

Abstract
The study aims to evaluate the potency of two hundred natural antiviral phytocompounds against the active site of the Severe Acquired Respiratory Syndrome - Coronavirus − 2 (SARS-CoV-2) Main-Protease (Mpro) using AutoDock 4.2.6. The three- dimensional crystal structure of the Mpro (PDB Id: 6LU7) was retrieved from the Protein Data Bank (PDB), the active site was predicted using MetaPocket 2.0. Food and Drug Administration (FDA) approved viral protease inhibitors were used as standards for comparison of results. The compounds theaflavin-3-3’-digallate, rutin, hypericin, robustaflavone, and (-)-solenolide A with respective binding energy of −12.41 (Ki = 794.96 pM); −11.33 (Ki = 4.98 nM); −11.17 (Ki = 6.54 nM); −10.92 (Ki = 9.85 nM); and −10.82 kcal/mol (Ki = 11.88 nM) were ranked top as Coronavirus Disease − 2019 (COVID-19) Mpro inhibitors. The interacting amino acid residues were visualized using Discovery Studio 3.5 to elucidate the 2-dimensional and 3-dimensional interactions. The study was validated by i) re-docking the N3-peptide inhibitor-Mpro and superimposing them onto co-crystallized complex and ii) docking decoy ligands to Mpro. The ligands that showed low binding energy were further predicted for and pharmacokinetic properties and Lipinski’s rule of 5 and the results are tabulated and discussed. Molecular dynamics simulations were performed for 50 ns for those compounds using the Desmond package, Schrödinger to assess the conformational stability and fluctuations of protein-ligand complexes during the simulation. Thus, the natural compounds could act as a lead for the COVID-19 regimen after in-vitro and in- vivo clinical trials.
Communicated by Ramaswamy H. Sarma
Keywords: Antiviral phytocompounds, SARS-CoV-2, main-protease, COVID-19, decoy ligands, pharmacokinetic properties, molecular dynamics simulations
Graphical Abstract

1. Introduction
The 21st century has encountered with deadly viral diseases viz., SARS-CoV-1 and MERS coronaviruses, avian and swine influenza, Ebola, Zika, and hantaviruses (Dawood et al., 2009; Grubaugh et al., 2018; Ksiazek et al., 2003; Mesch & Schwirian, 2019; Zaki et al., 2012). The novel COVID-19 originated from Wuhan, China in December 2019. The World Health Organization (WHO) declared the disease as a public pandemic emergency on March 11, 2020, as causative for the Severe Acquired Respiratory Syndrome (SARS-CoV-2) (Andersen et al., 2020; Zhang et al., 2020). The disease outbreak has affected more than 16.09 million people globally (as of 27th July, 2020) with a casualty of 6,46,384 people from 216 countries and a case fatality rate of 4.01% declared by the WHO. The current global hotspot for coronavirus outbreak includes nations like the USA, Brazil, India, Russia, South Africa, Mexico, Peru, Chile, Spain, the United Kingdom, Iran, etc. India reported a total of 1.43 million cases with a casualty of more than 32,000 people as of end-July, 2020. After days of research, the scientists identified that the beta family of coronaviruses (SARS-CoV-2) is responsible for the respiratory disease, which showed only 4% genome variability compared to the Bat SARS coronavirus. The SARS-CoV-2, which shares a close relation to the SARS group of viruses are considered to be zoonotic. Studies also report the presence of SARS-CoV-2 in the lung tissue samples of pangolin in China that also claimed to be a potential host for Coronavirus and Sendai viruses (Liu et al., 2019). Pangolin-CoV isolated from dead Malayan pangolin matched 91.02% and 90.55% similarities in genome sequences with SARS-CoV-2 and BatCoV RaTG13, respectively (Zhang et al., 2020). The virus was suspected to be transmitted from Bats to an intermediate host and then to the humans, because of the difference in the Ribosome Binding Sites between the two viruses (Lillie et al., 2020).
The SARS-CoV-2 belongs to the beta-family of the coronaviruses. It is a single-stranded RNA virus encapsulated by a lipid membrane and spike-like protein S, membrane protein M, Nucleo-Capsid N protein, and envelope E protein. The S spike protein is responsible for the adhesion onto the Angiotensin-Converting Enzyme 2 (ACE2) receptor of mammalian lung cells causing the release of endogenous viral RNA genetic material into the host cell (Bosch et al., 2003; Ge et al., 2013; Han et al., 2006). The main-protease called 3CLpro (3- chymotrypsin-like protease) helps in the replication of the viruses and thus becomes an important antiviral drug target. Protease inhibitors are effective in blocking the coronavirus replication and proliferation by interfering with the post-translational processing of essential viral polypeptides (Zhavoronkov et al., 2020). Liu et al. crystallized the main-protease (PDB Id: 6LU7) from the SARS-CoV-2, which provided an opportunity to combat the disease by manifesting as a drug target. Mpro of SARS-CoV-2 shares 96% homology with the SARS-CoV-1, with its dimer-like structure present in with a peptide-like inhibitor as a co-crystallized structure, N3-Mpro (Xu et al., 2020). The crystal structure of Mpro is a monomer (molecular formula: C1499H2318N402O445S22) and possesses a molecular mass of 33.79 KDa with 306 amino acids and a resolution of 2.16 Å. The 3CLpro referred to as Nsp5, shares similar amino acid residues CYS 145 and His 41 with SARS-CoV-1 protease that makes it to be an essential antiviral target leading to the development of small-molecule inhibitors. The protease possesses three domains, namely, I (8-101 residues), II (102-184 residues), and III (201-303 residues) with 185-200 amino acid residues linking domain I and II forming a loop (Yang et al., 2003). To initiate a therapeutic strategy, it is necessary to observe the conservation of the sequences among SARS-CoV and other CoV, which was the protease (Hatada et al., 2020). The main-protease 3CLpro proteolytically cleaves the transcribed viral polypeptide at 11 different sites that facilitate the viral replication. By inhibiting the activity of Mpro, the viral replication inside the host gets blocked (Boopathi et al., 2020; Islam et al., 2020).
People infected with coronavirus currently treated with a cocktail of medications used for influenza virus, HIV, and other severe respiratory illnesses, as there are no effective treatment methods to date. Drug repurposing sought to reduce death rates worldwide. Medications like convalescent plasma therapy (da Silva, 2020), dexamethasone, remdesivir, hydroxychloroquine, chloroquine, azithromycin, and vitamin D are used. Current prevention strategies include the use of hand sanitizers, soaps to dissolve virus lipid coat, face masks, self-isolation, and social distancing (Peng et al., 2020). It takes about 4-6 months to develop a commercially available vaccine and strain-specific antiviral agents. Many institutes and companies succeeded in clinical trials for vaccine development (Jackson et al., 2020). Current treatment strategies include orotracheal intubation, invasive, and non-invasive ventilation support in critical cases (Ñamendys-Silva, 2020). Formulation of improvised antiviral drugs is essential to counteract the viral-escape mutants. The use of antiviral agents from natural sources helps in improving the use of broad-spectrum drugs. It can be effective against novel viruses and helps build resistance. Hydroxychloroquine, an antimalarial drug has inhibited SARS-CoV-2 viral infection under in-vitro conditions (Boopathi et al., 2020; Liu et al., 2020).
Mythological and culturally, there has been evidence of reliability on the medicinal plants for health care. With thousands of currently available natural drugs isolated from plants, the latter can produce a large number of secondary metabolites such as flavonoids, terpenoids, alkaloids tannins, etc. in response to abiotic stress. These metabolites exhibit different biological activities such as anti-inflammatory, anti-bacterial, antiviral, anti-cancer, etc. that has been under extensive research for ages. For several years, the in-vitro and in- vivo proved the exhibition of antiviral properties by several plants and their associated compounds (Zakaryan et al., 2017). For instance, apigenin is active against Herpes Simplex Virus (HSV-1 and HSV-2), Luteolin showing antiviral effects against the SARS-CoV, rhesus rotavirus, and Japanese encephalitis virus (JEV), kaempferol, and myricetin as a chemical inhibitor against the SARS-CoV (Yi et al., 2004). The therapeutic drug discovery is a time-consuming, and expensive process wherein the initial screening of large libraries of potential drugs against the diseases (Gupta et al., 2020; Sarma et al., 2020). With recent advances, these tools have gained phenomenal acceleration in lead identification and optimization. These computational tools enable the visualization of the ligand-target interaction (molecular docking) and the identification of the compounds that bind more efficiently with the target (Zoete et al., 2009).
Over 30 different docking programs are currently available, of which the most widely used is the AutoDock. The aim of molecular docking involves the binding mode identification, for which there is a need for a search algorithm to perform native protein-ligand interaction simulations (Sousa et al., 2006). AutoDock 4.2.6 works on the principle of the Lamarckian Genetic Algorithm (LGA) which is a combination of the Genetic Algorithm (GA), Monte Carlo simulation, and hybrid local search Genetic Algorithm (Forli et al., 2016; Ibrahim et al., 2020; Morris et al., 1998). This algorithm helps us in the comprehensive study of protein-ligand conformations based on lowest-binding energies (Kumar et al., 2018). Development of bioinformatics and computational biology has led to several add on features such as overcoming the previous drawbacks such as the identification of inhibitory constants for the docked conformations.
Molecular dynamics (MD) simulation is an essential tool that aid in the study of macromolecules like nucleosomes (Roccatano et al., 2007), ribosomes (Brandman et al., 2012), membrane proteins, organic solids, proteins-ligand complexes, etc. and has evolved rapidly over the last 4 decades due to advances in force fields, thanks to the development of quantum physics and computational chemistry. The simulation is widely used in the analysis of the structure to function relationship of protein and protein-ligand complexes. The current generation molecular dynamics mimic the actual biological systems (Hospital et al., 2015) with a potential of simulation up to 5,00,000 atoms and their behaviour in the order of nanoseconds with appropriate system configurations using high-speed supercomputers. It takes thousand to several million steps and involves intra and interatomic interactions simulated simultaneously for which supercomputers play a vital role in attaining so. It is very essential to study the simulation in the order of shortest duration preferably femtoseconds since the structural and functional properties of biomolecules concerning to nano and microseconds (Hollingsworth & Dror, 2018). Optimized Potentials for Liquid Simulations 3 (OPLS), a force field developed at Purdue University is very similar to AMBER (Jorgensen and Tirado-Rives et al., 1988). The OPLS3 force field is more accurate than other small molecule force fields due to its fitting on to torsional parameters. MD simulation is very helpful in analyzing the conformational stability and dynamics of the protein and protein-ligand complexes at different nanosecond time intervals, fluctuations, and their deviations from the reference structure that will be discussed in the study.
Hence, this research work scintillates on the development of antiviral lead molecules from natural compounds against Mpro of SARS-CoV-2 in-silico tools and study their pharmacokinetic and toxicity properties. This can be accomplished by inhibiting the active site of Mpro thereby preventing the adhesion and entry of viral RNA into host cells. The work aims to compare the binding energies of natural compounds with the FDA approved protease inhibitors and the drugs currently repurposed against COVID-19. The MD simulation will help analyze insights of stability and interactions of the docked complexes. The work also discusses the future prospectus anticipated to be carried out soon. The schematic of the workflow is shown in Figure 1.
Figure 1.

Work flow of the study.
2. Materials and methods
The following software were used in the present study: i) AutoDock 4.2.6, ii) Python 3.8.2, iii) MGLTools 1.5.4 iv) Discovery Studio 3.5, v) UCSF Chimera 1.13.1, vi) PyMOL 2.3, vii) LigPlot + v.2.2, viii) Java Platform SE binary version 8, and ix) Toxicity Estimation Software Tool 4.2.1 by the Environmental Protection Agency (EPA). ChemDraw online server was used to draw the two-dimensional structure of ligands (https://chemdrawdirect.perkinelmer.cloud/js/sample/index.html).
2.1. System information
The following were the system properties with which the study was conducted. Processor: Intel CORE i3-7100U CPU @ 2.40 GHz processor, system memory: 4 GB RAM, system type: 64-bit operating system, Windows 10 as Operating System. These requirements were prescribed in the software manual for the compatibility of the above-mentioned software. The molecular dynamics simulations were performed in HP z238 microtower workstation with i7 processor and system memory 8 GB RAM in Ubuntu 18 Operating System. These system requirements were prescribed for the compatibility by the software owners.
2.2. Ligand preparation
Two hundred natural antiviral compounds were collected through a literature survey and databases and retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) (Bekhit & Bekhit, 2014). Canonical Smiles of ligands were retrieved, converted to protein data bank format, and used for docking (Table S1). FDA approved protease inhibitors were retrieved by the same procedure and used for docking. These were used as standards for comparison. Moreover, several drugs that are repurposed and used as ‘drug of choice’ against the Mpro were also docked and compared.
Table 1.
Active site of Mpro predicted using MetaPocket 2.0.
| S. No | Amino acid | Residue number |
|---|---|---|
| 1 | Threonine | 24 |
| 2 | Threonine | 25 |
| 3 | Threonine | 26 |
| 4 | Leucine | 27 |
| 5 | Histidine | 41 |
| 6 | Valine | 42 |
| 7 | Cysteine | 44 |
| 8 | Threonine | 45 |
| 9 | Serine | 46 |
| 10 | Glutamic acid | 47 |
| 11 | Aspartic acid | 48 |
| 12 | Methionine | 49 |
| 13 | Leucine | 50 |
| 14 | Asparagine | 51 |
| 15 | Proline | 52 |
| 16 | Tyrosine | 54 |
| 17 | Tyrosine | 118 |
| 18 | Asparagine | 119 |
| 19 | Phenylalanine | 140 |
| 20 | Leucine | 141 |
| 21 | Asparagine | 142 |
| 22 | Glycine | 143 |
| 23 | Serine | 144 |
| 24 | Cysteine | 145 |
| 25 | Histidine | 163 |
| 26 | Histidine | 164 |
| 27 | Methionine | 165 |
| 28 | Glutamic acid | 166 |
| 29 | Leucine | 167 |
| 30 | Proline | 168 |
| 31 | Histidine | 172 |
| 32 | Aspartic acid | 187 |
| 33 | Arginine | 188 |
| 34 | Glutamine | 189 |
| 35 | Threonine | 190 |
| 36 | Alanine | 191 |
| 37 | Glutamine | 192 |
| 38 | Alanine | 193 |
2.3. Protein preparation
The 3-dimensional structure of Mpro (PDB Id: 6LU7) responsible for Severe Acquired Respiratory Syndrome was retrieved from the Protein Data Bank database (https://www.rcsb.org/) (Figure 2). The alpha helices denoted in red; beta-sheets in yellow and turns and loops in green colour. The protein was in a complex with a peptide inhibitor. Water molecules, inhibitor, and other heteroatoms from the protein removed using and used for docking.
Figure 2.

Three-dimensional structure of Main-protease (PDB Id: 6LU7).
2.4. Determining the active site
The active site is the target for an enzyme’s inhibition. The active site of the protease was predicted using the MetaPocket 2.0 online server (https://projects.biotec.tu-dresden.de/metapocket/) (Huang, 2009; Zhang et al., 2011). The processed protein data bank file without heteroatoms was uploaded and the top result from the best 3 (based on the z-score) potential ligand-binding sites was chosen for docking. Table 1 shows the active site of the protease used in the study. The amino acid residues predicted were then compared with the amino acids in the active site of the N3 inhibitor-Mpro co-crystallized complex. This was done by manually opening the co-crystallized complex in the LigPlot + v.2.2 (Wallace et al., 1995) and the interacting residues were identified and were very similar to the ones predicted by MetaPocket 2.0 server, hence proving the active site. Only after this step, molecular docking was succeeded.
2.5. Molecular docking using AutoDock 4.2.6
AutoDock 4.2.6 was downloaded from ‘The Scripps Research Institute’ official website (http://autodock.scripps.edu/) along with other supporting software viz., Python 3.8.2 and MGLTools 1.5.4. Docking of ligands and Mpro was performed indigenously by docking ‘one ligand at a time to the protein’ manually using AutoDock 4.2.6 (Morris et al., 2009). It is free and considered one of the most reliable software for molecular docking by the scientific community (Ravindranath et al., 2015).
2.5.1. Initializing and preparation of PDBQT files
Before docking, the starting directory was set to the desired folder. The processed protein molecule was imported into the AutoDock 4.2.6 workspace. The polar hydrogen atoms were added; the Kollman and Gasteiger charges were computed for the protein. The protein was then saved in PDBQT format that was then used as the target. The ligand was imported into the workstation; the torsion tree was defined by choosing the root; the number of rotatable bonds was identified and saved in PDBQT format. The ligand and protein were imported in PDBQT format into the workspace for further simulation process.
2.5.2. Grid parameters
The active site predicted matched the 2-dimensional LigPlot of co-crystallized protease with a peptide inhibitor. This was done to ensure the ligand exactly binds to the active site of the protease. Assigning the grid parameters is the most important step in molecular docking since it navigates the ligand to the binding site of the protease. Grid spacing was set to 0.375 Å (default). Center grid box values were set to x = −13.677, y = 12.737, and z = 70.782 with offset values −18.806, 8.167, and 43.083 respectively. The number of grid points along the x, y, and z dimensions was set as 70 × 76 x 78. The total grid points per map were 431893. These parameters were set to cover the entire 3-dimensional active site of the protease. The output was saved in the grid parameter file (GPF) file format. The grid size and coordinates fixed was very similar to the ones reported by Odhar et al. and Yu et al. (Odhar et al., 2020; Yu et al., 2020).
2.5.3. Running AutoGrid and AutoDock
The AutoGrid was executed by providing the AutoGrid executable and GPF files as input and converted to the grid log file (GLG). The grid was then launched. After the successful execution of AutoGrid, the genetic algorithm was set to default and is as follows: i) the number of GA runs: 10; ii) population size: 150; iii) the number of energy evaluations: 2.5 million (2.0 Å clustered tolerance); and iv) the number of generations: 27000. The Lamarckian genetic algorithm was used and the output was saved in docking parameter file (DPF) file format. The AutoDock was executed by providing the AutoDock executable and DPF files as input, converted to the docking log file (DLG) and docking was launched. The final DLG file contained essential details viz., top ten free binding energy energies for every run and inhibitory constant. The results were analyzed; ranked based on their binding energies; saved in PDBQT format; the lowest binding energy complex was saved in PDB format for further analysis.
2.6. Visualizing interactions
Discover Studio 3.5 from Biovia, LigPlot + v.2.2, UCSF Chimera 1.13.1, and PyMOL 2.3 were used to visualize and study the 2-dimensional, 3-dimensional, and surface annotation of ligand interaction with the protein.
2.7. Docking validation
The docking procedure was validated using two methods viz., i) The N3 peptide inhibitor from the Mpro was removed and re-docked into the active site using AutoDock 4.2.6 (Al-Khodairy et al., 2013). It was done manually by opening the co-crystallized complex in a notepad, removing the inhibitor heteroatoms from the Mpro, and pasting it into a new notepad and saved as an inhibitor in PDB file format. The same protocol including the grid parameters was unchanged in the process. It was done to ensure the inhibitor binds exactly to the active site cleft and must show less deviation compared to the actual co-crystallized complex. The re-docked complex was then superimposed on to the reference co-crystallized complex using PyMOL 2.3 and the root mean square deviation (RMSD) was calculated and the 2-dimensional image showing the superimposed amino acid residues were highlighted using LigPlot v.2.2 software and ii) Decoy ligands similar to N3 peptide inhibitor were obtained from DUD-E online server (http://dude.docking.org/) (Mysinger et al., 2012) and docked against the active site of Mpro. Decoys are compounds that are similar in physical properties with respect to the reference ligand that might not bind effectively to a protein. It was done to enhance ligand enrichment, which is essential to assess the docking procedure and to eliminate false positives.
These were done to validate the docking procedure to ensure the validation of docking.
2.8. Pharmacokinetic properties and Lipinski’s rule of 5
Pharmacokinetic properties are essential in determining the absorption, distribution, metabolism, and excretion of drug molecules. This prediction was done using admetSAR (http://lmmd.ecust.edu.cn/admetsar1/; http://lmmd.ecust.edu.cn/admetsar2/) versions 1 and 2 and SwissADME (http://www.swissadme.ch/) (Cao et al., 2012; Daina et al., 2014; 2017; Daina & Zoete, 2016). Lipinski’s oral drug likeliness properties were predicted using the PubChem database. This includes i) Molecular weight (<500 Daltons), ii) Number of hydrogen bond donors (<5), iii) Number of hydrogen bond acceptors (<10), iv) Log P (<5), and v) Molar refractivity (<140) (Lipinski, 2000; 2004). The toxicity properties of ligands were assessed through the EPA’s Toxicity Estimation Software Tool 4.2.1 software (Benfenati et al., 2009). The pharmacokinetic properties were predicted for the top 10 ligands that showed the best binding energy and the drugs that are currently investigated for a potential cure against SARS-CoV-2.
2.9. Molecular dynamics simulations
Molecular dynamics simulations were performed using the Desmond package for the following complexes i) Free-protein, ii) Protein co-crystallized with N3 peptide inhibitor, and iii) Top three best low binding energy ligand-protein complexes after docking. All the complexes were solvated individually by placing them in an explicit water box of size 10 Å with a single-point charge (SPC) water model TIP3P with periodic boundary condition (PBC). The OPLS3e force field was used (Harder et al., 2016; Sarma et al., 2020) to model the protein and ligand, Na+, and Cl- ions were added to make the total charge of system neutral. Subsequently, the system was energy minimized for 2000 steps before a production run of 50 ns. After minimization, the complex was further subjected to the production run at the NPT ensemble. The system was slowly heated to maintain a temperature of 300 K and pressure by using the Nose-Hoover thermostatic algorithm and The Martina-Tobias-Klein method. Particle-Mesh Ewald (PME) method was utilized to calculate long-range electrostatic interactions keeping a grid spacing of 0.8 Å. The Simulation Interaction Diagram tool implemented in the Desmond package was used to analyze the detailed interactions between the ligand and protein. The results were analyzed in terms of protein and ligand RMSD and root mean square fluctuation (RMSF) values with respect to the reference. The same procedure was reported by Pant et al. (Pant et al., 2020).
3. Results and discussion
Details of ligands with best conformations are listed in Table 2. From the work, the natural compounds have shown the potential to inhibit the virulence of viral Mpro (Table 3). This was compared with the available data from the PDBsum web page. The LigPlot interactions from PDBsum significantly match with the interacting residues in the present study.
Table 2.
Details of top 10 natural compounds with high binding energies.
| S. No | Compound | IUPAC name | Structure | Chemical formula |
|---|---|---|---|---|
| 1 | Theaflavin-3-3′-digallate | [3-hydroxy-5-oxo-4-(3,4,5-trihydroxybenzoyl)oxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-yl] 3,4,5-trihydroxybenzoate | 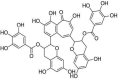 |
C43H32O20 |
| 2 | Rutin | 2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one | 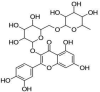 |
C27H30O16 |
| 3 | Hypericin | 9,11,13,16,18,20-hexahydroxy-5,24-dimethyloctacyclo[13.11.1.12,10. 03,8.04,25.019,27.021,26.014,28]octacosa-1(26),2,4(25),5,8,10,12,14(28),15(27), 16,18,20,23-tridecaene-7,22-dione | 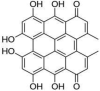 |
C30H16O8 |
| 4 | Robustaflavone | 6-[5-(5,7-dihydroxy-4-oxochromen-2-yl)-2-hydroxyphenyl]-5,7-dihydroxy-2-(4-hydroxyphenyl) chromen-4-one | 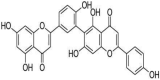 |
C30H18O10 |
| 5 | (-)-Solenolide A | [(1S,2S,3R,4R,7R,8S,10Z,12S,13S,14R,16S,17S,18R)-12-acetyloxy-8-chloro-2,3-dihydroxy-4,13,18-trimethyl-9-methylidene-5-oxo-6,15-dioxatetracyclo[11.5.0.03,7.014,16]octadec-10-en-17-yl] hexanoate | 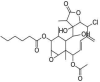 |
C28H39ClO9 |
| 6 | Rhusflavone | 6-[5,7-dihydroxy-2-(4-hydroxyphenyl)-4-oxo-2,3-dihydrochromen-8-yl]-5,7-dihydroxy-2-(4-hydroxyphenyl)-2,3-dihydrochromen-4-one | 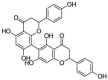 |
C30H22O10 |
| 7 | Ginkgetin | 5,7-dihydroxy-8-[5-(5-hydroxy-7-methoxy-4-oxochromen-2-yl)-2-methoxyphenyl]-2-(4-hydroxyphenyl)chromen-4-one | 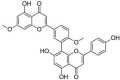 |
C32H22O10 |
| 8 | Rhinacanthin E | Dimethyl (2E)-2-(1,3-benzodioxol-5-ylmethylidene)-3-[(7-methoxy-1,3-benzodioxol-5-yl) methyl]butanedioate | 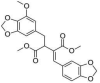 |
C23H22O9 |
| 9 | Sorbarin | 5,6-dihydroxy-2-(4-hydroxyphenyl)-7-[(2S,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl] oxychromen-4-one |  |
C21H20O10 |
| 10 | Betulinic acid | (1R,3aS,5aR,5bR,7aR,9S,11aR,11bR,13aR,13bR)-9-hydroxy-5a,5b,8,8,11a-pentamethyl-1-prop-1-en-2-yl-1,2,3,4,5,6,7,7a,9,10,11,11b,12,13, 13a,13b-hexadecahydrocyclopenta[a]chrysene-3a-carboxylic acid |  |
C30H48O3 |
Table 3.
Interactions and binding energies of top 10 conformations of ligands with Mpro within 3 Å.
| S. No | Compound | Binding energy (kcal/mol) |
Interacting amino acid residues | No. of hydrogen bonds | Hydrogen bond interactions |
|---|---|---|---|---|---|
| 1 | Theaflavin-3-3′- digallate | −12.41 | ARG 188, ASN 142, ASP 187, CYS 145, GLN 189, GLU 166, GLY 143, HIS 41, HIS 164, LEU 27, LEU 141, MET 49, MET 165, PRO 168, SER 144, THR 25, THR 26, THR 190, TYR 54 | 7 | CYS 145, HIS 41, HIS 164, THR 26, TYR 54 |
| 2 | Rutin | −11.33 | ALA 191, ARG 188, ASN 142, ASP 187, CYS 145, GLN 189, GLN 192, GLU 166, HIS 41, HIS 163, HIS 164, HIS 172, LEU 141, LEU 167, MET 165, PHE 140, PRO 168, SER 144, THR 190, TYR 54 | 10 | CYS 145, GLN 192, GLU 166, HIS 163, HIS 164, PHE 140, TYR 54 |
| 3 | Hypericin | −11.17 | ASN 142, ARG 188, CYS 145, GLN189, GLN 192, GLU 166, GLY 143, HIS 41, HIS 164, LEU 141, LEU 167, MET 165, PHE 140, PRO 168, SER 144 | 6 | ASN 142, CYS 145, GLU 166, HIS 164 |
| 4 | Robustaflavone | −10.92 | ALA 191, ASN 142, CYS 145, GLN 189, GLN 192, GLU 166, GLY 143, HIS 41, HIS 163, HIS 164, HIS 172, LEU 27, LEU 141, MET 165, PRO 168, SER 144, THR 25, THR 26, THR 190 | 2 | HIS 163, THR 26 |
| 5 | (-)-Solenolide A | −10.81 | ALA 191, ARG 188, ASN 142, ASP 187, CYS 145, GLN 189, GLN 192, GLU 166, GLY 143, HIS 41, HIS 163, HIS 164, HIS 172, LEU 141, LEU 167, MET 49, MET 165, PHE 140, PRO 168, SER 144, THR 190, TYR 54 | 0 | – |
| 6 | Rhusflavone | −10.77 | ALA 191, ARG 188, ASP 187, CYS 145, GLN 189, GLN 192, GLU 166, HIS 41, HIS 163, HIS 164, HIS 172, LEU 141, LEU 167, MET 165, PHE 140, PRO 168, SER 144, THR 190, TYR 54 | 4 | CYS 145, GLN 189, GLN 192, HIS 163 |
| 7 | Ginkgetin | −10.47 | ARG 188, ASN 142, ASP 187, CYS 145, GLN 189, GLU 166, GLY 143, HIS 41, HIS 163, HIS 164, HIS 172, LEU 27, LEU 141, MET 165, PHE 140, SER 144, THR 25, THR 26, TYR 54 | 3 | ASN 142, HIS 163, THR 26 |
| 8 | Rhinacanthin E | −10.43 | ARG 188, ASN 142, ASP 187, CYS 44, CYS 145, GLN 189, GLU 166, GLY 143, HIS 41, HIS 164, LEU 27, LEU 141, MET 49, MET 165, PRO 52, SER 144, THR 25, THR 26, TYR 54 | 0 | – |
| 9 | Sorbarin | −10.02 | ALA 191, ARG 165, ASN 142, ASP 187, CYS 145, GLN 189, GLN 192, GLU 166, GLY 143, HIS 41, LEU 27, LEU 167, MET 49, MET 165, PRO 168, SER 144, THR 25, THR 26, THR 190, TYR 54 | 4 | CYS 145, GLN 192, THR 26 |
| 10 | Betulinic acid | −9.99 | ASN 142, ASP 187, CYS 145, GLN 189, GLU 166, GLY 143, HIS 41, HIS 164, MET 49, MET 165, THR 25, THR 26 | 2 | GLU 166, THR 26 |
Theaflavin-3-3′-digallate, a natural phenolic compound had the best binding conformation with the protease with a binding energy of −12.41 kcal/mol followed by rutin, hypericin, robustaflavone, and (−)-solenolide A with −11.33, −11.17, −10.92, and −10.81 kcal/mol respectively (Table 3) which were comparable with the FDA approved viral protease inhibitors (Table 4).
Table 4.
Binding energy of FDA approved protease inhibitors.
| S. No | Drug | Binding energy (kcal/mol) |
|---|---|---|
| 1 | Atazanavir | −13.24 |
| 2 | Saquinavir | −12.74 |
| 3 | Darunavir | −12.50 |
| 4 | Ritonavir | −11.15 |
| 5 | Indinavir | −11.07 |
| 6 | Amprenavir | −10.38 |
| 7 | Nelfinavir | −10.02 |
| 8 | Fosamprenavir | −9.77 |
| 9 | Lopinavir | −9.70 |
| 10 | Tipranavir | −9.13 |
Lesser the binding energy, the greater the binding efficiency, hence augmented inhibition. Drugs namely atazanavir (−13.24 kcal/mol) (Figures 3 & 4), saquinavir (-12.74 kcal/mol), darunavir (−12.50 kcal/mol), and ritonavir (−11.15 kcal/mol) exhibited the best binding conformations with Mpro in the current study.
Figures 3 and 4.

3 D and 2 D interaction of atazanavir with Mpro (−13.24 kcal/mol).
Since the motive of the study was to find a cure from natural plant metabolites, these FDA drugs were used as a comparison. Theaflavin-3-3′-digallate is formed due to the oxidative condensation of flavonoids epigallocatechin 3-gallate and epicatechin 3-gallate during fermentation and is rich in natural sources including citrus fruits like orange, lemon, sweet lime and black tea (Ding et al., 2017; 2020; Schuck et al., 2008). The compound has proven potential as antiviral, antioxidants, and anticancer agents through apoptosis (De Oliveira et al., 2015; Hibasami et al., 2004). The compound interacted with 19 amino acid residues in the active site of the protease with 7 hydrogen bonds within 3 Å with 5 amino acid residues viz., CYS 145, HIS 41, HIS 164, THR 26, TYR 54 residues (Figures 5 and 6). Similar interacting residues were reported by (Khan et al., 2020). Greater the number of hydrogen bonds, the higher the binding efficiency and inhibition (Kumar et al., 2015). Figure 7 denotes the surface image active site cleft of the Mpro with theaflavin-3-3′-digallate bound to it. The in-silico study predicted an inhibition constant (Ki) value of 794.96 pM of theaflavin-3-3′-digallate as effective against viral protease (Table S1). Inhibition constant value is the half-maximum inhibition of an enzyme by a chemical compound and is used to estimate the potential of substrate/inhibitor in enhancing/inhibiting the biological and function of enzymes (He et al., 2016). Compounds with an inhibition constant less than 100 µM are considered to be potential inhibitors whereas inhibition constant greater than 100 µM are non-potent inhibitors (Zheng & Polli, 2010).
Figures 5 and 6.

3 D and 2 D interaction of theaflavin-3-3’-digallate with Mpro (−12.41 kcal/mol).
Figure 7.

Surface image of theaflavin-3-3’-digallate in the Mpro active site cleft.
Rutin, also known as rutoside is a flavonoid glycoside formed by combining disaccharide rutinose and flavonol quercetin abundantly found in citrus fruits has interacted with 20 amino acid in the active site of Mpro. Apart from this, the bioflavonoid has also formed 10 hydrogen bonds with 7 residues, namely, CYS 145, GLN 192, GLU 166, HIS 163, HIS 164, PHE 140, and TYR 54 (Figures 8 and 9) (Das et al., 2020). Rutin has a predicted inhibition constant of 4.98 nM at 298 K.
Figures 8 and 9.

3 D and 2 D interaction of rutin with Mpro (−11.33 kcal/mol).
Hypericin is an anthraquinone predominantly an active component of Hypericum perforatum, a flowering plant belonging to Hypericaceae family was effective against viral protease with a binding energy of −11.17 kcal/mol. The plant is well known for its activity against several viruses namely, influenza A, herpes simplex virus, bronchitis virus, and human immune viruses (Chen et al., 2019; Pu et al., 2009). Hypericin interacted with 15 amino acids in the active site with 6 hydrogen bonds with ASN 142, CYS 145, GLU 166, HIS 164 amino acid residues (Figures 10 and 11) with predicted Ki value of 6.54 nM. CYS 145 and MET 165 formed alkyl and π-alkyl bonds with hypericin. Islam et al. reported the antiviral potential of hypericin with a binding energy −10.70 kcal/mol against Mpro at the active site with a π-alkyl interaction with CYS 145 (Islam et al., 2020) similar to the present study.
Figures 10 and 11.

3 D and 2 D interaction of hypericin with Mpro (−11.17 kcal/mol).
Robustaflavone, a natural flavonoid formed by combining two molecules of apigenin through oxidative coupling forming hydrogen bond between C3 and C6 carbon atoms of hydroxyphenyl and chromene ring respectively (Lin et al., 2000; Xu et al., 2009; Zengion & Yarnell, 2011). Robustaflavone in conformation with protease has binding energy −10.92 kcal/mol and interacted with 19 residues and 2 hydrogen bonds with HIS 163 and THR 26. The biflavonoid compound has proven to be potential against hepatitis B infections. Apart from its antiviral property, the biflavonoid also has been reported as free radical scavengers (Zembower et al., 1998).
Figure 12 shows the LigPlot image of Mpro with an N3 peptide (02 J-ALA-VAL-LEU-PJE-010) inhibitor. The inhibitor formed 8 hydrogen bond interactions with PHE 140, GLY 143, HIS 163, HIS 164, GLU 166, GLN 189 and THR 190 residues of protease which denote the active site of the enzyme and is comparable with 2-dimensional and 3-dimensional structure of Mpro in complex with natural inhibitors in the present study. It is evident from the present study that confirms hydrogen bonds with the same amino acid residues. Apart from hydrogen bonds, other interactions like van der Waals, alkyl, carbon-hydrogen, sigma, and π-π bonds were found. Hydrogen bonds are the strongest bonds bonded to end OH- ions of the ligand to amino acids of the protein. More the number of hydroxyl groups in a ligand, more the number of hydrogen bonds, hence higher the binding efficiency (Khodarahmi et al., 2015).
Figure 12.

Two-dimensional LigPlot image of N3-Mpro complex from PDBsum.
There has been no reported case for in-silico elucidation of bioactive compounds against SARS-CoV-2 protease. However, studies have been carried out using SARS-CoV-1 Mpro and SARS-CoV-2 RNA polymerases. Chowdhury et al. reported the antiviral property of polyphenolic compounds from black tea namely theaflavin, theaflavin-3′-monogallate, and theaflavin-3-3′-digallate during the early onset of hepatitis C viral infections under in-vitro conditions. Their study reported that theaflavin and its derivatives could inhibit viral replication and cell-to-cell proliferation (Chowdhury et al., 2018). They reported an EC50 value of 17.89, 4.08, and 2.02 μM respectively had a significant impact against viral replication (Chowdhury et al., 2018). Chen et al. screened 720 natural compounds using HPLC protease assay to combat against 3 C-like protease of SARS- CoV-1 and reported two compounds tannic acid and 3-isotheaflavin-3-gallate with IC50 values 3 µM and 7 µM respectively inhibited the enzyme’s virulence which is comparable with the present study (Chen et al., 2005). Lung et al. reported the efficiency of flavonoid theaflavin and its derivatives against modelled SARS-CoV-2, SARS-CoV-2, and MERS-CoV RNA polymerases by inhibiting transcription process. The best docking conformation was attained with binding energy −9.11 kcal/mol for SARS-CoV-2, SARS-CoV-1 for −8.03 kcal/mol, and −8.26 kcal/mol for MERS-CoV (Lung et al., 2020). Jo et al. reported the antiviral activity of 64 flavonoid metabolites against SARS-CoV-1 Mpro 3CLpro using Schrödinger software. They reported the maximum binding energy of −9.26 kcal/mol with the flavonoid herbacetin and kaempferol with −8.52 kcal/mol (Jo et al., 2020). The present study reported a maximum of −12.41 kcal/mol against the protease. Elfiky et al. studied the effectiveness of FDA approved antiviral compounds against modelled RNA polymerases of SARS-CoV-2 virus using the SWISS-MODEL and simulated using AutoDock Vina. Setrobuvir exhibited the best inhibition with the energy of −9.3 kcal/mol followed by ribavirin and tenofovir (Elfiky, 2020). Rutin is proven to possess antiviral property against hepatitis B, hepatitis C, herpes, orthomoxyvirus, and retroviruses (Carvalho et al., 2013; Ganeshpurkar & Saluja, 2017). Chen et al. studied the in-vitro and in-vivo antiviral potential of hypericin against bronchitis virus on CEK cells. Hypericin was extracted using ethyl acetate solvent from flowers and leaves of Hypericum perforatum. mRNA expression of interleukin-6, tumour necrosis factor and nuclear factor kappa beta of bronchitis virus were reduced. Other factors such as MDA5, INF-α, IFN-β, and mitochondrial antiviral signalling gene expressions were augmented significantly and inhibiting the viral proliferation and neutralizing its potential (Chen et al., 2019).
Pillayar et al. used chemical small molecules to inhibit protease 3CLpro of SARS-CoV-1, similar to Mpro of SARS-CoV-2. Interestingly 3CLpro (SARS-CoV-2) contains similar amino acid residues at their active sites and small molecule inhibitors from their study interacted with same amino acid residues concerning the present study viz., CYS 145, GLN 189, GLN 192, GLU 166, HIS 41, HIS 164, LEU 167, MET 165, PRO 168, SER 144, THR 25, and THR 190 thereby proving that the present study has inhibited the viral protease significantly (Pillaiyar et al., 2016). Shie et al. studied a diverse library of synthesized aniline against SARS-CoV-1 3CL protease. 2- chloro-4-nitroaniline, l-phenylalanine, and 4-(dimethylamino) benzoic acid showed the highest inhibition potential with Ki = 0.03 mµM binding with CYS 145 containing thiol group in the active site (Shie et al., 2005). Lalani and Poh studied the in-vitro antiviral potency of flavonoids and their derivatives against enteroviral protease from the Enterovirus EV-A71 strain. Compounds namely, penduletin, quercetin, baicalein, and kaempferol exhibited best antiviral potentials with IC50 values less than 10 µM (Lalani & Poh, 2020). Lin et al. studied the efficiency of robustaflavone from Rhus succedanea as hepatitis B non-nucleoside inhibitor viral replication by inhibiting the RNA polymerase with an EC50 value of 0.25 µM (Lin et al., 1997). Dang et al. reported the antiviral property of (-)-solenolide A isolated from marine ear shell Haliotis laevigata against herpes simplex (Dang et al., 2011). Hayashi et al. extracted biflavonoid ginkgetin from Cephalotaxus drupacea and tested against the herpes simplex virus and was found to inhibit the viral replication with an IC50 value of 0.91 µg/mL and inhibited the early transcription genes and protein (Hayashi et al., 1992). Ngoc et al. studied the antiviral properties of rhinacanthone, racemate, rhinacanthins C, D, E, N, and Q from the root extract of Rhinacanthus nasutus against CVB3, HR1BV, and PR8 infected cells. 1 D, 2 D-NMR, and Mass spectrometry were used to determine their structures. Rhinacanthins inhibited all three infected cell lines with an IC50 value of 0.03 to 23.7 µM (Ngoc et al., 2019). Wen et al. in their research work reported that savinin and betulinic acid competitively inhibited SARS-CoV-1 protease 3CLpro with Ki value of 9.1 ± 2.4 µM and 8.2 ± 0.7 µM (Wen et al., 2007). The present study reported a Ki value of 47.91 nM for betulinic acid against Mpro of SARS-CoV-1.
Table 5 shows the binding energies of drugs that are currently repurposed against the SARS-CoV-2 viral infections. These drugs were docked against SARS-CoV-2 Mpro (PDB Id: 6LU7) to analyze and compare their efficiencies with the potential antiviral phytocompounds reported in the present study. The same docking procedure was followed including the grid parameters. The following is the increasing order of binding energies dexamethasone (−7.77 kcal/mol)< hydroxychloroquine (−7.28 kcal/mol)<festinavir (−7.15 kcal/mol)<oseltamivir (−7.01 kcal/mol)<chloroquine (−6.93 kcal/mol)<remdesivir (−6.77 kcal/mol)<azithromycin (−6.14 kcal/mol)<favipiravir (−4.78 kcal/mol). From the above-docked results, dexamethasone, a corticosteroid, according to WHO, had a significant reduction in death rate in COVID-19 patients showed the best binding conformation interacting within the active site cleft with 6 hydrogen bond interactions with ASN 142, CYS 145, HIS 163, LEU 141, PHE 140, and SER 144 within 3 Å. The antimalarial drug, hydroxychloroquine was repurposed as a drug against COVID-19, however the exact mechanism of action is not known some studies report a possible alteration in the pH in the cell membrane surface (Liu et al., 2020; Singh et al., 2020). Hydroxychloroquine has ranked second in the present study in terms of binding energy forming 2 hydrogen bonds. Hagar et al. studied the in-silico elucidation of hydroxychloroquine and remdesivir against Mpro of SARS-CoV-2 using AutoDock 4.2 and reported a slightly higher binding energy of −6.06 and −4.96 kcal/mol forming polar hydrogen bonds with ALA 191, GLN 192, GLU 166 and GLN 189, GLU 166 (Hagar et al., 2020). Beura and Chetti performed a pharmacophore study that reported a binding energy of −3.48 and −3.27 kcal/mol for hydroxychloroquine and chloroquine which is higher in energy compared to the present study, however, no interactions were formed with the amino acids in the active site (Beura & Prabhakar, 2020). Festinavir, an HIV nucleotide reverse transcriptase inhibitor drug was found to possess binding energy of −7.15 kcal/mol against the Mpro in the present study. Muralidharan et al. performed the in-silico blind docking of lopinavir, oseltamivir, and ritonavir against the Mpro (PDB Id: 6LU7) using AutoDock and reported binding energy of −4.10 kcal/mol, −4.65 kcal/mol, and −5.11 kcal/mol respectively, however only ritonavir was found to interact with the amino acids in the active site viz., GLN 189, GLU 166, HIS 41, MET 165, and PRO 168 (Muralidharan et al., 2020). The reported results were quite contradictory to results reported in the present study viz., lopinavir (-9.70 kcal/mol), oseltamivir (−7.01 kcal/mol), and ritonavir (−11.15 kcal/mol), however these drugs exactly bound to the active site residues. Beck et al. studied the in-silico computational analysis of 3411 drugs against SARS-CoV-2 3CLpro, 2′-O-ribose methyltransferase, 3′-to-5′ exonuclease, endoRNAse, helicase, and RNA-dependent RNA polymerase using AutoDock Vina. They reported the following results similar to the current study i.e. i) atazanavir, a retrovirus targeting drug was found to be the best drug candidate against 3CLpro with an inhibitory potential 94.94 nM (7.40 kcal/mol), followed by ii) remdesivir (113.13 nM) (-6.40 kcal/mol), iii) efavirenz (199.17 nM) (-5.40 kcal/mol), iv) ritonavir (204.05 nM) (-6.80 kcal/mol), and v) dolutegravir (336.91 nM) (-7.20 kcal/mol) against the 3CLpro (Beck et al., 2020). The present study also reported the best drug to be atazanavir. A recent research work by Yu et al. reported the antiviral activity of remdesivir, chloroquine, ribavirin, and luteolin against Mpro (PDB Id: 6LU7), of which ribavirin and luteolin reported best binding conformations interacting with active site cleft with CYS 145, GLN 189, LEU 4, LEU 27, THR 25, THR 26, VAL 3, and ASN 142 forming covalent hydrogen bonds and π-cation interactions (Yu et al., 2020).
Table 5.
Binding energies of drugs repurposed against COVID-19.
| S. No | Drugs | Binding energy (kcal/mol) | Hydrogen bonds |
|---|---|---|---|
| 1 | Dexamethasone | −7.77 | 6 |
| 2 | Hydroxychloroquine | −7.28 | 2 |
| 3 | Festinavir | −7.15 | 3 |
| 4 | Oseltamivir | −7.01 | 5 |
| 5 | Chloroquine | −6.93 | 1 |
| 6 | Remdesivir | −6.77 | 5 |
| 7 | Azithromycin | −6.14 | 3 |
| 8 | Favipiravir | −4.78 | 7 |
3.1. Docking validation
3.1.1. Re-docking and superimposition
The re-docking was done to examine the docking procedure and efficiencies. The same methodology that was used previously was used in the re-docking process. The peptide inhibitor bound exactly to the active site with good binding energy of −8.12 kcal/mol. ALA 191, ASN 142, CYS 145, GLN 189, HIS 41, LEU 50, LEU 141, MET 49, PRO 168, and THR 190 are the interacting amino acids (Data not shown) in the active site pocket and totally 8 hydrogen bonds were formed with a threshold distance of 3.00 Å. The re-docked complex was then superimposed on to the native co-crystallized N3-Mpro from PDB using PyMOL and a low RMSD of 0.615 Å was observed (Figure 13). Gentile et al. performed the re-docking of N3 peptide inhibitor against SARS-CoV-2 Mpro using AutoDock 4 and observed binding energy of −11.00 kcal/mol and superimposed the re-docked complex onto co-crystallized N3-Mpro complex with RMSD of 0.254 Å were reported (Gentile et al., 2020). The complex was then found to interact with the same amino acid residues compared to the ones reported in the present study. Mirza and Froeyen performed similar re-docking of N3-Mpro of SARS-CoV-2 and observed covalent interactions with the CYS 145 and HIS 41 residues and the peptide inhibitor with a low RMSD after superimposing onto native complex (Mirza & Froeyen, 2020). Andrade et al. in their in-silico research against SARS-CoV-2 Mpro re-docked the N3-Mpro and observed an RMSD of 1.94 Å after superimposing onto the native co-crystallized complex. Similar to the present study, they reported the contribution of CYS 145 and HIS 41 amino acids in the active site with the peptide inhibitor (Andrade et al., 2020).
Figure 13.

Superimposition of re-docked N3-Mpro (blue) onto co-crystallized complex (red) in the active site using PyMOL (RMSD = 0.625 Å).
The re-docked complex was then superimposed onto the native co-crystallized N3-Mpro using LigPlot + v.2.2 interestingly re-docked complex was superimposed completely onto the native co-crystallized complex without any adjustments. All the atoms of amino acids of both the complexes were superimposed without any constraints. On the whole, there were a total of 14 amino acid residues superimposed. The superimposed 2-dimensional structure is shown in Figure 14, the superimposed amino acids of the complexes are encircled in red. This partially proved the efficiency and validity of the docking protocol (Joshi et al., 2020).
Figure 14.

Re-docked N3-Mpro onto co-crystallized complex using LigPlot + v.2.2 showing superimposed amino acids (red circle).
3.1.2. Docking decoy ligands
Decoy are compounds similar to the active ligands in physical properties like molecular weight, log P values, topological surface area, hydrogen bond donors, etc. (Verdonk et al., 2004). but chemically different from them. This is based on the fact to overcome false positives and enhancing ligand enrichment (Kirchmair et al., 2008). Decoy ligands are assumed that they do not bind to the target molecule (Cereto-Massagué et al., 2012) because the chemical properties are the ones that influences the interactions between a ligand and a target, physical similarities have nothing to do with (Huang et al., 2006). This is an easy technique to assess the protocol and efficiency of the AutoDock 4.2.6 software. Docking a ligand to a target must show high binding energy in comparison with N3-Mpro re-docked complex to prove the docking efficiency. A total of 51 decoy ligands similar to N3 peptide inhibitor were retrieved from the DUD-E web server along with binding energies are reported in Table 6, their binding energies remained between −8.23 to −2.00 kcal/mol (Table 6). Fifty decoy ligands out of fifty-one showed high binding energy compared to the re-docked Mpro-N3 complex, in other words only one decoy ligand had low binding energy (−8.23 kcal/mol) and ranked higher compared to the re-docked complex (−8.12 kcal/mol). Only a negligible quantity of decoy was found to have greater binding affinity than the re-docked complex, hence this confirms the docking efficiency and protocol.
Table 6.
Binding energies of decoy ligands against Mpro.
| S. No | Decoy ligands in smiles format | Binding energy (kcal/mol) |
|---|---|---|
| 1 | Cc1cc(nc(n1)NS(=O)(=O)c2ccc(cc2)NC(=O)CCCCC(=O)Nc3ccc(cc3)S(=O)(=O)[N-]c4nc(cc(n4)C)C)C | −8.23 |
| 2 | Cc1c(c2cc(ccc2[nH]1)OC)CC(=O)Oc3ccc(cc3)C(=O)OCCN4CC[NH+](CC4)CCN5CCOCC5 | −8.11 |
| 3 | COC(=O)[C@H]1CCCN1C(=O)C2 = C[C@H]([C@H]([C@@H](C2)OC(=O)NCCCl)OC(=O)NCCCl)NC(=O)Nc3ccccc3Br | −8.05 |
| 4 | CCOC(=O)Oc1c(cc(cc1OC)C(=O)O[C@@H]2C[C@@H]3C[NH+]4CCc5c6ccc(cc6[nH]c5[C@@H]4C[C@@H]3[C@@H] ([C@@H]2OC)C(=O)OC)OC)OC | −7.99 |
| 5 | c1cc-2c(cc1S(=O)(=O)NCCCCCC(=O)O)C(=O)c3c2ccc(c3)S(=O)(=O)NCCCCC C(=O)[O-] | −7.93 |
| 6 | CCOC(=O)N1CCc2c(sc(c2C(=O)N)NC(=O)c3ccc(cc3)S(=O)(=O)N(CCOC)CCOC)C1 | −7.90 |
| 7 | c1ccc(cc1)OCC[NH+]2CCN(CC2)C[C@@H](COc3ccc(cc3)OC[C@@H](CN4CCN(CC4)CCOc5ccccc5)O)O | −7.90 |
| 8 | c1cc(cnc1)[C@H](C[NH2+]CCc2ccc(cc2)[N-]S(=O)(=O)c3ccc(cc3)n4c(=O)n(nn 4)CCCC5CCCC5)O | −7.85 |
| 9 | c1cnc(nc1)NS(=O)(=O)c2ccc(cc2)NC(=O)CCSCCC(=O)Nc3ccc(cc3)S(=O)(=O)[N-]c4ncccn4 | −7.68 |
| 10 | c1cc(c(cc1C/C(=N/O)/C(=O)NCCSSCCNC(=O)/C(=N\O)/Cc2ccc(c(c2)Br)[O-]) Br)O | −7.65 |
| 11 | CCOC(=O)COc1ccc(cc1OC)C = NNC(=O)C(=O)N/N = C/c2ccc(c(c2)OC)OCC(=O)OCC | −7.63 |
| 12 | c1cc(ccc1C(=O)NNC(=O)CCCN2C(=O)/C(=C\3/C(=O)N(C(=S)S3)CCCC(=O)NNC(=O)c4ccc(cc4)[N+](=O)[O-])/ SC2 = S)[N+](=O)[O-] | −7.60 |
| 13 | c1ccn2c(c1)nc(c(c2 = O)/C = C\3/C(=O)N(C(=S)S3)CCCCCCCCCCC(=O)[O-])NC COCCO | −7.59 |
| 14 | CC(=O)O[C@@H]1CC[C@@]2([C@H](C1)CC[C@@H]3[C@@H]2[C@@H](C[C@@]4([C@@H]3CC[C@@H]4/C(=N\NC(=O)N)/CO[C@@H]5[C@H]([C@@H]([C@H]([C@@H](O5)C(=O)OC)OC(=O)C)OC(=O)C)OC(=O)C)C)O)C | −7.56 |
| 15 | CCOc1cc(ccc1OC(=O)c2cccc(c2)OC)/C = N/NC(=O)c3c(n(nn3)c4c(non4)N)COc5ccc(cc5)F | −7.54 |
| 16 | CCN(CC)S(=O)(=O)c1ccc(cc1)C(=O)NCc2nnc(n2c3ccccc3OC)SCC(=O)Nc4nnc(s4)SCC | −7.51 |
| 17 | c1cc(ccc1C(=O)Nc2nnc(s2)CCOCCOCCc3nnc(s3)NC(=O)c4ccc(cc4)Cl)Cl | −7.30 |
| 18 | CC(=O)OC[C@H]([C@]1(CC[C@@H]2[C@@]1(C[C@@H]([C@@H]3[C@@H]2CC[C@@H]4[C@@]3(CC[C@H](C4)O[C@H]5 [C@@H]([C@H]([C@@H]([C@H](O5)C(=O)OC)OC(=O)C)OC(=O)C)OC(=O)C)C)O)C)O)OC(=O)C | −7.27 |
| 19 | COc1ccc(cc1OC)CNc2nc(n(n2)C(=O)COc3ccc(cc3)Cl)NCc4ccc(c(c4)OC)OC | −7.04 |
| 20 | c1cc(cnc1)[C@H](CNCCc2ccc(cc2)[N-]S(=O)(=O)c3ccc( cc3)n4c(=O)n(nn4)CC CC5CCCC5)O | −6.96 |
| 21 | Cc1ccc(cc1)S(=O)(=O)N(CCN(CCO)S(=O)(=O)c2ccc(cc2)C)CCN(CCO)S(=O)(=O)c3ccc(cc3)C | −6.94 |
| 22 | COC(=O)c1cc(c(nc1C/C(=N\NC(=S)N)/C(=N/c2ccc(c(c2)Cl)Cl)/[O-])C/C(=N /NC(=S)N)/C(=N/c3ccc(c(c3)Cl)Cl)/[O-])C(=O)OC | −6.86 |
| 23 | CCN(CC)C(=O)CSc1nc2ccsc2c(=O)n1CCCC(=O)NCCc3ccc(cc3)S(=O)(=O)N | −6.85 |
| 24 | CCCCOc1ccc(cc1)C(=O)NCc2nnc(n2c3ccc(cc3)Br)SCC(=O)Nc4nnc(s4)SCC | −6.66 |
| 25 | c1ccc(cc1)OCCN2CC[NH+](CC2)C[C@H](COc3ccc(cc3)OC[C@H](CN4CCN(CC4)CCOc5ccccc5)O)O | −6.65 |
| 26 | c1ccc(c(c1)C(=O)NNC(=O)CCCCCN2C(=O)/C(=C\3/C(=O)N(C(=S)S3)CCCCCC(=O)NNC(=O)c4ccccc4[O-])/SC2 = S)O | −6.59 |
| 27 | Cc1c(c(sc1C(=O)OC)NC(=O)CCCS(=O)(=O)c2nc(cc(n2)C(F)(F)F)c3ccc(c(c3)OC)OC)C(=O)N | −6.58 |
| 28 | C/C(=C\CC[C@](C)(C = C)O[C@H]1[C@@H]([C@H]([C@@H]([C@H](O1)COC(=O)/C(=C/CC[C@](C)(C = C)O)/C)O)OC(=O)/C(=C/CC[C@@] (C)(C = C)O)/C)O)/C(=O)O | −6.55 |
| 29 | CCOC(=O)[C@@](C(F)(F)F)(NC(=O)Nc1ccc(cc1)S(=O)(=O)[N-]c2nnc(s2)SCC )OCC(F)(F)F | −6.46 |
| 30 | Cc1cc(no1)C(=O)N[C@@H](C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](C[C@H]2CCNC2 = O)/C = C/C(=O)OC | −6.34 |
| 31 | COc1cc(cc(c1OC)OC)/C = C/C(=O)O[C@H]2[C@@H]([C@H](O[C@@]2(CO)O[C@@H]3[C@@H]([C@H]([C@@H]([C@H](O3)COC (=O)c4ccc(cc4)O)O)O)O)CO)O | −6.32 |
| 32 | CC(=O)OC/C(=N\NC(=O)N)/[C@]1(CC[C@H]2[C@]1(CC(=O)[C@@H]3[C@@H]2CC[C@@H]4[C@@]3(CC[C@H](C4)O[C@H]5[C@@H]([C@H] ([C@@H]([C@@H](O5)C(=O)OC)OC(=O)C)OC(=O)C)OC(=O)C)C)C)O | −6.00 |
| 33 | CCN(CC)S(=O)(=O)c1ccc(cc1)C(=O)NCc2nnc(n2c3ccccc3OC)SCC(=O)Nc4ccc(cc4)OC | −5.86 |
| 34 | COc1c(c(cc(c1)/C = [NH+]/CCCN2CCN(CC2)CCC/[NH+]=C/c3cc(c(c(c3)OC)OC)OC)OC)OC | −5.83 |
| 35 | C/C(=C\CC[C@](C)(C = C)O[C@H]1[C@@H]([C@H]([C@@H]([C@H](O1)CO)OC(=O)/C(=C/CC[C@](C)(C = C)O)/C)OC(=O)/C(=C/CC[C@@](C) (C = C)O)/C)O)/C(=O)O | −5.65 |
| 36 | CC(=O)O[C@@H]1[C@H]([C@@H](O[C@@H]([C@@H]1O)O[C@@H](C/N = C/c2c3ccccc3ccc2O)[C@@H](CO)O[C@@H](C/ N = C/c4c5ccccc5ccc4[O-])O) CO)O | −5.59 |
| 37 | CCOC(=O)c1ccc(cc1)NC(=O)CSc2nnc(n2c3cc(ccc3OC)OC)CNC(=O)c4ccc(cc4)S(=O)(=O)N(C)C | −5.53 |
| 38 | C/C(=N\O)/C(NCCC(CCNC(/C(=N/O)/C)(C)C)CCNC(=O)CCCC(=O)Oc1c(c(c(c(c1F)F)F)F)F)(C)C | −5.52 |
| 39 | CCCCC/C = C\C/C = C\C = C/C = C\[C@H]([C@H](CCCC(=O)OC)O)SC[C@](C(=O)NCC(=O)[O-])([NH3+])N1[C@@H](CCC1 = O)C(=O)[O-] | −5.49 |
| 40 | C/C = C(\C[C@@H](N(Cc1c[nH+]c(nc1N)C)C = O)O)/SS/C(=C\C)/C[C@@H](N(Cc2cnc(nc2N)C)C = O)O | −5.47 |
| 41 | c1c(csc1COc2c3c(nc(n2)N)n(cn3)CCCCCCCCO[C@@H]4[C@H]([C@@H]([C@@H]([C@@H](O4)CO)O)O)O)Br | −5.17 |
| 42 | CCO[P@@](=O)(CCCCCCCCCC(=O)NCCCCCNC(=O)CCCC[C@@H]1[C@@H]2[C@@H](CS1)NC(=O)N2)[O-] | −4.87 |
| 43 | c1ccc(c(c1)C(=O)[O-])NC(=O)CCCCCN2C(=O)/C(=C\3/C(=O)N(C(=S)S3)CCC CCC(=O)Nc4ccccc4C(=O)[O-])/SC2 = S | −4.87 |
| 44 | C/C = C(\C[C@H](N(Cc1cnc(nc1N)C)C = O)O)/SS/C(=C\C)/C[C@@H](N(Cc2cnc(nc2N)C)C = O)O | −4.70 |
| 45 | c1cc(c(cc1C[C@H](C(=O)[O-])OC(=O)[C@@H]2[C@@H]([C@@H]([C@@H] 2C(=O)O[C@@H](Cc3ccc(c(c3)O)O)C(=O) [O-])c4ccc(c(c4)O)O)c5ccc(c(c5)O) O)O)O | −4.60 |
| 46 | CC1(c2ccccc2[N+](=C1/C = C/C = C/3\C(c4ccccc4N3CCCCC(=O)NCCOCCOCC[NH2+]CCO[C@@H]5[C@H]([C@H]([C@@H]([C@@H](O5)CO)O) O)O)(C)C)C)C | −4.42 |
| 47 | CC(C)(C)OC(=O)C[NH+](CC(=O)OC(C)(C)C)[C@@H](Cc1ccc(cc1)N)CN(CC(=O)OC(C)(C)C)CC(=O)OC(C)(C)C | −4.29 |
| 48 | COc1cc(c(c(c1OC)OC)S(=O)(=O)NCCOc2ccccc2)S(=O)(=O)NCCOc3ccccc3 | −3.80 |
| 49 | CC[C@H]([C@](C)(/C = C(\C)/C(=O)[C@H](C)C[C@@](C)([C@@H]([C@@H](C)[C@@H]([C@@H](C)C(=O)[O-])O[C@H]1C[C@@]([C@H] ([C@@H](O1) C)O)(C)OC)O[C@H]2[C@@H]([C@H](C[C@H](O2)C)[NH+](C)C)O)OC)O)O | −3.55 |
| 50 | CCCCCCCCCC(=O)O[C@@H]1[C@H]([C@@H]([C@@H](O[C@@H]1O[C@@]2([C@@H]([C@@H]([C@@H](O2)CO)O)O)CO)CO)OC(=O)C(C)C)OC (=O)C(C)C | −3.08 |
| 51 | CCCCCCCCCCCCCCCCO[C@H]1[C@@H]([C@H]([C@@H]([C@H](O1)CO)O[C@@H]2[C@@H]([C@H]([C@@H]([C@H](O2)CO)O)O)O)O)O | −2.00 |
3.2. Pharmacokinetic prediction and lipinski’s rule of 5
Pharmacokinetic and Lipinski properties of the top 10 best potential antiviral ligands were predicted, studied, and tabulated (Tables 7–9). It is clear that except (-)-solenolide A and rhinacanthin E, none penetrated the Blood-brain barrier. Mitochondria is the subcellular location for the distribution of all the compounds after entering the human body.
Table 7.
Pharmacokinetic properties of top ten ligands with high binding energies.
| S. No | Compound | GI absorption | BBB permeation | Caco-2 permeability | P-glycoprotein substrate | P-glycoprotein inhibitor | Subcellular localization | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Log KpSkin permeation (cm/s) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Theaflavin-3-3′- digallate | Yes | No | No | No | Yes | Mitochondria | No | No | No | No | No | −9.31 | |
| 2 | Rutin | Yes | No | No | No | No | Mitochondria | No | No | No | No | No | −10.26 | |
| 3 | Hypericin | Yes | No | No | No | No | Mitochondria | Yes | Yes | Yes | No | Yes | −5.32 | |
| 4 | Robustaflavone | Yes | No | No | No | Yes | Mitochondria | Yes | Yes | Yes | No | Yes | −6.01 | |
| 5 | (-)-Solenolide A | Yes | Yes | No | Yes | Yes | Mitochondria | No | No | No | No | No | −7.32 | |
| 6 | Rhusflavone | Yes | No | No | No | Yes | Mitochondria | Yes | Yes | Yes | No | Yes | −6.42 | |
| 7 | Ginkgetin | Yes | No | No | No | Yes | Mitochondria | Yes | Yes | Yes | No | Yes | −5.72 | |
| 8 | Rhinacanthin E | Yes | Yes | Yes | No | Yes | Mitochondria | Yes | Yes | Yes | No | Yes | −6.45 | |
| 9 | Sorbarin | Yes | No | No | No | No | Mitochondria | No | No | No | No | No | −8.22 | |
| 10 | Betulinic acid | Yes | No | No | No | No | Mitochondria | No | No | No | No | No | −3.26 | |
Table 8.
Drug likeliness of top 10 ligands with high binding energies.
| S. No | Compound | Lipinski | Ghose | Veber | Egan | Muegge | Bioavailability score |
|---|---|---|---|---|---|---|---|
| 1 | Theaflavin-3-3′-digallate | No | No | No | No | No | 0.17 |
| 2 | Rutin | No | No | No | No | No | 0.17 |
| 3 | Hypericin | No | No | No | No | No | 0.17 |
| 4 | Robustaflavone | No | No | No | No | No | 0.17 |
| 5 | (-)-Solenolide A | Yes | No | Yes | No | Yes | 0.55 |
| 6 | Rhusflavone | No | No | No | No | No | 0.17 |
| 7 | Ginkgetin | Yes | No | No | No | No | 0.55 |
| 8 | Rhinacanthin E | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 9 | Sorbarin | Yes | Yes | No | No | No | 0.55 |
| 10 | Betulinic acid | Yes | No | Yes | No | No | 0.56 |
Table 9.
Toxicity properties of top 10 ligands with high binding energies.
| S. No | Compound | AMES toxicity | Acute oral toxicity (kg/mol) | Carcinogenicity | Hepatotoxicity | Tetrahymena Pyriformis
Toxicity pIGC50, µg/L |
Rat acute toxicity LD50, mol/kg | Biodegradation |
|---|---|---|---|---|---|---|---|---|
| 1 | Theaflavin-3-3′-digallate | No | 2.952 | No | Yes | 0.653 | 2.669 | No |
| 2 | Rutin | No | 2.593 | No | Yes | 1.200 | 2.593 | No |
| 3 | Hypericin | No | 1.010 | No | Yes | 2.175 | 2.687 | No |
| 4 | Robustaflavone | No | 1.529 | No | Yes | 1.143 | 3.123 | No |
| 5 | (-)-Solenolide A | No | 4.951 | No | Yes | 0.847 | 2.566 | No |
| 6 | Rhusflavone | No | 2.712 | No | Yes | 0.917 | 3.643 | No |
| 7 | Ginkgetin | No | 2.492 | No | Yes | 1.046 | 2.898 | No |
| 8 | Rhinacanthin E | No | 2.185 | No | Yes | 0.809 | 3.045 | No |
| 9 | Sorbarin | No | 2.669 | No | Yes | 1.027 | 2.545 | No |
| 10 | Betulinic acid | No | 3.382 | No | No | 1.575 | 3.891 | No |
(-)-Solenolide A, ginkgetin, rhinacanthin E, sorbarin, and betulinic acid satisfied their properties according to Lipinski’s rule of 5 (Table 10). Lipinski rule is considered one of the essential criteria to predict the oral drug likeliness of a drug (Benet et al., 2016). To enhance the predictions of drug likeliness, Ghose, Veber, Egan, and Muegge filters have been used in the study. Rhinacanthin E with bioavailability score of 0.55 is the only compound that satisfied all the rules used to predict the drug likeliness. Theaflavin-3-3′-digallate, rutin, hypericin, and robustaflavone did not follow any of the rules for oral drug likeliness, however, these lead molecules upon in-vitro, in-vivo, and clinical trials, can be administered through other routes of drug administration namely, intravenous, intranasal, intraperitoneal, and subcutaneous. None of the drugs are either mutagenic or carcinogenic. Every compound is hepatotoxic if administered above prescribed limits.
Table 10.
Lipinski properties of ligands.
| S. No | Compound | Molecular weight in g/mol (<500 Da) | Log P (<5) | H-bond donor (<5) | H-bond acceptor (<10) | Molar refractivity (<130) |
|---|---|---|---|---|---|---|
| 1 | Theaflavin-3-3′-digallate | 868.7 | 0 | 13 | 20 | 0 |
| 2 | Rutin | 610.5 | 0 | 10 | 16 | 0 |
| 3 | Hypericin | 504.4 | 1.15 | 6 | 10 | 49.73 |
| 4 | Robustaflavone | 538.5 | −0.02 | 6 | 10 | 121.82 |
| 5 | (-)-Solenolide A | 555.1 | −0.69 | 2 | 9 | 107.71 |
| 6 | Rhusflavone | 542.5 | −0.22 | 6 | 10 | 122.19 |
| 7 | Ginkgetin | 566.5 | −1.04 | 4 | 10 | 124.72 |
| 8 | Rhinacanthin E | 442.4 | 1.74 | 0 | 9 | 85.23 |
| 9 | Sorbarin | 432.4 | −1.91 | 6 | 10 | 89.83 |
| 10 | Betulinic acid | 456.7 | 0.42 | 2 | 3 | 98.49 |
Theaflavin derivatives, theaflavin (TF1), theaflavin-3-monogallate (TF2A), theaflavin-3′-monogallate (TF2B), and theaflavin-3-3′-digallate (TF3) extracted from black tea, was proven to be an anti-oxidant, anti-cancer, anti-inflammatory, and antiviral agents. Determining the anti-HSV1-1 effect on Vero and A549 cells, TF3 showed the best activity among other theaflavin derivatives with a half-maximum effective concentration (EC50 = 20 µM) and selective index (SI = 4). The cytotoxicity of TF3 was evaluated via MTS assay showing non-toxicity up to 75 µM, with a hindrance in cell proliferation at the rate of 2.75%. The CC50 value was obtained as 112.5 µM, moreover, TF3 showed the best antiviral potency when compared to other derivatives (De Oliveira et al., 2005). Rutin, water-insoluble ubiquitous flavonoid was studied in-vitro regarding its potent antiviral ability against the Dengue virus type-2 (DENV-2). The 50% cytotoxicity value of the compound via MTT assay was evaluated as >1000 µg/mL (CC50). Thus, showing no significant cytotoxicity against Vero cells (Keivan et al., 2011; Silva et al., 2011). Hypericin, with effective inhibitory activity against Retrovirus, had a low in-vitro cytotoxic activity with the concentration enough to show antiviral effect in Friend virus-induced murine tissue culture. With prolonged exposure at a dose of <50µg/mL, for almost 9 days, the viability of the culture was retained along with the proliferation rate (Meruelo et al., 1988). Rhusflavone exhibited significant antiviral activities against the Influenza virus, measles, and HSV-2 viruses with SI values of 9.3, 8, and >6.4. The lower EC50 value of the rhusflavone against these viruses indicates high potency at low concentrations. Robustaflavone has a significantly higher SI value (438) against the Influenza B virus and slightly moderate activity against influenza virus B and found to be the most potent antiviral drug among the screened compounds (Lin et al., 1999). Robustaflavone was reported against HIV-1 reverse transcriptase (RT), with IC50 values of 65 mM, and proved to be an efficient drug component (Perez, 2003). Ginkgetin had lower cytotoxicity and was found to be a potent inhibitor against the influenza virus even at lower concentrations when used as a conjugate with sialic acid (Miki et al., 2007). Ginkgetin caused a dose-dependent inhibition of virus replication with a 50% cytotoxic activity at 12.8 pg/mL and 50% anti-HSV-1 activity at 0.91ug/ml, the therapeutic index 14.1. Ginkgetin also showed inhibitory effects against HSV type 2 and human cytomegalovirus with therapeutic indices of 13.8 and 11.6, respectively. Ginkgetin had a weak viricidal activity against HSV-1 at more than 5 µg/mL. Both adsorptions of HSV-1 to host cells and virus penetration into cells were unaffected by this agent. Ginkgetin suppressed viral protein synthesis when added at various steps of HSV-1 replication and exerted strong inhibition of transcription of the immediate-early genes (Hayashi et al., 1992). Rhinacanthin E exhibited significant antiviral activity against influenza type A virus (Flu-A) when tested in a hemadsorption-inhibition assay and in viral cytopathic effect assay. Under the assay conditions used, the compound exhibited some cytotoxicity, but at concentrations well above the antiviral endpoints (Kernan et al., 1997). Rhinacanthin E exhibits antiviral activity against the influenza PR8 virus with CC50 >50 (Ngoc et al., 2019). Betulinic acid, a natural triterpene is associated with anti-cancer, anti-bacterial, and anti-malarial properties. On estimating the anti-viral herpesviral activity, it was found that the betulinic acid showed compelling inhibition, with IC50 = 1.6 µM. The cytotoxicity examined in Vero cells was found to be CC50 greater than 100 µM (Visalli et al., 2015). Thus, natural phytocompounds are not naturally occurring and reported negligible toxicity when tested in-vitro, hence could be a promising drug candidate and can be tested in-vitro then in-vivo.
3.3. Pharmacokinetics of drugs that are currently repurposed against COVID-19
Pharmacokinetic properties of the drug that are currently repurposed against COVID-19 is reported in table 11. Of these drugs, except favipiravir, festinavir, and remdesivir, the rest of the other drugs are approved by the FDA. None of these drugs were carcinogenic. No drugs inhibited the cytochromes. Azithromycin and remdesivir were P-glycoprotein inhibitors.
Table 11.
Pharmacokinetic properties of drugs currently repurposed.
| S. No | Compound | GI absorption | BBB permeation | Caco-2 permeability | P-glycoprotein substrate | P-glycoprotein inhibitor | Subcellular localization | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Log KpSkin permeation (cm/s) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Dexamethasone | Yes | Yes | Yes | Yes | No | Mitochondria | No | No | No | No | No | −7.32 |
| 2 | Hydroxychloroquine | Yes | Yes | Yes | Yes | No | Lysosomes | No | No | No | No | No | −5.81 |
| 3 | Festinavir | Yes | Yes | No | No | No | Mitochondria | No | No | No | No | No | −8.32 |
| 4 | Oseltamivir | Yes | Yes | No | Yes | No | Mitochondria | No | No | No | No | No | −7.42 |
| 5 | Chloroquine | Yes | Yes | Yes | Yes | No | Lysosomes | No | No | No | No | No | −4.96 |
| 6 | Remdesivir | Yes | Yes | No | Yes | Yes | Lysosomes | No | No | No | No | No | −8.62 |
| 7 | Azithromycin | No | No | No | Yes | Yes | Lysosomes | No | No | No | No | No | −8.01 |
| 8 | Favipiravir | Yes | Yes | No | No | No | Mitochondria | No | No | No | No | No | −7.66 |
Drugs dexamethasone, hydroxychloroquine, oseltamivir, and chloroquine satisfied all the drug likeliness properties. Festinavir and favipiravir did not satisfy the Ghose filter respectively. Azithromycin and remdesivir did not satisfy any of the drug likeliness properties (Table 12). The Lipinski’s rule of 5 is one the essential criteria for oral drug likeliness, that was not satisfied by remdesivir and azithromycin with 2 violations each.
Table 12.
Drug likeliness of drugs currently repurposed.
| S. No | Compound | Lipinski | Ghose | Veber | Egan | Muegge | Bioavailability score |
|---|---|---|---|---|---|---|---|
| 1 | Dexamethasone | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 2 | Hydroxychloroquine | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 3 | Festinavir | Yes | No | Yes | Yes | Yes | 0.55 |
| 4 | Oseltamivir | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 5 | Chloroquine | Yes | Yes | Yes | Yes | Yes | 0.55 |
| 6 | Remdesivir | No | No | No | No | No | 0.17 |
| 7 | Azithromycin | No | No | No | No | No | 0.17 |
| 8 | Favipiravir | Yes | No | Yes | Yes | No | 0.55 |
Hydroxychloroquine and chloroquine share similarities with each other and found to be AMES toxic, from the two, chloroquine is more toxic compared to hydroxychloroquine, hence hydroxychloroquine has the greater therapeutic ratio (Table 13) (Wallace et al., 2012). These two drugs in S(+) hydroxychloroquine and S(+) chloroquine forms had significantly contributed renal failure at high dosage compared to S(-) hydroxychloroquine and S(-) chloroquine forms (Salazar-Bookaman et al., 1994). The dosage of chloroquine is often given lower when compared to hydroxychloroquine. Chloroquine, favipiravir, festinavir, and remdesivir were reported to be hepatotoxic. Azithromycin, an antibiotic was found to be toxic causing phospholipidosis during clinical trials in rats (30 mg/kg dose) was reported (Patel et al., 2019). None of the natural compounds did report any toxicity and mutagenic property. Lethal doses (LD50) of all the natural compounds were higher when compared to chemical drugs, which denotes that even at a higher dosage, natural compounds are less toxic compared to chemically synthesized drugs. Thus, chemical drugs are toxic from the pharmacokinetic predictions compared to natural compounds, moreover, natural compounds have shown potential against several diseases with the least side effects (Ardalan & Rafieian-Kopaei, 2013).
Table 13.
Toxicity properties of drugs currently repurposed.
| S. No | Compound | AMES toxicity | Acute oral toxicity (kg/mol) | Carcinogenicity | Hepatotoxicity | Tetrahymena Pyriformis
Toxicity pIGC50, µg/L |
Rat acute toxicity LD50, mol/kg | Biodegradation |
|---|---|---|---|---|---|---|---|---|
| 1 | Dexamethasone | No | 2.486 | No | No | 0.737 | 2.148 | No |
| 2 | Hydroxychloroquine | Yes | 2.665 | No | No | 1.818 | 2.634 | No |
| 3 | Festinavir | No | 2.025 | No | Yes | 0.925 | 2.090 | No |
| 4 | Oseltamivir | No | 2.713 | No | No | 0.276 | 2.269 | No |
| 5 | Chloroquine | Yes | 2.684 | No | Yes | 1.891 | 2.954 | No |
| 6 | Remdesivir | No | 3.428 | No | Yes | 1.645 | 2.717 | No |
| 7 | Azithromycin | No | 3.202 | No | No | 0.524 | 2.542 | No |
| 8 | Favipiravir | No | 1.780 | No | Yes | 0.675 | 2.126 | No |
3.4. Molecular dynamics simulations
To investigate the stability and dynamics of the free protein (PDB Id: 6LU7), protein-N3 co-crystallized complex, and top 3 ligand-protein complexes that showed high affinity low binding energy were simulated for 50 ns to analyse the RMSD and RMSF (Figure 15 (i to x)). Lower the RMSD, greater the stability of protein (Aier et al., 2016). Mpro of Covid-19 was considered as control in our MD simulation calculations, protein and protein-ligand co-crystallised complex of Mpro were found to be stable within 50 ns of simulation. The RMSD of free protein was found to increase initially and reached 2.25 Å at 20 ns and later the RMSD decreased slightly to 2 Å and remained constant for the remaining simulation and no significant deviation in the RMSF of each amino acid residue measured with respect to Cα carbon atom was observed during the simulation (Figure 15(i)). The highest fluctuation was observed in the ASP 155 with RMSF of 2 Å possibly due its high flexibility due to the formation of β-turn in the protein structure at that position (Huang et al., 2003) (Figure 15(ii)).
Figure 15.

Molecular dynamics RMSD and RMSF of free Mpro (i, ii); N3-Mpro co-crystallized complex (iii, iv); theaflavin-3-3’-digallte (v, vi); rutin (vii, viii); hypericin (ix, x).
For the MD of protein and ligand complexes, the response was studied in terms of protein and ligand RMSD values that is generally used to measure the scalar distance for the protein (Cα backbone) and ligand throughput the trajectory. For the protein-N3 co-crystallized complex, the protein attained a maximum RMSD value of 3.1 Å at 35 ns and decreased further increase in the time up to 50 ns and became stable. For the same complex, the N3 ligand underwent fluctuations initially and reached 4.5 Å and became stable touched 4.0 Å at 50 ns (Figure 15(iii)). The RMSF of the complex remained less than 2 Å throughout the simulation (Figure 15(iv)). In comparison with control, three of our test complexes viz., theflavin-3-3-digallate, rutin, and hypericin performed well and were stable.
Theaflavin-3-3-digallate-protein also performed well, and there was no much fluctuation observed in the complex during MD simulation. The protein RMSD remained stable, and the maximum deviation was observed at 37 ns (2.5 Å) that later got stabilized (Figure 15(v)). The ligand RMSD was found maximum at 13 ns and further got stable for the rest of the simulation time. The maximum fluctuation in RMSF was observed in the residue GLY 275 probably (Figure 15(vi)) due to its high flexibility because of the proton side chain and backbone φ and ψ angles responsible for the degree of rotation (Koča et al., 1994).
In the rutin-protein complex, the protein was stable till 35 ns within 2.4 Å, then fluctuated slightly and became stabilized after 45 ns remained within ∼2 Å. The ligand showed a little fluctuation but got stable till 10 ns and then got a sharp jump in RMSD at 42 ns which further came down (Figure 15(vii)). The RMSF of the rutin-protein complex was stable with fewer fluctuations. Among the three test complexes, hypericin-protein showed a little fluctuation in RMSD. The highest RMSF of 2.55 Å was observed towards the terminal end of the protein, SER 301, a flexible amino acid (Figure 15(viii)).
For the hypericin-protein complex, the protein RMSD fluctuated initially at attained peak at 14 ns with 2.7 Å and got stabilized for rest of the simulation. The ligand RMSD was stable throughout the simulation and fluctuated slightly after 45 ns and was stabilized on reaching 50 ns (Figure 15 (ix)). Hypericin fluctuated throughout the simulation was reported by Islam et al. (Islam et al., 2020). Similar to theaflavin-3-3′-digallate, the RMSF values during the simulation was observed highest in GLY 275 however, the complex was stable (Figure 15(x)).
Protein-ligand interactions provided a better insight of simulation, elucidated the conformational stability, and correlated well. CYS 145, HIS 41, and GLU 166 were among the top critically important residue which was observed to play a predominant role in ligand binding (Figure 16(i–viii)). CYS 145 worked as a nucleophile and has its major role in the formation of a covalent bond with the ligand. However, we were not focussing on covalent inhibition in the present work. In the N3-Mpro complex, GLU 166 donated side chain hydrogen bonds 54%, 67%, and 68% of the time to nitrogen (lone pair electrons), oxygen, and hydrogen atoms of the inhibitor (Figure 16 (i)). THR 190 formed bonds with N-H 94% of the time with the ligand. GLY 143 and SER 144 donated their side chain hydrogen bonds 32% and 51% of the time with a hydroxyl group on the ligand. Lowering the contact strength further less than 50%, water momentarily starts to form bridge interactions. A few hydrophobic interactions were formed with the MET 49, CYS 145, MET 165, and PRO 168 residues (Figure 16(ii)). All three test complexes were found to interact with HIS 41 and GLU 166 with an average occupancy of more than 50% throughout the simulation. Theaflavin-3-3-digallate was found to interact with GLU 166 and HIS 41 with more than 80% and along with these two, it was also able to engage ASP 187 with 86% and a bridge between ligand-water-ASN 119 and THR 26 with 81% and 79% (Figure 16(iii)). Moreover, water molecules started to interact with the residues. Hydrophobic interactions were formed with MET 49 and CYS 145 residues (Figure 16(iv)).
Figure 16.

Protein-ligand contact plots and ligand-protein interaction residues of co-crystallized complex (i, ii); theaflavin-3-3’-digallate (iii, iv); rutin (v, vi); hypericin (vii, viii).
Rutin and hypericin were able to form intramolecular bonds throughout the simulation. Rutin formed a stable interaction with GLU 166 and was found to interact with the hydroxyl group for 98% and donated its side chain to oxygen tom for 66% of the time (Figure 16(v)). THR 190 and HIS 41 residues donated their hydrogen bond side chains 71% and 54% of the time with a hydroxyl group in the ligand. GLN 189 interacted 47% with an oxygen atom of the ligand. MET 49, MET 165, and PRO 168 formed hydrophobic bonds (Figure 16(vi)). Hypericin was found to form a water network with GLU 166 which was stable for 33% and oxygen atom with HIS 41 for 84% throughout the simulation (Figure 16(vii)). Similar to other compounds, hydrophobic interactions were formed by CYS 145, MET 165, LEU 167, and PRO 168 residues (Figure 16(viii)). In summary, many hydrogen bonds were formed between GLU 166, HIS 41, and the protein was observed, thus contributing to high binding affinity. Comparing the results with our Control (PDB Id: 6LU7), theaflavin-3-3′-digallate was found to be more stable and could be further explored for in-vitro or in-vivo studies.
4. Conclusion
Till today there is no cure for SARS-CoV-2 disease, research ongoing in developing lead molecules and precursors that could act as potential antiviral drugs against the disease. The motive of the present study was to discover natural compounds that could be potential antiviral agents to inhibit the viral SARS-CoV-2 Mpro (PDB Id: 6LU7), thereby neutralizing its virulence. Two hundred potential antiviral natural compounds previously reported were surveyed from the literature and databases. The active site of the protease was determined using the MetaPocket 2.0 online server. Molecular docking was done using AutoDock 4.2.6 along with supporting software and Discovery Studio 3.5 to elucidate the interactions between the ligands and Mpro. Out of two hundred compounds docked, the top ten compounds with high binding energies were reported. Theaflavin-3-3′-digallate (-12.41 kcal/mol), rutin (-11.33 kcal/mol), hypericin (-11.17 kcal/mol), robustaflavone (-10.92 kcal/mol), and (-)-solenolide A (-10.81 kcal/mol) have shown potential to inhibit the protease. The study was compared by docking Mpro with FDA approved viral protease inhibitors. Drugs like atazanavir (-13.24 kcal/mol), saquinavir (-12.74 kcal/mol), and darunavir (-12.50 kcal/mol) were found to inhibit the protease very effectively and the interactions were compared with natural compounds. The study also analyzed the efficiency of drugs that are currently repurposed against COVID-19 using docking studies. Pharmacokinetics and toxicity properties of natural compounds and drugs repurposed against COVID-19 were reported and studied. Even though FDA approved drugs possessed high binding energies, the number of hydrogen bonds formed with Mpro were found to be less when compared to the hydrogen bonds formed with natural compounds used in the study. Natural compounds like flavonoids, terpenoids, alkaloids, phenolics, tannins, and saponins are plant metabolites and did not possess any mutagenic and carcinogenic properties. There are least or no side effects caused by natural compounds. (-)-Solenolide A, ginkgetin, rhinacanthin E, sorbarin, and betulinic acid satisfied the Lipinski rule for oral drug likeliness. Rhinacanthin E satisfied all the filters used for assessing the drug likeliness. Molecular dynamics were performed for 50 ns to assess the stability and flexibility using the Desmond package, Schrödinger and the results reported that the protein and ligand complexes were stable throughout the simulation. On the whole, phytocompounds that are a part of our day-to-day diet were found to be very potent antiviral candidates against COVID-19, hence one could prevent the infections using these. This research could act as a road map for the discovery of natural antiviral compounds. Thus, in-silico studies provided rapid and comprehensive insights into results in screening a library of compounds. Future studies will focus on the other virulent protein of SARS-CoV-2 though in-silico studies. In-vitro and in-vivo clinical trials with the top compounds reported in the study that has shown potential towards inhibiting the protease. Advancements in bio nanotechnology have led to the development of targeted drug delivery systems using nano-synthesized metal, metal oxide, and polymeric nanoparticle carriers. Nanomaterials have enhanced electrical, electrical, optical, physical, and chemical properties, high surface area, and permeability. Future studies will also focus on natural compounds as capping and reducing agents onto metal nanoparticles which will surely provide positive insights towards the cure of infection.
Supplementary Material
Acknowledgements
The authors sincerely thank Vels Institute of Science, Technology and Advanced Studies for the successful completion of the research work, Mr. Suyash Pant, National Institute of Pharmaceutical Education and Research, Kolkata, West Bengal, and Kumaun University, Bhimtal campus for performing Molecular Dynamics Simulations. We are also grateful to Dr. Priyanka Purkayastha, Senior Data Engineer, FedEx, the Netherlands to help attain publishable status and reviewers for providing valuable suggestions.
Glossary
Abbreviations
- µM
micromolar
- nM
nanomolar
- pM
picomolar
- µg
microgram
- pg
picogram
- mµM
millimicron molar
- 3CLpro
3-chymotrypsin-like protease
- ACE-2
Angiotensin-converting enzyme-2
- BatCoV RaTG13
Bat Corona Virus
- COVID-19
Corona Virus Disease-2019
- FDA
Food and Drug Administration
- HPLC
High Performance Liquid Chromatography
- HSV-1, HSV-2
Herpes Simplex Virus-1, 2
- IC50
Inhibitory Constant 50
- LD50
Lethal Dosage
- JEV
Japanese Encephalitis Virus
- OPLS
Optimized Potential for Liquid Simulations
- AMBER
Assisted Model Building with Energy Refinement
- kcal/mol
Kilocalories per mole
- Ki
Inhibition constant
- LGA
Lamarckian Genetic Algorithm
- GA
Genetic Algorithm
- Mpro
Main-Protease
- NMR
Nuclear; Magnetic Resonance
- Nsp5
Non-structural protein 5
- PDB
Protein Data Bank
- RNA
Ribonucleic Acid
- SARS-CoV-1
Severe Acquired Respiratory Syndrome-Corona Virus-1
- SARS-CoV-2
Severe Acquired Respiratory Syndrome-Corona Virus-2
- WHO
World Health Organization
- GPF
Grid Parameter File
- GLG
Grid Log File
- DPF
Docking Parameter File
- DLG
Docking Log File
- MD
Molecular Dynamics; Single-Point Charge
- SPC; PBC
Periodic Boundary Conditions
- PME
Particle -Mesh Ewald
- RMSD
Root Means Square Deviation
- RMSF
Root Mean Square Fluctuation
- TF1
Theaflavin
- TF2A
Theaflavin-3-monogallate
- TF2B
Theaflavin-3’-monogallate
- TF3
Theaflavin-3-3’-digallate
- HIV
Human Immuno Virus
- MTT
(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- SI
Selective Index
Disclourse statement
The authors declare a conflict of interest as none.
References
- Aier, I., Varadwaj, P. K., & Raj, U. (2016). Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Scientific Reports, 6(1), 34984.Mismatch] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khodairy, F. M., Khan, M. K., Kunhi, M., Pulicat, M. S., Akhtar, S., & Arif, J. M. (2013). In Silico prediction of mechanism of Erysolin-induced apoptosis in human breast cancer cell lines. American Journal of Bioinformatics Research, 3, 62–71. [Google Scholar]
- Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature Medicine, 26(4), 450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardalan, M. R., & Rafieian-Kopaei, M. (2013). Is the safety of herbal medicines for kidneys under question? Journal of Nephropharmacology, 2(2), 11–12. [PMC free article] [PubMed] [Google Scholar]
- Beck, B. R., Shin, B., Choi, Y., Park, S., & Kang, K. (2020). Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Computational and Structural Biotechnology Journal, 18, 784–790. 10.1016/j.csbj.2020.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekhit, A. E., & Bekhit, A. A.. Natural antiviral compounds. In Studies in natural products chemistry. 2014 Jan 1. (Vol. 42, pp. 195–228). Elsevier. [Google Scholar]
- Benet, L. Z., Hosey, C. M., Ursu, O., & Oprea, T. I. (2016). BDDCS, the rule of 5 and drugability . Advanced Drug Delivery Reviews, 101, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati, E., Benigni, R., Demarini, D. M., Helma, C., Kirkland, D., Martin, T. M., Mazzatorta, P., Ouédraogo-Arras, G., Richard, A. M., Schilter, B., Schoonen, W. G. E. J., Snyder, R. D., & Yang, C. (2009). Predictive models for carcinogenicity and mutagenicity: Frameworks, state-of-the-art, and perspectives. Journal of Environmental Science and Health. Part C, Environmental Carcinogenesis & Ecotoxicology Reviews, 27(2), 57–90. [DOI] [PubMed] [Google Scholar]
- Beura, S., & Prabhakar, C. (2020). In-silico strategies for probing chloroquine based inhibitors against SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 29, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch, B. J., van der Zee, R., de Haan, C. A., & Rottier, P. J. (2003). The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. Journal of Virology, 77(16), 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman, R., Brandman, Y., & Pande, V. S. (2012). A-site residues move independently from P-site residues in all-atom molecular dynamics simulations of the 70S bacterial ribosome. PloS One, 7(1), e29377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, D., Wang, J., Zhou, R., Li, Y., Yu, H., & Hou, T. (2012). ADMET evaluation in drug discovery. 11. PharmacoKinetics Knowledge Base (PKKB): A comprehensive database of pharmacokinetic and toxic properties for drugs. Journal of Chemical Information and Modeling, 52(5), 1132–1137. [DOI] [PubMed] [Google Scholar]
- Carvalho, O. V., Botelho, C. V., Ferreira, C. G. T., Ferreira, H. C. C., Santos, M. R., Diaz, M. A. N., Oliveira, T. T., Soares-Martins, J. A. P., Almeida, M. R., & Silva, A. (2013). In vitro inhibition of canine distemper virus by flavonoids and phenolic acids: Implications of structural differences for antiviral design. Research in Veterinary Science, 95(2), 717–724. [DOI] [PubMed] [Google Scholar]
- Cereto-Massagué, A., Guasch, L., Valls, C., Mulero, M., Pujadas, G., & Garcia-Vallvé, S. (2012). DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics (Oxford, England)), 28(12), 1661–1662. [DOI] [PubMed] [Google Scholar]
- Chen, C. N., Lin, C. P., Huang, K. K., Chen, W. C., Hsieh, H. P., Liang, P. H., & Hsu, J. T. (2005). Inhibition of SARS-CoV 3C-like protease activity by Theaflavin-3,3'-digallate (TF3) ). Evidence-Based Complementary and Alternative Medicine :Ecam, 2(2), 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., Muhammad, I., Zhang, Y., Ren, Y., Zhang, R., Huang, X., Diao, L., Liu, H., Li, X., Sun, X., Abbas, G., & Li, G. (2019). Antiviral activity against infectious bronchitis virus and bioactive components of Hypericum perforatum L. Frontiers in Pharmacology, 10, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury, P., Sahuc, M. E., Rouillé, Y., Vandeputte, A., Brodin, P., Goswami, M., Bandyopadhyay, T., Dubuisson, J., & Seron, K. (2018). Theaflavins, polyphenols of black tea, inhibit entry of hepatitis C virus. BioRxiv, 325126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva, J. A. (2020). Convalescent plasma: A possible treatment of COVID-19 in India. Medical Journal Armed Forces India, 76(2), 236–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina, A., & Zoete, V. (2016). A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules . ChemMedChem., 11(11), 1117–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2014). iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach . Journal of Chemical Information and Modeling, 54(12), 3284–3301. Dec 22 [DOI] [PubMed] [Google Scholar]
- Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang, V. T., Benkendorff, K., & Speck, P. (2011). In vitro antiviral activity against herpes simplex virus in the abalone Haliotis laevigata. Journal of General Virology, 92(3), 627–637. 10.1099/vir.0.025247-0 [DOI] [PubMed] [Google Scholar]
- Das, S., Sarmah, S., Lyndem, S., & Roy, S. (2020). A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 30, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood, F. S., Jain, S., Finelli, L., Shaw, M. W., Lindstrom, S., Garten, R. J., Gubareva, L. V., Xu, X., Bridges, C. B., & Uyeki, T. M. (2009). Emergence of a novel swine-origin influenza A (H1N1) virus in humans. The New England Journal of Medicine, 360(25), 2605–2615. 10.1056/NEJMoa0903810 [DOI] [PubMed] [Google Scholar]
- De Oliveira, A., Prince, D., Lo, C. Y., Lee, L. H., & Chu, T. C. (2015). Antiviral activity of theaflavin digallate against herpes simplex virus type 1. Antiviral Research, 118, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y., Chen, B., Gao, Z., Suo, H., & Xiao, H. (2017). Pre-treated theaflavin-3,3'-digallate has a higher inhibitory effect on the HCT116 cell line . Food & Nutrition Research, 61(1), 1400340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y., Chen, B., Suo, H., & Tong, H. (2020). The enzyme‐oriented regulation of theaflavin‐3, 3′‐digallate synthesis and the accurate determination of its yield. International Journal of Food Science & Technology, 55(4), 1531–1538. 10.1111/ijfs.14429 [DOI] [Google Scholar]
- Elfiky, A. A. (2020). Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS- CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sciences, 253, 117592. 10.1016/j.lfs.2020.117592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., & Olson, A. J. (2016). Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols, 11(5), 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshpurkar, A., & Saluja, A. K. (2017). The pharmacological potential of rutin. Saudi Pharmaceutical Journal : SPJ: The Official Publication of the Saudi Pharmaceutical Society, 25(2), 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, X.-Y., Li, J.-L., Yang, X.-L., Chmura, A. A., Zhu, G., Epstein, J. H., Mazet, J. K., Hu, B., Zhang, W., Peng, C., Zhang, Y.-J., Luo, C.-M., Tan, B., Wang, N., Zhu, Y., Crameri, G., Zhang, S.-Y., Wang, L.-F., Daszak, P., & Shi, Z.-L. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor . Nature, 503(7477), 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile, D., Patamia, V., Scala, A., Sciortino, M. T., Piperno, A., & Rescifina, A. (2020). Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Marine Drugs, 18(4), 225. 10.3390/md18040225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh, N. D., Faria, N. R., Andersen, K. G., & Pybus, O. G. (2018). Genomic insights into Zika virus emergence and spread. Cell, 172(6), 1160–1162. [DOI] [PubMed] [Google Scholar]
- Gupta, M. K., Vemula, S., Donde, R., Gouda, G., Behera, L., & Vadde, R. (2020). In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. Journal of Biomolecular Structure and Dynamics, 13, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar, M., Ahmed, H. A., Aljohani, G., & Alhaddad, O. A. (2020). Investigation of some antiviral N-heterocycles as COVID 19 drug: Molecular docking and DFT calculations. International Journal of Molecular Sciences, 21(11), 3922. Jan 10.3390/ijms21113922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, D. P., Penn-Nicholson, A., & Cho, M. W. (2006). Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology, 350(1), 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder, E., Damm, W., Maple, J., Wu, C., Reboul, M., Xiang, J. Y., Wang, L., Lupyan, D., Dahlgren, M. K., Knight, J. L., Kaus, J. W., Cerutti, D. S., Krilov, G., Jorgensen, W. L., Abel, R., & Friesner, R. A. (2016). OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. Journal of Chemical Theory and Computation, 12(1), 281–296. 12 10.1021/acs.jctc.5b00864 [DOI] [PubMed] [Google Scholar]
- Hatada, R., Okuwaki, K., Mochizuki, Y., Fukuzawa, K., Komeiji, Y., Okiyama, Y., & Tanaka, S. (2020). Fragment molecular orbital based interaction analyses on COVID-19 main protease- inhibitor N3 complex (PDB ID: 6LU7). ChemRxiv. ( 10.26434/chemrxiv.11988120.v1) [DOI] [PubMed] [Google Scholar]
- Hayashi, K. Y., Hayashi, T., & Morita, N. (1992). Mechanism of action of the antiherpesvirus biflavone ginkgetin. Antimicrobial Agents and Chemotherapy, 36(9), 1890–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y., Zhu, Q., Chen, M., Huang, Q., Wang, W., Li, Q., Huang, Y., & Di, W. (2016). The changing 50% inhibitory concentration (IC50) of cisplatin: A pilot study on the artifacts of the MTT assay and the precise measurement of density-dependent chemoresistance in ovarian cancer. Oncotarget, 7(43), 70803–70821. Oct 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibasami, H., Jin, Z. X., Yoshioka, K., Ina, K., & Ohnishi, K. (2004). Human colon cancer cells undergo apoptosis by theaflavin digallate, epigallocatechin gallate, and oolong tea polyphenol extract. Journal of Herbs, Spices & Medicinal Plants, 10(4), 29–38. 10.1300/J044v10n04_04 [DOI] [Google Scholar]
- Hollingsworth, S. A., & Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron, 99(6), 1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital, A., Goñi, J. R., Orozco, M., & Gelpí, J. L. (2015). Molecular dynamics simulations: Advances and applications. Advances and Applications in Bioinformatics and Chemistry: AABC, 8, 37–47. 10.2147/AABC.S70333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, B. (2009). MetaPocket: A meta approach to improve protein ligand binding site prediction. Omics: A Journal of Integrative Biology, 13(4), 325–330. [DOI] [PubMed] [Google Scholar]
- Huang, F., & Nau, W. M. (2003). A conformational flexibility scale for amino acids in peptides. Angewandte Chemie (International ed. in English)), 42(20), 2269–2272. [DOI] [PubMed] [Google Scholar]
- Huang, N., Shoichet, B. K., & Irwin, J. J. (2006). Benchmarking sets for molecular docking. Journal of Medicinal Chemistry, 49(23), 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim, M. T., Uzairu, A., Shallangwa, G. A., & Uba, S. (2020). In-silico activity prediction and docking studies of some 2, 9-disubstituted 8-phenylthio/phenylsulfinyl-9h-purine derivatives as Anti-proliferative agents. Heliyon, 6(1), e03158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, R., Parves, M. R., Paul, A. S., Uddin, N., Rahman, M. S., Mamun, A. A., Hossain, M. N., Ali, M. A., & Halim, M. A A. (2020). Molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, L. A., Anderson, E. J., Rouphael, N. G., Roberts, P. C., Makhene, M., Coler, R. N., McCullough, M. P., Chappell, J. D., Denison, M. R., Stevens, L. J., Pruijssers, A. J. & An, (2020). mRNA vaccine against SARS-CoV-2—Preliminary report. New England Journal of Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo, S., Kim, S., Shin, D. H., & Kim, M. S. (2020). Inhibition of SARS-CoV 3CL protease by flavonoids. Journal of Enzyme Inhibition and Medicinal Chemistry, 35(1), 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, W. L., & Tirado-Rives, J. (1988). The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. Journal of the American Chemical Society, 110(6), 1657–1666. [DOI] [PubMed] [Google Scholar]
- Joshi, T., Joshi, T., Sharma, P., Chandra, S., & Pande, V. (2020). Molecular docking and molecular dynamics simulation approach to screen natural compounds for inhibition of Xanthomonas oryzae pv. Oryzae by targeting peptide deformylase. Journal of Biomolecular Structure and Dynamics, 1–8. [DOI] [PubMed] [Google Scholar]
- Keivan, Z., Teoh, B. T., Sam, S. S., Wong, P. F., Mustafa, M. R., & AbuBakar, S. (2014). In vitro antiviral activity of fisetin, rutin and naringenin against dengue virus type-2. Journal of Medicinal Plants Research, 8(6), 307–309. 10.5897/JMPR11.1046 [InsertedFromOnline [DOI] [Google Scholar]
- Kernan, M. R., Sendl, A., Chen, J. L., Jolad, S. D., Blanc, P., Murphy, J. T., Stoddart, C. A., Nanakorn, W., Balick, M. J., & Rozhon, E. J. (1997). Two new lignans with activity against influenza virus from the medicinal plant Rhinacanthus nasutus. Journal of Natural Products, 60(6), 635–637. [DOI] [PubMed] [Google Scholar]
- Khan, S. A., Ashraf, Z. K., Uddin, S., & Ul-Haq, R. (2020). Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 11, 1–10. [DOI] [PubMed] [Google Scholar]
- Khodarahmi, G., Asadi, P., Farrokhpour, H., Hassanzadeh, F., & Dinari, M. (2015). Design of novel potential aromatase inhibitors via hybrid pharmacophore approach: Docking improvement using the QM/MM method. RSC Advances, 5(71), 58055–58064. 10.1039/C5RA10097F [DOI] [Google Scholar]
- Kirchmair, J., Markt, P., Distinto, S., Wolber, G., & Langer, T. (2008). Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection-what can we learn from earlier mistakes? Journal of Computer-Aided Molecular Design, 22(3-4), 213–228. [DOI] [PubMed] [Google Scholar]
- Koča, J., Kříž, Z., & Carlsen, P. H. (1994). Computer study of conformational flexibility of 20 common amino acids. Journal of Molecular Structure: Theochem, 306(2-3), 157–164. 10.1016/0166-1280(94)80036-7 [DOI] [Google Scholar]
- Ksiazek, T. G., Erdman, D., Goldsmith, C. S., Zaki, S. R., Peret, T., Emery, S., Tong, S., Urbani, C., Comer, J. A., Lim, W., Rollin, P. E., Dowell, S. F., Ling, A.-E., Humphrey, C. D., Shieh, W.-J., Guarner, J., Paddock, C. D., Rota, P., Fields, B., … Anderson, L. J. (2003). A novel coronavirus associated with severe acute respiratory syndrome. The New England Journal of Medicine, 348(20), 1953–1966. [DOI] [PubMed] [Google Scholar]
- Kumar, K. S., Rao, A. L., & Rao, M. B. (2018). Design, synthesis, biological evaluation and molecular docking studies of novel 3-substituted-5-[(indol-3-yl) methylene]-thiazolidine-2, 4-dione derivatives. Heliyon, 4(9), e00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P., Choonara, Y. E., & Pillay, V. (2014). In silico affinity profiling of neuroactive polyphenols for post-traumatic calpain inactivation: A molecular docking and atomistic simulation sensitivity analysis. Molecules, 20(1), 135–168. 10.3390/molecules20010135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani, S., & Poh, C. L. (2020). Flavonoids as antiviral agents for Enterovirus A71 (EV-A71). Viruses, 12(2), 184. 10.3390/v12020184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie, P. J., Samson, A., Li, A., Adams, K., Capstick, R., Barlow, G. D., Easom, N., Hamilton, E., Moss, P. J., Evans, A., & Ivan, M. (2020). Novel coronavirus disease (Covid-19): The first two patients in the UK with person to person transmission. Journal of Infection. 10.1016/j.jinf.2020.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, L. C., Kuo, Y. C., & Chou, C. J. (2000). Cytotoxic biflavonoids from Selaginella delicatula. Journal of Natural Products, 63(5), 627–630. [DOI] [PubMed] [Google Scholar]
- Lin, Y. M., Flavin, M. T., Schure, R., Chen, F. C., Sidwell, R., Barnard, D. I., Huffmann, J. H., & Kern, E. R. (1999). Antiviral activities of biflavonoids. Planta Medica, 65(2), 120–125. 10.1055/s-1999-13971 [DOI] [PubMed] [Google Scholar]
- Lin, Y. M., Zembower, D. E., Flavin, M. T., Schure, R. M., Anderson, H. M., Korba, B. E., & Chen, F. C. (1997). Robustaflavone, a naturally occurring biflavanoid, is a potent non-nucleoside inhibitor of hepatitis B virus replication in vitro. Bioorganic & Medicinal Chemistry Letters, 7(17), 2325–2328. 10.1016/S0960-894X(97)00422-8 [DOI] [Google Scholar]
- Lipinski, C. A. (2000). Drug-like properties and the causes of poor solubility and poor permeability. Journal of Pharmacological and Toxicological Methods, 44(1), 235–249. 10.1016/S1056-8719(00)00107-6 [DOI] [PubMed] [Google Scholar]
- Lipinski, C. A. (2004). Lead- and drug-like compounds: the rule-of-five revolution . Drug Discovery Today. Technologies, 1(4), 337–341. [DOI] [PubMed] [Google Scholar]
- Liu, K., Tang, M., Liu, Q., Han, X., Jin, H., Zhu, H., Li, Y., He, L., Ji, H., & Zhou, B. (2020). Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discovery, 6(1), 1–4. 10.1038/s41421-020-0156-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P., Chen, W., & Chen, J. P. (2019). Viral metagenomics revealed sendai virus and coronavirus infection of Malayan Pangolins (Manis javanica). Viruses, 11(11), 979. Nov 10.3390/v11110979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lung, J., Lin, Y. S., Yang, Y. H., Chou, Y. L., Shu, L. H., Cheng, Y. C., Liu, H. T., & Wu, C. Y. (2020). The potential chemical structure of anti‐SARS‐CoV‐2 RNA‐dependent RNA polymerase. Journal of Medical Virology, 92(6), 693–697. 10.1002/jmv.25761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meruelo, D., Lavie, G., & Lavie, D. (1988). Therapeutic agents with dramatic antiretroviral activity and little toxicity at effective doses: Aromatic polycyclic diones hypericin and pseudohypericin. Proceedings of the National Academy of Sciences of the United States of America, 85(14), 5230–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesch, G. S., & Schwirian, K. P. (2019). Vaccination hesitancy: Fear, trust, and exposure expectancy of an Ebola outbreak. Heliyon, 5(7), e02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki, K., Nagai, T., Suzuki, K., Tsujimura, R., Koyama, K., Kinoshita, K., Furuhata, K., Yamada, H., & Takahashi, K. (2007). Anti-influenza virus activity of biflavonoids . Bioorganic & Medicinal Chemistry Letters, 17(3), 772–775. [DOI] [PubMed] [Google Scholar]
- Mirza, M. U., & Froeyen, M. (2020). Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. Journal of Pharmaceutical Analysis, 10(4), 320–328. https://doi.org/ 10.1016/j.jpha2020.04.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., & Olson, A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry, 19(14), 1639–1662. [DOI] [Google Scholar]
- Morris, G. M., Huey, R., & Olson, A. J. (2008). Using autodock for ligand‐receptor docking. Current Protocols in Bioinformatics, 24(1), 8–14. 10.1002/0471250953.bi0814s24 [DOI] [PubMed] [Google Scholar]
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan, N., Sakthivel, R., Velmurugan, D., & Gromiha, M. M. (2020). Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. Journal of Biomolecular Structure and Dynamics, 14, 1–6. [DOI] [PubMed] [Google Scholar]
- Mysinger, M. M., Carchia, M., Irwin, J. J., & Shoichet, B. K. (2012). Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. Journal of Medicinal Chemistry, 55(14), 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ñamendys-Silva, S. A. (2020). Respiratory support for patients with COVID-19 infection. The Lancet. Respiratory Medicine, 8(4), e18. 10.1016/S2213-2600(20)30110-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoc, T. M., Phuong, N. T., Khoi, N. M., Park, S., Kwak, H. J., Nhiem, N. X., Trang, B. T., Tai, B. H., Song, J. H., Ko, H. J., & Kim, S. H. (2019). A new naphthoquinone analogue and antiviral constituents from the root of Rhinacanthus nasutus. Natural Product Research, 33(3), 360–366. [DOI] [PubMed] [Google Scholar]
- Odhar, H. A., Ahjel, S. W., Albeer, A. A., Hashim, A. F., Rayshan, A. M., & Humadi, S. S. (2020). Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation, 16(3), 236–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant, S., Singh, M., Ravichandiran, V., Murty, U. S., & Srivastava, H. K. (2020). Peptide-like and small-molecule inhibitors against Covid-19. Journal of Biomolecular Structure and Dynamics, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A., Hoffman, E., Ball, D., Klapwijk, J., Steven, R. T., Dexter, A., Bunch, J., Baker, D., Murnane, D., Hutter, V., Page, C., Dailey, L. A., & Forbes, B. (2019). Comparison of oral, intranasal and aerosol administration of amiodarone in rats as a model of pulmonary phospholipidosis. Pharmaceutics, 11(7), 345. 10.3390/pharmaceutics11070345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, X., Xu, X., Li, Y., Cheng, L., Zhou, X., & Ren, B. (2020). Transmission routes of 2019-nCoV and controls in dental practice. International Journal of Oral Science, 12(1), 9–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, R. M. (2003). Antiviral activity of compounds isolated from plants. Pharmaceutical Biology, 41(2), 107–157. 10.1076/phbi.41.2.107.14240 [DOI] [Google Scholar]
- Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y., & Jung, S. H. (2016). An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy . Journal of Medicinal Chemistry, 59(14), 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu, X. Y., Liang, J. P., Wang, X. H., Xu, T., Hua, L. Y., Shang, R. F., Liu, Y., & Xing, Y. M. (2009). Anti-influenza A virus effect of Hypericum perforatum L. extract. Virologica Sinica, 24(1), 19–27. 10.1007/s12250-009-2983-x [DOI] [Google Scholar]
- Ravindranath, P. A., Forli, S., Goodsell, D. S., Olson, A. J., & Sanner, M. F. (2015). AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Computational Biology, 11(12), e1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccatano, D., Barthel, A., & Zacharias, M. (2007). Structural flexibility of the nucleosome core particle at atomic resolution studied by molecular dynamics simulation. Biopolymers, 85(5-6), 407–421. [DOI] [PubMed] [Google Scholar]
- Salazar-Bookaman, M. M., Wainer, I., & Patil, P. N. (1994). Relevance of drug-melanin interactions to ocular pharmacology and toxicology. J Ocul Pharmacol, 10(1), 217–239. [DOI] [PubMed] [Google Scholar]
- Sarma, P., Shekhar, N., Prajapat, M., Avti, P., Kaur, H., Kumar, S., Singh, S., Kumar, H., Prakash, A., Dhibar, D. P., & Medhi, B. (2020). In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). Journal of Biomolecular Structure and Dynamics, 16, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck, A. G., Ausubel, M. B., Zuckerbraun, H. L., & Babich, H. (2008). Theaflavin-3,3'-digallate, a component of black tea: an inducer of oxidative stress and apoptosis. Toxicology in Vitro: An International Journal Published in Association with Bibra, 22(3), 598–609. [DOI] [PubMed] [Google Scholar]
- Shie, J. J., Fang, J. M., Kuo, C. J., Kuo, T. H., Liang, P. H., Huang, H. J., Yang, W. B., Lin, C. H., Chen, J. L., Wu, Y. T., & Wong, C. H. (2005). Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. Journal of Medicinal Chemistry, 48(13), 4469–4473. [DOI] [PubMed] [Google Scholar]
- Silva Andrade, B., Ghosh, P., Barh, D., Tiwari, S., José Santana Silva, R., Rodrigues de Assis Soares, W., Silva Melo, T., Santos Freitas, A., González-Grande, P., Sousa Palmeira, L., Carlos Junior Alcantara, L., Giovanetti, M., Góes-Neto, A., & Ariston de Carvalho Azevedo, V. (2020). Computational screening for potential drug candidates against the SARS-CoV-2 main protease. F1000Research, 9(514), 514. 4 10.12688/f1000research.23829.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, A. R., Morais, S. M., Marques, M. M., Lima, D. M., Santos, S. C., Almeida, R. R., Vieira, I. G., & Guedes, M. I. (2011). Antiviral activities of extracts and phenolic components of two Spondias species against dengue virus. Journal of Venomous Animals and Toxins Including Tropical Diseases, 17(4), 406–413. [Google Scholar]
- Singh, A. K., Singh, A., Shaikh, A., Singh, R., & Misra, A. (2020). Chloroquine and hydroxychloroquine in the treatment of COVID-19 with or without diabetes: A systematic search and a narrative review with a special reference to India and other developing countries. Diabetes & Metabolic Syndrome, 14(3), 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa, S. F., Fernandes, P. A., & Ramos, M. J. (2006). Protein-ligand docking: current status and future challenges. Proteins, 65(1), 15–26. [DOI] [PubMed] [Google Scholar]
- Verdonk, M. L., Berdini, V., Hartshorn, M. J., Mooij, W. T., Murray, C. W., Taylor, R. D., & Watson, P. (2004). Virtual screening using protein-ligand docking: Avoiding artificial enrichment. Journal of Chemical Information and Computer Sciences, 44(3), 793–806. [DOI] [PubMed] [Google Scholar]
- Visalli, R. J., Ziobrowski, H., Badri, K. R., He, J. J., Zhang, X., Arumugam, S. R., & Zhao, H. (2015). Ionic derivatives of betulinic acid exhibit antiviral activity against herpes simplex virus type-2 (HSV-2), but not HIV-1 reverse transcriptase. Bioorganic & Medicinal Chemistry Letters, 25(16), 3168–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, A. C., Laskowski, R. A., & Thornton, J. M. (1995). LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein engineering, design and selection. Protein Engineering, 8(2), 127–134. [DOI] [PubMed] [Google Scholar]
- Wallace, D. J., Gudsoorkar, V. S., Weisman, M. H., & Venuturupalli, S. R. (2012). New insights into mechanisms of therapeutic effects of antimalarial agents in SLE. Nature Reviews. Rheumatology, 8(9), 522–533. [DOI] [PubMed] [Google Scholar]
- Wen, C.-C., Kuo, Y.-H., Jan, J.-T., Liang, P.-H., Wang, S.-Y., Liu, H.-G., Lee, C.-K., Chang, S.-T., Kuo, C.-J., Lee, S.-S., Hou, C.-C., Hsiao, P.-W., Chien, S.-C., Shyur, L.-F., & Yang, N.-S. (2007). Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. Journal of Medicinal Chemistry, 50(17), 4087–4095. [DOI] [PubMed] [Google Scholar]
- Xu, G. H., Ryoo, I. J., Kim, Y. H., Choo, S. J., & Yoo, I. D. (2009). Free radical scavenging and antielastase activities of flavonoids from the fruits of Thuja orientalis. Archives of Pharmacal Research, 32(2), 275–282. [DOI] [PubMed] [Google Scholar]
- Xu, X., Chen, P., Wang, J., Feng, J., Zhou, H., Li, X., Zhong, W., & Hao, P. (2020). Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Science China Life Sciences. Science China. Life Sciences, 63(3), 457–460. Mar- [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H., Yang, M., Ding, Y., Liu, Y., Lou, Z., Zhou, Z., Sun, L., Mo, L., Ye, S., Pang, H., Gao, G. F., Anand, K., Bartlam, M., Hilgenfeld, R., & Rao, Z. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences of the United States of America, 100(23), 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, L., Li, Z., Yuan, K., Qu, X., Chen, J., Wang, G., Zhang, H., Luo, H., Zhu, L., Jiang, P., Chen, L., Shen, Y., Luo, M., Zuo, G., Hu, J., Duan, D., Nie, Y., Shi, X., Wang, W., … Xu, X. (2004). Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. Journal of Virology, 78(20), 11334–1133 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, R., Chen, L., Lan, R., Shen, R., & Li, P. (2020). Computational screening of antagonist against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. International Journal of Antimicrobial Agents, 56(2), 106012. 10.1016/j.ijantimicag.2020.106012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakaryan, H., Arabyan, E., Oo, A., & Zandi, K. (2017). Flavonoids: Promising natural compounds against viral infections. Archives of Virology, 162(9), 2539–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki, A. M., Van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D., & Fouchier, R. A. (2012). Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. The New England Journal of Medicine, 367(19), 1814–1820. [DOI] [PubMed] [Google Scholar]
- Zembower, D. E., Lin, Y. M., Flavin, M. T., Chen, F. C., & Korba, B. E. (1998). Robustaflavone, a potential non- nucleoside anti-hepatitis B agent. Antiviral Research, 39(2), 81–88.- 10.1016/S0166-3542(98)00033-3 [DOI] [PubMed] [Google Scholar]
- Zengion, A. H., & Yarnell, E. (2011). Herbal and nutritional supplements for painful conditions. InPain Procedures in Clinical Practice 2011 Jan 1. (pp. 187–204). Hanley & Belfus. [Google Scholar]
- Zhang, D. H., Wu, K. L., Zhang, X., Deng, S. Q., & Peng, B. (2020). In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. Journal of Integrative Medicine, 18(2), 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T., Wu, Q., & Zhang, Z. (2020). Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Current Biology: Cb, 30(7), 1346–1351.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z., Li, Y., Lin, B., Schroeder, M., & Huang, B. (2011). Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics (Oxford, England)), 27(15), 2083–2088. [DOI] [PubMed] [Google Scholar]
- Zhavoronkov, A., Aladinskiy, V., Zhebrak, A., Zagribelnyy, B., Terentiev, V., Bezrukov, D. S., Polykovskiy, D., Shayakhmetov, R., Filimonov, A., Orekhov, P., & Yan, Y. (2020). Potential COVID-2019 3C-like protease inhibitors designed using generative deep learning approaches. Insilico Medicine Hong Kong Ltd A, 307, E1. [Google Scholar]
- Zheng, X., & Polli, J. (2010). Identification of inhibitor concentrations to efficiently screen and measure inhibition Ki values against solute carrier transporters. European Journal of Pharmaceutical Sciences, 41(1), 43–52. - 10.1016/j.ejps.2010.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete, V., Grosdidier, A., & Michielin, O. (2009). Docking, virtual high throughput screening and in silico fragment-based drug design . Journal of Cellular and Molecular Medicine, 13(2), 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.