

Abstract
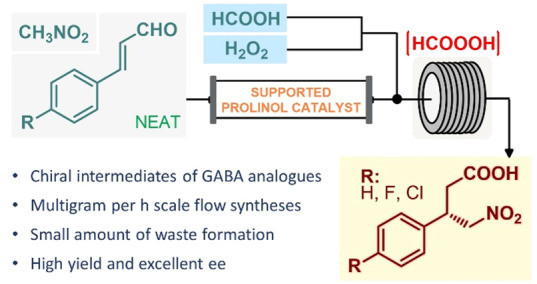
The two-step flow asymmetric synthesis of chiral γ-nitrobutyric acids as key intermediates of the GABA analogues baclofen, phenibut, and fluorophenibut is reported on a multigram scale. The telescoped process comprises an enantioselective Michael-type addition facilitated by a polystyrene-supported heterogeneous organocatalyst under neat conditions followed by in situ-generated performic acid-mediated aldehyde oxidation. Simple access to valuable optically active substances is provided with key advances in terms of productivity and sustainability compared to those of previous batch approaches.
3-Substituted derivatives of γ-aminobutyric acid (GABA) are of outstanding pharmaceutical relevance.1 For example, baclofen is an antispastic drug that is broadly applied as a muscle relaxant in the case of certain types of spasticity.2 Another GABA-derived medication, phenibut, exhibits anxiolytic effects and is used to treat anxiety and insomnia.3 Most similar to the closely related fluorophenibut, these compounds act as potent GABAB receptor agonists (Scheme 1).4 The asymmetric synthesis of these chiral GABA analogues has attracted a great deal of interest. The corresponding chiral center is typically introduced by using chiral auxiliaries, by chemical and biocatalytic resolution, or by means of asymmetric hydrogenation and enzymatic desymmetrization strategies.5 Current synthetic routes often involve catalytic enantioselective conjugate additions, which provide more direct access to the chiral key intermediates using inexpensive and readily available achiral components.6 From a practical and environmental perspective, the application of chiral organocatalysts in related syntheses is exceptionally attractive.7 However, in many cases, the low productivity of the asymmetric key step and the high cost of the homogeneous chiral catalyst decrease the practical value for potential manufacturing purposes.8
Scheme 1. Synthetic Strategy toward GABA Derivatives.
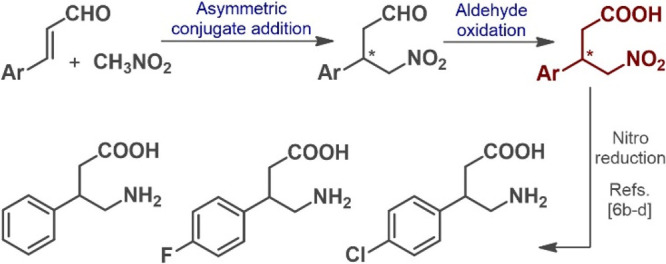
The asymmetric Michael-type addition of nitromethane to α,β-unsaturated aldehydes offers practical access to γ-nitroaldehydes as critical chiral precursors,9 which can be transformed into the desired GABA analogues via subsequent oxidation and reduction steps (Scheme 1). Considering that chemical processes that reduce solvent usage directly impact environmental performance, cost, and health issues,10 we envisioned that the enantioselective key step may be achieved under neat conditions in the presence of a robust solid-supported organocatalyst that enables cost-efficient and scalable continuous flow operation with facile product isolation.11 While telescoped flow processes for the multistep syntheses of active pharmaceutical ingredients (APIs) and related compounds offer numerous benefits over conventional batch protocols,12 continuous flow multistep strategies are scarcely exploited for the enantioselective synthesis of chiral APIs and their advanced intermediates.13 By merging the targeted organocatalytic asymmetric conjugate addition with the subsequent aldehyde oxidation in a telescoped flow process, we anticipated a simple and sustainable entry to optically active γ-nitrobutyric acids as key intermediates of the GABA analogues phenibut, fluorophenibut, and baclofen. The results are presented herein.
Our study was initiated by step-by-step continuous flow method development. For the organocatalytic asymmetric conjugate addition, a polystyrene-supported cis-4-hydroxydiphenylprolinol TBS ether (1) was selected as a catalyst (Figure 1A), which was chosen on the basis of our earlier results on a solvent-free asymmetric Michael-type addition of dimethyl malonate leading to a key chiral intermediate of a well-known antidepressant.13b Catalyst 1 was developed as an improved version of classical trans-diphenylprolinol analogues,14 but it has not yet been exploited for conjugate additions of nitromethane to α,β-unsaturated aldehydes. Because of the beneficial cis arrangement and the TBS protecting group,14a,14b high catalytic activity and high robustness were expected even under demanding conditions, such as continuous neat reactions.
Figure 1.
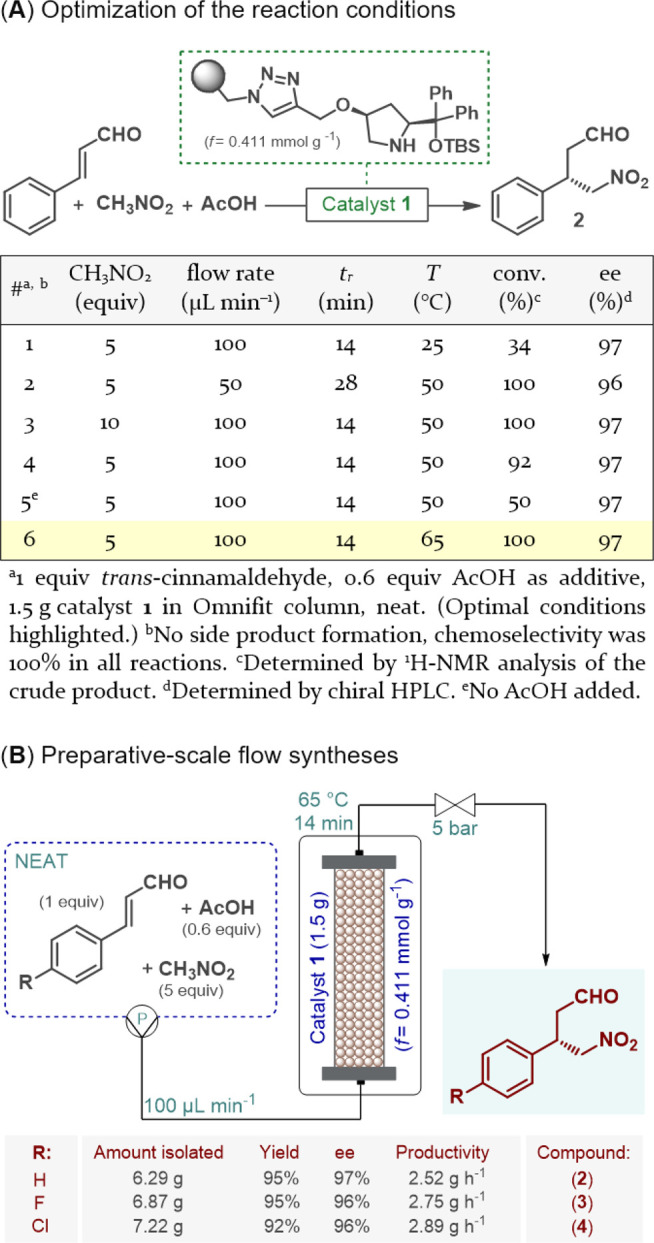
Continuous flow enantioselective synthesis of γ-nitroaldehydes as chiral intermediates of GABA analogues.
To test our hypothesis, catalyst 1 was placed in an Omnifit glass column, and the effects of different reaction conditions were carefully examined using the trans-cinnamaldehyde–nitromethane conjugate addition as a model. To our delight, the reaction performed well under neat conditions, despite the fact that such conjugate additions with polystyrene-supported organocatalysts are typically executed in dichloromethane as optimal swelling media.11c It was observed that the rate of the reaction could remarkably be improved at higher temperatures without a loss of ee and that 0.6 equiv of acetic acid was required as an additive to reach high conversions.9 As a result of the parameter optimization, at a flow rate of 100 μL min–1 (14 min residence time) and 65 °C we managed to reduce the nitromethane excess to 5 equiv and reached full conversion and 97% ee without side product formation (Figure 1A and Tables S1–S4). With the optimal reaction conditions in hand, the preparative capabilities of the flow method were next explored utilizing the appropriate cinnamaldehyde derivatives as substrates to yield chiral intermediates of phenibut, fluorophenibut, and baclofen (Figure 1B). For each substrate, a 2.5 h scale-out was performed with the same batch of the catalyst being utilized for all three experiments. Gratifyingly, high yields (up to 95%) and excellent ee’s (up to 97%) were achieved for each reaction, and chiral γ-nitroaldehydes 2–4 were obtained on a multigram scale (6.29, 6.87, and 7.22 g, respectively) without the need for chromatographic purification, after excess nitromethane and residual AcOH were removed by simple evaporation. The large-scale syntheses offered productivities of ≥2.52 g h–1 and resulted in a cumulative turnover number (TON) of 158 for the whole experiment. Importantly, the flow processes involved a small amount of waste formation as indicated by E factors of 1.45, 1.33, and 1.25 for the syntheses of 2–4, respectively.
After having established a robust flow protocol for the asymmetric synthesis of γ-nitroaldehydes 2–4, we next turned our attention to the subsequent aldehyde oxidations. To avoid large amounts of inorganic waste formation and possible precipitation issues, we did not consider, for example, Pinnick–Kraus oxidations,15 which were frequently utilized in earlier batch syntheses of GABA derivatives.6d,9a In searching for an appropriate oxidation method, we aimed for a clean, selective, and sustainable flow protocol.16
Following unsuccessful trials on catalyst-free and platinum-catalyzed aerobic aldehyde oxidations (see the Supporting Information for details),17 a novel flow method was attempted by using N-hydroxyphthalimide (NHPI) as a homogeneous oxidation catalyst in the presence of molecular O2 as the oxidant.18 The reaction was investigated in nitromethane so that it would be compatible with the organocatalytic conjugate addition upon telescoping. Following parameter optimization (Tables S8 and S9), quantitative and selective oxidation of 2 was achieved by using the setup depicted in Figure 2. Under optimal oxidation conditions, intermediate 2 was employed at a concentration of 2.3 M, which was similar to the aldehyde concentration determined in the appropriate organocatalytic key step (Figure 1B).
Figure 2.
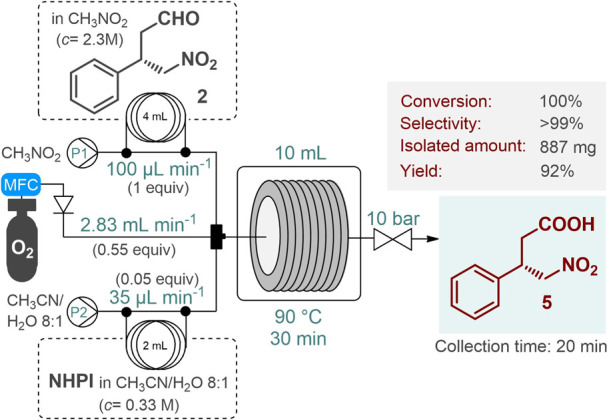
Continuous flow NHPI-catalyzed aerobic oxidation of chiral γ-nitroaldehyde 2 (MFC, mass flow controller).
Percarboxylic acids are well-known as oxidizing agents; however, their tendency toward explosive decomposition poses a significant safety hazard.19 Due their small internal dimensions and the minimal amounts of hazardous material that accumulated, continuous flow reactors are capable of safe operation within such potentially explosive regimes.20 Peracid-mediated aldehyde oxidation has not yet been reported under flow conditions. However, taking into account the low molecular weight, low cost, and environmentally benign nature of simpler percarboxylic acids in addition to the facile experimental setup (i.e., elimination of gas handling), some of these substances were investigated as oxidizing agents to yield the targeted γ-nitrobutyric acids. After promising preliminary results with a commercially available peracetic acid solution (Table S10), aldehyde oxidation was next attempted with performic acid generated in situ from formic acid as a benign precursor.21 To this end, the solution of aldehyde 2 in formic acid was combined with a stream of aqueous H2O2, and the oxidizing agent was formed and consumed continuously within the closed environment of a reaction coil (see the Supporting Information for details). (To minimize the hazard of peracid accumulation, H2O2 and the substrate were streamed at flow rates that corresponded to 1 equiv of each.) During the oxidation of aldehyde 2, it was observed that at temperatures of ≤50 °C, a significant amount of perhydrate (5a) formation occurred as side reaction, indicating the presence of unreacted H2O2 and thus incomplete peracid formation (Table S11). When the reaction temperature was increased to 100 °C, quantitative and selective carboxylic acid formation was detected.22 To match the oxidation with the organocatalytic key step during telescoping, the flow setup was modified (Figure 3A). Neat formic acid, an aqueous H2O2 solution, and the substrate in nitromethane at a concentration of 2.3 M were fed via separate streams. The effects of individual flow rates were explored to determine that a residence time of 15 min in combination with a formic acid excess of 5 equiv is sufficient to quantitatively and selectively perform the oxidation (Figure 3A and Table S12). Gratifyingly, the optimal conditions proved to be applicable to all three chiral aldehydes in preparative-scale oxidations, and the desired γ-nitrobutyric acids (5–7) were obtained in excellent yields and in multigram quantities after simple evaporation (Figure 3B).
Figure 3.
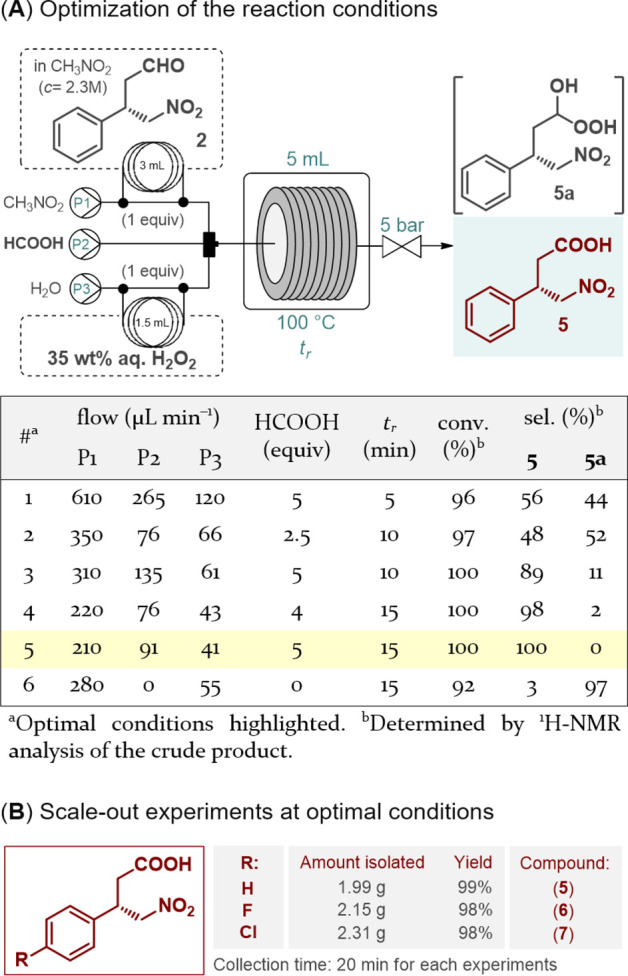
Continuous flow oxidation of chiral γ-nitroaldehydes with in situ-generated performic acid.
After step-by-step reaction method development and optimization, the combination of the organocatalytic asymmetric conjugate addition and the subsequent aldehyde oxidation into an uninterrupted flow sequence was targeted. With regard to the oxidation step, the process with in situ-generated performic acid was chosen instead of the NHPI-catalyzed reaction, which would involve gas handling and chromatographic purification to remove the homogeneous catalyst from the product. Moreover, to introduce the NHPI catalyst, an organic solvent (acetonitrile) would be required, whereas generation of the peracid demands only formic acid and an aqueous H2O2 solution as inexpensive and environmentally benign components.
The optimal conditions determined during the step-by-step experiments served as a basis in setting up the telescoped flow sequence, as shown in Figure 4. The organocatalytic asymmetric Michael-type addition of nitromethane to the appropriate cinnamaldehyde derivative took place under neat conditions while the reactants were being passed through a heated column filled with catalyst 1, similar to the stepwise process. Neat formic acid and an aqueous H2O2 solution were introduced as separate feeds at flow rates that corresponded to 1 equiv of H2O2 and 5 equiv of acid with respect to the aldehyde stream. The reaction mixture exiting the catalyst column was mixed with the combined formic acid/H2O2 stream, and the resulting solution was finally directed through a heated reaction coil where simultaneous performic acid generation and aldehyde oxidation took place. The telescoped system proved to be stable in reactions of all three substrates and was run for 1 h in each case after reaching steady state. The targeted γ-nitrobutyric acids were obtained in high yields (up to 96%) and in excellent ee’s (up to 97%) comparable to those of the stepwise processes. Multigram quantities of compounds 5–7 (2.77, 2.94, and 3.14 g, respectively) were achieved in a highly productive manner without the need for chromatographic purification. For the uninterrupted two-step sequences, E factors of only 2.44, 2.25, and 2.13 were obtained in cases of 5–7, respectively.
Figure 4.

Continuous flow synthesis of chiral γ-nitrobutyric acids as key intermediates of GABA analogues via a telescoped organocatalytic conjugate addition–aldehyde oxidation sequence.
In summary, a telescoped continuous flow process was developed for the asymmetric synthesis of γ-nitrobutyric acids, which are advanced chiral intermediates of the GABA analogues baclofen, phenibut, and fluorophenibut. The process ensured access to the targeted optically active substances directly from readily available cinnamaldehyde derivatives and nitromethane via telescoped enantioselective conjugate addition–aldehyde oxidation sequence. The asymmetric key step was achieved under neat conditions in the presence of a polystyrene-supported cis-4-hydroxydiphenylprolinol as a heterogeneous organocatalyst. The subsequent aldehyde oxidation was accomplished by taking advantage of the safe utilization of performic acid generated in situ using formic acid as a benign precursor. Valuable γ-nitrobutyric acids, which can readily be transformed to the appropriate GABA analogues by well-established nitro reduction methods,6b−6d,23 were obtained in excellent yields and in high ee’s. With a cumulative residence time of only 29 min, the sequential flow protocol enabled significant chemical intensification and offered productivities up to 3.14 g h–1 of pure products after simple isolation. Importantly, the methodology ensured high chemo- and stereoselectivities and generated a small amount of waste as demonstrated by E factors of ∼2 for the two-step processes.
Acknowledgments
S.B.Ö. acknowledges the Austrian Science Fund (FWF) for financial support through Project M 2413-B21. The authors are grateful to Prof. Wolfgang Kroutil and Dr. Michael Fuchs (Institute of Chemistry, University of Graz) for their support in the enantiomeric excess determinations.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.0c03100.
Synthetic procedures, additional reaction data, compound characterization data, copies of NMR spectra, and HPLC chromatograms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bryans J. S.; Wustrow D. J. Med. Res. Rev. 1999, 19, 149.. [DOI] [PubMed] [Google Scholar]
- Kent C. N.; Park C.; Lindsley C. W. ACS Chem. Neurosci. 2020, 11, 1740. 10.1021/acschemneuro.0c00254. [DOI] [PubMed] [Google Scholar]
- Lapin I. CNS Drug Rev. 2001, 7, 471. 10.1111/j.1527-3458.2001.tb00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino G.; Macchiarulo A.; Entrena Guadix A.; Pellicciari R. J. Med. Chem. 2001, 44, 1827. 10.1021/jm0100133. [DOI] [PubMed] [Google Scholar]
- For a review, see:; a Ramesh P.; Suman D.; Reddy K. S. N. Synthesis 2018, 50, 211. 10.1055/s-0036-1590938. [DOI] [Google Scholar]; For selected examples, see:; b Zhang Q.; Wu Z.-M.; Liu S.; Tang X.-L.; Zheng R.-C.; Zheng Y.-G. Org. Process Res. Dev. 2019, 23, 2042. 10.1021/acs.oprd.9b00285. [DOI] [Google Scholar]; c Winkler C. K.; Clay D.; Davies S.; O’Neill P.; McDaid P.; Debarge S.; Steflik J.; Karmilowicz M.; Wong J. W.; Faber K. J. Org. Chem. 2013, 78, 1525. 10.1021/jo302484p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mukherjee H.; Martinez C. A. ACS Catal. 2011, 1, 1010. 10.1021/cs2002466. [DOI] [Google Scholar]; e Fryszkowska A.; Fisher K.; Gardiner J. M.; Stephens G. M. Org. Biomol. Chem. 2010, 8, 533. 10.1039/B919526B. [DOI] [PubMed] [Google Scholar]; f Camps P.; Muñoz-Torrero D.; Sánchez L. Tetrahedron: Asymmetry 2004, 15, 2039. 10.1016/j.tetasy.2004.05.021. [DOI] [Google Scholar]; g Thakur V. V.; Nikalje M. D.; Sudalai A. Tetrahedron: Asymmetry 2003, 14, 581. 10.1016/S0957-4166(03)00024-7. [DOI] [Google Scholar]; h Hoge G. J. Am. Chem. Soc. 2003, 125, 10219. 10.1021/ja034715o. [DOI] [PubMed] [Google Scholar]; i Mazzini C.; Lebreton J.; Alphand V.; Furstoss R. Tetrahedron Lett. 1997, 38, 1195. 10.1016/S0040-4039(97)00062-2. [DOI] [Google Scholar]; j Allan R. D.; Bates M. C.; Drew C. A.; Duke R. K.; Hambley T. W.; Johnston G. A. R.; Mewett K. N.; Spence I. Tetrahedron 1990, 46, 2511. 10.1016/S0040-4020(01)82032-9. [DOI] [Google Scholar]
- For selected examples, see:; a Ishitani H.; Kanai K.; Yoo W.-J.; Yoshida T.; Kobayashi S. Angew. Chem., Int. Ed. 2019, 58, 13313. 10.1002/anie.201906349. [DOI] [PubMed] [Google Scholar]; b Biewenga L.; Saravanan T.; Kunzendorf A.; van der Meer J.-Y.; Pijning T.; Tepper P. G.; van Merkerk R.; Charnock S. J.; Thunnissen A.-M. W. H.; Poelarends G. J. ACS Catal. 2019, 9, 1503. 10.1021/acscatal.8b04299. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Veverková E.; Bilka S.; Baran R.; Šebesta R. Synthesis 2016, 48, 1474. 10.1055/s-0035-1560420. [DOI] [Google Scholar]; d Hayashi Y.; Sakamoto D.; Okamura D. Org. Lett. 2016, 18, 4. 10.1021/acs.orglett.5b02839. [DOI] [PubMed] [Google Scholar]; e Leyva-Pérez A.; García-García P.; Corma A. Angew. Chem., Int. Ed. 2014, 53, 8687. 10.1002/anie.201403049. [DOI] [PubMed] [Google Scholar]; f Feu K. S.; de la Torre A. F.; Silva S.; de Moraes Junior M. A. F.; Corrêa A. G.; Paixão M. W. Green Chem. 2014, 16, 3169. 10.1039/C4GC00098F. [DOI] [Google Scholar]; g Tsakos M.; Kokotos C. G.; Kokotos G. Adv. Synth. Catal. 2012, 354, 740. 10.1002/adsc.201100636. [DOI] [Google Scholar]; h Jensen K. L.; Poulsen P. H.; Donslund B. S.; Morana F.; Jørgensen K. A. Org. Lett. 2012, 14, 1516. 10.1021/ol3002514. [DOI] [PubMed] [Google Scholar]; i Ogawa T.; Mouri S.; Yazaki R.; Kumagai N.; Shibasaki M. Org. Lett. 2012, 14, 110. 10.1021/ol202898e. [DOI] [PubMed] [Google Scholar]; j Wang Y.; Li P.; Liang X.; Zhang T. Y.; Ye J. Chem. Commun. 2008, 1232. 10.1039/b717000a. [DOI] [PubMed] [Google Scholar]
- Krištofíková D.; Modrocká V.; Mečiarová M.; Šebesta R. ChemSusChem 2020, 13, 2828. 10.1002/cssc.202000137. [DOI] [PubMed] [Google Scholar]
- Reyes-Rodríguez G. J.; Rezayee N. M.; Vidal-Albalat A.; Jørgensen K. A. Chem. Rev. 2019, 119, 4221. 10.1021/acs.chemrev.8b00583. [DOI] [PubMed] [Google Scholar]
- a Gotoh H.; Ishikawa H.; Hayashi Y. Org. Lett. 2007, 9, 5307. 10.1021/ol702545z. [DOI] [PubMed] [Google Scholar]; b Palomo C.; Landa A.; Mielgo A.; Oiarbide M.; Puente Á.; Vera S. Angew. Chem., Int. Ed. 2007, 46, 8431. 10.1002/anie.200703261. [DOI] [PubMed] [Google Scholar]
- a Bryan M. C.; Dunn P. J.; Entwistle D.; Gallou F.; Koenig S. G.; Hayler J. D.; Hickey M. R.; Hughes S.; Kopach M. E.; Moine G.; Richardson P.; Roschangar F.; Steven A.; Weiberth F. J. Green Chem. 2018, 20, 5082. 10.1039/C8GC01276H. [DOI] [Google Scholar]; b Prat D.; Wells A.; Hayler J.; Sneddon H.; McElroy C. R.; Abou-Shehada S.; Dunn P. J. Green Chem. 2016, 18, 288. 10.1039/C5GC01008J. [DOI] [Google Scholar]
- For reviews on flow enantioselective catalysis, see:; a De Risi C.; Bortolini O.; Brandolese A.; Di Carmine G.; Ragno D.; Massi A. React. Chem. Eng. 2020, 5, 1017. 10.1039/D0RE00076K. [DOI] [Google Scholar]; b Yu T.; Ding Z.; Nie W.; Jiao J.; Zhang H.; Zhang Q.; Xue C.; Duan X.; Yamada Y. M. A.; Li P. Chem. - Eur. J. 2020, 26, 5729. 10.1002/chem.201905151. [DOI] [PubMed] [Google Scholar]; c Rodríguez-Escrich C.; Pericàs M. A. Chem. Rec. 2019, 19, 1872. 10.1002/tcr.201800097. [DOI] [PubMed] [Google Scholar]; d Atodiresei I.; Vila C.; Rueping M. ACS Catal. 2015, 5, 1972. 10.1021/acscatal.5b00002. [DOI] [Google Scholar]; e Rodríguez-Escrich C.; Pericàs M. A. Eur. J. Org. Chem. 2015, 2015, 1173. 10.1002/ejoc.201403042. [DOI] [Google Scholar]; f Puglisi A.; Benaglia M.; Chiroli V. Green Chem. 2013, 15, 1790. 10.1039/c3gc40195b. [DOI] [Google Scholar]; g Tsubogo T.; Ishiwata T.; Kobayashi S. Angew. Chem., Int. Ed. 2013, 52, 6590. 10.1002/anie.201210066. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Fülöp Z.; Szemesi P.; Bana P.; Éles J.; Greiner I. React. Chem. Eng. 2020, 5, 1527. 10.1039/D0RE00273A. [DOI] [Google Scholar]; b Bloemendal V. R. L. J.; Janssen M. A. C. H.; van Hest J. C. M.; Rutjes F. P. J. T. React. Chem. Eng. 2020, 5, 1186. 10.1039/D0RE00087F. [DOI] [Google Scholar]; c Gérardy R.; Emmanuel N.; Toupy T.; Kassin V.-E.; Tshibalonza N. N.; Schmitz M.; Monbaliu J.-C. M. Eur. J. Org. Chem. 2018, 2018, 2301. 10.1002/ejoc.201800149. [DOI] [Google Scholar]; d Porta R.; Benaglia M.; Puglisi A. Org. Process Res. Dev. 2016, 20, 2. 10.1021/acs.oprd.5b00325. [DOI] [Google Scholar]; e Gutmann B.; Cantillo D.; Kappe C. O. Angew. Chem., Int. Ed. 2015, 54, 6688. 10.1002/anie.201409318. [DOI] [PubMed] [Google Scholar]; f Baumann M.; Baxendale I. R. Beilstein J. Org. Chem. 2015, 11, 1194. 10.3762/bjoc.11.134. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selected recent examples, see:; g França A. d. S.; Leão R. A. C.; de Souza R. O. M. A. J. Flow Chem. 2020, 10, 563. 10.1007/s41981-020-00098-2. [DOI] [Google Scholar]; h Chatterjee S.; Guidi M.; Seeberger P. H.; Gilmore K. Nature 2020, 579, 379. 10.1038/s41586-020-2083-5. [DOI] [PubMed] [Google Scholar]; i Russell M. G.; Jamison T. F. Angew. Chem., Int. Ed. 2019, 58, 7678. 10.1002/anie.201901814. [DOI] [PubMed] [Google Scholar]; j Kassin V.-E. H.; Gérardy R.; Toupy T.; Collin D.; Salvadeo E.; Toussaint F.; Van Hecke K.; Monbaliu J.-C. M. Green Chem. 2019, 21, 2952. 10.1039/C9GC00336C. [DOI] [Google Scholar]; k Ziegler R. E.; Desai B. K.; Jee J.-A.; Gupton B. F.; Roper T. D.; Jamison T. F. Angew. Chem., Int. Ed. 2018, 57, 7181. 10.1002/anie.201802256. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Lin H.; Dai C.; Jamison T. F.; Jensen K. F. Angew. Chem., Int. Ed. 2017, 56, 8870. 10.1002/anie.201703812. [DOI] [PubMed] [Google Scholar]; m Ishitani H.; Kanai K.; Saito Y.; Tsubogo T.; Kobayashi S. Eur. J. Org. Chem. 2017, 2017, 6491. 10.1002/ejoc.201700998. [DOI] [Google Scholar]; n Borukhova S.; Noël T.; Hessel V. ChemSusChem 2016, 9, 67. 10.1002/cssc.201501367. [DOI] [PubMed] [Google Scholar]; o Ghislieri D.; Gilmore K.; Seeberger P. H. Angew. Chem., Int. Ed. 2015, 54, 678. 10.1002/anie.201409765. [DOI] [PubMed] [Google Scholar]
- a Ishitani H.; Furiya Y.; Kobayashi S. Chem. - Asian J. 2020, 15, 1688. 10.1002/asia.202000065. [DOI] [PubMed] [Google Scholar]; b Ötvös S. B.; Pericàs M. A.; Kappe C. O. Chem. Sci. 2019, 10, 11141. 10.1039/C9SC04752B. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rossi S.; Porta R.; Brenna D.; Puglisi A.; Benaglia M. Angew. Chem., Int. Ed. 2017, 56, 4290. 10.1002/anie.201612192. [DOI] [PubMed] [Google Scholar]; d Ogasawara S.; Hayashi Y. Synthesis 2017, 49, 424. 10.1055/s-2016-0036-1588899. [DOI] [Google Scholar]; e Tsubogo T.; Oyamada H.; Kobayashi S. Nature 2015, 520, 329. 10.1038/nature14343. [DOI] [PubMed] [Google Scholar]
- a Lai J.; Sayalero S.; Ferrali A.; Osorio-Planes L.; Bravo F.; Rodríguez-Escrich C.; Pericàs M. A. Adv. Synth. Catal. 2018, 360, 2914. 10.1002/adsc.201800572. [DOI] [Google Scholar]; b Arenas I.; Ferrali A.; Rodríguez-Escrich C.; Bravo F.; Pericàs M. A. Adv. Synth. Catal. 2017, 359, 2414. 10.1002/adsc.201700120. [DOI] [Google Scholar]; c Donslund B. S.; Johansen T. K.; Poulsen P. H.; Halskov K. S.; Jørgensen K. A. Angew. Chem., Int. Ed. 2015, 54, 13860. 10.1002/anie.201503920. [DOI] [PubMed] [Google Scholar]; d Alza E.; Sayalero S.; Kasaplar P.; Almaşi D.; Pericàs M. A. Chem. - Eur. J. 2011, 17, 11585. 10.1002/chem.201101730. [DOI] [PubMed] [Google Scholar]; e Alza E.; Sayalero S.; Cambeiro X. C.; Martin-Rapun R.; Miranda P. O.; Pericas M. A. Synlett 2011, 2011, 464. 10.1055/s-0030-1259528. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Alza E.; Pericàs M. A. Adv. Synth. Catal. 2009, 351, 3051. 10.1002/adsc.200900817. [DOI] [Google Scholar]
- a Bal B. S.; Childers W. E.; Pinnick H. W. Tetrahedron 1981, 37, 2091. 10.1016/S0040-4020(01)97963-3. [DOI] [Google Scholar]; b Kraus G. A.; Taschner M. J. J. Org. Chem. 1980, 45, 1175. 10.1021/jo01294a058. [DOI] [Google Scholar]
- For reviews of continuous flow oxidation chemistries, see:; a Hone C. A.; Kappe C. O. Top. Curr. Chem. 2019, 377, 2. 10.1007/s41061-018-0226-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gemoets H. P. L.; Su Y.; Shang M.; Hessel V.; Luque R.; Noel T. Chem. Soc. Rev. 2016, 45, 83. 10.1039/C5CS00447K. [DOI] [PubMed] [Google Scholar]; c Gavriilidis A.; Constantinou A.; Hellgardt K.; Hii K. K.; Hutchings G. J.; Brett G. L.; Kuhn S.; Marsden S. P. React. Chem. Eng. 2016, 1, 595. 10.1039/C6RE00155F. [DOI] [Google Scholar]; d Mallia C. J.; Baxendale I. R. Org. Process Res. Dev. 2016, 20, 327. 10.1021/acs.oprd.5b00222. [DOI] [Google Scholar]
- a Durndell L. J.; Isaacs M. A.; Li C. e.; Parlett C. M. A.; Wilson K.; Lee A. F. ACS Catal. 2019, 9, 5345. 10.1021/acscatal.9b00092. [DOI] [Google Scholar]; b Durndell L. J.; Cucuzzella C.; Parlett C. M. A.; Isaacs M. A.; Wilson K.; Lee A. F. Catal. Today 2019, 333, 161. 10.1016/j.cattod.2018.02.052. [DOI] [Google Scholar]; c Vanoye L.; Wang J.; Pablos M.; Philippe R.; Bellefon C. d.; Favre-Réguillon A. Org. Process Res. Dev. 2016, 20, 90. 10.1021/acs.oprd.5b00359. [DOI] [Google Scholar]; d Vanoye L.; Aloui A.; Pablos M.; Philippe R.; Percheron A.; Favre-Réguillon A.; de Bellefon C. Org. Lett. 2013, 15, 5978. 10.1021/ol401273k. [DOI] [PubMed] [Google Scholar]
- a Dai P.-F.; Qu J.-P.; Kang Y.-B. Org. Lett. 2019, 21, 1393. 10.1021/acs.orglett.9b00101. [DOI] [PubMed] [Google Scholar]; b Vanoye L.; Abdelaal M.; Grundhauser K.; Guicheret B.; Fongarland P.; De Bellefon C.; Favre-Réguillon A. Org. Lett. 2019, 21, 10134. 10.1021/acs.orglett.9b04193. [DOI] [PubMed] [Google Scholar]
- Renz M.; Meunier B. Eur. J. Org. Chem. 1999, 1999, 737.. [DOI] [Google Scholar]
- For reviews of safety aspects of continuous flow reactors, see:; a Gutmann B.; Kappe C. O. J. Flow Chem. 2017, 7, 65. 10.1556/1846.2017.00009. [DOI] [Google Scholar]; b Kockmann N.; Thenée P.; Fleischer-Trebes C.; Laudadio G.; Noël T. React. Chem. Eng. 2017, 2, 258. 10.1039/C7RE00021A. [DOI] [Google Scholar]; c Movsisyan M.; Delbeke E. I. P.; Berton J. K. E. T.; Battilocchio C.; Ley S. V.; Stevens C. V. Chem. Soc. Rev. 2016, 45, 4892. 10.1039/C5CS00902B. [DOI] [PubMed] [Google Scholar]
- a Mata A.; Cantillo D.; Kappe C. O. Eur. J. Org. Chem. 2017, 2017, 6505. 10.1002/ejoc.201700811. [DOI] [Google Scholar]; b Ebrahimi F.; Kolehmainen E.; Oinas P.; Hietapelto V.; Turunen I. Chem. Eng. J. 2011, 167, 713. 10.1016/j.cej.2010.08.091. [DOI] [Google Scholar]; For a recent review of chemical generators in continuous flow, see:; c Dallinger D.; Gutmann B.; Kappe C. O. Acc. Chem. Res. 2020, 53, 1330. 10.1021/acs.accounts.0c00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- At this temperature, any incidental unreacted peracid residue should safely decompose before leaving the reaction coil.
- For nitro reductions, see:; a Orlandi M.; Brenna D.; Harms R.; Jost S.; Benaglia M. Org. Process Res. Dev. 2018, 22, 430. 10.1021/acs.oprd.6b00205. [DOI] [Google Scholar]; b Porta R.; Puglisi A.; Colombo G.; Rossi S.; Benaglia M. Beilstein J. Org. Chem. 2016, 12, 2614. 10.3762/bjoc.12.257. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Orlandi M.; Tosi F.; Bonsignore M.; Benaglia M. Org. Lett. 2015, 17, 3941. 10.1021/acs.orglett.5b01698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.