

Abstract
Metabolic activation of the carcinogenic tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and N’-nitrosonornicotine (NNN) results in formation of reactive electrophiles that modify DNA to produce a variety of products including methyl, 4-(3-pyridyl)-4-oxobutyl (POB)-, and 4-(3-pyridyl)-4-hydroxybutyl adducts. Among these are adducts such as 7-POB-deoxyguanosine (N7POBdG) which can lead to apurinic/apyrimidinic (AP) sites by facile hydrolysis of the base-deoxyribonucleoside bond. In this study, we used a recently developed highly sensitive mass spectrometric method to quantitate AP sites by derivatization with O-(pyridin-3-yl-methyl)hydroxylamine (PMOA) (detection limit, 2 AP sites per 108 nucleotides). AP sites were quantified in DNA isolated from tissues of rats treated with NNN and NNK and from human lung tissue and leukocytes of cigarette smokers and non-smokers. Rats treated with 5 or 21 mg/kg bw NNK for four days by s.c. injection had 2–6 and 2–17 times more AP sites than controls in liver and lung DNA (p<0.05). Increases in AP sites were also found in liver DNA of rats exposed for 10 and 30 weeks (p<0.05), but not for 50 and 70 weeks to 5 ppm NNK in their drinking water. Levels of N7POBG were significantly correlated with AP sites in rats treated with NNK. In rats treated with 14 ppm (S)-NNN in their drinking water for 10 weeks, increased AP site formation compared to controls was observed in oral and nasal respiratory mucosa DNA (p<0.05). No significant increase in AP sites was found in human lung and leukocyte DNA of cigarette smokers compared to non-smokers, although AP sites in leukocyte DNA were significantly correlated with urinary levels of the NNK metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL). This is the first study to use mass spectrometry based methods to examine AP site formation by carcinogenic tobacco-specific nitrosamines in laboratory animals, and to evaluate AP sites in DNA of smokers and non-smokers.
Graphical Abstract
INTRODUCTION
Apurinic/apyrimidinic (AP) sites are common types of DNA damage. AP sites can be formed spontaneously by hydrolysis of the N-glycosidic bond of deoxyribonucleosides, or during excision repair of DNA exposed to UV radiation, oxidizing species, or alkylating agents, in which N-glycosylases recognize and excise aberrant bases.1 AP sites are mainly repaired by base excision repair (BER): an AP site is incised by AP endonucleases forming a single-nucleotide gap, and then DNA polymerase β (β-pol) and DNA ligase (ligase I or IIIyXRCC1) can insert the missing nucleotide and connect the DNA strands.2,3 However, a certain fraction of AP sites may escape repair or be falsely repaired, causing G→T and G→C transversion mutations, chromosome aberrations, or transcription errors, which may be associated with cancer and degenerative disorders.4,5
The tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and N’-nitrosonornicotine (NNN) are considered carcinogenic to humans by the International Agency for Research on Cancer.6 Metabolic activation of NNK and NNN results in formation of reactive electrophiles that modify DNA to produce a variety of products including methyl, 4-(3-pyridyl)-4-oxobutyl (POB)-, and 4-(3-pyridyl)-4-hydroxybutyl (PHB)- adducts that have been previously characterized.7–9 Among these are adducts such as 7-POB-deoxyguanosine (N7POBdG) that can result in apurinic/apyrimidinic (AP) sites by facile hydrolysis of the base-deoxyribonucleoside bond (Scheme 1). This step may initiate the carcinogenic process.10 The quantification of AP sites may provide a general measurement of DNA exposure to mutagens.11
Scheme 1.
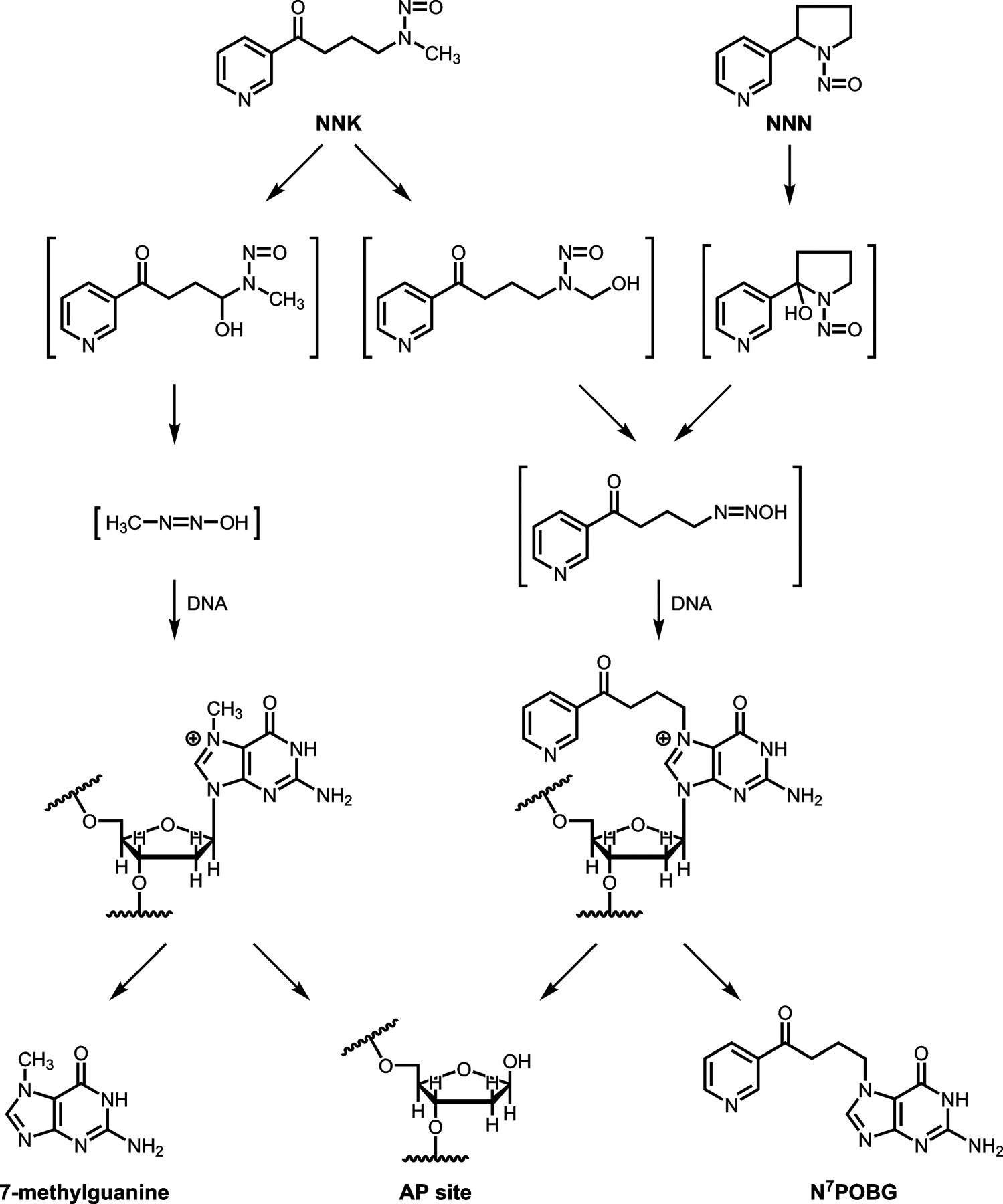
Metabolic activation of NNK and NNN to intermediates and products that could result in the production of AP sites in DNA.
Several approaches have been used for the quantitation of AP sites in genomic DNA, including radiometric labeling of the AP site with 14C-labeled reagents,12,13 32P-postlabeling,14 fluorescent probe tagging,11 ELISA assays,2,15,16 and an aldehyde reactive probe.4,17 More recent studies have been developing mass spectrometric methods to quantify AP sites. These studies use a derivatization reaction of the ring-opened aldehydic form of the deoxyribosyl moiety with certain agents, such as pentafluorophenylhydrazine (PFPH),18 O-4-nitrobenzylhydroxylamine (NBHA),19 semicarbazide,20 or a large molecule hydroxylamine reagent.21 Chen et al. have developed a highly sensitive mass spectrometric method to quantitate AP sites using O-(pyridin-3-yl-methyl)hydroxylamine (PMOA) achieving a limit of detection of 2 AP sites per 108 nucleotides (nts).22 In this method, AP sites are derivatized before DNA isolation to limit the formation of artifacts during DNA processing as shown in Supporting Information (SI) Scheme S1.
In this study we applied this newly developed mass spectrometric method22 to quantify AP sites in genomic DNA extracted from 1) lung and liver of rats treated with NNK or lung and liver homogenate reacted with 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc); 2) six different tissues of rats treated with (S)-NNN; and 3) lung tissue and leukocytes from cigarette smokers and non-smokers. Two major POB-DNA adducts — N7POBG and O2POB-thymidine (O2POBdT) — were measured along with AP sites in these samples and the relationship between POB-DNA adducts and AP sites was investigated. This is the first study to investigate AP sites induced by tobacco-specific nitrosamines in vivo under various conditions, and in cigarette smokers and non-smokers.
MATERIAL AND METHODS
Caution: NNK, (S)-NNN, and NNKOAc are carcinogenic. They should be handled in a well-ventilated hood with extreme care and with personal protective equipment.
Material and Chemicals
(3S,4R)-3,4,5-Trihydroxypentanal,O-pyridin-3-yl-methyl oxime (PMOA-dR) and [13C5]PMOA-dR were obtained as described previously.22 N7POBG and O2POBdT were synthesized previously by our group.23,24 Deoxyguanosine (dG) was obtained from Chem-Impex Int’l Inc. (Wood Dale, IL). [13C1015N5]dG was procured from Cambridge Isotope Laboratories (Tewksbury, MA). 4-[(Acetoxymethyl)nitrosamino]-1-(3-pyridyl)-1-butanone (NNKOAc) was purchased from Toronto Research Chemicals (Toronto, Candada). Porcine liver esterase was obtained from MyBioSource (San Diego, CA). Reagents and enzymes for DNA isolation were obtained from Qiagen Sciences (Germantown, MD). All other chemicals and solvents were purchased from Sigma-Aldrich Chemical Co. (Milwaukee, WI) or Fisher Scientific (Fairlawn, NJ).
Animal experiments
These studies were approved by the University of Minnesota Institutional Animal Care and Use Committee. Male F344 rats were purchased from Charles River Laboratories (Kingston, NY) and housed two rats per microisolator cage with Harlan Teklad ¼ in. corn cob bedding (Harlan, Indianapolis, IN) in the Research Animal Resources facility under the following conditions: 20–24 °C, 30–70% relative humidity, and 12/12h light/dark cycle.
In vitro studies.
Rat liver or lung tissue (~100 mg) was homogenized with a Tissue Ruptor (Qiagen Sciences, Germantown, MD) and incubated with 0 (control), 2, 12, 49, or 243 μM NNKOAc (n=3 per dose group per tissue) for 1.5 h at 37 °C, in the presence of porcine liver esterase (5.5 U).
NNK short time exposure.
Each rat was given a subcutaneous injection (s.c.) of either 0.4 mL of saline (control) or 0.4 mL of saline containing 5.2 mg (0.025 mmol)/kg body weight (bw) NNK or 20.7 mg (0.1 mmol)/kg bw NNK for 4 consecutive days (n=5 rats per group)25. They were sacrificed 4 h after the final dose.
NNK 70 week exposure.
Rats were obtained at age 6 weeks. After acclimating for one week, they were treated chronically with 5 ppm of NNK in their drinking water for 10, 30, 50, and 70 weeks (n = 3 per group).26 Control animals had no NNK in their drinking water. Further details have been described.26 Each rat tissue was analyzed in triplicate.
(S)-NNN exposure.
Rats were obtained at 6 weeks of age. After 1 week of acclimation, rats were maintained on tap water and NIH-07 diet or given (S)-NNN (14 ppm) in their drinking water for 10 weeks. The rats were then humanely euthanized.
Human samples
These studies were approved by the University of Minnesota Institutional Review Board.
Lung.
The samples were collected for a previous study.27 Thirty-seven lung tissue samples (from 20 smokers, 17 non-smokers) were obtained from the Cancer Center Tissue Procurement Facility (TPF). Subject information is shown in SI Table S1. The samples were from the margins of tumors and histologically confirmed as normal tissue. They were obtained at surgery, immediately frozen in liquid N2, and stored at −80 °C until DNA isolation. Urine samples were also obtained from some subjects just prior to surgery. They were analyzed for cotinine and in some cases for the NNK metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) using methods described previously.28
Leukocytes.
Blood was collected from 26 smokers and 26 non-smokers participating in an observational study carried out in Washington, DC and Minneapolis, MN. The study was approved by the Georgetown University Institutional Review Board and the University of Minnesota Institutional Review Board. The study had a repeated measures design to characterize three groups: current smokers, former smokers, and never smokers, at monthly intervals over 12 weeks. Volunteers were recruited from prior studies; local print, radio and internet advertisements; and snowball sampling. Samples from current smokers and never smokers were used here.
Eligible participants included adults (age 18–70 years) in good physical and mental health as evidenced by a medical history with no unstable medical conditions. Current smokers were required to have a stable smoking pattern of at least ten cigarettes per day and a smoking history of at least ten years. Never smokers were required to have smoked 100 cigarettes or less in their lifetime. Participants were excluded if they reported consuming >21 drinks per week or there was an active infection such as influenza, cold, or respiratory infection; current oral cavity or respiratory disease, such as cancer, chronic obstructive pulmonary disease, radiation therapy to the lung, or general anesthesia within six months; uncontrolled hypertension; treatment with nicotine replacement therapy within six months; treatment with antidepressants or drugs used for smoking cessation within six months; a psychiatric disorder or any other reason that precluded understanding of the informed consent; a pregnancy plans to become pregnant in the next three months or currently breastfeeding; a reported clinical diagnosis of an immune system disease such as lupus, multiple sclerosis, rheumatoid arthritis, hepatitis or HIV; a reported diagnosis of cancer, past or present, excluding non-melanoma skin cancers; or a reported diagnosis of diabetes, kidney, or liver disease. In addition, smokers were excluded if they reported using any combination of other tobacco products, such as cigars, pipes or smokeless tobacco, more than three times per week in the past six months or marijuana in the past six months. Never smokers were excluded if they used other tobacco products, such as cigars, pipes, smokeless tobacco, or marijuana in the last five years.
Participants attended an orientation visit and four clinical visits approximately four weeks apart (baseline, 4, 8, and 12 weeks). In addition to other biologic samples, a fasting blood sample (eight hours) was collected at each visit. Blood components were separated and stored in a −70°C freezer. Only the baseline samples were used here.
Samples were also obtained from the Tobacco Research Programs Repository, University of Minnesota (5 smokers and 5 nonsmokers) with similar selection criteria. Leukocytes were isolated from blood using a method previously reported.29 Urinary NNAL concentration was analyzed for smokers using a previously reported method.28 All leukocyte donor information are shown in SI Table S2.
Derivatization of AP Sites in Mammalian Tissue and DNA Isolation
Freshly frozen rat or human lung tissue (~100 mg) was homogenized in HE lysis buffer [50 mM HEPES buffer and 10 mM EDTA (pH 8.0)]. The tissue homogenate was then centrifuged at 3000g for 10 min at 4 °C, and the nuclear pellet was resuspended in HE buffer (1mL).
Leukocytes were thawed in ice and 3 volumes of red blood cell (RBC) lysis solution were added. The mixed solution was vortexed and incubated at room temperature for 5 min, then centrifuged at 3000 g for 10 min at 4 °C. The supernatant was removed and the leukocyte pellet with ~50 μL residual liquid was resuspended in 1mL HE buffer.
To the suspension of the above rat or human tissues, proteinase K (40 μL) and 10% sodium dodecyl sulfate (SDS) (120 μL) were added, and the solution was incubated in a water bath at 37 °C for 1.5 h. PMOA (60 μL of 100 mM) was added to the nuclear lysate and incubated at 37 °C for 1.5 h. Butyraldehyde (60 μL, 1 M in 50% isopropanol/H2O ) was then added to quench the remaining PMOA by further incubation for 10 min. The samples were kept on ice for 5 min, and then 500 μL of the Puregene Protein Precipitation solution was added. The tubes were vortexed thoroughly and centrifuged at 3800g for 10 min. The supernatant was transferred to new 5 mL Eppendorf tubes, and the nucleic acids were precipitated with 0.1 volume of NaCl solution (5 M) and 2 mL of isopropanol. The DNA was washed with 70% ethanol in 5 mM HEPES buffer (pH 8.0) and dried under vacuum centrifugation. The derivatized DNA was reconstituted with 600 μL of HE buffer and incubated at 37 °C with RNase A (300 μg) and RNase T1 (1 μg) for 1.5 h, and then DNA was reprecipitated with 50 μL 5M NaCl and 1 mL isopropanol, washed twice with 70% ethanol in 5 mM HEPES buffer (pH 8.0) and dried under vacuum centrifugation.
Two experiments were done to test whether the first 1.5 h SDS incubation without adding PMOA introduces extra AP sites. As shown in SI Table S3, when PMOA was added along with SDS at the same time and incubated for 3 h, 24% more AP sites were found, which may be due to the longer incubation time (3 h vs. 1.5 h). However, when PMOA and SDS were incubated for 1.5 h, the AP sites decreased by 39% due probably to the failure to completely digest histones and release all DNA for derivatization.22
Enzymatic Digestion of AP Sites in DNA.
[13C5]PMOA-dR (0.5 pmol), [13C1015N5]dG (14 nmol), [pyridine-D4]N7POBG (0.5 pmol), and [pyridine-D4]O2POBdT (0.5 pmol) were added to the DNA prior to enzymatic digestion. The DNA samples were incubated at 37 °C with DNase I (0.2 U) and nuclease P1 (>0.2 U) for 3 h, followed by phosphodiesterase I (4 μU) and alkaline phosphatase (12 U) for 15 h. After DNA digestion, the samples were filtered with a 10K filter to remove the enzymes. Five percent of the filtrate was taken to measure dG using liquid chromatography - tanden mass spectrometry (LC−MS/MS). The rest was further purified by solid phase extraction (SPE).
SPE Method.
After DNA digestion, the derivatized AP sites were enriched by SPE with a Strata-X polymeric cartridge (30 mg, 33 μm) preconditioned with CH3OH, followed by H2O. The cartridge was washed with 5% CH3OH in 0.5 mM NH4HCO3 (3 mL), and the PMOA-dR was eluted from the cartridge with 90% CH3OH in 0.5 mM NH4HCO3 (1 mL). The eluents were dried by vacuum centrifugation, reconstituted in 50 μL of 2.5 mM NH4HCO3, and transferred to LC autosampler vials for LC−MS/MS analysis.
The derivatization, digestion, and SPE purification followed a previous publication with slight modifications.22 The experimental flow chart is shown in SI Figure S1.
LC−MS/MS Method for Quantitation of PMOA-dR and N7POBG in DNA.
Quantitation of PMOA-dR and N7POBG were conducted in the same run with a Linear Trap Quadropole (LTQ) Orbitrap Velos instrument (Thermo Scientific, Waltham, MA) interfaced with a Dionex Ultimate 3000 UHPLC system (NCS-3500RS pump and WPS-3000PL autosampler) using nanoelectrospray ionization. The analysis was performed using a nano column (75 μm i.d., 20 cm length, 10 μm orifice) prepared by packing a commercially available fused-silica emitter (New Objective, Woburn, MA) with Luna C18-bonded separation media (5 μm particles with 100 Å pores, Phenomenex, Torrance, CA). The mobile phases consisted of 5 mM NH4OAc (A) and CH3CN (B), using a linear gradient at a flow rate of 300 nL/min with increasing B from 2 to 15% over 14.5 min, followed by ramping to 98% B within 1 min, and holding at this composition for 3 min. The gradient was then returned to 2% B in 4 min, and the system was re-equilibrated at this mobile phase composition at 1000 nL/min before the next injection. For injections, a 5 μL injection loop was used and the sample (4 μL) was loaded onto the nano column with a 1000 nL/min flow at the initial conditions (2% B) for 5.5 min. The nanoelectrospray source voltage was set at 2.2 kV. The capillary temperature was 350 °C, and the S-Lens RF level was set at 40%. The analysis was performed using accurate mass Extracted Ion Chromatograms (EIC) of the following ions: fragments m/z 92.0495 [C6H6N]+, 108.0444 [C6H6NO]+, 110.0600 [C6H8NO]+, 125.0709 [C6H9N2O]+ from parent ion m/z 241.1 for PMOA-dR and corresponding fragments m/z 92.0495, 108.0444, 110.0600, and 125.0709 from parent ion m/z 246.1 for [13C5]PMOA-dR, and fragment m/z 148.0757 [C9H10NO]+ from parent ion m/z 299.1 for N7POBG and corresponding fragment m/z 152.10080 from parent ion m/z 303.1 for [pyridine-D4]N7POBG with a mass tolerance of 5 ppm. The scan events were performed using higher-energy collisional dissociation (HCD) fragmentation with a normalized collision energy of 60% for PMOA-dR and 30% for N7POBG, isolation widths of 2 Da for both analytes and internal standards, and product ion spectra acquisition at a resolution of 30 000.
Analysis of O2POBdT by LC−MS/MS.
Analysis of O2POBdT was performed on a TSQ Vantage triple-quadrupole mass spectrometer (Thermo Scientific, Waltham, MA) interfaced with a Dionex Ultimate 3000 UHPLC system (NCS-3500RS pump and WPS-3000PL autosampler). Analysis was performed on a Luna C18 column (5 μm particles with 100 Å pores, 250 mm length× 0.5 mm diameter, Phenomenex) at a flow rate of 10 μL/min at room temperature. Sample injection volume was 4 μL. The mobile phases consisted of 5 mM NH4OAc (A) and CH3CN (B) with a linear gradient from 3 to 40% B over a period of 20 min, followed by ramping to 90% B within 1 min and holding at this composition for 4 min. The gradient was then returned to 3% B in 1 min followed by 10 min re-equilibration. The Electrospray Ionization (ESI) source was operated in positive ion mode, monitoring m/z 390 [M+H]+→148 [C9H10NO]+ for O2POBdT and corresponding transitions m/z 394→152 for [pyridine-D4]O2POBdT. The collision gas was Ar at 0.3 mTorr with a collision energy of 15 V. The ion source parameters were set as follows: spray voltage 2600 V, S-lens 85 V, capillary temperature 300 °C, sheath gas pressure 25. The quadrupoles Q1 and Q3 were operated at an isolation width of 0.7 Da. Scan width was set at 0.1 m/z with a scan time of 0.15 s.
Analysis of dG by LC−MS/MS.
Analysis of dG was performed with the same instrument, mobile phases, column, flow rate, column temperature and injection volume as the analysis of O2POBdT but using a different LC gradient program. The 5% DNA hydrolysis solution filtrate was diluted 10 times before injection onto the column, resulting in 6–200 pmol dG and 14 pmol [13C1015N5]dG on column. The LC program was a linear gradient from 3 to 13% CH3CN over a period of 14 min, followed by ramping to 90% CH3CN within 1 min and holding at this composition for 2 min. The gradient was then returned to 3% CH3CN in 1 min followed by 10 min re-equilibration. The first 11 min of the eluants were diverted to waste. The ESI source was also operated in positive ion mode, monitoring m/z 268 [M+H]+→152 [C5H6N5O]+ for dG and the corresponding transition m/z 283→162 for [13C1015N5]dG. The other mass spectrometer parameters were the same as described for O2POBdT. The measurement of dG was used to calculate total nucleotides, assuming that dG accounts for 21% of the total nucleotides in human and rat DNA.30 The measured dG levels are shown in SI Tables S1, S2 and S4.
The chromatograms resulting from the above analyses are shown in SI Figure S2. PMOA-dR and [13C5]PMOA-dR have E and Z isomers but were integrated as a single peak.
Data Analysis.
Descriptive statistical analyses were completed with Microsoft Excel 2013. Student’s t test (type 1, one tailed, equal variance) and the Mann-Whitney U test (one tailed, equal variance) were both used to compare the exposed and control groups and to compare smokers with non-smokers. The Mann-Whitney U test requires a sample size not less than 5, so the mean of each tissue in the NNN control group was used as the fifth sample to conduct this test. The same results were achieved with both tests for all the comparisons except the AP site comparison between exposed and control groups in esophageal mucosa of NNN treated rats, which was significant in the Mann-Whitney U test but not in Student’s t test. Therefore, the results were interpreted based on Student’s t test unless specified. Regression analysis was used to investigate the correlation between AP sites and POB DNA adducts, subject age, urinary cotinine or NNAL, and the correlation between AP sites in different tissues.
RESULTS
AP site formation from acute exposure to NNK
Rats treated with 5.2 mg/kg bw (0.025 mmol/kg bw) or 20.7 mg/kg bw (0.1 mmol/kg bw) NNK daily for four days by s.c. injection had accumulated total doses of 20.8 mg/kg and 82.8 mg/kg of NNK. Levels of AP sites and POB-DNA adducts are shown in Figure 1. Increases in AP sites were observed in both exposure groups in liver and lung (p<0.05). In liver, the average AP sites (± standard deviation) in controls were 2.23 ± 0.52 AP sites/107 nts, consistent with the levels from a recent study using a similar method.22 Liver DNA from the exposed rats had AP sites 6 times those of controls in the 5.2 mg/kg NNK exposure group, and 16.5 times those of controls in the 20.7 mg/kg NNK exposure group. AP sites in the lung of control rats were similar to those in the liver of control rats. AP sites in the lung of exposed groups were 1.6 and 2.3 fold those of lung controls. Boturyn et al. have exposed rats to a single dose of NNK (150 mg/kg bw) for 16 h and found a 3-fold increase of AP sites (relative to controls) after heating of the modified DNA.11
Figure 1.
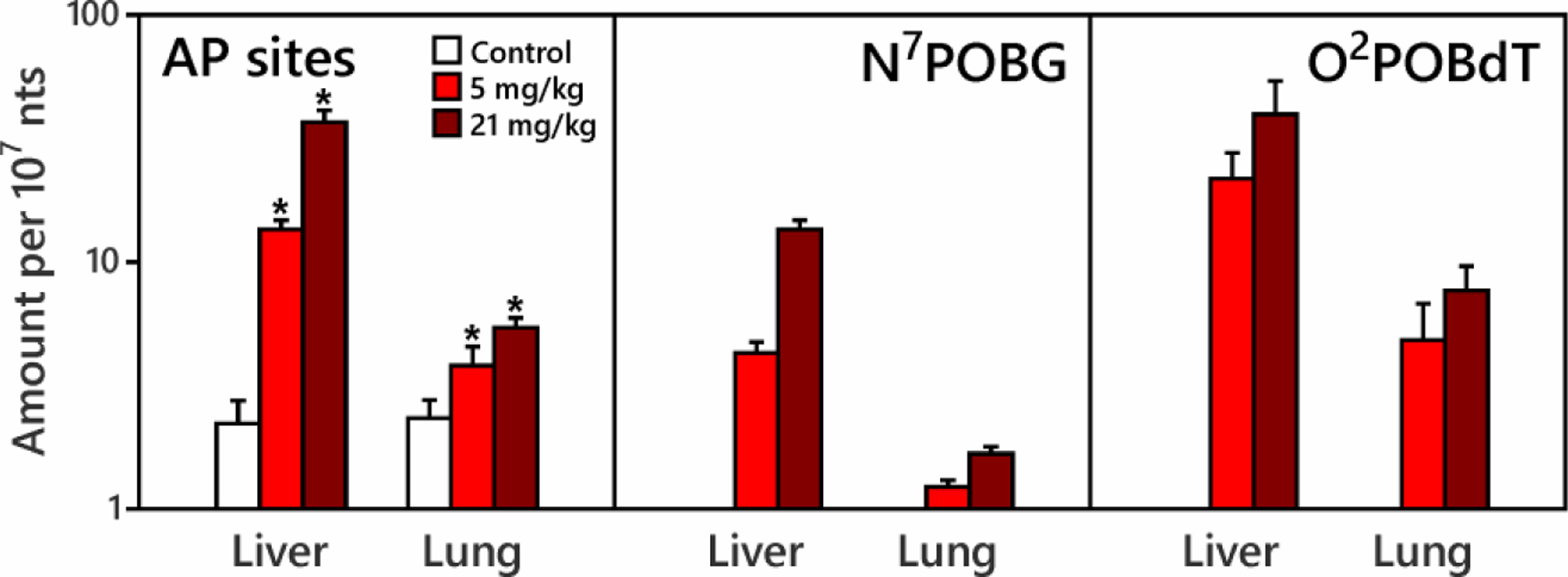
AP sites, N7POBG and O2POBdT in lung and liver of control rats and rats treated with 5.2 mg/kg or 20.7 mg/kg NNK for 4 days by subcutaneous injection (total doses 20.8 and 82.8 mg/kg). Rats were sacrificed four h after the final dose. Increases in AP sites were observed in both exposure groups in liver and lung (*indicates exposure is significantly higher than control at 95% confidence interval). N7POBG and O2POBdT were not detected in the control tissues.
O2POBdT levels in liver or lung DNA were 2 to 4 times higher than those of N7POBG at each dose. Amounts of O2POBdT and N7POBG in liver were 3 to 7 times higher than those in lung, and were consistent with those measured previously25 as shown in SI Table S5a. N7POBG and O2POBdT in both liver and lung were significantly correlated with increased AP site levels (p<0.05) (SI Figure S3). AP site levels were 2–4 times those of N7POBG and 0.5 to 1.5 times those of O2POBdT.
Given the above results, we conducted an in vitro experiment by exposing rat liver and lung homogenates to different doses of NNKOAc, a chemically activated form of NNK. Significant increases in AP sites were observed in liver and lung incubated with 49 μM and 243 μM of NNKOAc (p<0.05) (Figure 2). N7POBG and O2POBdT levels increased with increasing amounts of NNKOAc. The levels of both adducts were significantly correlated with the levels of AP sites (r>0.98, p<0.001).
Figure 2.
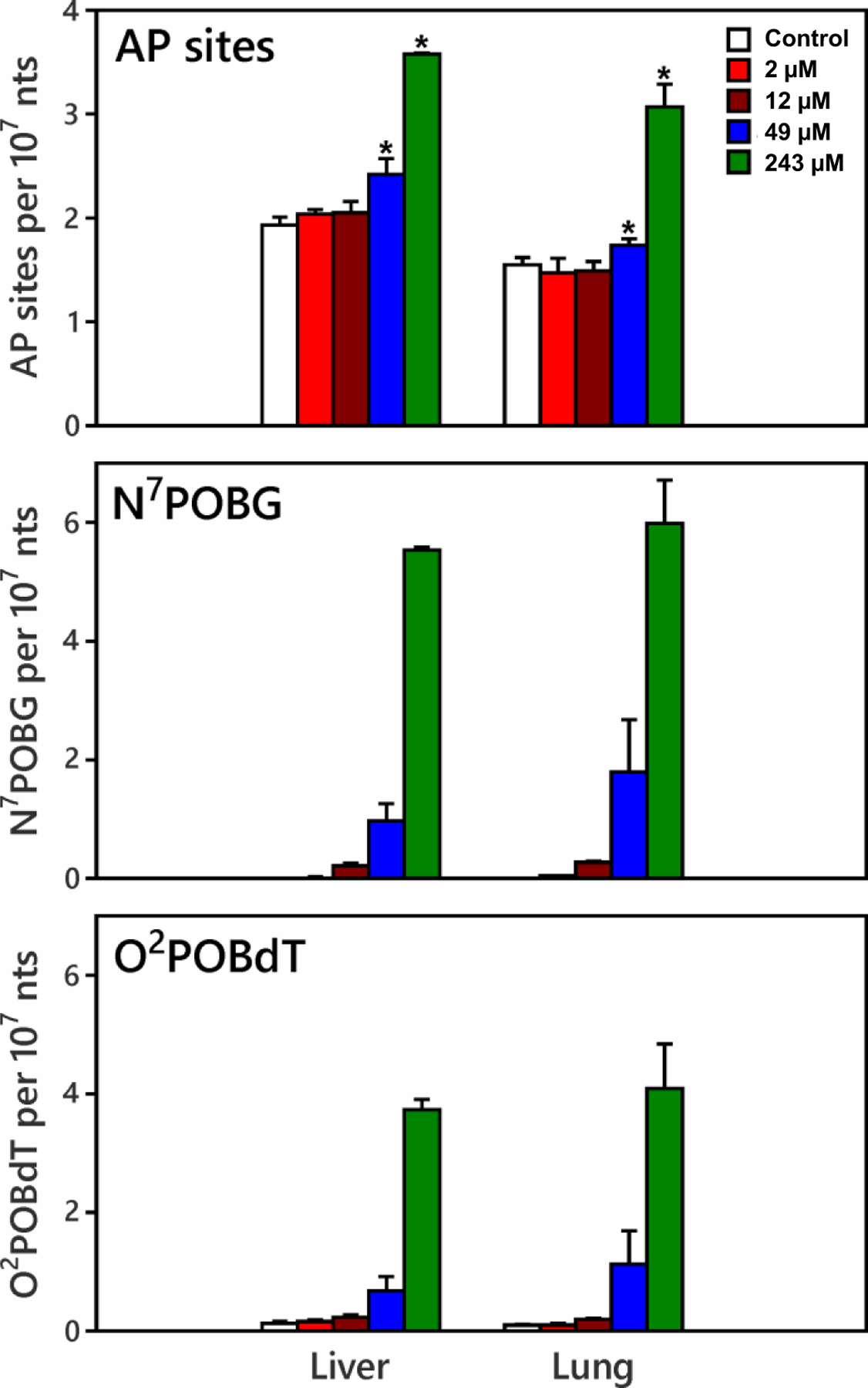
Dependence of AP site, N7POBG and O2POBdT formation on NNKOAc concentration. Liver or lung homogenates (~100 mg) were incubated with 0 (control), 2 12, 49, and 243 μM NNKOAc for 1.5 h. Esterase was added to catalyze the release of NNK-OH from NNKOAc. Each concentration level was performed in triplicate. Relative to controls, significant increases in AP sites were observed in liver and lung incubated with 49 μM and 243 μM of NNKOAc (p<0.05).
AP site formation from chronic exposure to NNK
Based on the results of AP site formation in rats exposed for a short time to high doses of NNK, we further investigated AP site formation in rats exposed chronically to a low dose of NNK. Rats were treated with 5 ppm NNK in their drinking water for 10, 30, 50 or 70 weeks (total doses of approximately 19, 44, 68 and 97 mg/kg NNK, respectively26). Increases in AP sites (compared to controls) were found in the liver DNA of rats exposed for 10 and 30 weeks as shown in Figure 3 (p<0.05). The differences between levels in the exposed and control rats were 0.45 AP sites/107 nts at 10 weeks, and 0.38 AP sites/107 nts at 30 weeks; this difference was more significant at 10 weeks (p=0.0003) than at 30 weeks (p=0.028). AP site levels returned to control levels at the 50 and 70 week time points (<0.10 AP sites/107 nts between exposed and control rats).
Figure 3.
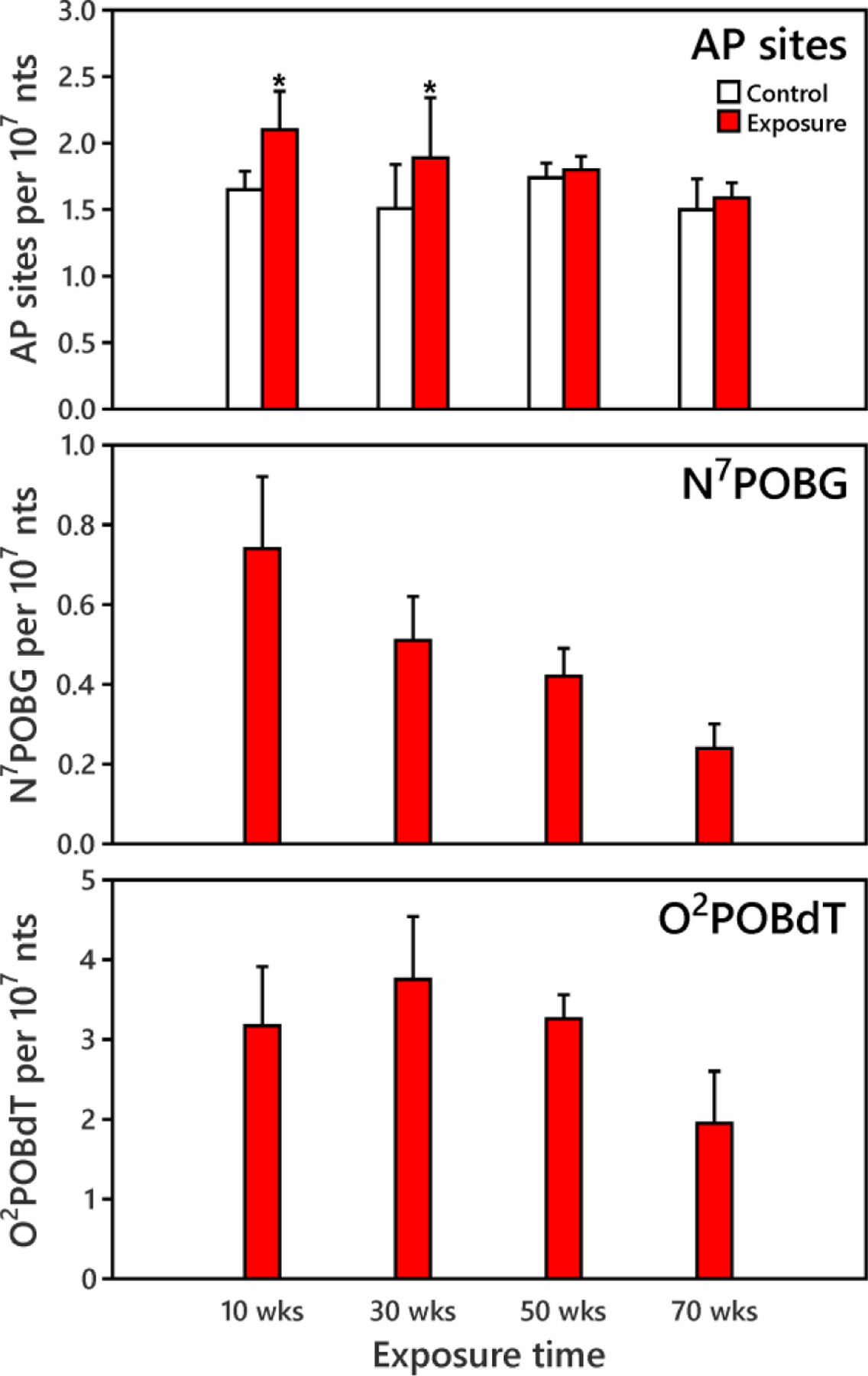
Dependence of AP site, N7POBG and O2POBdT formation on exposure time in rat liver. Rats were treated with 5 ppm NNK in their drinking water for 10, 30, 50 or 70 weeks (total doses of 19, 44, 68 and 97 mg/kg NNK, respectively26). Increases in AP sites were found in liver of rats exposed for 10 and 30 weeks (*indicates exposure is significantly higher than control at 95% confidence interval). AP sites and N7POBG amounts decreased with increased exposure time. N7POBG and O2POBdT were not detected in the control tissues.
N7POBG and O2POBdT were detected in liver DNA of the exposed rats at each time point (Figure 3). N7POBG levels were 0.74 ± 0.18 per 107 nts at 10 weeks and decreased to one third of that value at 70 weeks; while O2POBdT increased 20% from 10 weeks to 30 weeks, then decreased. These trends are similar to those reported previously in lung DNA from the same group of rats,26 and to those in rats exposed to 10 ppm NNK in the drinking water for 20 weeks,31 as shown in SI Table S5b. Levels of both adducts were significantly correlated with increased AP sites (p<0.05) and the correlation was slightly stronger for N7POBG (r=0.45, p=0.006) than for O2POBdT (r=0.35, p=0.035) as shown in SI Figure S4. AP site levels were 2–3 times those of N7POBG and 0.5 to 1 times those of O2POBdT.
AP site formation in different tissues of NNN exposed rats
The predominant form of NNN in tobacco is (S)-NNN.32 We treated rats with 14 ppm (S)-NNN in their drinking water for 10 weeks and determined AP site formation in DNA of 6 different tissues (Figure 4). Data from 4 control rats and 5 rats exposed to NNN showed that the variation of AP sites among tissues (41 ± 12%) was larger than among rats (16 ± 10%). In the six tissues measured among control rats, lowest levels of AP sites were formed in lung DNA (1.25 ± 0.41 per 107 nts) while the highest were in the oral mucosa (3.12 ± 0.46 per 107 nts). Increased AP site formation was observed in NNN exposed oral and nasal respiratory mucosa (p<0.02), which had 1.24 and 0.42 per 107 nts more AP sites than controls. A marginally significant (in Student’s t test) or significant (in Mann-Whitney U test) increase of AP sites was found in DNA from NNN exposed esophageal mucosa (p=0.08 for Student’s t test, p<0.05 for Mann-Whitney U test), which had 1.11 per 107 nts more AP sites than controls. There were no significantly increased AP sites found in DNA of NNN exposed nasal olfactory mucosa, lung or liver compared to controls (p>0.1).
Figure 4.
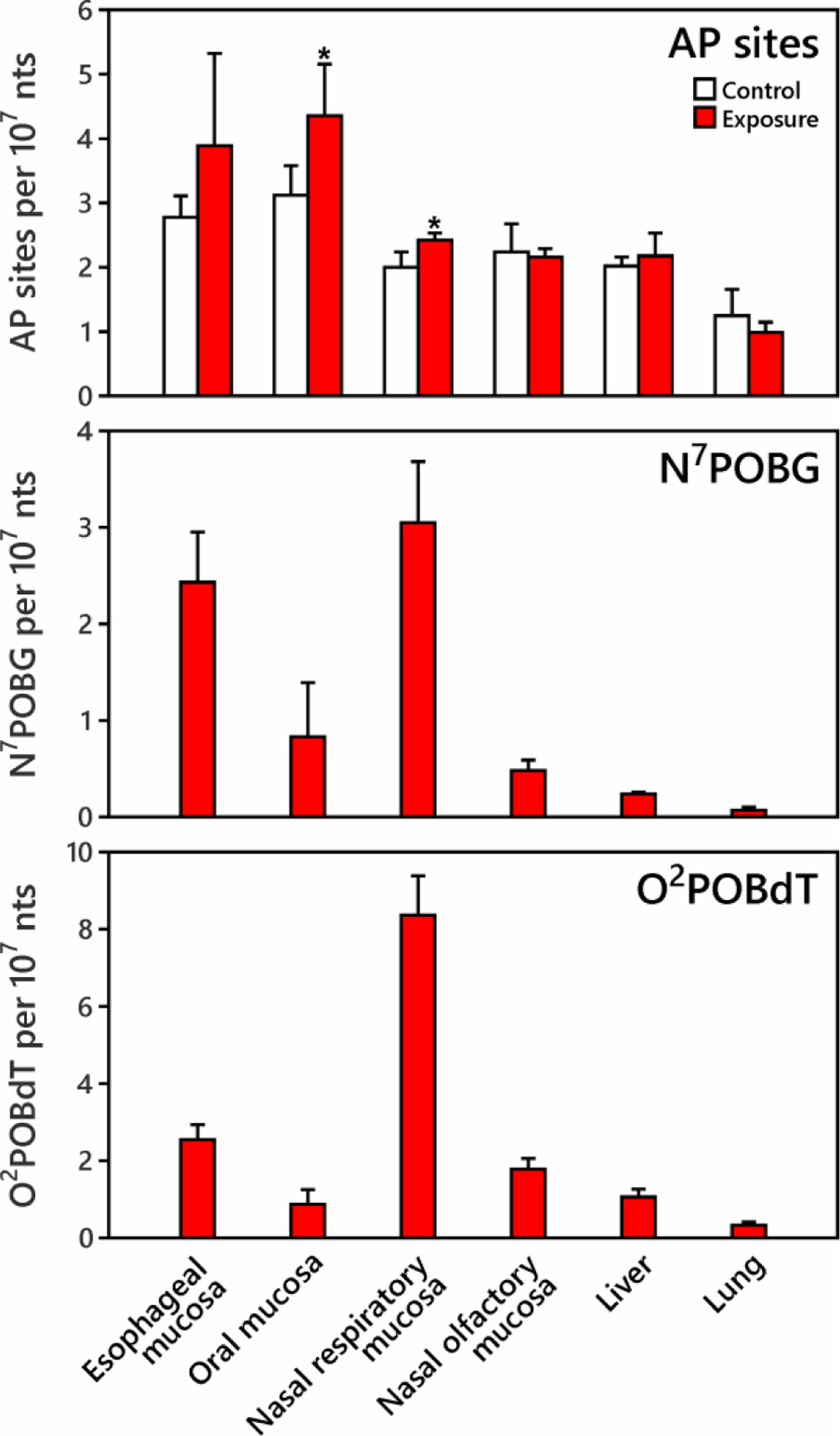
AP sites, N7POBG and O2POBdT formation in tissues of rats treated with 14 ppm (S)-NNN in drinking water for 10 weeks (n=4 for control, n=5 for exposure). Increased AP site formation was observed in oral and nasal respiratory mucosa (*indicates exposure is significantly higher than control at 95% confidence interval). N7POBG and O2POBdT were not detected in the control tissues.9
The highest levels of N7POBG and O2POBdT were found in NNN exposed nasal respiratory and esophageal mucosa, followed by oral and nasal olfactory mucosa. O2POBdT levels were significantly higher than those of N7POBG in most exposed tissues except in esophageal and oral mucosa, where levels of N7POBG and O2POBdT were similar. These results are similar to those of previous studies of DNA adduct formation in NNN exposed rats9,33 as shown in SI Table S6. Unlike in the NNK-treated rats, the levels of N7POBG and O2POBdT did not correlate with the levels of AP sites in individual or in combined NNN exposed tissues (p>0.05).
AP site formation in human samples
The increased AP sites observed in some tissues of NNK and NNN exposed rats encouraged us to examine the difference in AP site formation between cigarette smokers and non-smokers. To determine whether our AP site measurements in human tissues would be consistent with those obtained in rat tissues, we measured AP sites in lung tissue from untreated rats along with the human samples. The rat lung AP sites ranged from 1.43 ± 0.09 to 1.95 ± 0.19 per 107 nts (Table S3), which were consistent with our measurements in the rat studies described above.
Human lung tissue samples were obtained at surgery from 37 subjects, who gave urine samples just before the surgery. We divided these samples into smokers and non-smokers based on urinary cotinine and NNAL measurements (SI Table S1). All self-reported current smokers, and those self-reported previous smokers using nicotine replacement but with NNAL detected in urine were assigned as smokers; those who had cotinine levels <8 ng/mL and who had quit smoking for >10 years were assigned as non-smokers. All subjects had AP sites detected. The average (± SD) AP sites in smokers were 2.88 ± 1.39 per 107 nts, not significantly different from non-smokers (2.65 ± 1.29 AP sites/107 nts) as shown in SI Figure S5 (p=0.303). This level is one order of magnitude higher than Acr-dG (sum of α-Acr-dG and γ-Acr-dG, DNA adducts formed by acrolein)34 and total methyl DNA phosphate adducts,35 and four orders of magnitude higher than BPDE-N2-dG (a DNA adduct formed by benzo[a]pyrene)36 determined in human lung tissue in our earlier studies (Figure 5 and SI Table S7). AP sites in lung did not correlate with the cotinine or NNAL levels in smokers (p>0.05).
Figure 5.
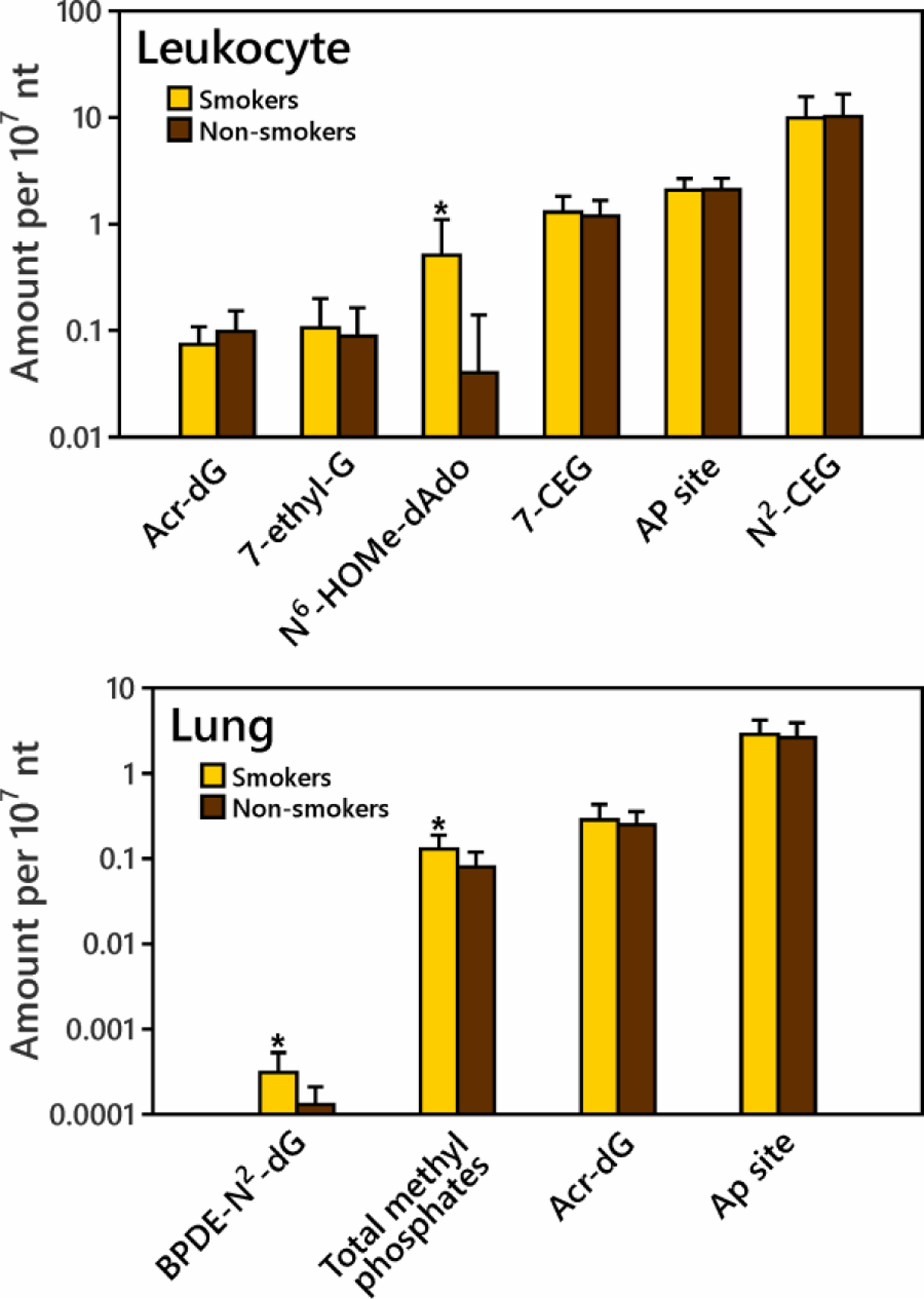
Comparison of average DNA adduct levels and AP site levels in leukocyte and lung DNA of smokers and non-smokers. Detailed information and abbreviations are presented in SI Table S7. The DNA adduct and AP site data are arranged from low to high in both leuckocytes and lung. For N6-HOMe-dAdo in leukocyte DNA and BPDE-N2-dG and total methyl phosphates in lung DNA, smokers had significantly higher amounts than non-smokers (indicated by *).
Next, AP sites were analyzed in leukocyte DNA from 31 self-reported current smokers and 31 non-smokers. Smoking status was confirmed by carbon monoxide measurement (> 5 ppm). AP sites were detected in DNA of all subjects (SI Table S2). The AP sites in smokers and non-smokers were 2.07 ± 0.60 per 107 nts and 2.10 ± 0.59, respectively (p=0.414, SI Figure S5). AP site levels were in the same order of magnitude as N2-(1’-carboxyethyl)guanine (N2-CEG) and 7-(2’-carboxyethyl)guanine (7-CEG),37 and higher than N6-hydroxymethyldeoxyadenosine (N6-HOMe-dAdo),29 7-ethyl-G,38 and Acr-dG39 determined in leukocyte DNA in our previous studies, as shown in Figure 5 and SI Table S7. In this study, we measured urinary NNAL in most smokers (0.17 to 2.47 pmol/mL urine, SI Table S2), which confirmed the NNK exposure in these smokers. AP sites in smokers were significantly correlated with their urinary NNAL levels (Figure 6B, p=0.005). We also divided the smokers and non-smokers based on gender, race and age. No significant differences were found between gender or race subgroups (p>0.1, SI Table S8). Increased AP sites were found in the older age group (age 31+) compared to the younger age group (age 20–30) (SI Table S8). AP sites in non-smokers, but not in smokers, significantly correlated with age (Figure 6A, p=0.037). A previous study also found significantly higher AP sites in leukocytes of older donors (age 58–75) than in leukocytes of younger donors (age 21–33).2
Figure 6.
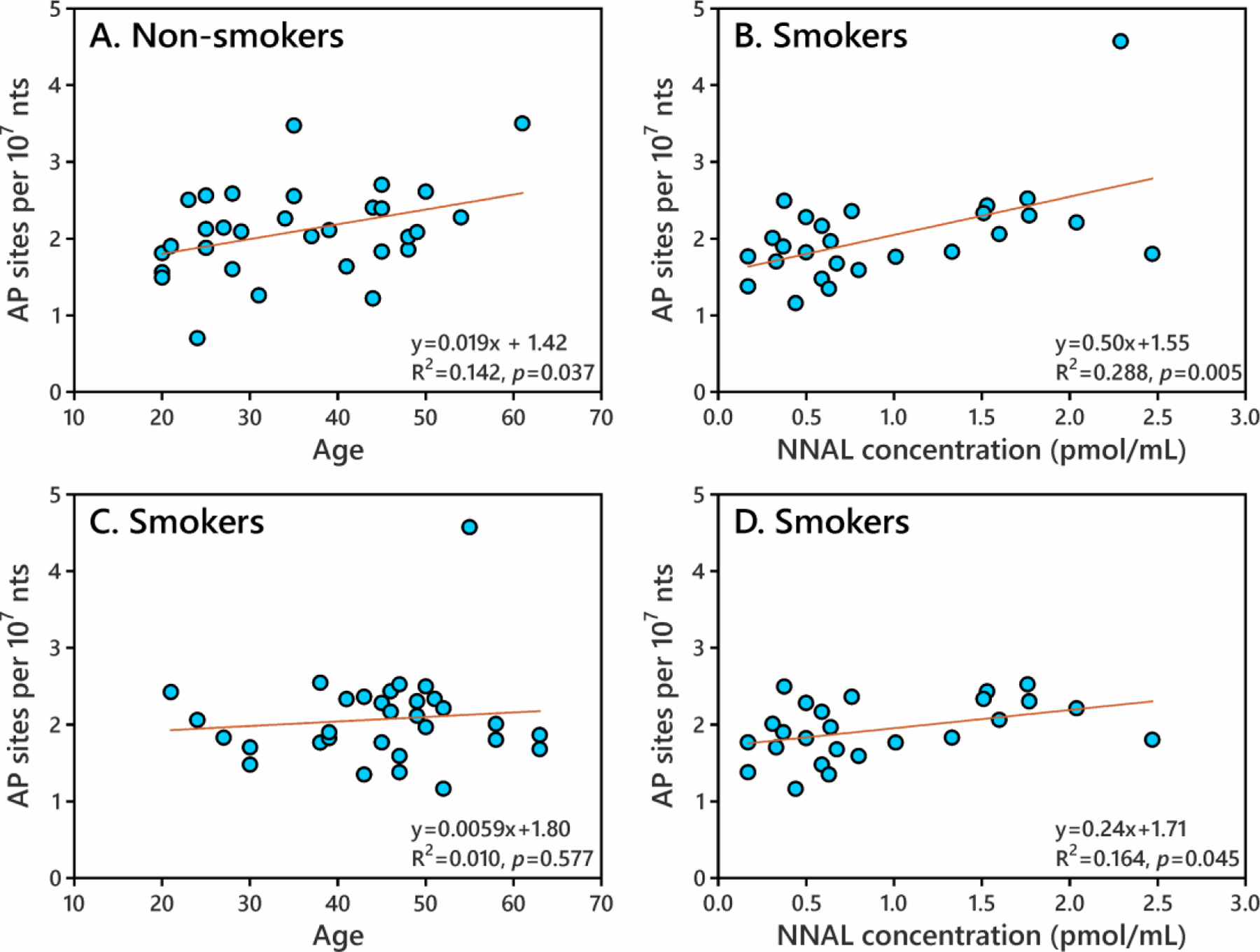
The dependence of human leukocyte AP site formation on subject age (A, C) or urinary NNAL concentration in smokers (B, D). AP sites in non-smokers significantly increased with age (p=0.037, A). AP sites in smokers significantly increased with urinary NNAL concentration with all data (p=0.005, B) or with data excluding an outlier (NNAL 2.29 pmol/mL, AP site 4.57 per 107 nts) (p=0.045, D).
DISCUSSION
This is the first study to use a highly sensitive and validated mass spectrometric method22 to quantify AP sites in the DNA of smokers and non-smokers and to relate this to the effects of the tobacco-specific nitrosamines NNK and NNN as observed in rats. The ability of NNK and NNN to induce AP sites in rats was clearly demonstrated. We also observed a significant relationship between levels of the NNK metabolite NNAL in the urine of cigarette smokers and AP sites in their leukocyte DNA. Since the dose of NNK in smokers is far less than in our rats, the correlation of NNAL with AP sites probably results from NNAL acting as a biomarker for multiple DNA damaging constituents of tobacco smoke. However, we did not observe a difference in AP sites between smokers and non-smokers, possibly because the study was relatively small and humans vary widely in their uptake and metabolism of tobacco smoke carcinogens.
Obvious dose-response relationships between NNK/NNKOAc exposure and AP site formation (Figures 1 and 2) and significant correlations between AP sites and POB-DNA adducts were found in rat lung and liver DNA upon exposure to NNK (SI Figure S3 and S4). NNKOAc formed POB adducts and AP sites, but not methyl-DNA adducts (SI Scheme S2). These results imply the formation of AP sites from the excision of POB-DNA adducts, most likely from the major adduct N7POBdG quantified here, although we cannot exclude the contribution of 7-methyl-dG from NNK which can depurinate but was not quantified (Scheme 1). The correlation between AP sites and O2POBdT may be attributed to the correlation between N7POBG and O2POBdT (SI Figure S4), both of which were formed from α-hydroxylation of NNK.
AP sites are one of the most frequent lesions in DNA, and can be formed spontaneously or by the removal of damaged or inappropriate bases by DNA N-glycosylases.1 AP sites in the control tissues of rats probably came from spontaneous base hydrolysis while in the exposed tissues they were from both spontaneous base hydrolysis and aberrant base incision by N-glycosylases. AP sites serve as key intermediates in the excision of damaged DNA bases and restoration of normal DNA structure.40 BER is an important pathway for the repair of nitrosamine derived DNA damage, and POB adducts serve as substrates for BER glycosylases.41 The POB adducts measured in this study are aberrant bases attached to DNA before enzyme hydrolysis during the experimental procedures. The resulting AP sites from excision and the POB adducts may coexist in a steady state. In the short time NNK exposed rat tissues in this study (Figure 1 and Figure 3 at 10 weeks), there were consistent ratios of ~3:1 between AP sites and N7POBG and ~1:1 between AP sites and O2POBdT. Other adducts, e.g. 7-methyl-dG and O2-POB-deoxycytidine (O2POBdC), may also contribute to the AP sites in the NNK exposed rats.11
In the NNN exposed rat tissues, there was no significant correlation between AP sites and N7POBG or O2POBdT. The sample size in this part of our study was relatively small (n=5 for each tissue), which might have masked a significant relationship. It is also possible that AP sites and N7POBG or O2POBdT hadn’t reached a steady state in the NNN exposed tissues after 10 weeks of treatment, so that a higher NNN dose or longer exposure time may be required. The formation of other NNN-DNA adducts, such as O2POBdC, or those formed by 5′-hydroxylation, may play a more significant role than N7POBG and O2POBdT in the production of AP sites. NNN-DNA adducts from 5′-hydroxylation may surpass those from 2′-hydroxylation,42 but were not quantified here.
N7POBG and O2POBdT were not detected in any human lung or leukocyte DNA samples in this study. However, 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB) releasing DNA adducts (indicator of POB DNA adduct formation) have been reported in the lung and other tissues of smokers and non-smokers, and they were significantly higher in smokers than in non-smokers.43–45 Even though POB adducts were not detected, urinary NNAL was measured and it correlated with AP sites in smokers’ leukocyte DNA (Figure 6B). Even after removing an outlier (NNAL 2.29 pmol/mL and AP site 4.57 per 107 nts), a significant relationship was still observed (p=0.045, Figure 6D). Previous prospective molecular epidemiology studies have found significant associations between urinary NNAL and increased lung cancer risk,46 implying a possible link between AP sites and lung cancer. In cell lines, NNK exposure induced AP sites and increased the activity of c-Myc, a proto-oncogene ubiquitously expressed in both small cell and non-small cell lung cancer cells.47
It is of interest to compare levels of various DNA adducts in smokers and non-smokers to those of the AP sites quantified here (Figure 5 and Table S7). Among DNA adducts quantified by mass spectrometry in our studies, BPDE-N2-dG in lung DNA and N6-HOMe-dAdo in leukocyte DNA, resulting from exposures to benzo[a]pyrene and formaldehyde, respectively, were higher in smokers than in non-smokers,29,36 but their levels were much lower than the AP sites (Figure S5) and are thus unlikely to contribute to the AP sites measured here. Total methyl DNA phosphate adducts from exposure to methylating carcinogens were also higher in smokers than in non-smokers.35 The methyl DNA phosphate adducts are methylated on phosphates rather than bases and are unlikely to contribute to AP site formation. Acr-dG adducts from acrolein exposure have been quantified in both human lung and leukocyte DNA in our previous studies,34,39 and were also considerably lower in both sources of DNA than AP sites. Acr-dG levels in lung were 2–3 times higher than those in leukocytes; AP sites were also significantly higher in lung than in leukocytes (p=0.0003, Figure S5)). N2-CEG and 7-CEG in leukocytes resulting from reactions of DNA with methyl glyoxal and an unknown carboxyethylating agent, respectively,37,48 were present in comparable levels to the AP sites in our study (Figure 5). There was no correlation of N2-CEG or 7-CEG with AP sites in matched leukocytes (n=29) (data not shown). Levels of 7-ethyl-G, a possible degradation product of 7-CEG,38 were one order of magnitude lower than those of AP sites.
Aging may play some role in affecting AP sites and DNA adduct formation. AP site formation decreased with time in the livers of rats exposed to NNK, and this was also the case for POB-DNA adducts (Figure 3). Similar decreases in adduct formation were seen in lung DNA of these NNK-treated rats.26 Significant decreases of keto aldehyde formation resulting from α-methylene hydroxylation of NNK (compared with controls) with time have also been observed in rats receiving NNK for 12 and 20 weeks.49 Thus, chronic NNK treatment may inhibit its metabolic activation, possibly due to inflammation, leading to lower levels of DNA adduct formation and consequent lower AP sites. The older rats — 50 and 70 weeks — also may have had lower N-glycosylase activity resulting in a lower extents of POB-DNA adduct excision and AP site formation. A previous study found that AP sites in senescent cells are less likely to be induced than in young cells by oxidizing and alkylating agents because N-glycosylases decline in activity with age.2
In human leukocytes of this study, AP sites significantly increased with subject age (p<0.05, Figure 6A) in non-smokers, which may be due to an increase in endogenous oxidative insults and a decrease in AP site repair (decline of AP endonuclease or β-pol, which are downstream of N-glycosylase) with age.2 This trend was not found in smokers because they clustered at older age; the age of our smokers (45 ± 10 years) was significantly higher than that of the non-smokers (35 ± 11 years). It seems that aging in non-smokers and NNK exposure (NNAL) in smokers decrease the AP site difference between non-smokers and smokers. The failure to see AP site differences in lung DNA between smokers and non-smokers may be due in part to the fact that the subjects in both groups were older (mean ages 61 ± 8 vs. 66 ± 9), and the aging influence may be more significant than that of smoke components. A previous study found that older age group (age 65) abolished the adaptive chromosomal repair mechanism in response to increased X-ray dose in humans.50
AP sites together with DNA adducts may relate to tumor formation in target tissues of NNK. We found increased AP sites in both liver and lung of NNK exposed rats (Figure 1). NNK is a powerful pulmonary carcinogen51 but it can also induce hepatic tumors when given by injection at total doses comparable to those used here.25,52 NNK acute exposure in our study induced more AP sites (3 to 6 times higher) and N7POBG and O2POBdT in the liver than in the lung. In a previous study, NNK exposure produced more N7-methylguanine (7-mGua) and O6-methylguanine (O6-mGua),52 and more 7-mGua and POB adducts at high doses (150–5000 μg/kg/day),53 in liver than in lung. However, at lower NNK doses (<150–300 μg/kg/day), more 7-mGua and POB-DNA adducts were produced in lung than in liver.53 NNK chronic treatment (5 ppm) induced more N7POBG and O2POBdT at 10–70 weeks (SI Table S5b), more POB adducts at 1–20 weeks, and more O6-mGua at 5–20 weeks,54 in lung than in liver. We don’t have AP site data in the lung DNA of the rats treated chronically with 5 ppm NNK, but it seems that AP site formation varies in the same way as DNA adduct levels in lung and liver, and thus likely relates to metabolic activation of NNK in specific tissues. The higher concentration of DNA adducts and AP sites in lung than in liver at low NNK exposure is due to the presence of high affinity P450 isozymes in lung but not in liver.53,55 The lower concentration of these adducts in lung than in liver at high NNK doses may be attributed to the saturation of P450 isozymes in the lung. In the in vitro experiment, we didn’t see tissue differences of AP site and POB adduct formation (Figure 2) because NNKOAc does not need to be activated by P450 enzymes.
Increased AP sites were also formed in target tissues of NNN. In (S)-NNN treated rats, significant increases in AP sites were found in the oral and nasal respiratory mucosa, and marginally significantly (or significantly based on Mann-Whitney U test) increased AP sites were found in the esophageal mucosa (Figure 4). There were some technical difficulties in isolating esophageal mucosa, which may have weakened the AP site differences in esophagus between exposed and control rats. Higher levels of N7POBG and O2POBdT were found in oral, nasal and esophageal tissues than in the other tissues. In our previous studies, all rats treated with 14 ppm (S)-NNN had oral cavity tumors and esophageal tumors.9,56 Six of 20 rats treated with (S)-NNN were observed with adenomas in the nasal respiratory epithelium.9 Increased AP site formation may relate to these tumors.
CONCLUSIONS
We measured AP sites in living cells using a mass spectrometric approach, which provided better sensitivity and specificity than previous methods. The acute s.c. injection and in vitro NNKOAc exposure studies demonstrated a dose-response relationship between NNK exposure and AP site formation in rat liver and lung. In the chronic administration study, modest but significant time dependent AP site formation was observed in the liver of rats treated with NNK. Target tissues of NNN, nasal and oral mucosa, had increased AP sites in the (S)-NNN exposed rats. Thus AP sites could play some role in NNK and NNN carcinogenicity. The relatively small sample size may have limited our ability to observe a difference in AP site formation in the lung and leukocytes of cigarette smokers and non-smokers, but we did observe a correlation between urinary NNAL and AP site formation in leukocytes of smokers. This study creates a new dimension in our understanding of the possible mechanisms by which tobacco-specific nitrosamines induce cancer. Further research with larger sample sizes comparing AP site formation in people who use tobacco products to non-users is needed.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the U.S. National Cancer Institute [CA-081301 to S.S.H., P01 CA-160032 to R.J.T]. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, supported in part by Cancer Center Support Grant CA-077598. We thank Sarah Reisinger and Professors Peter G. Shields and Dorothy Hatsukami for providing leukocyte samples from smokers and non-smokers participating in an observational study supported in part by the U.S. National Cancer Institute [N01 PC-064402]. We thank Laura Maertens for providing leukocyte samples from the Tobacco Research Programs Repository, Bob Carlson for editorial support, Yupeng Li for teaching how to isolate the rat tissues, and Steven Carmella and Guang Cheng for help obtaining the human samples. We also thank Nicole Thomson from the Sharon E. Murphy laboratory in the Masonic Cancer Center for measurements of urinary cotinine and NNAL.
Footnotes
SUPPORTING INFORMATION
Supporting Information is available at CRT online.
REFERENCES
- 1.Boiteux S, and Guillet M (2004) Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 3, 1–12. [DOI] [PubMed] [Google Scholar]
- 2.Atamna H, Cheung I, and Ames BN (2000) A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proc. Natl. Acad. Sci. U.S.A 97, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahlmann HA, Vaidyanathan VG, and Sturla SJ (2009) Investigating the biochemical impact of DNA damage with structure-based probes: abasic sites, photodimers, alkylation adducts, and oxidative lesions. Biochemistry 48, 9347–9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakamura J, and Swenberg JA (1999) Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 59, 2522–2526. [PubMed] [Google Scholar]
- 5.Boysen G, Pachkowski BF, Nakamura J, and Swenberg JA (2009) The formation and biological significance of N7-guanine adducts. Mutat. Res 678, 76–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hecht SS (2014) It Is time to regulate carcinogenic tobacco-specific nitrosamines in cigarette tobacco. Cancer Prev. Res 7, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lao Y, Villalta PW, Sturla SJ, Wang M, and Hecht SS (2006) Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol 19, 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Wang M, Villalta PW, Lindgren BR, Lao Y, and Hecht SS (2009) Quantitation of pyridyloxobutyl DNA adducts in nasal and oral mucosa of rats treated chronically with enantiomers of N’-nitrosonornicotine. Chem. Res. Toxicol 22, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao L, Balbo S, Wang M, Upadhyaya P, Khariwala SS, Villalta PW, and Hecht SS (2013) Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)- or (S)-N’-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol 26, 1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hecht SS (1998) Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol 11, 559–603. [DOI] [PubMed] [Google Scholar]
- 11.Boturyn D, Constant J-F, Defrancq E, Lhomme J, Barbin A, and Wild CP (1999) A simple and sensitive method for in vitro quantitation of abasic sites in DNA. Chem. Res. Toxicol 12, 476–482. [DOI] [PubMed] [Google Scholar]
- 12.Zhou X, Liberman RG, Skipper PL, Margolin Y, Tannenbaum SR, and Dedon PC (2005) Quantification of DNA strand breaks and abasic sites by oxime derivatization and accelerator mass spectrometry: application to gamma-radiation and peroxynitrite. Anal. Biochem 343, 84–92. [DOI] [PubMed] [Google Scholar]
- 13.Talpaert-Borle M, and Liuzzi M (1983) Reaction of apurinic/apyrimidinic sites with [14C]methoxyamine. A method for the quantitative assay of AP sites in DNA. Biochim. Biophys. Acta 740, 410–416. [DOI] [PubMed] [Google Scholar]
- 14.Weinfeld M, Liuzzi M, and Paterson MC (1990) Response of phage T4 polynucleotide kinase toward dinucleotides containing apurinic sites: design of a 32P-postlabeling assay for apurinic sites in DNA. Biochemistry 29, 1737–1743. [DOI] [PubMed] [Google Scholar]
- 15.Chen B-X, Kubo K, Ide H, Erlanger BF, Wallace SS, and Kow YW (1992) Properties of a monoclonal antibody for the detection of abasic sites, a common DNA lesion. Mutation Research/DNA Repair 273, 253–261. [DOI] [PubMed] [Google Scholar]
- 16.Kow YW, and Dare A (2000) Detection of abasic sites and oxidative DNA base damage using an ELISA-like assay. Methods 22, 164–169. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura J, Walker VE, Upton PB, Chiang S-Y, Kow YW, and Swenberg JA (1998) Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 58, 222–225. [PubMed] [Google Scholar]
- 18.Li J, Leung EM, Choi MM, and Chan W (2013) Combination of pentafluorophenylhydrazine derivatization and isotope dilution LC-MS/MS techniques for the quantification of apurinic/apyrimidinic sites in cellular DNA. Anal Bioanal Chem 405, 4059–4066. [DOI] [PubMed] [Google Scholar]
- 19.Roberts KP, Sobrino JA, Payton J, Mason LB, and Turesky RJ (2006) Determination of apurinic/apyrimidinic lesions in DNA with high-performance liquid chromatography and tandem mass spectrometry. Chem. Res. Toxicol 19, 300–309. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Chan HW, and Chan W (2016) Facile formation of a DNA adduct of semicarbazide on reaction with apurinic/apyrimidinic sites in DNA. Chem. Res. Toxicol 29, 834–840. [DOI] [PubMed] [Google Scholar]
- 21.Rahimoff R, Kosmatchev O, Kirchner A, Pfaffeneder T, Spada F, Brantl V, Müller M, and Carell T (2017) 5-formyl- and 5-carboxydeoxycytidines do not cause accumulation of harmful repair intermediates in stem cells. J. Am. Chem. Soc 139, 10359–10364. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Yao L, Brown C, Rizzo CJ, and Turesky RJ (2019) Quantitation of apurinic/apyrimidinic sites in isolated DNA and in mammalian tissue with a reduced level of artifacts. Anal. Chem 91, 7403–7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sturla SJ, Scott J, Lao Y, Hecht SS, and Villalta PW (2005) Mass spectrometric analysis of relative levels of pyridyloxobutylation adducts formed in the reaction of DNA with a chemically activated form of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol 18, 1048–1055. [DOI] [PubMed] [Google Scholar]
- 24.Upadhyaya P, Sturla SJ, Tretyakova N, Ziegel R, Villalta PW, Wang M, and Hecht SS (2003) Identification of adducts produced by the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem. Res. Toxicol 16, 180–190. [DOI] [PubMed] [Google Scholar]
- 25.Wang M, Cheng G, Villalta PW, and Hecht SS (2007) Development of liquid chromatography electrospray ionization tandem mass spectrometry methods for analysis of DNA adducts of formaldehyde and their application to rats treated with N-nitrosodimethylamine or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol 20, 1141–1148. [DOI] [PubMed] [Google Scholar]
- 26.Balbo S, Johnson CS, Kovi RC, James-Yi SA, O’Sullivan MG, Wang M, Le CT, Khariwala SS, Upadhyaya P, and Hecht SS (2014) Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis 35, 2798–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Villalta PW, Wang M, and Hecht SS (2007) Detection and quantitation of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human lung by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol 20, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecht SS, Carmella SG, Chen M, Koch JFD, Miller AT, Murphy SE, Jensen JA, Zimmerman CL, and Hatsukami DK (1999) Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Res. 59, 590–596. [PubMed] [Google Scholar]
- 29.Wang M, Cheng G, Balbo S, Carmella SG, Villalta PW, and Hecht SS (2009) Clear differences in levels of a formaldehyde-DNA adduct in leukocytes of smokers and nonsmokers. Cancer Res. 69, 7170–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bansal M (2003) DNA structure: Revisiting the Watson–Crick. Curr. Sci 85, 557. [Google Scholar]
- 31.Lao Y, Yu N, Kassie F, Villalta PW, and Hecht SS (2007) Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol 20, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stepanov I, Yershova K, Carmella S, Upadhyaya P, and Hecht SS (2012) Levels of (S)-N’-Nitrosonornicotine in U.S. Tobacco Products. Nicotine Tob. Res 15, 1305–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Villalta PW, Upadhyaya P, and Hecht SS (2016) Analysis of O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2’-deoxyguanosine and other DNA adducts in rats treated with enantiomeric or racemic N’-nitrosonornicotine. Chem. Res. Toxicol 29, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J, Balbo S, Villalta PW, and Hecht SS (2019) Analysis of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human lung DNA from smokers and nonsmokers. Chem. Res. Toxicol 32, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma B, Villalta PW, Hochalter JB, Stepanov I, and Hecht SS (2019) Methyl DNA phosphate adduct formation in lung tumor tissue and adjacent normal tissue of lung cancer patients. Carcinogenesis 40, 1387–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villalta PW, Hochalter JB, and Hecht SS (2017) Ultrasensitive high-resolution mass spectrometric analysis of a DNA adduct of the carcinogen benzo[a]pyrene in human lung. Anal. Chem 89, 12735–12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng G, Reisinger SA, Shields PG, Hatsukami DK, Balbo S, and Hecht SS (2020) Quantitation by liquid chromatography-nanoelectrospray ionization-high resolution tandem mass spectrometry of DNA adducts derived from methyl glyoxal and carboxyethylating agents in leukocytes of smokers and non-smokers. Chem. Biol. Interact 327, 109140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balbo S, Villalta PW, and Hecht SS (2011) Quantitation of 7-ethylguanine in leukocyte DNA from smokers and nonsmokers by liquid chromatography–nanoelectrospray-high resolution tandem mass spectrometry. Chem. Res. Toxicol 24, 1729–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S, Balbo S, Wang M, and Hecht SS (2011) Analysis of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human leukocyte DNA from smokers and nonsmokers. Chem. Res. Toxicol 24, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu W, C. W, Li Y, Somoza E, Liu L,. (2012) In vivo quantification of abasic sites for efficacious evaluation of DNA-targeted chemotherapies. J. Cancer Sci. Ther S5, 1–7. [Google Scholar]
- 41.Peterson LA (2010) Formation, repair, and genotoxic properties of bulky DNA adducts formed from tobacco-specific nitrosamines. J Nucleic Acids 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zarth AT, Upadhyaya P, Yang J, and Hecht SS (2016) DNA adduct formation from metabolic 5’-hydroxylation of the tobacco-specific carcinogen N’-nitrosonornicotine in human enzyme systems and in rats. Chem. Res. Toxicol 29, 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foiles PG, Akerkar SA, Carmella SG, Kagan M, Stoner GD, Resau JH, and Hecht SS (1991) Mass spectrometric analysis of tobacco-specific nitrosamine-DNA adducts in smokers and nonsmokers. Chem. Res. Toxicol 4, 364–368. [DOI] [PubMed] [Google Scholar]
- 44.Hölzle D, Schlöbe D, Tricker AR, and Richter E (2007) Mass spectrometric analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in human lung. Toxicology 232, 277–285. [DOI] [PubMed] [Google Scholar]
- 45.Ma B, Ruszczak C, Jain V, Khariwala SS, Lindgren B, Hatsukami DK, and Stepanov I (2016) Optimized liquid chromatography nanoelectrospray–high-resolution tandem mass spectrometry method for the analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in human oral cells. Chem. Res. Toxicol 29, 1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan J-M, Butler LM, Stepanov I, and Hecht SS (2014) Urinary tobacco smoke-constituent biomarkers for assessing risk of lung cancer. Cancer Res. 74, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin Z, May WS, Gao F, Flagg T, and Deng X (2006) Bcl2 suppresses DNA repair by enhancing c-Myc transcriptional activity. J. Biol. Chem 281, 14446–14456. [DOI] [PubMed] [Google Scholar]
- 48.Cheng G, Wang M, Villalta PW, and Hecht SS (2010) Detection of 7-(2’-carboxyethyl)guanine but not 7-carboxymethylguanine in human liver DNA. Chem. Res. Toxicol 23, 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Staretz ME, Koenig LA, and Hecht SS (1997) Effects of long term dietary phenethyl isothiocyanate on the microsomal metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F344 rats. Carcinogenesis 18, 1715–1722. [DOI] [PubMed] [Google Scholar]
- 50.Gadhia RK. (1998) Possible age-dependent adaptive response to a low dose of X-rays in human lymphocytes. Mutagenesis 13, 151–152. [DOI] [PubMed] [Google Scholar]
- 51.Hoffmann D, Rivenson A, Amin S, and Hecht SS (1984) Dose-response study of the carcinogenicity of tobacco-specific N-nitrosamines in F344 rats. J. Cancer Res. Clin. Oncol 108, 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hecht SS, Trushin N, Castonguay A, and Rivenson A (1986) Comparative tumorigenicity and DNA methylation in F344 rats by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N-nitrosodimethylamine. Cancer Res. 46, 498–502. [PubMed] [Google Scholar]
- 53.Murphy SE, Palomino A, Hecht SS, and Hoffman D (1990) Dose-response study of DNA and hemoglobin adduct formation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 50, 5446–5452. [PubMed] [Google Scholar]
- 54.Upadhyaya P, Lindgren BR, and Hecht SS (2009) Comparative levels of O6-methylguanine, pyridyloxobutyl-, and pyridylhydroxybutyl-DNA adducts in lung and liver of rats treated chronically with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Drug Metab. Dispos 37, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belinsky SA, White CM, Devereux TR, Swenberg JA, and Anderson MW (1987) Cell selective alkylation of DNA in rat lung following low dose exposure to the tobacco specific carcinogen 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 47, 1143–1148. [PubMed] [Google Scholar]
- 56.Balbo S, James-Yi S, Johnson CS, O’Sullivan MG, Stepanov I, Wang M, Bandyopadhyay D, Kassie F, Carmella S, Upadhyaya P, and Hecht SS (2013) ( S )-N’-Nitrosonornicotine, a constituent of smokeless tobacco, is a powerful oral cavity carcinogen in rats. Carcinogenesis 34, 2178–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.