

Abstract
The COVID-19 epidemic has put an enormous burden on the health-care system and the economy. The virus has very high infectivity and is crippling in patients developing severe disease. The disease caused by this infective agent, a novel RNA coronavirus (SARS-CoV-2), was named by the World Health Organization as COVID-19. SARS-CoV-2 usually enters the human body from the respiratory tract and gradually causes systemic disease. The disease is mild in 81% and severe in the balance. The virus causes multiorgan damage and primarily damages airway epithelium, small intestine epithelium, and vascular endothelium, which are organs with high angiotensin-converting enzyme (angiotensin-converting enzyme-2 [ACE2] expression). The most affected organ is the lungs, and the cardiovascular system follows it closely. Symptomatic hypoxic patients are initially treated with oxygen supplementation, but those with severe hypoxia need mechanical ventilation support. Patients with COVID-19 infection present as two phenotypes. The ventilation strategy should be based on the phenotype. The disease causes major hemodynamic disturbances in its invasion of the cardiovascular system. Strict personal protection protocols are needed to ensure the safety of health-care workers and nosocomial spread.
Keywords: COVID-19, hemodynamic management COVID, respiratory management COVID
In December 2019, multiple cases of viral pneumonia were reported in the city of Wuhan of Hubei Province of China. A novel RNA coronavirus (SARS-CoV-2) was found to be causing this unusual respiratory illness. The World Health Organization named this disease as COVID-19 (an acronym for “coronavirus disease 2019”). The COVID-19 infection has an unusual property to spread like wildfire in humans. Within a few days, this virus spread across the entire world thanks to stupendous multi-international travel in this globalized world. The knowledge of the clinical features, pathogenesis, and management of COVID-19 disease is still limited. The entire world is currently working on developing a clear understanding of the disease and its management. The issues covered by this review are evolving, and there are multiple publications in contemporary literature on the subject matter. The limitation of this review is that this manuscript only covers the literature published until May 10, 2020, and much new knowledge may evolve by the time this gets published.
COVID-19 usually enters the human body from the respiratory tract and gradually causes systemic disease. The virus results in multiorgan dysfunction and failure. The disease is mild in 81% and severe in the balance. Respiratory failure, septic shock, and multiorgan dysfunction are seen in 5% of the positive cases and result in a fatality in half of such cases.[1] The most affected organ is the lungs, and the cardiovascular system follows it closely. Other organs developing significant dysfunction are the kidney and intestines. Lifesaving measures, such as ventilation support, are most pertinent in this subset of patients. This review is focused on the respiratory and cardiovascular effects of COVID-19 infection, its pathogenesis, and the management of these systems.
Pathogenesis of Respiratory Involvement
Classic COVID-19 infection has three clinical stages: stage 1—an asymptomatic incubation period; stage 2—a symptomatic period with the presence of virus; and stage 3—severe respiratory symptomatic stage. The patient may not be infective in the first stage but is very infective in the other two, especially so in the third stage as the viral load is high.[2] The asymptomatic carriers are responsible for the volatile spread of the disease, as they unconsciously spread the disease in the community. A flow chart displaying the progression of the respiratory features, in the different stages of COVID-19 infection, is shown in Figure 1.
Figure 1.
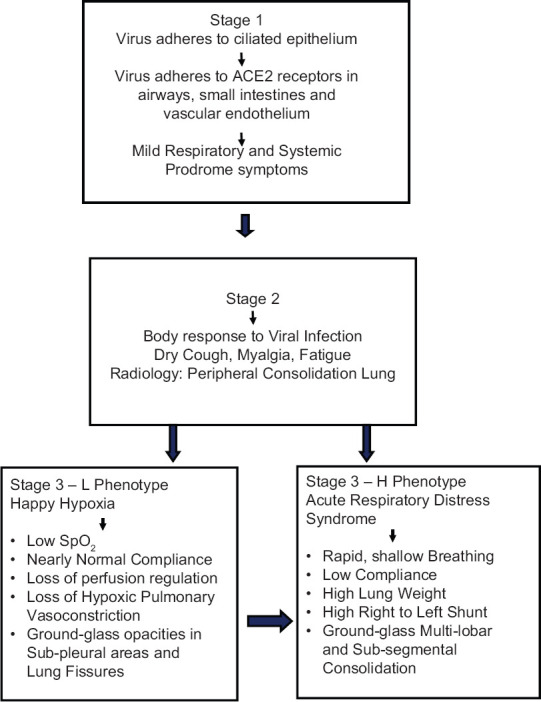
Respiratory features of different stages of COVID-19 infection
Stage 1 (asymptomatic stage)
The inhaled COVID-19 virus binds to nasal epithelial cells and starts replicating. Data indicate that coronavirus infections primarily affect the ciliated cells in the conducting airways.[3] After the inoculation of the virus, most victims have an incubation period associated with mild and nonspecific symptoms such as malaise, fever, and dry cough. During the first clinical phase, COVID-19 multiplies and primarily resides in the respiratory system of the host. The virus binds to the target cells hijacking the ACE2 receptor.[4] ACE2 receptors are present in all tissues. However, airway epithelium, small intestine epithelium, and vascular endothelium have a high ACE2 expression.[5] On aerosol inoculation, the infection presents with mild respiratory and systemic symptoms, due to the affinity of the coronavirus for ACE2 receptors. Nasal swabs are more sensitive than pharyngeal swabs to detect the virus at this stage. The real-time quantitative polymerase chain reaction value for the viral RNA can predict the viral load and help forecast its infectivity and clinical course. However, it is essential to remember that even individuals with low viral loads are infectious.[6]
Stage 2 (upper airway and conducting airway response)
The COVID-19 infection induces a biphasic immune response. During the first two clinical stages, there is an adaptive immune response to eliminate the virus and to avert disease progression to severe stages. A therapeutic strategy focused on boosting immunity is vital in this phase. To develop an endogenous protective immune response, the host should be in good health and possess a genetic background that elicits specific immunity.[7]
In the second clinical stage of established pulmonary disease, viral multiplication occurs, and there is respiratory inflammation. Patients commonly present with dry cough, fever, myalgia, or fatigue. Some patients may have sputum production, headache, hemoptysis, and diarrhea. When the protective immune response is impaired, the virus propagates, destroying the affected tissues. Lung inflammation is mediated by proinflammatory macrophages and granulocytes. Antigen presentation stimulates the body's humoral and cellular immunity, and this immune response is mediated by virus-specific B and T cells.[8] In about 80% of the infected patients, the disease is mild and restricted to the upper and conducting airways. These individuals need monitoring, isolation, and are managed with conservative symptomatic therapy.[9]
Stage 3 (hypoxia, ground-glass infiltrates, and progression to adult respiratory distress syndrome)
About 20% of the infected patients progress to stage 3 disease and develop pulmonary infiltrates. In this stage, the virus infiltrates the gas exchange units of the lung and infects alveolar type II cells. Initially, the peripheral and subpleural units are involved, and the cells undergo apoptosis and die.[10] Some develop very severe disease. The fatality rate is about 2% as per initial estimates, but this rate varies markedly with age and comorbidity.[9]
Studies have shown that inflammatory cytokines and biomarkers such as interleukin (IL)-2, IL-6, IL-7, granulocyte-colony-stimulating factor, macrophage inflammatory protein 1-α, tumor necrosis factor-α, C-reactive protein (CRP), ferritin, and d-dimer to be significantly elevated in the patients with severe disease. COVID-19 causes diffuse alveolar damage by the formation of fibrin-rich hyaline membranes and a few multinucleated giant cells.[11] The healing leads to more severe scarring and fibrosis than other forms of adult respiratory distress syndrome (ARDS).
Pulmonary postmortem findings of 38 COVID-19 cases from Italy revealed exudative and proliferative diffuse alveolar disease with capillary congestion, desquamation of pneumocytes, hyaline membrane formation, interstitial edema, hyperplasia of pneumocytes, platelet–fibrin thrombi, and interstitial infiltration by macrophages. Viral particles were seen in the cytoplasm of pneumocytes.[12] Recent autopsies have shown that the lungs are filled with clear liquid jelly material.[11] The high levels of inflammatory cytokines in the lungs of COVID-19 induce the formation of hyaluronan-synthase-2 in lung alveolar epithelial cells and fibroblasts.[13] Although the composition of the jelly has not yet been determined, it is likely to be hyaluronan, which has an ability to absorb water up to 1000 times its molecular weight.[7]
Aggressive viral replication causes massive epithelial and endothelial cell death and vascular leakage, triggering the production of breezy proinflammatory cytokines and chemokines.[14] Loss of pulmonary ACE2 function is thought to cause acute lung injury by the renin–angiotensin system dysfunction, which amplifies inflammation and vascular permeability.[15] Breakneck viral multiplication, cell damage, ACE2 receptor downregulation, and antibody-dependent enhancement initiate an aggressive host inflammatory response to COVID-19, which is responsible for the violent cytokine storm. The cytokine storm triggers a violent autoimmune attack, causes ARDS and multiple organ failure.[11] Uncontrolled pulmonary inflammation leads to case fatality.
In stage 2 of the disease, proinflammatory CD4 T cells and cytotoxic granules CD8 T cells are increasingly mobilized, imply antiviral immune responses, and hyperactivation of T cells. However, in stage 3, peripheral CD4 and CD8 T cells show reduction in activity.[11] A strong innate and acquired immune response and epithelial regeneration are required for recovery. The elderly/debilitated/immunocompromised has a lesser immune response and reduced ability to repair the damaged epithelium and is thus more at risk. The elderly also have impaired mucociliary clearance, allowing the virus to spread more readily.[16] The therapy of cytokine storm is still evolving and multiple agents are under evaluation. The review of different therapeutic modalities was not in the scope of this article, and thus the details of the trials underway are not presented.
Clinically, the COVID-19 disease presents with impressive nonuniformity in patients. The disease has two distinct phenotypes. Nearly 50% of COVID-19 pneumonia patients have distinctive features of severe hypoxemia with near normal-respiratory system compliance, something never seen in severe ARDS.
The type-L phenotype patients have low elastance, low ventilation to perfusion ratio, low lung weight, and low ability to recruit nonaerated segments. This phenotype displays normal breathing but has low oxygen saturations (“silent hypoxemia” or “happy hypoxic”).[17] Patients with type-L COVID-19 pneumonia present with near-normal compliance, indicating that the aeration in the lung is nearly normal. Hypoxemia results from the loss of regulation of perfusion and the loss of hypoxic vasoconstriction. The pulmonary artery pressure is near normal. On computerized tomography (CT) scan, ground-glass densities are primarily located subpleural and along the lung fissures.[17]
The type-H phenotype patients have high elastance, high right-to-left shunt, high lung weight, and high ability to recruit nonaerated segments. These patients are extremely dyspneic and display the features of ARDS patients, that is, short/rapid breathing and cyanosis.[17] Patients with type-H COVID-19 pneumonia have low lung compliance due to edema. The dependent lung regions are not aerated due to the increased edema and superimposed pressure. There is right-to-left shunting as the nonperfused zones of the lung receive a fraction of cardiac output. Quantitative analysis of the CT scan reveals an increase in lung weight, and the lung weight reflects the severity of the ARDS.[18]
Chest X-ray reveals bilateral peripheral focal or multifocal infiltrates or white opacities, suggestive of viral pneumonia [Figure 2]. Subpleural ground-glass opacities in the lower lobes unilaterally or bilaterally appear within 0–4 days of development of symptoms and may even be seen in asymptomatic patients. CT images reveal characteristic fluid containing multilobar and subsegmental areas of consolidation, with ground-glass appearance with a crazy-paving pattern [Figure 3].[2,19] Although experience with ultrasonography is limited, a small report suggests that pleural-line irregularities, a “B-line” pattern, and consolidations are suggestive of COVID-19.[20] There is increasing leukopenia (especially lymphocytopenia) and thrombocytopenia. There is also a rise in the levels of transaminases, lactate dehydrogenase, creatinine, creatinine kinase, d-dimer, and other markers of systemic inflammation.[11] Serum procalcitonin is low to normal in most cases of COVID-19 pneumonia.[21]
Figure 2.
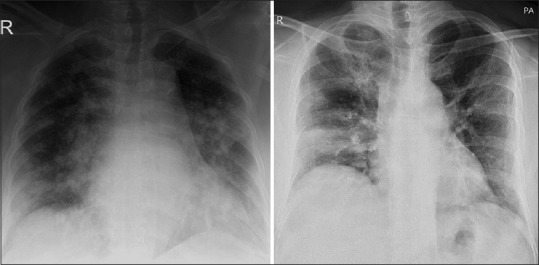
Chest X-ray of two patients revealing bilateral peripheral focal or multifocal infiltrates or white opacities
Figure 3.
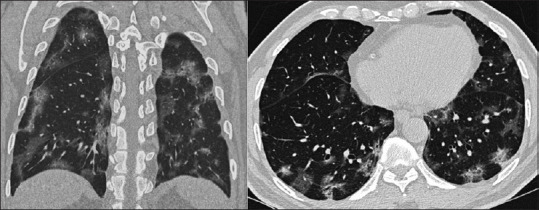
High-resolution computerized tomographic longitudinal and axial scan images of a patient revealing characteristic fluid containing multilobar and subsegmental areas of consolidation, with ground-glass appearance with a crazy-paving pattern
Pathogenesis of Cardiovascular Involvement
Cardiac involvement is frequent in patients with COVID-19 disease. The salient features of the cardiovascular involvement are shown in Table 1. Patients with cardiac risk factors and cardiac disease are more vulnerable to develop COVID-19 and have worse clinical outcomes.[22] In hospitalized COVID-19 patients, 14–16% had baseline cardiovascular disease, and 15–32% had hypertension.[2,23,24] The ACE2 receptors act as an entry point to the myocardial cells causing direct injury in them. The severe cytokine surge results in injury to multiple organs, including cardiac myocytes. Increased metabolic demand associated with systemic infection and decreased supply, due to the ongoing hypoxia, results in myocardial damage.[25]
Table 1.
Cardiovascular effects of COVID-19 infection
| System | Effects |
|---|---|
| Cardiac | Myocardial injury by hypoxia; cytokines; coronary spasm |
| Coronary artery disease-exacerbation of existing disease by plaque rupture and by disturbance of coronary demand/supply | |
| Myocarditis with low left ventricular ejection fraction | |
| Pulmonary edema due to fluid retention | |
| Drug therapy-induced cardiotoxicity | |
| Arrhythmia-dyselectrolytemia, neurohumoral, hypoxia, inflammatory toxins | |
| Vascular | Hypercoagulopathy, venous thromboembolism |
| Pulmonary | Pulmonary artery hypertension mediated right heart failure |
In patients with COVID-19, myocardial injury results from plaque rupture, cytokine storm, hypoxia, coronary spasm, thrombi, or direct endothelial injury.[26] Systemic inflammation and catecholamine surge inherent in COVID-19 increases plaque rupture vulnerability and may lead to the acute coronary syndrome. Electrolyte disturbance, especially hypokalemia, can trigger hemodynamically significant arrhythmias in patients with underlying cardiac disease.[25] Patients with hypertension on ACE inhibitors express excess ACE receptors, and it was postulated that they might be at an increased risk of contracting COVID-19. However, most cardiac professional bodies (American College of Cardiology, American Heart Association, Heart Failure Society of America) have recommended the continuation of prior ACE inhibitors or angiotensin receptor blockers.[27] Magnetic resonance imaging has demonstrated myocardial edema in COVID-19 patients.[28]
Many cardiovascular complications such as myocardial injury, myocarditis, arrhythmias, and venous thromboembolism (VTE) are associated with COVID-19 infection. A meta-analysis of six studies, including 1527 patients with COVID-19, reported cardiac injury in 8% of the patients. An increase in troponin levels has been reported in 12% of COVID-19 patients. Cardiac injury is exceedingly prevalent in COVID-19. It may present as an elevation of cardiac biomarkers and electrocardiographic/echocardiographic abnormalities. The measure of troponin elevation correlates with the degree of high-sensitivity CRP elevation, and dynamic increases in troponin are associated with higher mortality.[29] This is associated with more severe disease and is an indicator of poor prognosis. Cohort studies estimate cardiovascular involvement in 7–17% of hospitalized patients, in about 22% in patients admitted to the ICU, and in 59% among those who died.[2,23,30] This rise can be attributed to virus-induced myocardial cell death and myocardial supply/demand imbalance secondary to the cytokine storm.[31]
A total of 18 COVID-19 patients in a New York center had an ST-segment elevation suggestive of acute myocardial infarction.[32] Of these, 56% had ST-segment elevation at the time of presentation, and the others developed ST-segment elevation during hospitalization. Seventy-eight percent of these patients had focal ST-segment elevation [36% having a normal left ventricular ejection fraction (LVEF) and 57% having reduced LVEF]. The balance 22% of patients had diffuse ST-segment elevation (75% had a normal LVEF while one patient had a 10% LVEF with global hypokinesia). Fifty percent of patients were subjected to coronary angiography, and out of this, 67% had an obstructive disease. Seventy-two percent of these patients died in the hospital.[32]
Cardiac arrhythmia was noted in 44.4% of cases in COVID-19 patients in ICU and 6.9% in non-ICU cases. Atrial arrhythmia has been reported in 17.7% of patients in a recent report from New York.[33] The arrhythmia may be attributed to metabolic disturbances, hypoxia, neurohormonal, or inflammatory stress. Malignant tachyarrhythmia, with raised troponin levels, are indicative of myocarditis.[34] Heart failure was observed in 23.0% of COVID-19 patients and was seen in 51.9% of patients who did not survive. It is still not clear whether heart failure results from the exacerbation of preexisting left ventricular dysfunction, new cardiomyopathy, or right heart failure precipitated by severe parenchymal lung disease/ARDS.[31,35]
In patients with cardiogenic pulmonary edema, brain natriuretic peptide levels and echocardiography help guide therapy. The presence/absence of a concomitant cardiogenic component indicates whether venovenous or venoarterial cannulation is preferred for extracorporeal membrane oxygenation (ECMO).[36] COVID-19 patients are at an increased risk of VTE. In addition to disseminated intravascular coagulation, critically ill patients with prolonged immobilization are inherently at a high risk for VTE. Vascular inflammation contributes to hypercoagulability and endothelial dysfunction. Microvascular thrombosis has been hypothesized to be irresponsible for hypoxemia. Some autopsies have demonstrated microvascular thrombosis and hemorrhage in pulmonary vessels.[37]
Several potential drug therapies are under evaluation. Some of the drugs under evaluation exhibit critical cardiovascular side effects. There is a higher risk of QTc prolongation, and a potential risk for torsade-de-pointes, when a combination of azithromycin and hydroxychloroquine is administered, especially in the presence of hypokalemia. Protease inhibitors, lopinavir/ritonavir, have been shown to prolong PR and QT intervals leading to AV blocks. Ribavirin is sometimes combined with lopinavir/ritonavir, increasing their levels and may precipitate toxicity. Remdesivir, being studied in COVID-19, has been reported to precipitate hypotension and bradycardia. Corticosteroids should be avoided in patients with COVID-19 because they cause fluid retention, electrolyte derangement, and hypertension.[38] The Surviving Sepsis guidelines, however, suggest the use of low-dose corticosteroids in patients with refractory shock, as the time to resolution of shock is shorter with corticosteroid therapy.[39]
Respiratory Management
Most patients with mild disease may not have indications for hospitalization and can be isolated in nontraditional settings or at home. Regular remote monitoring for symptoms is needed but no therapy is required. Only symptomatic treatment, such as antipyretics for fever, is required in patients with mild COVID-19. The flow chart of the guiding principles of respiratory management is shown in Figure 4.
Figure 4.
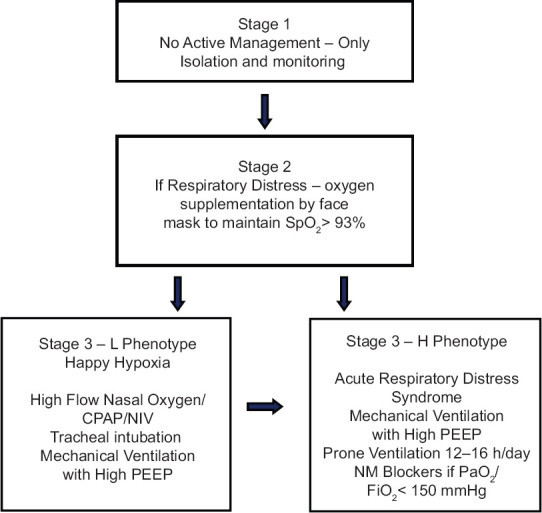
Respiratory management of different stages of COVID-19 infection
Supplemental oxygen therapy must be administered to the following category of patients: severe acute respiratory infection, obstructed or absent breathing, respiratory distress, central cyanosis, shock, coma or convulsions, respiratory distress, and hypoxemia or shock. The oxygen flow rate must be titrated to reach target SpO2≥93%. If the patient is critical, 10–15 L/min oxygen (FiO20.60–0.95) must be administered by a face mask with a reservoir bag. Once a patient is stable, the target is >90% SpO2 in nonpregnant adults and ≥92–95% in pregnant patients.
A type-L patient responds well to oxygen therapy, if not yet breathless. If type-L patients develop dyspnea, noninvasive options should be considered, such as high flow nasal oxygen (HFNO), continuous positive airway pressure (CPAP), or noninvasive ventilation (NIV). Active monitoring for the clinical signs of excessive inspiratory effort should be performed at this stage. As surrogate measures for work of breathing, swings of central venous pressure must be observed and inspiratory esophageal pressure must be monitored.[17,40] Swings in pleural pressure may exacerbate lung injury. NIV is, however, associated with high failure rates, aerosol generation, and delays intubation in a patient who needs prolonged respiratory support.[17] Consider helmet-based ventilation as it is less likely to leak and with it, one can increase air pressure to deliver NIV.[41]
Early tracheal intubation may avert the transition to type-H phenotype. Tracheal intubation should be performed as the risk of lung injury increases. While performing tracheal intubation, appropriate infection control precautions must be ensured. The patient must be preoxygenated with 100% FiO2 for 5 min via a face mask with a reservoir bag, rebreathing circuit, HFNO, or NIV. Rapid-sequence intubation should be performed by an experienced physician, preferably using a video laryngoscope. Posttracheal intubation and deep sedation, hypercapnic patients can be ventilated with tidal volumes >6 mL/kg (up to 8–9 mL/kg).[17] Deep sedation may be required to control respiratory drive and achieve tidal volume targets. Plateau pressure must be maintained <30 cmH2O. Permissive hypercapnia is permitted.[42]
Although one school of thought recommends the early institution of mechanical ventilation, another school of thought is against early mechanical ventilation. It is the opinion of the second group that many known or suspected COVID-19 with respiratory distress may improve with CPAP or NIV and not need tracheal intubation at all. Additionally, unnecessary intubation and ventilation of one patient might deny what might be a lifesaving treatment for another patient in resource-limited settings.[43]
A rise in the swings in the esophageal pressures to >15 cmH2O indicates a transition from the type-L to type-H phenotype. Type-H patients are identified best by CT imaging. Experience with ultrasound is limited and not yet recommended to stage lung changes.[20] Bronchoscopy is aerosol generating and hence not recommended as a tool for evaluation. If CT imaging is not feasible, surrogate clinical markers like compliance and the ability to recruit nonaerated lung segments can help identify the change in phenotype.[17] Type-H patients should be treated on lines of severe ARDS, including high peak end-expiratory pressure (PEEP) compatible with hemodynamics, prone positioning, and extracorporeal support. Prone positioning can be used as a rescue to improve stress and strain redistribution.[17] The application of prone ventilation is strongly recommended but requires additional human resources, protocols, and expertise for its safe performance.[42] For COVID-19 patients, prone ventilation for 12–16 h/day is recommended.[43]
High PEEP may decrease the swings in pleural pressure and prevent exacerbation of lung injury but may adversely affect patient hemodynamics in patients with normal compliance. PEEP should be titrated to strike a balance between its benefits (reducing trauma due to atelectasis and improving alveolar recruitment) versus risks (air trapping leading to lung injury and high pulmonary vascular resistance).[43]
In ARDS, recruitment maneuvers (RM) are performed by intermittent delivery of 30–40 cmH2O CPAP, incremental increase in PEEP, or using high drive pressure. A recent trial of high PEEP and prolonged high-pressure RM showed harm, suggesting that it should be avoided.[44] Incremental PEEP RM is not recommended as a technique, in case RM are needed.
Neuromuscular blockade by administration of a continuous infusion should not be routinely used in moderate–severe ARDS (PaO2/FiO2<150 mmHg). It is used as a last resort in gravely ill patients with very low compliance, deranged hemodynamics, ventilator desynchronization despite sedation, inability to achieve desired tidal volume, or refractory hypoxemia or hypercapnia.[43]
Patient disconnection from the ventilator results in a loss of PEEP. It may induce atelectasis, apart from the venting of the patient's expired air from the ventilator circuit and its dispersal into the ICU atmosphere. For airway suctioning, endotracheal tubes with subglottic suction or in-line suction catheters should be used to avoid disconnection for tracheal suction. Whenever a disconnection is required, the endotracheal tube should be clamped to prevent the dispersion of expired air. During the disconnection, breathing gas delivery from the ventilator must be put on hold.
Mechanically ventilated patients with refractory hypoxemia (despite optimization of ventilation, rescue therapies, and prone ventilation) venovenous EMCO must be considered. The patient must be shifted to an ECMO center if the facility for ECMO is not available. EMCO is resource-intensive and requires experienced centers/health-care workers and infrastructure. The use of ECMO should be considered only in carefully selected salvageable patients with severe ARDS.[45]
After recovery, weaning of the patient has to be done following a protocol advocated in standard guidelines.[46] Safe tracheal extubation protocols must be followed to ensure minimal spillage of secretions, aerosol generation, and dispersion of the aerosol/vented breathing gases.[47]
Cardiovascular Management
Direct evidence on patients with COVID-19 and shock is not established as yet. The recommendations of different academic bodies are based on indirect evidence and experience from critically ill patients in sepsis.[39] The reports of the prevalence of shock in patients with COVID-19 are highly variable. A recent report from New York has reported that nearly all patients receiving invasive mechanical ventilation need vasopressor support (95.4%).[33] Early reports from China describe an incidence of vasopressor support in 20–35% among patients in the ICU, and it was related to the patient population studied, the severity of the disease, and the definition of shock considered.[2,34]
A conservative fluid management strategy is recommended to ensure adequate tissue perfusion. To assess fluid responsiveness, the surviving sepsis guidelines for COVID-19 patients in shock recommend the use of dynamic parameters like skin temperature, capillary refilling time, and/or serum lactate measurement rather than static parameters. The guidelines also recommend using a conservative fluid strategy. The preferred fluid should be buffered/balanced crystalloids, and colloids use is not recommended.[39] The above guidelines recommend norepinephrine as the first-line vasoactive agent, as it is the most widely studied vasoactive agent with a low inferred risk of adverse effects. If norepinephrine is not available, either vasopressin or epinephrine must be administered as the first-line vasoactive agent as no clear evidence of harm has been demonstrated with the use of both these agents in trials.[39] Vasopressin may be added as a second-line agent if a target mean arterial pressure of 60–65 mmHg cannot be achieved by norepinephrine alone.
The American Society of Hematology guidelines recommend that all hospitalized COVID-19 patients should receive pharmacologic VTE prophylaxis with low-molecular weight heparin or fondaparinux, unless contraindicated. Therapeutic anticoagulation is recommended for patients with very high levels of d-dimer, abnormal coagulation parameters, in cytokine storm syndrome, and/or multiorgan failure.[37]
Conclusion
The COVID-19 epidemic has put an enormous burden on the health-care system and the economy. The virus has very high infectivity and is crippling in patients developing severe disease. The virus causes multiorgan damage and primarily damages airway epithelium, small intestine epithelium, and vascular endothelium, which are organs with high ACE2 expression. Symptomatic hypoxic patients are treated with oxygen supplementation initially, but severely symptomatic patients need mechanical ventilation support. The patients with COVID-19 infection present have two phenotypes, and the ventilation strategy should be based on the phenotype. The disease causes major hemodynamic disturbances in its invasion of the cardiovascular system. Strict personal protection protocols are needed to ensure the safety of health-care workers and nosocomial spread.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Novel Coronavirus Pneumonia Emergency Response Epidemiology Team. The Epidemiological Characteristics of an Outbreak of 2019 Novel Coronavirus Diseases (COVID-19) - China, 2020 China CDC Weekly Published February 1, 2020. [Last accessed on 2020 May 01]. Available from: http://weeklychinacdccn/en/article/id/e53946e2-c6c4-41e9-9a9b-fea8db1a8f51 . [PMC free article] [PubMed]
- 2.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–9. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sims AC, Baric RS, Yount B, Burkett SE, Collins PL, Pickles RJ. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: Role of ciliated cells in viral spread in the conducting airways of the lungs. J Virol. 2005;79:15511–24. doi: 10.1128/JVI.79.24.15511-15524.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J Virol. 2020;94:e00127–20. doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mason RJ. Pathogenesis of COVID-19 from a cell biology perspective. Eur Respir J. 2020;55:2000607. doi: 10.1183/13993003.00607-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020;27:1451–4. doi: 10.1038/s41418-020-0530-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. 2020;10:102–8. doi: 10.1016/j.jpha.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA. 2020;323:1239–42. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 10.Qian Z, Travanty EA, Oko L, Edeen K, Berglund A, Wang J, et al. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am J Respir Cell Mol Biol. 2013;48:742–8. doi: 10.1165/rcmb.2012-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420–2. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carsana L, Sonzogni A, Nasr A, Rossi R, Pellegrinelli A, Zerbi P, et al. Pulmonary post-mortem findings in a large series of COVID-19 cases from Northern Italy medRxiv. 2020 doi: 10.1016/S1473-3099(20)30434-5. doi: 101101/2020041920054262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell TJ, Brand O, Morgan DJ, Salek-Ardakani S, Jagger C, Fujimori T, et al. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol. 2019;80:14–28. doi: 10.1016/j.matbio.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang M. Cell Pyroptosis, a Potential Pathogenic Mechanism of 2019-nCoV Infection (January 29, 2020) [Last accessed on 2020 May 04]. Available at SSRN: http://dxdoiorg/102139/ssrn3527420 .
- 15.Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. 2008;93:543–8. doi: 10.1113/expphysiol.2007.040048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho JC, Chan KN, Hu WH, Lam WK, Zheng L, Tipoe GL, et al. The effect of aging on nasal mucociliary clearance, beat frequency, and ultrastructure of respiratory cilia. Am J Respir Crit Care Med. 2001;163:983–8. doi: 10.1164/ajrccm.163.4.9909121. [DOI] [PubMed] [Google Scholar]
- 17.Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L, et al. COVID-19 pneumonia: Different respiratory treatments for different phenotypes? Intensive Care Med. 2020;14:1–4. doi: 10.1007/s00134-020-06033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maiolo G, Collino F, Vasques F, Rapetti F, Tonetti T, Romitti F, et al. Reclassifying acute respiratory distress syndrome. Am J Respir Crit Care Med. 2018;197:1586–95. doi: 10.1164/rccm.201709-1804OC. [DOI] [PubMed] [Google Scholar]
- 19.Pan F, Ye T, Sun P, Gui S, Liang B, Li L, et al. Time course of lung changes on chest CT during recovery from 2019 novel coronavirus (COVID-19) Radiology. 2020;295:715–721. doi: 10.1148/radiol.2020200370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng QY, Wang XT, Zhang LN Chinese Critical Care Ultrasound Study Group (CCUSG) Findings of lung ultrasonography of novel corona virus pneumonia during the 2019-2020 epidemic. Intensive Care Med. 2020;46:849–50. doi: 10.1007/s00134-020-05996-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J Heart Lung Transplant. 2020;39:405–7. doi: 10.1016/j.healun.2020.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basu-Ray I, Soos MP. Cardiac Manifestations of Coronavirus (COVID-19) StatPearls [Internet] Treasure Island (FL): StatPearls Publishing; 2020. [Last accessed on 2020 May 04]. Available from: https://wwwncbinlmnihgov/books/NBK556152/report=printable . [PubMed] [Google Scholar]
- 23.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020:e200950. doi: 10.1001/jamacardio.2020.0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong TY, Redwood S, Prendergast B, Chen M. Coronaviruses and the cardiovascular system: Acute and long-term implications. Eur Heart J. 2020;41:1798–800. doi: 10.1093/eurheartj/ehaa231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavazzi G, Pellegrini C, Maurelli M, Belliato M, Sciutti F, Bottazzi A, et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail. 2020;22:911–5. doi: 10.1002/ejhf.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.American College of Cardiology. HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19. [Last accessed on 2020 May 14]. Available from: https://wwwaccorg/latest-in-cardiology/articles/2020/03/17/08/59/hfsa-acc-aha-statement-addresses-concerns-re-using-raas-antagonists-in-covid-19 .
- 28.Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020 doi: 10.1001/jamacardio.2020.1096. doi: 101001/jamacardio 20201096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020:e201017. doi: 10.1001/jamacardio.2020.1017. doi: 101001/jamacardio20201017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet. 2020;395:1054–62. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Driggin E, Madhavan MV, Bikdeli B, Chuich T, Laracy J, Bondi-Zoccai G, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. J Am Coll Cardiol. 2020;75:2352–71. doi: 10.1016/j.jacc.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bangalore S, Sharma A, Slotwiner A, Yatskar L, Harari L, Shah B, et al. ST-segment elevation in patients with Covid-19 - A case series. N Engl J Med. 2020 doi: 10.1056/NEJMc2009020. NEJMc2009020 doi: 101056/NEJMc2009020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goyal P, Choi JJ, Pinheiro LC, Schenck EJ, Chen R, Jabri A, et al. Clinical characteristics of Covid-19 in New York City. N Engl J Med. 2020 doi: 10.1056/NEJMc2010419. NEJMc2010419 doi: 101056/NEJMc2010419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475–81. doi: 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buzon J, Roignot O, Lemoine S, Perez P, Kimmoun A, Levy B, et al. Takotsubo cardiomyopathy triggered by influenza a virus. Intern Med. 2015;54:2017–9. doi: 10.2169/internalmedicine.54.3606. [DOI] [PubMed] [Google Scholar]
- 36.MacLaren G, Fisher D, Brodie D. Preparing for the most critically ill patients with COVID-19: The potential role of extracorporeal membrane oxygenation. JAMA. 2020 Feb 19; doi: 10.1001/jama.2020.2342. doi: 101001/jama 20202342 Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 37.American Society of Hematology. COVID-19 and VTE/Anticoagulation: Frequently Asked Questions (Version 21; last updated 2020 Apr 17) [Last accessed on 2020 May 15]. Available from: https://wwwhematologyorg/covid-19/covid-19-and-vte-anticoagulation .
- 38.Lee N, Allen Chan KC, Hui DS, Ng EK, Wu A, Chiu RW, et al. Effects of early corticosteroid treatment on plasma SARS-associated Coronavirus RNA concentrations in adult patients. J Clin Virol. 2004;31:304–9. doi: 10.1016/j.jcv.2004.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alhazzani W, Møller MH, Arabi YM, Loeb M, Ng Gong M, Fan E, et al. Surviving Sepsis Campaign: Guidelines on the management of critically ill adults with Coronavirus Disease 2019 (COVID-19) Intensive Care Med. 2020;46:854–87. doi: 10.1007/s00134-020-06022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walling PT, Savege TM. A comparison of oesophageal and central venous pressures in the measurement of transpulmonary pressure change. Br J Anaesth. 1976;48:475–9. doi: 10.1093/bja/48.5.475. [DOI] [PubMed] [Google Scholar]
- 41.Patel BK, Wolfe KS, MacKenzie EL, Salem D, Esbrook CL, Pawlik AJ, et al. One-year outcomes in patients with acute respiratory distress syndrome enrolled in a randomized clinical trial of helmet versus facemask noninvasive ventilation. Crit Care Med. 2018;46:1078–84. doi: 10.1097/CCM.0000000000003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.World Health Organization. Clinical management of severe acute respiratory infection (SARI) when COVID-19 disease is suspected: interim guidance. World Health Organization; 2020. [Last accessed on 2020 July 10]. https://appswhoint/iris/handle/10665/331446 . [Google Scholar]
- 43.Arulkumaran N, Brealey D, Howell D, Singer M. Use of non-invasive ventilation for patients with COVID-19: A cause for concern?? Lancet Respir Med. 2020 doi: 10.1016/S2213-2600(20)30181-8. S2213-2600(20)30181-8. doi: 101016/S2213-2600(20)30181-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Writing Group for the Alveolar Recruitment for Acute Respiratory Distress Syndrome Trial (ART) Investigators. Effect of lung recruitment and titrated positive end-expiratory pressure (PEEP) vs low PEEP on mortality in patients with acute respiratory distress syndrome: A randomized clinical trial. JAMA. 2017;318:1335–45. doi: 10.1001/jama.2017.14171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.European Society of Intensive Care Medicine and the Society of Critical Care Medicine. COVID-19 Ventilation Clinical Practice Guidelines (ESICM, 2020) Medscape. 2020. Apr 07, [Last accessed on 2020 May 06]. Available from: https://referencemedscapecom/viewarticle/928191 .
- 46.European Society of Intensive Care Medicine Academy. Mechanical Ventilation-COVID-19. [Last accessed on 2020 May 09]. Available from: https://academy.esicm.org/course/view.php?id=293 .
- 47.D'Silva DF, McCulloch TJ, Lim JS, Smith SS, Carayannis D. Extubation of patients with COVID-19? Br J Anaesth. 2020 doi: 10.1016/j.bja.2020.03.016. S0007-0912(20) 30172-0. doi: 10.1016/j.bja.2020.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]