

Abstract
Interaction of transforming growth factor-β (TGFβ)-induced canonical signaling with the noncanonical kinase cascades regulates glomerular hypertrophy and matrix protein deposition, which are early features of glomerulosclerosis. However, the specific target downstream of the TGFβ receptor involved in the noncanonical signaling is unknown. Here, we show that TGFβ increased the catalytic loop phosphorylation of platelet-derived growth factor receptor β (PDGFRβ), a receptor tyrosine kinase expressed abundantly in glomerular mesangial cells. TGFβ increased phosphorylation of the PI 3-kinase–interacting Tyr-751 residue of PDGFRβ, thus activating Akt. Inhibition of PDGFRβ using a pharmacological inhibitor and siRNAs blocked TGFβ-stimulated phosphorylation of proline-rich Akt substrate of 40 kDa (PRAS40), an intrinsic inhibitory component of mTORC1, and prevented activation of mTORC1 in the absence of any effect on Smad 2/3 phosphorylation. Expression of constitutively active myristoylated Akt reversed the siPDGFRβ-mediated inhibition of mTORC1 activity; however, co-expression of the phospho-deficient mutant of PRAS40 inhibited the effect of myristoylated Akt, suggesting a definitive role of PRAS40 phosphorylation in mTORC1 activation downstream of PDGFRβ in mesangial cells. Additionally, we demonstrate that PDGFRβ-initiated phosphorylation of PRAS40 is required for TGFβ-induced mesangial cell hypertrophy and fibronectin and collagen I (α2) production. Increased activating phosphorylation of PDGFRβ is also associated with enhanced TGFβ expression and mTORC1 activation in the kidney cortex and glomeruli of diabetic mice and rats, respectively. Thus, pursuing TGFβ noncanonical signaling, we identified how TGFβ receptor I achieves mTORC1 activation through PDGFRβ-mediated Akt/PRAS40 phosphorylation to spur mesangial cell hypertrophy and matrix protein accumulation. These findings provide support for targeting PDGFRβ in TGFβ-driven renal fibrosis.
Keywords: kidney, diabetic nephropathy, Akt (protein kinase B), mTOR complex (mTORC), PDGFRβ, diabetic nephropathy, transforming growth factor-β (TGFβ)
Chronic kidney disease affects 10% of the world's population, including 20 million Americans, and causes increased risk of cardiovascular diseases and loss of renal function, leading to end-stage renal disease with significant public health costs (1, 2). Thus, understanding the progression of the disease process is of prime importance for its prevention and arrest. The pathologic correlates of progressive renal function impairment include renal structural changes with initial renal hypertrophy, especially glomerular hypertrophy, that leads to hyperfiltration and microalbuminuria, followed by accumulation of matrix proteins and a greater degree of proteinuria (3, 4). A significant pathologic characteristic of chronic kidney disease is glomerulosclerosis. One-third of the cell population in the glomerulus is made up of mesangial cells, which have the capacity to communicate with the endothelial cells; they can also regulate glomerular filtration because of their contractile nature (5, 6). Transforming growth factor-β (TGFβ) contributes to glomerulosclerosis and albuminuria during progressive kidney injury, especially in diabetic kidney disease (4, 7). Liver-specific TGFβ transgenic mice with increased circulating levels of cytokine developed mesangial expansion with augmented glomerular immune deposits and matrix proteins (8). In the disease milieu or in vitro when mesangial cells are exposed to TGFβ, they undergo hypertrophy and acquire a myofibroblast-like phenotype to synthesize larger amounts of matrix proteins (9, 10). Also, anti-TGFβ antibody decreases mesangial and fibrotic protein expression in rodent models of nephropathy in type 1 and type 2 diabetes where TGFβ contributes to the pathology (11, 12).
The dimeric active TGFβ directly binds to TGFβ receptor II, which is a constitutively active kinase. After TGFβ binding, TGFβRI is recruited into the complex and undergoes phosphorylation by TGFβRII. Activated TGFβRI then binds and phosphorylates the receptor-specific Smad, Smad 2, and Smad 3, which form a complex with a common Smad, Smad 4, for translocation to the nucleus and regulation of gene transcription (13). Apart from this canonical signaling, TGFβ also initiates other noncanonical signal transduction pathways involving mitogen-activated protein kinases, PI 3-kinase/Akt/mTORC1, and Rho GTPase, which contribute to the renal complications under disease conditions (13–17).
Among many hormones, growth factors, and cytokines, platelet-derived growth factor (PDGF) contributes to significant injury in different renal, especially glomerular, diseases (5). Four different isoforms of PDGF (A, B, C, and D) form a total of five homo- or heterodimers (18). These dimers bind to three dimeric PDGF receptors (PDGFRs), αα, ββ, and αβ, with variable affinity (19). For example, whereas PDGF AA and CC show affinity for PDGFRα, CC also binds to PDGFRαβ. PDGF DD binds to PDGFRαβ at a low affinity, but it interacts with PDGFRββ with significantly higher affinity. On the other hand, PDGF AB and BB bind to both receptors. All these PDGF dimers and three dimeric receptors are expressed in mesangial cells (19). In glomerulonephritis, PDGF BB, CC, and DD play prominent roles and stimulate proliferation of mesangial cells, whereas PDGF AA does not (5, 20–22). The importance of PDGF BB in glomerular mesangial cell biology is established because mice deficient in either the B-chain or its receptor β lack these glomerular cells (23, 24). In fibrotic disorders such as diabetic nephropathy where TGFβ contributes to the pathology, expression of PDGF BB and PDGFRβ is increased, especially in mesangial cells (25, 26). Whether PDGFRβ contributes to the signal transduction of TGFβ in mesangial cell pathology through a cross-talk has not been investigated. In our studies, we show that TGFβ activates PDGFRβ to initiate Akt signaling to inactivate PRAS40, which results in the increased mTORC1 activity necessary for mesangial cell hypertrophy and expression of matrix proteins fibronectin and collagen I (α2).
Results
Activation of TGFβRI stimulates PDGFRβ autophosphorylation in mesangial cells
Previously, we demonstrated that TGFβ increases the tyrosine phosphorylation of several proteins, including a protein at 190 kDa to induce noncanonical signaling in mesangial cells (15). To characterize the tyrosine kinase involved in the action of TGFβ, we considered PDGFRβ because it is a receptor tyrosine kinase, it is one of most abundant receptors expressed in mesangial cells, and it elicits pathology in glomerulosclerosis (5, 19, 27). Because activation of PDGFRβ requires its phosphorylation at Tyr-857 in the activation loop, we examined this in glomerular mesangial cells. TGFβ increased the phosphorylation of PDGFRβ in a time-dependent manner (Fig. 1A). Because TGFβ utilizes its type I serine/threonine kinase receptor for its signal transduction, we used a specific TGFβRI inhibitor, SB-431542 (SB), to test its involvement in PDGFRβ phosphorylation. Similar to the effect on phosphorylation of Smad 2 and Smad 3, SB significantly inhibited TGFβ-stimulated PDGFRβ Tyr-857 phosphorylation (Fig. 1, B–D). PDGFRβ undergoes autophosphorylation at this site (13). Therefore, we used a specific PDGFRβ ATP competitive inhibitor, JNJ-10198409 (JNJ), in mesangial cells. JNJ blocked the autophosphorylation of PDGFRβ in response to TGFβ (Fig. 1E). Importantly, JNJ did not have any effect on phosphorylation of Smad 2 and Smad 3 in response to TGFβ (Fig. 1, F and G). Similarly, siRNAs against PDGFRβ to block its expression (Fig. 1H) showed no effect on TGFβ-induced phosphorylation of Smad 2 and Smad 3 (Fig. 1, I and J). These data indicate that TGFβ activates PDGFRβ; however, PDGFRβ does not control the canonical Smad 2/3 phosphorylation by TGFβRI.
Figure 1.

TGFβ receptor activates PDGFRβ. A, TGFβ increases activating phosphorylation of PDGFRβ at Tyr-857. Mesangial cells were incubated with 2 ng/ml TGFβ for the indicated duration. The cleared cell lysates were immunoblotted with phospho-PDGFRβ (Tyr-857) and PDGFRβ antibodies. B–J, mesangial cells were treated with 5 µm SB (panels B–D) or 0.1 µm JNJ (panels E–G) or transfected with siRNAs against PDGFRβ or scrambled RNA (panels H–J) prior to incubation with 2 ng/ml TGFβ for 2 h. The cell lysates were immunoblotted against the indicated antibodies. Molecular weight markers are shown in the left margins.
TGFβ uses PDGFRβ for Akt kinase activation
We and others demonstrated previously that TGFβ regulates PI 3-kinase–dependent Akt activation (15, 28, 29). For activation of PI 3-kinase, the SH2 domain of the lipid kinase has to interact with the tyrosine phosphorylated proteins (30). In the case of activated PDGFRβ, PI 3-kinase associates with the phosphorylated Tyr-751 (31). Therefore, we determined the phosphorylation of PDGFRβ at this site in mesangial cells. TGFβ increased the phosphorylation of PDGFRβ at Tyr-751 in a time-dependent manner similar to its catalytic loop Tyr-857 phosphorylation (Fig. 2A). Both TGFβRI and PDGFRβ inhibitors SB and JNJ significantly blocked the phosphorylation of PDGFRβ at Tyr-751 (Fig. 2, B and C). Activation of PI 3-kinase results in the phosphorylation of its downstream target Akt at the catalytic loop Thr-308 and hydrophobic motif Ser-473 sites (32). Therefore, to determine the activation of PI 3-kinase, we measured the phosphorylation of these Akt residues. TGFβ increased the phosphorylation of both these residues in a time-dependent manner (Fig. 2D). JNJ significantly inhibited the TGFβ-induced phosphorylation of Akt at both of these sites (Fig. 2E). Because phosphorylation of Akt increases its kinase activity, we determined the phosphorylation of one of its endogenous substrates, glycogen synthase kinase 3-β (GSK3β). JNJ significantly inhibited GSK3β phosphorylation by TGFβ (Fig. 2F). To confirm this observation, we used siRNAs to down-regulate PDGFRβ in mesangial cells. siRNAs against PDGFRβ inhibited the TGFβ-induced phosphorylation of Akt and its endogenous substrate GSK3β (Fig. 2, G and H).
Figure 2.

TGFβ-stimulated PDGFRβ activates Akt signaling. A and D, TGFβ increases phosphorylation of PDGFRβ at the PI 3-kinase binding site Tyr-751 to increase Akt phosphorylation. Mesangial cells were incubated with 2 ng/ml TGFβ for the indicated duration. The cleared cell lysates were immunoblotted with indicated antibodies. B, C, and E–H, mesangial cells were treated with 5 µm SB (panel B) or 0.1 µm JNJ (panels C, E, and F) or transfected with siRNAs against PDGFRβ or scrambled RNA (panels G and H) prior to incubation with 2 ng/ml TGFβ for 2 h. The cell lysates were immunoblotted against the indicated antibodies. Molecular weight markers are shown in the left margins.
PDGFRβ regulates TGFβ-induced mTORC1 activation
We and others demonstrated that TGFβ regulates mTORC1 activity (14, 16, 33). The exclusive mTORC1 subunit PRAS40 acts as an inhibitor of mTORC1 activity (34). Similarly, the GTPase-activating protein tuberin inhibits mTORC1 by acting on the Rheb GTPase (35). When phosphorylated by Akt, both of these proteins undergo inactivation (36–38). We determined the effect of PDGFRβ inhibition on the phosphorylation of these proteins. JNJ significantly inhibited the TGFβ-stimulated phosphorylation of PRAS40 and tuberin (Fig. 3, A and B). Similarly, PDGFRβ siRNAs blocked phosphorylation of PRAS40 and tuberin in response to TGFβ (Fig. 3, C and D). Because phosphorylation/inactivation of PRAS40 and tuberin activate mTORC1, we determined activity of the latter by using phosphorylation of its substrates S6 kinase and 4EBP1. We also examined the phosphorylation of ribosomal protein S6 (rps6), which undergoes phosphorylation upon S6 kinase activation. TGFβ increased the phosphorylation of S6 kinase and 4EBP1 at mTORC1 sites and rps6 at S6 kinase sites. Both JNJ and siPDGFRβ markedly inhibited phosphorylation at these sites by TGFβ (Fig. 3, E–J).
Figure 3.

TGFβ-stimulated PDGFRβ regulates mTORC1signaling. Mesangial cells were treated with 0.1 µm JNJ (panels A, B, E, G, and I) or transfected with siRNAs against PDGFRβ or scramble RNA (C, D, F, H, and J) prior to incubation with 2 ng/ml TGFβ. The cleared cell lysates were immunoblotted with indicated antibodies.
PDGFRβ-independent signaling by TGFβ
Along with Akt/mTORC1, TGFβ also increases phosphorylation/activation of extracellular signal-regulated kinase (ERK)1/2 and STAT3 (39, 40). TGFβ increased phosphorylation of ERK1/2 and STAT3 with similar kinetics (Fig. 4, A and B) as autophosphorylation of PDGFRβ (Fig. 1A). We examined whether activation of PDGFRβ is necessary for ERK1/2 and STAT3 phosphorylation. Interestingly, incubation of mesangial cells with JNJ did not have any effect on TGFβ-induced phosphorylation of ERK1/2 and STAT3 (Fig. 4, C and D). Similarly, siRNAs against PDGFRβ did not inhibit phosphorylation of ERK1/2 and STAT3 in response to TGFβ (Fig. 4, E and F). These results indicate that PDGFRβ is involved in TGFβ-induced Akt/mTORC1 activation but not in the regulation of ERK1/2 and STAT3.
Figure 4.

PDGFRβ does not regulate TGFβ-induced ERK1/2 and STAT3 phosphorylation. A and B, mesangial cells were incubated with 2 ng/ml TGFβ for the indicated periods of time. C–F, mesangial cells were treated with 0.1 µm JNJ (panels C and D) or transfected with siRNAs against PDGFRβ (panels E and F) prior to incubation with TGFβ (2 ng/ml) for 2 h. The cell lysates were immunoblotted with the indicated antibodies.
TGFβ regulates PDGF B expression
Our results above demonstrate that TGFβ activates PDGFRβ. To determine the mechanism of activation of PDGFRβ, we considered the expression of the PDGFRβ ligand PDGF B, which forms BB homodimer to activate the receptor (41–43). TGFβ time-dependently increased the expression of PDGF B in mesangial cells (Fig. 5A). These results indicate that a TGFβ-induced increase in PDGF expression may regulate PDGFRβ activation. We tested this hypothesis. We used siRNAs against PDGF B. siPDGF B inhibited TGFβ-stimulated phosphorylation of PDGFRβ (Fig. 5B). Also, TGFβ-induced phosphorylation of Akt was inhibited by siRNAs against PDGF B (Fig. 5C). These data conclusively demonstrate that PDGF B regulates the activation of PDGFRβ and the downstream activation of Akt/mTORC1 by TGFβ. We tested this notion by using PDGF BB ligand directly in mesangial cells. PDGF BB increased the phosphorylation of PDGFRβ in a time-dependent manner (Fig. 6A) similar to the kinetics of PDGF B expression by TGFβ (Fig. 5A). Also, PDGF BB enhanced the phosphorylation of Akt (Fig. 6B), resulting in phosphorylation of PRAS40 and tuberin (Fig. 6, C and D), which led to the activation of mTORC1 as judged by phosphorylation of S6 kinase, rps6, and 4EBP1 (Fig. 6, E–G). Furthermore, increased phosphorylation of PDGFRβ was observed when PDGF BB ligand was added as compared with TGFβ alone (Fig. S1A). No further increase in phosphorylation was found when both ligands were added (Fig. S1A). Importantly, this increased PDGFRβ phosphorylation resulted in a similar increase in Akt phosphorylation leading to mTORC1 activation (Fig. S1, B–G). Similar to the observations with TGFβ, PDGFRβ inhibitor JNJ blocked PDGF BB–stimulated phosphorylation of Akt, PRAS40, and tuberin (Fig. S2, A–C). JNJ also blocked activation of mTORC1 in response to PDGF BB (Fig. S2, D–F).
Figure 5.

TGFβ increases PDGF B expression to augment activating phosphorylation of PDGFRβ and Akt. A, mesangial cells were incubated with TGFβ (2 ng/ml) for the indicated period of time. B and C, mesangial cells were transfected with siPDGF B or scrambled RNA prior to incubation with TGFβ (2 ng/ml) for 2 h. The cell lysates were immunoblotted with the indicated antibodies.
Figure 6.

PDGF increases activating phosphorylation of PDGFRβ leading to Akt/mTORC1 activity. Mesangial cells were incubated with 20 ng/ml PDGF for indicated period of time. The cell lysates were immunoblotted with the indicated antibodies.
PDGF regulates TGFβ signaling
The above results show that TGFβ utilizes PDGF ligand to activate PDGFRβ. We examined whether there is a cross-talk between PDGF and TGFβ. Incubation of mesangial cells with PDGF increased the expression of TGFβ in a time-dependent manner (Fig. 7A). Inhibition of PDGFRβ with JNJ blocked PDGF-stimulated TGFβ expression (Fig. 7B). To test if PDGF signals through TGFβ, we examined phosphorylation of Smads. PDGF increased phosphorylation of Smad 2 and Smad 3, and the TGFβ receptor kinase inhibitor SB blocked these phosphorylations (Fig. 7, C and D). Interestingly, TGFβ antibody inhibited PDGF-stimulated Smad 2 and Smad 3 phosphorylation (Fig. 7, E and F). These data demonstrate the presence of a positive feedback loop between TGFβ and PDGF.
Figure 7.

PDGF increases expression of TGFβ to induce its signaling. A, mesangial cells were incubated with PDGF (20 ng/ml) for the indicated period of time. B–F, mesangial cells were treated with 0.1 µm JNJ (panel B) or 5 µm SB (panels C and D) or 1 μg/ml IgG or TGFβ antibody (panels E and F) prior to incubation with PDGF for 2 h. The cell lysates were immunoblotted with the indicated antibodies.
PRAS40 regulates TGFβ-induced mTORC1 activity downstream of PDGFRβ
Our results above show that TGFβ-induced PDGFRβ activation contributes to phosphorylation of Akt and PRAS40, resulting in mTORC1 activation (Fig. 2 and Fig. 3). To determine whether Akt-mediated phosphorylation of PRAS40 regulates the activation of mTORC1 downstream of PDGFRβ, we transfected mesangial cells with siRNAs against PDGFRβ, along with constitutively active myristoylated Akt (myr-Akt) and the non-phosphorylatable mutant PRAS40 T246A. Expression of myr-Akt alone increased the phosphorylation of S6 kinase, rps6, and 4EBP1, similar to TGFβ treatment (data not shown). However, constitutively active Akt kinase reversed the inhibition of phosphorylation of S6 kinase, rps6, and 4EBP1 by siPDGFRβ in the presence of TGFβ (Fig. 8, A–C; compare 4th lanes with 3rd lanes). Importantly, the phospho-deficient mutant of PRAS40 abrogated the reversal of phosphorylation of these mTORC1 and S6 kinase substrates by constitutively active Akt (Fig. 8, A–C; compare 5th lanes with 4th lanes). These results conclusively demonstrate that Akt-mediated phosphorylation-dependent inactivation of PRAS40 downstream of PDGFRβ regulates TGFβ-induced mTORC1 activation in mesangial cells.
Figure 8.

PDGFRβ regulates TGFβ-induced Akt/PRAS40-mediated mTORC1 activation. A, mesangial cells were transfected with siRNAs against PDGFRβ or scrambled RNA or HA-tagged myr-Akt and PRAS40 (Thr-246A) mutant expression vectors as indicated. Transfected cells were incubated with 2 ng/ml TGFβ. The cleared cell lysates were immunoblotted with indicated antibodies.
Activation of Akt kinase by PDGFRβ regulates TGFβ-induced mesangial cell hypertrophy and matrix protein expression
We demonstrated that TGFβ-stimulated Akt kinase regulates mesangial cell hypertrophy and expression of matrix proteins (14, 44). Because PDGFRβ contributes to the activation of Akt by TGFβ, we investigated the involvement of this receptor tyrosine kinase in mesangial cell hypertrophy. JNJ significantly inhibited TGFβ-induced protein synthesis and hypertrophy of mesangial cells (Fig. 9, A and B). Similarly, siRNAs against PDGFRβ attenuated both these phenomena (Fig. 9, C and D). Because PRAS40 phosphorylation by Akt regulates its downstream signaling, we determined the involvement of Akt. Expression of constitutively active myr-Akt restored the siPDGFRβ-mediated inhibition of mesangial cell protein synthesis and hypertrophy in the presence of TGFβ (Fig. 9, E and F). Furthermore, expression of phospho-deficient PRAS40 T246A significantly prevented the myr-Akt-mediated reversal of siPDGFRβ-induced inhibition of protein synthesis and hypertrophy (Fig. 9, E and F).
Figure 9.

PDGFRβ regulates TGFβ-induced protein synthesis and hypertrophy of mesangial cells. Mesangial cells were treated with 0.1 µm JNJ (panels A and B) or transfected with siRNAs against PDGFRβ or scrambled RNA (C and D) along with myr-Akt and PRAS40 (Thr-246A) as indicated (E and F) prior to incubation with 2 ng/ml TGFβ for 24 h. Protein synthesis and hypertrophy were measured as described in “Experimental procedures.” Mean ± S.D. of triplicate (A–D) or quadruplicate (E and F) measurements is shown. In panels A–D, *p < 0.0001–0.002 versus control; **p < 0.001–0.006 versus TGFβ. In panels E and F, *p < 0.0001 versus control; **p < 0.01 versus TGFβ; #p < 0.01 versus TGFβ + siPDGFRβ; @p < 0.01 versus TGFβ + siPDGFRβ + myr-Akt.
TGFβ regulates renal fibrosis, including glomerulosclerosis, by increasing the synthesis and deposition of matrix proteins fibronectin and collagen (3). TGFβ increased the expression of fibronectin and collagen I (α2) in mesangial cells (44–46). Both JNJ and siRNA against PDGFRβ inhibited TGFβ-stimulated expression of these matrix proteins (Fig. 10, A–D). We showed above that TGFβ-induced expression of PDGF B regulates activation of PDGFRβ and Akt kinase (Fig. 5). Therefore, we tested the effect of siPDGF B on expression of fibronectin and collagen I (α2). Inhibition of PDGF B blocked fibronectin and collagen I (α2) expression in response to TGFβ (Fig. 10, E and F). To further evaluate the mechanism of matrix expression, we determined the role of Akt because it is activated downstream of TGFβ by PDGFRβ activation (Fig. 2D, Fig. 6B, and Fig. S1B). We used an Akt inhibitor, MK, which blocked Akt phosphorylation by TGFβ (Fig. S3A). MK inhibited TGFβ-induced expression of fibronectin and collagen I (α2) (Fig. S3, B and C). We showed above that siPDGFRβ blocked TGFβ-induced expression of fibronectin and collagen I (α2) (Fig. 10, C and D). Expression of myr-Akt reversed the siPDGFRβ-mediated inhibition of TGFβ-stimulated expression of both of these matrix proteins (Fig. 10, G and H; compare 4th lanes with 3rd lanes). Importantly, PRAS40 T246A phospho-deficient mutant inhibited this myr-Akt effect on fibronectin and collagen I (α2) expression (Fig. 10, G and H; compare 5th lanes with 4th lanes). Together, these data show a significant role of Akt downstream of PDGFRβ in mesangial cell matrix protein expression. Furthermore, our results demonstrate a critical role for the Akt and its substrate PRAS40 in PDGFRβ-mediated signaling by TGFβ in mesangial cell pathology.
Figure 10.

PDGFRβ regulates TGFβ-induced fibronectin and collagen I (α2). Mesangial cells treated with JNJ (panels A and B) or transfected with siRNAs against PDGFRβ or scrambled RNA (panels C and D) or siRNAs against PDGF B (panels E and F) or myr-Akt and PRAS40 (panels G and H) and incubated with TGFβ for 24 h. The cleared cell lysates were immunoblotted with the indicated antibodies.
PDGFRβ-stimulated mTORC1 regulates TGFβ-induced mesangial cell hypertrophy and matrix protein expansion
We demonstrated previously that activation of mTORC1 by TGFβ contributes to mesangial cell hypertrophy and matrix protein expression (14, 46, 47). Our results above show a role of PDGFRβ in TGFβ-stimulated mTORC1 activation in mesangial cells (Fig. 3). Therefore, we examined whether mTORC1 downstream of PDGFRβ regulates mesangial cell hypertrophy. To test this, we used a vector expressing raptor, which is a constitutive component of mTORC1 and is essential for its kinase activity (48, 49). Expression of raptor in mesangial cells increased mTORC1 activity as judged by phosphorylation of S6 kinase, rps6, and 4EBP1 (Fig. S4, A–C). Raptor reversed the siPDGFRβ-mediated inhibition of protein synthesis and hypertrophy of mesangial cells in the presence of TGFβ (Fig. 11, A and B). Similarly, raptor restored the inhibition of fibronectin and collagen I (α2) by siPDGFRβ in the TGFβ-stimulated cells (Fig. 11, C and D). To confirm these observations, we used a mutant of mTOR, which delivers hyperactive mTORC1 (49, 50). Expression of this mutant increased the phosphorylation of S6 kinase, rps6, and 4EBP1 in the absence of any effect on phosphorylation of Akt at Ser-473 in mesangial cells (Fig. S5, A–D), indicating that it acts as hyperactive mTORC1 and not mTORC2. Expression of this hyperactive mTORC1 reversed the siPDGFRβ-mediated inhibition of protein synthesis, hypertrophy, and matrix proteins fibronectin and collagen I (α2) expression (Fig. S6, A–D). These results show a positive role of mTORC1 downstream of PDGFRβ in the TGFβ-induced mesangial cell pathology.
Figure 11.

PDGFRβ-stimulated mTORC1 regulates TGFβ-induced protein synthesis, hypertrophy, and matrix proteins fibronectin and collagen I (α2) expression. Mesangial cells were transfected with siRNAs against PDGFRβ or scrambled RNA along with Myc-tagged raptor expression vector as indicated. Transfected cells were incubated with 2 ng/ml TGFβ. In panels A and B, respectively, protein synthesis and hypertrophy of mesangial cells were measured as described in the experimental procedures. Mean ± S.D. of triplicate measurements is shown. *p < 0.008 (panel A) and *p < 0.0001 (panel B) versus control; **p < 0.013 (panel A) and **p < 0.002 (panel B) versus TGFβ; #p < 0.005 (panels A) and #p < 0.004 (panel B) versus TGFβ + siPDGFRβ. In panels C and D, the cleared cell lysates were immunoblotted with the indicated antibodies.
Phosphorylation of PDGFRβ in the kidneys of type 1 diabetic OVE26 mice and streptozotocin-induced rats
Diabetic nephropathy is a fibrotic disorder. In this disease, mesangial expansion proceeds to glomerular hypertrophy and accumulation of matrix proteins, which lead to glomerulosclerosis (51). Hyperglycemia produces multiple fibrotic growth factors, including TGFβ, which contribute to the pathogenesis of diabetic nephropathy (4, 51). Our results described above show a role of PDGFRβ in TGFβ-induced mesangial cell hypertrophy and matrix protein expression. To investigate the in vivo relevance of our results, we used OVE26 type 1 diabetic mice. We demonstrated recently that the blood glucose levels in 3-month-old OVE26 mice are significantly increased and are associated with renal hypertrophy (52). Renal cortical lysates were used to examine the expression of TGFβ. In diabetic renal cortical samples, a significant increase in TGFβ was detected (Fig. 12, A and B). This increased TGFβ correlated with an increase in phosphorylation of Smad 2 and Smad 3, two downstream TGFβ receptor-specific Smads (Fig. 12, C–E). Furthermore, diabetic mice showed a significantly increased expression of PDGF B (Fig. 12, F and G). The level of phosphorylation of PDGFRβ at Tyr-857, which is required for its tyrosine kinase activity, was significantly enhanced (Fig. 12, H and I). Also, the level of phosphorylation at Tyr-751 of PDGFRβ was elevated in the diabetic renal cortex (Fig. 12, J and K). Our results above show that PDGFRβ is necessary for TGFβ-induced mTORC1 activation (Fig. 3). We determined the phosphorylation of S6 kinase, rps6, and 4EBP1 as readout of mTORC1 activation in renal cortical lysates. A significant increase in phosphorylation of S6 kinase, rps6, and 4EBP1 was observed in the diabetic mice, indicating activation of mTORC1 (Fig. 12, L–R). To confirm these observations in mice, we used a streptozotocin (STZ)-induced rat model of type 1 diabetes, which showed early pathologic changes of diabetic nephropathy (44). From kidney cortex, we prepared glomeruli, which contains the mesangial cells. Glomerular lysates were used to determine the expression of TGFβ. In the diabetic glomeruli, significantly increased TGFβ was detected, which correlated with enhanced Smad 2 and Smad 3 phosphorylation (Fig. S7, A–E). Furthermore, increased expression of PDGF B and phosphorylation of PDGFRβ were observed in the diabetic glomeruli (Fig. S7, F–K). Additionally, diabetic rat glomeruli showed activation of mTORC1 as judged by phosphorylation of S6 kinase, rps6, and 4EBP1 (Fig. S7, L–R). These results show that activation of PDGFRβ is associated with increased expression of TGFβ in the diabetic kidney.
Figure 12.

Increased level of TGFβ is associated with activation of PDGFRβ and mTORC1 signaling in OVE26 mice renal cortexes. Renal cortical lysates from control and OVE26 diabetic mice were immunoblotted with indicated antibodies. Bottom panels in each blot represent quantification of protein bands. Mean ± S.D. of 3–4 animals is shown. *p < 0.001–0.03 versus control animals.
Discussion
We report activation of PDGFRβ by TGFβ via expression of PDGF, which also contributes to TGFβ signaling in glomerular mesangial cells (Fig. 13). We show that TGFβ-induced Akt kinase activation requires PDGFRβ tyrosine kinase activity. Furthermore, we provide novel evidence that Akt-mediated phosphorylation of PRAS40 by TGFβ is mediated by PDGFRβ and is required for mesangial cell hypertrophy and matrix protein expression. Finally, we show activation of PDGFRβ and mTORC1 in kidneys of diabetic mice that are undergoing matrix expansion.
Figure 13.
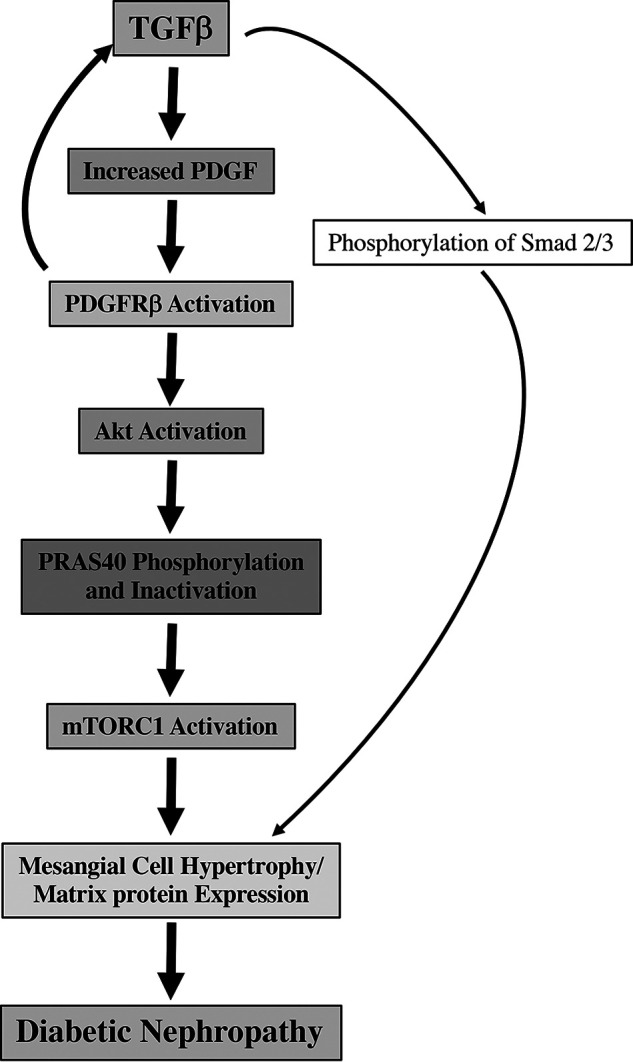
Schematic summarizing our results. Thin arrows indicate canonical TGFβ signaling. Bold arrows show noncanonical signal transduction described in the present study.
A role of TGFβ in human chronic kidney disease was established decades ago (53, 54). Also, in the animal models of renal fibrosis such as diabetic nephropathy, increased expression of TGFβ promotes the pathology (4, 17, 55). Similarly, in a rat model of glomerulonephritis, increased expression of TGFβ promotes fibrosis (56). Administration of anti-TGFβ antibody ameliorates the disease progression (57). Similarly, a murine monoclonal TGFβ antibody reduced matrix expansion and disease-associated histological changes in adriamycin- and podocyte ablation–induced nephropathies in mice (58). Furthermore, in diabetic nephropathy associated with STZ-induced type 1 diabetes in mice and with type 2 diabetic db/db mice, neutralization of TGFβ with anti-TGFβ antibody ameliorates the glomerular hypertrophy and matrix protein expansion (11, 12). A hypomorphic mouse with TGFβ expression at 10% the level of that in WT showed significant reduction in glomerular filtration rate and albuminuria. In fact, 3-fold overexpression of TGFβ in mice causes glomerulosclerosis and albuminuria (59). More recently, administration of a soluble TGFβ receptor II by a gene therapy protocol to mice with renal fibrosis, where TGFβ contributes to the pathology, showed significant beneficial effects (60). These results conclusively demonstrate a significant pathologic role of TGFβ in renal diseases, suggesting that blocking of TGFβ may be beneficial in renal fibrosis. However, the lack of progress is due to the concerns that inhibition of TGFβ may enhance the risk of autoimmune disease (17). Because TGFβ has multiple roles in maintaining various homeostatic processes, direct targeting of this cytokine may elicit multi-organ side effects. Thus, an alternative therapeutic strategy to inhibit pathologic actions of TGFβ in glomerulosclerosis needs to be identified.
The role of receptor tyrosine kinases in renal disease is established (61). Both positive and negative regulatory roles of epidermal growth factor receptor (EGFR) in renal pathology have been reported. For example, in ischemic kidney injury, activation of EGFR ameliorates the disease progression (62). Furthermore, in an EGFR mutant mouse with significantly reduced tyrosine kinase activity, tubular damage was more severe with increased apoptosis after nephrotoxic injury than in the WT mice, demonstrating a protective role of EGFR (63). On the other hand, a pathologic role of this receptor in TGFβ-mediated renal fibrosis was reported (17). EGFR is abundantly expressed in the renal proximal tubular epithelial cells and interstitial fibroblasts (62). Inhibition of EGFR and proximal tubule-specific deletion of this receptor tyrosine kinase ameliorate TGFβ-mediated renal fibrosis (64, 65). Also, involvement of Src tyrosine kinase has been reported in angiotensin II–induced kidney fibrosis (66). Thus, a role of tyrosine kinases in renal fibroblasts and tubular epithelial cells is established for induction of fibrosis. In the present study, we found that TGFβ activates the PDGFRβ in the renal mesangial cells. These results are in contrast to the observations of a recent report in which TGFβ did not have any effect on PDGFRβ phosphorylation in fibroblasts, suggesting cell-specificity for this interaction (67). In fact, we showed that TGFβ increased the expression of PDGF, which binds to the PDGFRβ to induce autophosphorylation and its activation. Addition of PDGF directly to mesangial cells also increased phosphorylation/activation of PDGFRβ.
Our data in renal mesangial cells suggest that PDGFRβ may serve as the initiator of TGFβ noncanonical signaling. We and others previously reported that both Smad 3 and PI 3-kinase/Akt signaling regulate mesangial cell pathology in response to TGFβ (15, 29, 45, 68). PI 3-kinase activation requires its association with tyrosine phosphorylated proteins (69). In the case of PDGFRβ, the specific residue was identified as Tyr-751 (31). We showed that TGFβ increased the tyrosine phosphorylation of this residue, which was sensitive to the inhibition of PDGFRβ tyrosine kinase activity, demonstrating autophosphorylation by PDGFRβ. These results indicate that TGFβ may activate PI 3-kinase via this direct interaction mechanism with PDGFRβ. In fact, we found that TGFβ-stimulated phosphorylation of Akt, a downstream target of active PI 3-kinase, depended upon PDGFRβ and its tyrosine kinase activity. This observation was similar to that obtained with direct addition of PDGF to mesangial cells. Interestingly, we found that PDGF increased the expression of TGFβ to induce Smad 2/3 phosphorylation. These results identify a positive feedback loop between TGFβ and PDGF signaling in mesangial cells (Fig. 13)
Recently, we and others demonstrated that mTORC1 controls renal cell pathology in rodent models of fibrosis where TGFβ plays important role (70–76). Three protein subunits of mTORC1, raptor, deptor, and PRAS40, regulate the activity of this kinase complex (48). Both nutrients and growth factors use independent mechanisms for mTORC1 activation. Amino acids promote formation of GTP-bound Rag proteins via Ragulator to recruit mTORC1 to GTP-bound Rheb for its activation (77). During growth factor receptor tyrosine kinase stimulation of cells, activated Akt kinase phosphorylates the raptor binding protein, PRAS40, which under basal state inhibits the substrate recruitment to mTORC1. Akt-mediated phosphorylation of PRAS40 results in its inactivation to increase the mTORC1 activity (34). For example, inactivation of PRAS40 in HeLa cells increased mTORC1 activity, leading to inhibition of their apoptosis. Interestingly, rapamycin did not reverse this anti-apoptotic effect, indicating that the effect of PRAS40 does not involve inhibition of mTORC1 (78). In 293 cells, PRAS40 inactivation showed inhibition of mTORC1 activity (79). These results are similar to those observed in Drosophila eyes where the hypomorphic allele of lobe, the fly ortholog of PRAS40, resulted in inhibition of mTORC1 and reduced eye size (80, 81). We reported previously that TGFβ stimulates phosphorylation of PRAS40 in mesangial cells (46). Now we show that PDGFRβ tyrosine kinase is required for this phosphorylation of PRAS40 by TGFβ, similar to PDGF addition. In contrast to the results in 293 cells and in Drosophila described above, phosphorylation of PRAS40 by PDGFRβ-mediated activated Akt kinase enhances mTORC1 activity in mesangial cells by both TGFβ and PDGF.
Induction of mesangial cell hypertrophy and matrix protein accumulation correlate with increased expression of TGFβ in rodent models of fibrosis (17, 51, 82). Also, renal cells derived from TGFβ null mice show impaired hypertrophic response and fibronectin expression (83). Similarly, glomerular hypertrophy and mesangial matrix expansion are reduced in TGFβ receptor II heterozygous mice models of diabetes (84). We demonstrated previously that TGFβ regulates mesangial cell hypertrophy and fibronectin expression (14, 46, 47). A role of Akt kinase was initially identified in cell-size control in the fruit fly. Overexpression of Akt in the imaginal disc of Drosophila increases the cell size (85). Similarly, transgenic mice expressing constitutively active Akt in the heart show cardiomyocyte hypertrophy (86). Also, Akt kinase activity is required for TGFβ-induced mesangial cell hypertrophy (44). We showed that expression of both fibronectin and collagen I (α2) by TGFβ was meditated by Akt kinase. However, the specific substrate that regulates this TGFβ response is not clear. In mouse hearts, Akt-mediated phosphorylation of PRAS40 increases mTORC1 activity to induce pathological hypertrophy and fibrosis (87). We reported the requirement of mTORC1 in TGFβ-induced mesangial cell hypertrophy and matrix protein expression (14, 46, 47). Now we demonstrate that Akt-phosphorylated PRAS40 is required for mesangial cell hypertrophy and fibronectin and collagen I (α2) expression in response to TGFβ. In fact, we provide evidence that PDGFRβ tyrosine kinase downstream of TGFβRI acts as a mediator of these phenomena. To our knowledge, this is the first demonstration of the involvement of PDGFRβ tyrosine kinase in TGFβ-stimulated Akt/PRAS40/mTORC1 signaling that contributes to mesangial cell hypertrophy and matrix protein expansion. Furthermore, our results demonstrate an in vivo relevance showing association of increased TGFβ expression with activation of PDGFRβ and mTORC1 in the renal cortex and glomeruli of diabetic mice and rats, respectively. Thus, in TGFβ-regulated fibrotic renal diseases such as diabetic nephropathy, attacking PDGFRβ may provide an attractive therapeutic option that bypasses the detrimental effects of directly targeting TGFβ.
Experimental procedures
Reagents
TGFβ1 and PDGF BB were obtained from R&D Systems. Cell culture materials, including Opti-MEM medium, were purchased from Thermo Fisher Scientific. PMSF, Nonidet P-40, Na3VO4, protease inhibitor mixture, JNJ, β-actin (catalog no. A2066, lot 3082M4781) and fibronectin (catalog no. F3648, lot 103M4818V) antibodies were purchased from Sigma. The antibodies against phospho-PDGFRβ (Tyr-857) (catalog no. 3270S, lot 3), phospho-PDGFRβ (Tyr-751; catalog no. 3161S, lot 7), phospho-Smad 3 (Ser-423/425; catalog no. 9520S, lot 15), Smad 3 (catalog no. 9513S, lot 2), phospho-Smad 2 (Ser-465/467; catalog no. 3108S, lot 2), Smad 2 (catalog no. 5339S, lot 6), phospho-Akt (Ser-473; catalog no. 9271S, lot 14), phospho-Akt (Thr-308; catalog no. 9275S, lot 26), Akt, (catalog no. 9272S, lot 28) phospho-GSK3β (Ser-9; catalog no. 9323S, lot 9), GSK3β (catalog no. 12456S, lot 3), phospho-PRAS40 (Thr-246; catalog no. 2997S, lot 12), PRAS40 (catalog no. 2691S, lot 8), phospho-tuberin (catalog no. 3611S, lot 6), phospho-4EBP1 (Thr-37/46) (catalog no. 9459S, lot 10), phospho-4EBP1 (Ser-65; catalog no. 9456S, lot 5), 4EBP1 (catalog no. 9452S, lot 10), phospho-S6 kinase (Thr-389; catalog no. 9205S, lot 16), S6 kinase (catalog no. 9202S, lot 20), phospho-rps6 (Ser-240/244; catalog no. 5364S, lot 5), rps6 (catalog no. 2217S, lot 7), phospho-STAT3 (Tyr-705; catalog no. 9131S, lot 18), mTOR (catalog no. 3270S, lot 3), phospho-ERK1/2 (Thr-202/Tyr-204; catalog no. 9101S, lot 28), and ERK1/2 (catalog no. 9102S, lot 19) were obtained from Cell Signaling Technology. PDGFRβ (catalog no. 2972S, lot 6), tuberin (SC-893, lot K1703), STAT3 (catalog no. SC-482, lot E176), and Myc (catalog no. SC-40, lot A1201) antibodies and pooled siRNAs against PDGFRβ were purchased from Santa Cruz Biotechnology. Collagen I (α2) antibody (catalog no. 14695-1-AP) was obtained from Proteintech. PDGF B antibody was purchased from Millipore (catalog no. 06-127, lot 2585791). HA antibody (catalog no. MMS-101R-500, lot D13FF01646) was obtained from Babco (Princeton, NJ). TGFβ antibody (catalog no. ab27969, lot GR66929-7) was purchased from Abcam. The PVDF membrane to transfer proteins was obtained from PerkinElmer. The transfection reagent FuGENE HD was purchased from Promega. HA-tagged myr-Akt, PRAS40 (Thr-246A), and Myc-tagged raptor expression plasmids have been described previously (49, 88, 89). The mTOR mutant expression vector (SL1 + I2017T), which renders hyperactive mTORC1 activity, has been described (49, 50).
Cell culture
Human mesangial cells were originally prepared by Abboud and co-workers (90) from glomeruli isolated by differential sieving. The resuspended glomeruli were digested with collagenase. Mesangial cells were cultured from outgrowths of collagenase-treated glomeruli and characterized (90). The frozen cells were thawed and grown in DMEM in the presence of 10% FBS as described previously (47, 91–93). The cells were used between passages 9 and 12. Before performing experiments, the cells were grown in complete medium until they reached confluency. The cell monolayer was then starved in serum-free medium for 24 h prior to incubation with TGFβ (2 ng/ml) for indicated periods of time.
Animals
FVB mice overexpressing calmodulin transgene in the β-cell of pancreas, called OVE26 mice, were used and purchased from The Jackson Laboratory. At 3 days of age the OVE26 mice are hyperglycemic (94). At 2 months of age, these mice develop pathologies of diabetic nephropathy, including significant renal hypertrophy, glomerular hypertrophy, and albuminuria (52, 95). The control FVB and diabetic OVE26 mice had free access to food and water. At 3 months of age, the animals were sacrificed, the kidneys were removed, and renal cortical tissues were isolated and frozen as described previously (52).
To induce diabetes in rats (Sprague Dawley; 200–250 g), 55 mg/kg of body weight STZ in sodium citrate buffer (pH 4.5) was injected by tail vein. The blood glucose was measured after 24 h. The animals had free access to water and food. At 5 days after STZ injection, the rats were euthanized and the kidneys were removed to isolate cortical sections. Glomeruli were prepared from the kidney cortexes by a differential sieving technique as described (90). Both mice and rats were kept at the University of Texas Health Science Center animal facility. The University of Texas Health Science Center Institutional Animal Care and Use Committee approved both protocols.
Preparation of cell, cortical, and glomerular lysates
At the end of the incubation period, the mesangial cell monolayer was washed twice with PBS. The monolayer was then harvested in radioimmune precipitation assay buffer (RIPA; 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1 mm Na3VO4, 1% Nonidet P-40, 1 mm PMSF, and 0.1% protease inhibitor mixture). The renal cortexes from control and diabetic mice and the glomeruli from the control and STZ-induced diabetic rats were harvested in the same RIPA buffer. The cells, cortexes, and glomeruli were lysed in RIPA at 4 °C for 30 min. After the incubation, the cell, cortical, and glomerular extracts were centrifuged at 12,000 × g for 30 min at 4 °C. The cleared supernatant was collected as cell, cortical, and glomerular lysates. Protein concentration was determined.
Immunoblotting
Cell lysates containing equal amounts of protein were separated by SDS-PAGE. The proteins were transferred to PVDF membrane. For immunoblotting, the membrane containing the proteins was incubated with indicated primary antibody at 4 °C. The primary antibody dilutions used for immunoblotting were 1:1000. The membrane was washed, followed by incubation with horseradish peroxidase-conjugated secondary antibody (1:10,000). The membrane was then incubated with enhanced chemiluminescence reagent to develop a specific protein band, which was visualized by exposing the membrane to X-ray film (91, 96).
Transfection
The semiconfluent mesangial cell monolayer was washed with PBS. Opti-MEM was added. The expression vector or siRNAs against PDGFRβ were transfected in Opti-MEM with FuGENE HD as described previously (91). After 24 h, the cells were starved as described above prior to adding TGFβ.
Measurement of protein synthesis
Protein synthesis was determined as described previously (14, 89, 91). Briefly, during the last four hours of incubation with TGFβ, the cells were incubated with 1 µCi of [35S]methionine as described before (44). At the end of incubation, the cells were lysed in RIPA as described above and the protein was estimated. Equal amount of protein was spotted onto 3MM filter paper and washed in boiling 10% trichloroacetic acid containing 0.1 gm/l methionine for 1 min. The filters were then dried and the radioactivity was counted using scintillation fluid.
Measurement of hypertrophy
The cells were trypsinized in the medium at the end of the incubation and counted using a hemocytometer (91, 96). The cells were then gently centrifuged at 1,000 × g for 5 min at 4 °C. The cell pellet was washed with PBS and resuspended in RIPA buffer to lyse, as described above. Protein concentration was measured in the cell lysate and the ratio of total protein to cell number was determined. The increase in the ratio was considered as cell hypertrophy (44, 89).
Statistics
Prism GraphPad software was used to determine the significance of the data. Analysis of variance followed by Tukey's multiple comparison test was used. A p-value of <0.05 was considered significant (97).
Data availability
All data are contained within the article and in the supporting information.
Supplementary Material
This article contains supporting information.
Author contributions—S. M. and F. D. data curation; S. M., F. D., and G. G.-C. formal analysis; G. G.-C. conceptualization; G. G.-C. supervision; G. G.-C. funding acquisition; G. G.-C. writing-original draft; G. G.-C. project administration; G. G.-C. writing-review and editing; B. S. K. analyzed in vivo data, took part in the discussion of the data, and edited the manuscript; N. G.-C. took part in discussion of the results and provided intellectual input.
Funding and additional information—This work was supported by the Department of Veterans Affairs Biomedical Laboratory Research and Development Service Merit Review Award 2I01 BX000926 to G. G.-C. G. G.-C. is a recipient of Research Career Scientist Award IK6BX00361 from the Department of Veterans Affairs Biomedical Laboratory Research and Development Service.
Conflict of interest—The authors declare that they do not have any conflicts of interest with the contents of the article.
- TGFβ
- transforming growth factor-β
- PDGF
- platelet-derived growth factor
- PDGFR
- PDGF receptor
- PI
- phosphatidylinositol
- PRAS40
- proline-rich Akt substrate of 40 kDa
- mTORC
- mTOR complex
- Akt
- protein kinase B
- TGFβR
- TGFβ receptor
- SB
- SB-431542
- JNJ
- JNJ-10198409
- SH
- src homology
- GSK
- glycogen synthase kinase
- 4EBP
- eukaryotic translation initiation factor 4E-binding protein
- rps6
- ribosomal protein S6
- ERK
- extracellular signal-regulated kinase
- STZ
- streptozotocin
- EGFR
- epidermal growth factor receptor
- RIPA
- radioimmune precipitation assay buffer.
References
- 1. Collins A. J., Foley R. N., Chavers B., Gilbertson D., Herzog C., Johansen K., Kasiske B., Kutner N., Liu J., St Peter W., Guo H., Gustafson S., Heubner B., Lamb K., Li S., et al. (2012) United States renal data system 2011 annual data report: atlas of chronic kidney disease & end-stage renal disease in the United States. Am. J. Kidney Dis. 59, A7, e1–e420 10.1053/j.ajkd.2011.11.015 [DOI] [PubMed] [Google Scholar]
- 2. Go A. S., Chertow G. M., Fan D., McCulloch C. E., and Hsu C. Y. (2004) Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 351, 1296–1305 10.1056/NEJMoa041031 [DOI] [PubMed] [Google Scholar]
- 3. Vega G., Alarcón S., and San Martin R. (2016) The cellular and signalling alterations conducted by TGF-β contributing to renal fibrosis. Cytokine 88, 115–125 10.1016/j.cyto.2016.08.019 [DOI] [PubMed] [Google Scholar]
- 4. Kanwar Y. S., Sun L., Xie P., Liu F. Y., and Chen S. (2011) A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 6, 395–423 10.1146/annurev.pathol.4.110807.092150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abboud H. E. (2012) Mesangial cell biology. Exp. Cell Res. 318, 979–985 10.1016/j.yexcr.2012.02.025 [DOI] [PubMed] [Google Scholar]
- 6. Schlondorff D. (1987) The glomerular mesangial cell: an expanding role for a specialized pericyte. FASEB J. 1, 272–281 10.1096/fasebj.1.4.3308611 [DOI] [PubMed] [Google Scholar]
- 7. Kanwar Y. S., Wada J., Sun L., Xie P., Wallner E. I., Chen S., Chugh S., and Danesh F. R. (2008) Diabetic nephropathy: mechanisms of renal disease progression. Exp. Biol. Med. 233, 4–11 10.3181/0705-MR-134 [DOI] [PubMed] [Google Scholar]
- 8. Kopp J. B., Factor V. M., Mozes M., Nagy P., Sanderson N., Bottinger E. P., Klotman P. E., and Thorgeirsson S. S. (1996) Transgenic mice with increased plasma levels of TGF-β 1 develop progressive renal disease. Lab. Invest. 74, 991–1003 [PubMed] [Google Scholar]
- 9. Kagami S., Border W. A., Miller D. E., and Noble N. A. (1994) Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J. Clin. Invest. 93, 2431–2437 10.1172/JCI117251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riser B. L., Denichilo M., Cortes P., Baker C., Grondin J. M., Yee J., and Narins R. G. (2000) Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 11, 25–38 [DOI] [PubMed] [Google Scholar]
- 11. Sharma K., Jin Y., Guo J., and Ziyadeh F. N. (1996) Neutralization of TGF-β by anti-TGF-β antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 45, 522–530 10.2337/diabetes.45.4.522 [DOI] [PubMed] [Google Scholar]
- 12. Ziyadeh F. N., Hoffman B. B., Han D. C., Iglesias-De La Cruz M. C., Hong S. W., Isono M., Chen S., McGowan T. A., and Sharma K. (2000) Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. U. S. A. 97, 8015–8020 10.1073/pnas.120055097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moustakas A., and Heldin C. H. (2009) The regulation of TGFβ signal transduction. Development 136, 3699–3714 10.1242/dev.030338 [DOI] [PubMed] [Google Scholar]
- 14. Das F., Ghosh-Choudhury N., Mahimainathan L., Venkatesan B., Feliers D., Riley D. J., Kasinath B. S., and Choudhury G. G. (2008) Raptor-rictor axis in TGFβ-induced protein synthesis. Cell Signal. 20, 409–423 10.1016/j.cellsig.2007.10.027 [DOI] [PubMed] [Google Scholar]
- 15. Ghosh Choudhury G., and Abboud H. E. (2004) Tyrosine phosphorylation-dependent PI 3 kinase/Akt signal transduction regulates TGFβ-induced fibronectin expression in mesangial cells. Cell Signal. 16, 31–41 10.1016/S0898-6568(03)00094-9 [DOI] [PubMed] [Google Scholar]
- 16. Lamouille S., and Derynck R. (2007) Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451 10.1083/jcb.200611146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meng X. M., Nikolic-Paterson D. J., and Lan H. Y. (2016) TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 10.1038/nrneph.2016.48 [DOI] [PubMed] [Google Scholar]
- 18. Fredriksson L., Li H., and Eriksson U. (2004) The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 15, 197–204 10.1016/j.cytogfr.2004.03.007 [DOI] [PubMed] [Google Scholar]
- 19. Boor P., Ostendorf T., and Floege J. (2014) PDGF and the progression of renal disease. Nephrol. Dial. Transplant. 29, i45–i54 10.1093/ndt/gft273 [DOI] [PubMed] [Google Scholar]
- 20. van Roeyen C. R., Ostendorf T., and Floege J. (2012) The platelet-derived growth factor system in renal disease: an emerging role of endogenous inhibitors. Eur. J. Cell Biol. 91, 542–551 10.1016/j.ejcb.2011.07.003 [DOI] [PubMed] [Google Scholar]
- 21. Choudhury G. G., Biswas P., Grandaliano G., Fouqueray B., Harvey S. A., and Abboud H. E. (1994) PDGF-mediated activation of phosphatidylinositol 3 kinase in human mesangial cells. Kidney Int. 46, 37–47 10.1038/ki.1994.242 [DOI] [PubMed] [Google Scholar]
- 22. Choudhury G. G., Grandaliano G., Jin D. C., Katz M. S., and Abboud H. E. (2000) Activation of PLC and PI 3 kinase by PDGF receptor alpha is not sufficient for mitogenesis and migration in mesangial cells. Kidney Int. 57, 908–917 10.1046/j.1523-1755.2000.00907.x [DOI] [PubMed] [Google Scholar]
- 23. Levéen P., Pekny M., Gebre-Medhin S., Swolin B., Larsson E., and Betsholtz C. (1994) Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 8, 1875–1887 10.1101/gad.8.16.1875 [DOI] [PubMed] [Google Scholar]
- 24. Lindahl P., Hellström M., Kalén M., Karlsson L., Pekny M., Pekna M., Soriano P., and Betsholtz C. (1998) Paracrine PDGF-B/PDGF-Rβ signaling controls mesangial cell development in kidney glomeruli. Development 125, 3313–3322 [DOI] [PubMed] [Google Scholar]
- 25. Langham R. G., Kelly D. J., Maguire J., Dowling J. P., Gilbert R. E., and Thomson N. M. (2003) Over-expression of platelet-derived growth factor in human diabetic nephropathy. Nephrol. Dial. Transplant 18, 1392–1396 10.1093/ndt/gfg177 [DOI] [PubMed] [Google Scholar]
- 26. Uehara G., Suzuki D., Toyoda M., Umezono T., and Sakai H. (2004) Glomerular expression of platelet-derived growth factor (PDGF)-A, -B chain and PDGF receptor-α, -β in human diabetic nephropathy. Clin. Exp. Nephrol. 8, 36–42 10.1007/s10157-003-0265-8 [DOI] [PubMed] [Google Scholar]
- 27. Soriano P. (1994) Abnormal kidney development and hematological disorders in PDGF β-receptor mutant mice. Genes Dev. 8, 1888–1896 10.1101/gad.8.16.1888 [DOI] [PubMed] [Google Scholar]
- 28. Bakin A. V., Tomlinson A. K., Bhowmick N. A., Moses H. L., and Arteaga C. L. (2000) Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275, 36803–36810 10.1074/jbc.M005912200 [DOI] [PubMed] [Google Scholar]
- 29. Runyan C. E., Schnaper H. W., and Poncelet A. C. (2004) The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-β1. J. Biol. Chem. 279, 2632–2639 10.1074/jbc.M310412200 [DOI] [PubMed] [Google Scholar]
- 30. McGlade C. J., Ellis C., Reedijk M., Anderson D., Mbamalu G., Reith A. D., Panayotou G., End P., Bernstein A., and Kazlauskas A. and (1992) SH2 domains of the p85 α subunit of phosphatidylinositol 3-kinase regulate binding to growth factor receptors. Mol. Cell Biol. 12, 991–997 10.1128/mcb.12.3.991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valius M., and Kazlauskas A. (1993) Phospholipase C-γ 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell 73, 321–334 10.1016/0092-8674(93)90232-F [DOI] [PubMed] [Google Scholar]
- 32. Manning B. D., and Toker A. (2017) AKT/PKB signaling: navigating the network. Cell 169, 381–405 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Das F., Bera A., Ghosh-Choudhury N., Abboud H. E., Kasinath B. S., and Choudhury G. G. (2014) TGFβ-induced deptor suppression recruits mTORC1 and not mTORC2 to enhance collagen I (α2) gene expression. PLoS One 9, e109608 10.1371/journal.pone.0109608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., and Sabatini D. M. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 10.1016/j.molcel.2007.03.003 [DOI] [PubMed] [Google Scholar]
- 35. Inoki K., Li Y., Xu T., and Guan K. L. (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Inoki K., Li Y., Zhu T., Wu J., and Guan K. L. (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657 10.1038/ncb839 [DOI] [PubMed] [Google Scholar]
- 37. Manning B. D., Tee A. R., Logsdon M. N., Blenis J., and Cantley L. C. (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162 10.1016/S1097-2765(02)00568-3 [DOI] [PubMed] [Google Scholar]
- 38. Potter C. J., Pedraza L. G., and Xu T. (2002) Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 4, 658–665 10.1038/ncb840 [DOI] [PubMed] [Google Scholar]
- 39. Heldin C. H., and Moustakas A. (2016) Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 8, a022053 10.1101/cshperspect.a022053 [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu S., Mao Y., Wu J., Feng J., Li J., Wu L., Yu Q., Zhou Y., Zhang J., Chen J., Ji J., Chen K., Wang F., Dai W., Fan X., et al. (2020) TGF-β/Smad and JAK/STAT pathways are involved in the anti-fibrotic effects of propylene glycol alginate sodium sulphate on hepatic fibrosis. J. Cell. Mol. Med. 24, 5224–5237 10.1111/jcmm.15175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bruna A., Darken R. S., Rojo F., Ocaña A., Peñuelas S., Arias A., Paris R., Tortosa A., Mora J., Baselga J., and Seoane J. (2007) High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–160 10.1016/j.ccr.2006.11.023 [DOI] [PubMed] [Google Scholar]
- 42. Charbonneau M., Lavoie R. R., Lauzier A., Harper K., McDonald P. P., and Dubois C. M. (2016) Platelet-derived growth factor receptor activation promotes the prodestructive invadosome-forming phenotype of synoviocytes from patients with rheumatoid arthritis. J. Immunol. 196, 3264–3275 10.4049/jimmunol.1500502 [DOI] [PubMed] [Google Scholar]
- 43. Heldin C. H., and Westermark B. (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 79, 1283–1316 10.1152/physrev.1999.79.4.1283 [DOI] [PubMed] [Google Scholar]
- 44. Mahimainathan L., Das F., Venkatesan B., and Choudhury G. G. (2006) Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes 55, 2115–2125 10.2337/db05-1326 [DOI] [PubMed] [Google Scholar]
- 45. Kato M., Putta S., Wang M., Yuan H., Lanting L., Nair I., Gunn A., Nakagawa Y., Shimano H., Todorov I., Rossi J. J., and Natarajan R. (2009) TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell Biol. 11, 881–889 10.1038/ncb1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dey N., Ghosh-Choudhury N., Kasinath B. S., and Choudhury G. G. (2012) TGFβ-stimulated microRNA-21 utilizes PTEN to orchestrate AKT/mTORC1 signaling for mesangial cell hypertrophy and matrix expansion. PLoS One 7, e42316 10.1371/journal.pone.0042316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Das F., Ghosh-Choudhury N., Bera A., Dey N., Abboud H. E., Kasinath B. S., and Choudhury G. G. (2013) Transforming growth factor β integrates Smad 3 to mechanistic target of rapamycin complexes to arrest deptor abundance for glomerular mesangial cell hypertrophy. J. Biol. Chem. 288, 7756–7768 10.1074/jbc.M113.455782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das F., Ghosh-Choudhury N., Dey N., Mandal C. C., Mahimainathan L., Kasinath B. S., Abboud H. E., and Choudhury G. G. (2012) Unrestrained mammalian target of rapamycin complexes 1 and 2 increase expression of phosphatase and tensin homolog deleted on chromosome 10 to regulate phosphorylation of Akt kinase. J. Biol. Chem. 287, 3808–3822 10.1074/jbc.M111.246397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohne Y., Takahara T., Hatakeyama R., Matsuzaki T., Noda M., Mizushima N., and Maeda T. (2008) Isolation of hyperactive mutants of mammalian target of rapamycin. J. Biol. Chem. 283, 31861–31870 10.1074/jbc.M801546200 [DOI] [PubMed] [Google Scholar]
- 51. Kato M., and Natarajan R. (2014) Diabetic nephropathy–emerging epigenetic mechanisms. Nat. Rev. Nephrol. 10, 517–530 10.1038/nrneph.2014.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bera A., Das F., Ghosh-Choudhury N., Mariappan M. M., Kasinath B. S., and Ghosh Choudhury G. (2017) Reciprocal regulation of miR-214 and PTEN by high glucose regulates renal glomerular mesangial and proximal tubular epithelial cell hypertrophy and matrix expansion. Am. J. Physiol. Cell Physiol. 313, C430–C447 10.1152/ajpcell.00081.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sharma K., Ziyadeh F. N., Alzahabi B., McGowan T. A., Kapoor S., Kurnik B. R., Kurnik P. B., and Weisberg L. S. (1997) Increased renal production of transforming growth factor-β1 in patients with type II diabetes. Diabetes 46, 854–859 10.2337/diabetes.46.5.854 [DOI] [PubMed] [Google Scholar]
- 54. Yoshioka K., Takemura T., Murakami K., Okada M., Hino S., Miyamoto H., and Maki S. (1993) Transforming growth factor-β protein and mRNA in glomeruli in normal and diseased human kidneys. Lab. Invest. 68, 154–163 [PubMed] [Google Scholar]
- 55. Border W. A., and Noble N. A. (1994) Transforming growth factor β in tissue fibrosis. N. Engl. J. Med. 331, 1286–1292 10.1056/NEJM199411103311907 [DOI] [PubMed] [Google Scholar]
- 56. Okuda S., Languino L. R., Ruoslahti E., and Border W. A. (1990) Elevated expression of transforming growth factor-β and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J. Clin. Invest. 86, 453–462 10.1172/JCI114731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Border W. A., Okuda S., Languino L. R., Sporn M. B., and Ruoslahti E. (1990) Suppression of experimental glomerulonephritis by antiserum against transforming growth factor β 1. Nature 346, 371–374 10.1038/346371a0 [DOI] [PubMed] [Google Scholar]
- 58. Liang X., Schnaper H. W., Matsusaka T., Pastan I., Ledbetter S., and Hayashida T. (2016) Anti-TGF-β antibody, 1D11, ameliorates glomerular fibrosis in mouse models after the onset of proteinuria. PLoS One 11, e0155534 10.1371/journal.pone.0155534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hathaway C. K., Gasim A. M., Grant R., Chang A. S., Kim H. S., Madden V. J., Bagnell C. R. Jr, Jennette J. C., Smithies O., and Kakoki M. (2015) Low TGFβ1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc. Natl. Acad. Sci. U. S. A. 112, 5815–5820 10.1073/pnas.1504777112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Davidsohn N., Pezone M., Vernet A., Graveline A., Oliver D., Slomovic S., Punthambaker S., Sun X., Liao R., Bonventre J. V., and Church G. M. (2019) A single combination gene therapy treats multiple age-related diseases. Proc. Natl. Acad. Sci. U. S. A. 116, 23505–23511 10.1073/pnas.1910073116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu F., and Zhuang S. (2016) Role of receptor tyrosine kinase signaling in renal fibrosis. Int. J. Mol. Sci. 17, 972 10.3390/ijms17060972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zeng F., Singh A. B., and Harris R. C. (2009) The role of the EGF family of ligands and receptors in renal development, physiology and pathophysiology. Exp. Cell Res. 315, 602–610 10.1016/j.yexcr.2008.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang Z., Chen J. K., Wang S. W., Moeckel G., and Harris R. C. (2003) Importance of functional EGF receptors in recovery from acute nephrotoxic injury. J. Am. Soc. Nephrol. 14, 3147–3154 10.1097/01.asn.0000098681.56240.1a [DOI] [PubMed] [Google Scholar]
- 64. Chen J., Chen J. K., Nagai K., Plieth D., Tan M., Lee T. C., Threadgill D. W., Neilson E. G., and Harris R. C. (2012) EGFR signaling promotes TGFβ-dependent renal fibrosis. J. Am. Soc. Nephrol. 23, 215–224 10.1681/ASN.2011070645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Samarakoon R., Dobberfuhl A. D., Cooley C., Overstreet J. M., Patel S., Goldschmeding R., Meldrum K. K., and Higgins P. J. (2013) Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 25, 2198–2209 10.1016/j.cellsig.2013.07.007 [DOI] [PubMed] [Google Scholar]
- 66. Liu N., Wang L., Yang T., Xiong C., Xu L., Shi Y., Bao W., Chin Y. E., Cheng S. B., Yan H., Qiu A., and Zhuang S. (2015) EGF receptor inhibition alleviates hyperuricemic nephropathy. J. Am. Soc. Nephrol. 26, 2716–2729 10.1681/ASN.2014080793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Porsch H., Mehić M., Olofsson B., Heldin P., and Heldin C.-H. (2014) Platelet-derived growth factor β-receptor, transforming growth factor β type I receptor, and CD44 protein modulate each other's signaling and stability. J. Biol. Chem. 289, 19747–19757 10.1074/jbc.M114.547273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Das F., Ghosh-Choudhury N., Venkatesan B., Li X., Mahimainathan L., and Choudhury G. G. (2008) Akt kinase targets association of CBP with SMAD 3 to regulate TGFβ-induced expression of plasminogen activator inhibitor-1. J. Cell. Physiol. 214, 513–527 10.1002/jcp.21236 [DOI] [PubMed] [Google Scholar]
- 69. Yuan T. L., and Cantley L. C. (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510 10.1038/onc.2008.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kasinath B. S., Feliers D., Sataranatarajan K., Ghosh Choudhury G., Lee M. J., and Mariappan M. M. (2009) Regulation of mRNA translation in renal physiology and disease. Am. J. Physiol. Renal Physiol. 297, F1153–F1165 10.1152/ajprenal.90748.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sakaguchi M., Isono M., Isshiki K., Sugimoto T., Koya D., and Kashiwagi A. (2006) Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem. Biophys. Res. Commun. 340, 296–301 10.1016/j.bbrc.2005.12.012 [DOI] [PubMed] [Google Scholar]
- 72. Sataranatarajan K., Mariappan M. M., Lee M. J., Feliers D., Choudhury G. G., Barnes J. L., and Kasinath B. S. (2007) Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. Am. J. Pathol. 171, 1733–1742 10.2353/ajpath.2007.070412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Eid A. A., Ford B. M., Bhandary B., de Cassia Cavaglieri R., Block K., Barnes J. L., Gorin Y., Choudhury G. G., and Abboud H. E. (2013) Mammalian target of rapamycin regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes 62, 2935–2947 10.2337/db12-1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Inoki K., Mori H., Wang J., Suzuki T., Hong S., Yoshida S., Blattner S. M., Ikenoue T., Rüegg M. A., Hall M. N., Kwiatkowski D. J., Rastaldi M. P., Huber T. B., Kretzler M., Holzman L. B., et al. (2011) mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Invest. 121, 2181–2196 10.1172/JCI44771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen J. K., Chen J., Neilson E. G., and Harris R. C. (2005) Role of mammalian target of rapamycin signaling in compensatory renal hypertrophy. J. Am. Soc. Nephrol. 16, 1384–1391 10.1681/ASN.2004100894 [DOI] [PubMed] [Google Scholar]
- 76. Gödel M., Hartleben B., Herbach N., Liu S., Zschiedrich S., Lu S., Debreczeni-Mór A., Lindenmeyer M. T., Rastaldi M. P., Hartleben G., Wiech T., Fornoni A., Nelson R. G., Kretzler M., Wanke R., et al. (2011) Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Invest. 121, 2197–2209 10.1172/JCI44774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zoncu R., Efeyan A., and Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Thedieck K., Polak P., Kim M. L., Molle K. D., Cohen A., Jenö P., Arrieumerlou C., and Hall M. N. (2007) PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One 2, e1217 10.1371/journal.pone.0001217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fonseca B. D., Smith E. M., Lee V. H., MacKintosh C., and Proud C. G. (2007) PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J. Biol. Chem. 282, 24514–24524 10.1074/jbc.M704406200 [DOI] [PubMed] [Google Scholar]
- 80. Chern J. J., and Choi K. W. (2002) Lobe mediates Notch signaling to control domain-specific growth in the Drosophila eye disc. Development 129, 4005–4013 [DOI] [PubMed] [Google Scholar]
- 81. Wang Y. H., and Huang M. L. (2009) Reduction of Lobe leads to TORC1 hypoactivation that induces ectopic Jak/STAT signaling to impair Drosophila eye development. Mech. Dev. 126, 781–790 10.1016/j.mod.2009.08.005 [DOI] [PubMed] [Google Scholar]
- 82. Kato M., and Natarajan R. (2012) MicroRNA circuits in transforming growth factor-β actions and diabetic nephropathy. Semin. Nephrol. 32, 253–260 10.1016/j.semnephrol.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen S., Hoffman B. B., Lee J. S., Kasama Y., Jim B., Kopp J. B., and Ziyadeh F. N. (2004) Cultured tubule cells from TGF-β1 null mice exhibit impaired hypertrophy and fibronectin expression in high glucose. Kidney. Int. 65, 1191–1204 10.1111/j.1523-1755.2004.00492.x [DOI] [PubMed] [Google Scholar]
- 84. Kim H. W., Kim B. C., Song C. Y., Kim J. H., Hong H. K., and Lee H. S. (2004) Heterozygous mice for TGF-βIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int. 66, 1859–1865 10.1111/j.1523-1755.2004.00959.x [DOI] [PubMed] [Google Scholar]
- 85. Verdu J., Buratovich M. A., Wilder E. L., and Birnbaum M. J. (1999) Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat. Cell Biol. 1, 500–506 10.1038/70293 [DOI] [PubMed] [Google Scholar]
- 86. Shioi T., McMullen J. R., Kang P. M., Douglas P. S., Obata T., Franke T. F., Cantley L. C., and Izumo S. (2002) Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell Biol. 22, 2799–2809 10.1128/mcb.22.8.2799-2809.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Völkers M., Doroudgar S., Nguyen N., Konstandin M. H., Quijada P., Din S., Ornelas L., Thuerauf D. J., Gude N., Friedrich K., Herzig S., Glembotski C. C., and Sussman M. A. (2014) PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol. Med. 6, 57–65 10.1002/emmm.201303183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ghosh Choudhury G., Lenin M., Calhaun C., Zhang J.-H., and Abboud H. E. (2003) PDGF inactivates forkhead family transcription factor by activation of Akt in glomerular mesangial cells. Cell Signal. 15, 161–170 10.1016/S0898-6568(02)00057-8 [DOI] [PubMed] [Google Scholar]
- 89. Dey N., Ghosh-Choudhury N., Das F., Li X., Venkatesan B., Barnes J. L., Kasinath B. S., and Ghosh Choudhury G. (2010) PRAS40 acts as a nodal regulator of high glucose-induced TORC1 activation in glomerular mesangial cell hypertrophy. J. Cell. Physiol. 225, 27–41 10.1002/jcp.22186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shultz P. J., DiCorleto P. E., Silver B. J., and Abboud H. E. (1988) Mesangial cells express PDGF mRNAs and proliferate in response to PDGF. Am. J. Physiol. 255, F674–F684 10.1152/ajprenal.1988.255.4.F674 [DOI] [PubMed] [Google Scholar]
- 91. Das F., Maity S., Ghosh-Choudhury N., Kasinath B. S., and Ghosh Choudhury G. (2019) Deacetylation of S6 kinase promotes high glucose-induced glomerular mesangial cell hypertrophy and matrix protein accumulation. J. Biol. Chem. 294, 9440–9460 10.1074/jbc.RA118.007023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Das F., Ghosh-Choudhury N., Bera A., Kasinath B. S., and Choudhury G. G. (2013) TGFβ-induced PI 3 kinase-dependent Mnk-1 activation is necessary for Ser-209 phosphorylation of eIF4E and mesangial cell hypertrophy. J. Cell. Physiol. 228, 1617–1626 10.1002/jcp.24327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Choudhury G. G., Biswas P., Grandaliano G., and Abboud H. E. (1993) Involvement of PKC-alpha in PDGF-mediated mitogenic signaling in human mesangial cells. Am. J. Physiol. 265, F634–F642 10.1152/ajprenal.1993.265.5.F634 [DOI] [PubMed] [Google Scholar]
- 94. Epstein P. N., Overbeek P. A., and Means A. R. (1989) Calmodulin-induced early-onset diabetes in transgenic mice. Cell 58, 1067–1073 10.1016/0092-8674(89)90505-9 [DOI] [PubMed] [Google Scholar]
- 95. Zheng S., Noonan W. T., Metreveli N. S., Coventry S., Kralik P. M., Carlson E. C., and Epstein P. N. (2004) Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes 53, 3248–3257 10.2337/diabetes.53.12.3248 [DOI] [PubMed] [Google Scholar]
- 96. Das F., Ghosh-Choudhury N., Dey N., Bera A., Mariappan M. M., Kasinath B. S., and Ghosh Choudhury G. (2014) High glucose forces a positive feedback loop connecting Akt kinase and FoxO1 transcription factor to activate mTORC1 kinase for mesangial cell hypertrophy and matrix protein expression. J. Biol. Chem. 289, 32703–32716 10.1074/jbc.M114.605196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Maity S., Das F., Ghosh-Choudhury N., Kasinath B. S., and Ghosh Choudhury G. (2019) High glucose increases miR-214 to power a feedback loop involving PTEN and the Akt/mTORC1 signaling axis. FEBS Lett. 593, 2261–2272 10.1002/1873-3468.13505 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article and in the supporting information.