 6748–62. [DOI] [PubMed] [Google Scholar]
- 57.Gebler JC; Woodside AB; Poulter CD, Dimethylallyltryptophan Synthase - an Enzyme-Catalyzed Electrophilic Aromatic-Substitution. J. Am. Chem. Soc 1992, 114 (19), 7354–7360. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.