

Abstract
Chaperones are essential components of the protein homeostasis network. There is a growing interest in optimizing chaperone function, but exactly how to achieve this aim is unclear. Here, using a model chaperone, the bacterial protein Spy, we demonstrate that substitutions that alter the electrostatic potential of Spy's concave, client-binding surface enhance Spy's anti-aggregation activity. We show that this strategy is more efficient than one that enhances the hydrophobicity of Spy's surface. Our findings thus challenge the traditional notion that hydrophobic interactions are the major driving forces that guide chaperone–substrate binding. Kinetic data revealed that both charge- and hydrophobicity-enhanced Spy variants release clients more slowly, resulting in a greater “holdase” activity. However, increasing short-range hydrophobic interactions deleteriously affected Spy's ability to capture substrates, thus reducing its in vitro chaperone activity toward fast-aggregating substrates. Our strategy in chaperone surface engineering therefore sought to fine-tune the different molecular forces involved in chaperone–substrate interactions rather than focusing on enhancing hydrophobic interactions. These results improve our understanding of the mechanistic basis of chaperone–client interactions and illustrate how protein surface–based mutational strategies can facilitate the rational improvement of molecular chaperones.
Keywords: molecular chaperone, Spy, protein engineering, protein aggregation, electrostatic interaction, hydrophobic interaction, kinetics, protein folding, conformational change, chaperone-substrate interaction
Molecular chaperones are evolutionarily conserved, intricate molecular machines designed to combat protein folding problems (1, 2). Many are up-regulated under stress conditions and act not only to prevent protein aggregation, but also to assist in protein refolding. Some even facilitate the disaggregation of damaged proteins to allow them to refold (3). Chaperones also function during de novo protein folding, in part by decreasing nonnative interactions between folding intermediates and thus acting to minimize aggregation and promote productive folding (4, 5). Although chaperone action is spatially and temporally complex, the fundamental mechanism by which chaperones recognize their substrates must ultimately be controlled by basic intermolecular forces. Because the exposure of hydrophobic segments on the surface of unfolded or partially folded proteins is critical for their aggregation (6–8), it has been assumed that molecular chaperones recognize and bind these hydrophobic regions on their substrates to protect them from aggregation (9–11). One very widely cited review for instance makes the statement that chaperones “typically recognize hydrophobic amino acid side chains exposed by nonnative proteins” (9). Very similar statements are present in many other reviews; sheer repetition has made the vital nature of hydrophobicity in chaperone action almost axiomatic.
However, in addition to hydrophobic interactions, it is important to consider that electrostatic interactions can also play an important role in the initial binding of chaperones to unfolded substrates (12–14). Electrostatic forces can speed association reactions and have been recognized as a dominant long-range force in mediating protein-protein associations (15). High-speed binding events are often correlated with favorable electrostatic interactions (16, 17). Kinetic studies of the GroEL-barnase interaction, for instance, revealed that substrate association is strongly dependent on ionic strength (18). This dependence is evident only in the early stages of complex formation. Thus, for this chaperone-substrate interaction, an initial, transient ionic interaction exists before any hydrophobic binding occurs (18). Similarly, client binding to the ATP-independent chaperone Spy is also extremely sensitive to ionic strength; as the ionic strength increases, the rate of association decreases (19). When assayed in vivo, the activity of Spy also shows a strong dependence on salt concentration, further demonstrating the important role that electrostatic interactions play in Spy chaperone activity (20).
Detailed knowledge of how chaperones interact with substrates can guide efforts to optimize the performance of molecular chaperones. However, unlike with enzymes, where substrate specificity has frequently been altered by directed evolution and protein design, efforts to alter the specificity or efficiency of molecular chaperones have rarely been successfully undertaken (21). This is in part due to the lack of detailed information about substrate-chaperone interactions, because chaperones are often promiscuous and because precise substrate-chaperone binding sites are often poorly defined (12, 22). Fortunately, directed evolution techniques offer an opportunity for altering a chaperone's binding properties even in the absence of detailed knowledge of the chaperone's binding sites and mechanism. These types of experiments can provide a unique perspective in efforts to understand the working mechanisms of chaperones, for example, through the analysis of how beneficial mutations contribute to function (23, 24). Previously, using genetic selections to stabilize unstable variants of immunity protein 7 (Im7, a 10-kDa Escherichia coli protein with a well-characterized folding pathway (25)), we succeeded in isolating Spy variants that were able to increase the stability of these destabilized Im7 mutants. Most of these appear to work by expanding hydrophobic regions on the concave surfaces of Spy, where the client protein is believed to bind (26). We wondered, however, if increasing surface hydrophobicity is the only avenue for improving a chaperone's function or if nature can provide other solutions.
Here, using an improved in vivo selection, we discovered new point mutations that enhance Spy's activity. Some of these new variants act not by increasing hydrophobicity but instead by increasing the positive charges on Spy's concave surface, allowing Spy to capture its substrates more quickly. Combining these positively charged point mutations further enhanced Spy's anti-aggregation activity; however, combining the previously discovered hydrophobicity-enhancing mutations did not. By performing kinetic measurements on these hydrophobically enhanced mutants, we were able to attribute this inability to further increase Spy's anti-aggregation activity to a considerable reduction in the rates at which they bind to the substrate. Additionally, we demonstrated that rapid capture of the substrate is more important than longer substrate binding in enhancing Spy's ability to suppress rapid aggregation. Despite these mechanistic differences, we found that the performance of Spy variants in vivo is largely determined by their expression levels, suggesting that instead of relying on a single trait, nature appears to balance multiple activity determinants to optimize Spy performance.
Results
Isolation and initial characterization of new Spy variants with enhanced chaperone activities
To identify the determinants of Spy chaperone activity, we performed directed evolution to enhance Spy activity using our previously established approach (26) but with an important modification. Instead of subjecting the spy gene to random point mutagenesis, the Spy-encoding library was prepared with a DNA shuffling-based method called “random insertional-deletional strand exchange mutagenesis” (RAISE) (Fig. S1A) (27). This was done, in part, to overcome the codon bias of the error-prone PCR approach that was used to generate the previous spy mutant library (28, 29). The selection relies on Spy's ability to stabilize the L53A I54A mutant of Im7 inserted into the β-lactamase stability biosensor (30). Improved folding of Im7 L53A I54A allows the two split fragments of β-lactamase to re-associate and restore its enzymatic activity. Selection for penicillin resistance of the cells co-expressing the library and the β-lactamase (βla)-Im7 L53A I54A stability biosensor allowed us to isolate 42 Spy variants (Fig. S1B). Although 27 (64%) of these Spy variants contained known activity-enhancing mutations (26) : (Q100L, H96L, Q49L, L32P, and Q25R), we obtained six new gain-of-function mutations, including four isolated as single substitutions (F29V, L32Q, G48A, and Q52R) and two as high-frequency mutations (H65Y and H96R) that were identified in combination with other amino acid substitutions (Fig. 1A and Fig. S2).
Figure 1.

Characterization of the new gain-of-function variants in the chaperone Spy. A, distribution of the beneficial mutations on Spy's sequence, with the secondary structures annotated. Variants identified in this study (but not in the previous work) are shown in orange, those obtained in both studies are shown in black, and those only isolated in the previous work are shown in gray. B, all newly isolated Spy variants exhibited enhanced in vivo chaperone activities (n = 3, Student's t test). *p < 0.05; **p < 0.01; ***p < 0.001. The variants' in vivo activities are expressed as normalized MIC (MICnormal = MICmutant/MICWT) toward penicillin in strains expressing the Spy variants and the βla-Im7 L53A I54A folding biosensor containing a destabilized Im7 variant. C, all newly obtained Spy variants showed higher activity in preventing the aggregation of DTT-reduced α-LA (α-LA agg. Prev) compared with WT (n = 3, Student's t test). **p < 0.01; ***p < 0.001. Assay buffer contained 100 mm NaCl. D, variants with higher affinity (lower Kd) toward Im7 H40W L53A I54A as measured by bio-layer interferometry exhibit higher in vivo chaperone activity toward Im7 L53A I54A. E, thermodynamic stabilities of the Spy variants determined by urea-induced denaturation at 25 °C indicate that only three new variants (F29V, L32Q, and H96R) and the Q100L control are destabilized compared with WT. ΔGNU is the free energy difference between the unfolded and folded states. Error bars in (B), (C), and (D) (for MICnormal), S.D. of three independent measurements whose data points are shown; error bars in (D) (for Kd) and (E) are fitting errors.
To verify that the high-frequency mutations (H65Y and H96R) are able to exert a positive effect on their own and that the six new mutations (F29V, L32Q, G48A, Q52R, H65Y, and H96R) act to enhance the activity of Spy as a chaperone, we reintroduced the individual mutations into the spy gene on pCDFTrc-Spy and assayed for penicillin resistance of cells co-expressing the βla-Im7 L53A I54A biosensor (26). As expected, we found that the relative minimal inhibitory concentrations (MICs) of cells expressing the Spy variants were higher than the MIC of cells expressing WT Spy (Fig. 1B), verifying that all these mutations are on their own sufficient to enhance the antibiotic resistance of the biosensor. Next, to test whether the newly selected Spy variants are generally more active toward other substrates, we purified these Spy variant proteins and assayed for their abilities to prevent the aggregation of DTT-reduced α-lactalbumin (α-LA) and heat-denatured malate dehydrogenase (MDH). All six of these variants increased Spy activity on α-LA and four showed increased activity toward MDH (Fig. 1C and Fig. S3A), indicating that beneficial mutations selected in vivo on one substrate (Im7 L53A I54A) also function better in vitro toward at least one other substrate. Overall, activity-enhancing variants obtained in the current and previous selection were localized to three regions of Spy: the disordered N terminus, the flexible linker between helix 1 and 2, and the regions near the junction between helix 2 and 3, and helix 3 and 4 (Fig. 1A). Most of these mutants were found in regions that, when changed, presumably minimally perturb Spy's overall structure. These mutations may affect local flexibility, which if true would be consistent with our previous observations that flexibility is important for Spy's action (26).
Our previous result revealed that tighter binding to substrates, slower substrate release rates, and increased structural flexibility all contributed to enhancing Spy's activity (26). To explore whether the new Spy variants used similar strategies to improve their function, we conducted thermodynamic and kinetic characterization of these new variants. Consistent with our previous findings, we observed a correlation between the dissociation constants (Kds) of the six new Spy variants toward the substrate Im7 H40W L53A I54A and their abilities to stabilize Im7 L53A I54A in vivo (Fig. 1D). However, unlike with our previous observations in which 70% of the mutants decreased the thermodynamic stability of Spy, only half of our new Spy variants showed dramatically decreased thermodynamic stabilities (F29V, L32Q, and H96R); the stabilities of the other three variants essentially remained unaltered (Fig. 1E). In addition, all the new Spy variants had similar secondary structures to the WT protein as measured by circular dichroism (CD) at 25 °C (Fig. S3B). Therefore, the activity-increasing mechanisms for half of the new variants (G48A, Q52R, and H65Y) cannot be easily explained by a decrease in stability or a change in overall secondary structure. These results motivated us to conduct a more detailed investigation in the hope of uncovering additional factor(s) that help determine Spy's anti-aggregation activity.
Increased hydrophobicity at the concave surface contributes to the increased chaperone activities of some Spy variants
Previously, we proposed that increasing surface hydrophobicity is beneficial because three of the activity-enhancing variants that we had previously isolated (Q49L, H96L, and Q100L) are located immediately adjacent to the two predominantly hydrophobic patches (P1 and P2) on the concave surface of Spy (26). The His-65 and Gly-48 residues in the newly isolated mutants are also located near these patches (Fig. 2A), suggesting that the activity of the H65Y and G48A variants might be explained by a similar mechanism. To evaluate the extent to which these mutations change Spy's hydrophobicity, we performed 4,4′-bis-1-anilinonaphthalene-8-sulfonate (bis-ANS) titration on these two variants and WT Spy. Bis-ANS is an environmentally sensitive fluorescent probe, the quantum yield of which increases upon binding to hydrophobic regions of proteins (31). In addition to the Q49L, H96L, and Q100L “super Spy” variants, our assay also intended to include a “nonsuper Spy” control with one of its hydrophobic residues located near Spy's hydrophobic patches replaced by a hydrophilic serine residue. To identify the hydrophobic residue that tolerates the serine substitution, we surveyed all the hydrophobic residues located in the two main hydrophobic patches (P1 and P2) and one hydrophobic residue near P1, which are L34S, I42S, M46S, M53S, M64S, I68S, M85S, M93S, M97S, and I103S (Fig. S4A). We found that the M53S mutation had less impact on the overall protein stability than all the other substitutions so we used it as the nonsuper Spy control (Fig. S4B). When co-expressed with the βla-Im7 L53A I54A biosensor, the M53S mutant decreased the MIC to ∼70% of the antibiotic resistance of the strain expressing WT Spy (Fig. S5A). At each bis-ANS concentration tested, all variants except M53S produced fluorescent signals higher than that of WT, indicating that the surface hydrophobicity of all variants except the control M53S was increased (Fig. 2B). Using reverse titration, we determined the stoichiometry of bis-ANS binding and found that all variants except M53S bound more bis-ANS molecules than the WT (Fig. 2B and Fig. S6), a further indication that these super Spy mutants had enhanced surface hydrophobicity. In addition, we observed a good correlation between the bis-ANS binding capacities and the anti-aggregation activities of the variants (Fig. 2C).
Figure 2.
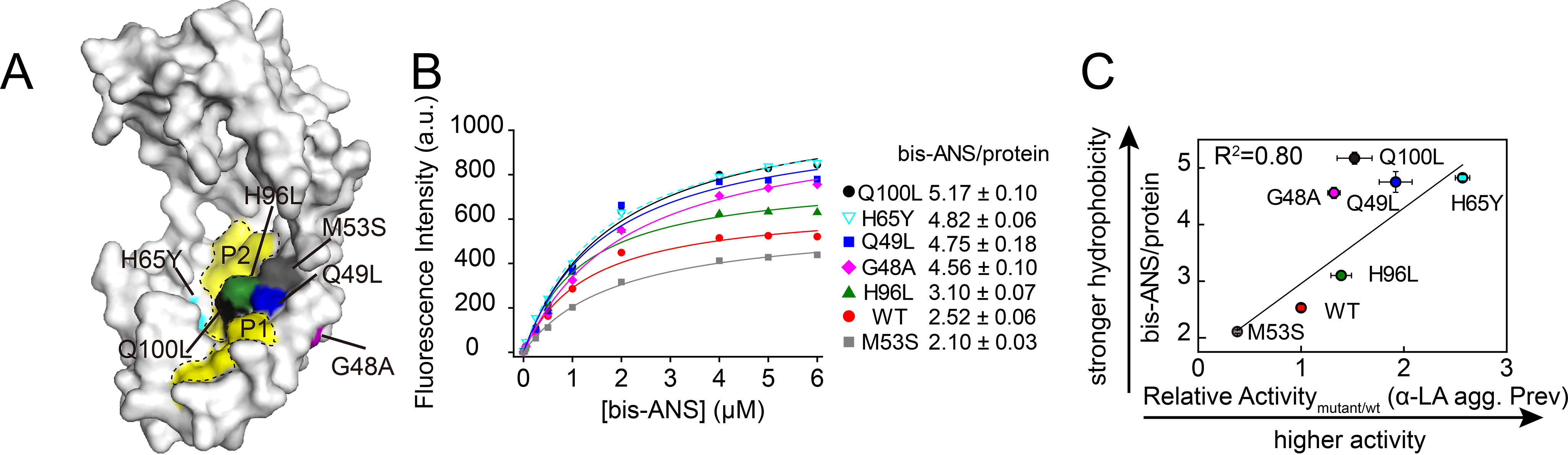
Activity-enhancing mechanism of H65Y and G48A variants may be explained by increased surface hydrophobicity. A, surface representation of Spy (PDB: 3O39) with the two main hydrophobic patches (P1 and P2) on the concave surface colored in yellow and the mutated sites in various colors. B, Spy variants at 0.5 µm were titrated with up to 6 µm bis-ANS in HN buffer (40 mm HEPES and 150 mm NaCl, pH 7.5) and the fluorescence signals were plotted against bis-ANS concentration. The number of bis-ANS molecules bound by each Spy molecule at saturation was obtained after a reverse titration (Fig. S6). C, the relationship between the number of bis-ANS molecules bound by each Spy molecule at saturation and the anti-aggregation activities toward α-LA (α-LA agg. Prev) of WT Spy and its variants. A–C, the same colors represent the same Spy variants in all three panels with red, magenta, blue, gray, cyan, olive, and black indicating WT, G48A, Q49L, M53S, H65Y, H96L, and Q100L, respectively. Error bars in (B), curve fitting errors; error bars in (C), S.D. of three independent measurements.
For the M53S control mutant, we found that its chaperone activity also decreased in vitro, although its stability and secondary structure were not fundamentally altered compared with the WT protein (Fig. S4B and Fig. S5, B and C). Thus, the decrease in M53S's activity might be attributed to the reduction in surface hydrophobicity. Together, these results suggest that Spy's activity can be correlated to its accessible hydrophobic surface area.
Optimizing surface charge confers Spy with higher activity than maximizing surface hydrophobicity
It is unlikely that the enhanced activities of the other two variants, Q52R and H96R, are attributable to increased surface hydrophobicity because the mutant amino acid is charged. Spy has an overall positively charged concave surface (Fig. 3A). This feature allows Spy to use long-range electrostatic interactions to form the initial complex with its substrates (19). Because Q52R and H96R both increase the positive charge on the mutated sites, we suspected that they might function by enhancing these initial electrostatic interactions between Spy and its substrates. To test this hypothesis, we increased the concentration of salt in the α-LA aggregation assay. We found that Q52R and H96R remained substantially more active than WT Spy in 100 mm salt but no longer showed substantial enhancement in 150 mm salt (Fig. 1C and Fig. 3, B and C). This result suggests that the activity enhancement of these two variants emanates from enhanced electrostatic interactions with substrates. We wondered if two other positions, which are adjacent to Spy's main hydrophobic areas and also to sites of super Spy substitutions, namely, Gln-49 and Gln-100, could also tolerate positively charged mutations. To address this question, we constructed the Q49R and Q100R variants and assayed their anti-aggregation activity toward α-LA, which is negatively charged overall (Fig. 3A) but presumably presents both hydrophobic and charged surfaces upon unfolding. Consistent with the idea that electrostatic interactions are important for Spy-substrate interaction, we found that the chaperone activities of these variants toward α-LA were also improved relative to WT Spy (Fig. 3D). Additionally, whereas Q49R was almost as active as the previously isolated super Spy variant Q49L, the activities of the Q100R and H96R variants were even higher than their corresponding previously isolated leucine-substituted super Spy variants (Fig. 3D).
Figure 3.

Charge-enhanced Spy variants show increased activity. A, surface charge distribution of Spy (PDB: 3O39) and α-LA (PDB: 1F6R). The concave surface of Spy is dominated by positive charges (blue), whereas the overall surface of native α-LA is negatively charged (red). The electrostatic surface potential was calculated in PyMol with the Adaptive Poisson-Boltzmann Solver tools 2.1 plugin. A color scale for the charge distribution from –8 to 8 was used. B and C, anti-aggregation activities of Spy variants Q52R and H96R in the presence of 100 mm (B) or 150 mm NaCl (C). Representative curves from duplicate measurements are shown (B–C). D, both leucine-substituted (yellow) and arginine-substituted (blue) Spy variants increase their abilities to prevent α-LA aggregation (α-LA agg. Prev). However, the charge-enhanced variants are in general more active than their corresponding hydrophobically enhanced counterparts (n = 3, Student's t test). ***p < 0.001; ns, not significant. E, introducing negative charges on the concave surface of Spy results in decreased chaperone activity. Chaperone activities of glutamate-substituted Spy variants at residues Q49, H96, and Q100 were assayed for their abilities to prevent aggregation of DTT-reduced α-LA (α-LA agg. Prev). Glutamate-substituted Spy variants showed decreased chaperone activities compared with Spy WT (n = 3, Student's t test). ***p < 0.001. Error bars, S.D. of three independent measurements whose data points are shown.
We then wondered if combinations of activity-enhancing mutations, particularly those with the same type of substitution, could result in a chaperone with further enhanced activity. Indeed, the combination of H96R and Q100R resulted in a roughly 2-fold increase in Spy activity compared with the single variants, indicating that the effects of these two arginine substitutions are additive (Fig. 3D). However, the incorporation of a third arginine substitution (Q49R) to form the triple variant Spy Q49R H96R Q100R had negligible effects. We next tested whether activity enhancement was specific to the positive charge on the arginine residues by mutating Gln-49, His-96, and Gln-100 to glutamic acid, whose side chain is similar to that of glutamine except that it bears a negative charge. We found that the chaperone activities of these more negatively charged variants were substantially decreased compared with those of WT (Fig. 3E), even though the in vitro stabilities of Q49E and H96E were almost unchanged from that of the WT (Fig. S7, A and B). Q100E, despite having a 9 °C decrease in the temperature at the midpoint of the unfolding curve, is 94% folded at 25 °C, the temperature at which the activity assay was conducted (Fig. S7C). This result indicates that the observed activity enhancement for the arginine-substituted variants depends on the positive nature of the charge.
In contrast, combining leucine substitutions produced very different results: the combination of H96L and Q100L, with or without the additional Q49L, decreased activities to WT Spy levels (Fig. 3D). The effect of these combinations is somewhat surprising given the common assumption that chaperones are primarily attracted to their substrates via hydrophobic interactions, an assumption that might lead one to naively think that increasing surface hydrophobicity would always be advantageous for a chaperone. The nonadditive effect of the leucine substitutions instead suggests that surface hydrophobicity has been optimized rather than maximized by evolution, as is common for many traits that are subject to evolutionary tradeoffs.
Decreased substrate association rates hinder chaperone activities for hydrophobically enhanced variants
Previous work revealed that electrostatic interactions dominate the early stage of Spy-substrate complex formation, whereas hydrophobic interactions dominate the later stage to further stabilize the complex (19). Our observations about the effects of various combinations of mutations on activity could be interpreted to indicate that optimizing chaperone-client interactions at early stages of the interaction is more important for aggregation prevention than optimizing interactions that occur at later stages. To test whether the behavior of our newly isolated Spy variants is consistent with these findings, we determined the thermodynamic and kinetic parameters of their interaction with reduced, carboxymethylated α-LA using a bio-layer interferometry–based association/dissociation assay at the physiological salt concentration (150 mm NaCl). We found that the arginine-substituted and leucine-substituted Spy variants all bound to α-LA more tightly than did WT Spy, and the combination of either type of mutation resulted in even higher affinities. The leucine-substituted variants had ∼2-fold higher affinities (Kds) than the corresponding arginine-substituted variants, with H96L being the only exception in that it bound α-LA as tightly as did H96R (Fig. 4A).
Figure 4.

Kinetic parameters characterizing the interaction of Spy variants and α-LA. The dissociation constants Kd (A), dissociation rate constants koff (B), and association rate constants kon (C) of leucine-substituted (yellow) and arginine-substituted (blue) Spy variants toward reduced, carboxymethylated α-LA (unfolded) were determined by bio-layer interferometry. Generally, leucine-substituted variants bind substrate tighter (A), recognize substrate slower (B), and release substrate slower (C) than their corresponding arginine-substituted counterparts (n = 2, Student's t test). *p < 0.05; **p < 0.01; ns, not significant. Error bars, S.D. of two independent measurements whose data points are shown.
We observed a similar trend with the substrate release rate (i.e. the dissociation rate, koff); all Spy variants had decreased koff values relative to WT, with the leucine-substituted variants exhibiting lower values than the corresponding arginine-substituted ones (Fig. 4B). This trend was also observed when assaying two other substrates, Im7 H40W L53A I54A and α-casein (Fig. S8). Previously, based on the observation that the substrate release rate was less affected by ionic strength than the binding process, it was concluded that substrate release is controlled mainly by hydrophobic forces, whereas electrostatic interactions contribute more to the initial stages of complex formation than to substrate release (19). Our new observation that the leucine-substituted variants hold the substrate longer than their arginine-substituted counterparts do is consistent with this conclusion. Furthermore, when increasing the temperature of kinetic assays from 25 °C to 32 °C, we found an increase in the koff rates for all the variants tested (Fig. S9). As temperature increases, electrostatic interactions become strengthened due to reduced solvent screening (32, 33), which then will tend to decrease the koff values if the substrate release process is also primarily governed by electrostatic interactions. Our observed koff increase instead argues the involvement of hydrophobic forces, the strength of which peaks at around 16 °C and diminishes as the temperature is raised or lowered relative to this temperature (34). Thus, the increased koff rates from 25 °C to 32 °C is consistent with the conclusion that the client release process is dominated by hydrophobic interactions.
To effectively prevent aggregation, Spy must recognize the aggregation-prone species quickly before they can form aggregation nuclei. We found that the three arginine-substituted variants (Q49R, H96R, and Q100R) had slightly increased association rates (kon), but when these mutations were combined, the kon rates decreased to that of the WT protein (for H96R Q100R (2R)) or even slightly below WT (for Q49R H96R Q100R (3R)) (Fig. 4C). In contrast, only one leucine-substituted variant (Q49L) acted to increase kon, whereas the H96L Q100L (2L) or Q49L H96L Q100L (3L) substitutions decreased kon by over 2-fold, which might account for the reduced anti-aggregation activities of these variants. These results also suggest that the kon value must be kept above a specific threshold to allow for effective substrate capture. The 2L and 3L variants with rate constants of ∼105 m−1s−1 are at the lower end of the diffusion-controlled regime (the “basal” value for diffusion-controlled association rate constants ranges from 105 to 106 m−1s−1) (15). To go beyond the basal diffusion-controlled rate constant, intermolecular forces must be active during the diffusion process leading to the formation of a transient complex (15). Long-range electrostatic interactions are thought to be the main type of interaction capable of speeding up the formation of the initial complex and may therefore help explain why the charge-enhanced single arginine-substituted variants have accelerated substrate association rates and enhanced activities. In contrast, short-range forces, including hydrophobic and van der Waals interactions, are relatively ineffective in affecting diffusion-limited interactions and contribute much less to rate enhancements compared with long-range electrostatic interactions (15). Our kinetic results thus provide a plausible explanation as to why the more hydrophobic 2L and 3L variants showed no enhanced anti-aggregation activity toward α-LA.
To further examine the involvement of electrostatic interactions in the binding reaction of these Spy variants, we repeated the kinetic measurements at different ionic strengths. We saw a decrease in the kon rates of Spy WT and the Q49R, H96R, and Q100R mutants as the salt concentration increased from 150 mm to 250 mm (Fig. S10A). The Q49L, H96L, 2R, and 3R mutants were less sensitive to changes in salt concentration from 150 mm to 200 mm, but all showed a decrease in kon rates at 250 mm salt. Interestingly, the Q100L, 2L, and 3L mutants showed a different trend in kon rates: their kon rates increased with increasing ionic strength (Fig. S10A). This trend is not surprising given that higher ionic strength is known to contribute to stronger hydrophobic interactions (35, 36). Thus, our results suggest that higher ionic strength increases the kon rates of the leucine-substituted Spy variants, possibly by enhancing their hydrophobic interactions with α-LA. On the other hand, consistent with previous reports that electrostatic interactions also contribute to the maintenance of Spy-substrate complexes (19, 20), we found that the koff rates of all Spy variants increased with increasing salt concentrations, which resulted in their overall lower affinities at higher ionic strengths (Fig. S10, B and C). These kinetic measurements confirmed the involvement of both electrostatic and hydrophobic interactions in Spy's substrate binding and release and also revealed the interplay of different molecular forces as ionic strength changes.
Enabling rapid substrate association is likely to be more effective than attempting to decrease substrate dissociation rates in preventing aggregation
To determine whether the apparently important role of fast association in preventing aggregation can be generalized to other substrate proteins, we measured the anti-aggregation activity of Spy toward another substrate, MDH.
We used two methods to denature MDH: 8 m urea and heat. We did this to test different types of unfolding intermediates that aggregate at different rates. We reasoned that a rapidly aggregating MDH unfolding intermediate would be more dependent on chaperone capture to delay the formation of the aggregation nuclei than a slowly aggregating intermediate. If fast association is indeed a key factor in preventing aggregation, given the marked difference in the kon rates, we would expect to observe a more pronounced difference in the activities of arginine- or leucine-substituted Spy variants in preventing aggregation of fast-aggregating MDH unfolding intermediates compared with their activities toward slow-aggregating MDH intermediates. We found that MDH that had been chemically denatured using 8 m urea aggregated very rapidly at both 32 °C and 25 °C because the T50 (times to reach half of the maximum aggregation signal) of MDH alone at these temperatures were short: 8 s at 32 °C and 15 s at 25 °C (Fig. 5, A and B). However, when the incubation temperature was further decreased to 22 °C, the aggregation of urea-denatured MDH was significantly slowed; the T50 was extended to 56 s. This indicated to us that reduction in temperature could change the aggregating kinetics of urea-denatured MDH and could change the rapid-aggregating nature of urea-denatured MDH at high temperatures to a more slowly aggregating behavior (Fig. 5C). Heat-denatured MDH in the absence of Spy aggregated at 43 °C (T50 = 201 s) even slower than did urea-denatured MDH at 22 °C (T50 = 56 s) (Fig. 5D). Using these methods, we could observe two rapidly aggregating species (urea-denatured MDH at 32 °C and 25 °C) and two slowly aggregating models (urea-denatured MDH at 22 °C and heat-denatured MDH at 43 °C).
Figure 5.
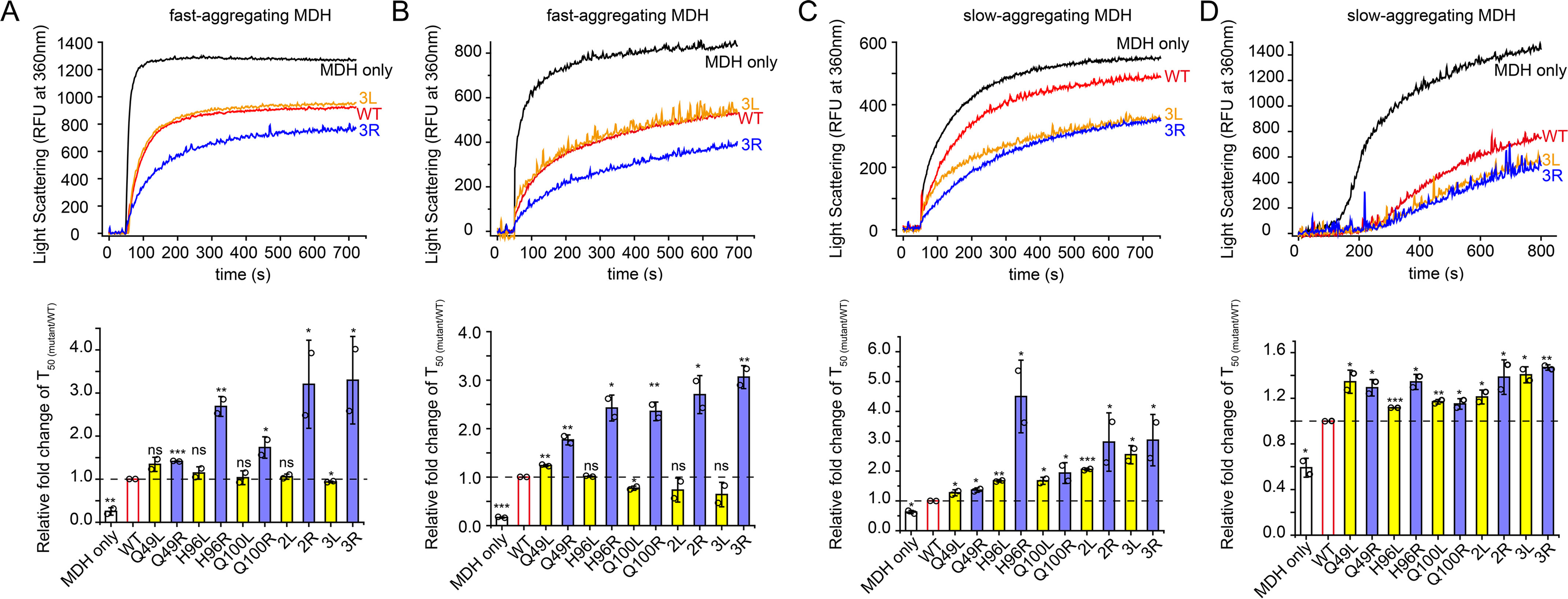
Anti-aggregation activities of leucine-substituted and arginine-substituted Spy variants toward MDH. A–C, MDH (80 µm) denatured in 8 m urea was diluted 160-fold into 40 mm HEPES-KOH buffer (pH 7.5) in the presence or absence of 0.05 µm Spy or different Spy variants at 32 °C (A), 25 °C (B), or 22 °C (C), and aggregation was monitored by light scattering at 360 nm. D, aggregation of 0.5 µm MDH denatured by heat at 43 °C in the absence or presence of 0.25 µm Spy WT or different Spy variants was monitored by light scattering at 360 nm. The upper panels in A–D, representative aggregation curves of MDH in the absence or presence of Spy WT or 3L/3R from duplicate measurements. The lower panel in A–D, relative anti-aggregation activities of Spy variants to Spy WT are expressed as the relative fold change of T50 (mutant/WT). T50 is the time to reach half of the maximum aggregation signal and is obtained by fitting the aggregation curves with the Hill equation with three parameters. For fast-aggregating MDH denatured by urea at 32 °C (A) or 25 °C (B), the arginine-substituted variants (blue) still show enhanced activities; however, the leucine-substituted variants (yellow) are only as active as the WT (red) (n = 2, Student's t test). For slow-aggregating MDH denatured by urea at 22 °C (C) or by heat at 43 °C (D), all arginine-substituted variants (blue) and leucine-substituted variants (yellow) performed better than Spy WT (n = 2, Student's t test). *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. Error bars, S.D. of two independent measurements.
We found that for rapidly aggregating MDH, all leucine-substituted Spy variants, including the 2L and 3L variants, displayed equal or slightly worse activities than did WT Spy, whereas all arginine-substituted Spy variants, including the 2R and 3R variants, performed better than did WT Spy (Fig. 5, A and B and Fig. S11, A and B). This result is consistent with the observations made with α-LA aggregation, which is also rapid (Fig. 3D). For slowly aggregating MDH, almost all leucine-substituted variants were more active than WT, and the Q49L, Q100L, and 3L variants were nearly as active as their corresponding arginine-substituted variants (Fig. 5, C and D and Fig. S11, C and D). Taking all these results into account, we conclude that for relatively slow-aggregating MDH, the formation of a more stable Spy-MDH complex still allows for the productive inhibition of aggregation. However, if one is seeking to prevent problematically fast aggregation, then promoting rapid substrate association rates may be more impactful than attempting to stabilize the complex.
Activities of the charge-enhanced Spy variants are limited by decreased protein levels in vivo
Our in vitro data showed that the charge-enhanced variants at Gln-49, His-96, and Gln-100 were more effective than the WT or even the hydrophobicity-enhanced variants at inhibiting the aggregation of fast-aggregating substrates. This result led us to wonder why the Q49R and Q100R variants or their combinations did not emerge during our selections. To answer this question, to systematically analyze the effects of chaperone action of these two residues that are clearly vital for chaperone action, and to better tie together our in vivo and in vitro results, we mutated Gln-100 to each of the other 19 amino acids and assayed the chaperone activities of these variants in vivo using the βla-Im7 L53A I54A stability biosensor. Unexpectedly given our current interpretation based on in vitro assays, but consistent with our previous interpretations, we found that the antibiotic resistance of the strains expressing these Spy variants was reasonably well-correlated with the hydropathicity of the substituting residue (Fig. 6A). There was also a correlation, although weaker, between the hydropathicity of the substituting residue and the amount of βla-Im7 L53A I54A (Fig. 6B). These results indicate that there is a relationship between the hydropathicity of the substituting residue and the stabilization it confers. We then examined the steady-state levels of each Spy variant and found that many of the variants with hydrophilic substitutions had overall lower levels compared with the variants substituted with hydrophobic amino acids (Fig. 6, C and D). In particular, the Q100R mutant, which had the lowest MIC value, was also expressed in very low amounts. Therefore, the in vivo performance of charge-enhanced Spy variants was probably impaired by their steady-state levels. This indicates that these variants may have in vivo folding problems and thus be more prone to proteolysis in vivo.
Figure 6.

Lower steady-state levels of Spy variants harboring hydrophilic mutations at Gln-100 may explain their decreased in vivo activities. A, relative MICs of Spy variants compared with WT Spy were plotted against the amino acids' hydropathy index. B, amounts of βla-Im7 L53A I54A in strains expressing different Spy variants (normalized to the βla-Im7 L53A I54A amount in the strain expressing WT Spy) were plotted against the amino acids' hydropathy index. C, steady-state levels of each Spy variant and the βla-Im7 L53A I54A fusion protein co-expressed in the same strain were analyzed by Western blotting with anti-Spy and anti-TEM-1 β-lactamase antibodies, respectively. Endogenous DnaK was used as a loading control. D, amounts of different Spy variants (normalized to the amount of WT Spy) were plotted against the amino acids' hydropathy index. A, B, and D, hydropathy index is according to Zviling (57); yellow dots, amino acids with a hydropathy index above 0; gray dots, amino acids with a hydropathy index below 0. Error bars in (A), S.D. of three independent measurements; error bars in (D), S.D. of two technical repeats.
Discussion
A single protein can undergo an almost bewildering variety of conformational changes during the folding process. This, combined with the fact that most chaperones assist in the folding of a broad variety of substrates, makes understanding the nature of chaperone-substrate interaction difficult from both a theoretical and practical standpoint, but understanding this interaction is vital because it represents one of the core problems in chaperone biology. It is understandable that scientists have attempted to simplify this problem by using the following, quite attractive, logic: folding and unfolding proteins display hydrophobic regions, the hydrophobic effect drives such regions together, which should cause proteins to aggregate, and chaperones prevent aggregation; ergo, chaperones recognize hydrophobic regions. Early attempts at solving this problem by mapping chaperone specificity using peptide display approaches provided some evidence for this model, but close examination of even this early data revealed a more complex picture: the recognition site for Hsp70, though it does contain hydrophobic residues, also contains positive charge (37). The commonly held view that molecular chaperones bind their substrates primarily via hydrophobic interactions was thus too simple a picture even in the early days of chaperone biology (5, 38). Another somewhat neglected point concerning protein aggregation is that in addition to being triggered by exposure of sticky hydrophobic surface patches, self-association of native-like structures can also result in aggregation (39). Both theoretical and experimental evidences also suggest that electrostatic interactions can drive aggregation (40, 41). Though electrostatic repulsion between protein molecules disfavors self-association, asymmetric distribution of surface charge can allow for alignment of patches with complementary charges, which increases a protein's aggregation propensity (39). Recapitulating these overlooked facts will help us to achieve our long-term goal, i.e. to understand protein folding in sufficient detail so that it can be manipulated in vivo.
Our mechanistic characterization of activity-enhanced Spy variants enabled us to delineate the particular contributions that various intermolecular forces play in contributing to Spy's anti-aggregation activity (Fig. 7). This has allowed us to gain a much more nuanced view of chaperone-substrate interaction than the current view, where hydrophobic interactions are thought to dominate. We found that Spy variants possessing discrete surface properties have very different effects on their ability to prevent aggregation; specifically, Spy variants with more positively charged concave surfaces are able to interact with aggregation-prone species quickly and retain these substrates on their surfaces for a relatively longer period of time because of slower dissociation rates. This results in less aggregation-dependent precipitation. These results contradict the widely held assumption that chaperones and their clients generally interact through hydrophobic interactions. Hydrophobically enhanced variants were also able to reduce the formation of aggregates because of their property of only allowing for the very slow release of their substrates. Hydrophobic substitutions may be effective as long as their rate of binding is above some threshold value that allows for effective substrate capture and these substitutions do not substantially change the distribution of the original electrostatic potential on the concave surface of Spy.
Figure 7.

Mechanism of the charge-enhanced or hydrophobicity-enhanced Spy variants in preventing client aggregation. For fast-aggregating clients (upper panel), both WT Spy and the charge-enhanced variants are capable of rapidly binding unfolded clients, but the latter also releases the substrate more slowly, resulting in very few aggregates. In contrast, although the hydrophobicity-enhanced variants are capable of forming tighter complexes with clients, their slow association rates limit their ability to prevent rapid aggregation. For slow-aggregating clients (lower panel), the very slow dissociation rates of the hydrophobicity-enhanced variants make up for their shortage in recognizing clients, resulting in almost equally holding activities as the charge-enhanced variants. Boxed regions indicate the processes more impactful in determining the aggregation of different types of clients.
There are additional indications that chaperone function is not solely dependent on hydrophobic interactions. For instance, it is known that GroEL preferentially recognizes amphipathic regions within its substrate proteins (42). The related eukaryotic chaperonin TRiC was also found to rely on a combinatorial substrate-recognition mechanism that involves a distinct, evolutionarily conserved pattern of polar and hydrophobic residues. Similar to Spy, charged and polar residues on TRiC contribute to enhancing the association rates, and hydrophobic residues are responsible for decreasing the dissociation rates (43). Likewise, the binding sites of the intrinsically disordered protein Tau on the chaperone Hsp90 are composed of scattered patches that have both hydrophobic and negative charge characteristics, allowing the chaperone to make a large number of low-affinity contacts with various substrates (44). The electrostatic interactions between Skp and its outer membrane protein substrates were found to be important for substrate delivery and folding into the liposomes in vitro (45). It has been shown that trigger factor recognizes the peptide segments enriched in aromatic and basic residues within its substrate proteins (46). In addition, SecB also appears to recognize both hydrophobic and positively charged regions, allowing it to deliver substrates with different hydrophobicity and electrostatic potential to SecA for translocation (47, 48). Thus, many molecular chaperones appear to have a bipartite nature associated with their substrate binding sites. This may enable their adaptation to the different structural properties present in their diverse substrates and thus facilitate promiscuous substrate recognition. Our previous and current work suggests that Spy also possesses this bipartite nature and optimizing either hydrophobic or electrostatic properties can convert it into a more efficient chaperone toward different in vitro substrates.
Engineering molecular chaperones, especially improving their anti-aggregation or disaggregation activities, is important because it may provide crucial insights that could be helpful in treating several age-related neurodegenerative diseases whose hallmarks include toxic protein misfolding and aggregation (21, 49). One successful example of engineering to modify aggregation activity is the optimization of the Hsp104 chaperone, which disaggregates and eradicates preformed fibrils of many amyloid proteins (24, 50–52). Improved variants identified in an in vivo screen against α-synuclein and other amyloid proteins in yeast revealed a hot spot: Ala-503; substitution with any residue except proline at this site made Hsp104 more active (24). However, directed evolution of chaperones to improve their general activity is not entirely straightforward. For example, the GroEL chaperonin appears to possess a narrow mutational window; mutations that made it better at folding GFP decreased its ability to fold its native substrates (23). Our work with Spy also revealed that mutants obtained in an in vivo screen against one type of substrate might not work efficiently toward another type of substrate. We saw this with α-LA and chemically denatured MDH whose in vivo aggregation is difficult to model because these substrates aggregate very rapidly in vitro, so rapidly that chaperones are unable to act on them. Multiple factors can contribute to chaperone activity. These factors may already have been optimized for each chaperone by evolution, and improving on this evolutionary balance may be difficult. However, knowledge is power, and more detailed knowledge about chaperone-substrate interactions will undoubtedly be helpful in designing chaperones.
In this study, we have presented a genetic means of modulating chaperone activity. The observation that chaperones can act by modifying the surface electrostatic potential and hydrophobic properties suggests that even evolutionarily optimized chaperones can be further improved to more effectively coordinate substrate binding and release. Although our findings here are based on the optimization of Spy, the binary nature of many chaperone surfaces supports similar optimization strategies for these chaperones, including ATP-dependent ones such as Hsp90. To this end, we believe that our approach, along with insights gained from other successful engineering endeavors, such as improving overall structural flexibility (26), enhancing ATPase activity (23), and combining functional domains derived from different molecular chaperones (53), will provide a powerful means of enhancing chaperone activity. We expect that as more key mechanistic and functional insights into the details of chaperone-substrate interaction emerge, the field of rational chaperone design will advance and will eventually be able to support the development of therapeutic chaperones targeting neurodegenerative diseases.
Experimental procedures
RAISE library construction and selection
Two independent RAISE libraries, each with ∼5 × 104 clones, were constructed as previously described with the following modifications (27). 10 μg of the PCR product of the spy gene was digested by 0.1 units of DNaseI in a 20-μl reaction at 37 °C for either 25 s or 18 s to generate a random fragment mixture, with a range of 75 to 150 bp or 100 to 300 bp, respectively. Subsequently, 2 pmol of the digested fragments were 3′-tailed with random nucleotides by 2.5 units of terminal deoxynucleotidyl transferase enzyme in a 20-μl reaction at 37 °C for 1 h. Then, 1 pmol of 3′-tailed fragments were assembled into full-length spy via self-priming PCR mediated by 2 units of Deep VentR (exo-) polymerase in a 20-μl reaction. Finally, the mutated spy libraries were amplified with primers complementary to the end of the spy gene, ligated in the pCDF vector, and transformed into E. coli strain Trans1T1.
Library selection was conducted by transforming the pCDFTrc-Spy library into a spy and ampC null strain HW41, which also contained the pBR322 bla::GSlinker Im7 L53A I54A vector. Subsequently, the transformants were plated on agar plates supplemented with 3 mg/ml penicillin and 0.1 mm isopropyl β-d-thiogalactopyranoside, and resistant colonies were picked for sequencing after overnight incubation at 37 °C.
Mutagenesis and protein purification
Mutations to spy were generated via the QuikChange site-directed mutagenesis method (Agilent, catalog no. 200518). All Spy variants were purified according to Quan et al. (26) with the following modifications. After initial purification with immobilized nickel-nitrilotriacetic acid affinity chromatography, His6-SUMO-Spy was eluted with 300 mm imidazole and digested by ubiquitin-like-specific protease 1 in solution while dialyzing against buffer A (20 mm HEPES and 0.5 mm EDTA, pH 7.5) before undergoing cation exchange chromatography. Protein concentrations were determined by absorbance at 280 nm, and all proteins were aliquoted into 1.5-ml Eppendorf tubes and stored at –80 °C prior to use.
Chaperone activity assay in vivo
Spot titer experiments were performed to quantify the relative in vivo chaperone activities of Spy variants. Generally, cells expressing each Spy variant and the βla-Im7 L53A I54A sensor were grown to mid-log phase, serially diluted, and plated onto agar plates containing increasing concentrations of penicillin V (0–3500 mg/ml) and 0.1 mm isopropyl β-d-thiogalactopyranoside. Then, the relative MIC (MICmutant/MICWT) of Spy variants toward penicillin V was calculated as described previously (26, 30).
α-LA aggregation assay
Bovine α-LA, type III (Sigma-Aldrich) was used as a model substrate to quantify the specific in vitro holding activities of Spy variants as described previously (26). Aggregation of α-LA (50 μm) was initiated by the addition of 20 mm DTT in a buffer containing 50 mm sodium phosphate, 100 mm sodium chloride, and 5 mm EDTA at pH 6.9. The light scattering of α-LA in the absence or presence of Spy was monitored at 360 nm using a Synergy HTX Multi-Mode Microplate Reader with a 5-min detection period for 10 h at room temperature. For relative activity quantification, a standard curve was produced by plotting the slopes calculated from light scattering data between 120 and 180 min against 22 different Spy WT:α-LA ratios. Then, slopes of light scattering determined at the same time range for different Spy variants:α-LA ratios (0.075, 0.1, 0.15, and 0.2) were calculated to quantify the relative activity according to the standard curve. The final average relative activities of Spy variants were generated from eight relative activity values (four concentrations in duplicate).
MDH aggregation assay
For thermal-denatured MDH aggregation, 0.5 μm MDH was added into 43 °C pre-warmed 40 mm HEPES-KOH, pH 7.5 buffer in the presence or absence of 0.25 μm Spy or Spy variants under stirring. MDH aggregation was monitored by light scattering at 360 nm using a Lumina fluorescence spectrometer supplied with a Peltier temperature controller (Thermo Fisher Scientific).
For chemically denatured MDH aggregation, MDH (denatured in 8 m urea for 1 h at 25 °C) was diluted into 40 mm HEPES-KOH, pH 7.5 buffer with a final concentration of 0.5 μm in the presence or absence of 0.05 μm Spy or Spy variants under stirring. Light scattering at 360 nm was recorded with a Lumina fluorescence spectrometer at 22, 25, and 32 °C.
Bio-layer interferometry
Kinetics of Spy variants toward biotinylated and carboxymethylated α-LA and biotinylated Im7 H40W L53A I54A were measured with an Octet RED96 system (ForteBio, Menlo Park, CA). Carboxymethylated α-LA was prepared to obtain a stable, totally unfolded substrate as previously described (54). The carboxymethylated α-LA and Im7 H40W L53A I54A were biotinylated with EZ-link NHS-biotin (Thermo Fisher Scientific) with a 1:1 molar ratio of biotin to protein. 20 μg/ml biotinylated and carboxymethylated α-LA or biotinylated Im7 H40W L53A I54A was immobilized to the streptavidin sensor, whereas different concentrations (0.25, 0.5, 1, 1.5, and 2 μm) of Spy WT and variants in 40 mm HEPES and 150 mm NaCl, pH 7.5 were added to a 96-well plate (for carboxymethylated α-LA, the assay buffer contains 150–250 mm NaCl). One kinetic cycle at a defined Spy concentration included the following steps: ligand binding for 25 min, wash for 5 min, equilibrium for 1 min, association for 10 min, and dissociation for 10 min. Curve fitting and calculation of association rate constants (kon), dissociation rate constants (koff), and dissociation constants (Kd) were performed as described previously (26).
Thermostability measurement
For urea-induced unfolding of WT Spy and variants, ellipticity at 226 nm was monitored for each protein over increasing concentrations of urea (0–9 m) with a Chirascan circular dichroism (CD) spectrometer (Applied Photophysics Corp., Surrey, UK). All proteins were brought to a concentration of 0.1 mg/ml and incubated in denaturation buffer (30 mm diglycine, pH 8.0, and 0.1 M NaCl) supplemented with different concentrations of urea for 30 min before measurement. The Gibbs free energy of unfolding of each protein was determined via the linear extrapolation method; entire unfolding curves were fitted by the method of Santoro and Bolen using Origin 8.0 software (OriginLab) as described previously (55).
The thermal unfolding of WT Spy and variants was followed by monitoring the ellipticity at 222 nm over the temperature range of 20–75 °C, with an interval of 1 °C and a heating rate of 1.5 °C/min. The thermal unfolding curves were fitted with the Gibbs-Helmholtz equation using Origin 8.0 software (OriginLab) and the transition temperature at the midpoint of the unfolding curve for each protein was calculated as previously described (56).
Bis-ANS fluorescence
Bis-ANS was dissolved in HN buffer (40 mm HEPES-NaOH and 150 mm NaCl, pH 7.5) to obtain a 7 mm stock. To estimate the number of bis-ANS bound to each Spy molecule, 0.5 μm Spy variants in HN buffer were titrated with bis-ANS over a range of concentrations (0–6 μm). The emission of bis-ANS at 493.4 nm was recorded with an excitation wavelength of 395 nm. The observed fluorescence intensity was plotted as a function of bis-ANS concentration, and the curves were fitted with the hyperbolic function shown in Equation 1,
where Fobs is the recorded bis-ANS fluorescence signal, Fmax is the maximal fluorescence reached upon saturation of the complex, [A] is the concentration of bis-ANS, and K is the apparent binding constant of bis-ANS. To obtain the stoichiometry of ANS binding, a reverse titration was performed using a fixed bis-ANS concentration of 1 μm and increasing concentrations of Spy or Spy variants (0–3 μm) (Fig. S6). The reverse titration curves were fitted with Equation 2,
where fobs is the recorded bis-ANS fluorescence signal in the reverse titration, fmax is the maximal ANS fluorescence when saturated with Spy, [S] is the concentration of Spy, and K′ is the apparent binding constant of Spy. Then, the stoichiometry of bis-ANS was calculated according to Equation 3,
where n is the stoichiometry of bis-ANS bound to one Spy and [S0] is the Spy concentration used in the first titration experiment (0.5 μm).
Statistical analysis
Appropriate two-tailed unpaired student's t test was employed to compare two independent groups. p values less than 0.05 were considered significant. Statistical analysis was conducted with GraphPad Prism software (version 5.0).
Data availability
All data are contained with this article and in the supporting information.
Supplementary Material
Acknowledgments
We thank Kevin Wu, Philipp Koldewey, Frederick Stull, and Scott Horowitz for useful discussions and critical suggestions for the article.
This article contains supporting information.
Author contributions—W. H. and S. Q. conceptualization; W. H., J. Z., V. S., and L. W. data curation; W. H., J. Z., and V. S. formal analysis; W. H., J. Z., V. S., and L. W. investigation; W. H., J. Z., and V. S. methodology; W. H. and V. S. writing-original draft; J. C. A. B. and S. Q. supervision; J. C. A. B. and S. Q. project administration; J. C. A. B. and S. Q. writing-review and editing; S. Q. funding acquisition.
Funding and additional information—This work was supported by the National Natural Science Foundation of China Grants 31661143021 and 31400664 (to S. Q.), the Fundamental Research Funds for the Central Universities Grant 22221818014 (to S. Q.), and the Research Program of State Key Laboratory of Bioreactor Engineering (to S. Q.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- Im7
- immunity protein 7
- RAISE
- random insertional-deletional strand exchange mutagenesis
- MIC
- minimal inhibitory concentration
- α-LA
- α-lactalbumin
- MDH
- malate dehydrogenase
- bis-ANS
- 4,4′-bis-1-anilinonaphtalene-8-sulfonate
- Kd
- dissociation constant
- kon
- association rate constant
- koff
- dissociation rate constant
- βla
- β-lactamase-matrix
- T50
- time to reach half of the maximum aggregation signal
- SUMO
- small ubiquitin-like modifier.
References
- 1. Feldman D. E., and Frydman J. (2000) Protein folding in vivo: the importance of molecular chaperones. Curr. Opin. Struct. Biol. 10, 26–33 10.1016/S0959-440X(99)00044-5 [DOI] [PubMed] [Google Scholar]
- 2. Balchin D., Hayer-Hartl M., and Hartl F. U. (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- 3. Hartl F. U., and Hayer-Hartl M. (2009) Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581 10.1038/nsmb.1591 [DOI] [PubMed] [Google Scholar]
- 4. Maier T., Ferbitz L., Deuerling E., and Ban N. (2005) A cradle for new proteins: trigger factor at the ribosome. Curr. Opin. Struct. Biol. 15, 204–212 10.1016/j.sbi.2005.03.005 [DOI] [PubMed] [Google Scholar]
- 5. Hartl F. U., and Hayer-Hartl M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858 10.1126/science.1068408 [DOI] [PubMed] [Google Scholar]
- 6. Chiti F., Taddei N., Baroni F., Capanni C., Stefani M., Ramponi G., and Dobson C. M. (2002) Kinetic partitioning of protein folding and aggregation. Nat. Struct. Biol. 9, 137–143 10.1038/nsb752 [DOI] [PubMed] [Google Scholar]
- 7. Eichner T., Kalverda A. P., Thompson G. S., Homans S. W., and Radford S. E. (2011) Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 41, 161–172 10.1016/j.molcel.2010.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bolognesi B., Kumita J. R., Barros T. P., Esbjorner E. K., Luheshi L. M., Crowther D. C., Wilson M. R., Dobson C. M., Favrin G., and Yerbury J. J. (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 5, 735–740 10.1021/cb1001203 [DOI] [PubMed] [Google Scholar]
- 9. Hartl F. U., Bracher A., and Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 10. Walter S., and Buchner J. (2002) Molecular chaperones–cellular machines for protein folding. Angew. Chem. Int. Ed. 41, 1098–1113 10.1002/1521-3773(20020402)41:7<1098::AID-ANIE1098>3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- 11. Pelham H. R. (1986) Speculations on the functions of the major heat shock and glucose-regulated proteins. Cell 46, 959–961 10.1016/0092-8674(86)90693-8 [DOI] [PubMed] [Google Scholar]
- 12. Koldewey P., Horowitz S., and Bardwell J. C. A. (2017) Chaperone-client interactions: non-specificity engenders multifunctionality. J. Biol. Chem. 292, 12010–12017 10.1074/jbc.R117.796862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark P. L., and Elcock A. H. (2016) Molecular chaperones: providing a safe place to weather a midlife protein-folding crisis. Nat. Struct. Mol. Biol. 23, 621–623 10.1038/nsmb.3255 [DOI] [PubMed] [Google Scholar]
- 14. Liu W., Bratko D., Prausnitz J. M., and Blanch H. W. (2003) Electrostatic interactions between peptides and the molecular chaperone DnaK. J. Phys. Chem. B 107, 11563–11569 10.1021/jp035872c [DOI] [Google Scholar]
- 15. Schreiber G., Haran G., and Zhou H. X. (2009) Fundamental aspects of protein-protein association kinetics. Chem. Rev. 109, 839–860 10.1021/cr800373w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Radíc Z., Kirchhoff P. D., Quinn D. M., McCammon J. A., and Taylor P. (1997) Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin. J. Biol. Chem. 272, 23265–23277 10.1074/jbc.272.37.23265 [DOI] [PubMed] [Google Scholar]
- 17. Alsallaq R., and Zhou H. X. (2008) Electrostatic rate enhancement and transient complex of protein-protein association. Proteins 71, 320–335 10.1002/prot.21679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perrett S., Zahn R., Stenberg G., and Fersht A. R. (1997) Importance of electrostatic interactions in the rapid binding of polypeptides to GroEL. J. Mol. Biol. 269, 892–901 10.1006/jmbi.1997.1081 [DOI] [PubMed] [Google Scholar]
- 19. Koldewey P., Stull F., Horowitz S., Martin R., and Bardwell J. C. A. (2016) Forces driving chaperone action. Cell 166, 369–379 10.1016/j.cell.2016.05.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee C., Kim H., and Bardwell J. C. A. (2018) Electrostatic interactions are important for chaperone-client interaction in vivo. Microbiology 164, 992–997 10.1099/mic.0.000676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mack K. L., and Shorter J. (2016) Engineering and evolution of molecular chaperones and protein disaggregases with enhanced activity. Front. Mol. Biosci. 3, 8 10.3389/fmolb.2016.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saibil H. (2013) Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14, 630–642 10.1038/nrm3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J. D., Herman C., Tipton K. A., Gross C. A., and Weissman J. S. (2002) Directed evolution of substrate-optimized GroEL/S chaperonins. Cell 111, 1027–1039 10.1016/S0092-8674(02)01198-4 [DOI] [PubMed] [Google Scholar]
- 24. Jackrel M. E., and Shorter J. (2014) Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis. Model. Mech. 7, 1175–1184 10.1242/dmm.016113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Capaldi A. P., Kleanthous C., and Radford S. E. (2002) Im7 folding mechanism: misfolding on a path to the native state. Nat. Struc. Biol. 9, 209–216 10.1038/nsb757 [DOI] [PubMed] [Google Scholar]
- 26. Quan S., Wang L., Petrotchenko E. V., Makepeace K. A., Horowitz S., Yang J., Zhang Y., Borchers C. H., and Bardwell J. C. (2014) Super Spy variants implicate flexibility in chaperone action. elife 3, e01584 10.7554/eLife.01584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujii R., Kitaoka M., and Hayashi K. (2006) RAISE: a simple and novel method of generating random insertion and deletion mutations. Nucleic Acids Res. 34, e30 10.1093/nar/gnj032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin-Goerke J. L., Robbins D. J., and Burczak J. D. (1997) PCR-based random mutagenesis using manganese and reduced dNTP concentration. BioTechniques 23, 409–412 10.2144/97233bm12 [DOI] [PubMed] [Google Scholar]
- 29. Vanhercke T., Ampe C., Tirry L., and Denolf P. (2005) Reducing mutational bias in random protein libraries. Anal. Biochem. 339, 9–14 10.1016/j.ab.2004.11.032 [DOI] [PubMed] [Google Scholar]
- 30. Foit L., Morgan G. J., Kern M. J., Steimer L. R., von Hacht A. A., Titchmarsh J., Warriner S. L., Radford S. E., and Bardwell J. C. (2009) Optimizing protein stability in vivo. Mol. Cell 36, 861–871 10.1016/j.molcel.2009.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farris F. J., Weber G., Chiang C. C., and Paul I. C. (1978) Preparation, crystalline structure, and spectral properties of the fluorescent probe 4,4'-bis-1-phenylamino-8-naphthalenesulfonate. J. Am. Chem. Soc. 100, 4469–4474 10.1021/ja00482a026 [DOI] [Google Scholar]
- 32. Okazaki K., Sato T., and Takano M. (2012) Temperature-enhanced association of proteins due to electrostatic interaction: a coarse-grained simulation of actin-myosin binding. J. Am. Chem. Soc. 134, 8918–8925 10.1021/ja301447j [DOI] [PubMed] [Google Scholar]
- 33. Xia Y. L., Sun J. H., Ai S. M., Li Y., Du X., Sang P., Yang L. Q., Fu Y. X., and Liu S. Q. (2018) Insights into the role of electrostatics in temperature adaptation: a comparative study of psychrophilic, mesophilic, and thermophilic subtilisin-like serine proteases. Rsc. Adv. 8, 29698–29713 10.1039/C8RA05845H [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schellman J. A. (1997) Temperature, stability, and the hydrophobic interaction. Biophys. J. 73, 2960–2964 10.1016/S0006-3495(97)78324-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Z. Y., Yang Y. L., Tang X. Z., Chen Y. J., and You Y. (2015) Effects of ionic strength on chemical forces and functional properties of heat-induced myofibrillar protein gel. Food. Sci. Technol. Res. 21, 597–605 10.3136/fstr.21.597 [DOI] [Google Scholar]
- 36. Scott A. M., Antal C. E., and Newton A. C. (2013) Electrostatic and hydrophobic interactions differentially tune membrane binding kinetics of the C2 domain of protein kinase C α. J. Biol. Chem. 288, 16905–16915 10.1074/jbc.M113.467456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clerico E. M., Tilitsky J. M., Meng W. L., and Gierasch L. M. (2015) How Hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J. Mol. Biol. 427, 1575–1588 10.1016/j.jmb.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bukau B., Deuerling E., Pfund C., and Craig E. A. (2000) Getting newly synthesized proteins into shape. Cell 101, 119–122 10.1016/S0092-8674(00)80806-5 [DOI] [PubMed] [Google Scholar]
- 39. Dindo M., Conter C., and Cellini B. (2017) Electrostatic interactions drive native-like aggregation of human alanine:glyoxylate aminostransferase. FEBS J. 284, 3739–3764 10.1111/febs.14269 [DOI] [PubMed] [Google Scholar]
- 40. Majhi P. R., Ganta R. R., Vanam R. P., Seyrek E., Giger K., and Dubin P. L. (2006) Electrostatically driven protein aggregation: β-lactoglobulin at low ionic strength. Langmuir 22, 9150–9159 10.1021/la053528w [DOI] [PubMed] [Google Scholar]
- 41. Calamai M., Taddei N., Stefani M., Ramponi G., and Chiti F. (2003) Relative influence of hydrophobicity and net charge in the aggregation of two homologous proteins. Biochemistry 42, 15078–15083 10.1021/bi030135s [DOI] [PubMed] [Google Scholar]
- 42. Li Y. L., Gao X. F., and Chen L. L. (2009) GroEL recognizes an amphipathic helix and binds to the hydrophobic side. J. Biol. Chem. 284, 4324–4331 10.1074/jbc.M804818200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Joachimiak L. A., Walzthoeni T., Liu C. W., Aebersold R., and Frydman J. (2014) The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell 159, 1042–1055 10.1016/j.cell.2014.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karagöz G. E., Duarte A. M. S., Akoury E., Ippel H., Biernat J., Luengo T. M., Radli M., Didenko T., Nordhues B. A., Veprintsev D. B., Dickey C. A., Mandelkow E., Zweckstetter M., Boelens R., Madl T., et al. (2014) Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 156, 963–974 10.1016/j.cell.2014.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McMorran L. M., Bartlett A. L., Huysmans G. H. M., Radford S. E., and Brockwell D. J. (2013) Dissecting the effects of periplasmic chaperones on the in vitro folding of the outer membrane protein PagP. J. Mol. Biol. 425, 3178–3191 10.1016/j.jmb.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patzelt H., Rüdiger S., Brehmer D., Kramer G., Vorderwülbecke S., Schaffitzel E., Waitz A., Hesterkamp T., Dong L., Schneider-Mergener J., Bukau B., and Deuerling E. (2001) Binding specificity of Escherichia coli trigger factor. Proc. Natl. Acad. Sci. U. S. A. 98, 14244–14249 10.1073/pnas.261432298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Knoblauch N. T., Rüdiger S., Schönfeld H. J., Driessen A. J., Schneider-Mergener J., and Bukau B. (1999) Substrate specificity of the SecB chaperone. J. Biol. Chem. 274, 34219–34225 10.1074/jbc.274.48.34219 [DOI] [PubMed] [Google Scholar]
- 48. Huang C., Rossi P., Saio T., and Kalodimos C. G. (2016) Structural basis for the antifolding activity of a molecular chaperone. Nature 537, 202–206 10.1038/nature18965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Balch W. E., Morimoto R. I., Dillin A., and Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 10.1126/science.1141448 [DOI] [PubMed] [Google Scholar]
- 50. Sweeny E. A., and Shorter J. (2016) Mechanistic and structural insights into the prion-disaggregase activity of Hsp104. J. Mol. Biol. 428, 1870–1885 10.1016/j.jmb.2015.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shorter J., and Lindquist S. (2004) Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304, 1793–1797 10.1126/science.1098007 [DOI] [PubMed] [Google Scholar]
- 52. Lo Bianco C., Shorter J., Régulier E., Lashuel H., Iwatsubo T., Lindquist S., and Aebischer P. (2008) HsP104 antagonizes α-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Invest. 118, 3087–3097 10.1172/JCI35781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Geitner A. J., and Schmid F. X. (2012) Combination of the human prolyl isomerase FKBP12 with unrelated chaperone domains leads to chimeric folding enzymes with high activity. J. Mol. Biol. 420, 335–349 10.1016/j.jmb.2012.04.018 [DOI] [PubMed] [Google Scholar]
- 54. Schechter Y., Patchornik A., and Burstein Y. (1973) Selective reduction of cystine 1-8 in α-lactalbumin. Biochemistry 12, 3407–3413 10.1021/bi00742a007 [DOI] [PubMed] [Google Scholar]
- 55. Grimsley G. R., Trevino S. R., Thurlkill R. L., and Scholtz J. M. (2013) Determining the conformational stability of a protein from urea and thermal unfolding curves. Curr. Protoc. Protein Sci. 71, 28.4.1–28.4.14 10.1002/0471140864.ps2804s71 [DOI] [PubMed] [Google Scholar]
- 56. Greenfield N. J. (2006) Analysis of the kinetics of folding of proteins and peptides using circular dichroism. Nat. Protoc. 1, 2891–2899 10.1038/nprot.2006.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zviling M., Leonov H., and Arkin I. T. (2005) Genetic algorithm-based optimization of hydrophobicity tables. Bioinformatics 21, 2651–2656 10.1093/bioinformatics/bti405 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained with this article and in the supporting information.