

Abstract
Light-driven biocatalysis in recombinant cyanobacteria provides highly atom-efficient cofactor regeneration via photosynthesis, thereby remediating constraints associated with sacrificial cosubstrates. However, despite the remarkable specific activities of photobiocatalysts, self-shading at moderate-high cell densities limits efficient space-time-yields of heterologous enzymatic reactions. Moreover, efficient integration of an artificial electron sink into the tightly regulated network of cyanobacterial electron pathways can be highly challenging. Here, we used C=C bond reduction of 2-methylmaleimide by the NADPH-dependent ene-reductase YqjM as a model reaction for light-dependent biotransformations. Time-resolved NADPH fluorescence spectroscopy allowed direct monitoring of in-cell YqjM activity and revealed differences in NADPH steady-state levels and oxidation kinetics between different genetic constructs. This effect correlates with specific activities of whole-cells, which demonstrated conversions of >99%. Further channelling of electrons toward heterologous YqjM by inactivation of the flavodiiron proteins (Flv1/Flv3) led to a 2-fold improvement in specific activity at moderate cell densities, thereby elucidating the possibility of accelerating light-driven biotransformations by the removal of natural competing electron sinks. In the best case, an initial product formation rate of 18.3 mmol h–1 L–1 was reached, allowing the complete conversion of a 60 mM substrate solution within 4 h.
Keywords: light-driven biotransformations, electron channeling, NADPH fluorescence, photocatalysis, photosynthesis, flavodiiron proteins, cyanobacteria, Synechocystis
Genetically engineered photoautotrophic organisms have emerged as possible hosts for the future production of biobased chemicals devoid of land-use conflicts for agricultural purposes.1,2 Algae and cyanobacteria have been applied for the production of food additives3,4 and feed,5 with metabolic engineering further expanding their potential for the production of various chemicals such as terpenoids,6 alcohols,7,8 and sugars.9,10
The utilization of photoautotrophs for biotechnology provides several advantages such as the capacity to fix carbon dioxide and utilize photosynthetically derived redox power. Furthermore, photosynthetic cells offer specific benefits including an NADPH-based metabolism11 and in situ oxygen production.12,13 However, despite these benefits, inadequate light availability limits the growth of suspension cultures to a few grams per liter.14 This occurs due to self-shading—a phenomenon where growing cells conceal each other, thereby leading to uneven and inadequate light distribution.15 Consequently, a considerable increase of photoautotrophic production rates is crucial to compete with highly optimized heterotrophic production systems.
Using the photosynthetic pool of reducing equivalents, in the form of reduced ferredoxin (Fd) or NADPH for reductive biotransformations (Figure 1), allows one to overcome the poor atom economy (i.e., conversion efficiency of a chemical process accounting for all atoms involved) associated with the use of sacrificial organic cosubstrates, such as hydrogen,16 which is a limitation commonly associated with biocatalytic redox reactions. The work by Kuk et al. serves as an example where water was used as an electron donor to produce NADH for the reduction of CO2 to methanol.17 Furthermore, the work of Nakamura et al. and Hölsch et al. helped establish the concept of using a photosynthetically derived pool of reductants with the use of wildtype cyanobacteria for the reduction of aryl ketones.18−20
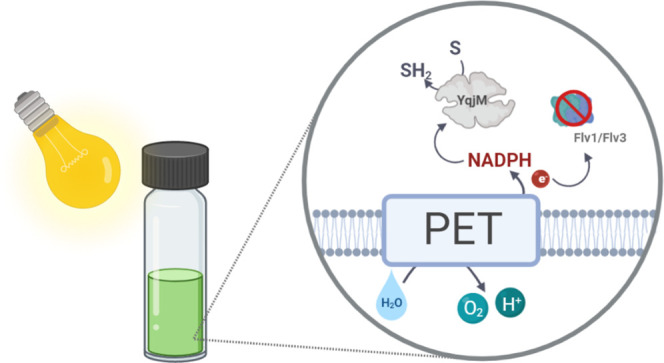
Figure 1.

Simplified representation of the photosynthetic electron transport (PET) chain in Synechocystis depicting the introduction of the heterologous ene-reductase, YqjM (PDB ID: 1Z41).22 NADPH, a primary photosynthetic reductant, mainly produced via linear PET, involves three major photosynthetic complexes, PSII, Cytochrome (Cyt) b6f, and PSI. PSII drives water oxidation on the luminal side of the thylakoid membrane using light energy. The electrons extracted from water travel via several redox cofactors to plastoquinone (PQ) pool, Cytb6f, plastocyanin/Cytc6, PSI and end up reducing Fd. Fd is eventually oxidized by Fd-NADP+ reductase (FNR) to generate NADPH. Depicted above are examples of naturally occurring electron sinks such as the bidirectional hydrogenase (HOX), type I NADH dehydrogenase (NDH1), and flavodiiron proteins. YqjM constitutes a strong, yet tunable electron sink which can be suddenly activated through the introduction of a substrate, 2-methylmaleimide (1a) that is converted to 2-methylsuccinimide (1b). Other abbreviations PQH2: plastoquinol, PC: plastocyanin. Image created using BioRender.
We have previously expanded this approach with a proof-of-concept study in Synechocystis sp. PCC 6803 (hereafter Synechocystis or Syn) by introducing the NAD(P)H dependent ene-reductase YqjM21,22 from Bacillus subtilis. With this system, we achieved product formation rates of 10 mM h–1 for the conversion of 2-methylmaleimide (1a) to 2-methylsuccinimide (1b).11 Light-driven whole-cell biotransformations with other oxidoreductases, such as monooxygenases, alcohol dehydrogenases, and imine reductases, have also been reported by us and others.13,23−25 Thus far, the highest specific activities reported in Synechocystis are those of the ene-reductase YqjM, indicating that it behaves as a strong electron sink drawing photosynthetically derived electrons.
In Synechocystis, the photosynthetic reducing power is required for a wide range of metabolic processes such as carbon fixation and the assimilation of nitrate and sulfur.26,27 However, if the supply of energy-rich electrons exceeds demand, the photosynthetic apparatus is protected by alternative electron sinks like flavodiiron proteins (FDPs). These proteins transfer excess electrons from NAD(P)H28,29 or reduced Fd30−32 to oxygen, thus generating a water–water cycle.33 The Flv1/Flv3 hetero-oligomer is, therefore, indispensable for the survival of cyanobacteria and green algae under fluctuating light intensities.34,35 Under specific conditions, these proteins act as a very strong electron sink, redirecting up to 40–60% of electrons originating from photosystem (PS) II water-splitting to oxygen.36,37 Under controlled laboratory conditions, these photoprotective processes may no longer be needed, potentially opening the possibility of redirecting electrons toward a desired process and thus alleviating the effects of self-shading.
Previously, enhanced activity of a heterologous cytochrome P450 monooxygenase was demonstrated through the disruption of a competing pathway by deleting a type I NADH dehydrogenase complex (NDH-1) subunit in Synechococcus sp. PCC 7002.38 Similar methods have also been reported for other purposes including the production of sucrose and photohydrogen.39,40
Herein, we investigate the effect of the NADPH-pool as a possible bottleneck for light-driven biotransformations using the stereoselective reduction of 1a catalyzed by the ene-reductase YqjM as a model. The extent to which productivity and space-time-yields can be accelerated under self-shading conditions, where NADPH production may be compromised, is also studied by the deletion of the Flv1/Flv3 oligomer.
Results
Promoter Engineering Is Effective for Productivity Enhancement
In previous work,23 the effect of the promoter used for the expression of a heterologous gene of an imine reductase proved to be crucial for an activity increase of the whole-cell cyanobacterial biocatalyst. We, therefore, investigated the effects of three different promoters in order to elucidate their effects on the expression of YqjM and its activity. The inducible, Zn2+-dependent promoter Pzia41,42 was expected to produce lower amounts of protein in comparison to the light-induced promoter PpsbA2 and the partially light-regulated promoter Pcpc. Wildtype Synechocystis and transgenic strains constructed in this work and previous work11 were used (SI, Table S1). We were able to detect intracellular in vitro YqjM activities in Syn::PpsbA2YqjM and Syn::PcpcYqjM; however, no such activity was detected in Syn::PziaYqjM (Figure 2a). This observation was further sustained by SDS-PAGE analysis which yielded discernible YqjM bands under the control of PpsbA2 and Pcpc but not Pzia (SI, Figure S1). Next, we investigated the YqjM-mediated biotransformation of 1a in the engineered whole cells with these different promoters to assess specific initial activities.
Figure 2.

YqjM-mediated biotransformations of the substrate 1a (10 mM) using whole Synechocystis cells under the control of different promoters after cultivation under standard conditions. (for more details please refer to the Materials and Methods section). [a] shows in vitro quantified active YqjM activity, relative to total protein, in strains carrying Pcpc::YqjM and PpsbA2::YqjM cassettes (this activity could not be determined in strains with the Pzia::YqjM cassette). [b] and [c] show specific whole cell activities relative to cell density and chlorophyll a (chla) content, respectively. [d] shows conversion levels of 1a to the product 1b. Reactions were performed at a light intensity of 150 μE m–2 s–1. Data includes values from biological replicates (N = 3) ± SD. P values were calculated using Welch’s t test (*P < 0.05, **P < 0.001) and their exact values, where asterisks are placed, can be found in the raw data files.
Using cells grown under standard conditions (described in the Materials and Methods section), Syn::PcpcYqjM and Syn::PpsbA2YqjM both catalyzed the conversion of 10 mM 1a to completion within 1 h. In these whole-cell biotransformations, Syn::PcpcYqjM exhibited a 1.3-times (±0.16) faster initial activity compared with Syn::PpsbA2YqjM based on dry cell weight (DCW) and chlorophyll a (chla) content (Figure 2b,c). This difference, however, did not correspond with the 1.7-times (±0.45) higher amount of active enzyme in the crude cell extract determined in an in vitro activity assay (Figure 2a). Although this difference is not statistically significant, we nonetheless believe that this suggests that whole-cell biotransformations are limited by factors other than the intracellular concentration of the oxidoreductase. As the availability of the redox factor is an obvious limiting factor, we set out to measure the kinetic parameters of the enzyme toward both the substrate and NADPH and determine a possible reducing effect of the biotransformation on intracellular NADPH.
NADPH Is a Limiting Factor for the YqjM Reaction
In order to investigate the possible limiting role of the redox cofactor on the enzymatic reaction, we sought to determine the kinetic parameters of the reductive and oxidative half reactions in presteady state measurements of purified YqjM in stop-flow experiments. First, the reduction of the flavin cofactor was determined as a function of the concentration of either NADPH or NADH. In both cases, we observed saturation behavior with a maximal velocity of kred of 12.58 ± 1.06 s–1 and 0.97 ± 0.03 s–1 for NADPH and NADH, respectively (Figure 3a,b). From these data, we deduced KD-values of 39.8 ± 7.1 and 140.4 ± 24.5 μM for NADPH and NADH, respectively—these values clearly show that NADPH is the preferred substrate. Second, the oxidative rate of YqjM was determined using 1a as an electron-accepting substrate (Figure 3c). In contrast to the reductive half reaction, the observed rate of oxidation increased linearly with higher substrate concentrations reaching a kobs of 500 s–1 at 250 μM 1a. Higher concentrations of this substrate could not be analyzed because faster rates could not be reliably measured in our experimental setup.
Figure 3.

Determination of the reductive rate of purified YqjM with [a] NADPH and [b] NADH as reducing agents. [c] Determination of the oxidative rate of YqjM with 1a as substrate and 1b as reduced product. [a], [b], and [c] were determined using purified enzyme, and their corresponding equations are shown. Panel [d] depicts the decay constant b of Syn::PcpcYqjM and Syn::PpsbA2YqjM with different 1a concentrations up to 1 mM; these values were determined in whole-cells using fluorescence spectroscopy. Data plotted is derived from biological replicates (N = 3) ± SD. Panel [e] shows the reductive and oxidative half reactions of YqjM.
The kinetics revealed that 1a is an extremely efficient substrate for YqjM, where at concentrations of 250 μM, the observed kobs of 500 s–1 shows that the oxidative half-reaction is at least 50-fold faster than the reductive half-reaction (Figure 3c). Our measurements also reveal that for concentrations ≥500 μM NADPH, cofactor reduction occurs at maximal velocity (Figure 3a). Thus, an intracellular NADPH concentration higher than 500 μM, would not lead to an increase in cellular turnover. However, if NADPH is depleted and drops significantly below 200 μM, a strong effect on kobs can be observed (Figure 3a).
The data presented clearly indicate that the reductive half reaction is the rate limiting step for enzymatic activity, which strengthens the assumption that the supply of reduced nicotinamide cofactors might be a limiting factor for light-driven ene-reduction. In order to exclude substrate limitation, we also conducted whole cell NADPH decay measurements (described in the following section) using different substrate concentrations and found saturation above 0.5 mM (Figure 3d), which ruled out a limiting role of substrate transport and supported our conclusions from the enzyme kinetics. Therefore, the supply of NADPH appeared to be a limiting factor for the ene-reduction.
Heterologous Ene-Reductions Decrease Intracellular NADPH Levels
To the best of our knowledge, NAD(P)H-availability in light-driven biotransformations has never been investigated before. The fast C=C double-bond reduction by YqjM in recombinant Synechocystis is a suitable model for investigations.
Fast changes (∼s) in the redox state of the intracellular NADP pool can be monitored by pulse amplitude modulated (PAM) fluorescence spectroscopy. We applied this spectroscopic tool to compare the NADPH steady-state levels and oxidation kinetics after light-to-dark transition between the WT and the three engineered strains (Syn::PziaYqjM, Syn::PpsbA2YqjM and Syn::PcpcYqjM) in the presence and absence of 1a (Figure 4a). WT cells showed a steady-state fluorescence level during illumination with actinic light for 20 s that corresponds to constant NADP+ reduction by the PET and concomitant NADPH reoxidation by downstream processes such as CO2 fixation as well as nitrate and sulfur assimilation. The relatively small NADP+ pool (∼8 NADP+ per PSI according to Kauny and Sétif43) is highly reduced under these conditions because of efficient PET. After switching off actinic light, the NADPH fluorescence signal rapidly decays (∼3 s) to a temporary minimum that reflects a more oxidized NADP+ pool. The addition of 1a to WT cells changes the steady-levels or decay kinetics only slightly (Figure 4b,c), which indicates minimal background reactions and excludes interference of the fluorescence assay with the model substrate, 1a, for YqjM-dependent reactions.
Figure 4.

NADPH-fluorescence measurements after cultivation at a light intensity of 50 μE m–2 s–1 showing [a] the experimental NADPH decay in (i) Syn WT, (ii) Syn::PziaYqjM, (iii) Syn::PpsbA2YqjM and (iv) Syn::PcpcYqjM cells (OD750 = 1). The gray lines [control] indicate decays in the absence of substrate, the blue [2MM] lines show decays in the presence of the substrate 1a. The time 0 ms indicates the point at which the lights are switched off. Curves were normalized to zero at 3 s. [b] depicts the steady-state values during illumination of the strains. [c] shows the decay constants of the respective strains with and without the addition of 1a (1 mM). Bars include data from three measurements ± SD with individual values depicted. P values were calculated using Welch’s t test (**P < 0.01, ***P < 0.001). Exact P values where asterisks are placed can be found in the raw data files.
Heterologous expression of YqjM under the control of the inducible Pzia promoter revealed no statistically significant difference in the NADPH steady-levels or decay kinetics with or without the addition of 1a, which is consistent with the low intracellular enzyme concentration and slow whole-cell activity of the corresponding cells (Figure 2). Conversely, the strength of the PpsbA2 and Pcpc promoters, which led to high intracellular levels of YqjM and fast conversion activity of the corresponding cells (Figure 2), is also reflected by the NADPH fluorescence measurements (Figure 4). The addition of 1a (1 mM) lowered the steady-state NADPH fluorescence level in the light by ∼30% in both mutant lines due to increased NADPH consumption by YqjM. The effect might be more complex, as the dark state, which we used for normalization of the data in all graphs, might not reflect a fully oxidized NADP+ pool and may also change because of 1a related NADPH consumption in the dark. However, the prompt and fast fluorescence decay upon light-to-dark transition indicates that NADPH is mainly responsible for the signal. This fluorescence decay is ∼3 times faster in the Syn::PpsbA2YqjM and Syn::PcpcYqjM cells after the addition of 1a, which also indicates an efficient flux of NADPH to YqjM.
2-Methylmaleimide Toxicity Is Mitigated by YqjM Expression
Maleimides are known to form stable thioether bonds with free sulfhydryl groups of cysteine amino acid residues. As several vital photosynthetic proteins contain cysteine,44 thioesterisation potentially compromises their function. Therefore, we investigated the effects of 1a on the photosynthetic machinery of Syn WT, Syn::PcpcYqjM, ΔFlv1, and ΔFlv1::PcpcYqjM. The effective yields of PSI and PSII (Y(I) and Y(II), respectively) were assessed by probing whole cell chlorophyll fluorescence indicative for PSII and determining absorbance of the PSI reaction center P700 (Figure 5, Figure S4). Syn WT and the mutant strains Syn::PcpcYqjM, ΔFlv1, and ΔFlv1::PcpcYqjM demonstrated comparable Y(I) (Figure 5a, without 1a) and Y(II) (Figure 5b, without 1a) yields in the absence of 1a. Conversely, in the presence 10 mM 1a, the WT and ΔFlv1 demonstrated lower Y(II) (Figure 5, with 1a) under illumination, compared to the cells incubated without the substrate. Importantly, this Y(II) decrease was rescued by the introduction of YqjM to the WT and ΔFlv1 strains. Notably, ΔFlv1::PcpcYqjM displayed a somewhat higher Y(II) compared to Syn::PcpcYqjM. Compared to cells kept in preconditions without 1a, the WT and ΔFlv1 exposed to the substrate had markedly higher Y(I) in far red light but a relatively unchanged yield in red light; while Syn::PcpcYqjM and ΔFlv1::PcpcYqjM had comparable Y(I) regardless of the presence or absence of 1a. These results indicated that 1a has adverse and toxic effects on the photosynthetic machinery, especially PSII, and that the introduction of YqjM mitigates these negative effects by rapidly converting intracellular 1a to the product 1b.
Figure 5.

Yield of [a] PSI and [b] PSII without and with 1a (10 mM). Colored bar represents light conditions during the respective times (black: dark, dark red: far red light, red: red light). Values plotted represent averages derived from biological replicates (N = 2) ± SD. [c] Shows a schematic representation of the viability tests performed on Syn WT and Syn::PcpcYqjM with the original images presented in Figure S5 of the SI. For the tests, either the substrate or product were added at final concentrations of 10, 25, 50, and 100 mM. Cells used for the viability test were cultured under standard cultivation conditions (60 μE m–2 s–1, 30 °C) with the reactions themselves carried out with a light intensity of 150 μE m–2 s–1.
In order to translate these observations to cell viability and growth, viability assays in liquid cultures using 10, 25, 50, and 100 mM of 1a and 1b were conducted on Syn WT and Syn::PcpcYqjM. In all tests, the product 1b did not show toxicity as cells tolerated all tested concentrations; growing back on solid media even after 24 h of exposure (Figure 5c,d). As expected, 1a was much more toxic; as evidenced by the lack of WT growth after exposure to concentrations over 25 mM for 30 min (Figure 5c). In contrast, recombinant cells could be recultivated, albeit in a reduced fashion, after exposure to concentrations >25 mM for a period of 1 h (Figure 5c). The tolerance of the recombinant Syn::PcpcYqjM cells was also reflected by their activity, as complete conversions of 10 mM within 1 h, 25 mM within 4 h, and 50 mM within 7 h were possible (SI, Figure S2). Viability assays using Syn::PpsbA2YqjM can be found in the Supporting Information (SI, Figure S5).
Disruption of the Flv1/Flv3 Electron Valve Enhances YqjM Activity
The kinetic parameters of the enzyme (Figure 3) show clearly that the reductive-step is the rate limiting step of the YqjM-catalyzed ene-reduction. Hence, we assumed that by removing competing electron sinks, more electrons may be funnelled toward YqjM, leading to accelerated reaction rates in the whole-cell biotransformations. Potential electron channelling toward heterologous YqjM was done through the use of Synechocystis mutants lacking Flv1 and Flv3 which are known to work as a heterodimer to protect PSI.34,45
The activities of YqjM expressed in Flv1 and Flv3 deletion mutants were initially evaluated using cells, at a final OD750 ∼ 10, that were cultivated under standard cultivation conditions (60 μE m–2 s–1, 30 °C). Under those growth conditions, no significant differences in product formation rates and specific activities between Syn::PcpcYqjM and ΔFlv1::PcpcYqjM was observed (SI, Figure S6). We, therefore, speculated that effects—if any—would become apparent for cells cultured at higher light intensities due to the natural role of flavodiiron proteins, that are involved in photoprotection and acclimation.
The results presented (Figure 6) were obtained using cells cultivated at increased light intensities of 150 μE m–2 s–1 with the biotransformations themselves conducted in a photobioreactor46 with light intensities within the same range.
Figure 6.

Comparison of [a (i)] product formation rate and [a (ii)] specific whole cell activity normalized to cell density of Syn::PcpcYqjM) and the corresponding deletion mutants, ΔFlv1::PcpcYqjM and ΔFlv3::PcpcYqjM, carried out at an OD750 ∼10. Panel [b] presents active intracellular YqjM concentrations (relative to total protein), determined in vitro, between the three strains expressing YqjM under control of the Pcpc promoter.[c] shows examples of product formation over time at an OD750 ∼ 10, the values within the first 5–10 min were used for calculations whenever relevant. The effects of reducing the cell density used for biotransformations on [d (i)] whole cell specific activities normalized to cell density and [d (ii)] product formation rate are shown for the strains Syn::PcpcYqjM and ΔFlv1::PcpcYqjM and is compared to the values derived at an OD750 of 10. In all cases, the starting concentration of 1a was ∼10 mM. All bars represent data generated from reactions stemming from biological replicates (N = 3) with individual values depicted. Error bars represent SD. P-values were calculated using Welch’s t test and represent comparisons between Syn::PcpcYqjM and respective deletion mutant (***P < 0.001, **P < 0.01, *P <0.05). Exact P-values where asterisks are placed can be found in the raw data files.
We observed that the gene deletions, which prevent the formation of the Flv1/Flv3 heterodimer, led to improvements in the in vivo YqjM-mediated reduction of 1a at final cell densities where significant self-shading is expected (OD750 ∼10, chla = ∼ 60 μg mL–1). Specifically, the product formation rate of ΔFlv1::PcpcYqjM and ΔFlv3::PcpcYqjM resulted in a 2.70- and 2-fold improvement compared to Syn::PcpcYqjM, respectively (Figure 6a (i)). This effect held true when activities were normalized to cell density (Figure 6a (ii)) and to the content of the pigment chla (SI, Figure S7).
In order to rule out differences in enzymatic expression being responsible for improved activity, we determined active intracellular YqjM concentrations in an NADPH assay after cell disruption (for more details, refer to the Materials and Methods). These tests confirmed that YqjM levels, relative to total protein, did not significantly differ between Syn::PcpcYqjM and any of the Flv deletion mutants (Figure 6b); thus, effectively ruling out enzymatic expression differences as the cause of activity enhancement. The dark control reactions performed, however, indicate that sugars naturally present in the strains fuel at least a part of the reaction, albeit not to completion. We also noted that this dark activity was slightly more pronounced in both ΔFlv mutants. This may be attributed to an increased oxidative glycolytic metabolism brought about by the deletions which have been previously reported to occur in both Synechocystis39 and Chlamydomonas reinhardtii.(47)
The formation of the product, 1b, across the first 30 min of biotransformations is shown in Figure 6c. At a starting substrate concentration of 10 mM, all strains achieve full conversion within 1h; at higher concentrations, however, the enhanced ΔFlv1::PcpcYqjM activity was more pronounced. Using ΔFlv1::PcpcYqjM, 25 mM and 50 mM of 1a were converted completely within 1.5 and 3.5 h, respectively (SI, Figure S8) while Syn::PcpcYqjM required between 3 and 4 h and 5–7 h to convert 25 and 50 mM, respectively (SI, Figure S2).
Next, we sought to investigate the effects of lowering the cell density at which biotransformations are conducted on the product formation rate and whole-cell specific activities using Syn::PcpcYqjM and ΔFlv1::PcpcYqjM. As foreseen, productivities are highest at low cell densities (OD750 ∼ 2) where less self-shading occurs (Figure 6d (i)). At moderate cell densities (OD750 ∼ 10), however, the productivity of Syn::PcpcYqjM is negatively affected. This decrease in productivity appears to be indicative of NADPH limitations that arise due to light attenuation. Our results suggest that ΔFlv1::PcpcYqjM may alleviate these limitations as evidenced by a 2-fold improvement in specific activity. This improvement translated into a remarkable increase of product formation rate from 7.2 mM h–1 to 18.3 mM h–1(Figure 6d (ii)).
In order to demonstrate the effectiveness of the engineered cells for biocatalytic applications, we applied ΔFlv1::PcpcYqjM at cell densities of 2.4 gDCW L–1 (OD750 ∼10) to convert higher concentrations of 1a to the product 1b. We noted that ΔFlv1::PcpcYqjM could convert ∼60 mM of substrate in 4 h and 83.4 mM in 6 h, representing conversions of 100% and 94%, respectively, when a substrate feeding approach was followed. Collectively, these results indicate that the deletion of competing NADPH-consuming pathways represents a feasible approach to improve heterologous redox reactions.
In order to further validate our results, we sought to try substrates other than 1a under the same conditions described. We noted that, indeed, the activity-enhancing effects we report for ΔFlv1::PcpcYqjM are only apparent for fast reactions where we assume NADPH limitation occurs. For the substrate 2-methylcyclohexenone (5a) almost no differences in reaction rates (∼ 2 mM h–1 and 15 U gDCW–1) under both dark and light conditions (Figure 7) were observed, and thus, the Flv-deletion mutant does not enhance the reaction. In contrast, the other substrates all had faster reaction rates with the ΔFlv1::PcpcYqjM strain with a range of activities varying from 100 to 132 U gDCW–1. This shows that the rate-enhancing effect of the Flv-deletion applies for fast biotransformations. The fact that the conversion of 5a is not significantly accelerated indicates that here, the photosynthetic electron supply is sufficient to fuel this reaction.
Figure 7.

Evaluation of [a] product formation rate and [b] whole cell specific activity (normalized to dry cell weight) of YqjM using different substrates 2a–5a known to be accepted by YqjM using Syn::PcpcYqjM and ΔFlv1::PcpcYqjM. Biotransformations were carried out at an OD750 ∼10 and 10 mM substrate in a photobioreactor at an intensity of 150 μE m–2 s–1 and in the dark. All bars represent data generated from reactions stemming from biological replicates with individual values depicted. Error bars represent SD. P-values were calculated using Welch’s t test or Mann–Whitney. (*P <0.05). Exact P-values where asterisks are placed can be found in the raw data files. (N = 2–3).
Discussion
Here we demonstrate that the combination of different engineering strategies like promoter engineering and the simultaneous deletion of electron sinks has the potential to overcome NADPH limitation in light-driven biocatalysis. Moreover, it is shown, for the first time, that determining in vivo NADPH fluorescence is a valuable tool when engineering light-fueled biotransformations in cyanobacteria.
The utilization of photoautotrophic cyanobacteria and other algal strains in biotechnology holds the potential to expand the repertoire of environmentally friendly industrial processes. However, various obstacles have to be overcome. It is clear that sufficient light availability is a critical factor for cyanobacterial growth48 and for high productivity of NADPH-dependent heterologous reactions.49 However, feasible catalytic applications of cyanobacteria would require cell densities higher than a few grams per liter to obtain industrially relevant space-time-yields. Under these conditions, the limited availability of light quickly turns into a major bottleneck; the production of photosynthetic reducing equivalents (e.g., NADPH) is compromised, thus leading to an insufficient electron supply for any fast whole-cell redox reaction. As such, various engineering approaches have been focused on genetic manipulation of cofactor metabolism to increase the availability of reducing equivalents.50,51
In this work, the use of NADPH fluorescence for the characterization of a light-driven biotransformation with a recombinant NADPH dependent enzyme in vivo is shown. It has been previously applied to elucidate dynamic changes in intracellular NADPH levels and to demonstrate the interplay between light and dark reactions of cyanobacteria,52 the activity of the Calvin–Benson cycle upon decreasing inorganic carbon availability,53 the induction of ferredoxin-NAD(P)H reductase (FNR) at dark-to-light transitions and relative in vivo NADPH pool size,43 or to determine reaction partners of Flv1/Flv3.31,32 It is important to note that, although intracellular NADH may contribute to in vivo NADPH fluorescence, it is unlikely to be of significant value given that NADH concentrations are not expected to undergo dramatic light-dependent changes within the ms time-frames reported.43,52
Using the model ene-reductase YqjM, we show that the implementation of a strong heterologous electron sink together with elimination of a natural electron outlet can significantly improve photosynthesis-driven bioproduction processes. The integration of YqjM in Synechocystis can be used to quantify additional NADPH consumption by substrate depletion and product formation.11 With a specific activity of 680 μmol mgchla–1 h–1 (OD750 ∼2, chla ∼ 6 μg mL–1) Syn::PcpcYqjM constitutes a relatively strong and tunable electron sink that deviates at least ∼60% of photosynthetically produced NADPH. This is based on Kauny and Sétif’s estimation that the NADPH pool in Synechocystis has a photoproduction rate of 530–1079 μmol mgchla–1 h–1 (determined at a density equaling 2.5 μg mL–1 chla).43
We believe that the main reaction highlighted in this work, the stereoselective reduction of the substrate 1a, is not only a model for highly selective biotransformations but may also be applied to probe NADPH in photoautotrophs to study other pathways that deviate a large percentage of photosynthetic reducing equivalents. This approach could even be partnered with existing screening technologies, such as CRISPRi-based screenings.54
Other reported rates for recombinant whole-cell biotransformations in Synechocystis using heterologously produced oxidoreductases range from 5.5 U gDCW–1 for the monooxygenase AlkBGT,55 5.7 U gDCW–1 for the monooxygenase CHMO,25 20 U gDCW–1 for an imine reductase23 to 39 U gDCW–1 for a P450 monooxygenase.13 In comparison, the YqjM rates we report are consistently higher than 50 U gDCW–1. We assume that activities beyond this would inflict severe NADPH-supply limitations and would, therefore, be a general problem for the acceleration of light-driven whole cell biotransformations. In order to alleviate some of this limitation, we sought to increase the NADPH available for enzymatic redox reactions by the deletion of the Flv1/Flv3 oligomer. By following this approach, we were able to successfully increase the specific activity of the ene-reduction of 1a to ∼150 U gDCW–1 or 300 μmol mg(Chl)–1 h–1 at a cell density of 2.4 gDCW L–1 (OD750 ∼10) where self-shading reduces light availability and causes light fluctuations within the vessel. This is key to consider as most bioreactors often experience light and dark zones56,57 and the optimization of photoautotrophs within these constraints can be industrially and economically relevant. In fact, the improved activity we reported at 2.4 gDCW L–1, does fall in line with the supposed role of the Flv1/Flv3 oligomer as a strong electron sink under fluctuating light.34 It must be noted, however, that the ΔFlv mutants may be more light sensitive under specific conditions.31,58 Nonetheless, engineering heterologous strong electron sinks (e.g., YqjM) should overcome the imbalance between energy input and electron outlet eliminating oxidative stress and feedback reactions.
The product formation rate and specific activities we report with ΔFlv1::PcpcYqjM, represent, to our knowledge, the fastest overall rates reported for a light-powered biotransformation. Additionally, we assume that FDPs represent a significant source of NADPH oxidation59 and, as a result, their removal would favor a higher YqjM activity. Nonetheless, the product formation rate of 18.3 mM h–1 that we report of at an optical density of 10 compares favorably to rates obtained by using hydrogen16 or catalytic water-oxidation in vitro as electron source.17
We would like to note that although the deletion of either Flv1 or Flv3 as a competing electron sink seemingly confirms the limiting role of NADPH availability for the biotransformation, their physiological effects may be more complex. The exact redox partner of Flv1/Flv3, for instance, is as of yet not fully elucidated. Recently, the work from two different laboratories31,32 identified Fd as the electron donor of FDPs in an in vivo study. However, other recent in vitro findings by Brown et al.29 show that both Flv1 and Flv3 homodimers exhibit binding with NAD(P)H to catalyze the protective oxygen reduction reaction with efficiencies of 105 M–1 s–1.
In conclusion, our findings confirm that the supply of reduced redox cofactors for light-driven biotransformations in cyanobacteria is a limiting factor for the production of chemicals. This is further exacerbated by the reduction in photosynthetic activity due to lower light availability and fluctuating light, respectively, that cannot be avoided at higher cell densities. Nevertheless, the removal of natural electron sinks such as FDPs that are dispensable under controlled cultivation conditions appears to be a highly promising strategy to alleviate the effects of “self-shading”, which will be crucial for development of light-driven catalysts that can compete with current, cosubstrate-dependent systems.
Materials and Methods
Chemicals and Reagents
The substrate, 2-methylmaleimide (1a), and its product, 2-methylsuccinimide (1b) were obtained as white powder from the manufacturer Chiracon GmbH (Luckenwalde, Germany). All other materials were purchased from Sigma-Aldrich (Steinheim, Germany) or Carl Roth (Karlsruhe, Germany) unless otherwise indicated.
Strains and Plasmid
A list of all strains and plasmids used in this study is given in Table S1.
Plasmid Construction
FastCloning was used to clone the integrative plasmids SynRekB_PcpcYqjM and SynRekB_PziaYqjM. The genome of Synechocystis was isolated and used as the template to amplify the sequences for the promoters and their upstream regions. 628 bp upstream to the cpcB gene were amplified for the Pcpc promoter as previously reported. The cloned sequence for the Pzia promoter includes the repressor gene ziaR. The primers used for the cloning of these vectors are given in Table S2.
Construction of Synechocystis Mutants
All transformed strains in this work were generated by homologous recombination using plasmids described in Table S1, targeting the gene locus slr0168. WT and mutant strains were cultured at 30 °C in liquid BG-11 medium (pH = 8) and in an atmosphere of 50% humidity and atmospheric CO2 levels. Cultures were agitated by a rotary shaker at 140 rpm under white fluorescent light (60 μE m–2 s–1). Cells were grown to their midexponential phase (OD750 0.5–1) and washed with fresh BG-11, and subsequent natural transformation was carried out by mixing plasmid DNA (2–5 μg) with cells in a sterile tube (1.5 mL). This mixture was shaken in darkness (140 rpm) for a period of 5 h after which cells were plated on a sterile nitrocellulose membrane (GE Healthcare, catalog number: 11314874) placed on antibiotic free BG-11 plates. After 24 h, the membrane was transferred to a plate with low concentrations of the required antibiotics (10–25 μg mL–1) until colonies were discernible (10–21 days). As Synechocystis is polyploid in nature, complete segregation of the mutant alleles was achieved by restreaking the colonies onto plates supplemented with gradually increasing levels of antibiotics (50–150 μg mL–1). Segregation was confirmed via colony PCR. The used primers are listed in SI, Table S2.
Standard Cultivation and Maintenance Conditions
Seed cultures in liquid and on plates were maintained in a plant growth chamber (SWGC-1000, WISD lab instruments) under atmospheric carbon dioxide conditions, fitted with white fluorescent lamps that provide 24 h illumination at an average intensity falling within 40–60 μE m–2 s–1. The chamber was set to a temperature of 30 °C and humidity levels of 50%. Cells were cultured in 50, 100, or 300 mL Erlenmeyer flasks (working volumes: 25, 50, and 100 mL, respectively) on rotary shakers fitted within the chamber (140 rpm). Kanamycin (50 μg/mL) was used as selective antibiotic for all strains containing an expression cassette for YqjM. Additionally, ΔFlv1 and ΔFlv3 variants were supplemented with chloramphenicol (10 μg/mL) or spectinomycin (30 μg/mL), respectively.
Higher Light Cultivation Conditions
Synechocystis was cultivated in Erlenmeyer flasks (300 mL, working volume: 100 mL) in standard BG-11 containing the respective antibiotics at room temperature. Cultures were agitated on a rotary shaker (140 rpm) and illuminated following a constant light regime by a tunable LED lamp (CellDEG, Berlin, Germany) emitting mostly blue and red light (intensity of ∼150 μE m–2 s–1), spectra can be found in the SI (Figure S9). Cultures were grown until they reached an OD750 of 2–2.5, which typically required between 6–7 days. A graphical depiction of this particular set up is shown in the SI (Figure S10).
The light intensity was measured with a LI-250A light meter (LICOR Biosciences, Hamburg, Germany) and a submersible spherical micro quantum sensor, US-SQS/L (Walz, Effeltrich, Germany). A graphical depiction of light intensity measurement using both sensors and a table showing the values is shown in the SI (Figure S11, Table S3).
Quantification of Chlorophyll a Content
The content of chla was determined using methanol extraction as described previously. This was done for all strains discussed in this work and was used to generate line graphs to determine the relationship between the total chla content and the corresponding optical density (OD750). These graphs are presented in the SI (Figure S12).
Quantification of the Dry Cell Weight
Cells were grown to an OD750 of 2–2.5 and harvested by centrifugation. The cell pellet was resuspended in BG-11 medium to yield different optical densities. Samples from these (1 mL) were centrifuged, and the pellets were washed twice with Milli Q water before being dried in an incubator (60 °C). The resulting dried cell mass was weighed, and corresponding values were plotted against its respective OD750. A line graph depicting this is provided in the SI (Figure S13).
Light-Driven Biotransformations
After cultivation at standard or higher light conditions, cells were harvested by centrifugation (24 °C, 25 min, 3220g) and the resulting pellet was resuspended in fresh BG-11 to reach the desired optical density (measured at 750 nm). Whole-cell biotransformations were performed at 30 °C in 5 mL glass vials (working volume 1 mL) and were started by the addition of 1a–5a in the respective concentration. The vials were placed into a bioreactor equipped with LED lamps (average intensity 150 μE m–2 s–1) and were agitated (140 rpm). Multiple samples were taken as required at different time-points and typically included samples at the start (t0) and after 5, 10, 15, 30, 60, and 120 min with further sampling if necessary. The samples were directly frozen in liquid nitrogen to quench the reaction. Relevant control reactions were conducted in BG-11, under dark conditions and with WT cells—these are shown in the SI (Figures S14, S15). All substrate and product stocks 1b were prepared in BG-11.
Cell Disruption
Cells (1 mL, OD750 ≈ 20) were centrifuged (3 min, 4 °C, 16 000g), and the pellet was washed by resuspending it in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH = 7.4, 2 mL). After subsequent centrifugation (5 min, 4 °C, 16 000g), the supernatant was discarded, and the cells were resuspended in PBS (200 μL). The proteinase inhibitor aminocaproic acid (1 mM) and glass beads (diameter 0.25 mm) were added. Subsequently, cells were disrupted by vortexing (4 times x 30 s) with cooling breaks on ice (2 min) in between. After centrifugation (5 min, 4 °C, 16 000g), PBS (100 μL) was added, and the resulting blue colored cell lysate was carefully separated from the glass beads. The protein concentration of the crude cell extract was determined with BCA assay (ThermoFisher Scientific, catalog number: 23227) using BSA solution (25–2000 μg/mL) as a standard for calibration. Cell lysate was used for the in vitro NADPH assay and SDS-PAGE analysis.
In Vitro NAD(P)H Assay
Potassium buffer (20 mM, pH = 6.5) was degassed by sparging with N2 for 10 min. 1a (1 mM) and NAD(P)H (125 μM) were mixed with buffer in a cuvette. The reaction was started by the addition of cell lysate (100 μL) and the absorption at 340 nm was monitored for 3 min. An example of the raw data generated is given in the SI along with the determination of the respective extinction coefficients (SI, Figures S16, S17). Control reactions without the addition of substrates were performed for every lysate sample to subtract background reactions. The activity in mU mgtotal protein–1 was then calculated from the slope and extinction coefficient for NAD(P)H, and the protein concentrations determined with the BCA assay.
Viability Assay
Toxicity effects of 1a and 1b were investigated in the relevant time span for biotransformations as described previously. Substrate and product were added at final concentrations of 10, 25, 50, and 100 mM. To the respective time points, samples (100 μL) were taken and washed twice with BG-11 (500 μL). Cells were pelleted by centrifugation and streaked with a toothpick.
Quantitative GC-FID Analysis
GC-FID (GC-2010 Plus, Shimadzu, Japan) equipped with a ZB-5 column was used to analyze samples taken from biotransformations of 1a–4a. 5a and 5b were analyzed on GC-FID (GC-2030, Shimadzu, Japan) equipped with a hydrodex β-TBDAc column. Sample preparation was carried out by organic phase extraction using ethyl-acetate for compounds 1–5. 1-Decanol (2 mM) was used as an internal standard. Detailed description of the utilized separation method is available in the SI (Table S4). Additionally, a sample chromatogram and calibration for the substrate (1a) and product (1b) can be found (SI, Figure S18, S19).
In Vivo NADPH Decay Measurements
NADPH fluorescence measurements were performed using the NADPH/9-AA module of a DUAL-PAM 100 device (Walz, Germany) following the general set up and software settings described in the work of Kauny and Sétif. Samples for measurements were prepared by growing cultures (50 mL) until an OD750 of 1 was reached (30 °C, light intensity: 50 μE m–2 s–1). Cultures (20 mL) were harvested by centrifugation (4000g, 10 min, room temperature) and the sedimented cells were washed once with fresh BG-11 medium then resuspended to reach the final chlorophyll a concentration (2.5 μg mL–1). Then, cells were incubated at growth conditions for 30 min prior to the measurements. For measurements, cells from each culture (3 mL) were preincubated in a quartz cuvette at 30 °C under actinic light (AL) with an intensity of ∼120 μE m–2 s–1 and were alternated with dark periods using a regimen of 30 s on, 10 s off. Directly afterward, FastKinetics were measured using a 20 s Clock interval starting a trigger run that was as follows: AL off with a delay of 100 ms, AL on at 3000 ms, AL stays on after each triggered run. NADPH ML frequency was 5000 Hz during illumination and 100 Hz in darkness. A total of 20 measurements were averaged to obtain a sufficient signal-to-noise ratio. Samples were measured with or without the addition 1a (30 μL, 100 mM, final concentration 1 mM). If substrate was added, cells were mixed by pipetting and incubated under illumination (20 s) before starting the measurement. Data was normalized to 0 at the end of dark incubation. Steady-state fluorescence during illumination was determined by averaging the signal within the −100 ms delay in the beginning. The NADPH decay rate constant [b] was determined by fitting the decay part of the curves in the SigmaPlot software using the function: f = y0 + a – b * x.
Determination of Photosynthetic Parameters
Chlorophyll fluorescence and P700 redox kinetics in intact cells were determined by a pulse amplitude-modulated fluorometer (Dual-PAM-100, Walz, Germany). Prior to the activity measurements, chlorophyll a concentration was adjusted to 15 μg mL–1, and cells were exposed to 400 μE m–2 s–1 white light in AlgaeTRON AG130 cool-white LED chambers (PSI Instruments, Czech Republic) for 15 min (in the presence or absence of 10 mM 1a). Then, the sample cells were transferred to the sample holder of the instrument and kept in darkness for 1–2 min. During the measurements, multiple turnover saturating pulses (5000 μE m–2 s–1, 500 ms) were applied under far red (720 nm, 40 W m–2) and actinic red (635 nm, 50 μE m–2 s–1) illumination. Yields of PSI and PSII were calculated as described previously.60,61
Determination of YqjM Kinetic Parameters
In order to determine the reductive rate of YqjM, the rate of cofactor reduction was determined in an anaerobic glovebox using a stopped flow device equipped with a photomultiplier. Prior to the measurements, samples were flushed with nitrogen and incubated in the glovebox. A final concentration of 25 μM YqjM was shot against 30–1000 μM of NADPH or NADH using Tris-HCl (50 mM, pH 7.5) as a reaction buffer and the rapid reactions were recorded at 452 nm at 25 °C. The observed rates of cofactor reduction were plotted against the final concentration of NADPH or NADH to determine kred and KD.
The oxidative rate of YqjM was determined by the same device, anaerobic conditions were achieved as described above. For this experiment, the enzyme was reduced by adding approximately equimolar amounts of NADPH to the YqjM solution until approximately a 90% reduced flavin spectrum was observed. This reduced protein sample (final concentration: 20 μM YqjM) was then shot against a solution of 1a in buffer (final concentration of 25–500 μM) and the rapid reactions were recorded at 452 nm at 25 °C in Tris-HCl (50 mM, pH 7.5). In this setup, no saturation was observed, because the reaction progressed too rapidly to be measured at high substrate concentations; therefore, only the biomolecular rate kox of 2.08 ± 0.02 * 106 M–1 s–1 was determined.
Steady-state kinetics (SI, Figure S3) were determined by following the oxidation of 100 μM NADPH at 340 nm in Tris-HCl (50 mM, pH 7.5) at 25 °C using 100 nm of YqjM and 25–5000 μM 1a. Stock solutions of 1a were prepared in the assay buffer.
Statistical Analysis of Experimental Data
Unless otherwise indicated, statistical testing was performed using the appropriate tools in GraphPad Prism version 8.0. Unpaired Welch’s t test or Mann–Whitney was utilized for the analysis of two groups where data followed Gaussian distribution or violated it, respectively. The assumption of normality was assessed using Shapiro–Wilk’s test. For comparisons including three groups, one-way analysis of variance (ANOVA) was used, and the following pairwise comparisons were carried out using Tukey’s posthoc. The samples which were chosen for analysis were derived from a minimum of three independent experiments and cultivations. Data are presented as bar-graphs, line-graphs or in-text showing the mean ± SD as indicated. Wherever applicable, significance reported is based on an alpha value of 0.05 and is always two-sided.
Acknowledgments
The authors thank the Austrian Science Funds (FWF, P31001-B29), Deutsche Bundesstiftung Umwelt (DBU, AZ 33741/01-32) and the NordForsk Nordic Center of Excellence “NordAqua” (#82845) for financial support. We also thank Chiracon GmbH for the synthesis and provision of key compounds used in this study. Furthermore, we thank Dr. Elia Calderini (TU Graz) and Dr. Dmytro Neshchadin (TU Graz) for support and measurements of light spectra.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c02601.
Author Contributions
# L.A.-C. and H.C.B. contributed equally to this work
Author Contributions
R.K. and M.M.N. conceived the project and main conceptual ideas. L.A-C. and H.C.B. equally contributed to the development of the project, performed measurements, analyzed results, drafted the manuscript and designed figures. D.S. performed photosynthetic activity experiments, N.G.D-N performed NADPH decay measurements, K.K.F.B contributed to YqjM activity measurements under standard conditions and S.W determined the kinetics of YqjM experimentally. P.M supervised YqjM kinetics experiments and Y.A. supervised photosynthetic activity experiments and provided valuable insights. All authors discussed results, contributed to the writing of the manuscript and provided critical commentary.
The authors declare no competing financial interest.
Supplementary Material
References
- Claassens N. J.; Sousa D. Z.; dos Santos V. A. P. M.; de Vos W. M.; van der Oost J. Harnessing the Power of Microbial Autotrophy. Nat. Rev. Microbiol. 2016, 14 (11), 692–706. 10.1038/nrmicro.2016.130. [DOI] [PubMed] [Google Scholar]
- Janssen P. J. D.; Lambreva M. D.; Plumeré N.; Bartolucci C.; Antonacci A.; Buonasera K.; Frese R. N.; Scognamiglio V.; Rea G.. Photosynthesis at the Forefront of a Sustainable Life. Front. Chem. 2014, 2 ( (36), ). 10.3389/fchem.2014.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y.; Yoshino T.; Matsunaga T.; Matsumoto M.; Tanaka T. Marine Microalgae for Production of Biofuels and Chemicals. Curr. Opin. Biotechnol. 2018, 50, 111–120. 10.1016/j.copbio.2017.11.018. [DOI] [PubMed] [Google Scholar]
- Chew K. W.; Yap J. Y.; Show P. L.; Suan N. H.; Juan J. C.; Ling T. C.; Lee D.-J.; Chang J.-S. Microalgae Biorefinery: High Value Products Perspectives. Bioresour. Technol. 2017, 229, 53–62. 10.1016/j.biortech.2017.01.006. [DOI] [PubMed] [Google Scholar]
- Yarnold J.; Karan H.; Oey M.; Hankamer B. Microalgal Aquafeeds as Part of a Circular Bioeconomy. Trends Plant Sci. 2019, 24 (10), 959–970. 10.1016/j.tplants.2019.06.005. [DOI] [PubMed] [Google Scholar]
- Lauersen K. J.; Baier T.; Wichmann J.; Wördenweber R.; Mussgnug J. H.; Hübner W.; Huser T.; Kruse O. Efficient Phototrophic Production of a High-Value Sesquiterpenoid from the Eukaryotic Microalga Chlamydomonas reinhardtii. Metab. Eng. 2016, 38, 331–343. 10.1016/j.ymben.2016.07.013. [DOI] [PubMed] [Google Scholar]
- Liu X.; Miao R.; Lindberg P.; Lindblad P. Modular Engineering for Efficient Photosynthetic Biosynthesis of 1-Butanol from CO2 in Cyanobacteria. Energy Environ. Sci. 2019, 12 (9), 2765–2777. 10.1039/C9EE01214A. [DOI] [Google Scholar]
- Chin T.; Okuda Y.; Ikeuchi M. Sorbitol Production and Optimization of Photosynthetic Supply in the Cyanobacterium Synechocystis PCC 6803. J. Biotechnol. 2018, 276–277, 25–33. 10.1016/j.jbiotec.2018.04.004. [DOI] [PubMed] [Google Scholar]
- Knoot C. J.; Ungerer J.; Wangikar P. P.; Pakrasi H. B. Cyanobacteria: Promising Biocatalysts for Sustainable Chemical Production. J. Biol. Chem. 2018, 293 (14), 5044–5052. 10.1074/jbc.R117.815886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch F.; Luo Q.; Lu X.; Hagemann M. Inactivation of Invertase Enhances Sucrose Production in the Cyanobacterium Synechocystis sp. PCC 6803. Microbiology 2018, 164 (10), 1220–1228. 10.1099/mic.0.000708. [DOI] [PubMed] [Google Scholar]
- Köninger K.; Gómez Baraibar Á.; Mügge C.; Paul C. E.; Hollmann F.; Nowaczyk M. M.; Kourist R. Recombinant Cyanobacteria for the Asymmetric Reduction of C = C Bonds Fueled by the Biocatalytic Oxidation of Water. Angew. Chem., Int. Ed. 2016, 55 (18), 5582–5585. 10.1002/anie.201601200. [DOI] [PubMed] [Google Scholar]
- Hoschek A.; Schmid A.; Bühler B. In Situ O2 Generation for Biocatalytic Oxyfunctionalization Reactions. ChemCatChem 2018, 10 (23), 5366–5371. 10.1002/cctc.201801262. [DOI] [Google Scholar]
- Hoschek A.; Toepel J.; Hochkeppel A.; Karande R.; Bühler B.; Schmid A. Light-dependent and Aeration-independent Gram-scale Hydroxylation of Cyclohexane to Cyclohexanol by CYP450 Harboring Synechocystis sp. PCC 6803. Biotechnol. J. 2019, 14 (8), 1800724 10.1002/biot.201800724. [DOI] [PubMed] [Google Scholar]
- Wen X.; Du K.; Wang Z.; Peng X.; Luo L.; Tao H.; Xu Y.; Zhang D.; Geng Y.; Li Y. Effective Cultivation of Microalgae for Biofuel Production: A Pilot-Scale Evaluation of a Novel Oleaginous Microalga Graesiella sp. WBG-1. Biotechnol. Biofuels 2016, 9, 123. 10.1186/s13068-016-0541-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubble D. S.; Harper D. M. Impact of Light Regimen and Self-Shading by Algal Cells on Primary Productivity in the Water Column of a Shallow Tropical Lake (Lake Naivasha, Kenya). Lakes Reservoirs 2001, 6 (2), 143–150. 10.1046/j.1440-1770.2001.00133.x. [DOI] [Google Scholar]
- Lauterbach L.; Lenz O.; Vincent K. A. H2-Driven Cofactor Regeneration with NAD(P)+ -Reducing Hydrogenases. FEBS J. 2013, 280 (13), 3058–3068. 10.1111/febs.12245. [DOI] [PubMed] [Google Scholar]
- Kuk S. K.; Singh R. K.; Nam D. H.; Singh R.; Lee J.-K.; Park C. B. Photoelectrochemical Reduction of Carbon Dioxide to Methanol through a Highly Efficient Enzyme Cascade. Angew. Chem., Int. Ed. 2017, 56 (14), 3827–3832. 10.1002/anie.201611379. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; Yamanaka R. Light Mediated Cofactor Recycling System in Biocatalytic Asymmetric Reduction of Ketone. Chem. Commun. 2002, (16), 1782–1783. 10.1039/b203844g. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; Yamanaka R. Light-Mediated Regulation of Asymmetric Reduction of Ketones by a Cyanobacterium. Tetrahedron: Asymmetry 2002, 13 (23), 2529–2533. 10.1016/S0957-4166(02)00683-3. [DOI] [Google Scholar]
- Hölsch K.; Havel J.; Haslbeck M.; Weuster-Botz D. Identification, Cloning, and Characterization of a Novel Ketoreductase from the Cyanobacterium Synechococcus sp. Strain PCC 7942. Appl. Environ. Microbiol. 2008, 74 (21), 6697–6702. 10.1128/AEM.00925-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesic M.; Fernández-Fueyo E.; Hollmann F. Characterization of the Old Yellow Enzyme Homolog from Bacillus Subtilis (YqjM). ChemistrySelect 2017, 2 (13), 3866–3871. 10.1002/slct.201700724. [DOI] [Google Scholar]
- Kitzing K.; Fitzpatrick T. B.; Wilken C.; Sawa J.; Bourenkov G. P.; Macheroux P.; Clausen T. The 1.3 Å Crystal Structure of the Flavoprotein YqjM Reveals a Novel Class of Old Yellow Enzymes. J. Biol. Chem. 2005, 280 (30), 27904–27913. 10.1074/jbc.M502587200. [DOI] [PubMed] [Google Scholar]
- Büchsenschütz H. C.; Vidimce-Risteski V.; Eggbauer B.; Schmidt S.; Winkler C. K.; Schrittwieser J. H.; Kroutil W.; Kourist R. Stereoselective Biotransformations of Cyclic Imines in Recombinant Cells of Synechocystis Sp. PCC 6803. ChemCatChem 2020, 12 (3), 726–730. 10.1002/cctc.201901592. [DOI] [Google Scholar]
- Sengupta A.; Sunder A. V.; Sohoni S. V.; Wangikar P. P. The Effect of CO2 in Enhancing Photosynthetic Cofactor Recycling for Alcohol Dehydrogenase Mediated Chiral Synthesis in Cyanobacteria. J. Biotechnol. 2019, 289, 1–6. 10.1016/j.jbiotec.2018.11.002. [DOI] [PubMed] [Google Scholar]
- Böhmer S.; Köninger K.; Gómez-Baraibar Á.; Bojarra S.; Mügge C.; Schmidt S.; Nowaczyk M.; Kourist R. Enzymatic Oxyfunctionalization Driven by Photosynthetic Water-Splitting in the Cyanobacterium Synechocystis sp. PCC 6803. Catalysts 2017, 7 (8), 240. 10.3390/catal7080240. [DOI] [Google Scholar]
- Esteves-Ferreira A. A.; Inaba M.; Fort A.; Araújo W. L.; Sulpice R. Nitrogen Metabolism in Cyanobacteria: Metabolic and Molecular Control, Growth Consequences and Biotechnological Applications. Crit. Rev. Microbiol. 2018, 44 (5), 541–560. 10.1080/1040841X.2018.1446902. [DOI] [PubMed] [Google Scholar]
- Zhang C.-C.; Zhou C.-Z.; Burnap R. L.; Peng L. Carbon/Nitrogen Metabolic Balance: Lessons from Cyanobacteria. Trends Plant Sci. 2018, 23 (12), 1116–1130. 10.1016/j.tplants.2018.09.008. [DOI] [PubMed] [Google Scholar]
- Vicente J. B.; Gomes C. M.; Wasserfallen A.; Teixeira M. Module Fusion in an A-Type Flavoprotein from the Cyanobacterium Synechocystis Condenses a Multiple-Component Pathway in a Single Polypeptide Chain. Biochem. Biophys. Res. Commun. 2002, 294 (1), 82–87. 10.1016/S0006-291X(02)00434-5. [DOI] [PubMed] [Google Scholar]
- Brown K. A.; Guo Z.; Tokmina-Lukaszewska M.; Scott L. W.; Lubner C. E.; Smolinski S.; Mulder D. W.; Bothner B.; King P. W. The Oxygen Reduction Reaction Catalyzed by Synechocystis sp. PCC 6803 Flavodiiron Proteins. Sustain. Energy Fuels 2019, 3 (11), 3191–3200. 10.1039/C9SE00523D. [DOI] [Google Scholar]
- Santana-Sanchez A.; Solymosi D.; Mustila H.; Bersanini L.; Aro E.-M.; Allahverdiyeva Y. Flavodiiron Proteins 1–to-4 Function in Versatile Combinations in O2 Photoreduction in Cyanobacteria. eLife 2019, 8, e45766 10.7554/eLife.45766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkanen L.; Santana Sánchez A.; Ermakova M.; Rögner M.; Cournac L.; Allahverdiyeva Y. Functional Redundancy between Flavodiiron Proteins and NDH-1 in Synechocystis sp. PCC 6803. Plant J. 2020, 103, 1460–1476. 10.1111/tpj.14812. [DOI] [PubMed] [Google Scholar]
- Sétif P.; Shimakawa G.; Krieger-Liszkay A.; Miyake C. Identification of the Electron Donor to Flavodiiron Proteins in Synechocystis sp. PCC 6803 by in Vivo Spectroscopy. Biochim. Biophys. Acta, Bioenerg. 2020, 1861 (10), 148256. 10.1016/j.bbabio.2020.148256. [DOI] [PubMed] [Google Scholar]
- Helman Y.; Tchernov D.; Reinhold L.; Shibata M.; Ogawa T.; Schwarz R.; Ohad I.; Kaplan A. Genes Encoding A-Type Flavoproteins Are Essential for Photoreduction of O2 in Cyanobacteria. Curr. Biol. 2003, 13 (3), 230–235. 10.1016/S0960-9822(03)00046-0. [DOI] [PubMed] [Google Scholar]
- Allahverdiyeva Y.; Mustila H.; Ermakova M.; Bersanini L.; Richaud P.; Ajlani G.; Battchikova N.; Cournac L.; Aro E.-M. Flavodiiron Proteins Flv1 and Flv3 Enable Cyanobacterial Growth and Photosynthesis under Fluctuating Light. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (10), 4111–4116. 10.1073/pnas.1221194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokel M.; Johnson X.; Peltier G.; Aro E.-M.; Allahverdiyeva Y. Hunting the Main Player Enabling Chlamydomonas reinhardtii Growth under Fluctuating Light. Plant J. 2018, 94 (5), 822–835. 10.1111/tpj.13897. [DOI] [PubMed] [Google Scholar]
- Helman Y.; Barkan E.; Eisenstadt D.; Luz B.; Kaplan A. Fractionation of the Three Stable Oxygen Isotopes by Oxygen-Producing and Oxygen-Consuming Reactions in Photosynthetic Organisms. Plant Physiol. 2005, 138 (4), 2292–2298. 10.1104/pp.105.063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdiyeva Y.; Ermakova M.; Eisenhut M.; Zhang P.; Richaud P.; Hagemann M.; Cournac L.; Aro E.-M. Interplay between Flavodiiron Proteins and Photorespiration in Synechocystis sp. PCC 6803. J. Biol. Chem. 2011, 286 (27), 24007–24014. 10.1074/jbc.M111.223289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berepiki A.; Gittins J. R.; Moore C. M.; Bibby T. S. Rational Engineering of Photosynthetic Electron Flux Enhances Light-Powered Cytochrome P450 Activity. Synth. Biol. 2018, 3 (1), ysy009. 10.1093/synbio/ysy009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel K.; Patrikainen P.; Nagy C.; Fitzpatrick D.; Pope N.; Aro E.-M.; Kallio P. Redirecting Photosynthetic Electron Flux in the Cyanobacterium Synechocystis sp. PCC 6803 by the Deletion of Flavodiiron Protein Flv3. Microb. Cell Fact. 2019, 18 (1), 189. 10.1186/s12934-019-1238-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokel M.; Nagy V.; Tóth S. Z.; Kosourov S.; Allahverdiyeva Y. Elimination of the Flavodiiron Electron Sink Facilitates Long-Term H2 Photoproduction in Green Algae. Biotechnol. Biofuels 2019, 12, 280. 10.1186/s13068-019-1618-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund E.; Liang F.; Lindberg P. Evaluation of Promoters and Ribosome Binding Sites for Biotechnological Applications in the Unicellular Cyanobacterium Synechocystis Sp. PCC 6803. Sci. Rep. 2016, 6, 36640. 10.1038/srep36640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelwell C.; Robinson N. J.; Turner-Cavet J. S. An SmtB-like Repressor from Synechocystis PCC 6803 Regulates a Zinc Exporter. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (18), 10728–10733. 10.1073/pnas.95.18.10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauny J.; Sétif P. NADPH Fluorescence in the Cyanobacterium Synechocystis Sp. PCC 6803: A Versatile Probe for in vivo Measurements of Rates, Yields and Pools. Biochim. Biophys. Acta, Bioenerg. 2014, 1837 (6), 792–801. 10.1016/j.bbabio.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Zaffagnini M.; Fermani S.; Marchand C. H.; Costa A.; Sparla F.; Rouhier N.; Geigenberger P.; Lemaire S. D.; Trost P. Redox Homeostasis in Photosynthetic Organisms: Novel and Established Thiol-Based Molecular Mechanisms. Antioxid. Redox Signaling 2019, 31 (3), 155–210. 10.1089/ars.2018.7617. [DOI] [PubMed] [Google Scholar]
- Allahverdiyeva Y.; Isojärvi J.; Zhang P.; Aro E.-M. Cyanobacterial Oxygenic Photosynthesis Is Protected by Flavodiiron Proteins. Life 2015, 5 (1), 716–743. 10.3390/life5010716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyza Özgen F.; Runda M. E.; Burek B. O.; Wied P.; Bloh J. Z.; Kourist R.; Schmidt S. Artificial Light-harvesting Complexes Enable Rieske Oxygenase Catalyzed Hydroxylations in Non-photosynthetic Cells. Angew. Chem., Int. Ed. 2020, 59 (10), 3982–3987. 10.1002/anie.201914519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saroussi S.; Karns D. A. J.; Thomas D. C.; Bloszies C.; Fiehn O.; Posewitz M. C.; Grossman A. R. Alternative Outlets for Sustaining Photosynthetic Electron Transport during Dark-to-Light Transitions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (23), 11518–11527. 10.1073/pnas.1903185116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P. E.; Leister D.. Cyanobacteria as an Experimental Platform for Modifying Bacterial and Plant Photosynthesis. Front. Bioeng. Biotechnol. 2014, 2. 10.3389/fbioe.2014.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebesta J.; Peebles C. AM. Improving Heterologous Protein Expression in Synechocystis Sp. PCC 6803 for Alpha-Bisabolene Production. Metab. Eng. Commun. 2020, 10, e00117 10.1016/j.mec.2019.e00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf W.; Schlebusch M.; Hüge J.; Kopka J.; Hagemann M.; Forchhammer K. Metabolic Changes in Synechocystis PCC6803 upon Nitrogen-Starvation: Excess NADPH Sustains Polyhydroxybutyrate Accumulation. Metabolites 2013, 3 (1), 101–118. 10.3390/metabo3010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Zhang F.; Meng H.; Zhang Y.; Li Y. Introducing Extra NADPH Consumption Ability Significantly Increases the Photosynthetic Efficiency and Biomass Production of Cyanobacteria. Metab. Eng. 2016, 38, 217–227. 10.1016/j.ymben.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Mi H.; Klughammer C.; Schreiber U. Light-Induced Dynamic Changes of NADPH Fluorescence in Synechocystis PCC 6803 and Its NdhB-Defective Mutant M55. Plant Cell Physiol. 2000, 41 (10), 1129–1135. 10.1093/pcp/pcd038. [DOI] [PubMed] [Google Scholar]
- Holland S. C.; Kappell A. D.; Burnap R. L. Redox Changes Accompanying Inorganic Carbon Limitation in Synechocystis sp. PCC 6803. Biochim. Biophys. Acta, Bioenerg. 2015, 1847 (3), 355–363. 10.1016/j.bbabio.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Yao L.; Shabestary K.; Björk S. M.; Asplund-Samuelsson J.; Joensson H. N.; Jahn M.; Hudson E. P. Pooled CRISPRi Screening of the Cyanobacterium Synechocystis sp PCC 6803 for Enhanced Industrial Phenotypes. Nat. Commun. 2020, 11 (1), 1666. 10.1038/s41467-020-15491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoschek A.; Bühler B.; Schmid A. Stabilization and Scale-up of Photosynthesis-driven Ω-hydroxylation of Nonanoic Acid Methyl Ester by Two-liquid Phase Whole-cell Biocatalysis. Biotechnol. Bioeng. 2019, 116 (8), 1887–1900. 10.1002/bit.27006. [DOI] [PubMed] [Google Scholar]
- Heining M.; Sutor A.; Stute S. C.; Lindenberger C. P.; Buchholz R. Internal Illumination of Photobioreactors via Wireless Light Emitters: A Proof of Concept. J. Appl. Phycol. 2015, 27 (1), 59–66. 10.1007/s10811-014-0290-x. [DOI] [Google Scholar]
- Barbosa M. J.; Janssen M.; Ham N.; Tramper J.; Wijffels R. H. Microalgae Cultivation in Air-Lift Reactors: Modeling Biomass Yield and Growth Rate as a Function of Mixing Frequency. Biotechnol. Bioeng. 2003, 82 (2), 170–179. 10.1002/bit.10563. [DOI] [PubMed] [Google Scholar]
- Hackenberg C.; Engelhardt A.; Matthijs H. C. P.; Wittink F.; Bauwe H.; Kaplan A.; Hagemann M. Photorespiratory 2-Phosphoglycolate Metabolism and Photoreduction of O2 Cooperate in High-Light Acclimation of Synechocystis sp. Strain PCC 6803. Planta 2009, 230 (4), 625–637. 10.1007/s00425-009-0972-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulychev A. A.; Cherkashin A. A.; Muronets E. M.; Elanskaya I. V. Photoinduction of Electron Transport on the Acceptor Side of PSI in Synechocystis PCC 6803 Mutant Deficient in Flavodiiron Proteins Flv1 and Flv3. Biochim. Biophys. Acta, Bioenerg. 2018, 1859 (10), 1086–1095. 10.1016/j.bbabio.2018.06.012. [DOI] [PubMed] [Google Scholar]
- Klughammer C.; Schreiber U. Complementary PS II Quantum Yields Calculated from Simple Fluorescence Parameters Measured by PAM Fluorometry and the Saturation Pulse Method. PAM Appl. Notes 2008, 1, 27–35. [Google Scholar]
- Klughammer C.; Schreiber U. Saturation Pulse Method for Assessment of Energy Conversion in PS I. PAM Application Notes 2008, 1, 11–14. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.