

ABSTRACT
It is well established that 5-methylcytosine (5mC) in genomic DNA of mammalian cells can be oxidized into 5-hydroxymethylcytosine (5hmC) and other derivates by DNA dioxygenase TETs. While conversion of 5mC to 5hmC plays an important role in active DNA demethylation through further oxidation steps, a certain proportion of 5hmCs remain in the genome. Although 5hmCs contribute to the flexibility of chromatin and protect bivalent promoters from hypermethylation, the direct effect of 5hmCs on gene transcription is unknown. In this present study, we have engineered a zinc-finger protein-based P16-specific DNA dioxygenase (P16-TET) to induce P16 hydroxymethylation and demethylation in cancer cells. Our results demonstrate, for the first time, that although the hydroxymethylated P16 alleles retain transcriptionally inactive, hydroxymethylation could increase the susceptibility of reactivation of methylated P16 alleles.
KEYWORDS: P16 gene, CpG islands, methylation, hydroxymethylation, epigenetic editing
Introduction
It is well known that ten-eleven translocation methylcytosine dioxygenases (TET1/2/3) oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) in the genome [1–4]. Oxidation of 5mC leads to active DNA demethylation, while a certain proportion of 5hmC sites remain in the genome and serves as a regulatory function [5–9]. It has been reported that the levels of 5hmC of some genes is positively correlated with increased gene expression [8], it is unclear whether 5hmC itself contributes to the reactivation of gene transcription.
Typical bisulphite-based assays cannot discriminate 5mCs from 5hmCs. The classic term ‘DNA methylation’ is in fact total DNA methylation, which includes true methylation and hydroxymethylation of genes. Total methylation of the CpG island (CGI) flanking the transcription start site (TSS) of the P16 gene (CDKN2A) has been shown to be prevalent in human cancer and precancerous tissues [10,11] and is linked to increased cancer development from epithelial dysplasia in many organs [12–18]. P16 methylation (P16M) not only directly inactivates P16 transcription [19] but also represses ANRIL transcription [20]. Our recent study demonstrates the presence of dense 5hmCs in the P16 exon-1 CGI in HCT116 cells, and no mRNA transcripts from the hydroxymethylated P16 (P16H) alleles were detected in the cells [21,22]. P16H was detected in 9.3% of human oral epithelial dysplasia (OED) tissues, and the malignant transformation risk was similar between P16M-positive OED patients with and without P16H [23]. It is a fundamental question in epigenetic research to clarify whether hydroxymethylation of TSS-CGIs affects transcriptional activation of genes including P16.
To elucidate the possible role of P16H, we constructed an epigenetic editing tool, P16-specific TET1 (P16-TET), and a transcription editing tool, P16-specific artificial transcription factor (P16-ATF) [24] to induce P16H and reactivate P16M in cancer cell lines. Our data showed, for the first time, that although stable P16H could not reactivate gene transcription, however, dense 5hmCs in TSS-CGIs could increase the susceptibility of reactivation of previously methylated P16 alleles.
Results
Induction of P16H by P16-TET
In order to study the effects of P16H on gene transcription, P16-specific dioxygenase P16-TET and its inactive mutant control vector were constructed by fusing an engineered P16 promoter-specific seven zinc finger protein (7ZFP-6I) [24] with the catalytic domain (CD) of human TET1 and integrated into the pcDNA3.1 expression vector (Figure 1a). H1299 cells were chosen because the epigenetic editing efficiency of the methylated P16 CGIs by the P16-specific transcription factor (P16-ATF; 7ZFP-6I-VP64) has been optimized [24]. As expected, qRT-PCR and immunofluorescence staining showed that the P16M alleles were reactivated in H1299 cells 6 days after transient transfection with the P16-TET vector (Figure 1b and c). P16 reactivation was not observed in the P16-TET mutant control cells. This confirmed that the genetic tool created P16-TET could reactive the P16 gene and be used in further studies.
Figure 1.
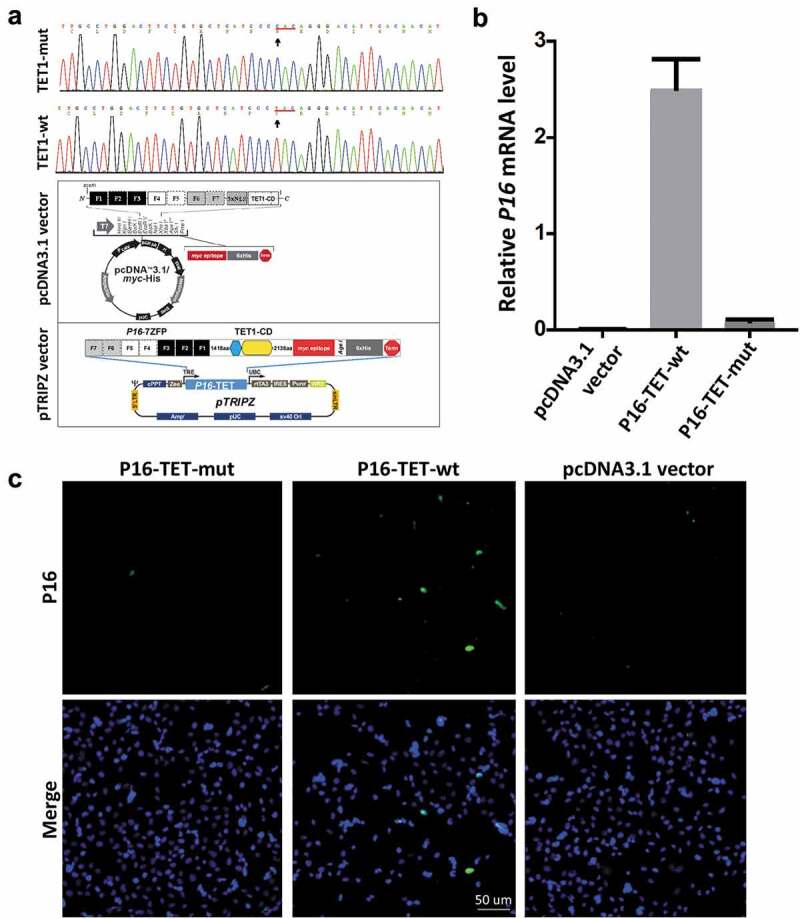
Reactivation of methylated P16 alleles in H1299 cells 6 days after P16-TET transient transfection. (a) Core sequences of the catalytic domain (CD) of the wildtype TET1 gene and its T > C H1671Y-mutant counterpart. The P16-TET pcDNA3.1 and pTRIPZ ‘Tet-on’ expression vectors are also illustrated and used in transient and stable transfection studies, respectively. (b) The results of qRT-PCR to detect the P16 mRNA levels in H1299 cells transiently transfected with the pcDNA3.1 empty control, P16-TET and its H1671Y mutant counterpart vectors. (c) Immunofluorescence staining with the mouse monoclonal antibody against the human P16 protein (Ventana Roche-E6H4, USA). These experiments were performed in triplicate and repeated at least one time. Bar: 50 μm.
To study the possible biological functions of P16-specific hydroxymethylation, the P16-TET coding sequence was cloned into the pTRIPZ lentivirus vector carrying a ‘Tet-on’ switch to inducibly control the gene expression of P16-TET (Figure 1a). In H1299 cells transfected with P16-TET, TET-Assisted Bisulphite (TAB) methylation-specific PCR (MSP) analysis showed P16H signals appeared 3 days after the induction of doxycycline (Dox; final conc. 0.25 μg/mL) (P16-TET&Dox_3d; Figure 2a, TAB-MSP), but did not appear in cells transfected with the empty vector (control cells with Dox treatment) (Vector&Dox_14d) or in P16-TET cells without Dox induction, in which only nonhydroxymethylated P16 alleles (P16N) were detected. In the MSP analysis, P16 unmethylated (or demethylated) alleles (P16U) were detectable in the P16-TET&Dox cells 3 days following Dox induction (Figure 2a, MSP). The bisulphite-denaturing high performance liquid chromatography (DHPLC) results showed a low P16U peak was detected beginning on the 14th day (Figure S1A, red-arrow). Two P16U clones were also observed on the 28th day from the bisulphite sequencing (Figure S1B, red-star). These results indicated that both P16H and P16U were induced in the P16-TET&Dox cells.
Figure 2.
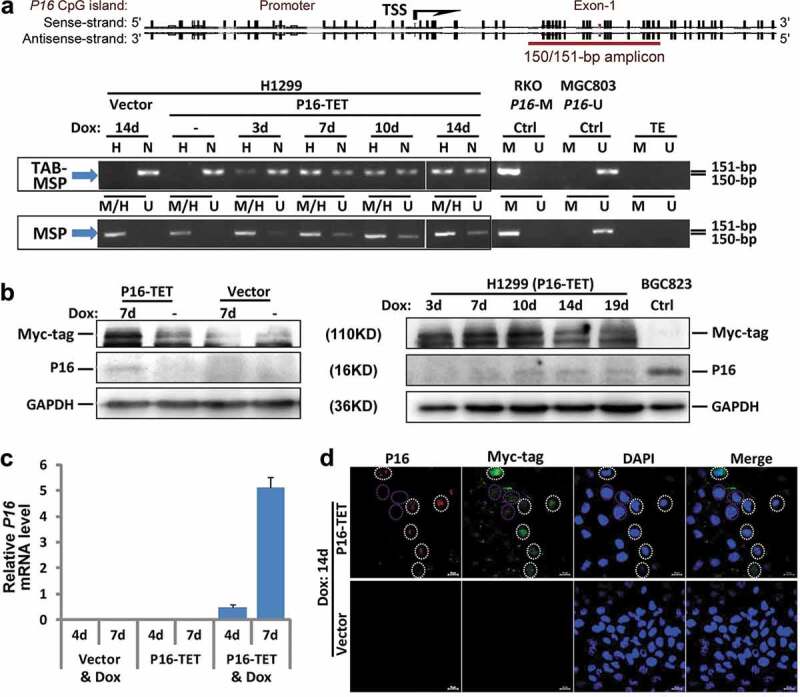
P16-TET induces hydroxymethylation of P16 CpG islands and reactivates expression of methylated P16 alleles in H1299 cells. (a) TAB-MSP analysis for detecting hydroxymethylated (H)- and nonhydroxymethylated (N)-P16 CpG alleles in H1299 cells stably transfected with P16-TET or empty control vector after doxycycline treatment. The MSP analysis results were also listed. Genomic DNA from RKO and BGC823 cells was used as P16M and P16U controls in the MSP assays, respectively. (b) Western blot analysis for detecting the P16 protein; Dox (±): with or without the doxycycline treatment (final conc. 0.25 μg/mL). Proteins from BGC823 cells were used as a P16U/active control. (c) qRT-PCR results for detecting P16 mRNA levels relative to Alu RNA levels; (d) Immunofluorescence confocal analysis for detecting P16 expression. P16-positive cells with P16-TET (Myc-tag) expression are highlighted with white dash cycles; P16-negative cells with P16-TET (Myc-tag) expression are highlighted with pink dash cycles. Bar: 30 μm.
Furthermore, Western blot revealed that P16 protein was detected in the P16-TET&Dox cells by the 7th day, but not in the Vector&Dox control cells (7d; Figure 2b). qRT-PCR showed weak reactivation of P16 transcription beginning on the 4th day (Figure 2c). Immunofluorescence confocal microscopy also confirmed the presence of P16 protein in the nuclei of H1299 cells (Figure 2d). In addition, the expression of control genes P15 and P14 was not affected, whereas the expression level of ANRIL, which is coordinately expressed with P16 [20], was increased (Figure S2). This suggests a high specificity for the zinc finger protein-based P16-TET to induce P16H and P16U.
Similar to what we observed in H1299, on the 7th day following Dox induction, transcriptional reactivation of P16 was also observed in P16-TET stably transfected gastric cancer AGS cells, in which P16 alleles are homogenously methylated (Figure 3a-e). Interestingly, P16H signals were observed in the TAB-DHPLC and TAB sequencing results in P16-TET AGS cells after Dox induction for 11 days (P16-TET&Dox_11d) (Figure 3b and d) and P16U signals were not detected in the bisulphite-DHPLC and bisulphite sequencing analyses (Figure 3a and c), indicating that hydroxymethylation occurred earlier than demethylation at P16 CGIs. A few baseline 5hmCs were also found in the P16 exon-1 antisense-strand of AGS mock control cells. Although weak P16 mRNA signals were detected in P16-TET AGS cells after Dox induction for 7 days and 11 days according to sensitive RT-PCR analysis (Figure 3e), P16 protein was not detected in these cells according to the insensitive Western blot analysis (figure 3f).
Figure 3.
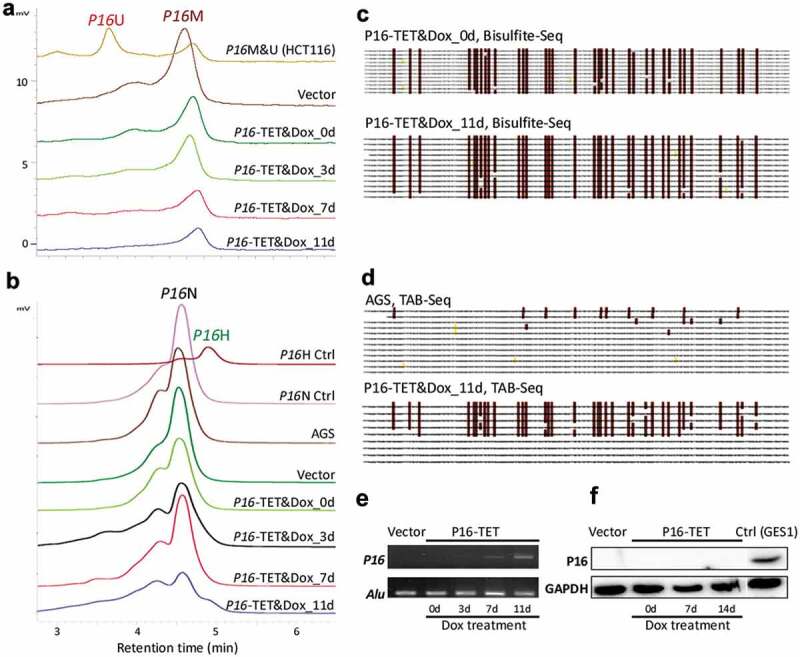
P16-TET induces hydroxymethylation of P16 CpG islands and reactivates expression of methylated-P16 alleles in AGS cells. (a) Bisulphite-DHPLC analysis for detecting methylated-P16 (P16M) and unmethylated-P16 (P16U) PCR products for the exon-1 antisense strand in P16-TET-transfected AGS cells with different doxycycline induction times. (b) The TAB-DHPLC analysis detected the hydroxymethylated P16 (P16H) PCR products and nonhydroxymethylated P16 (P16N) PCR products. (c and d) Bisulphite and TAB sequencing for detecting 5mC and 5hmC sites, respectively, in the same PCR products as were analysed by DHPLC. (e and f) The results of RT-PCR and Western blot analysis for detecting P16 reactivation in AGS cells.
Transcription silencing of P16 alleles by hydroxymethylation
To deduce whether DNA hydroxymethylation or demethylation contributes to P16 reactivation, we further analysed the hydroxymethylation status of P16 CGIs in cell subpopulations with strong, weak, and no P16 staining (P16(+), P16(±), and P16(-) that were sorted from P16-TET&Dox_21d H1299 cells (Figure 4a). Interestingly, P16H signal was detected only in the P16(-) subpopulation, but not in the P16(+) and P16(±) subpopulations in the TAB-MSP analysis (Figure 4b). TAB sequencing also showed dense 5hmCs among 3 of the 14 clones (21.4%) of the exon-1 antisense-strand TAB-PCR products from the P16(-) subpopulation, with an average hydroxymethylation density of 95.2% for these 3 clones (Figure 4c). The occurrence of 5hmCs in the promoter was not detected in the TAB-DHPLC and TAB sequencing results (data not shown).
Figure 4.
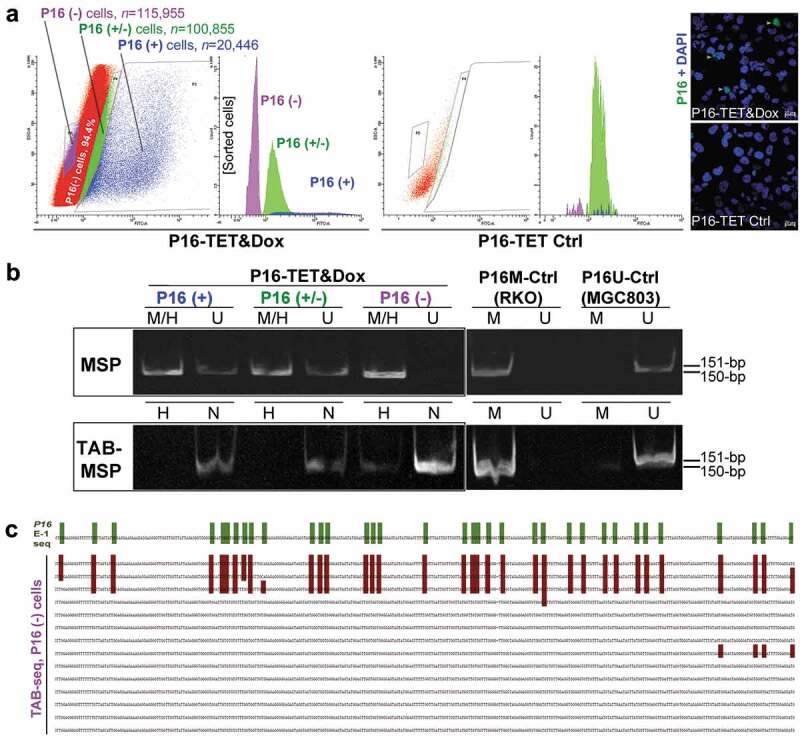
Characterization of P16H in FACS-sorted subpopulations of H1299 cells with various levels of of P16 expression reactivation. (a) FACS sorting of P16-TET stably transfected H1299 cells with and without Dox treatment. The confocal images of the P16 protein staining status are also attached. Bar: 30 μm. (b) Detection of the DNA hydroxymethylation status of P16 alleles in various FACS-sorted H1299 subpopulations with strong, weak, and no P16 immunostaining (P16(+)/(±)/(-)) in the TAB-MSP analysis. (c) The results of TAB sequencing for the P16 CpG islands in the P16-negative subpopulation.
The above results were further confirmed in AGS cells. P16 protein could not be detected in P16-TET&Dox AGS cells after Dox treatment for 11 days (figure 3f). To obtain a P16(+) AGS subpopulation by FACS, the DNA methyltransferase inhibitor 5-aza-deoxycytidine (DAC, final concentration 20 nmol/L) was used to increase the P16 protein level within P16-TET AGS cells. In the immunostaining cell analysis, nucleic P16 protein was detected in 3.5% of P16-TET AGS cells after DAC treatment for 10 days (P16-TET&DAC_10d, with baseline P16-TET expression without Dox induction), while nucleic P16 protein was detected in only 0.5% of the AGS cells treated with DAC alone (Figure S3). Next, the P16(+), P16(±), and P16(-) subpopulations were sorted from these P16-TET&DAC_10d AGS cells (Figure 5a). Once again, the P16H signal was detected only in the P16(-) subpopulation, and not in the P16(+) and P16(±) cells by the TAB-MSP and TAB-DHPLC assays (Figure 5b and c). In contrast, P16N signal was detected in all three subpopulations, and was confirmed by TAB sequencing. Dense 5hmCs were observed in the P16 exon-1 antisense-strand in the P16(-) subpopulation, but not in the P16(+) subpopulation (Figure 5d).
Figure 5.
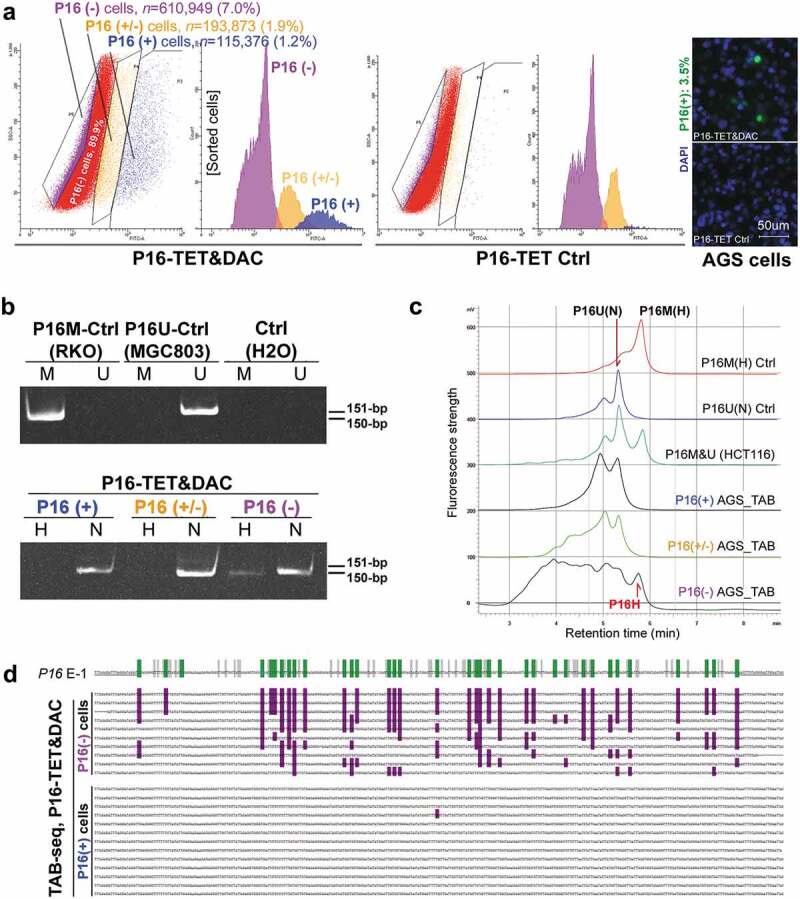
Characterization of P16H in FACS-sorted subpopulations of AGS cells with various levels of P16 expression reactivation. (a) FACS-sorting of P16-TET stably transfected AGS cells with and without DAC treatment. The confocal images of the P16 protein staining status are also attached. Bar: 50 μm. The sorted AGS cells not treated with doxycycline only expressed P16-TET at the baseline level. (b) Detection of the DNA hydroxymethylation status of P16 alleles in various FACS sorted AGS subpopulations with strong, weak, and no P16-immunostaining (P16(+)/(±)/(-)) in the TAB-MSP analysis. (c) The results of TAB-DHPLC for the P16 CpG islands in three subpopulations. (d) The results of TAB sequencing for the antisense strand of P16 exon-1 in P16(+) and P16(-) subpopulations.
Collectively, the above results indicate that P16H occurs only in P16(-) cells, and not in P16(+) and P16(±) cells, which suggests that P16H alleles are transcriptionally inactive.
P16 hydroxymethylation increases reactivation potential
While the wildtype P16 alleles are silenced by DNA methylation in HCT116 cells, the active P16 alleles containing a G-insertion in exon-1 that leads to frame-shift mutation and no P16 protein synthesis. It was reported that there were dense 5hmCs in the P16 exon-1 in HCT116 cells [21]. The results of our TAB-DHPLC and TAB-sequencing analyses further demonstrated that 5hmCs were enriched mainly in the antisense-strand of the wild-type P16 exon-1 in HCT116 cells (Figure S4). Thus, HCT116 cells were used to study possible functions of the endogenous hydroxymethylation of TSS-CGIs.
To study whether the occurrence of dense 5hmCs of the P16 exon-1 could affect reactivation potential, P16-ATF [24] and DAC (final concentration, 20 nmol/L) were used to reactivate the methylation-silenced wildtype P16 allele in HCT116 cells with and without siRNA-knockdown of TET1/2/3 genes, respectively. The mRNA levels of TET1/2/3 genes were significantly knocked down by these siRNAs, respectively (Figure 6a). While P16 protein staining signal was detected in 2.2% of the P16-ATF&DAC cells without siTET1/2/3 treatment, no P16 protein staining signal was detected in the P16-ATF&DAC cells with the siTET1/2/3 treatment (Figure 6b and S5). These results illustrate that blocking the natural occurrence of 5hmCs of the P16 exon-1 inhibits the reactivation of methylation-silenced wildtype P16 allele in HCT116 cells.
Figure 6.
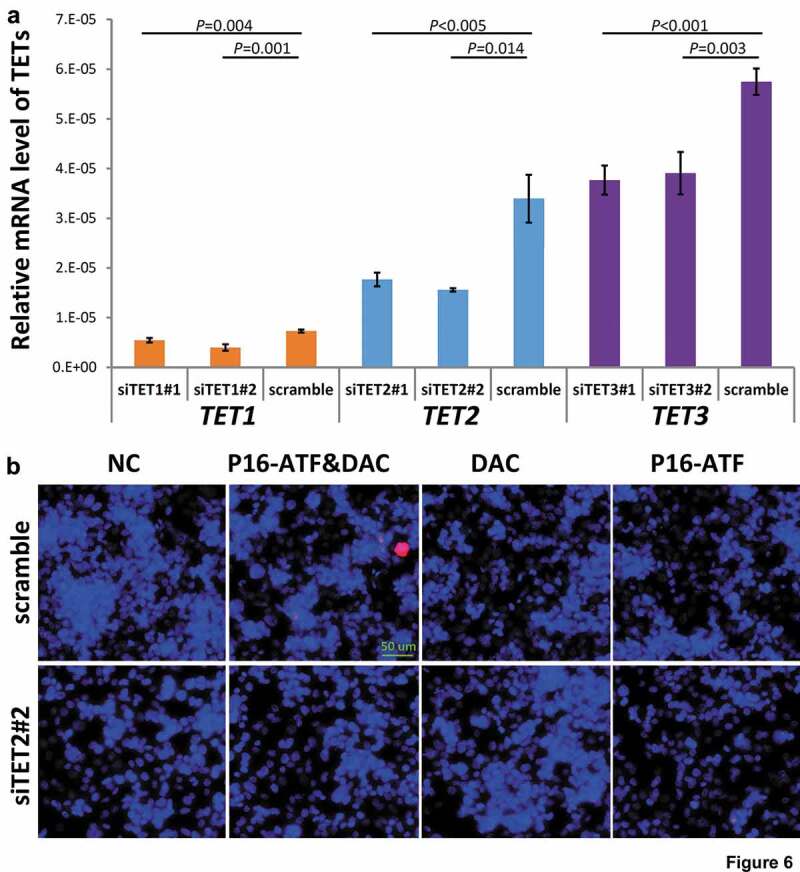
Effects of knockdown of TET1/2/3 on reactivation of the P16 gene by DNA methylation inhibitor DAC and P16-specific artificial transcription factor (P16-ATF) in HCT116 cells. After treated with the TET-specific siRNAs for 48 hrs, these cells were transiently transfected with the P16-ATF vector for 24 hrs, and then treated with DAC (final conc. 20 nmol/L) for 48 hrs. (a) The relative mRNA levels of TET1/2/3 in gene-specific siRNA treated cells in qRT-PCR ananlysis; (b) The confocal images of the P16 protein staining status for various group cells.
The inhibition of tumour growth by P16-TET
Although P16-TET did not affect the proliferation of H1299 cells in vitro (Figure 7a), our results of the IncuCyte ZOOM wound-scratch and transwell assays showed that P16-TET significantly inhibited H1299 cell migration (Figures S6A and S6B). In a rescue assay, P16 siRNA-knockdown significantly increased the migration of the P16-TET&Dox H1299 cells (Figure S6C), which further demonstrates the role of P16-TET in inhibiting cell migration through P16 reactivation.
Figure 7.
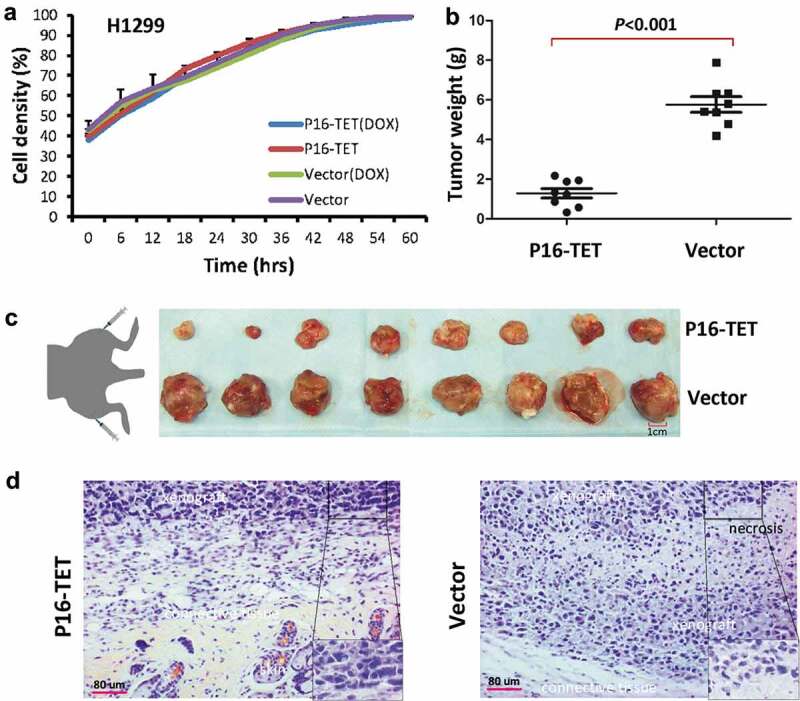
Effects of P16H on the proliferation of H1299 cells in vitro and in vivo. (a) Cell proliferation curves for H1299 cells with and without P16-TET expression in a live content kinetic imaging platform; (b) Comparison of weights of H1299 tumour xenografts with and without stable P16-TET transfection in SCID mice; (c and d) Images of xenografts on the 50th experimental day.
In the in vivo experiment, the average weight of tumour xenografts of the P16-TET stably transfected cells was significantly lower than that of the control cells in NOD-SCID mice (n = 8) on the 50th post-transplantation day (P < 0.001, Figure 7b and c). Morphologic differences were not observed between P16-TET and control vector xenografts (Figure 7d). These results were confirmed in a repeat experiment (Figure S7).
Discussion
DNA hydroxymethylomes at the base-resolution level have been analysed in embryonic stem cells, adult tissues, and tumours [25–31]. Many functions of DNA hydroxymethylation in the genome have been illustrated by TET-1/2/3 knockout studies [5–7,27,31,32]. However, the actual effect of hydroxymethylation of TSS-CGIs on gene transcription remains elusive. In the present study, we demonstrated that 5hmCs were enriched in the antisense-strand of the P16 exon-1 CGI. More importantly, this study showed for the first time that DNA hydroxymethylation itself could not reactivate P16 gene transcription. Instead, hydroxymethylation could increase reactivation potential of methylation-silenced P16 gene by DNA demethylation, which subsequently inhibited the migration and growth of cancer cells in vivo.
The global 5hmC levels are decreased in cancer genomes [29,30], probably through downregulation or mutation of the TET1/2/3 genes in cancer development [33–35]. However, certain proportion of 5hmCs remain in some cancer or precancer tissues [23]. Recently, we found that all P16 mRNA clones in the HCT116 cells were transcribed only from the mutant P16U alleles, and none from the methylated: hydroxymethylated (M:H) P16 alleles [21], and that both true P16M and P16H could similarly increase the risk for malignant transformation of oral epithelial dysplasia in a prospective study [23]. The findings of this present study further show that the P16-TET-induced hydroxymethylation of P16 alleles in both H1299 and AGS cells retained transcriptional silence, which may provide a possible mechanism to explain our observations.
It has been reported that triple knockout of TET-1/2/3 led to bivalent promoter hypermethylation in H1 cells [36]. We previously constructed a P16-ATF expression vector and fount that transient P16-ATF transfection combined with DAC (20 nmol/L) treatment could reactivate the methylated-P16 expression in H1299 cells, although the transient P16-ATF transfection or DAC treatment alone could not reactivate methylated-P16 gene expression [24]. This phenomena was also observed in the present study. Transient P16-ATF transfection and DAC combined treatment could reactivate the expression of wildtype methylated-P16 alleles in HCT116 cells. Interestingly, the reactivation of methylated-P16 alleles was dismissed in these cells when the 5hmCs in the P16 exon-1 was removed by siRNA knockdown of TET1/2/3 genes. These results unveil that 5hmCs in TSS-CGIs could increase the reactivation potential of methylated genes.
There are many differences between cell culture and animal models. Although the proliferation of H1299 cells that are stably transfected with P16-TET was not changed under in vitro culture conditions, the growth of xenograft tumours from these cells was obviously inhibited in host mice. The exact reasons leading to this difference are unknown; however, the reactivation of methylated P16 alleles via DNA demethylation by P16-TET may account for the growth inhibition in vivo. In the rescue assay, siRNA knockdown of P16-TET-reactivated P16 expression almost completely reversed the inhibition of P16-TET-induced cell migration. This further suggests that the inhibition of the cancer cell migration by P16-TET may be a P16-specific effect.
In conclusion, we found that although P16H alleles are transcriptionally inactive, hydroxymethylation could increase reactivate potential of the P16 gene in cancer cells.
Methods
Cell lines and culture
The colon cancer cell line HCT116 was purchased from the American Type Culture Collection (ATCC). The GC cell line AGS and the lung cancer cell line H1299 were kindly provided by Prof. Chengchao Shou from the Peking University Cancer Hospital and Institute. The colon cancer cell line, RKO was kindly provided by Prof. Guoren Deng from the University of California, San Francisco. RKO cells were cultured in DMED, AGS cells were cultured in F12 medium, HCT116 and H1299 cells were cultured in RPMI-1640, containing 10% FBS and 100 U/mL penicillin/streptomycin (Invitrogen, California, USA) at 37°C in a humidified incubator with 5% CO2.
These cell lines were tested and authenticated by Beijing JianLian Genes Technology Co., LTD before they were used in this study. STR patterns were analysed using a GoldeneyeTM20A STR Identifiler PCR Amplification Kit. Gene Mapper v3.2 software (ABI) was used to match the STR pattern with the ATCC online databases.
Characterization of 5mC and 5hmC sites in P16 CGIs
Total P16M was analysed using 150-bp regular methylation-specific PCR (MSP) targeted to the antisense strand of P16 exon-1 [37]. Genomic DNA of RKO and MGC803 cells were used as the P16M and P16U controls [15]. To selectively detect P16H, the genomic DNA (3 μg), spiked with M.sssI-methylated and 5hmC-containing λ-DNA controls, was modified using the TET-Assisted Bisulphite (TAB) Kit, according to manufacturer’s instructions (WiseGene, Cat# K001). During TAB-modification [38], 5mC was oxidized to 5caC, and both 5caC and unmethylated cytosine were subsequently converted to uracil through bisulphite-induced deamination, whereas 5hmC was protected from oxidation via 5hmC-specific β-glucosylation [25]. The conversion rates of unmethylated cytosine, 5mCs, and 5hmCs in the bisulphite-/TAB-treated λ-DNA controls were 100%, 99.7%, and 1.5%, respectively (Figure S8). P16H was analysed using the TAB-MSP.
We amplified the sense- and antisense-strands of the P16 promoter and exon-1 CGIs in HCT116 cells using a corresponding CpG-free primer set, respectively (Figure S4A; 402-bp and 369-bp for the sense-strand of the P16 promoter and exon-1, 367-bp and 392-bp for the antisense-strand of the P16 promoter and exon-1). The proportion of hydroxymethylated sense- and antisense-strands of the P16 promoter and exon-1 CGIs were analysed using DHPLC and clone sequencing, respectively [39,40]. The adjusted ratio of the peak height for the hydroxymethylated region to that of the unmethylated region was used to represent the P16H proportion that was adjusted. The ratio of the P16M peak height to the P16U peak height for P16-hemimethylated HCT116 cells was used as a reference. The sequences of the universal primers used to amplify these fragments are listed in Table 1.
Table 1.
Sequences of oligonucleotides used as primers in various PCR-based assays.
| Gene name | Entrez gene ID | Assay | Oligo name | Primer sequence (5ʹ→3ʹ) | Product size (bp) | PCR Tm (°C) |
|---|---|---|---|---|---|---|
| P16 | 1029 | qRT-PCR | P16-F | gctgcccaacgcaccgaata | 180 | 58 |
| P16-R | accaccagcgtgtccaggaa | |||||
| DHPLC/Seq | P16-E1F | tttttagaggatttgagggatagg | 392 | 57 | ||
| P16-E1R | ctacctaattccaattcccctacaaactt | |||||
| P16-E1SF | gttgtagattttttatttatttggat | 369 | 56 | |||
| P16-E1SR | tccccttacctaaaaaaatacc | |||||
| P16-PF | ttggtagttaggaaggttgtat | 367 | 55 | |||
| P16-PR | tttagaggatttgagggatagg | |||||
| P16-PSF | gttttttaaattttttggagggat | 402 | 55 | |||
| P16-PSR | ttggtgttatagggaaagtatgg | |||||
| MSP-M/H | P16-MF | ttattagagggtggggcggatcgc | 150 | 62 | ||
| P16-MR | gaccccgaaccgcgaccgtaa | |||||
| MSP-U/N | P16-UF | ttattagagggtggggtggattgt | 151 | 62 | ||
| P16-UR | caaccccaaaccacaaccataa | |||||
| ANRIL | NR_003529 | qRT-PCR | E3-E4R | cagcagaaggtgggcagcagat | 145 | 64 |
| E3-E4F | ttcctcgacagggcaggcaggt | |||||
| P15 | 1030 | qRT-PCR | P15-qF | agtcaaccgtttcgggaggcg | 168 | 58 |
| P15-qR | accaccagcgtgtccaggaag | |||||
| P14 | 1029 | qRT-PCR | P14-qF | gccaggggcgcccgccgctg | 236 | 62 |
| P14-qR | ggcccggtgcagcaccacca | |||||
| ALU | qRT-PCR | ALU-qF | gaggctgaggcaggagaatcg | 54 | ||
| ALU-qR | gtcgcccaggctggagtg | |||||
| GAPDH | 2597 | (q)RT-PCR | GAPDH-F | gaaggtgaaggtcggagt | 226 | 62 |
| GAPDH-R | gaagatggtgatgggatttc | |||||
| λ-DNA | 5hmC-ctrl | PCR | 5hmC-F | ggagttggtatgtagggtagaaagg | 202 | 55 |
| 5hmC-R | attcactctctcacctactctct | |||||
| 5mC-ctrl | PCR | 5mC-F | tttgggttatgtaagttgattttatg | 296 | 55 | |
| 5mC-R | caccctacttactaaaatttacacc |
Construction of expression vectors and transfection
To construct the P16-specific DNA dioxygenase (P16-TET) expression vector, an SP1-like engineered seven-zinc finger protein (7ZFP-6I) [19] that can specifically bind to the 21-bp fragment (5ʹ-gaggaaggaaacggggcgggg-3ʹ, including an Sp1-binding site) within the human P16 core promoter [24,41], was fused with the catalytic domain (CD: 1418–2136 aa) of human TET1 (NM_030625.2) [42] and inserted into a pcDNA3.1b vector and then used in transient transfection assays. An inactive P16-TET mutant containing an H1671Y mutation in the CD domain vector was also constructed and used as a negative control vector (Figure 1a). The P16-TET sequence was further integrated into the expression-controllable pTRIPZ vector carrying a ‘Tet-on’ switch (Open Biosystem, USA) [19]. Purified P16-TET pTRIPZ plasmid was mixed with VSVG and Δ8.9 (Addgene, USA) to prepare lentivirus transfection particles. The fresh lentivirus particles were used to stably infect AGS and H1299 cells containing homogenously methylated P16 CpG islands. Doxycycline (Dox; optimized final conc. 0.25 μg/mL) was added to the medium to induce P16-TET expression as we described previously [19].
P16-specific siRNAs (5ʹ-ccgua aaugu ccauu uauatt-3ʹ and 5ʹ–uauaa augga cauuu acggtt-3ʹ) and TET-specific siRNAs for TET1 (#1: 5ʹ-gccau cagau cugua agaatt-3ʹ and 5ʹ-uucuu acaga ucuga uggctt-3ʹ; #2: 5-gaagc ccaca guugu aagutt-3ʹ and 5ʹ-acuua caacu guggg cuuctt-3ʹ), TET2 (#1: 5ʹ-gccag uaaac uagcu gcaatt-3ʹ and 5ʹ-uugca gcuag uuac uggctt-3ʹ; #2: 5ʹ-ccauc acaau ugcuu cuuutt-3ʹ and 5ʹ-aaaga agcaa uugug auggtt-3), and TET3 (#1: 5ʹ-ggaaa uaaag gcugg ugaatt-3ʹ and 5ʹ-uucac cagcc uuuau uucctt-3ʹ; #2: 5ʹ-gccug ugguu ccucc ugaatt-3ʹ and 5ʹ-uucag gagga accac aggctt-3ʹ) were synthesized (GenePharma, Shanghai) and used to transiently transfect cells at a final concentration of 1.0 μg/mL. Two scrambled siRNAs (5ʹ-uucuc cgaac guguc acgutt-3ʹ and 5ʹ-acgug acacg uucgg agaatt-3ʹ) were used as negative controls (NC).
Treatment of 5ʹ-aza-deoxycytidine (DAC)
The AGS cells were treated with DAC (final concentration 20 nmol/L; Abcam ab120842, Cambridge, UK) for 7 days in the P16-immunostaining assay or 10 days prior to FACS sorting. The HCT116 cells were treated with DAC (20 nmol/L) for 48 hrs and used in P16-ATF transient transfection experiment as previously described [24].
Extraction of RNA and quantitative RT-PCR (qRT-PCR)
Cells were harvested when they reached a confluency of approximately 70%. Total RNA was extracted by TRIzol (Invitrogen, California, USA). The cDNA was reverse-transcribed using the ImProm-IITM Reverse Transcription System (A3800; Promega). The expression levels of the ANRIL, P16, P15, P14, and TET-1/2/3 genes were analysed by quantitative RT-PCR using the corresponding primer sets (Table 1), as previously described [20]. Power SYBR Green PCR Master Mix (Fermentas, Canada) was used in the qRT-PCR analyses (ABI-7500FAST). The relative mRNA level was calculated based on the average Ct value of the target gene and the Alu reference [2−(Cttarget_gene−CtAlu)] [43].
Western blot and confocal microscopy analysis of the P16 expression status
The P16 mRNA and protein levels in the cells were analysed as previously described [19]. Rabbit monoclonal antibody against human P16 protein (ab108349, Abcam, Britain) was used in the Western blot assay, and mouse monoclonal antibody against the human P16 protein (Ventana Roche-E6H4, USA) was used in the immunostaining assay. These experiments were performed in triplicate and repeated at least one time.
Cell FACS sorting
The P16-TET stably transfected H1299 cells (treated with doxycycline for 21 days) and AGS cells (treated with 5-aza-deoxycytidine for 10 days) were fixed with methanol, permeabilized with 0.1% Tween-20 in PBS, pretreated with 10% foetal bovine serum and 0.3 M glycine in PBS, and were then stained with the mouse monoclonal antibody against the human P16 protein (Ventana Roche-E6H4, USA) and the FITC-tagged secondary antibody. The P16-staining cell population proportion was determined using an immuno-fluorescence confocal microscope. These cells were sorted by FACS and divided into three subpopulations, strong-, weak-, and non-P16-staining, using P16-TET H1299 cells without doxycycline treatment or AGS cells without DAC treatment as P16 protein negative controls. According to the confocal analysis results, we setup the cut-off value to sort definite and indefinite P16 protein positive (P16(+) and P16(±)) cell subpopulations. The strong and weak FITC-staining cells were called as the P16(+) and P16(±) subpopulations, respectively. The sorting experiment was repeated for at least one time for each cell line.
IncuCyte ZOOM and transwell migration tests
The long-term live content kinetic imaging platform (IncuCyte Zoom, Essen BioSci, USA) was used to dynamically detect the proliferation and migration of live cancer cells. The phase object confluence (%) was used to generate a cell proliferation curve. The relative wound density, a measure (%) of the density of the wound region relative to the density of the cell region, was used as the metric for cell migration. The transwell migration test was repeated at least two times in triplicate as previously described [19].
Xenografts in SCID mice
Cells stably transfected with the P16-TET vector were induced with 0.25 μg/mL doxycycline for 7 days and then subcutaneously injected into one lower limb of each NOD-SCID mouse (105 cells/injection; female, 5 weeks old, 10 ~ 20 g, purchased from Beijing Huafukang Biotech). The negative control cells stably transfected with the empty pTRIPZ vector were simultaneously injected into the opposite side of each mouse. These mice were given distilled, sterile water containing 2 μg/mL doxycycline and were sacrificed on the 50th post-transplantation day. The xenografts were weighed and histologically confirmed [19]. Two repeat experiments were performed.
Statistical analysis
Student’s t-test was used for statistical analysis. All P-values were two-sided, and a P-value of <0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank Mr. Jordan M. Grainger (Predoctoral student, Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, Minnesota, USA), Dr. Huidong Shi (Cancer Center, Georgia Medical College, Augusta, USA), and Mr. James Hyun (Pharmacology Department, University of Illinois, Chicago, USA) for English language editing.
Funding Statement
This work was supported by the National Natural Science Foundation of China under Grant number 81672770; Beijing Municipal Commission of Health and Family Planning under Grant number PXM2018_026279_000005, Beijing Municipal Administration of Hospital Clinical Medical Development of Special Funding Support under Grant number XM201303.
Author contributions
PL and YG: elucidated the biological function of P16 hydroxymethylation; SQ: discovered P16 hydroxymethylation; XH: demonstrated the strand-bias distribution of 5hmCs in the P16 alleles; CC, Z-mL, and BZ: constructed the P16-specific oxygenase; LG performed the animal experiments; BZ performed immunostaining and cell sorting assays and codesigned the study; ZL and JZ: carried out other experiments; DD: designed the study, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics approval and consent to participate
The Peking University Cancer Hospital and Institutional Review Boards approved this study. Ethical approval for the animal experiments was obtained.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kriaucionis S, Heintz N.. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gu TP, Guo F, Yang H, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. [DOI] [PubMed] [Google Scholar]
- [6].Hackett JA, Sengupta R, Zylicz JJ, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pastor WA, Pape UJ, Huang Y, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ficz G, Branco MR, Seisenberger S, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. [DOI] [PubMed] [Google Scholar]
- [9].Yu M, Hon GC, Szulwach KE, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Merlo A, Herman JG, Mao L, et al. 5ʹ CpG island methylation is associated with transcriptional silencing of the tumor-suppressor p16/cdkn2/mts1 in human cancers. Nat Med. 1995;1:686–692. [DOI] [PubMed] [Google Scholar]
- [11].Herman JG, Merlo A, Mao L, et al. Inactivation of the cdkn2/p16/mts1 gene is frequently associated with aberrant dna methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- [12].Sun Y, Deng DJ, You WC, et al. Methylation of p16 CpG islands associated with malignant transformation of gastric dysplasia in a population-based study. Clin Cancer Res. 2004;10:5087–5093. [DOI] [PubMed] [Google Scholar]
- [13].Belinsky SA, Liechty KC, Gentry FD, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006;66:3338–3344. [DOI] [PubMed] [Google Scholar]
- [14].Hall GL, Shaw RJ, Field EA, et al. p16 promoter methylation is a potential predictor of malignant transformation in oral epithelial dysplasia. Cancer Epidemiol Biomarkers Prev. 2008;17:2174–2179. [DOI] [PubMed] [Google Scholar]
- [15].Cao J, Zhou J, Gao Y, et al. Methylation of p16 CpG Island associated with malignant progression of oral epithelial dysplasia: a prospective cohort study. Clin Cancer Res. 2009;15:5178–5183. [DOI] [PubMed] [Google Scholar]
- [16].Jin Z, Cheng Y, Gu W, et al. A multicenter, double-blinded validation study of methylation biomarkers for progression prediction in Barrett’s esophagus. Cancer Res. 2009;69:4112–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu HW, Liu XW, Dong GY, et al. P16 methylation as an early predictor for cancer development from oral epithelial dysplasia: a double-blind multicentre prospective study. EBioMedicine. 2015;2:432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gao H-E, Zhang Y, Zhou J, et al. Association between p16 methylation and malignant transformation of gastric dysplasia. Chin J Can Prev Treat. 2017; 24:431–436. [Google Scholar]
- [19].Cui C, Gan Y, Gu L, et al. P16-specific DNA methylation by engineered zinc finger methyltransferase inactivates gene transcription and promotes cancer metastasis. Genome Biol. 2015;16:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gan Y, Ma W, Wang X, et al. Coordinate transcription of ANRIL and P16Genes silenced by DNA methylation. Chin J Cancer Res. 2018;30:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Qin SS, Li Q, Zhou J, et al. Homeostatic maintenance of allele-specific p16 methylation in cancer cells accompanied by dynamic focal methylation and hydroxymethylation. Plos One. 2014;9:E97785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qin S, Zhang B, Tian W, et al. Kaiso mainly locates in the nucleus in vivo and binds to methylated, but not hydroxymethylated DNA. Chin J Cancer Res. 2015;27:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu HW, Liu ZJ, Liu XW, et al. A similar effect of P16 hydroxymethylation and true-methylation on the prediction of malignant transformation of oral epithelial dysplasia: observation from a prospective study. BMC Cancer. 2018;18:918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang B, Xiang S, Zhong Q, et al. The p16-specific reactivation and inhibition of cell migration through demethylation of CpG Islands by engineered transcription factors. Hum Gene Ther. 2012;23:1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sun Z, Terragni J, Jolyon T, et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang T, Wu H, Li Y, et al. Subtelomeric hotspots of aberrant 5-hydroxymethylcytosine-mediated epigenetic modifications during reprogramming to pluripotency. Nat Cell Biol. 2013;15:700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim M, Park YK, Kang TW, et al. Dynamic changes in DNA methylation and hydroxymethylation when hES cells undergo differentiation toward a neuronal lineage. Hum Mol Genet. 2014;23:657–667. [DOI] [PubMed] [Google Scholar]
- [28].Wen L, Li X, Yan L, et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014;15:R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen K, Zhang J, Guo Z, et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res. 2016;26:103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li X, Liu Y, Salz T, et al. Whole-genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver. Genome Res. 2016;26:1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang Y, Chavez L, Chang X, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2014;111:1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Uribe-Lewis S, Stark R, Carroll T, et al. 5-hydroxymethylcytosine marks promoters in colon that resist DNA hypermethylation in cancer. Genome Biol. 2015;16:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Carella A, Tejedor JR, García MG, et al. Epigenetic downregulation of TET3 reduces genome-wide 5hmC levels and promotes glioblastoma tumorigenesis. Int J Cancer. 2019. DOI: 10.1002/ijc.32520. [DOI] [PubMed] [Google Scholar]
- [34].Liu J, Jiang J, Mo J, et al. Global DNA 5-hydroxymethylcytosine and 5-formylcytosine contents are decreased in the early stage of hepatocellular carcinoma. Hepatology. 2019;69(1):196–208. [DOI] [PubMed] [Google Scholar]
- [35].Kudo Y, Tateishi K, Yamamoto K, et al. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012;103(4):670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Verma N, Pan H, Doré LC, et al. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat Genet. 2018;50:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Herman JG, Graff JR, Myöhänen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yu M, Hon GC, Szulwach KE, et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 2012;7:2159–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Deng DJ, Deng GR, Smith MF, et al. Simultaneous detection of CpG methylation and single nucleotide polymorphism by denaturing high performance liquid chromatography. Nucleic Acids Res. 2002;30:13E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luo DY, Zhang BZ, Lv LB, et al. Methylation of CpG islands of p16 associated with progression of primary gastric carcinomas. Lab Invest. 2006;86:591–598. [DOI] [PubMed] [Google Scholar]
- [41].Zhang B, Xiang S, Yin Y, et al. C-terminal in Sp1-like artificial zinc-finger proteins plays crucial roles in determining their DNA binding affinity. BMC Biotechnol. 2013;13:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Guo JU, Su Y, Zhong C, et al. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zheng X, Zhou J, Zhang B, et al. Critical evaluation of Cbx7 downregulation in primary colon carcinomas and its clinical significance in Chinese patients. BMC Cancer. 2015;15:1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.