

Within mammalian brain circuits, activity-dependent synaptic adaptations, such as synaptic scaling, stabilize neuronal activity in the face of perturbations. Stability afforded through synaptic scaling involves uniform scaling of quantal amplitudes across all synaptic inputs formed on neurons, as well as on the postsynaptic side. It remains unclear whether activity-dependent uniform scaling also operates within peripheral circuits.
Keywords: Drosophila neuromuscular junction, firing rates, homeostatic plasticity, motor patterns, quantal content, synaptic scaling
Abstract
Within mammalian brain circuits, activity-dependent synaptic adaptations, such as synaptic scaling, stabilize neuronal activity in the face of perturbations. Stability afforded through synaptic scaling involves uniform scaling of quantal amplitudes across all synaptic inputs formed on neurons, as well as on the postsynaptic side. It remains unclear whether activity-dependent uniform scaling also operates within peripheral circuits. We tested for such scaling in a Drosophila larval neuromuscular circuit, where the muscle receives synaptic inputs from different motoneurons. We used motoneuron-specific genetic manipulations to increase the activity of only one motoneuron and recordings of postsynaptic currents from inputs formed by the different motoneurons. We discovered an adaptation which caused uniform downscaling of evoked neurotransmitter release across all inputs through decreases in release probabilities. This “presynaptic downscaling” maintained the relative differences in neurotransmitter release across all inputs around a homeostatic set point, caused a compensatory decrease in synaptic drive to the muscle affording robust and stable muscle activity, and was induced within hours. Presynaptic downscaling was associated with an activity-dependent increase in Drosophila vesicular glutamate transporter expression. Activity-dependent uniform scaling can therefore manifest also on the presynaptic side to produce robust and stable circuit outputs. Within brain circuits, uniform downscaling on the postsynaptic side is implicated in sleep- and memory-related processes. Our results suggest that evaluation of such processes might be broadened to include uniform downscaling on the presynaptic side.
SIGNIFICANCE STATEMENT To date, compensatory adaptations which stabilise target cell activity through activity-dependent global scaling have been observed only within central circuits, and on the postsynaptic side. Considering that maintenance of stable activity is imperative for the robust function of the nervous system as a whole, we tested whether activity-dependent global scaling could also manifest within peripheral circuits. We uncovered a compensatory adaptation which causes global scaling within a peripheral circuit and on the presynaptic side through uniform downscaling of evoked neurotransmitter release. Unlike in central circuits, uniform scaling maintains functionality over a wide, rather than a narrow, operational range, affording robust and stable activity. Activity-dependent global scaling therefore operates on both the presynaptic and postsynaptic sides to maintain target cell activity.
Introduction
Changes in neuronal activity can alter synaptic strength and refine synaptic connections as part of normal developmental and behavioral plasticity (Flavell and Greenberg, 2008). Abnormal levels of activity could disrupt normal activity-dependent synaptic modifications, leading to pathophysiological imbalances in activity within neural circuits (Turrigiano, 2008; Swann and Rho, 2014; Whitt et al., 2014). Neurons have however evolved the capacity to functionally adapt and maintain stable activities in the face of perturbations (Turrigiano, 2011; Vitureira et al., 2012). The most intensely studied adaptation, which stabilizes neuronal activity within mammalian brain circuits, is synaptic scaling (Turrigiano et al., 1998). It operates globally by uniformly scaling quantal amplitudes across all synaptic inputs formed on a neuron, through changes in postsynaptic receptor numbers (O'Brien et al., 1998; Turrigiano et al., 1998). Synaptic scaling causes compensatory changes in excitatory synaptic drive to the neuron in a direction that enables its firing rate, which has strayed out of set point range, to return back within range (Turrigiano and Nelson, 2004). Uniform scaling is also proposed to maintain the relative differences in synaptic weights among inputs, thereby maintaining the ability of neural circuits to appropriately process and interpret divergent inputs with physiological relevance (Turrigiano, 2008). Considering that maintenance of stable activity is imperative for the robust function of the nervous system as a whole, we tested whether activity-dependent uniform scaling could also operate within peripheral circuits.
Peripheral circuits also exhibit compensatory adaptations and activity-dependent synaptic modifications. One such circuit is the Drosophila larval neuromuscular circuit which drives crawling (Atwood and Karunanithi, 2002; Newman et al., 2017; Clark et al., 2018). This is a converging circuit, where different glutamatergic motoneurons form discrete neuromuscular junctions (NMJs) on each muscle (see Fig. 1) (Hoang and Chiba, 2001). The highly active 1b and the less active 1s motoneurons form NMJs on virtually all larval muscles (Chouhan et al., 2010; Newman et al., 2017) (see Fig. 1, middle). Compensatory adaptations observed at those NMJs are mainly those which preserve strong muscular excitation by homeostatically maintaining synaptic drive to the muscle. They include presynaptic homeostatic potentiation, presynaptic homeostatic depression (PHD), and synaptic homeostasis through structural compensations (Stewart et al., 1996; Petersen et al., 1997; Davis and Goodman, 1998; Daniels et al., 2004; Frank et al., 2006; Gavino et al., 2015). Those adaptations are recruited through changes at synapses themselves, rather than through changes in neuronal and muscle activities (firing rates), and are therefore not regarded as activity-dependent (Turrigiano, 1999; Davis and Muller, 2014). It is unclear whether there are activity-dependent adaptations that manifest at NMJs, involving uniform scaling, to afford robust muscle activity.
Figure 1.

Third instar larval NMJ preparation. Experimental third instar larval NMJ preparation (left). Diagram represents the larval brain (CNS) and the commonly studied muscles (2, 6, and 7 in segment A3). The 1b and 1s motoneurons innervating muscle 2 (left) form a converging neuromuscular circuit (middle). 1s activity was increased. A live preparation showing the 1b (red) and 1s (yellow) synaptic boutons, which form NMJs with the muscle (right). The mitochondrial dye, 4-Di-2-Asp (5 μm; red), was used to visualize the boutons. On muscle 2, RRAGal4 drives gene expression in only the 1s motoneuron, as shown by the GFP expression in only the 1s boutons (yellowish green; produced by the merger of the red and green channels). Scale bar, 10 µm.
Activity-dependent synaptic modifications, which appear within the larval neuromuscular circuit following long-term increases in neuronal activity, have been associated with synapse formation and maturation, and with synaptic modifications correlated to information storage (Martin and Kandel, 1996; Thomas and Sigrist, 2012; Harris and Littleton, 2015). Such work used fly strains where activity was chronically increased in all neurons or motoneurons (Budnik et al., 1990; Zhong et al., 1992; Davis et al., 1996; Schuster et al., 1996; Mosca et al., 2005). With current knowledge regarding activity-dependent compensatory adaptations, we wondered whether such synaptic modifications were fully or partly compensatory to afford robust muscle activity. We also wondered whether compensatory adaptations could be recruited by increasing the activity of only one of the two motoneuron types.
Using genetic manipulations, we transformed the activity of the 1s motoneuron, innervating muscle 2, from being lowly active to being highly active (see Fig. 1, middle). We then assessed whether that transformation recruited compensatory adaptations by testing for alterations in muscle activity and NMJ properties formed by the genetically manipulated 1s and the unmanipulated 1b motoneurons (see Fig. 1, right). We uncovered a compensatory adaptation, which we termed “presynaptic downscaling,” which uniformly downscaled 1b and 1s evoked neurotransmitter release to afford robust and stable muscle activity. Activity-dependent uniform scaling can therefore manifest also within the periphery and on the presynaptic side.
Materials and Methods
Drosophila strains
Fly stocks were reared at 25°C, relative humidity of 70%. The Gal4/UAS system was used to drive transgene expression in selected tissues. The following Gal4 stocks were used: (A) RRAGal4, UAS-mCD8-GFP (RRA; third chromosome): to drive transgene expression in the 1s motoneuron innervating muscle 2 (Fujioka et al., 2003), provided by Dr. Miki Fujioka; and (B) w-; P[tubP-Gal80ts]20; RRAGal4, UAS-mCD8-GFP (RRAGal80ts): to conditionally drive transgene expression in the 1s motoneuron innervating muscle 2 on exposure to elevated temperatures (Srinivasan et al., 2012), provided by Dr. Carsten Duch.
The following UAS stocks were used: (A) UAS-eagΔ932, UAS-SDN/+ (eagSh(DN); second chromosome): to elevate neuronal activity by driving expression of dominant-negative forms of ether-à-go-go and Shaker voltage-gated potassium channels (Wang et al., 2016), provided by Dr. Subhabrata Sanyal; (B) UAS-DVGLUT2 (DVGLUT; second chromosome): to overexpress the Drosophila vesicular glutamate transporter (DVGLUT) (Daniels et al., 2004), provided by Dr. Aaron DiAntonio; (C): UAS-DVGLUT RNAi (DVGLUT RNAi; second chromosome): to knock down endogenous levels of DVGLUT (Vienna Drosophila Resource Centre, transformant ID 104324); and (D) UAS-dTRPA1 (dTRPA1; second chromosome): using the heat-activated Drosophila transient receptor potential cation channel, dTRPA1, to conditionally elevated neuronal activity during exposure to elevated temperatures (Pulver et al., 2009). Transgenic lines were generated by crossing Gal4 virgin females with UAS males. The controls were Gal4/+ and UAS/+ lines. Experiments were conducted on muscle 2, segment 3 in wandering third instar larvae of either sex.
Conditional increase in 1s motoneuron activity
We used two methods to conditionally increase the activity of the 1s motoneuron innervating muscle 2 in the late stages of third instar larval development: (A) by using the TARGET gene expression system (McGuire et al., 2003; Srinivasan et al., 2012). Temperature-sensitive Gal80 (Gal80ts) was used to block Gal4 activity and prevent transgene expression until exposure to elevated temperature (McGuire et al., 2003). In RRAGal80ts>eagSh(DN) larvae, dominant-negative EAG and Sh were conditionally expressed in the 1s motoneuron upon exposure to 32°C. At 32°C, Gal80ts activity was blocked, enabling Gal4 to drive transgene expression. The transgenic and control lines were mated in vials and maintained at 19°C for 7 d. At 19°C, Gal4 activity was blocked by Gal80ts, preventing transgene expression. Three days after mating, the adult flies were removed from the vial. On the seventh night, the vials were transferred to 32°C for 36 h (between the mid and the late third instar developmental stages) to increase 1s activity (Srinivasan et al., 2012); (B) by using heat-activated dTRPA1 cation channels. At elevated temperature, the channels open and the large influx of cations depolarizes the membrane, causing the firing of action potentials (Pulver et al., 2009). In RRA> dTRPA1 larvae, dTRPA1 was expressed in only the 1s motoneuron. The transgenic and control lines were mated in vials and maintained at 19°C for 7 d. Three days after mating, the adult flies were removed from the vial. On the eighth night, the vial was transferred to 32°C for 12 h to increase 1s activity.
For both methods, control and transgenic lines were subjected to the same conditional transgene activation protocols and electrophysiological experiments were conducted on wandering third instar larvae straight after exposure to 32°C.
RT-PCR analysis
A quantitative RT-PCR assay was used to confirm knockdown of the VGLUT relative to that of the housekeeping gene Act88F (Ferguson et al., 2017). Act88F was first determined to be stably expressed across all experimental conditions (data not shown). Larvae were collected and snap frozen, and stored at −80°C. Five pools of five larvae (30 larvae total) were placed into a 1.5 ml Eppendorf tube. Total RNA was purified using TRIzol according to the manufacturer's protocols (Invitrogen), immediately after dissection. Total RNA was treated with DNase (Sigma Millipore) to eliminate genomic DNA; ∼0.5 μg of total RNA was reverse transcribed using random primers (Invitrogen) and reverse transcriptase (Invitrogen) according to the manufacturer's protocols. Gene expression was estimated with two technical replicates using a standard qPCR assay (Rohrscheib et al., 2015). Each qPCR mixture contained 12.5 μl of 2× SYBR premix (Invitrogen), 1 μl of forward primer, 1 μl of reverse primer, 100 ng of DNA, and H2O to a final volume of 25 μl. The expression of the two genes was estimated relative to Act88F using the δ-δ (where Ct is threshold cycle) Ct method (Pfaffl, 2001). Averages of expression were compared using unpaired t test.
Immunohistochemistry
The Bruchpilot (BRP) active zone marker (Developmental Studies Hybridoma Bank) was used to ascertain the number of active zones per bouton (Smith and Taylor, 2011). Motor nerve terminals were double-stained using HRP conjugated to Cy3, enabling determination of the number of boutons per nerve terminal (Hoang and Chiba, 2001). Imaging was performed on a 510 Meta confocal microscope (Carl Zeiss), and image processing was conducted using Fiji (ImageJ.) Bouton and active zone counts were conducted in seven different preparations for each genotype. The live staining of nerve terminals using the mitochondrial dye, 4-Di-2-Asp, was undertaken as previously described (Kurdyak et al., 1994).
Intracellular and focal macropatch recordings
Details of the methods and analysis for performing focal macropatch and intracellular recordings have been previously described (Macleod et al., 2006; Karunanithi et al., 2018). Recordings were conducted at room temperature (22°C) in HL6 hemolymph-like solution (Macleod et al., 2006) using [Ca2+]o = 1.0 mm, unless otherwise specified. Evoked responses were elicited by stimulating the 1b and 1s motor axons together at 1 Hz using suprathreshold stimulus strengths. The recorded signals were amplified under bridge/I-clamp mode. Following amplification, signals were filtered. For focal macropatch recordings, signals were high- and low-pass filtered at 1 Hz and 3 kHz, respectively. For intracellular recordings, signals were low-pass filtered at 3 kHz, and there was no high-pass filtering. The filtered signals were then digitized and stored on a computer. In all cases, sampling rates for digitization were set according the Nyquist sampling rate, where rates were set to at least twice the highest frequency component of the signal (∼0.3-0.4 kHz and 1-1.5 kHz for intracellular and focal macropatch recordings, respectively).
Intracellular recording
Intracellular recordings were used to record evoked excitatory junction potentials (EJPs), spontaneously occurring miniature excitatory junction potentials (mEJPs) and resting membrane potentials. Such recordings were also used to determine muscle input resistances and muscle membrane time constant. Intracellular recording electrodes (50-80 mΩ) were filled with a 2:1 mixture of 3 m potassium acetate to 3 m potassium chloride. The EJPs are compound responses produced on activating both 1b and 1s NMJs. Those compound responses are also referred to as synaptic drive in the text. Synaptic drive represents the total strength of synaptic activation of the postsynaptic cell. mEJPs were recorded in the absence of nerve stimulation for ∼1-2 min. For each genotype, recordings were made from at least eight different preparations; and in each experiment, 100 EJPs and 25-50 mEJPs were recorded. The recorded EJPs and mEJPs were digitized at a sampling rate of 40 kHz.
Focal macropatch recording
Drosophila larval muscles are multiply innervated and the 1b and 1s motoneurons form NMJs on virtually all larval muscles (Hoang and Chiba, 2001), generating different types of postsynaptic responses (Kurdyak et al., 1994; Lnenicka and Keshishian, 2000; Newman et al., 2017). To discriminate between the two response types at the level of individual boutons, we used the focal macropatch recording technique. Focal macropatching enables the recording of evoked excitatory junctional currents (EJCs) and spontaneously occurring miniature excitatory junctional currents (mEJCs) from individual 1b and 1s boutons (Heckmann and Dudel, 1998; Karunanithi et al., 2002, 2018, 2020; Pawlu et al., 2004; Kittel et al., 2006; Dawson-Scully et al., 2007). We also used focal macropatching because it satisfies the requirements for conducting proper quantal analysis, and those requirements have been previously discussed by Karunanithi et al. (2020). Focal macropatching was also used because we can detect individual action-potential evoked postsynaptic currents during high-frequency motor bursts, reflective of motoneuron firing properties (Fox et al., 2006) (see Motor pattern recordings from nerve and muscle during fictive locomotion section).
A brief account of the methods for conducting focal macropatching is provided as follows. The preparation was viewed with a 60× water immersion lens (NA 1.0) using Nomarski optics. Live images of the nerve terminals were captured using a low-light video camera and projected onto a computer monitor. Such a setup enabled selection of well-isolated, single 1b and 1s boutons for focal macropatch recordings and positioning of the focal macropatch electrode at the site of recording. 1b nerve terminals are composed of large synaptic boutons, whereas 1s nerve terminals are composed of small synaptic boutons, and these size differences are readily discerned under Nomarski optics. The focal macropatch electrodes possessed open tip diameters of ∼5 µm and were filled with HL6 solution. The open tip, when placed on the muscle to enclose the bouton from which recordings are conducted, does not exert pressure on the bouton during recordings (Karunanithi et al., 2002). The recorded signals were amplified using an Axoclamp 900A (under bridge/I-clamp mode) (Molecular Devices), and digitized and stored on a computer using the PowerLab 4/35 data acquisition system (ADInstruments). The recorded EJCs and mEJCs were digitized at a sampling rate of 40 kHz. In each experiment, recordings were made from either a single 1b or 1s bouton, or from two boutons of each type; mostly, the former. The assessment of the quality of postsynaptic current recordings, and the analysis and interpretation of the results have been previously described (Karunanithi et al., 2002, 2018). In experiments conducted in [Ca2+]o = 1.0 mm, we recorded and made measurements from 150-200 EJCs and 25-50 mEJCs in each experiment (Karunanithi et al., 2018). For each bouton type in each genotype, recordings were made from at least 10 different boutons, and each from a different preparation. In experiments conducted in [Ca2+]o = 0.5 mm, we used 150-200 stimuli and for each bouton type in each genotype, recordings were made from at least seven different boutons, and each from a different preparation.
It is important to note that the focal macropatch electrode does not form a seal with the larval muscle membrane, but forms a contact resistance which remains constant throughout recordings and appears not to affect current flow (Karunanithi et al., 2002). The sizes of quantal events were not therefore related to the contact resistance, and the observed 1b-1s differences in evoked and spontaneous responses appeared consistently different in recordings conducted by ourselves and others, providing a good comparison of relative 1b-1s EJC and mEJC amplitude differences among genotypes (Karunanithi et al., 2002; Pawlu et al., 2004; Dawson-Scully et al., 2007). Those 1b-1s evoked and spontaneous amplitude differences were also recently replicated using optical measurements (Newman et al., 2017; Li et al., 2018). It is also important to note that amplitude measurements of EJCs are not influenced by the timing of quantal release. That was indicated by the lack of correlation between the amplitudes and rise times of EJCs (Heckmann and Dudel, 1998).
The currents recorded using focal macropatching are referred to in millivolts (Karunanithi et al., 2018). This technique is a form of extracellular voltage recording that was originally pioneered by Bernard Katz and colleagues (Del Castillo and Katz, 1956). In this recording configuration, the recorded quantal events represent voltage changes in the extracellular solution. As the quantal current flows through the resistance of the extracellular solution, it produces a voltage drop across that resistance. The amplitude of the voltage drop is directly proportional to the amplitude of the quantal current because the resistance of the extracellular solution remains constant. The quantal events are therefore referred to as quantal currents, despite being measured in millivolts (Del Castillo and Katz, 1956).
Quantal analysis
The measured EJC and mEJC amplitudes were used to derive quantal size (q; average mEJC amplitude), average EJC amplitude, quantal content (QC; average EJC amplitude/average mEJC amplitude), and the quanta released per stimulus (Bademosi et al., 2018; Karunanithi et al., 2018). In each experiment, the quanta released per stimulus was calculated by dividing each EJC amplitude in the train by the average mEJC amplitude (Bademosi et al., 2018). The cumulative relative frequency histogram for each group represents the pooled data from all the experiments in that group. For each experiment, QC equals the average quanta released per stimulus.
At high [Ca2+]o (1 mm), neurotransmitter release can be described by the binomial model, where QC = p · n (Del Castillo and Katz, 1954; McLachlan, 1978). n equates to the average number of functionally active release sites/units and p equates to the average release probability of all the functionally active release sites/units. In each experiment, p was calculated using the following equation:
where q is the quantal size, S2 is the variance of the EJC amplitudes, and σ2 is the variance of the mEJC amplitudes (Bennett and Florin, 1974; McLachlan, 1978; Bennett et al., 1991). n was calculated by dividing QC by p.
In experiments that were conducted at low [Ca2+]o = 0.5 mm, neurotransmitter release can be described using the Poisson model. In this model for low neurotransmitter release (usually, when p < 0.3) (Bennett et al., 1991), QC was derived in each experiment using the method of failures (Del Castillo and Katz, 1954; McLachlan, 1978; Daniels et al., 2004):
where the Number of stimuli represents the number of times the nerve was stimulated during a low-frequency train of stimuli and the Number of failures represents the number of times the stimuli failed to produce an EJC. Although the parameter p does not hold any meaning in the Poisson model of neurotransmitter release, it was reported that, at the larval NMJ, the probability of observing the release of one packet (a quantum) of neurotransmitter during a train of stimuli reflected p (Paradis et al., 2001); one quantum being released by one vesicle. Under low release conditions, fewer stimuli in a train release a packet of transmitter. The probability of observing a quantum (p1) can be derived using the following equation (Katz, 1966):
Motor pattern recordings from nerve and muscle during fictive locomotion
We recorded centrally generated rhythmic motor bursts during fictive locomotion (Suster et al., 2004; Klose et al., 2005) to assess neuronal and muscle activity. Robust motor discharge was elicited in the third instar larval NMJ preparation by perfusing the bath with HL3 solution ([Ca2+]o = 1.0 mm) heated to 27°C (Suster et al., 2004; Klose et al., 2005). The CNS and the segmental nerves were left intact. Neuronal activity was assessed by recording the motor bursts from individual boutons using focal macropatch electrodes (Fox et al., 2006) (see Fig. 2A, left). In such recordings, signals were high- and low-pass filtered at 1 and 3 kHz, respectively, and then digitized at a sampling rate of 40 kHz. This method of recording assesses the EJCs resulting purely from action potential firing in the motoneuron, indicative of neuronal activity. Motoneuron activity was assessed by measuring the frequency of EJCs per burst (motoneuron firing rate). Recordings were made from at least five different boutons for each bouton type within each genotype. To determine the frequency of EJCs per burst, we first found the first-order derivative of the raw data files in LabChart Pro (version 8.1.5, ADInstruments). This accentuates the changes in the signal, making the EJCs within bursts easily detected. Figure 2A (right) compares the raw data (red traces) and the derivative (gray trace). The first-order derivative was calculated at each sampled point by determining the slope of a line fitted across a time window that was centered at that point (where the window width = 27 points on each side of the center point). The time window was determined empirically. These files were then converted into Axon PClamp files (Axon Binary Format: pClamp10), and Clampfit (version 10.5.0.9, Molecular Devices) was used to detect individual EJCs events using a threshold-based algorithm with a negative crossing threshold set at 2-3 times the SD of the baseline noise (see Fig. 2A, right, blue dots on gray trace). The baseline was determined from a region of ∼200 ms that was free of events. Once event detection was completed, we determined bursts of events using the Burst Analysis function in Clampfit. The minimum number of events in a burst was set at 5, and a maximal time between events in a burst (Burst delimiting interval) was set at 200-400 ms (both determined empirically to visually match the data; see Fig. 2). That analysis provided the frequency of EJCs per burst. Our estimates of control 1b and 1s motoneuron firing rates reported here are similar to those determined by others using a different method, supporting our findings (Chouhan et al., 2010).
Figure 2.
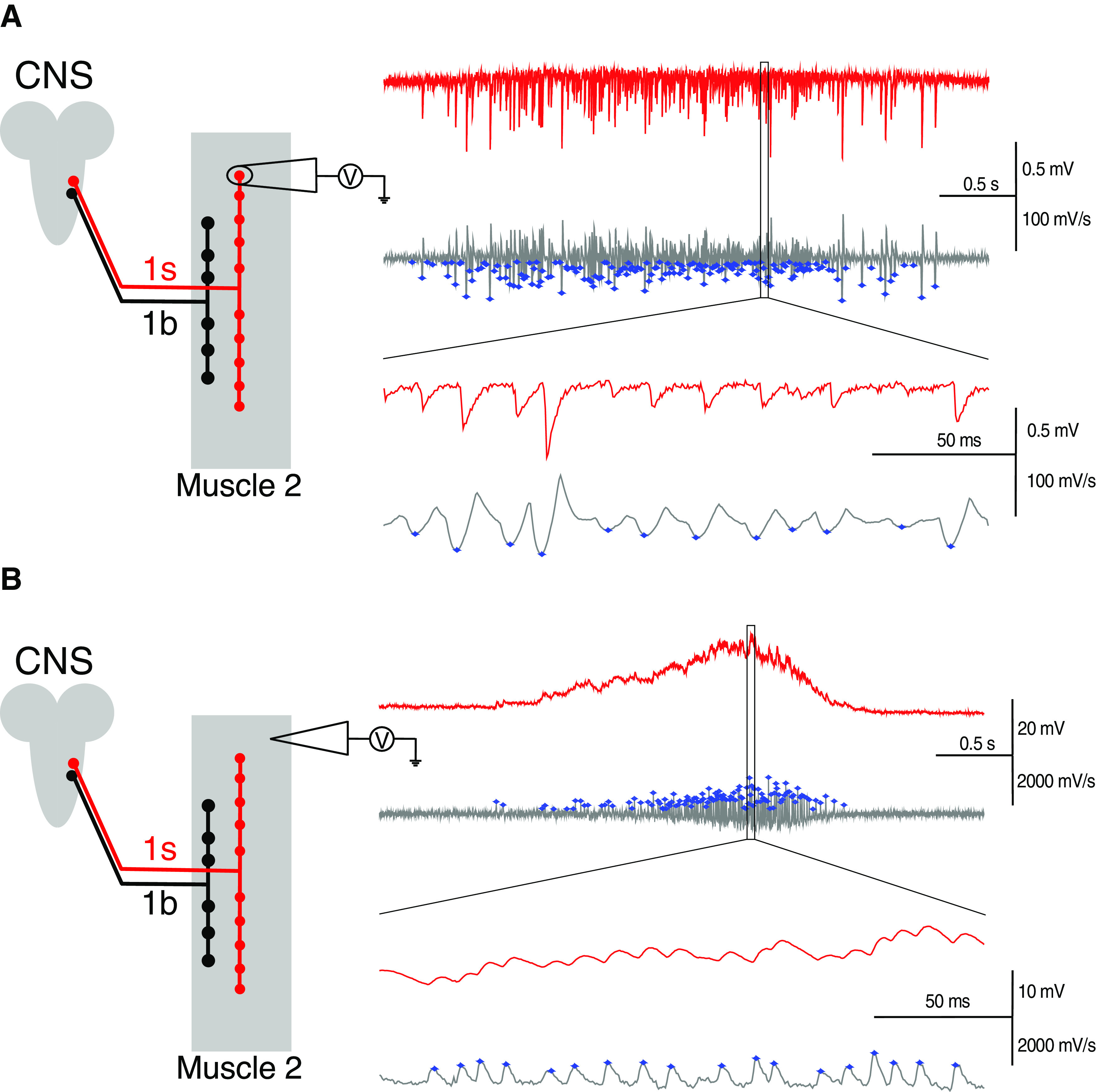
Determination of the neuronal and muscle firing rates within each motor burst. A, Left, Diagram represents the configuration for recording motor bursts from single synaptic boutons (which constitute the nerve terminal). The CNS was left intact. Right, Exemplary trace showing EJCs within a neuronal burst (red), and the first-order derivative of that raw data (gray) used for detecting the occurrence of EJCs within a burst. Blue asterisks on the gray traces indicate those events detected as EJCs. The selection box is magnified to show the clear detection of EJCs as events using the described settings. B, Left, Configuration for recording motor bursts from the muscle), presented as in A. Right, Same as in A, but for detecting the EJPs within a burst when recorded from muscle.
Motor discharge recorded from the muscle is influenced by the activities of the 1b and the 1s motoneurons (Chouhan et al., 2010; Newman et al., 2017), the properties of the NMJs, and the properties of the muscle that control its excitability (Paradis et al., 2001). Because of such influences, the motor discharge when recorded from muscle display different properties from those recorded from motoneurons. Motor discharges produce muscle contractions that drive larval locomotion, and the strengths of those contractions appear to be affected by the amplitude and frequency of EJPs within bursts (Paterson et al., 2010; Ormerod et al., 2015, 2016; Aponte-Santiago et al., 2020). Motor discharge was recorded from muscle 2, using intracellular electrodes (see Fig. 2B, left). In such recordings, signals were low-pass filtered at 3 kHz without any high-pass filtering, and then digitized at a sampling rate of 3 kHz. Muscle activity was determined by measuring the frequency of EJPs per burst (muscle firing rate) (see Fig. 2B, right). Recordings were made from at least 11 different preparations for each genotype. On the muscle side, the frequency of EJPs per burst in motor pattern recordings was determined using the same protocols used for determining the frequency of EJCs per burst, but with changes to some of the following parameters: (1) window width = 3 was applied when using the Derivative function in LabChart Pro; and (2) category = 1, positive-going was used when applying the Threshold Search function in Clampfit (raw data: red traces; derivative data: gray traces; detected events: blue dots; see Fig. 2B, right). Our estimates of the range of muscle firing rates in controls, which are reported here, are similar to those determined previously for muscle 6, supporting our findings (Barclay et al., 2002; Suster et al., 2004; Klose et al., 2005).
Experimental design and statistical analysis
Statistical and data analysis was performed using Excel (Microsoft) and Prism 6 (GraphPad) software. To assess statistical differences between two groups, the unpaired Student's t test was used. The nonparametric tests, one-way ANOVA and the Kolmogorov–Smirnov test, were used to assess statistical differences. In all cases, significance was set at p < 0.05. The Kolmogorov–Smirnov test was used to compare the shapes of standardized distributions. Distributions were standardized by subtracting the average and then normalizing to 1 SD (Frerking et al., 1995; Nusser et al., 1997; Karunanithi et al., 2002). The coefficient of variation (CV) was obtained by dividing the SD of a population by its average. Where possible, normalized parameters were provided in the main figures to improve readability. Parameters were normalized to their respective averages in RRA/+ controls. Throughout the text, we have used N rather than n to represent sample size because the latter, by convention, represents the number of active release sites in binomial analysis (see Quantal analysis section).
Results
Chronic increase in 1s activity caused a compensatory decrease in muscle activity
Within the larval neuromuscular circuit, we assessed the consequence to muscle activity on perturbing neuronal activity (neuronal firing rate) by chronically increasing 1s motoneuron activity (Fig. 1, middle). 1s activity was increased by using the RRAGal4 driver (Fujioka et al., 2003; Newman et al., 2017) to express dominant-negative forms of ether-à-go-go (EAG) and Shaker (Sh) potassium channels (Wang et al., 2016) throughout development into the 1s motoneuron innervating muscle 2 (RRA>eagSh(DN) larvae) (Fig. 1, right). We tested for increased 1s activity by recording centrally generated motor bursts which drive crawling. To distinguish between 1b and 1s activities, we used focal macropatch electrodes to record EJCs, which appeared at single 1b and 1s synaptic boutons (Fig. 2A, left). These EJCs occur in response to each presynaptic action potential, and therefore faithfully reflect motoneuron action potential discharge during bursts (Fox et al., 2006) (Fig. 3A, left). The frequency of EJCs per burst was representative of motoneuron firing rate (Fox et al., 2006), and we determined the mean firing rate across multiple bursts within each experiment. In RRA>eagSh(DN) larvae, we increased mean 1s firing rate by 89%, without any change in the mean 1b firing rate (Fig. 3A, right). There was, however, no change in the frequencies of 1b and 1s bursts (1b: RRA/+ = 0.063 + 0.009 Hz, N = 13; RRA>eagSh(DN) = 0.042 + 0.005 Hz, N = 10; p = 0.066; 1s: RRA/+ = 0.080 + 0.028 Hz, N = 6; RRA>eagSh(DN) = 0.084 + 0.025 Hz, N = 6; p = 0.929), but there was an increase in 1s burst duration (1b: RRA/+ = 3.51 + 0.056 s, N = 12; RRA>eagSh(DN) = 3.23 + 0.45 s, N = 12; p = 0.698; 1s: RRA/+ = 0.79 + 0.09 s, N = 8; RRA>eagSh(DN) = 3.74 + 1.09 s, N = 6; p = 0.0419).
Figure 3.

Increased 1s firing rate caused a compensatory decrease in muscle firing rate. A, Left, Exemplary traces of bursts of action potential-evoked 1b and 1s postsynaptic EJCs recorded from the indicated bouton and genotypes. Right, The graph of 1b and 1s mean motoneuron firing rates in individual experiments in the indicated genotypes. B, Left, Exemplary traces of motor bursts from the indicated genotypes, consisting of action potential-evoked EJPs. In those traces, the insets provide better time resolution of individual events (dotted) within the bursts over a 100 ms window (blue rectangles). As observed, burst duration increased in RRA>eagSh(DN) larvae (RRA/+ = 2.71 + 0.18 s, N = 12; RRA>eagSh(DN) = 3.95 + 0.39 s, N = 11; p = 0.0116) because of an increase in 1s burst duration. Right, Graph of the mean muscle firing rate in individual experiments in the indicated genotypes. C, Left, Exemplary traces of trains of motor bursts in muscle from the indicated genotypes. Right, Graph of the mean frequency of bursts in individual experiments in the indicated genotypes. *p < 0.05, #p < 0.0001. NS, Not significant, p > 0.05. Graphs represent individual data points. Error bars indicate average + SEM.
We next tested whether the near-doubling of 1s firing rate also resulted in a similar increase in muscle firing rate in RRA>eagSh(DN) larvae. We tested for that by recording the motor bursts which appeared in the muscle, following bursts of motoneuron action potentials, to produce the rhythmic waves of muscle contractions which drive larval crawling (Barclay et al., 2002; Suster et al., 2004; Klose et al., 2005). Using intracellular electrodes, we recorded the bursts of EJPs generated at NMJs (Figs. 2B, 3B, left) following bursts of 1b and 1s motoneuron action potentials (Fig. 3A, left). The frequency of EJPs per burst was representative of muscle firing rate (Pulver et al., 2009), and we calculated the mean firing rate across multiple bursts within each experiment. Surprisingly, the mean muscle firing rate was halved (decreased by 52%; Fig. 3B, right), compensating for the near-doubling of 1s firing rate. That decrease was not because of decreases in the passive muscle membrane properties, as indicated by no significant changes in the resting membrane potential (RRA/+ = −60.4 + 1.0 mV, N = 8; RRA>eagSh(DN) = −61.3 + 1.5 mV, N = 9; p = 0.6075), muscle input resistance (RRA/+ = 14.2 + 0.9 MOhm, N = 9; RRA>eagSh(DN) = 13.7 + 0.8 MOhm, N = 10; p = 0.666) and membrane time constant (RRA/+ = 25.7 + 1.2 ms, N = 9; RRA>eagSh(DN) = 25.6 + 0.5 ms, N = 8; p = 0.9031). The variation in mean muscle firing rates across experiments within each genotype (Fig. 3B, right), expressed as the CV (= SD/mean) (Hengen et al., 2013, 2016), was small (0.19) and similar to control (0.20), indicating that the mean firing rates within each genotype were maintained stably and tightly around set points (steady states). The frequency of bursts, which correspond to the rhythm frequency of peristaltic contractions driving larval crawling (Klose et al., 2005), was also decreased by 40.1% (Fig. 3C), representative of slower crawling. These results show that, when the muscle is overloaded with abnormally high motoneuron activity, compensatory adaptations strongly decrease muscle activity. We next elucidated those compensatory adaptations.
Muscle activity is decreased through a reduction in excitatory synaptic drive
In vertebrate skeletal muscles, action potential firing rates follow those in the innervating motoneurons because NMJs produce strong excitatory synaptic drive to the muscle fibers (Ouanounou et al., 2016). In RRA>eagSH(DN) larvae, synaptic drive to the muscle appears to be decreased because the mean muscle firing rate was decreased (Fig. 3B, right) despite an increase in mean 1s firing rate and no change in mean 1b firing rate (Fig. 3A, right). The relationship between muscle firing rate and excitatory synaptic drive is yet to be comprehensively assessed in this preparation. In neurons, however, firing rates are positively correlated with the degree of excitatory synaptic drive neurons receive, and changes in firing rates can therefore arise from changes in synaptic drive (Turrigiano and Nelson, 2000, 2004) (Fig. 4A). We therefore tested whether the decrease in mean muscle firing rate resulted from an activity-dependent decrease in excitatory synaptic drive.
Figure 4.

A decrease in synaptic drive afforded robust and stable muscle activity. A, The firing rate of neurons is positively correlated to the synaptic drive they receive. B, Left, Diagram represents the configuration for recording compound EJPs from the muscle using intracellular electrodes. Middle, Exemplary traces of EJPs from the indicated genotypes. Right, Mean EJP amplitudes in individual experiments in control and RRA>eagSh(DN) larvae. C, mEJP traces (left) and graph of mean mEJP amplitudes in individual experiments (right) for the indicated genotypes. When changes in EJP and mEJP amplitudes in RRA>eagSh(DN) larvae (EJP = 19.37 + 1.12 mV, N = 12; mEJP = 0.68 + 0.02 mV, N = 12) were compared with the UAS/+ control (EJP = 24.76 + 1.0 mV, N = 11; mEJP = 0.60 + 0.02 mV, N = 12), they yielded the same significant results (EJP: p = 0.0018; mEJP: p = 0.0024) as when compared with RRA/+ control (EJP = 27.04 + 0.78 mV, N = 11; mEJP = 0.55 + 0.78 mV, N = 12; comparisons shown in B, C). We also ruled out that synaptic physiology was unaffected by the position of transgene insertion and leaky transgene expression, on finding that there was no significant difference in EJP (p = 0.0883) and mEJP (p = 0.0592) amplitudes between the RRA/+ and UAS/+ controls. Subsequent comparisons of synaptic changes were therefore conducted using only RRA/+ as control. D, Motor nerve terminals double-stained with HRP (red, top panels) and BRP (green, bottom panels) antibodies in the indicated genotypes. Selected BRP-labeled boutons were similarly magnified to highlight the occurrence of the active zones on boutons (1b: *; 1s: #). Scale bar, 10 µm. Active zone numbers per bouton were determined by counting the BRP puncta per bouton for 1s (RRA/+ = 4.50 + 0.26, N = 81; RRA>eagSh(DN) = 6.70 + 0.42, N = 81; p < 0.0001), and 1b (RRA/+ = 10.57 + 0.59, N = 69; RRA>eagSh(DN) = 10.05 + 0.64, N = 94; p = 0.5578) boutons in both genotypes. E, No changes in absolute 1b and 1s nerve terminal bouton numbers (left), or when normalized to muscle surface area (MSA) (right). F, For each motoneuron type within each genotype, individual symbols in each column represent the motoneuron firing rates within individual bursts, pooled from all experiments (whereas in Fig. 3A, right, each symbol represents the mean value across multiple bursts in each experiment). Each column therefore displays the full range of firing rates recorded for each motoneuron type within each genotype. G, For each genotype, individual symbols in each column represent the muscle firing rates within individual bursts, pooled from all experiments (whereas in Fig. 3B, right, each symbol represents the mean value across multiple bursts in each experiment). Each column therefore displays the full range of muscle firing rates recorded for each genotype. The physiological range represents the full range of firing rates observed in control (gray shaded area). H, The CV of muscle firing rates in individual experiments for both genotypes. I, In RRA>eagSh(DN) larvae, the decrease in synaptic drive (black arrow) caused the decrease in mean muscle firing rate from the old set point (black dot) to the new set point (blue dot). The decrease in synaptic drive did not, however, displace firing rates out of physiological range (gray shaded area, as shown in G), or change the relative variation in firing rates around the downshifted set point (CV2, blue shaded area represents the mean value in H) compared with control (CV1, charcoal shaded area represents the mean value in H). Muscle activity therefore remained robust and stable. ***p < 0.001, #p < 0.0001. NS, Not significant, p > 0.05. Graphs represent individual data points. Error bars indicate average + SEM.
The mean peak amplitude of motor bursts recorded from muscles was decreased in RRA>eagSH(DN) larvae, consistent with a decrease in synaptic drive (RRA/+ = 22.15 + 1.3 mV, N = 11; RRA>eagSh(DN) = 13.66 + 1.37 mV, N = 11; p = 0.0002). Excitatory synaptic drive to the muscle was determined by recording the compound EJPs produced on activating both 1b and 1s NMJs in response to 1 Hz nerve stimulation (Fig. 4B, left and middle). We found that mean EJP amplitude was indeed decreased by 29.7% (Fig. 4B, right). That decrease could not be attributed to reduced neurotransmitter release: (1) from individual synaptic vesicles since mEJP amplitudes (produced by neurotransmitter release from individual vesicles) were larger than in control (Fig. 4C); and (2) from fewer 1b and 1s synaptic boutons because there were no changes in their numbers compared with control (Fig. 4D,E). Before presenting the analysis of the factors that decreased synaptic drive, it was imperative to address whether the decrease in muscle firing rate produced by the decrease in synaptic drive compromised muscle activity.
Decrease in synaptic drive affords robust and stable muscle activity
In RRA>eagSH(DN) larvae, we assessed whether the decrease in synaptic drive (Fig. 4B, right): (1) displaced muscle firing rates outside of the physiological range, compromising robust muscle activity; and (2) decreased the variation in muscle firing rates around the downshifted set point, potentially limiting the range of muscle contractions.
Robustness refers to maintaining functionality, whereas stability (homeostasis) refers to maintaining functionality around a steady state (or set point), against perturbations (Kitano, 2004, 2007). We tested for robustness by determining whether firing rates were maintained within the muscle's physiological range in RRA>eagSH(DN) larvae despite the downshift in the set point. We first determined the muscle's physiological range by pooling the firing rates from all experiments in control. Larval muscles produce wide variations in evoked muscle contractions to drive a range of movements, such as linear crawling, turning, and rolling (Ormerod et al., 2016; Clark et al., 2018). To produce such varied contractions, the range of firing rates of motoneurons and muscle is anticipated to be wide. In control, this was indeed the case for both 1b and 1s motoneurons (Fig. 4F, 1b: 7–146.6 Hz, black crosses; 1s: 7.6–53.6 Hz, red crosses) and muscle (Fig. 4G, ranging between 8.4 and 104.1 Hz and represented by the gray area). That range of muscle firing rates represents the physiological range (Fig. 4G, gray area).
In RRA>eagSH(DN) larvae, we determined whether muscle firing rates were displaced outside of the muscle's wide physiological range. We calculated the range of firing rates of motoneurons and muscle in that genotype. 1b and 1s motoneuron firing rates varied widely (Fig. 4F, 1b: 9–123.1 Hz, black circles; 1s: 7.8–94.2 Hz, red circles), but the range of muscle firing rates was, however, smaller than in control (Fig. 4G; ranging between 5.1 and 54.7 Hz). The muscle firing rates were not, however, displaced outside of the physiological range (gray area), indicating that muscle activity remained robust.
Because of the decrease in the range of muscle firing rates in RRA>eagSH(DN) larvae (Fig. 4G), we tested, in individual experiments, whether the relative variation in firing rates around the downshifted set point (Fig. 3B, right) was also decreased. Such a decrease could curtail the range of muscle contractions and in turn the range of movements. In each experiment, we determined the relative variation by calculating the CV. In both control and RRA>eagSH(DN) larvae, the CVs were similar and large (Fig. 4H; CV 0.20 was considered as small in Fig. 3B), showing that variations were similar and wide ranging around set points, and homeostatically maintained in RRA>eagSH(DN) larvae (Fig. 4H). Those results indicate that a wide range of contractions could still be produced as in controls, but those contractions may be weaker because of decreases in the magnitudes of firing rates (Fig. 3B, right). Our findings therefore indicate that the compensatory decrease in synaptic drive: (1) did not displace firing rates outside of the physiological range; and (2) did not disrupt the homeostatic maintenance of the wide variation in firing rates around the downshifted set point (Fig. 4I). Muscle activity therefore remained robust and stable.
Decrease in EJP amplitude resulted a from decrease in 1b EJC amplitude
We next elucidated the mechanisms that caused the compensatory decrease in synaptic drive, affording robust and stable muscle activity. Since the genetic manipulation was only to 1s motoneuron activity, we hypothesized that the decrease in compound EJP amplitude (Fig. 4B, right) resulted from a selective decrease in 1s rather than in 1b postsynaptic response amplitude. To discriminate between 1b and 1s postsynaptic responses, we used focal macropatch electrodes to record postsynaptic currents at the resolution of single boutons (Pawlu et al., 2004; Kittel et al., 2006; Karunanithi et al., 2018, 2020) (Fig. 5A). Measures of EJC and mEJC amplitudes from such recordings were used to derive quantal parameters to assess the release characteristics at the two bouton types (for further details, see Focal macropatch recording and Quantal analysis sections in the Materials and Methods) (Karunanithi et al., 2020). We recorded the evoked EJCs at 1 Hz nerve stimulation and the mEJCs that resulted when individual vesicles spontaneously released neurotransmitter (Fig. 5B). Despite an increase in 1s activity, 1s EJC amplitude was unchanged; and despite no alteration in 1b activity, 1b EJC amplitude decreased by 29.3% (Fig. 5C, EJC). The decrease in compound EJP amplitude therefore resulted from decreases in the amplitudes of the evoked postsynaptic currents at 1b boutons.
Figure 5.
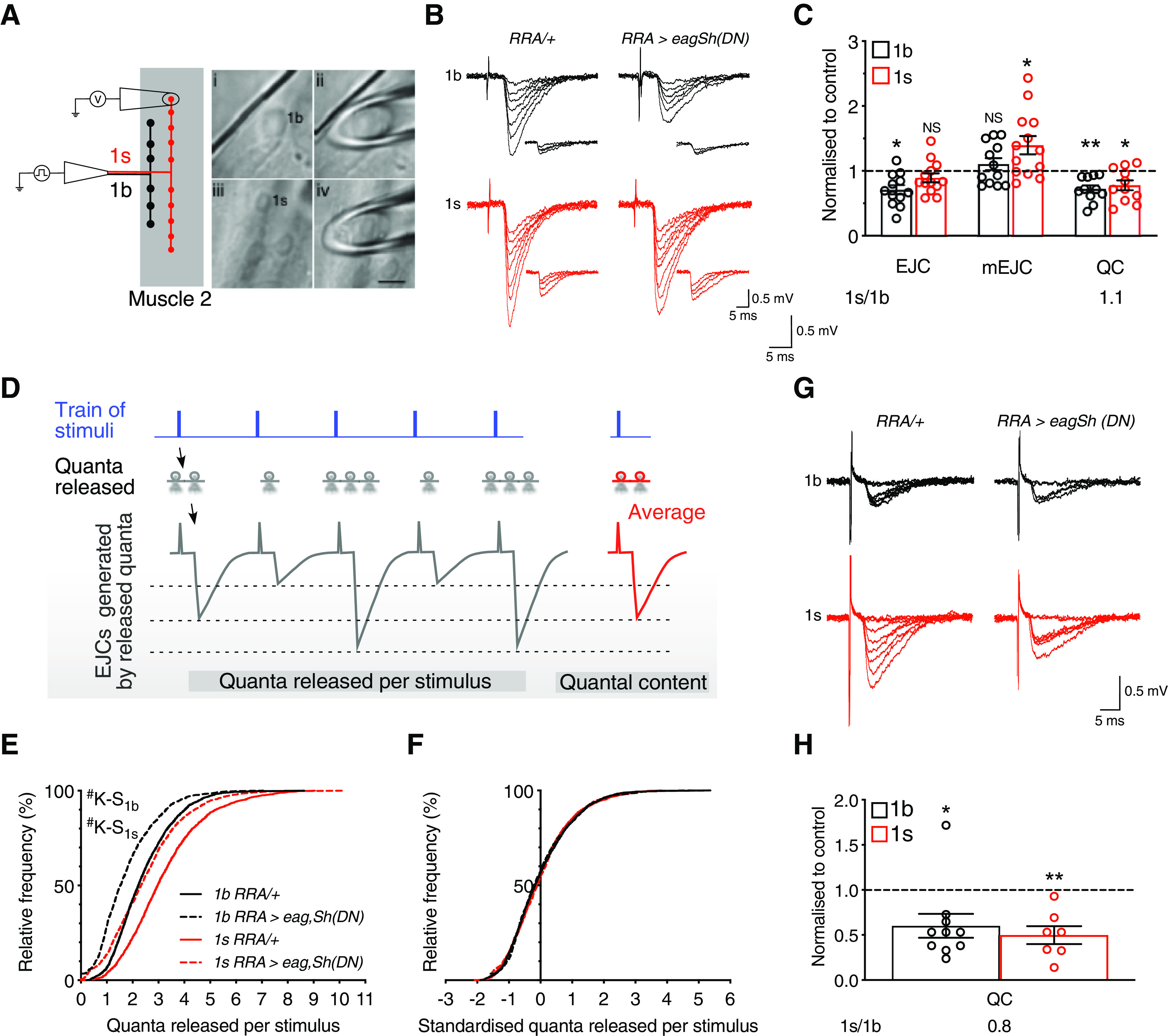
Presynaptic downscaling decreased synaptic drive. A, Left, Diagram represents the configuration for focal macropatch recordings from single boutons. Right, Electrode placement, enclosing either a 1b (Ai, Aii), or 1s (Aiii, Aiv) synaptic bouton (right). Scale bar, 5 µm. B, Exemplary traces of EJCs and mEJCs (inset, including scale bar), recorded from the indicated bouton types in the two genotypes. Experiments were performed in 1.0 mm [Ca2+]o. C, Normalized EJC and mEJC amplitudes, and QC. Those parameters were normalized to their respective averages in RRA/+ for EJC (1b: 0.57 + 0.05 mV, N = 15; 1s: 1.08 + 0.07 mV, N = 10; p < 0.0001), mEJC (1b: 0.22 + 0.01 mV, N = 15; 1s: 0.35 + 0.03 mV, N = 11; p = 0.0017), and QC (1b: 2.44 + 0.15, N = 14; 1s: 3.17 + 0.22, N = 11; p = 0.0115). Also shown is the QC 1s/1b ratio, equaling 1 in control. D, Illustration of the difference between quantal released per stimulus and QC. E, The cumulative frequency distributions of the quanta released per stimulus. F, The standardized cumulative frequency distributions of the quanta released per stimulus. G, H, QC is presented in C. QC was derived using the method of failures. All experiments performed in 0.5 mm [Ca2+]o. Exemplary traces of 1b and 1s EJCs recorded from the indicated genotypes (G). Normalized graph of QC as presented in C. Normalization was to their respective averages in RRA/+ (1b: 0.30 + 0.03, N = 9; 1s: 0.80 + 0.06, N = 7; p < 0.0001). Also shown on that normalized graphs are the QC 1s/1b ratios. *p < 0.05, **p < 0.01, #p < 0.0001. NS, Not significant, p > 0.05. Graphs represent individual data points. Error bars indicate average + SEM.
Average EJC amplitude is determined by quantal size (average amplitude of the postsynaptic response resulting from spontaneous neurotransmitter release from individual vesicles) and QC (average number of vesicles released per stimulus). The decrease in 1b EJC amplitude did not result from a decrease in quantal size (Fig. 5C, mEJC), but from a decrease in QC (by 28.1%; Fig. 5C, QC). Interestingly, 1s QC was also decreased by 22.5% (Fig. 5C, QC). However, 1s quantal size was increased by 28.4% (Fig. 5C, mEJC). The compensatory increase in 1s quantal size offset the decrease in 1s QC to homeostatically maintain 1s EJC amplitude near control level (Fig. 5C, EJC). Without that homeostatic compensation, the decrease in 1s QC would have decreased 1s EJC amplitude, further decreasing synaptic drive and potentially causing muscle firing rates to be displaced out of their physiological range (Fig. 4G,I). In summary, an increase in 1s firing rate caused both 1b and 1s QCs to be decreased. The decrease in 1b QC decreased 1b EJC amplitude, whereas the decrease in 1s QC did not decrease 1s EJC amplitude because of a compensatory increase in 1s quantal size. The decrease in synaptic drive therefore resulted from decreases in the strengths of the evoked postsynaptic currents at 1b boutons.
Presynaptic downscaling is a form of multiplicative scaling
A novel and important observation was that the 1b and 1s QCs were decreased proportionally (Fig. 5C, QC), indicating that their relative differences were maintained around a homeostatic set point (Turrigiano et al., 1998) (i.e., a similar 1s/1b ratio to control, Fig. 5C). This suggested that the 1b and 1s QCs were uniformly downscaled through multiplication by a constant (multiplicative scaling) (Turrigiano et al., 1998). We tested that idea by measuring the number of quanta released per stimulus during a train of stimuli.
During a train of nerve stimuli, the quanta released per stimulus represents the number of vesicles released by each stimulus, whereas QC represents the average number of vesicles released by a stimulus (Bademosi et al., 2018; Karunanithi et al., 2020) (Fig. 5D). For each bouton type within each genotype, we calculated the quanta released per stimulus during each train and constructed a cumulative distribution plot of this parameter by pooling the data from all experiments (Fig. 5E). The distributions in RRA>eagSH(DN) larvae for both bouton types were found to be significantly left-shifted from controls using the Kolmogorov–Smirnov nonparametric test, indicating that the decreases in QCs were because of each stimulus releasing fewer vesicles. If the left-shifted distributions arose through multiplicative scaling by a constant, the shapes of the standardized or scaled distributions should be indistinguishable from that of the control distributions. That was indeed the case for both bouton types in RRA>eagSH(DN) larvae, with no significant differences in the frequency distribution plots compared with controls (Fig. 5F; 1b: p > 0.1, 1s: p > 0.4). QC was therefore uniformly downscaled through multiplicative scaling at single boutons. We termed this process as “presynaptic downscaling.” “Presynaptic” indicated that the presynaptic side was affected. “Downscaling” indicated that QC was uniformly downscaled across 1b and 1s boutons so that their relative differences remained around a homeostatic set point (1s/1b ratio remaining close or equal to 1; Fig. 5C).
Finally, we used an alternative method, the method of failures, at low [Ca2+]o (0.5 mm), to test for presynaptic downscaling by deriving QC independently of EJC and mEJC amplitudes (see Materials and Methods). Consistently, both 1b and 1s QCs in RRA>eagSH(DN) larvae were uniformly downscaled by 39.9% and 50.2%, respectively, with the 1s/1b ratio remaining close to unity (0.8) (QC, Fig. 5G,H). In summary, activity-dependent presynaptic downscaling produced the decrease in synaptic drive.
Presynaptic downscaling is caused by a decrease in release probability
What caused presynaptic downscaling? In high [Ca2+]o, QC is influenced by n (average number of functionally active release sites) and p (average chance of a vesicle releasing neurotransmitter from an active release site following a stimulus) (Del Castillo and Katz, 1954; McLachlan, 1978). The model of quantal release from a bouton can be visualized as vesicles releasing neurotransmitter from a population of n active release sites, with each release site possessing an average probability, p (Fig. 6A). We found that presynaptic downscaling (Fig. 6B, QC) was not caused by decreases in n, but from decreases in p (Fig. 6B, p and n).
Figure 6.

Decreases in release probability caused presynaptic downscaling. A, Figure represents a presynaptic nerve terminal composed of a number of release sites (T-bars). Vesicles release neurotransmitter from a number of active release sites (*; n), and those active release sites release neurotransmitter with an average release probability p. B, Normalized QC, n, and p presented as in Figure 5C. Those parameters were normalized to their respective averages in RRA/+ for QC (1b: 2.44 + 0.15, N = 14; 1s: 3.17 + 0.22, N = 11; p = 0.0115), p (1b: 0.78 + 0.04, N = 13; 1s: 0.77 + 0.04, N = 9; p = 0.9658), and n (1b: 3.08 + 0.23, N = 12; 1s: 4.25 + 0.45, N = 10; p = 0.0383). C, Rank-ordered plot of p in RRA>eagSh(DN) larvae versus those in control. D, Rank-ordered plot of n as in C. E, Normalized QC and p1 presented as in B from experiments performed in 0.5 mm [Ca2+]o. Those parameters were normalized to their respective averages in RRA/+ for QC (1b: 0.30 + 0.03, N = 9; 1s: 0.80 + 0.06, N = 7; p < 0.0001) and p1 (1b: 0.22 + 0.01, N = 9; 1s: 0.35 + 0.01, N = 7; p < 0.0001). F, Rank-ordered plot of p1 in RRA>eagSh(DN) larvae versus those in control. G, Figure summarizes the concept of presynaptic downscaling. The increase in 1s firing rate causes the uniform downscaling of 1b and 1s QCs (downward arrows), where fewer vesicles release neurotransmitter because of decreases in release probabilities. Presynaptic downscaling causes the decrease in synaptic drive to afford robust and stabilize muscle activity. *p < 0.05, **p < 0.01. NS, Not significant, p > 0.05. Graphs represent individual data points. Error bars indicate average + SEM.
We next examined whether an increase in 1s firing rate differentially affected boutons exhibiting different p values. To do this, we rank-ordered, in ascending order, p collated from both 1b and 1s boutons in control and in RRA>eagSh(DN) larvae, and plotted them against one another (Karunanithi et al., 2020) (Fig. 6C). Points clustered close to the dashed line represent p values that were unaffected. We found that release probabilities at boutons exhibiting lower p values were decreased to a greater extent than at boutons with higher p values (Fig. 6C). Consistent with the grouped data (Fig. 6B), n was unaffected for all sites regardless of the rank of n (Fig. 6D). These results indicate that, regardless of bouton type, boutons displaying lower p values have their release probabilities decreased to a greater extent than those boutons displaying higher p values, following an increase in 1s firing rate.
At low [Ca2+]o, where evoked quantal release follows the Poisson model, the parameter p does not hold any meaning in that model (Katz, 1966; McLachlan, 1978). It was, however, shown that the parameter p1, the probability of observing a released quantum, which is the chance of observing the release of one packet of neurotransmitter following a stimulus, was reported to reflect release probability (Paradis et al., 2001) (see Materials and Methods). We used p1 to gauge for changes in release probability at low [Ca2+]o since our results at high [Ca2+]o indicated that the downscaling of QCs was because of decreases in p rather than in n (Fig. 6B). Presynaptic downscaling that was observed in low [Ca2+]o (QC in Figs. 5G,H, 6E) was found to be accompanied by a decrease in p1 (Fig. 6E, p1). As failure rates are reflective of QC and p1, we found that, in RRA>eagSh(DN) larvae, the higher percent failures corresponded with the lower values of QC and p1 for both bouton types (1b: RRA/+ = 74.1 + 1.8%, N = 9; RRA>eagSh(DN) = 83.9 + 3.0%, N = 10; p = 0.0139; 1s: RRA/+ = 45.4 + 2.8%, N = 7; RRA>eagSh(DN) = 68.3 + 5.3%, N = 7; p = 0.0037). As observed in high [Ca2+]o, boutons that displayed lower p1 values had that parameter decreased to a greater extent following an increase in 1s firing rate than in boutons displaying higher p1 values (Fig. 6F). Our findings therefore indicate that presynaptic downscaling resulted from a decrease in release probability (Fig. 6G).
A summary of the order of processes observed so far is as follows: chronic increase in 1s firing rate decreased 1b and 1s release probabilities, causing presynaptic downscaling (Fig. 6B, QC and p; Fig. 6E, QC and p1). The downscaling decreased the strengths of evoked postsynaptic currents at 1b boutons, but not at 1s boutons because of compensatory increases in 1s quantal size (Fig. 5C). The cumulative effect of presynaptic downscaling on the strengths of 1b and 1s evoked postsynaptic currents caused a decrease in synaptic drive to the muscle (Fig. 4B). That decrease caused a decrease in muscle firing rates (Fig. 3B, right), maintained muscle firing rates within physiological range (Fig. 4G), and homeostatically maintained the wide variation in firing rates around the downshifted set point (Fig. 4H). Muscle activity thereby remained robust and stable (Fig. 4G,I).
Presynaptic downscaling can be produced by an acute elevation in 1s activity
The above results demonstrate that presynaptic downscaling was induced by increasing 1s activity throughout development. Was presynaptic downscaling therefore a developmental compensation rather than a homeostatic compensation resulting from increased activity per se? To test this possibility, we acutely increased 1s activity by conditionally inducing the expression of dominant-negative forms of EAG and Sh in the 1s motoneuron using the TARGET gene expression system (McGuire et al., 2003) (Fig. 7A,B). Acutely increased 1s activity produced the following: (1) presynaptic downscaling and the maintenance of the relative QC differences around a set point (similar 1s/1b ratio as in control); and (2) decreases in p1 (QC and p1 in graph, Fig. 7B). Similar results were obtained using a different method to increase acutely 1s activity, by conditionally activating Drosophila TRPA1 (dTRPA1) cation channels (Pulver et al., 2009) (Fig. 7C,D). Presynaptic downscaling was therefore an adaptation resulting from a change in activity, rather than from a developmental compensation. That change in activity caused the decreases in release probabilities, as indicated by decreases in p1, to produce the downscaling.
Figure 7.

Acute increase in 1s activity caused presynaptic downscaling. A, B, 1s activity was acutely increased using the TARGET gene expression system. Exemplary 1b and 1s EJC traces for the indicated genotypes (A). Elevated temperature shift protocol for conditionally increasing neuronal activity (B, top). Shown are the developmental stages (E: embryo; L1-3: larval stages), the developmental stage of and the duration of temperature elevation, and the time of experimentation (B). In GFP-tagged RRAGal80ts>eagSh(DN) larvae, GFP fluorescence was only detected in motoneurons when larvae were exposed to 32°C, but not to 19°C (right), indicative of transgene activation at only the elevated temperature (B, right). Scale bar, 200 µm. Graph represents normalized QC and p1 for RRAGal80ts>eagSh(DN) larvae presented as in Figure 6E, and the QC 1s/1b ratio (B, bottom). Those parameters were normalized to their respective averages in RRAGal80ts/+ for QC (1b: 0.29 + 0.03, N = 12; 1s: 0.57 + 0.05, N = 11; p = 0.0004) and p1 (1b: 0.21 + 0.02, N = 12; 1s: 0.31 + 0.01, N = 11; p = 0.0002). The percentage of failures is inversely correlated with QC and p1 for both bouton types in RRAGal80ts>eagSh(DN) larvae (1b: RRAGal80ts/+ = 75.0 + 2.5%, N = 12; RRAGal80ts>eagSh(DN) = 89.3 + 1.7%, N = 12; p = 0.0002; 1s: RRAGal80ts/+ = 57.5 + 2.9%, N = 11; RRAGal80ts>eagSh(DN) = 79.8 + 3.0%, N = 12; p < 0.0001). Controls were subjected to the same temperature-shift protocol. C, D, Same as in A, B, following an acute increase in 1s activity in RRA>dTRPA1 larvae by using dTRPA1 channels. Those parameters were normalized to their respective averages in RRA/+ for QC (1b: 0.33 + 0.04, N = 12; 1s: 0.50 + 0.05, N = 11; p = 0.015) and p1 (1b: 0.23 + 0.02, N = 12; 1s: 0.29 + 0.02, N = 11; p = 0.0154). The percentage of failures is inversely correlated with QC and p1 for both bouton types in RRA>dTRPA1 larvae (1b: RRA/+ = 72.6 + 2.9%, N = 12; RRA>dTRPA1 = 84.2 + 1.2%, N = 12, p = 0.0023; 1s: RRA/+ = 61.6 + 2.9%, N = 11; RRA>dTRPA1 = 73.4 + 3.1%, N = 12, p = 0.0118). All experiments performed in 0.5 mm [Ca2+]o. *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.0001. Graphs represent individual data points. Error bars indicate average + SEM.
Activity-dependent increase in 1s DVGLUT expression can produce presynaptic downscaling
What presynaptic molecule(s) could induce presynaptic downscaling by responding to an increase in 1s activity? Previous work showed that, when the Drosophila vesicular glutamate transporter (DVGLUT) (Fig. 8A), which loads glutamate into synaptic vesicles, was overexpressed into both 1b and 1s motoneurons, it caused a homeostatic maintenance of the compound EJP amplitude through an increase in quantal size to compensate for the decrease in QC (Daniels et al., 2004; Gavino et al., 2015; Li et al., 2018). We observed phenotypic changes, similar to those described above, at 1s boutons following an increase in 1s activity, where the homeostatic maintenance of EJC amplitude occurred through an increase in quantal size to compensate for the decrease in QC (Fig. 5C). The similarity, therefore, of our results to previous results suggested that DVGLUT expression may have increased in the 1s motoneuron following an increase in 1s activity, resulting in presynaptic downscaling. Previous work at mammalian glutamatergic synapses supports the idea that vesicular glutamate transporter (VGLUT) expression can be altered through changes in activity (De Gois et al., 2005; Erickson et al., 2006). We therefore tested our ideas in the following ways. DVGLUT was overexpressed only into the 1s motoneuron (RRA>DVGLUT) (Fig. 8B), and then we used intracellular recordings of postsynaptic potentials from muscle 2 to test whether this manipulation produced the same phenotypic changes as those observed in RRA>eagSh(DN) larvae (Fig. 8C,D, gray traces and bar graphs). That was indeed the case, where we observed decreased compound EJP amplitude, increased mEJP amplitude, and decreased QC (Fig. 8C,D, red traces and bar graphs). By performing focal macropatch recordings of postsynaptic currents from single boutons at low [Ca2+]o (Fig. 8E), we next tested whether 1b and 1s QCs were also uniformly downscaled in RRA>DVGLUT larvae, like that observed in RRA>eagSh(DN) larvae (Fig. 6E). We observed the following at 1b and 1s boutons (RRA>DVGLUT; Fig. 8E,F): (1) uniform downscaling of QCs and maintenance of the relative 1b-1s QC difference around a set point (similar 1s/1b ratio to control); and (2) uniform decreases in p1, indicative of decreases in release probabilities (QC and p1 in Fig. 8F).
Figure 8.

Activity-dependent increase in 1s DVGLUT expression caused presynaptic downscaling. A, DVGLUT loads glutamate into synaptic vesicles. B, DVGLUT overexpression into the 1s motoneuron causes more of it to be incorporated into synaptic vesicles. C, D, Exemplary traces of EJPs and mEJPs from the indicated genotypes (C). Experiments were performed in 1.0 mm [Ca2+]o using HL3 solution. Normalized graph of EJP and mEJP amplitudes, and QC for the indicated genotypes (D). Those parameters were normalized to their respective averages in RRA/+ for EJP (22.17 + 1.51 mV, N = 10), mEJP (0.65 + 0.02 mV, N = 19), and QC (37.87 + 3.41, N = 10). E, F, Exemplary 1b and 1s EJC traces from the indicated genotypes (E). Normalized graph of QC, p1, and the QC 1s/1b ratio presented as in Figure 7B when DVGLUT was overexpressed or knocked down (F). Those parameters were normalized to their respective averages in RRA/+ for QC (1b: 0.30 + 0.03, N = 9; 1s: 0.80 + 0.06, N = 7; p < 0.0001) and p1 (1b: 0.22 + 0.01, N = 9; 1s: 0.35 + 0.01, N = 7; p < 0.0001). The percentage of failures is inversely correlated with the values of QC and p1 for both bouton types in both genotypes (1b: RRA/+ = 74.2 + 1.8%, N = 9; RRA>DVGLUT = 90.2 + 1.6%, N = 11, p < 0.0001; RRA>DVGLUT RNAi = 71.2 + 1.9%, N = 12, p = 0.2828; 1s: RRA/+ = 45.4 + 2.8%, N = 7; RRA>DVGLUT = 78.4.3 + 2.4%, N = 13, p < 0.0001; RRA>DVGLUT RNAi = 79.0 + 1.2%, N = 11, p < 0.0001). Experiments performed in 0.5 mm [Ca2+]o. G, DVGLUT RNAi expression in the 1s motoneuron significantly reduced DVGLUT mRNA expression. H, Figure represents the uniform downscaling of 1b and 1s QCs, where fewer vesicles release neurotransmitter, following overexpression of DVGLUT in only the 1s motoneuron. I, J, Shown is the hypothesis being tested, where presynaptic downscaling results from an increase in 1s DVGLUT expression following an increase in 1s motoneuron activity (I, top). Exemplary 1b and 1s EJC traces from the indicated genotypes when DVGLUT was overexpressed or was knocked down in RRA>eagSh(DN) larvae (I, bottom). Normalized graph of QC, p1, and QC 1s/1b ratio, presented as in F, when DVGLUT was overexpressed or was knocked down in RRA>eagSh(DN) larvae (J). Experiments performed in 0.5 mm [Ca2+]o. The percentage of failures is inversely correlated with the values of QC and p1 for both bouton types in both genotypes (1b: RRA/+ = 74.2 + 1.8%, N = 9; RRA>eagSh(DN), DVGLUT = 87.5 + 2.0%, N = 12, p < 0.0001; RRA>eagSh(DN), DVGLUT RNAi = 71.2 + 1.9%, N = 12, p = 0.6033; 1s: RRA/+ = 45.4 + 2.8%, N = 7; RRA> eagSh(DN),DVGLUT = 74.6 + 2.9%, N = 11, p < 0.0001; RRA>eagSh(DN), DVGLUT RNAi = 79.0 + 1.2%, N = 11, p < 0.0001), *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.0001. NS, Not significant, p > 0.05. Graphs represent individual data points. Error bars indicate mean + SEM.
If increasing 1s DVGLUT expression results in uniform downscaling, we tested whether decreasing DVGLUT expression prevented that downscaling. 1s DVGLUT expression was decreased in the 1s motoneuron using RNA interference (RRA>DVGLUT RNAi; Fig. 8G). Intracellular recordings of postsynaptic potentials from the muscle revealed that the phenotypic changes in EJP, mEJP, and QC in RRA>DVGLUT RNAi larvae were dissimilar to those seen in RRA>DVGLUT larvae (Fig. 8C,D, blue traces and bar graphs), indicating that uniform downscaling may not have resulted. This was indeed confirmed through focal macropatch recordings of postsynaptic currents from single boutons at low [Ca2+]o (QC, RRA>DVGLUT RNAi; Fig. 8E,F), where we observed the following: (1) only 1s QC was decreased, producing no uniform downscaling and no maintenance of the relative QC differences around a set point (dissimilar 1s/1b ratio to control); and (2) only 1s p1 was decreased (QC and p1 in Fig. 8F). Together, our findings show that increased DVGLUT expression in the 1s motoneuron resulted in uniform downscaling of QCs (Fig. 8H).
Increasing 1s activity (Fig. 6E) or increasing 1s DVGLUT expression (Fig. 8F) independently produced uniform downscaling. We therefore tested whether increased 1s activity produced presynaptic downscaling via an increase in 1s DVGLUT expression (schema, Fig. 8I, top). If true, overexpression of DVGLUT into the 1s motoneuron in the RRA>eagSh(DN) line (where 1s motoneuron activity was already elevated) should not cause any further changes, as when DVGLUT was overexpressed alone. We tested for that by conducting focal macropatch recordings of postsynaptic currents from single boutons at low [Ca2+]o (Fig. 8I). That was the case (compare RRA>eagSh(DN), DVGLUT in Fig. 8J with RRA>DVGLUT in Fig. 8F). We tested the same idea differently. If increased 1s DVGLUT expression resulted from increased 1s activity, knocking down DVGLUT expression in the 1s motoneuron in the RRA>eagSh(DN) line should not produce presynaptic downscaling, producing a similar result as when DVGLUT alone was knocked down. Again, this was the case (compare RRA>eagSh(DN), DVGLUT RNAi in Fig. 8J with RRA>DVGLUT RNAi in Fig. 8F). Increased 1s DVGLUT expression following increased 1s activity therefore results in presynaptic downscaling.
Discussion
Activity-dependent uniform scaling of a determinant of synaptic strength has previously been reported to occur only on the postsynaptic side within central circuits (O'Brien et al., 1998; Turrigiano et al., 1998). In this work, we show that such scaling of a determinant of synaptic strength can also occur on the presynaptic side within peripheral circuits. Presynaptic downscaling describes the uniform decreases in QCs across all inputs, maintaining their relative neurotransmitter release differences around a homeostatic set point. Presynaptic downscaling was induced by increasing the activity of only the 1s motoneuron but was manifest by decreasing release probabilities at both the manipulated 1s and the unmanipulated 1b boutons. Presynaptic downscaling decreased synaptic drive to the muscle: (1) stabilizing muscle firing rates around the downshifted set point and homeostatically maintaining the wide variation in firing rates around that set point; and (2) maintaining robust muscle activity by preventing muscle firing rates from being displaced outside of the physiological range. An activity-dependent increase in 1s DVGLUT expression resulted in presynaptic downscaling, adding support to the idea that regulation of VGLUT expression plays a role in the recruitment of presynaptic compensatory adaptations (Daniels et al., 2004; De Gois et al., 2005; Gavino et al., 2015). Figure 9 summarises our findings.
Figure 9.

Summary of how presynaptic downscaling affords robust and stable muscle activity. Left, 1b firing rate is normally higher than 1s firing rate (top). 1b and 1s neurotransmitter release (releasing vesicles), which are influenced by their release probabilities (p), regulate synaptic drive (EJP) to the muscle to control the muscle's firing rate. Muscle firing rates are maintained stably around a set point (black trace) and within physiological range (gray area), affording robust function. The relative variation in firing rates around the set point is wide (charcoal area). Right, The increase in 1s firing rate (top) produces an activity-dependent increase in 1s DVGLUT expression (red labels on 1s releasing vesicles), resulting in decreases in 1b and 1s release probabilities (p). Those decreases cause presynaptic downscaling (downward brown arrows). Presynaptic downscaling causes a decrease in synaptic drive (EJP). That decrease: (a) downshifts the mean muscle firing rate to a new set point (blue trace) without displacing firing rates outside of the physiological range (gray area); and (b) does not constrain the relative variation in firing rates around the new set point (light blue area), thereby homeostatically maintaining that variation to that observed in control. Presynaptic downscaling therefore afforded robust and stable muscle activity.
Set point variations, robustness, and stability
Our findings regarding muscle activity appear to fit within the framework of biological robustness, a concept in systems biology which accounts for variations in set points to maintain robust function against perturbations (Csete and Doyle, 2002; Kitano, 2004, 2007; Stelling et al., 2004). Within that framework, a robust system/component can operate stably at different set points (Kitano, 2004, 2007). That framework also accounts for set point shifts, as here, where excessive 1s neural drive to the muscle causes the muscle firing rate set point to downshift, without firing rates overshooting the physiological range (Fig. 4I) (Kitano, 2004; Giachello and Baines, 2015). Through such shifts, robust muscle activity is afforded to potentially generate a wide range of contractions to drive varied movements under adverse conditions.
Synaptic downscaling and presynaptic downscaling
In synaptic downscaling, which operates centrally on the postsynaptic side, the quantal amplitudes are uniformly downscaled across all inputs, decreasing synaptic drive to the postsynaptic neuron (Turrigiano et al., 1998; Turrigiano and Nelson, 2004). That decrease in synaptic drive affords stability by lowering firing rates back within the narrow set point range (Turrigiano et al., 1998; Turrigiano and Nelson, 2004). In presynaptic downscaling, QCs are uniformly downscaled across all glutamatergic inputs through decreases in release probabilities (although uniform downscaling of QCs could also occur through decreases in active release site numbers (Karunanithi et al., 2020)). The downscaling of QCs decreases synaptic drive, affording robust and stable muscle activity. Activity-dependent uniform scaling appears to therefore operate on both the presynaptic and postsynaptic sides by scaling different determinants of synaptic strength to maintain target cell firing rates within operational range. When the operational range is narrow, as in neurons, uniform downscaling stabilizes firing rates around a set point (Turrigiano et al., 1998; Turrigiano and Nelson, 2004). But when the range is wide, as in larval muscle, uniform downscaling stabilizes firing rates around a different set point without displacing firing rates outside of the physiological range, maintaining robust outputs. Activity-dependent uniform downscaling appears to therefore operate in different capacities in mammalian neurons and larval muscle.
Presynaptic downscaling induction
A difference between the induction of synaptic scaling centrally and of presynaptic downscaling peripherally is that the former can be produced cell-autonomously, but not the latter. Synaptic scaling can be produced by directly changing the activity of a postsynaptic neuron (Ibata et al., 2008). Such scaling can be swift, produced in an hour, as the signals activated in response to changes in activity directly affect the synapses formed on that postsynaptic neuron (Ibata et al., 2008). Presynaptic downscaling, on the other hand, is produced by increasing the activity of a single input neuron. Induction signals, once initiated in that input neuron, would need to travel anterograde to the target cell, and then retrograde from the target cell to the different synaptic inputs to produce uniform downscaling. Induction signals could also involve signaling between 1b and 1s inputs, possibly like those reported among mammalian glutamatergic inputs (Kullmann, 2000; Weber et al., 2016). Those signaling pathways suggest that presynaptic downscaling induction is a relatively slow (requiring more than an hour, Fig. 7) and nonautonomous process. Some candidate molecules carrying anterograde signals being vesicular glutamate and postsynaptic receptors, and retrograde signals could include Glass bottom boat (McCabe et al., 2003), Dystrophin (van der Plas et al., 2006), and Semaphorin (Davis and Muller, 2014; Orr et al., 2017). Molecules evidenced to mediate signaling among mammalian glutamatergic inputs include glutamate, calcium, protein kinase A, and neurotrophic factors (Colgan et al., 2018; Dittmer et al., 2019).
Our experiments argue for presynaptic downscaling resulting from increased 1s DVGLUT expression following increased 1s motoneuron activity (Fig. 8J). That order of process leading to presynaptic downscaling does not however imply direct causality among those steps. A number of intermediate steps may likely be involved, including retrograde signaling from the muscle (discussed above) to calibrate neurotransmitter release in order to prevent muscle activity from overshooting its physiological range. These possibilities need to be investigated in future work.
Preservation of synaptic diversity
Uniform scaling is proposed to maintain the relative differences among synapses around a set point to prevent disruption of mechanisms which rely on such differences being preserved (Turrigiano et al., 1998; Turrigiano, 2008). For example, in postsynaptic neurons within brain circuits, synaptic downscaling would enable synaptic inputs which underwent Hebbian potentiation to retain their stronger synaptic weighting relative to other inputs, while concurrently preventing such strengthening from causing runaway excitation (Turrigiano et al., 1998; Turrigiano, 2008). In the periphery, recent work at the larval NMJ shows that 1b synapses primarily drive larval muscle contractions during crawling, whereas 1s synapses drive intersegmental coordination of muscle contractions (Newman et al., 2017). Neurotransmitter release differences between 1b and 1s synapses underlie the differential execution of these two functions (Ashley and Budnik, 2017; Newman et al., 2017). Our findings therefore suggest that uniform downscaling could preserve the relative neurotransmitter release differences to enable 1b and 1s synapses to continue driving these two functions against perturbations. Uniform downscaling on the presynaptic or postsynaptic sides could therefore afford stability within circuits by maintaining the relative differences among synaptic inputs around a homeostatic set point.
PHD and presynaptic downscaling
Larval NMJs display forms of compensatory adaptations, which preserve strong muscular excitation in the face of perturbations. In those forms, EJP amplitude is homeostatically maintained through compensatory changes in both QC and quantal size (Davis, 2013; Davis and Muller, 2014). PHD is one such adaptation (Daniels et al., 2004; Gavino et al., 2015), where the increases in quantal sizes are offset by compensatory decreases in QCs at both 1b and 1s boutons (Li et al., 2018). PHD is induced by directly overexpressing DVGLUT into both 1b and 1s motoneurons (Daniels et al., 2004; Gavino et al., 2015), and as such, has not been shown to be induced by altered activity. By contrast, presynaptic downscaling causes a compensatory decrease in EJP amplitude (Fig. 4B) to afford robust and stable muscle activity. It results following an activity-dependent increase in only 1s DVGLUT expression, causing the uniform downscaling of both 1b and 1s QCs (Fig. 8J). PHD is therefore a compensatory adaptation that preserves muscular excitation by homeostatically maintaining EJP amplitude, whereas presynaptic downscaling is a compensatory adaptation which stabilizes muscle activity by decreasing EJP amplitude. The two different adaptations are produced by two different patterns of DVGLUT expression, but both involve uniform decreases in QCs across inputs. However, in the case of presynaptic downscaling, the uniform decreases are activity-dependent. In this preparation, compensatory adaptations therefore not only regulate muscle excitation, but also muscle activity.
Implications for central processes
Uniform downscaling has been implicated in processes related to sleep and memory. Work in mammalian brain circuits proposes that uniform downscaling of quantal amplitudes on the postsynaptic side renormalizes synapses during sleep following Hebbian potentiation throughout the day, and prevents runaway excitation following Hebbian potentiation during memory formation (Turrigiano et al., 1998; de Vivo et al., 2017; Diering et al., 2017; Turrigiano, 2017). Our finding suggests that the scope for evaluating such a process should be broadened to include any uniform downscaling on the presynaptic side, and questions whether such downscaling would involve changes in vesicular neurotransmitter transporter expression.
In future work, the molecular mechanisms underlying presynaptic downscaling will need to be elucidated, including identification of retrograde and anterograde signals, and the role of DVGLUT in such signaling. Assessments are also needed of how regulatory molecules, and changes in the activity profiles of the 1b motoneuron and both 1b and 1s motoneurons, contribute to robust and stable circuit function. Finally, detailed analysis of the frequency components within muscle bursts could provide better understanding of the mechanisms shaping muscle activity.
Footnotes
The authors declare no competing financial interests.
This work was supported by Australian Research Council Grants LE130100078 to S.K. and DP1093968 to B.v.S.; National Institutes of Health RO3 2951620 to S.K.; National Health and Medical Research Council APP568680 to P.G.N. and N.A.L., APP1103923 to B.v.S., APP1026310, APP1029672, APP1028887, APP1046090, APP1042416, APP1086851, and CDF1111940 to G.G.N.; Motor Neurone Disease Research Institute of Australia to P.G.N.; Griffith University to S.K.; and Australian Postgraduate Award to G.L.O. We thank Dr. Mani Ramaswami, Dr. Victor Anggono, Dr. Gregory T. Macleod, and Annette Vella for feedback on the manuscript; Dr. Kai Feng for help with confocal microscopy; Dr. Chelsie Rohrscheib for conducting the RT-PCR experiments; Dr. William D. Phillips for providing access to laboratory facilities; and Griffith University for loan of the electrophysiology equipment.
References
- Aponte-Santiago NA, Ormerod KG, Akbergenova Y, Littleton JT (2020) Synaptic plasticity induced by differential manipulation of tonic and phasic motoneurons in Drosophila. J Neurosci 40:6270–6288. 10.1523/JNEUROSCI.0925-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley J, Budnik V (2017) A tale of two inputs. Neuron 93:1245–1247. 10.1016/j.neuron.2017.03.013 [DOI] [PubMed] [Google Scholar]
- Atwood HL, Karunanithi S (2002) Diversification of synaptic strength: presynaptic elements. Nat Rev Neurosci 3:497–516. 10.1038/nrn876 [DOI] [PubMed] [Google Scholar]
- Bademosi AT, Steeves J, Karunanithi S, Zalucki OH, Gormal RS, Liu S, Lauwers E, Verstreken P, Anggono V, Meunier FA, van Swinderen B (2018) Trapping of syntaxin1a in presynaptic nanoclusters by a clinically relevant general anesthetic. Cell Rep 22:427–440. 10.1016/j.celrep.2017.12.054 [DOI] [PubMed] [Google Scholar]
- Barclay JW, Atwood HL, Robertson RM (2002) Impairment of central pattern generation in Drosophila cysteine string protein mutants. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 188:71–78. 10.1007/s00359-002-0279-9 [DOI] [PubMed] [Google Scholar]
- Bennett MR, Florin T (1974) A statistical analysis of the release of acetylcholine at newly formed synapses in striated muscle. J Physiol 238:93–107. 10.1113/jphysiol.1974.sp010512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Karunanithi S, Lavidis NA (1991) Probabilistic secretion of quanta from nerve terminals in toad (Bufo marinus) muscle modulated by adenosine. J Physiol 433:421–434. 10.1113/jphysiol.1991.sp018435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu CF (1990) Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci 10:3754–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Zhang J, Zinsmaier KE, Macleod GT (2010) Presynaptic mitochondria in functionally different motor neurons exhibit similar affinities for Ca2+ but exert little influence as Ca2+ buffers at nerve firing rates in situ. J Neurosci 30:1869–1881. 10.1523/JNEUROSCI.4701-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MQ, Zarin AA, Carreira-Rosario A, Doe CQ (2018) Neural circuits driving larval locomotion in Drosophila. Neural Dev 13:6. 10.1186/s13064-018-0103-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan LA, Hu M, Misler JA, Parra-Bueno P, Moran CM, Leitges M, Yasuda R (2018) PKCalpha integrates spatiotemporally distinct Ca(2+) and autocrine BDNF signaling to facilitate synaptic plasticity. Nat Neurosci 21:1027–1037. 10.1038/s41593-018-0184-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csete ME, Doyle JC (2002) Reverse engineering of biological complexity. Science 295:1664–1669. 10.1126/science.1069981 [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A (2004) Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci 24:10466–10474. 10.1523/JNEUROSCI.3001-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. (2013) Homeostatic signaling and the stabilization of neural function. Neuron 80:718–728. 10.1016/j.neuron.2013.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Goodman CS (1998) Synapse-specific control of synaptic efficacy at the terminals of a single neuron. Nature 392:82–86. 10.1038/32176 [DOI] [PubMed] [Google Scholar]
- Davis GW, Muller M (2014) Homeostatic control of presynaptic neurotransmitter release. Annu Rev Physiol 77:251–270. [DOI] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS (1996) Genetic dissection of structural and functional components of synaptic plasticity: III. CREB is necessary for presynaptic functional plasticity. Neuron 17:669–679. 10.1016/s0896-6273(00)80199-3 [DOI] [PubMed] [Google Scholar]
- Dawson-Scully K, Lin Y, Imad M, Zhang J, Marin L, Horne JA, Meinertzhagen IA, Karunanithi S, Zinsmaier KE, Atwood HL (2007) Morphological and functional effects of altered cysteine string protein at the Drosophila larval neuromuscular junction. Synapse 61:1–16. 10.1002/syn.20335 [DOI] [PubMed] [Google Scholar]
- De Gois S, Schafer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD (2005) Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci 25:7121–7133. 10.1523/JNEUROSCI.5221-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vivo L, Bellesi M, Marshall W, Bushong EA, Ellisman MH, Tononi G, Cirelli C (2017) Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355:507–510. 10.1126/science.aah5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B (1954) Quantal components of the end-plate potential. J Physiol 124:560–573. 10.1113/jphysiol.1954.sp005129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B (1956) Localization of active spots within the neuromuscular junction of the frog. J Physiol 132:630–649. 10.1113/jphysiol.1956.sp005554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, Huganir RL (2017) Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 355:511–515. 10.1126/science.aai8355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer PJ, Dell'Acqua ML, Sather WA (2019) Synaptic crosstalk conferred by a zone of differentially regulated Ca(2+) signaling in the dendritic shaft adjoining a potentiated spine. Proc Natl Acad Sci USA 116:13611–13620. 10.1073/pnas.1902461116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JD, De Gois S, Varoqui H, Schafer MK, Weihe E (2006) Activity-dependent regulation of vesicular glutamate and GABA transporters: a means to scale quantal size. Neurochem Int 48:643–649. 10.1016/j.neuint.2005.12.029 [DOI] [PubMed] [Google Scholar]
- Ferguson L, Petty A, Rohrscheib C, Troup M, Kirszenblat L, Eyles DW, van Swinderen B (2017) Transient dysregulation of dopamine signaling in a developing Drosophila arousal circuit permanently impairs behavioral responsiveness in adults. Front Psychiatry 8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME (2008) Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci 31:563–590. 10.1146/annurev.neuro.31.060407.125631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox LE, Soll DR, Wu CF (2006) Coordination and modulation of locomotion pattern generators in Drosophila larvae: effects of altered biogenic amine levels by the tyramine beta hydroxlyase mutation. J Neurosci 26:1486–1498. 10.1523/JNEUROSCI.4749-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW (2006) Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52:663–677. 10.1016/j.neuron.2006.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Borges S, Wilson M (1995) Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron 15:885–895. 10.1016/0896-6273(95)90179-5 [DOI] [PubMed] [Google Scholar]
- Fujioka M, Lear BC, Landgraf M, Yusibova GL, Zhou J, Riley KM, Patel NH, Jaynes JB (2003) Even-skipped, acting as a repressor, regulates axonal projections in Drosophila. Development 130:5385–5400. 10.1242/dev.00770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavino MA, Ford KJ, Archila S, Davis GW (2015) Homeostatic synaptic depression is achieved through a regulated decrease in presynaptic calcium channel abundance. Elife 4:e05473 10.7554/eLife.05473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachello CN, Baines RA (2015) Inappropriate neural activity during a sensitive period in embryogenesis results in persistent seizure-like behavior. Curr Biol 25:2964–2968. 10.1016/j.cub.2015.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KP, Littleton JT (2015) Transmission, development, and plasticity of synapses. Genetics 201:345–375. 10.1534/genetics.115.176529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann M, Dudel J (1998) Evoked quantal currents at neuromuscular junctions of wild type Drosophila larvae. Neurosci Lett 256:77–80. 10.1016/s0304-3940(98)00756-3 [DOI] [PubMed] [Google Scholar]
- Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG (2013) Firing rate homeostasis in visual cortex of freely behaving rodents. Neuron 80:335–342. 10.1016/j.neuron.2013.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengen KB, Torrado Pacheco A, McGregor JN, Van Hooser SD, Turrigiano GG (2016) Neuronal firing rate homeostasis is inhibited by sleep and promoted by wake. Cell 165:180–191. 10.1016/j.cell.2016.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang B, Chiba A (2001) Single-cell analysis of Drosophila larval neuromuscular synapses. Dev Biol 229:55–70. 10.1006/dbio.2000.9983 [DOI] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG (2008) Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57:819–826. 10.1016/j.neuron.2008.02.031 [DOI] [PubMed] [Google Scholar]
- Karunanithi S, Troup M, van Swinderen B (2018) Using Drosophila to understand general anesthesia: from synapses to behavior. Meth Enzymol 602:153–176. 10.1016/bs.mie.2018.02.003 [DOI] [PubMed] [Google Scholar]
- Karunanithi S, Marin L, Wong K, Atwood HL (2002) Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J Neurosci 22:10267–10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunanithi S, Cylinder D, Ertekin D, Zalucki OH, Marin L, Lavidis NA, Atwood HL, van Swinderen B (2020) Proportional downscaling of glutamatergic release sites by the general anesthetic propofol at Drosophila motor nerve terminals. eNeuro 7:ENEURO.0422-19.2020 10.1523/ENEURO.0422-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. (1966) Nerve, muscle and synapse. New York: McGraw-Hill. [Google Scholar]
- Kitano H. (2004) Biological robustness. Nat Rev Genet 5:826–837. 10.1038/nrg1471 [DOI] [PubMed] [Google Scholar]
- Kitano H. (2007) Towards a theory of biological robustness. Mol Syst Biol 3:137. 10.1038/msb4100179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ (2006) Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 312:1051–1054. 10.1126/science.1126308 [DOI] [PubMed] [Google Scholar]
- Klose MK, Chu D, Xiao C, Seroude L, Robertson RM (2005) Heat shock-mediated thermoprotection of larval locomotion compromised by ubiquitous overexpression of Hsp70 in Drosophila melanogaster. J Neurophysiol 94:3563–3572. 10.1152/jn.00723.2005 [DOI] [PubMed] [Google Scholar]
- Kullmann DM. (2000) Spillover and synaptic cross talk mediated by glutamate and GABA in the mammalian brain. Prog Brain Res 125:339–351. 10.1016/S0079-6123(00)25023-1 [DOI] [PubMed] [Google Scholar]
- Kurdyak P, Atwood HL, Stewart BA, Wu CF (1994) Differential physiology and morphology of motor axons to ventral longitudinal muscles in larval Drosophila. J Comp Neurol 350:463–472. 10.1002/cne.903500310 [DOI] [PubMed] [Google Scholar]
- Li X, Goel P, Wondolowski J, Paluch J, Dickman D (2018) A glutamate homeostat controls the presynaptic inhibition of neurotransmitter release. Cell Rep 23:1716–1727. 10.1016/j.celrep.2018.03.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lnenicka GA, Keshishian H (2000) Identified motor terminals in Drosophila larvae show distinct differences in morphology and physiology. J Neurobiol 43:186–197. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Chen L, Karunanithi S, Peloquin JB, Atwood HL, McRory JE, Zamponi GW, Charlton MP (2006) The Drosophila cacts2 mutation reduces presynaptic Ca2+ entry and defines an important element in Cav2.1 channel inactivation. Eur J Neurosci 23:3230–3244. 10.1111/j.1460-9568.2006.04873.x [DOI] [PubMed] [Google Scholar]
- Martin KC, Kandel ER (1996) Cell adhesion molecules, CREB, and the formation of new synaptic connections. Neuron 17:567–570. 10.1016/s0896-6273(00)80188-9 [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB (2003) The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39:241–254. 10.1016/S0896-6273(03)00426-4 [DOI] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL (2003) Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302:1765–1768. 10.1126/science.1089035 [DOI] [PubMed] [Google Scholar]
- McLachlan EM. (1978) The statistics of transmitter release at chemical synapses. Int Rev Physiol 17:49–117. [PubMed] [Google Scholar]
- Mosca TJ, Carrillo RA, White BH, Keshishian H (2005) Dissection of synaptic excitability phenotypes by using a dominant-negative Shaker K+ channel subunit. Proc Natl Acad Sci USA 102:3477–3482. 10.1073/pnas.0406164102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman ZL, Hoagland A, Aghi K, Worden K, Levy SL, Son JH, Lee LP, Isacoff EY (2017) Input-specific plasticity and homeostasis at the Drosophila larval neuromuscular junction. Neuron 93:1388–1404.e1310. 10.1016/j.neuron.2017.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M (1997) Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron 19:697–709. 10.1016/S0896-6273(00)80382-7 [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21:1067–1078. 10.1016/S0896-6273(00)80624-8 [DOI] [PubMed] [Google Scholar]
- Ormerod KG, Krans JL, Mercier AJ (2015) Cell-selective modulation of the Drosophila neuromuscular system by a neuropeptide. J Neurophysiol 113:1631–1643. 10.1152/jn.00625.2014 [DOI] [PubMed] [Google Scholar]
- Ormerod KG, LePine OK, Bhutta MS, Jung J, Tattersall GJ, Mercier AJ (2016) Characterizing the physiological and behavioral roles of proctolin in Drosophila melanogaster. J Neurophysiol 115:568–580. 10.1152/jn.00606.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr BO, Fetter RD, Davis GW (2017) Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 550:109–113. 10.1038/nature24017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouanounou G, Baux G, Bal T (2016) A novel synaptic plasticity rule explains homeostasis of neuromuscular transmission. Elife 5:e12190 10.7554/eLife.12190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW (2001) Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30:737–749. 10.1016/s0896-6273(01)00326-9 [DOI] [PubMed] [Google Scholar]
- Paterson BA, Anikin IM, Krans JL (2010) Hysteresis in the production of force by larval Dipteran muscle. J Exp Biol 213:2483–2493. 10.1242/jeb.043026 [DOI] [PubMed] [Google Scholar]
- Pawlu C, DiAntonio A, Heckmann M (2004) Postfusional control of quantal current shape. Neuron 42:607–618. 10.1016/s0896-6273(04)00269-7 [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19:1237–1248. 10.1016/S0896-6273(00)80415-8 [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulver SR, Pashkovski SL, Hornstein NJ, Garrity PA, Griffith LC (2009) Temporal dynamics of neuronal activation by Channelrhodopsin-2 and TRPA1 determine behavioral output in Drosophila larvae. J Neurophysiol 101:3075–3088. 10.1152/jn.00071.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrscheib CE, Bondy E, Josh P, Riegler M, Eyles D, van Swinderen B, Weible MW 2nd, Brownlie JC (2015) Wolbachia influences the production of octopamine and affects Drosophila male aggression. Appl Environ Microbiol 81:4573–4580. 10.1128/AEM.00573-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS (1996) Genetic dissection of structural and functional components of synaptic plasticity: II. Fasciclin II controls presynaptic structural plasticity. Neuron 17:655–667. 10.1016/s0896-6273(00)80198-1 [DOI] [PubMed] [Google Scholar]
- Smith R, Taylor JP (2011) Dissection and imaging of active zones in the Drosophila neuromuscular junction. J Vis Exp 50:2676 10.3791/2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Lance K, Levine RB (2012) Contribution of EAG to excitability and potassium currents in Drosophila larval motoneurons. J Neurophysiol 107:2660–2671. 10.1152/jn.00201.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelling J, Sauer U, Szallasi Z, Doyle FJ 3rd, Doyle J (2004) Robustness of cellular functions. Cell 118:675–685. 10.1016/j.cell.2004.09.008 [DOI] [PubMed] [Google Scholar]
- Stewart BA, Schuster CM, Goodman CS, Atwood HL (1996) Homeostasis of synaptic transmission in Drosophila with genetically altered nerve terminal morphology. J Neurosci 16:3877–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suster ML, Karunanithi S, Atwood HL, Sokolowski MB (2004) Turning behavior in Drosophila larvae: a role for the small scribbler transcript. Genes Brain Behav 3:273–286. 10.1111/j.1601-183X.2004.00082.x [DOI] [PubMed] [Google Scholar]
- Swann JW, Rho JM (2014) How is homeostatic plasticity important in epilepsy? Adv Exp Med Biol 813:123–131. [DOI] [PubMed] [Google Scholar]
- Thomas U, Sigrist SJ (2012) Glutamate receptors in synaptic assembly and plasticity: case studies on fly NMJs. Adv Exp Med Biol 970:3–28. 10.1007/978-3-7091-0932-8_1 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. (1999) Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci 22:221–227. 10.1016/s0166-2236(98)01341-1 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422–435. 10.1016/j.cell.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2011) Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 34:89–103. 10.1146/annurev-neuro-060909-153238 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. (2017) The dialectic of Hebb and homeostasis. Philos Trans R Soc Lond B Biol Sci 372:20160258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2000) Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol 10:358–364. 10.1016/s0959-4388(00)00091-x [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5:97–107. 10.1038/nrn1327 [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391:892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- van der Plas MC, Pilgram GS, Plomp JJ, de Jong A, Fradkin LG, Noordermeer JN (2006) Dystrophin is required for appropriate retrograde control of neurotransmitter release at the Drosophila neuromuscular junction. J Neurosci 26:333–344. 10.1523/JNEUROSCI.4069-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitureira N, Letellier M, Goda Y (2012) Homeostatic synaptic plasticity: from single synapses to neural circuits. Curr Opin Neurobiol 22:516–521. 10.1016/j.conb.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Martin S, Nguyen TH, Harper CB, Gormal RS, Martinez-Marmol R, Karunanithi S, Coulson EJ, Glass NR, Cooper-White JJ, van Swinderen B, Meunier FA (2016) Flux of signalling endosomes undergoing axonal retrograde transport is encoded by presynaptic activity and TrkB. Nat Commun 7:12976. 10.1038/ncomms12976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JP, Andrasfalvy BK, Polito M, Mago A, Ujfalussy BB, Makara JK (2016) Location-dependent synaptic plasticity rules by dendritic spine cooperativity. Nat Commun 7:11380. 10.1038/ncomms11380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt JL, Petrus E, Lee HK (2014) Experience-dependent homeostatic synaptic plasticity in neocortex. Neuropharmacology 78:45–54. 10.1016/j.neuropharm.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Budnik V, Wu CF (1992) Synaptic plasticity in Drosophila memory and hyperexcitable mutants: role of cAMP cascade. J Neurosci 12:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]