

Members of the arrestin superfamily have great propensity of self-association, but the physiological significance of this phenomenon is unclear. To determine the biological role of visual arrestin-1 oligomerization in rod photoreceptors, we expressed mutant arrestin-1 with severely impaired self-association in mouse rods and analyzed mice of both sexes.
Keywords: arrestin, cell death, oligomerization, photoreceptor, rod
Abstract
Members of the arrestin superfamily have great propensity of self-association, but the physiological significance of this phenomenon is unclear. To determine the biological role of visual arrestin-1 oligomerization in rod photoreceptors, we expressed mutant arrestin-1 with severely impaired self-association in mouse rods and analyzed mice of both sexes. We show that the oligomerization-deficient mutant is capable of quenching rhodopsin signaling normally, as judged by electroretinography and single-cell recording. Like wild type, mutant arrestin-1 is largely excluded from the outer segments in the dark, proving that the normal intracellular localization is not due the size exclusion of arrestin-1 oligomers. In contrast to wild type, supraphysiological expression of the mutant causes shortening of the outer segments and photoreceptor death. Thus, oligomerization reduces the cytotoxicity of arrestin-1 monomer, ensuring long-term photoreceptor survival.
SIGNIFICANCE STATEMENT Visual arrestin-1 forms dimers and tetramers. The biological role of its oligomerization is unclear. To test the role of arrestin-1 self-association, we expressed oligomerization-deficient mutant in arrestin-1 knock-out mice. The mutant quenches light-induced rhodopsin signaling like wild type, demonstrating that in vivo monomeric arrestin-1 is necessary and sufficient for this function. In rods, arrestin-1 moves from the inner segments and cell bodies in the dark to the outer segments in the light. Nonoligomerizing mutant undergoes the same translocation, demonstrating that the size of the oligomers is not the reason for arrestin-1 exclusion from the outer segments in the dark. High expression of oligomerization-deficient arrestin-1 resulted in rod death. Thus, oligomerization reduces the cytotoxicity of high levels of arrestin-1 monomer.
Introduction
Arrestin-1 [Arr1; (historic names are S-antigen, 48 kDa protein, visual, or rod arrestin)], a member of the arrestin family selectively expressed in the retina, is the key regulator of rhodopsin signaling in rod photoreceptors. Arr1 binds with high affinity to light-activated rhodopsin phosphorylated by rhodopsin kinase, which precludes light-dependent activation of the rod phosphodiesterase (Wilden et al., 1986). Reversible oligomerization of Arr1 was described before its function in photoreceptors was elucidated (Wacker et al., 1977). Analytical ultracentrifugation of purified arrestin-1 revealed monomer–dimer–tetramer equilibrium in solution (Schubert et al., 1999). Self-association via monomer–dimer–tetramer equilibrium was found to be a common feature of bovine, mouse, and human Arr1 (Hanson et al., 2008a; Kim et al., 2011), suggesting that it must be biologically important. However, the biological function of Arr1 oligomerization remains unclear. The identification of the key Arr1 residues involved in oligomerization provided the first opportunity to examine the potential biological significance of the Arr1 oligomerization in living animals. Here we used transgenic expression of an oligomerization-deficient mouse Arr1 in rods at varying levels, on the background of Arr1 knockout. We focused on addressing the following three questions regarding arrestin-1 self-association: (1) does it play a role in quenching rhodopsin signaling in vivo?; (2) does it affect arrestin-1 subcellular distribution in photoreceptors?; and (3) does it affect photoreceptor health and survival? Our data indicate that in vivo arrestin-1 self-association is not required for efficient rhodopsin quenching and does not affect its subcellular distribution in photoreceptors, but is necessary for rod functionality and longevity.
Materials and Methods
Ethics statement
Animal research was conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Vanderbilt University Institutional Animal Care and Use Committee/Office of Animal Welfare Assurance (protocol ID M1800048-00). The protocol for suction electrode recordings was approved by the Institutional Animal Care and Use Committee of the University of California, Davis.
Construction of oligomerization-deficient mouse Arr1 mutant and generation of transgenic mice
Double mutation F86A+F198A that suppresses self-association of mouse Arr1 (Kim et al., 2011) was introduced into mouse Arr1 cDNA (a gift from Dr. Cheryl Craft, University of Southern California) by PCR and confirmed by dideoxy sequencing. The coding sequence of F86A+F198A mutant (oligomerization-deficient Arr1, referred to as CM) with extended 5′- and 3′-UTRs followed by mp1 polyadenylation signal was placed under the control of the pRho4-1 rhodopsin promoter (Mendez et al., 2000) and was used to create several transgenic lines, as described previously (Nair et al., 2005; Song et al., 2009). Transgenic lines were bred onto an Arr1−/− background (Xu et al., 1997), and the levels of expression of arrestin and rhodopsin were determined by Western blot in retinal homogenates, as described previously (Song et al., 2009, 2011). Mice of both sexes were used.
Protein purification
Mouse Arr1 was expressed in Escherichia coli and purified by sequential heparin-Sepharose and Q-Sepharose chromatography, as described previously (Vishnivetskiy et al., 2014). Rhodopsin was purified from bovine retinas, as described previously (Vishnivetskiy et al., 2007).
Animals
Mice were maintained under controlled ambient illumination on a 12 h light/dark cycle unless otherwise specified. Genotype was determined by standard PCR-based technique, and the mice of all transgenic lines were regularly backcrossed to fresh breeders to maintain the genetic homogeneity of the colony. Mice of both sexes were used in the experiments, and the experimental groups were balanced by sex.
CM expression in the different transgenic lines
Whole mouse retina homogenate preparation
Dissected retinas were homogenized by pipetting and sonication (30 s at 15% maximum power; sonic dismembrator Model 500, Thermo Fisher Scientific) in 100 mm Tris-HCl, pH 8.0; 2 mm EDTA; 2 mm benzamidine; 1 mm PMSF (dissolved as 100× stock in DMSO). The homogenates were centrifuged for 3 min at 3000 rpm at 4°C in an Eppendorf 5417R centrifuge to pellet cell debris. Supernatant was collected, and protein concentration was measured by Bradford Assay (Bio-Rad). Protein was precipitated by the addition of nine volumes of methanol, washed with 90% methanol, and dissolved in SDS sample buffer.
Western blotting
The expression of Arr1 and rhodopsin in different lines was measured by quantitative Western blot, essentially as described previously (Song et al., 2011). Briefly, aliquots of whole retina homogenates containing 0.1–2.0 ng rhodopsin or 2–5 ng arrestin were subjected to SDS-PAGE along with four standards containing known amounts of the corresponding purified proteins in the same range. Rhodopsin and arrestin standards were supplemented with the same amount of protein from the retina of rhodopsin and arrestin knock-out mice, respectively, that was present in experimental samples. The proteins were transferred to an Immobilon-P (Millipore) membrane, which was blocked with 5% nonfat dry milk in TBS containing 0.1% Tween-20 (TBST) for 60 min at room temperature with gentle rocking. The membranes were rinsed with TBST, and arrestin and rhodopsin blots were incubated overnight at 4°C with gentle rocking with rabbit polyclonal anti-arrestin F431 antibody (1:10,000; Vishnivetskiy et al., 2014) or mouse monoclonal 4D2 anti-rhodopsin antibody (1:10,000; a gift from Dr. R.S. Molday; Department of Biochemistry and Molecular biology, the University of British Columbia, Canada), respectively, in TBST supplemented with 2% BSA. GAPDH and COX IV blots were incubated overnight at 4°C with appropriate primary antibodies (rabbit polyclonal COX IV antibody (1:1000; catalog #11–035-144, Jackson ImmunoResearch) and GAPDH (1:500; Thermo Fisher Scientific). The blots were washed three times with TBST and incubated 1 h at room temperature with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies (1:10,000; Jackson ImmunoResearch). After several washes, the bands were visualized with WestPico Pierce chemiluminescence reagent (Thermo Fisher Scientific) according to manufacturer instructions. Developed blots were exposed to Super RX X-ray film (Fujifilm) for appropriate periods to ensure that none of the bands to be used for quantification purposes are saturated on the film. The signals were quantified using VersaDoc with QuantityOne software (Bio-Rad), and the amount of arrestin and rhodopsin in experimental samples was quantified based on linear calibration curves using GraphPad Prism version 8.0.0 for Mac (GraphPad Software). Alternatively, the bands were directly detected using C-Digit (LI-COR) and Image Studio Digits software (version 1.14). Because rhodopsin always appears as a series of bands believed to represent monomers, dimers, trimers, and higher-order oligomers, the sum of the signal in all rhodopsin bands in each lane was used for quantification. The signal in corresponding areas of these “blank” lanes was subtracted from the signal in standards and all other samples. Each protein in every eye sample was quantified on two to three independent blots.
The analysis of retinal histology
At the indicated ages (from 6 to 52 weeks), mice were killed by an overdose of isoflurane, the eyes were enucleated and fixed in 4% paraformaldehyde at 4°C overnight, cryoprotected with 30% sucrose in PBS for 6 h, and frozen at −80°C. Sections (30 µm) were cut and mounted on poly-lysine-coated (0.1 mg/ml) slides. The sections were rehydrated for at least 40 min in PBS, pH 7.2, and were incubated for 10 min in PBS with 0.1% Triton X-100, washed twice for 5 min in PBS, and stained with Invitrogen NeuroTrace 500/525 green fluorescent Nissl (Thermo Fisher Scientific) in PBS (dilution, 1:100) for 20 min. The sections were then washed with PBS with 0.1% Triton X-100 for 10 min, twice with PBS (5 min each), and kept overnight at 4°C in PBS. Mounted sections were analyzed by confocal microscopy (LSM510 microscope, Zeiss). The outer nuclear layer (ONL) was visualized by fluorescence in FITC (green) channel, outer segments (OS) were viewed using differential interference contrast (DIC). Two-channel images were acquired for quantitative analysis. Nissl stains nucleic acids, providing a convenient method of prominently labeling DNA-rich nuclei in the outer nuclear layer and yielding the characteristic more diffuse staining of RNA-rich inner segments (ISs). It also helps to identify the outer segments that are essentially free of nucleic acids and therefore contain no Nissl stain, but are clearly visible in the DIC images.
The length of OS and the thickness of ONL were measured using NIS-Elements (Nikon Instruments). The retinas of five mice (three sections per mouse) for each genotype were used. Each side of the section was divided into three segments according to the distance from optical nerve: central, medium, and peripheral in both inferior and superior hemispheres, so that the whole length of the section was divided into six segments. The measurements were performed at five points spaced at equal distances within each segment. Average values of each parameter for individual animals was used to calculate the mean and SD for each genotype.
Immunohistochemistry
The eyes were carefully enucleated, immediately immersed into fixing solution [4% paraformaldehyde (Electron Microscopy Sciences) in 0.1 m PBS, pH 7.2] and incubated 24 h at 4°C in the dark (dark-adapted eyes) or in 4500 lux light (light-adapted eyes). Fixed eyes were rinsed three times with PBS, pH 7.2, cryoprotected overnight in 30% sucrose in PBS, and frozen at −80°C. The eyes were cut into 30-µm-thick sections. Free-floating sections were blocked for 1 h in PBS with 0.3% Triton X-100 and 5% BSA at room temperature and then incubated with rabbit polyclonal anti-arrestin antibody C10C10 (a gift from Narsing A. Rao, University of Southern California) in PBS with 0.03% Triton X-100, 1% BSA (integrated circuit buffer). The best for quantification staining was achieved after 48 h of incubation with the antibody at 1:1000 dilution for wild type (WT) and CM-77arr−/− and 5 d of incubation with the antibody at 1:4000 dilution for CM-292arr−/−, free-floating sections at 4°C, as judged by the intensity of staining of WT. After washing with PBS buffer, the sections were sequentially incubated with biotinylated anti-rabbit antibody (Vector Laboratories) and Invitrogen streptavidin-Alexa Fluor-488 antibody (Thermo Fisher Scientific) at a dilution of 1:1000 for 1 h each at room temperature. After the final wash with PBS buffer, the sections were mounted on Superfrost Plus Microslides (VWR) coverslip by using Invitrogen Antifade reagent (Thermo Fisher Scientific) and viewed by using the 40× 1.3 PlanNeoFluor objective lens of a Zeiss LSM 510 laser-scanning confocal microscope. The immunofluorescence was detected by excitation with a 488 nm laser line and an LP505 filter. The same settings for the acquisition of all images were used to enable subsequent quantitative analysis. DIC images were acquired in parallel to facilitate the identification of subcellular compartments. The total amount of fluorescence in the outer segments, inner segments, outer nuclear layer, and synaptic terminals was determined using NIS-Elements (Nikon Instruments) imaging software by creating a single-line fluorescence intensity profile across the photoreceptor layer and finding the area under the curve for respective photoreceptor region. The percentage of Arr1 in each region was calculated as a fraction of total area under the curve across the photoreceptor layer (see Fig. 7). The retinas of three to four mice (three sections per mouse) of both sexes for each genotype were used.
Figure 7.
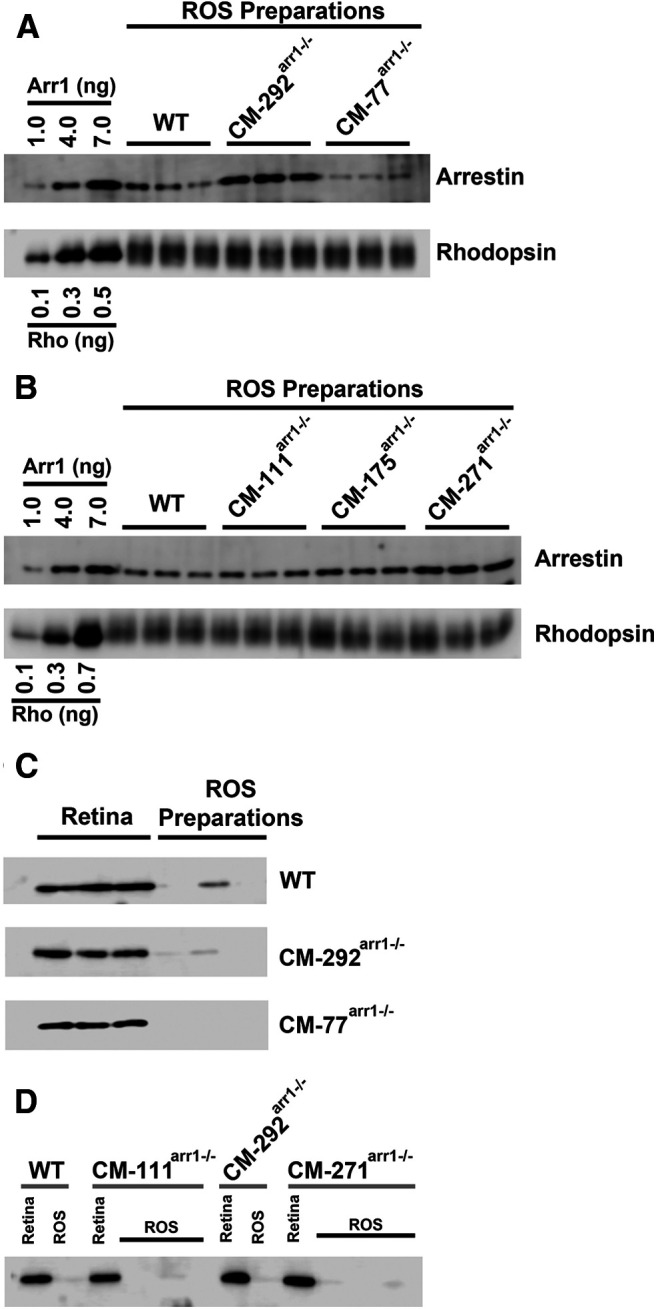
The expression of CM and levels in OSs in different transgenic lines. A, B, Western blot showing the levels of arrestin-1 in the rod outer segments in WT and different transgenic lines, with indicated amounts of corresponding purified proteins used as standards. Arr1 and rhodopsin were measured by quantitative Western blot, as described previously (Song et al., 2011). Three independent OS preparations used for each genotype are shown. The amounts of rhodopsin (Rho) were equalized to show the differences in Arr1 levels. C, D, To test the purity of the OS fraction and the extent of contamination with inner segment material, aliquots of whole retina (containing 30 ng of rhodopsin) and ROS homogenates (containing 150 ng of rhodopsin) from mice of indicated genotypes were run side by side on an SDS-PAGE gel. Western blots were developed with rabbit polyclonal antibody against mitochondrial marker COX IV (catalog #11-035-144, Jackson ImmunoResearch) and HRP-conjugated secondary anti-rabbit antibody (catalog #4844, Cell Signaling Technology). Only blots where calibration curves showed r2 > 0.95 were used.
Electroretinography
Electroretinograms (ERGs) were recorded from 7- to 8-week-old mice reared in 12 h light/dark cycle and dark-adapted overnight, as described previously (Lyubarsky et al., 2002; Song et al., 2009). Under a dim red light, mice were anesthetized by intraperitoneal injection of 15–20 ketamine, 6–8 xylazin, 600–800 mg/g body weight urethane in PBS. The pupils were dilated with 1% tropicamide in PBS. Eye electrodes made with a coiled 0.2 mm gold wire (catalog #D200, Diagnosys) were placed on the corneas of both eyes, a tungsten needle reference electrode was placed in the cheek, and a ground needle electrode was placed in the tail. ERGs were recorded using E2 Desktop visual electrophysiology system with software Espion Version 6.0.50 (Diagnosys). A Ganzfeld chamber was used to produce brief (from 20 µs to 1 ms) full-field flash stimuli. The light intensity was calibrated by the manufacturer and computer controlled. The mouse was placed on a heating plate connected to a temperature control unit to maintain the temperature at 37–38°C throughout the experiment.
Single-flash protocol
A series of stimuli from 0.00025 to 138 cd * s/m2 (or −3.6 to 2.15 log cd * s/m2, covering the range of 5.75 log units) in steps of 0.2 log units was presented in an ascending order. Sufficient time interval between flashes (from 20 s for low-intensity flashes up to 3 min for the highest intensity) was allowed for dark adaptation. To increase precision, the responses to dim flashes were recorded two to three times. The a-wave amplitude was measured from baseline to the a-wave trough, and the b-wave amplitude was measured from the a-wave trough to the b-wave peak. The retinal response from rods is called the scotopic ERG, which has no discernible a-wave and a slow b-wave.
Double-flash protocol
The double-flash recording was used to analyze the kinetics of recovery (Lyubarsky and Pugh, 1996). A test flash was delivered to suppress the circulating current of the rod photoreceptors. The recovery of this current was monitored by delivering a second (probe) flash. The time interval between the two flashes was varied from 50 to 2000 ms. The intensity of the test and probe flash was −0.4 and 0.65 log cd * s/m2, respectively, corresponding to ∼400 and ∼4500 photoisomerizations per rod (Lyubarsky et al., 2004). Sufficient time for dark adaptation was allowed between trials, as determined by the reproducibility of the response to the test flash. The response to a probe flash recorded without a preceding test flash was used to normalize the responses to probe flashes following a test flash. The normalized amplitude of the probe flash a-wave was plotted as a function of time between the two flashes. Instead of fitting the data points to a theoretical equation, which is based on certain assumptions that may not be correct for all of the genotypes, we fitted curves with polynomial nonlinear regression using GraphPad Prism version 8.0.0 for Mac (GraphPad Software) and considered R2 > 0.95 as a criterion for a good fit. The time interval necessary for half recovery was calculated from this fit (see Fig. 4).
Figure 4.

CM-expressing rod photoreceptors have normal flash responses. A, Suction electrode recordings of families of responses to flashes that ranged from 6 to 52,500 photons/µm2 with sequentially increased strength by factors of 2–4. Flashes were delivered at t = 0 s. B, Population average single-photon responses calculated from WT (n = 18) and CM77arr−/− (n = 17) rods. Light shading represents SEM. C, Relationship between the time that a bright flash response remained in saturation and the natural log of the number of photoexcited rhodopsins (R*) produced by the flash. The initial slope reflects the dominant time constant of recovery for bright flashes, which was the same for mutant and WT rods. Error bars represent the SEM.
Single-cell recording
Suction electrode recording was performed at the University of California, Davis, according to an approved protocol from the Institutional Animal Care and Use Committee and in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were maintained in 12 h cyclic lighting conditions and dark-adapted overnight before experiments. Suction electrode recordings were performed at 37°C in oxygenated, bicarbonate-buffered Locke's solution, as described previously (Gross et al., 2012). Briefly, the outer segment of an intact rod was drawn into a polished glass pipette filled with 140 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 3 mm HEPES, 0.2 mm EDTA, and 10 mm glucose, pH 7.4. The pipette and bath electrode were electrically connected to calomel half-cells by agar bridges, and the bath voltage was regulated by an active clamp circuit to minimize electrical artifacts. The membrane current was amplified by an Axopath 1B (Molecular Devices; filtered by an eight-pole Bessel filter with 30 Hz cutoff frequency, and digitized at 200 Hz using NiDAQ for Igor Pro (WaveMetrics).
The average single-photon response for each cell was estimated by variance-to-mean analysis of an ensemble of dim flash responses (at least 25 responses with amplitudes of <20% of the dark current), and the waveforms were averaged across cells of a given genotype. For analysis of saturating responses (see Fig. 4C), the time spent in saturation (Tsat) was defined as the time between the midpoint of the 10 ms flash and 10% recovery from saturation. One of the CM-77arr−/− rods with normal dim flash responses dramatically slowed when the flashes became saturating. This cell was excluded from the Tsat analysis presented in Table 1 (and see Fig. 4). Neither including nor excluding the cell resulted in a statistically significant difference between WT and mutant saturating flash recovery τD values.
Table 1.
Average kinetic parameters of WT and CM-77 light responses
| Dark current (pA) | SPR amplitude (pA) | Dim flash recovery (τrec) (ms) | Dim flash integration time (ms) | Flash sensitivity Io (photons μm−2) | Saturating flash recovery τD (ms) | Collecting area (μm2) | |
|---|---|---|---|---|---|---|---|
| WT | 11.8 ± 0.7 (18) | 0.55 ± 0.05 (18) | 149 ± 5.6 (18) | 187 ± 12 (18) | 46 ± 2.7 (18) | 163 ± 8.6 (18) | 0.42 ± 0.05 (18) |
| CM-77 | 12.9 ± 0.9 (17) | 0.52 ± 0.08 (17) | 174 ± 16 (17) | 246 ± 28 (17) | 52 ± 4.5 (17) | 200 ± 23 (16) | 0.45 ± 0.04 (17) |
For each, p > 0.05. Parameters were determined as described in the study by Gross et al. (2012).
Calculations of the concentration of CM monomers and dimers based on its total expression level
As CM dimerizes, but does not form tetramers, the calculations of the concentration of the monomer were based on the measured dimerization constant of this mutant [KD (dissociation constant) = 537 μm; Kim et al., 2011] and its measured (see Fig. 2) expression level. The concentration of WT Arr1 in the ISs and cell bodies of C57BL/6J mice (at 100% expression) is ∼2000 μm (Kim et al., 2011; Song et al., 2011). The distribution of CM in rods resembles that of WT Arr1 (see Fig. 2). Total CM concentration T = 2D + M, where D and M are the concentrations of dimer and monomer, respectively. By definition, the equilibrium KD of dimerization is KD = M2/D. As T = 2D + M, the concentration of the dimer D = (T – M)/2. Therefore, the concentration of the monomer is the positive root of the quadratic equation 2M2 + KD × M – KD x T = 0.
Figure 2.

Expression levels of CM in different transgenic lines. A, Western blot showing the levels of arrestin and rhodopsin in whole retina in WT mice (C57BL), and different transgenic lines, with indicated amounts of corresponding purified proteins used as standards. Arr1 and rhodopsin were measured by quantitative Western blot, as described previously (Song et al., 2011; for details, see Materials and Methods). Blots of representative samples (of three per genotype used) are shown. GAPDH was used as a loading control. B, Quantification of the expression of Arr1 and rhodopsin in whole mouse retina with indicated genotypes. Mean ± SD values are shown. Statistical significance of the differences (compared with WT) is shown, as follows: **p < 0.01; ***p < 0.001.
Dark and light adaptation and dark rearing
Mice were kept in a standard 12 h dark/light cycle. For ERG and single-cell recording, mice were dark adapted overnight (12–14 h). Dark-reared mice were kept in a ventilated light-proof box from birth. Ear punches for genotyping were taken from these mice at 21 d of age in dim red light. Food and water in cages kept in light-proof boxes were checked daily, cages were changed every week, all in dim red light.
Light adaptation was performed for 1 h in a box covered inside with reflective aluminum foil for 1 h at 4500 lux (model DLM2000 digital light meter, Mannix). Enucleated light-adapted eyes were fixed in ice-cold 4% paraformaldehyde in 0.1 m PBS, pH 7.2, in the same intense light for 1 h, and then overnight at 4°C in the refrigerator, and processed, as described above.
Arr1 fraction in dark-adapted OS
Mouse OSs were prepared by centrifugation on discontinuous OptiPrep (gradient medium; Sigma-Aldrich), as described previously (Song et al., 2011). The amounts of Arr1, rhodopsin (to calculate OS yield), and COX IV (to measure the OS contamination with IS material) in purified OSs (see Fig. 8) and total retina (see Fig. 2) were measured by quantitative Western blot. The fraction of Arr1 in the OSs was calculated using the total amount of Arr1 in the retina, the measured amount of Arr1 in purified OSs (corrected by yield based on rhodopsin, which localizes to the OS), and the amount of Arr1 in the OS fraction based on measured IS contamination and the fraction of Arr1 in dark-adapted ISs (determined by immunohistochemistry; see Fig. 7). Based on COX IV blots, IS contamination of the OS fraction was in the range of 0.6–2.5%. Three independent OS preps, each from two mice, were prepared for each line. Three measurements were used to calculate means and SDs shown in Table 2. Rhodopsin and Arr1 were quantified using calibration curves constructed by running known amounts of purified proteins on the same Western blot (see Figs. 2, 8).
Figure 8.
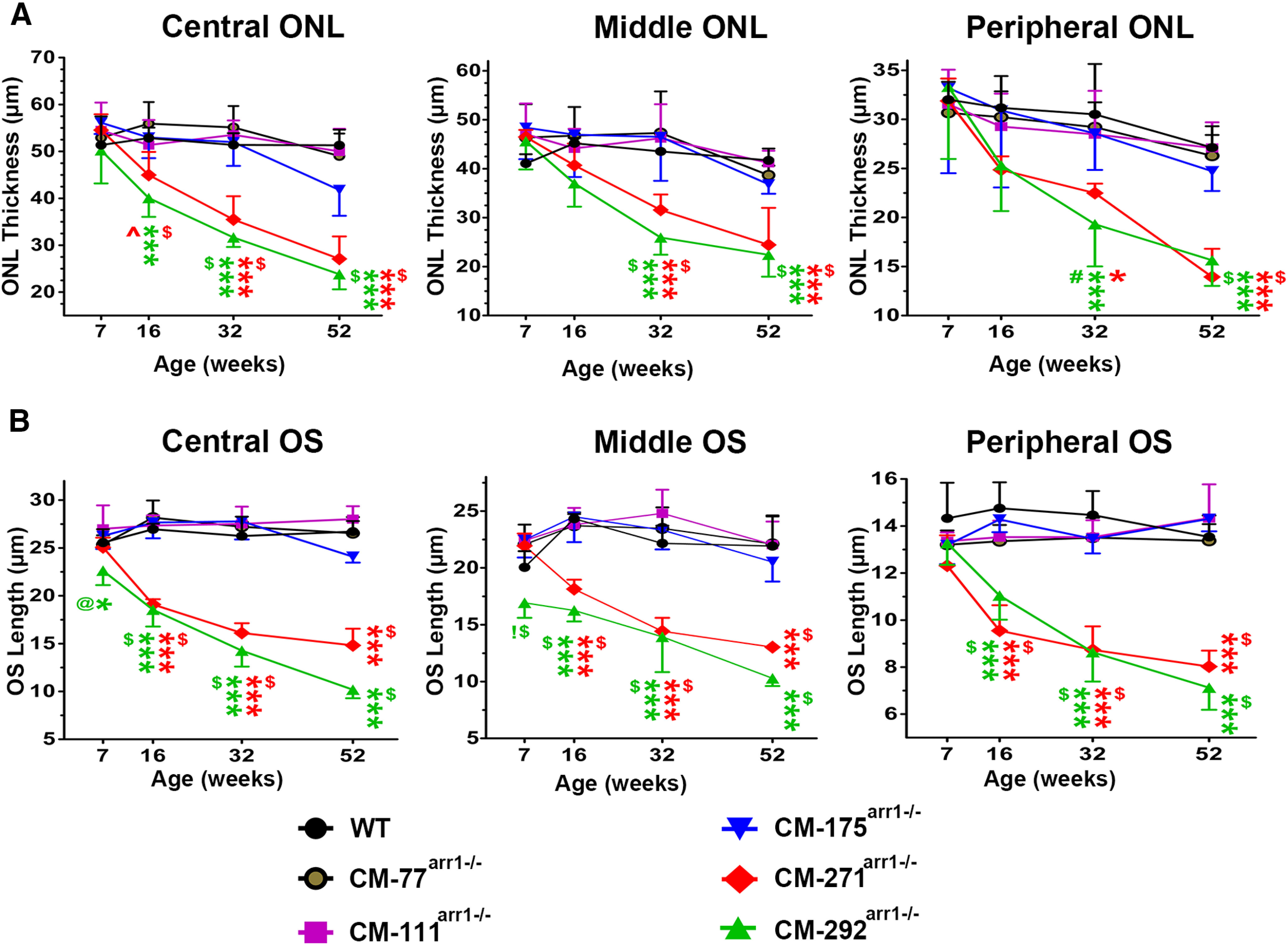
High expression of CM results in progressive loss of photoreceptor cells. A, The thickness of the ONL in the central, middle, and peripheral retina of mice at indicated ages. Two-way ANOVA with genotype and age as main factors revealed significant effects of genotype in the central (F(5,96) = 51, p < 0.0001), middle (F = 20.5, p < 0.0001), and peripheral (F = 11.77 p < 0.0001) retina. The effect of age was also significant in all retinal subdivisions (p < 0.001). The decline in the ONL thickness with age was more apparent in CM271arr−/− and CM292arr−/− lines, as evidenced by the significant genotype × age interaction (p < 0.0001 for the central and middle retina; p = 0.0012 for the peripheral retina). ***p < 0.001 to WT; $p < 0.001 to CM77arr−/−, CM111arr−/−, and CM175arr−/−; ∧p < 0.01 to CM77arr−/−, according to Bonferroni post hoc comparison for each retinal subdivision and age. The symbols are color coded to refer to CM271arr−/− or CM292arr−/− lines. N = 5 mice/genotype/age. B, The length of the OS in the central, middle, and peripheral retina of mice at indicated ages. Two-way ANOVA with genotype and age as main factors revealed significant effects of genotype in the central (F(5,96) = 224, p < 0.0001), middle (F = 104, p < 0.0001), and peripheral (F = 97 p < 0.0001) retina. The effect of age was significant in all retinal subdivisions (p < 0.0001; as was the genotype × age interaction, p < 0.0001). The statistical significance of the differences is shown, as follows: *p < 0.05, ***p < 0.001 to WT; $p < 0.001 to CM77arr−/−, CM111arr−/−, and CM175arr−/−; #p < 0.01 to CM77arr−/− and CM111arr−/−, and p < 0.05 to CM175arr−/−; @p < 0.01 to CM175arr−/− and CM111arr−/−; !p < 0.001 to CM271arr−/−, according to Bonferroni post hoc comparison following separate one-way ANOVA for each retinal subdivision and age. N = 5 mice/genotype/age. Representative sections showing ONL and OS in different lines are presented in Figures 9 and 10, respectively.
Table 2.
Arrestin-1 expression and distribution in dark-adapted rods
| Line | Arr1 expression (% of WT) | Fraction of Arr1 in the IS | Fraction of Arr1 in the OS |
|---|---|---|---|
| C57bl (WT) | 100 | 25.8 ± 1.9% | 3.26 ± 0.86% |
| CM-77arr−/− | 77 ± 3 | 20.5 ± 3.9% | 2.55 ± 0.69% |
| CM-292arr−/− | 292 ± 15 | 35.9 ± 2.7% | 3.05 ± 0.48% |
Experimental design and statistical analysis
GraphPad Prism version 8.0.0 for Mac (GraphPad Software) was used for statistical analysis. In all cases when a repeated-measures design was used, no sphericity was assumed and a Greenhouse–Geisser correction applied. In all analyses, the SDs in all groups were not statistically different as evidenced by the Brown–Forsythe test.
The histologic data on ONL thickness and OS length were analyzed separately for each retinal subdivision by two-way ANOVA with genotype and age followed by Bonferroni's post hoc comparison with correction for multiple comparisons to evaluate the effect of genotype across ages. This analysis was followed by one-way ANOVA with genotype as main factor separately for each age followed by Bonferroni's post hoc comparison.
The retinal histology in light- and dark-reared mice was analyzed by repeated-measures ANOVA with retinal subdivision (central, middle, peripheral) as the within-group factor and light as the between-group factor separately for each genotype and age. This analysis was supplemented by Bonferroni's multiple-comparison test to assess the effect of light.
For the a- and b-wave amplitudes, repeated-measures ANOVA, with genotype as the between-group factor and flash intensity steps as the within-group factor, was used. The ANOVA was followed by post hoc comparison with Dunnett's multiple-comparison test comparing the mutant genotypes with the WT. The time of half-recovery (Thalf) was analyzed using ANOVA with genotype as the main factor with post hoc comparison of means by Dunnett's test.
The light-dependent distribution of Arr1 measured by immunohistochemistry was analyzed by repeated-measures two-way ANOVA with genotype as the between-group factor and subcellular compartment as the within-group factor. The means of the mutant groups were compared with that of WT by multiple-comparison post hoc Dunnett's test. The amount of Arr1 in OSs detected by Western blot was analyzed by one-way ANOVA with genotype as the main factor followed by Bonferroni's multiple-comparison test to compare all means.
For suction electrode recordings, comparisons between genotypes for each measured parameter (time to peak, single photon response amplitude, and time constant of recovery) were made using a two-tailed Student's t test assuming equal variances. All error bars for single-cell recordings reflect the SEM. In all cases, p < 0.05 was considered significant.
Results
Expression levels of self-association-deficient arrestin-1
For transgenic expression, we chose the mutant mouse Arr1-(Phe86Ala, Phe198Ala) (Fig. 1A–C), which was previously shown to be oligomerization deficient (Kim et al., 2011). It has a dimerization constant almost 10 times higher than WT (537 vs 57.5 μm in WT) and no detectable tetramerization (Kim et al., 2011). Five founders that transduced the transgene to their progeny were bred onto an Arr1 knock-out (Arr1−/−) background (Xu et al., 1997), so that the mutant was the only form of Arr1 present in rods. This yielded five independent lines. Arr1 and rhodopsin expression levels in these lines were determined by Western blot in the whole retina homogenates (Fig. 2A,B). The data showed that created lines expressed self-association-deficient Arr1-(Phe86Ala, Phe198Ala) (CM) at different levels, from near-physiologic (CM-77arr−/−, 77 ± 3%; CM111arr−/−, 111 ± 6%) to supraphysiologic [CM175arr−/−, 175 ± 16% (p < 0.01); CM292arr−/−, 292 ± 15% (p < 0.001); CM271arr−/−, 271 ± 30% (p < 0.001); Arr1 expression in WT C57BL mice represents 100%]. Rhodopsin expression in all lines was not statistically different from WT (Fig. 2B).
Figure 1.

Arrestin-1 solution tetramer and mutations disrupting oligomerization. A, A model of the solution structure of bovine Arr1 tetramer (Hanson et al., 2008b) with protomers shown in different colors. B, One of the protomer–protomer interfaces with Phe197 of the two protomers shown as CPK model. C, Another protomer–protomer interface with Phe85 shown as CPK model. Note that Phe197 and Phe85 in bovine Arr1 are homologous to Phe86 and Phe198, respectively, in mouse Arr1. Double mutations F85A/F197A and F86A/F198A disrupt self-association of bovine and mouse Arr1, respectively (Kim et al., 2011). Images were created in DS ViewerPro 6.0.
Functional performance of photoreceptors expressing CM
The function of rods in mice expressing CM was first evaluated by ERG. The ERG data were statistically analyzed by two-way repeated-measures ANOVA. Genotype was the main variable of interest and was treated as a between-group factor. Flash intensity steps were treated as a within-group factor, since responses to different light intensities were recorded from the same mice. We found significant effect of genotype on the ERG a-wave (F(5,24) = 4.086, p = 0.008). As expected, a-wave was affected by flash intensity (F(3.019,72.46) =251, p < 0.0001). The highly significant genotype × flash intensity interaction (F(65,312) = 2.806, p < 0.0001) pointed to a change in the shape of the curve. The high expression of CM [lines CM-271arr−/− (p < 0.01) and CM-292arr−/− (p < 0.001)] reduces rod light response (ERG a-wave; Fig. 3A,B) even at 7–8 weeks, when OS length and ONL thickness in the retinas of these mice are essentially normal (see below), suggesting that the functionality of the rods in these lines is compromised even before histologic damage becomes detectable. Importantly, very high expression (∼220% of WT) of WT Arr1 did not change rod light sensitivity (Song et al., 2011), suggesting that this change is specific for rods expressing high levels of oligomerization-deficient mutant. The a-wave in lines expressing lower levels of CM (CM-77arr−/−, CM111arr−/−, and CM175arr−/−) was not statistically different from WT (Fig. 3A,B). The b-wave, reflecting the response of bipolar cells, was not statistically different from WT in any CM-expressing line (F(5,24) = 2.454, p = 0.0624 for genotype; Fig. 3A,B).
Figure 3.

Functional performance of rods in mice expressing different levels of CM. ERG responses to single flashes of increasing intensity in mice of indicated genotypes were recorded, as described in Materials and Methods. A, B, The a-wave (A) and b-wave (B) amplitudes were plotted as a function of flash intensity. Mean ± SEM values from five animals per genotype are shown. The a-wave responses of higher expressors were lower than those of WT mice. Data analysis by repeated-measures ANOVA with flash intensity as the within-group factor and genotype as the between-group factor revealed a highly significant effect of CM concentration on a-wave amplitude (p < 0.001). The genotype × flash intensity interaction was highly significant (p < 0.001), indicating differences in the shape of the curve. The mean values for the mutant mouse lines were compared with WT mice for every intensity using Dunnett's multiple post hoc comparison test. **p < 0.01, ***p < 0.001, compared with WT mice for all flash intensities ≥0. There were no significant differences in b-wave between any mutant line and WT mice. C, The normalized amplitude of the probe flash a-wave was plotted as a function of time elapsed after the first flash. The intensities of the first (desensitizing) and second (probe) flash were 0.4 and 0.65 log cd * s/m2, respectively. The interval between two flashes was varied from 50 to 2000 ms. To calculate the Thalf, recovery kinetics were fitted by polynomial nonlinear regression, with R2 > 0.95, as described in Materials and Methods. D, Calculated Thalf. The mean ± SD values for five animals per genotype are shown.
An important parameter that directly reflects Arr1 function is the Thalf. It is measured by ERG using a double-flash protocol (Song et al., 2009). The first flash desensitizes rods. The amplitude of the a-wave (expressed as a fraction of the response of dark-adapted rods to the flash of the same intensity) in response to the second flash, which is delivered at different time intervals after the first, is used as a measure of rod recovery. The absence of Arr1 (Xu et al., 1997), rhodopsin kinase (Chen et al., 1999a), phosphorylation sites on rhodopsin (Chen et al., 1995), or insufficient number of phosphorylation sites (Mendez et al., 2000) greatly slows down photoresponse recovery. We found that the effect of genotype, as detected by one-way ANOVA, was not significant (F(5,24) = 1.616, p = 0.194). Thalf values in CM-77arr−/−, CM-111arr−/−, and CM-175arr−/− lines did not differ from those in C57BL mice (Fig. 3C,D). Thalf in higher-expressing lines (CM-271arr−/− and CM-292arr−/−) showed a tendency of being somewhat longer, but the difference did not reach statistical significance (Fig. 3C,D). Thus, as measured by single- or double-flash ERG, the in vivo functional performance of rods that expressed Arr1 at near-physiological levels was not affected by oligomerization deficit, at least in 7- to 9-week-old animals.
In normal rods, Arr1 quenches rhodopsin activity in <40 ms (Gross and Burns, 2010), and so even 10-fold variations in arrestin binding to rhodopsin can be difficult to discern using ERG. To examine more subtle changes in rhodopsin deactivation that might arise from increased Arr1 monomer concentrations, we used suction electrode recordings from intact rods to measure the electrical responses to both dim and bright flashes. Single-cell recordings of rods of the CM-77arr−/− line with near-physiologic expression of CM revealed no statistically significant differences in kinetic parameters from WT rods by t test (Table 1). Most notably, CM-77arr−/− rods had normal light-driven changes in photocurrent amplitude (Fig. 4A, Table 1), including single-photon responses that were indistinguishable from those of WT C57BL rods (Fig. 4B, Table 1). In rods that underexpress WT Arr1 (Arr+/−), the single-photon responses are also indistinguishable from WT because of calcium feedback mechanisms, but bright flash responses that are not subject to dynamic calcium regulation remain in saturation for slightly longer times (Tsat) at a given flash strength (Gross and Burns, 2010). The consequence is a vertical offset in the Tsat versus flash strength plot, which can be used to calculate both increases and decreases in the active lifetime of rhodopsin of up to at least twofold (Gross et al., 2012). The saturating flash analysis of the CM-77arr−/− rods showed no such vertical offset (Fig. 4C), consistent with no change in rhodopsin lifetime arising from the profound shift in equilibrium favoring the monomer. These results support the conclusions that the Arr1 monomer rather than oligomer is the moiety responsible for quenching the activity of rhodopsin (Hanson et al., 2007b) and that Arr1 binding is not rate limiting for rhodopsin shutoff in dark-adapted rods (Gross and Burns, 2010).
Light-dependent arrestin-1 translocation
In the dark, Arr1 in rods is at disequilibrium (Peet et al., 2004), with the highest concentration in the ISs and the rest spread throughout cell bodies in the ONL. Two models were proposed to explain why only a small fraction of Arr1 is present in the OS in the dark. One model posits that Arr1 is mostly localized to the IS and cell bodies because of its low-affinity binding to microtubules (Nair et al., 2004, 2005), which are abundant in these compartments (Eckmiller, 2000) and represent the highest-affinity binding partner of Arr1 in the absence of light-activated rhodopsin (Nair et al., 2005). We showed that Arr1 monomers, dimers, and tetramers bind microtubules comparably (Hanson et al., 2007b). An alternative model explained Arr1 absence from the OSs in the dark by the size of the Arr1 oligomers, which are too big to fit between the discs, which greatly reduces the cytoplasmic volume available for their diffusion in the OSs (Najafi et al., 2012). The mouse lines expressing CM, which yields greatly increased concentration of the monomer, provided a perfect opportunity to test these hypotheses. The immunohistochemical detection of Arr1 in the CM-77arr−/− line, which is expected to have a 5.5-fold higher monomer concentration than WT C57BL mice, showed that in the dark CM is largely localized to the ISs, cell bodies, and synaptic terminals (Fig. 5), as in WT mice (Nair et al., 2005; Hanson et al., 2007a). The immunohistochemistry data for both dark- and light-adapted retinas were statistically analyzed by two-way repeated-measures ANOVA with genotype as a between-group factor and subcellular compartment as a within-group factor. The analysis of the Arr1 distribution in the dark-adapted retinas revealed no significant effect of genotype (F(2,8) = 2.542, p = 0.14). Post hoc comparison of means to WT by Dunnett's test showed no significant difference of Arr1 concentration in OSs in either genotype. The proportion of CM in the OS in the dark was equally low in all lines. Thus, even in the CM-292arr−/− line, the relative concentration of CM in OS was similar to that of WT Arr1 (p = 0.37; Fig. 5D). These data suggest that the size of the Arr1 oligomers does not determine its subcellular localization in rods in the dark. Surprisingly, in the CM-77arr−/− line there was a redistribution of CM between ISs and ONL, with a higher proportion of the CM detected in the ISs and reduced presence in the cell bodies (p = 0.0025 for IS and p = 0.0054 for ONL; Fig. 5D). Importantly, on light adaptation (1 h at 4500 lux; see Materials and Methods), CM translocated to the OS, as does WT Arr1 (Fig. 5A–C). Overall, the effect of genotype on the Arr1 distribution in light-adapted rods was not significant (two-way repeated-measures ANOVA genotype factor, F(2,9) = 0.146, p = 0.99; Fig. 5E). However, there was a highly significant genotype × subcellular compartment interaction (F(6,27) = 231, p < 0.0001), pointing to the differences in the subcellular distribution of Arr1 among the genotypes. That significant interaction was driven by the behavior of Arr1 in the high-expression line CM-292arr−/−, where the distribution of the mutant differed from both WT and CM-77arr−/− mice. A significantly smaller fraction of total mutant translocated to the OS (p < 0.001), with the rest remaining in the ONL (p < 0.001), IS (p < 0.05), and synaptic area (p < 0.01; Fig. 5E). This was expected, based on our previous finding that rhodopsin interacts with Arr1 at 1:1 ratio (Hanson et al., 2007a; Bayburt et al., 2011), so that Arr1 expressed in molar excess over rhodopsin (rhodopsin was expressed at essentially the same level in all lines; Fig. 2A,B) has no partner to translocate to and therefore remains in other cell compartments, where it was in the dark.
Figure 5.
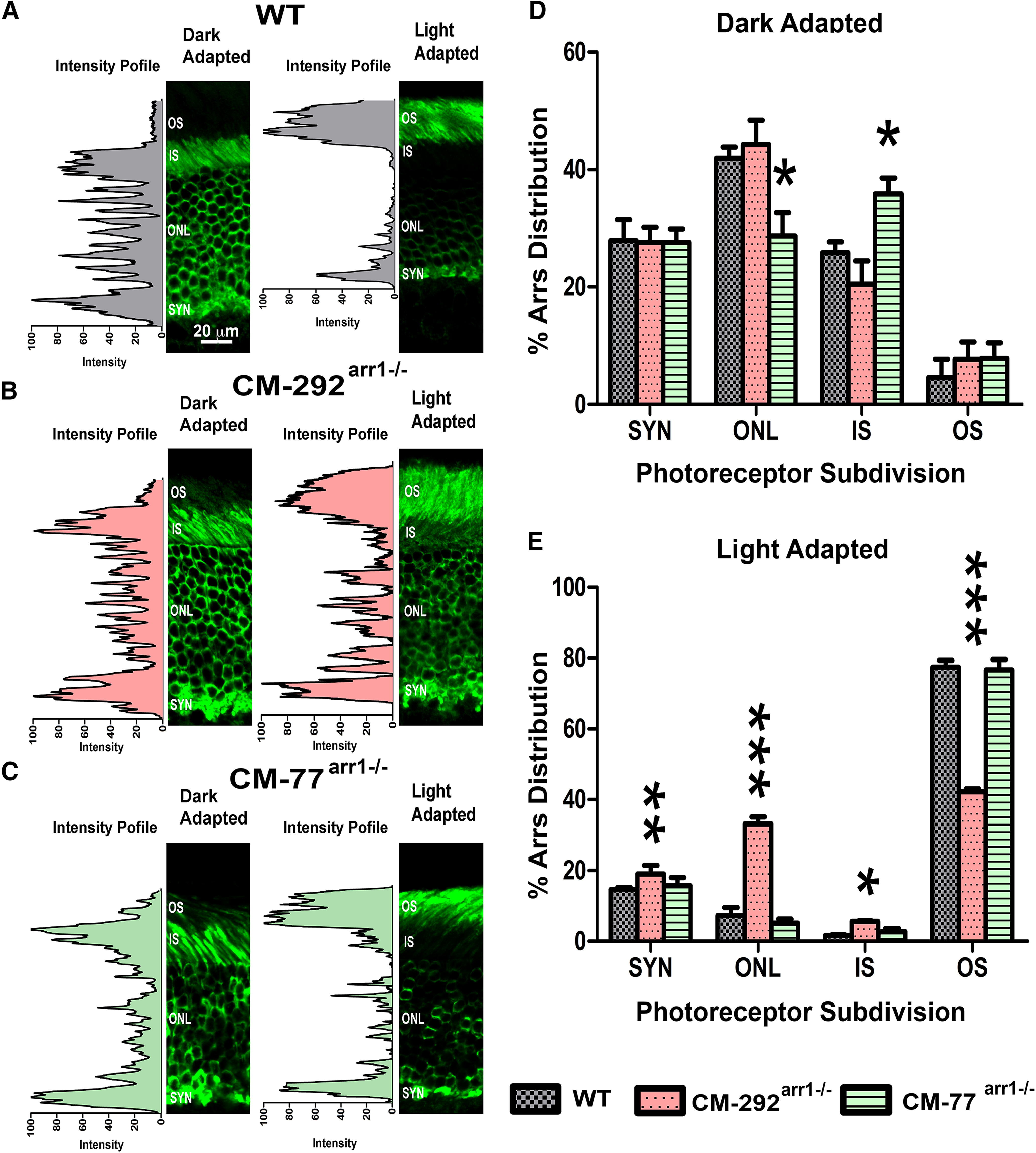
Arrestin-1 localization in the dark- and light-adapted rod photoreceptors. A–C, Mice with the indicated genotypes (A–C) were dark adapted overnight (DARK) or exposed to light (2700 lux for 1 h; LIGHT). Eyes were fixed and processed for Arr1 immunohistochemistry using C10C10 anti-arrestin-1 antibody (Knospe et al., 1988). The positions of the OSs, ISs, ONL, and synaptic layer (SYN) are indicated. D, E, The proportion of Arr1 localized in the OSs of dark-adapted (D) and light-adapted (E) mice was quantified by the intensity of Arr1 immunostaining (green) in three images per animal from three animals per genotype. Mean ± SD values are shown. The two-way ANOVA with genotype and rod compartment as main factors yielded no significant effect of genotype (p = 0.99) but a highly significant genotype × compartment interaction (p < 0.0001). *p < 0.05; **p < 0.01; ***p < 0.001, compared with the corresponding compartment in WT mice, as detected by independent multiple comparison of means with Dunnett's multiple-comparison tests.
To test our immunohistochemistry finding in dark-adapted mice using a more quantitative approach, we prepared OS from dark-adapted retinas of mice expressing different levels of CM and quantified CM content in the OS (using rhodopsin to calculate yield and mitochondrial marker COX IV to estimate OS contamination with ISs, which are particularly rich in mitochondria; Figs. 6A–C, 7A–D, Table 2). Complete data were collected for lines expressing the lowest (CM-77arr−/−) and the highest (CM-292arr−/−) levels of CM. No significant effect of genotype on the Arr1 concentration in OS was found (one-way ANOVA, F(2,6) = 0.828, p = 0.4815). The results confirmed that only a small fraction of CM in the dark-adapted rods is present in the OS, similar to WT Arr1 (Table 2). As the monomer concentrations in the OS and cell bodies of CM-292arr−/− mice is ∼12-fold higher than in WT, these data confirm that the size of the Arr1 oligomers does not play a role in its exclusion from the OS in dark-adapted rods.
Figure 6.
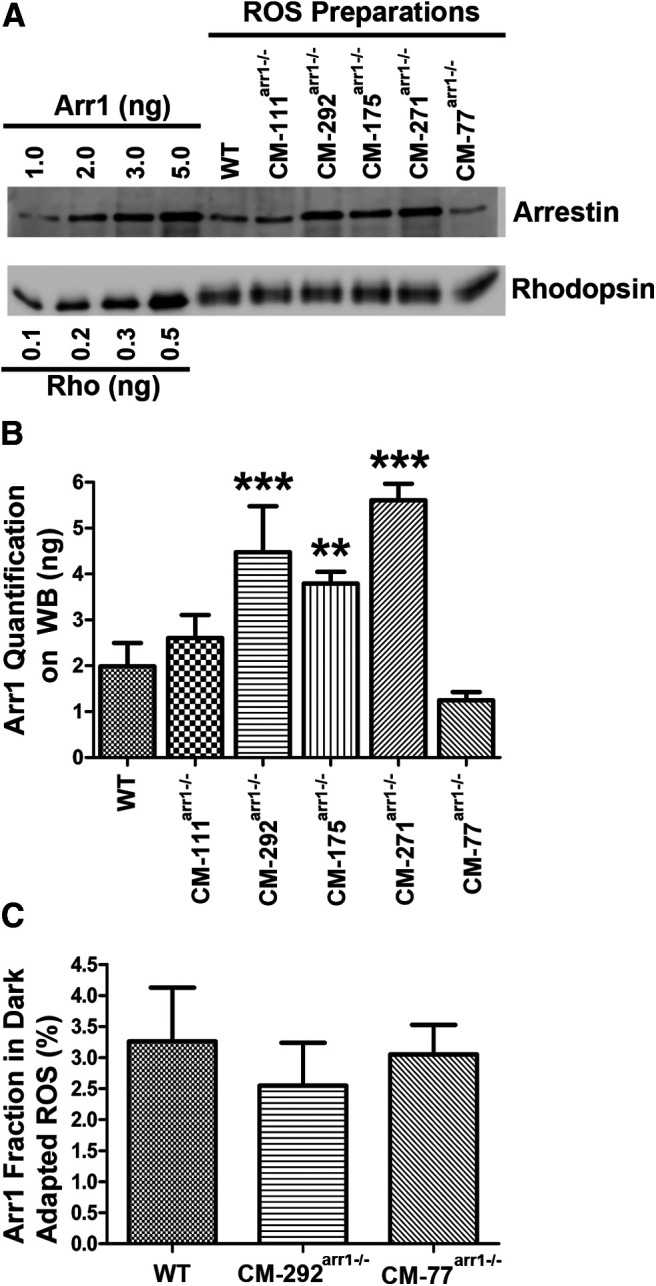
The expression of CM and its presence in the OS of dark-adapted rods. A, Western blot showing the levels of arrestin in the OS preparations in WT, and different transgenic lines, with indicated amounts of corresponding purified proteins used as standards. Arr1 and rhodopsin were measured by quantitative Western blot, as described previously (Song et al., 2011). Blots of representative samples (of three per genotype used) are shown. The amounts of rhodopsin were equalized to show the differences in Arr1 levels. B, Absolute amount of Arr1 in the OS of dark-adapted mice of indicated genotypes. Arr1 in each transgenic line was measured by quantitative Western blot in three independent OS preparations. Mean ± SD values are shown. Statistical significance of the differences is indicated, as follows (one-way ANOVA with genotype as the main factor followed by Bonferroni's multiple-comparison test): **p < 0.01; ***p < 0.001. C, The proportion of CM localized in rod OS of dark-adapted mice. The fraction of Arr1 in the dark-adapted OS was quantified by Western blot. The level of contamination by the IS material was determined by comparison of mitochondrial marker COX IV present in the whole retina and OS preparation. The fraction of Arr1 in the IS for correction purposes was determined by immunohistochemistry (Fig. 7) in three images per animal from three animals per genotype per dark condition. Mean ± SD values are shown. Additional data for CM-111arr−/−, CM-175arr−/−, and CM-271arr−/− lines are shown in Figure 7.
Health and survival of photoreceptors
Rods need very high expression of Arr1 to match rhodopsin levels, so WT mice express 8 Arr1 molecules per 10 rhodopsin molecules (Strissel et al., 2006; Hanson et al., 2007a; Song et al., 2011). Previously, we found that high expression of enhanced Arr1-3A mutant, which has a reduced ability to self-associate, is toxic for rods, causing the shortening of rod OSs and photoreceptor death (observed as thinning of the ONL; Song et al., 2013). This is in sharp contrast to WT Arr1, which is harmless at an equally high expression level (Song et al., 2011). Thus, we hypothesized that monomeric Arr1 is cytotoxic and that self-association of Arr1 is a cytoprotective mechanism (Song et al., 2009). However, the Arr1-3A mutant had other structural and functional features, such as the enhanced ability to bind unphosphorylated rhodopsin (Gurevich, 1998) and detached C-tail with exposed AP-2 binding site (Moaven et al., 2013), which could also be responsible for its toxicity.
Therefore, we compared the health and survival of rod photoreceptors in lines expressing CM, which has no apparent differences from WT Arr1 other than the greatly reduced ability to self-associate (Kim et al., 2011). We assessed the rod morphology by measuring the length of rod OS (Figs. 8A, 9) and thickness of the ONL (Figs. 8B, 10) up to 52 weeks. Two-way ANOVA with genotype and age as main factors yielded highly significant effects of genotype on the length of OS for all retinal subdivisions (F(5,96) = 224 for central, 104 for middle, and 96.7 for peripheral retina; p < 0.0001). Similarly, the thickness of ONL was significantly affected by genotype (F(5,96) = 51 for central, 20.5 for middle, and 11.8 for peripheral; p < 0.0001). We found age-dependent shortening of the OS and thinning of the ONL in the highest expressors (lines CM292arr−/−and CM271arr−/−; p < 0.001 at 16, 32, and 52 weeks in both cases), a tendency to slower rod demise in the CM175arr−/− mice with lower, but still supraphysiological, expression (which did not reach statistical significance), and no visible damage in lines expressing CM at subphysiological or nearly physiological levels (CM-77arr−/−, CM111arr−/−; Figs. 8–10). Damage in the highest expressors began earlier and was most pronounced in the central retina, but was also observed in other subdivisions (Figs. 8–10).
Figure 9.
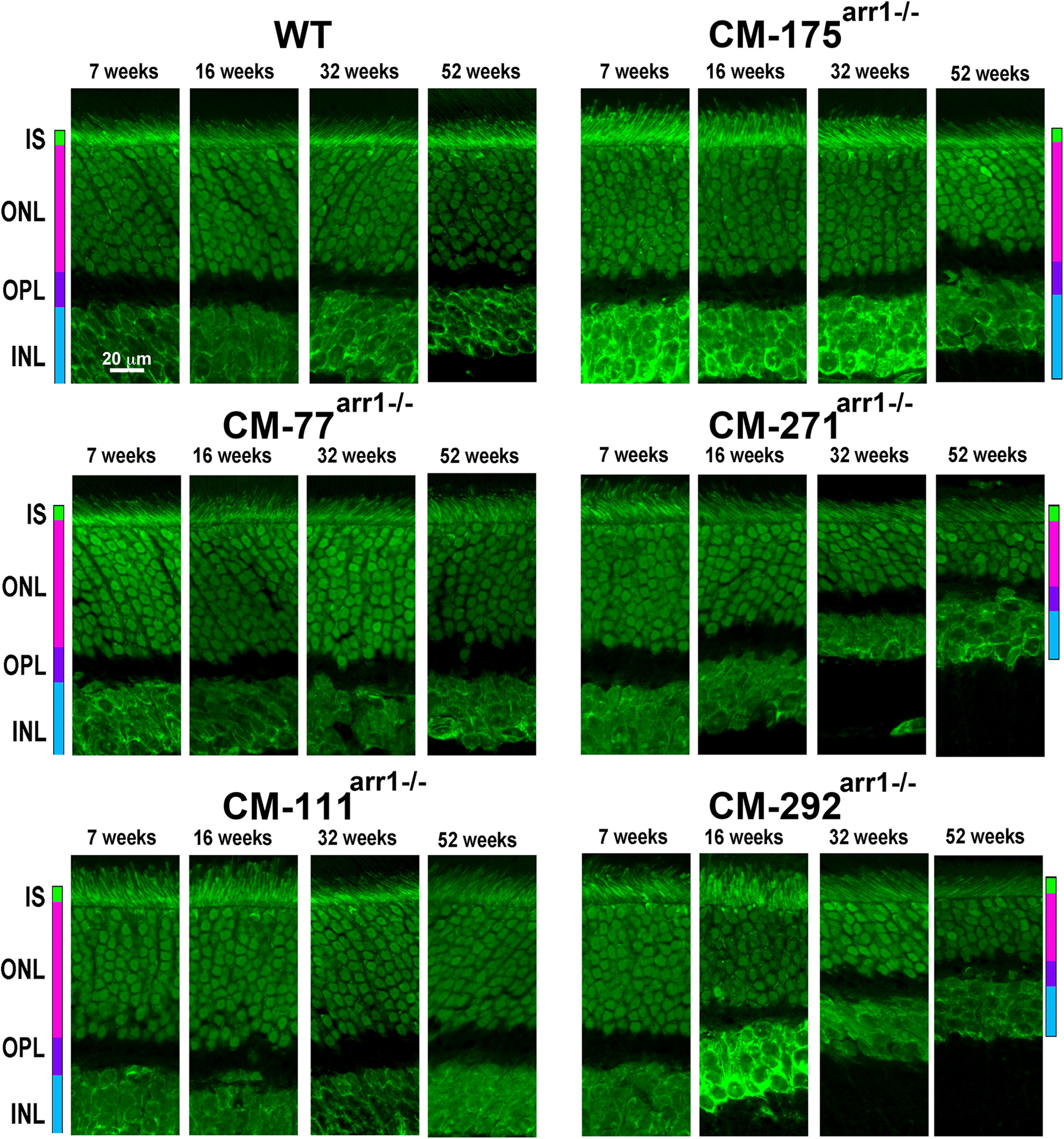
High expression of CM causes progressive loss of photoreceptor cells. Confocal images of middle retina sections of WT and mice expressing CM at indicated ages. ONL was visualized by staining with green fluorescent Nissl. OPL, Outer plexiform layer; INL, inner nuclear layer.
Figure 10.
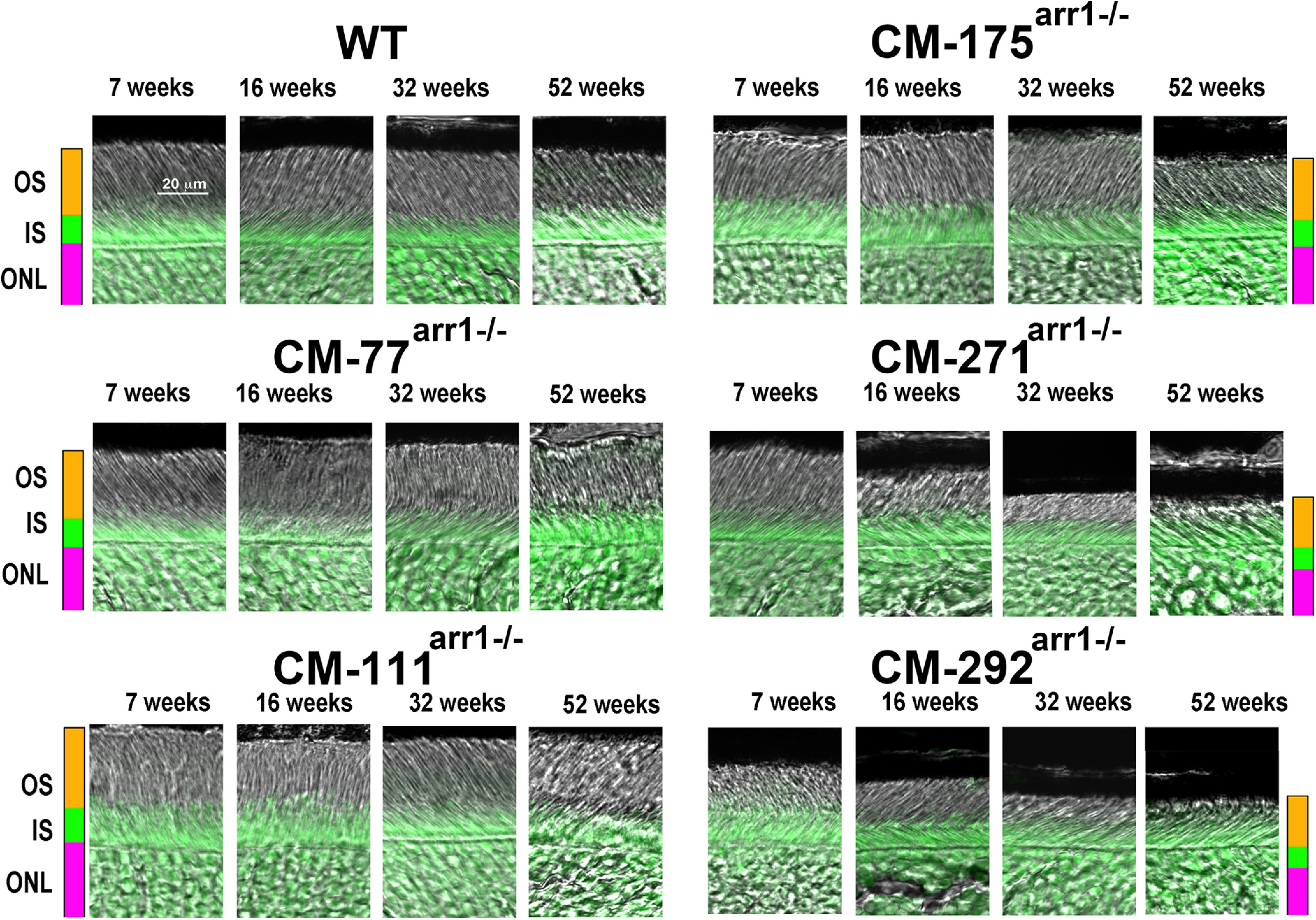
High expression of CM causes progressive shortening of rod OSs. Combined DIC and green fluorescent Nissl images of the middle retina sections of WT and mice expressing cmArr1 at indicated ages, enlarged to show OS more clearly. The positions of OSs, ISs, and ONL are shown on the left.
To determine whether the damage was light dependent, we compared CM292arr−/− mice reared in a normal 12 h light/dark cycle and in complete darkness, using Arr1−/− mice, where degeneration was shown to be light dependent (Chen et al., 1999b), as a control (Fig. 11A–D). Two-way repeated-measures ANOVA of the OS length in Arr1−/− mice yielded a significant effect of genotype for both 7 and 16 weeks of age (F(2,12) = 49.7 and 440, respectively; p < 0.0001). Similarly, in CM292arr−/− mice the effect of genotype was significant (7 weeks: F(2,12) = 9.4, p = 0.0035; 16 weeks: F(2,12) = 61.2, p < 0.0001). However, in contrast to Arr1−/− animals, where the shortening of the OS was completely prevented by dark rearing, the shortening of the OS in CM292arr−/− occurred regardless of environmental light (p < 0.001; Fig. 11A–D), similar to what we found in mice expressing Arr1-3A mutant partially deficient in self-association (Song et al., 2013). The damage tended to be more pronounced in the central retina, as evidenced by significant genotype × retinal subdivision (within-group factor) interactions [p < 0.0001 for all except for CM292arr−/− at 7 weeks (where it was not significant, p = 0.34)].
Figure 11.

The damage caused by supraphysiological expression of CM is light independent. A, B, Combined DIC and green fluorescent Nissl images of the retina sections of 7-week-old (A) and 16-week-old (B) mice of indicated genotypes, enlarged to show the OS more clearly. The positions of OSs, ISs, and ONL are shown on the left. C, D, The length of the OS measured in the central, middle, and peripheral retina were the average of inferior and superior hemispheres. Mean ± SEM values from five animals per genotype are shown. The length of OS was compared separately for each age by two-way repeated-measures ANOVA with retinal subdivision as the within-group factor and genotype as the between-group factor followed by Bonferroni post hoc test for each retinal subdivision. The CM-292arr1−/− mice were significantly different from WT mice regardless of light exposure at both ages across retinal subdivisions (p < 0.01 at 7 weeks; p < 0.0001 at 16 weeks). In contrast, Arr1−/− mice kept in a normal light cycle significantly differed from WT mice at both ages (p < 0.0001). The dark-reared Arr1−/− mice were no different from WT mice but were significantly different from light-exposed mice at both ages (p < 0.0001). Statistical significance of the differences is shown, as follows (color coded): *p < 0.05; ***p < 0.001 compared with WT mice; a p < 0.05, c p < 0.001 compared with Arr1−/− dark mice. Note that dark rearing preserves the OS in Arr1−/− mice, whereas the damage in CM-292Arr−/− animals is not affected by light.
Our studies with purified CM in vitro yielded a dimerization constant of 537 μm and did not detect tetramer formation (in contrast to WT mouse Arr1 with dimerization and tetramerization constants of ∼58 and ∼63 μm, respectively; Kim et al., 2011). Calculations based on these constants and expression levels in different lines (Fig. 2B) suggest that the absolute concentration of the mutant monomer in cell bodies and the IS in the dark, when the bulk of Arr1 is localized in these compartments (Nair et al., 2005; Hanson et al., 2007a), would be ∼524 μm in CM-77arr−/−, 651 μm in CM-111arr−/−, and ∼846 μm in CM-175arr−/−, and would reach ∼1082 μm in CM-271arr−/− and 1128 μm in CM-292arr−/− lines, compared with 95 μm in WT C57BL/6J mice (Kim et al., 2011). Rods apparently tolerated ∼500–650 µm, but higher levels were toxic (Fig. 3A). While the data confirm cytotoxicity of the monomer, it appears that at the normal level of Arr1 in WT cell bodies in the dark (∼2000 μm; Song et al., 2011) dimerization with a constant of ∼500 μm would be sufficient to protect rods, as it would yield an ∼590 μm monomer, which is slightly more than is present in the CM-77arr−/− mice and less than in CM-111arr−/− mice. It is worth noting that predicted concentrations of the dimer in CM lines would also far exceed ∼200 μm in WT photoreceptors (Kim et al., 2011), reaching ∼500 μm in CM-77arr−/−, ∼780 μm in CM111arr−/−, ∼1320 μm in CM175arr−/−, ∼2170 μm in CM292arr−/−, and ∼2350 μm in CM271arr−/− lines. Thus, the data are also consistent with the hypothesis that high Arr1 dimer concentrations, which are never reached in WT photoreceptors because of tetramer formation, could be harmful. However, the properties of human Arr1, which readily dimerizes but has a fairly high tetramerization constant (224 μm; Kim et al., 2011), suggest that the toxicity of the dimer is unlikely.
Discussion
The key biological function of Arr1 in photoreceptors is its binding to rhodopsin that is necessary for the rapid shutoff of the light response (Chen et al., 1995). Arr1 is known to oligomerize, and this ability is conserved in mouse, bovine, and human protein (Kim et al., 2011), suggesting its functional importance.
Two different crystallization studies yielded essentially the same tetramer of Arr1 (Granzin et al., 1998; Hirsch et al., 1999). Subsequent studies showed that the solution tetramer of Arr1 is structurally distinct from the crystal tetramer (Hanson et al., 2007b): Arr1 oligomerizes as a symmetrical diamond-shaped dimer of dimers (Hanson et al., 2008b; Fig. 1). Receptor binding elements on Arr1 were localized to the concave sides of both domains by mutagenesis (Vishnivetskiy et al., 2000, 2011; Hanson and Gurevich, 2006; Ostermaier et al., 2014; Peterhans et al., 2016) and different biophysical methods (Hanson et al., 2006; Zhuang et al., 2013). This localization was confirmed by the crystal structure of the Arr1 complex with rhodopsin (Kang et al., 2015; Zhou et al., 2017). In tetrameric Arr1, these receptor binding surfaces are shielded by sister protomers both in crystal (Granzin et al., 1998; Hirsch et al., 1999) and in solution (Hanson et al., 2008b). Direct measurements of interprotomer distances by double electron–electron resonance showed that only monomeric Arr1 binds rhodopsin (Hanson et al., 2007b), supporting an earlier hypothesis that Arr1 oligomers are storage forms (Schubert et al., 1999). However, all these studies were performed in vitro using purified proteins. The role of Arr1 oligomerization in quenching rhodopsin signaling in vivo in the native environment of a functional rod, where numerous other proteins are present, has never been tested. The elucidation of the structure of Arr1 solution tetramer and key residues involved in its oligomerization (Hanson et al., 2008b) allowed for the construction of Arr1 mutant (CM) with severely reduced ability to oligomerize (Kim et al., 2011), which preserved all other known functions (Kim et al., 2011). Here we used transgenic expression of CM to explore the biological role Arr1 oligomerization in living mice. As we were unable to determine Arr1 and CM oligomerization status inside rod photoreceptors in vivo, our conclusions are based on the assumption that CM mutant in vivo has the same oligomerization defect we found in vitro using purified proteins: it has an ∼10-fold higher dimerization constant than WT Arr1, and, in contrast to it, it does not form tetramers (Kim et al., 2011).
Our data show that CM quenches light-induced rhodopsin signaling normally, as determined by ERG and single-cell recordings (Figs. 3, 4). This is consistent with in vitro data that the Arr1 monomer is the species that binds rhodopsin (Hanson et al., 2007b). These data are the first in vivo evidence that the monomer is necessary and sufficient to bind and terminate rhodopsin signaling.
In dark-adapted rods, Arr1 is largely localized in rod IS and cell bodies, and translocates to the OS in bright light. The mechanism of Arr1 translocation is energy independent, suggesting that it moves by diffusion (Nair et al., 2005; Rosenzweig et al., 2007; Slepak and Hurley, 2008). In fact, it was calculated that the amount of energy that would be required for active transport of Arr1 is unrealistic (Gurevich et al., 2011). However, there is a controversy regarding the mechanisms that determine subcellular localization of Arr1 in rods. Our model posits that Arr1 localization depends on its interaction with its binding partners: rhodopsin in the light and microtubules in the dark (Nair et al., 2005). Upon illumination, rhodopsin becomes the highest-affinity binding partner, driving Arr1 into OS, whereas in the dark rhodopsin is inactive, allowing Arr1 to bind with lower affinity to microtubules abundant in the IS and ONL (Eckmiller, 2000). An alternative model posits that the exclusion of Arr1 from OS in the dark is because of it oligomerization: oligomers are simply too large to fit into spaces between disks (Najafi et al., 2012).
The availability of mice expressing CM that generates much higher steady-state levels of monomers allowed us to test these competing hypotheses directly. We found that only a small fraction of CM localizes to the OS in the dark regardless of its expression level, similar to WT Arr1 (Figs. 5, 6). This is inconsistent with the exclusion of Arr1 from the OS being because of the size of its oligomers (Najafi et al., 2012). Importantly, CM, just like WT Arr1, readily translocates to the OS on illumination (Fig. 5A–C,E), demonstrating that in vivo Arr1 monomer is the species that binds light-activated rhodopsin. Thus, our data suggest that Arr1 localization in the dark- and light-adapted photoreceptors is determined by its interaction with the highest-affinity partners available in each condition: activated phosphorylated rhodopsin in the OS in the light and microtubules in the dark (Nair et al., 2005).
The key difference between WT Arr1 and CM, both of which demonstrate the same rhodopsin binding specificity (Kim et al., 2011), is in their effect on rod health and survival. While WT Arr1 is harmless even at high supraphysiological expression levels (Song et al., 2011), CM at twofold to threefold higher than normal levels causes the shortening of the OS and photoreceptor death (Figs. 8–10). This effect is light independent (Fig. 11) and roughly proportional to its expression level (Figs. 8–10). Even before the damage becomes visible, the rods of higher expressor lines (CM-271arr−/− and CM-292arr−/−) do not perform as well as the rods of WT mice or CM lines with lower expression (Fig. 3).
Arr1 needs to be expressed at high levels in rods to quench rhodopsin signaling under bright illumination (Chen et al., 1999b). The apparent toxicity of monomeric or, possibly, dimeric Arr1 might explain robust self-association of Arr1 in evolutionary distant mammalian species, mice, cows, and humans (Kim et al., 2011). However, it appears to contradict structurally and functionally normal rods in CM-77arr−/− and CM-111arr−/− lines (Figs. 3, 4, 8), where the concentration of the monomer and dimer greatly exceeds that in WT animals. The contrast between WT and CM lines is particularly great in rod ISs and cell bodies in the dark, since when Arr1 concentrates in OS on light exposure, it does so via binding to rhodopsin as a monomer in all cases. Thus, it is conceivable that the excess of the monomer and/or dimer exerts its harmful effect not in the OS, but in the IS and/or cell bodies. As CM translocates to the OS in the light normally (Fig. 5A–C,E), its concentrations in the IS and cell bodies in mice expressing it at near-physiological levels are greatly reduced for ∼12 h every day. The amount of Arr1 that can translocate to the OS in the light is limited by the amount of rhodopsin in this compartment, as the stoichiometry of Arr1 binding to rhodopsin is 1:1 (Hanson et al., 2007a; Bayburt et al., 2011). Thus, in higher-expressing lines CM-271arr−/− and CM-292arr−/−, where Arr1 is in molar excess over rhodopsin, high monomer and dimer concentration in the ISs and cell bodies would persist throughout the light period, possibly causing sustained damage. The molecular mechanism whereby excessive levels of monomeric Arr1 harm photoreceptors, whereas oligomeric WT protein does not, remains to be elucidated.
To summarize, here we demonstrate that in living animals Arr1 self-association does not play a role in quenching light-induced rhodopsin signaling, does not affect subcellular distribution of Arr1 in rods, but prevents harmful effects of monomeric Arr1, thereby preserving long-term photoreceptor viability.
Footnotes
The authors declare no competing financial interests.
This research was supported by National Institutes of Health (NIH) Grants RO1-EY-011500 and R35-GM-122491 and the Cornelius Vanderbilt Chair (to V.V.G.); and NIH Grants EY-012155 and EY-027193 (to J.C.) and EY-014047 (to M.E.B.). We thank Drs. R.S. Molday and C.M. Craft for monoclonal anti-rhodopsin antibodies and mouse arrestin-1 cDNA, respectively; and Faiza Baameur for generating the clone with mouse Arr1-(F86A, F198A) under control of the rhodopsin promoter for transgenic expression.
References
- Bayburt TH, Vishnivetskiy SA, McLean M, Morizumi T, Huang C-c, Tesmer JJ, Ernst OP, Sligar SG, Gurevich VV (2011) Rhodopsin monomer is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem 286:1420–1428. 10.1074/jbc.M110.151043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI (1999a) Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci U S A 96:3718–3722. 10.1073/pnas.96.7.3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Simon MI, Matthes MT, Yasumura D, LaVail MM (1999b) Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest Ophthalmol Vis Sci 40:2978–2982. [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI (1995) Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science 267:374–377. 10.1126/science.7824934 [DOI] [PubMed] [Google Scholar]
- Eckmiller MS. (2000) Microtubules in a rod-specific cytoskeleton associated with outer segment incisures. Vis Neurosci 17:711–722. 10.1017/s0952523800175054 [DOI] [PubMed] [Google Scholar]
- Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, Büldt G (1998) X-ray crystal structure of arrestin from bovine rod outer segments. Nature 391:918–921. 10.1038/36147 [DOI] [PubMed] [Google Scholar]
- Gross OP, Burns ME (2010) Control of rhodopsin's active lifetime by arrestin-1 expression in mammalian rods. J Neurosci 30:3450–3457. 10.1523/JNEUROSCI.5391-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross OP, Pugh EN Jr, Burns ME (2012) Calcium feedback to cGMP synthesis strongly attenuates single-photon responses driven by long rhodopsin lifetimes. Neuron 76:370–382. 10.1016/j.neuron.2012.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV. (1998) The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem 273:15501–15506. 10.1074/jbc.273.25.15501 [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA, Gurevich EV (2011) The functional cycle of visual arrestins in photoreceptor cells. Prog Retin Eye Res 30:405–430. 10.1016/j.preteyeres.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Gurevich VV (2006) The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem 281:3458–3462. 10.1074/jbc.M512148200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A 103:4900–4905. 10.1073/pnas.0600733103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV (2007a) Each rhodopsin molecule binds its own arrestin. Proc Natl Acad Sci U S A 104:3125–3128. 10.1073/pnas.0610886104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, Klug CS, Hubbell WL, Gurevich VV (2007b) Structure and function of the visual arrestin oligomer. EMBO J 26:1726–1736. 10.1038/sj.emboj.7601614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Vishnivetskiy SA, Hubbell WL, Gurevich VV (2008a) Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry 47:1070–1075. 10.1021/bi7021359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Dawson ES, Francis DJ, Van Eps N, Klug CS, Hubbell WL, Meiler J, Gurevich VV (2008b) A model for the solution structure of the rod arrestin tetramer. Structure 16:924–934. 10.1016/j.str.2008.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB (1999) The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell 97:257–269. 10.1016/S0092-8674(00)80735-7 [DOI] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MHE, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, et al. (2015) Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature 523:561–567. 10.1038/nature14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Hanson SM, Vishnivetskiy SA, Song X, Cleghorn WM, Hubbell WL, Gurevich VV (2011) Robust self-association is a common feature of mammalian visual arrestin-1. Biochemistry 50:2235–2242. 10.1021/bi1018607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knospe V, Donoso LA, Banga JP, Yue S, Kasp E, Gregerson DS (1988) Epitope mapping of bovine retinal S-antigen with monoclonal antibodies. Curr Eye Res 7:1137–1147. 10.3109/02713688809001885 [DOI] [PubMed] [Google Scholar]
- Lyubarsky AL, Pugh EN Jr (1996) Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci 16:563–571. 10.1523/JNEUROSCI.16-02-00563.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubarsky AL, Lem J, Chen J, Falsini B, Iannaccone A, Pugh ENJ (2002) Functionally rodless mice: transgenic models for the investigation of cone function in retinal disease and therapy. Vision Res 42:401–415. 10.1016/S0042-6989(01)00214-0 [DOI] [PubMed] [Google Scholar]
- Lyubarsky AL, Daniele LL, Pugh ENJ (2004) From candelas to photoisomerizations in the mouse eye by rhodopsin bleaching in situ and the light-rearing dependence of the major components of the mouse ERG. Vision Res 44:3235–3251. 10.1016/j.visres.2004.09.019 [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J (2000) Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 28:153–164. 10.1016/s0896-6273(00)00093-3 [DOI] [PubMed] [Google Scholar]
- Moaven H, Koike Y, Jao CC, Gurevich VV, Langen R, Chen J (2013) Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc Natl Acad Sci USA U S A 110:9463–9468. 10.1073/pnas.1301126110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Kennedy MJ, Hurley JB, Gurevich VV, Slepak VZ (2004) Direct binding of visual arrestin to microtubules determines the differential subcellular localization of its splice variants in rod photoreceptors. J Biol Chem 279:41240–41248. 10.1074/jbc.M406768200 [DOI] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ (2005) Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron 46:555–567. 10.1016/j.neuron.2005.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi M, Maza NA, Calvert PD (2012) Steric volume exclusion sets soluble protein concentrations in photoreceptor sensory cilia. Proc Natl Acad Sci U S A 109:203–208. 10.1073/pnas.1115109109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermaier MK, Peterhans C, Jaussi R, Deupi X, Standfuss J (2014) Functional map of arrestin-1 at single amino acid resolution. Proc Natl Acad Sci USA U S A 111:1825–1830. 10.1073/pnas.1319402111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet JA, Bragin A, Calvert PD, Nikonov SS, Mani S, Zhao X, Besharse JC, Pierce EA, Knox BE, Pugh EN Jr (2004) Quantification of the cytoplasmic spaces of living cells with EGFP reveals arrestin-EGFP to be in disequilibrium in dark adapted rod photoreceptors. J Cell Sci 117:3049–3059. 10.1242/jcs.01167 [DOI] [PubMed] [Google Scholar]
- Peterhans C, Lally CC, Ostermaier MK, Sommer ME, Standfuss J (2016) Functional map of arrestin binding to phosphorylated opsin, with and without agonist. Sci Rep 6:28686. 10.1038/srep28686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig DH, Nair KS, Wei J, Wang Q, Garwin G, Saari JC, Chen CK, Smrcka AV, Swaroop A, Lem J, Hurley JB, Slepak VZ (2007) Subunit dissociation and diffusion determine the subcellular localization of rod and cone transducins. J Neurosci 27:5484–5494. 10.1523/JNEUROSCI.1421-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert C, Hirsch JA, Gurevich VV, Engelman DM, Sigler PB, Fleming KG (1999) Visual arrestin activity may be regulated by self-association. J Biol Chem 274:21186–21190. 10.1074/jbc.274.30.21186 [DOI] [PubMed] [Google Scholar]
- Slepak VZ, Hurley JB (2008) Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: interaction-restricted diffusion. IUBMB Life 60:2–9. 10.1002/iub.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV (2009) Enhanced arrestin facilitates recovery and protects rods lacking rhodopsin phosphorylation. Curr Biol 19:700–705. 10.1016/j.cub.2009.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV (2011) Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience 174:37–49. 10.1016/j.neuroscience.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Seo J, Baameur F, Vishnivetskiy SA, Chen Q, Kook S, Kim M, Brooks EK, Altenbach C, Hong Y, Hanson SM, Palazzo MC, Chen J, Hubbell WL, Gurevich EV, Gurevich VV (2013) Rapid degeneration of rod photoreceptors expressing self-association-deficient arrestin-1 mutant. Cell Signal 25:2613–2624. 10.1016/j.cellsig.2013.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY (2006) Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci 26:1146–1153. 10.1523/JNEUROSCI.4289-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez M-G, Gurevich VV (2000) An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J Biol Chem 275:41049–41057. 10.1074/jbc.M007159200 [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV (2007) Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem 282:32075–32083. 10.1074/jbc.M706057200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV (2011) Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem 286:24288–24299. 10.1074/jbc.M110.213835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Zhan X, Chen Q, Iverson TM, Gurevich VV (2014) Arrestin expression in E. coli and purification. Curr Protoc Pharmacol 67:2.11.11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker WB, Donoso LA, Kalsow CM, Yankeelov JA Jr, Organisciak DT (1977) Experimental allergic uveitis. Isolation, characterization, and localization of a soluble uveitopathogenic antigen from bovine retina. J Immunol 119:1949–1958. [PubMed] [Google Scholar]
- Wilden U, Hall SW, Kühn H (1986) Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci U S A 83:1174–1178. 10.1073/pnas.83.5.1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J (1997) Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature 389:505–509. 10.1038/39068 [DOI] [PubMed] [Google Scholar]
- Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, Yin Y, Pal K, Goswami D, White TA, Barty A, Latorraca NR, Chapman HN, Hubbell WL, Dror RO, Stevens RC, Cherezov V, Gurevich VV, Griffin PR, Ernst OP, et al. (2017) Identification of phosphorylation codes for arrestin recruitment by G protein–coupled receptors. Cell 170:457–469. 10.1016/j.cell.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T, Chen Q, Cho M-K, Vishnivetskiy SA, Iverson TM, Gurevich VV, Sanders CR (2013) Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc Natl Acad Sci U S A 110:942–947. 10.1073/pnas.1215176110 [DOI] [PMC free article] [PubMed] [Google Scholar]