

Graphical abstract

Keywords: COVID-19, Vaccine, Immuno-informatics, Epitope, Molecular dynamics simulation, Zidovudine
Highlights
-
•
404SKQLQQSMSSADS416 and 92RRIRGGDGKMKDL104 sequences of N-protein are predicted to induce B-cell memory response .
-
•
79SSPDDQIGY87 and 305AQFAPSASAFFGMSR319 are predicted to elicit T-cell-mediated immune response with accompanied memory.
-
•
305AQFAPSASAFFGMSR319 is predicted to cross-react with SARS-CoV1, MERS-CoV and hCoV−OC43.
-
•
Zidovudine triphosphate is a potential inhibitor of the N-terminal domain of SARS-CoV2 N-protein.
Abstract
The occurrence of the SARS-CoV2 infection has become a worldwide threat and the urgent need to discover therapeutic interventions remains paramount. The primary roles of the coronavirus nucleocapsid (N) protein are to interact with the viral genome and pack them into ribonucleoprotein complex. It also plays critical roles at many stages of the viral life cycle. Herein, we explore the N protein of SARS-CoV2 to identify promising epitope-based vaccine candidates and target the N-terminal domain of SARS-CoV2 N-protein for potential inhibitors using an integrative bioinformatics approach. We identified B-cell epitopes and T-cell epitopes that are non-toxic, non-allergenic, capable of inducing IFN-γ and structurally stable with high global population coverage of response. The 404SKQLQQSMSSADS416 and 92RRIRGGDGKMKDL104 sequences of N-protein were identified to induce B-cell immunity. We also identified 79SSPDDQIGY87 and 305AQFAPSASAFFGMSR319 as potential T-cell epitopes that form stable structures with human leucocyte antigens. We have also identified zidovudine triphosphate, an anti-HIV agent, as a potential inhibitor of the N-terminal domain of SARS-CoV2 N-protein based on docking and simulation analysis and should be considered for experimental validations. The findings of this study can help fast-track the discovery of therapeutic options to combat COVID-19.
1. Introduction
The global incidence of the coronavirus disease 2019 (COVID-19) has an estimated 20 million cases with over 740, 000 deaths as per current data available on 12th August 2020. The disease is caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV2), the newest member of the family Coronaviridae. SARS-CoV2, SARS-CoV1 and Middle East Severe Acute Respiratory Syndrome Coronavirus (MERS-CoV) belong to the genus Betacoronavirus, thus, share particular genomic and proteomic similarities. It has been reported that SARS-CoV2 genome shares approximately 80 % and 50 % similarity with SARS-CoV1 and MERS-CoV, respectively [1,2]. The structural proteins and enzymes of SARS-CoV2 are 90 % homologous to those of SARS-CoV1 and MERS-CoV [3]. The clinical symptoms at the early stages of the disease include fever, fatigue and dry cough [4,5]. Progression of the disease to a severe case is manifested as pneumonia and is usually accompanied by dyspnea and dry cough [6]. There are several complications associated with the SARS disease which include acute respiratory distress syndrome (ARDS), organ dysfunction, cardiac injury and systemic inflammation [4,6]. The structural proteins of SARS-CoV2 namely; spike (S) glycoprotein, envelope (E), membrane (M) and nucleocapsid (N) proteins, play crucial roles during entry and survival of the virus in the host cell. The primary roles of the coronavirus N protein are to interacts with the viral genome and pack them into ribonucleoprotein (RNP) structure [[7], [8], [9]]. Notwithstanding, the coronavirus N protein is multifunctional, having critical roles at many stages of the viral life cycle [10]. Notable among them are viral budding [10], viral assembly [11], regulation of host cell cycle [12] and regulation of viral mRNA replication [13,14]. Considering the roles of the N protein and its high expression during viral infections, the N protein can be a promising target for drug and vaccine development. Targeting the N-protein provides opportunities to interfere with coronavirus assembly, transcription and replication. With the progression of the COVID-19 pandemic, search for pharmaceutical options are mainly driven by drug repurposing to reduce the drug discovery timeline [15].
Considering the novelty and timing of COVID-19, research groups are beginning to understand aspects of the immunological response to the SARS-CoV2 to aid in the discovery of vaccines. The quest for safe and effective vaccines for COVID-19 has resulted in potential candidates with varying strategies and stages of developments. According to reports by the World Health Organization (WHO) on 10th August 2020, 28 vaccine candidates have been registered and are currently in clinical evaluations and 139 candidates in pre-clinical evaluations [16]. Many of these candidates are sub-unit vaccines, as they provide a more logistically workable approach [17] and are capable of inducing both cellular and humoral responses [18]. Majority of the candidates in clinical evaluations target the S protein, considering its immuno-dominant nature. However, there are reports that depict the N-protein as highly immunogenic with antigenic regions that are highly conserved across the coronaviruses [[19], [20], [21], [22], [23], [24]]. The conventional approaches for epitope-based vaccine identification are resource-demanding, thus, designing epitope-based vaccines using bioinformatics offer a safer, inexpensive and more effective approach. In this paper, we integrate immuno-informatics, structure-based drug discovery and other computational methods to identify potential epitope-based vaccine and drug candidates that target the N-protein of SARS-CoV2. The results reveal potential N-protein epitopes that are highly immunogenic and elicit B-cell immunity, T-cell immunity, virus-specific antibody neutralization and natural killer cell-mediated immunity. The anti-HIV agent, zidovudine triphosphate was identified to potentially inhibit the N-terminal domain of SARS-CoV2 N-protein based on binding energy and simulation analysis.
2. Methods
2.1. Immuno-informatics approach
2.1.1. Protein structure preparations
The SARS-CoV2 nucleocapsid protein (N-protein) sequence was fetched from the NCBI protein database (https://www.ncbi.nlm.nih.gov/protein/QIC53221.1) and human leucocyte antigen (HLA) protein sequences were retrieved from the UniProt database (https://www.uniprot.org/). We used the SWISS-MODEL server (https://www.swissmodel.expasy.org/) to generate structures of the SARS-CoV2 N-protein and the HLA proteins based on the co-ordinates of their respective homologous protein structures available at the protein data bank (https://www.rcsb.org/).
2.1.2. Epitope prediction and assessment
2.1.2.1. B-cell epitopes
The linear and discontinuous B-cell epitopes were predicted using Bepipred 2.0 (http://www.cbs.dtu.dk/services/BepiPred/index.php) and ElliPro (http://tools.iedb.org/ellipro/), respectively. We used the interface provided by the IEBD web server (http://www.iedb.org/) for the predictions. Bepipred 2.0 uses a Random Forest regression algorithm to predict the probability that a residue of the antigenic protein is a member of an epitope [25]. The algorithm considers several properties of the antigenic residues including surface accessibility, hydrophobicity, secondary structure and polarity [25]. ElliPro combines three algorithms to predict discontinuous epitopes from a protein structure [26]. The first algorithm reconfigures the shape and surface of the query protein structure into an ellipsoid. Based on the ellipsoid shape, another algorithm estimates the protrusion index (PI) of each residue such that when a particular residue is seen outside the ellipsoid shape after 70 % of the protein atoms are captured within the ellipsoid shape, then the PI of that residue is 0.7. The last algorithm then determines the discontinuous epitopes by clustering neighboring residues based on a PI cut-off.
2.1.2.2. T-cell epitopes
All T-cell epitopes were predicted using the IEBD web server (http://www.iedb.org/). The T-cell epitope predictions were based on the peptides affinity to major histocompatibility complexes (MHC) or HLA. Both class I MHC-binding peptides and class II MHC-binding peptides were predicted. For the class I MHC-binding peptides, a reference set consisting of several alleles each of HLA-A, HLA-B and HLA-C was used. Similarly, a reference set of MHC class II alleles was used for the prediction of class II MHC-binding peptides. The IEBD recommended prediction algorithms were employed in both cases.
2.1.2.3. Epitope assessment
The predicted epitopes were assessed based on toxicity, allergenicity and ability to induce IFN-γ. The toxicity of the peptides was predicted using a support vector machine-based algorithm developed by Gupta et al. [27]. We used AllerTop [28] to predict the antigenicity of each peptide. The ability of the epitopes to induce IFN-γ was predicted by IFNepitope [29] using the hybrid approach (motif and support vector machine).
2.1.2.4. Epitope modeling and stability studies
Structural configurations of the selected epitopes were modeled using PEP-FOLD 3 [30]. The peptide models were then simulated using GROMACS 2020.3, following the protocol described in section 2.1.7. A 20 ns run was performed for each epitope and their root mean square deviation was computed using the GROMACS analysis tool.
2.1.3. Population coverage analysis
The population coverage analysis tool available at IEBD (http://tools.iedb.org/population) was used to predict the percentage of individuals that would respond to the epitope(s) based on the HLA allele genotypic frequency within their ethnicities/areas. The frequency of HLA genotype within a population is provided by the Allele frequency database (http://www.allelefrequencies.net/).
2.1.4. Epitope conservation analysis
The occurrence of the epitope sequence in other related coronaviruses was analyzed using Clustal omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). The N-protein of SARS-CoV1, MERS-CoV, Human coronaviruses (hCoV) OC43 and NL63 were considered for the homology analysis, noting the conservation of the epitope sequences across the related coronaviruses.
2.1.5. Molecular docking of epitopes with HLA proteins
The best binding conformation of the epitope-HLA complex was determined with Cluspro 2.0 server [31] using the antibody model [32]. The Cluspro algorithm performs three steps in the protein-protein binding prediction [31]. The algorithm performs a rigid-body docking on a very large set of conformations using a Fast Fourier Transform (FFT)-dependent algorithm called PIPER [33,34]. The docked conformations are the clustered based on root mean square deviation to select representative conformations of lowest energy [31]. The last computational step is energy minimization of the top conformations from step 2 using CHARMM force field [35]. The server is available at https://cluspro.bu.edu/home.php.
2.1.6. Molecular dynamics simulation of complexes
The proteins were subjected to molecular dynamics simulations using GROMACS 2020.3 software suite [36] and CHARMM 2019 force filed. The structures were centered in a dodecahedron box, at least 1 nm from the edges and solvated with spc216 water model. The system was electrostatically neutralized with 0.154 M NaCl. In order to get rid of inappropriate geometry and steric hindrances, energy minimization was performed on the system using the steepest descent algorithm for 10,000 steps with a maximum force threshold of 100 kJ/mol/nm. Van der-Waals interactions were treated with a single cut-off of 1.216 nm and long-range electrostatics were treated with the Particle-Mesh Ewald (PME) method with 0.16 FFT grid spacing and 4th order B-spline interpolation. Neighbor search was performed every 20 steps using the grid method with Verlet cut-off scheme. Protein and non-protein components of the system were independently coupled to v-rescale thermostat and an isotropic Berendsen algorithm for pressure coupling. The pressure was maintained by weak coupling to a reference pressure of 1 bar, with an isothermal compressibility of 4.6 × 10−5 bar−1. The bond lengths within the protein were constrained using the LINCS algorithm. The system was subjected to a 400 ps NVT and NPT equilibration, followed by 200 ns simulation using the leap-frog integrator. The trajectories were analyzed with built-in GROMACS commands and all graphic data were generated with the GRACE tool (http://plasma-gate.weizmann.ac.il/Grace).
2.1.7. Immune simulation
The immune response that would be elicited by the predicted epitopes was simulated using the C-ImmSim server (http://150.146.2.1/C-IMMSIM/index.php). C-ImmSim is based on the models developed by Celada-Seiden [[37], [38], [39]] and operates as an agent-based model that uses machine learning techniques to simulate the immune response towards a particular pathogen. The players of the immune system are represented as agents and the interactions between the agents are guided by set of rules that emanate from present understanding of immunological principles. These principles that are incorporated into the C-ImmSim algorithm include MHC restriction [40], development of memory [41,42], clonal deletion theory [43], processing and presentation of antigens [[44], [45], [46]] among others [47]. The algorithm does well to mimic the immune system as it simultaneously simulates immunological processes in the bone marrow (production of lymphoid and myeloid cells), the thymus (T-cell education), and the lymphatic system (immune surveillance) [47]. Three injections were given according to the dose series of 0, 2 and 6 months. All simulation parameters were set at default with time steps set at 1, 180, and 360 (each time step represent 8 h in reality).
2.2. Structure-based drug discovery
2.2.1. Structure preparation
In our structure-based drug discovery approach, we targeted the N-terminal domain of SARS-CoV2 N-protein because the domain is reported to have binding pockets for ribonucleoside 5′- monophosphates such as adenosine 5′-monophosphate (AMP) [8]. We then retrieved an experimental structure of a related coronavirus in complex with AMP to aid in our analysis of the ligand’s binding site in SARS-CoV2 N-protein. The receptor (SARS-CoV2 N-protein) was energy minimized for 10,000 steps using GROMACS 2020.3 and the low-energy structure was used for the analysis.
2.2.2. AMP binding site analysis
The AMP binding site in the N-terminal domain of SARS-CoV2 N-protein was elucidated through alignment of protein structures using PyMol 2.2 [48]. The algorithm performs sequence alignment followed by a structural superposition, and then cycle refinement in order to reject structural outliers found during the fitting. Since we were also interested in the all-atom root mean square deviation, we turned off the refinement process.
2.2.3. Discovery of potential N-protein NTD inhibitors
The N-terminal domain of the SARS-CoV2 nucleocapsid protein was targeted for potential inhibitors using a library of approved antiviral drugs. The active site of the N-protein NTD accomodates nucleic acid substrates when packing the viral genome into a ribonucleoprotein complex. Targeting the N-protein can provide means to interrupt critical stages of the virus life cycle like virus assembly, transcription and replication processes. Thus, existing antiviral agents known to interfere with viral processes such as reverse transcription, virus integration, viral transcription and translation were considered because their mechanisms involve viral genetic materials. A set of 21 antiviral agents were randomly selected for this study. The selected ligands have a wide inhibitory spectrum including nucleoside analogues, nucleoside reverse transcriptase inhibitors, integrase inhibitors and acyclic guanosine analogues. The 3D structures of the drug candidates were generated with UCSF Chimera 1.14 [49] using their SMILE strings obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/). Ligands and receptor preparations were performed with AutoDock 4.2.6 tools [50]. AutoDock compatible structure files (pdbqt formats) were generated for both ligands and receptor after addition of hydrogen atoms and computation of Gasteiger charges. The Lamarckian genetic algorithm (LGA) was used and search parameters were set as follows; 50 GA runs, population size of 150, a maximum number of 25,000,000 energy evaluations, 27,000 number of generations, mutation rate of 0.02, and a crossover rate of 0.8. Grid was centered on the binding pocket of AMP and the size configured with dimensions 54, 52 and 48 along the x, y, and z-axis, respectively and 0.375 Å spacing. RMS cluster tolerance was set to 2.0 Å. We clustered the docking conformations based on root mean square deviation and a tolerance of 2.0 Å. The docked conformations were evaluated on the basis of binding energy and frequency of pose (cluster).
2.2.4. Binding energy analysis: MM/PBSA approach
The energy terms governing the protein-ligand interactions were analyzed using the MM-PBSA (Molecular Mechanics with Poisson–Boltzmann Surface Area) approach [51]. All energy terms were calculated by the g_mmpbsa 5.1.2 tool [52] based on simulation trajectories. We used GROMACS 5.1.5 (compatible with the g_mmpbsa tool) to run all the simulations following the protocol discussed in section 2.1.7 with slight modifications. We performed NVT and NPT equilibrations for 200 ps each under constraint conditions followed by 5 independent simulations each lasting for 1 ns with a sampling frequency of 10 ps. Frames from the last 500 ps of each run were used for the binding energy calculations.
2.2.5. Simulation of protein-ligand complexes
The behavior of the protein-ligand complexes at the atomistic level was elucidated with molecular dynamics simulation using the method described in section 2.1.7 with some modifications. Ligand topologies were generated by the Swiss Param server (http://www.swissparam.ch/). Each complex was simulated for 100 ns. RMSD calculation was performed with the 100 ns simulation trajectory while the last 80 ns simulation trajectory was used for all other protein-ligand complex analysis.
3. Results
3.1. Immuno-informatics approach
3.1.1. Prediction of B-cell epitopes
To determine the regions of SARS-CoV2 N-protein that may induce B-cell immunity, we predicted both linear and discontinuous B-cell epitopes using Bepipred 2.0 and Ellipro, respectively. The N-protein sequence obtained from the NCBI protein database (Accession number: QIC53221.1) was used for the prediction of the linear epitopes. The discontinuous epitopes, on the other hand, were predicted using a 3D structural conformation of the SARS-CoV2 N-protein. Structures of N-protein C-terminal domain and N-terminal domain were modeled as separate molecules using SWISS-MODEL (Fig. 1 ). The built structures were used to predict the discontinuous epitopes. Using a prediction cut-off of 0.5, the Bepipred 2.0 server predicted 11 candidates of varying residual length ranging from 6 to 52. The Ellipro server predicted 5 and 7 epitope candidates mapped to the C-terminal and N-terminal domains, respectively.
Fig. 1.

Structural models of N-protein and HLAs. (A) N-terminal domain of SARS-CoV2 N-protein (modeled based on PDB ID: 6M3M) (B) C-terminal domain of SARS-CoV2 N-protein based on PDB ID: 7C22 (C) HLA-A*03:01 based on PDB ID: 6O9B (D) HLA-B*35:01 based on PDB ID: 6AVG (E) HLA-DRB1*09:01 based on PDB ID: 1A6A. The β-sheets (blue), α-helix (red) and loops (green) are also shown.
3.1.2. Prediction of T-cell epitopes
We also predicted epitopes that may induce T-cell response against SARS-CoV2, based on ability to interact with specific HLA alleles. With over 3400 predicted MHC class I restricted epitopes, consideration was given to those below a percentile rank of 0.5 %. The percentile rank represents a randomly selected number of epitopes that are graded better than the candidate of interest, hence, a lower percentile rank is preferred.
3.1.3. Assessment of epitopes
As potential vaccine candidates, the predicted epitopes were profiled according to their toxicity, allergenicity and ability to induce IFN-γ. For the B-cell epitopes, preference was given to the non-toxins and non-allergen candidates with 8–11 amino acid residues. This resulted in 4 B-cell epitope candidates. The MHC-restricted epitopes that were predicted to be non-toxic, non-allergic and capable of inducing IFN-γ were considered for further analysis. This resulted in a total of 9 MHC-restricted epitopes. The sequences of the selected epitopes after the evaluation process are shown in Table 1 .
Table 1.
Evaluation and characterization of epitopes. The predicted HLA allele that binds well to the MHC-restricted epitopes are also provided.
| B-cell epitope | |||||||
| Candidate | Peptide | Length | Toxicity | Allergenicity | RMSD (nm) | ||
| 1 | AGLPYGANK | 9 | Non-toxin | Probable non-allergen | 0.47 | ||
| 2 | SKQLQQSMSSADS | 13 | Non-toxin | Probable non-allergen | 0.14 | ||
| 3 | RRIRGGDGKMKDL | 13 | Non-toxin | Probable non-allergen | 0.24 | ||
| 4 | LPQGTTLPKGF | 11 | Non-toxin | Probable non-allergen | 0.39 | ||
| MHC I | |||||||
| Peptide | length | Toxicity | Allergenicity | IFN-γ induction | RMSD (nm) | HLA allele | |
| 1 | KTFPPTEPK | 9 | Non-toxin | Probable non-allergen | Positive | 0.52 | HLA-A*11:01, HLA-A*03:01, HLA-A*30:01, HLA-A*31:01, HLA-A*68:01, HLA-A*32:01, HLA-A*30:02, HLA-A*33:01, HLA-B*57:01, HLA-B*58:01, HLA-A*26:01, HLA-A*01:01, HLA-B*15:01, HLA-B*07:02 |
| 2 | SSPDDQIGY | 9 | Non-toxin | Probable non-allergen | Positive | 0.26 | HLA-A*01:01, HLA-A*26:01, HLA-A*30:02, HLA-B*35:01, HLA-B*58:01, HLA-B*15:01, HLA-B*57:01, HLA-A*68:01, HLA-B*53:01, HLA-A*32:01, HLA-A*11:01, HLA-A*03:01, HLA-B*44:03, HLA-B*44:02, HLA-A*68:02, HLA-A*33:01 |
| 3 | SSPDDQIGYY | 10 | Non-toxin | Probable non-allergen | Positive | 0.35 | HLA-A*01:01, HLA-A*30:02, HLA-B*35:01, HLA-A*26:01, HLA-B*53:01, HLA-B*57:01, HLA-B*58:01, HLA-B*15:01, HLA-A*11:01, HLA-A*68:01, HLA-B*07:02, HLA-B*44:03, HLA-A*03:01, HLA-B*44:02, HLA-A*32:01, HLA-B*51:01 |
| 4 | VTPSGTWLTY | 10 | Non-toxin | Probable non-allergen | Positive | 0.3 | HLA-A*01:01, HLA-A*30:02, HLA-B*35:01, HLA-A*26:01, HLA-B*53:01, HLA-B*15:01, HLA-B*58:01, HLA-B*57:01, HLA-A*11:01, HLA-A*32:01, HLA-B*07:02, HLA-A*03:01, HLA-A*68:01, HLA-A*24:02, HLA-B*51:01, HLA-A*23:01, HLA-A*30:01, HLA-A*33:01, HLA-A*31:01, HLA-B*44:03, HLA-B*44:02 |
| MHC II | |||||||
| 1 | AQFAPSASAFFGMSR | 15 | Non-toxin | Probable non-allergen | Positive | 0.23 | HLA-DRB5*01:01, HLA-DRB5*01:01, HLA-DRB4*01:01, HLA-DRB4*01:01, HLA-DRB1*11:01, HLA-DRB1*12:01, HLA-DRB1*01:01 |
| 2 | IAQFAPSASAFFGMS | 15 | Non-toxin | Probable non-allergen | Positive | 0.24 | HLA-DQA1*01:01/DQB1*05:01, HLA-DQA1*03:01/DQB1*03:02, HLA-DRB1*11:01, HLA-DRB1*01:01, HLA-DRB3*01:01, HLA-DPA1*02:01/DPB1*14:01, HLA-DRB1*09:01, HLA-DRB1*12:01 |
| 3 | PQIAQFAPSASAFFG | 15 | Non-toxin | Probable non-allergen | Positive | 0.24 | HLA-DRB1*04:05, HLA-DRB1*04:01, HLA-DRB4*01:01, HLA-DQA1*04:01/DQB1*04:02, HLA-DRB3*01:01, HLA-DQA1*04:01/DQB1*04:02, HLA-DRB3*02:02, HLA-DRB1*15:01, HLA-DPA1*02:01/DPB1*05:01 |
| 4 | QIAQFAPSASAFFGM | 15 | Non-toxin | Probable non-allergen | Positive | 0.37 | HLA-DPA1*01:03/DPB1*02:01, HLA-DQA1*01:02/DQB1*06:02, HLA-DPA1*02:01/DPB1*01:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DQA1*03:01/DQB1*03:02, HLA-DRB5*01:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DRB5*01:01 |
| 5 | WPQIAQFAPSASAFF | 15 | Non-toxin | Probable non-allergen | Positive | 0.29 | HLA-DRB4*01:01, HLA-DPA1*02:01/DPB1*14:01, HLA-DRB4*01:01, HLA-DRB1*11:01, HLA-DRB1*04:01, HLA-DPA1*01:03/DPB1*02:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DRB5*01:01 |
RMSD was calculated after least square fitting of Cα atoms. The epitope candidates considered for further analysis are in bold.
3.1.4. Epitope stability studies
We performed molecular dynamics simulation for all the selected epitopes in order to inspect the stability of the peptides at the atomistic level. Prior to that, we built structural models for the epitopes using the PEP-FOLD3.5 server. The root mean square deviation (RMSD) from a 20 ns simulation was computed for each peptide and the results are shown in Table 1. The RMSD tool compares the simulating model to the initial structural configuration (reference structure) and computes the deviation between the two structures. The epitopes with low RMSD values were selected for further analysis.
3.1.5. Population coverage analysis of MHC restricted epitopes
The aim of the population coverage analysis was to predict the percentage of a population that would respond to the epitopes, considering the frequency of HLA genotypic traits in the population. In general, a higher fraction of the population was predicted to respond to the MHCI-restricted epitope than the MHCII-restricted epitopes. Considering the global population, 75.47 % of individuals would respond to the MHCI-restricted epitope while 56.23 % of the individuals would respond to the MHCII-restricted epitopes (Fig. 2 A and B). The lowest population coverage for the epitopes (both MHC I and II) was recorded in Central America and the highest recorded in East Asia (Fig. 2C). The South African population did not respond very well to the MHCII-restricted epitopes, although there was a significant population coverage in other regions of Africa. The best coverage of the MHCI-restricted epitope was observed in Europe, South Asia, Africa, North and Southern America and the Oceania.
Fig. 2.

Population coverage of MHC restricted epitope response. (A) Global population coverage of MHC I-restricted epitope (B) Global population of MHC II-restricted epitopes (C) Population coverage of MHC I and MHC II epitopes response in particular ethnicities across the world.
3.1.6. Epitope sequence conservation analysis
In order to check the cross-reactivity of the predicted epitopes across other related coronaviruses, a multiple sequence alignment study was performed using Clustal omega server. The N-protein sequence of SARS-CoV2 was aligned with N-protein sequences of SARS-CoV1, MERS-CoV, hCoV−OC43 and hCoV-NL63. The alignments of the protein sequences are shown Fig. 3 . Interestingly, the sequences of the MHCII-restricted epitopes were highly homologous to sequences of all the related coronaviruses except hCoV-NL63 and the MHCI-restricted epitope sequence was highly homologous to only the SARS-CoV1 N protein. The linear B-cell epitope, on other hand, shared low homology to N protein of SARS-CoV1 but none to the other related coronaviruses.
Fig. 3.

Multiple sequence alignment of SARS-CoV2 N-protein with other related coronaviruses. The sequences in the rectangular shape are peptide sequence of epitopes.
3.1.7. Structural studies of HLA-epitope complexes
The interaction between the selected epitopes and their respective HLA proteins were studied using docking and simulation analysis. We predicted the binding conformations of the T-cell epitopes to HLA alleles using the Cluspro 2 server. The best binding conformations of the T-cell epitopes to their respective HLA alleles are shown in Fig. 4 . We used HLA-A*03:01 and HLA-B*35:01 as the representative MHC class I proteins and HLA-DRB1*09:01 as the representative MHC class II protein. To further characterize the HLA-epitope interactions, we evaluated the structural properties and behavior of the complexes from a simulation trajectory using root mean square deviation, number of hydrogen bonds, and minimum distance. The structural properties and dynamics of the HLA-epitope complexes from 200 ns simulations are shown in Fig. 5 . The HLA-A*03:01-79SSPDDQIGY87 complex deviated less from its reference structure during the simulation than the HLA-B*35:01-79SSPDDQIGY87 complex (Fig. 5A) and a higher number of hydrogen bonds were formed between 79SSPDDQIGY87 and HLA-A*03:01 (Fig. 5C), suggesting a better interaction and a more stable complex when compared to the HLA-B*35:01-79SSPDDQIGY87 complex. The MHCII-restricted epitopes 305AQFAPSASAFFGMSR319, 304IAQFAPSASAFFGMS318 and 302PQIAQFAPSASAFFG316 were docked to HLA-DRB1*09:01 and simulated to study the epitope-HLA interactions. Although structural deviation was not significantly different for the MHCII-epitope complexes (Fig. 5B), the HLA-DRB1*09:01-305AQFAPSASAFFGMSR319 complex seems to form the highest number of hydrogen bonds among the MHCII-restricted epitopes with ≈11 hydrogen bonds (Fig. 5D). The results also suggest that the epitopes remained in close proximity to the HLA alleles with minimum distances ranging from 0.15 to 0.20 nm (Fig. 5E, F).
Fig. 4.

Predicted binding conformations of MHC-restricted epitopes to HLA proteins. (A) HLA-DRB1*09:01-AQFAPSASAFFGMSR complex (B) HLA-DRB1*09:01-IAQFAPSASAFFGMS complex (C) HLA-DRB1*09:01-PQIAQFAPSASAFFG complex (D) HLA-A*03:01-SSPDDQIGY complex (E) HLA-B*35:01-SSPDDQIGY complex. The 3D structures of the epitopes (red) and the surfaces of the HLA proteins (green) are shown.
Fig. 5.

Molecular dynamics simulation analysis of MHCI-restricted epitope complexes and MHC II-epitope complexes. The MHCI-restricted epitope (78SSPDDQIGY86) is complexed to either HLA-A*03:01 or HLA-B*35:01 and the MHCII-restricted epitopes (305AQFAPSASAFFGMSR319, 304IAQFAPSASAFFGMS318 and 302PQIAQFAPSASAFFG316) are complexed to HLA-DRB1*09:01. (A, B) RMSD. (C, D) number of hydrogen bonds. (E, F) Evolution of the minimum distance between epitope and HLA.
3.1.8. Simulation of immune response
We used the C-ImmSim server to simulate the immune response to the SARS-CoV2 N-protein in order to predict the immune response profile of the selected epitopes. The response elicited by antibodies, B-cells, T-cells and cytokines are shown in Fig. 6 . We simulated the response for a period of 1 year after three series of antigen injections. The second dose was given 2 months after the first and the third given in the 6th month (0, 2, 6 months). High levels of IgG + IgM immunocomplex and IgM were observed. The levels of these antibodies were low after the first injection but increased significantly following subsequent doses and this was accompanied by a concomitant reduction of antigen levels (Fig. 6A). The major cytokines observed were IFN- γ and IL-2 (Fig. 6B). Protection from B-cells and T-cells were notable along with development of immune memory. The immune memory developed by both B-cells and T-cells lasted for the period of the simulation (Fig. 6C, D). Other immune cells, including T-cytotoxic cells and natural killer cells, also elicited strong immune response to the antigen (Fig. 6E, F).
Fig. 6.

Simulation of immune responses to SARS-CoV2 N-protein. (A) Virus-specific antibody responses along with the antigen counts are shown. (B) The various cytokines released against the antigen. Insert plot shows danger signals and IL-2 levels. (C) The different forms of B-cells including the memory B-cells are shown. (D) T-helper cell counts and kinetics of memory T-cells (E) T-cytotoxic cell levels (F) Evolution of natural killer cells.
3.2. Structure-based drug design
3.2.1. Ligand binding site analysis
The N-terminal domain (NTD) of the nucleocapsid protein of the coronaviruses has a binding pocket for ribonucleoside 5′-monophosphates and forms complexes with them during the packing of viral RNA. While, the crystal structure of SARS-CoV2 NTD in complex with ribonucleoside 5′-monophosphates remains to be elucidated, the NTD of HCoV−OC43 N-protein in complex with adenosine 5′- monophosphate (AMP) has been solved and available at the PDB (ID: 4LI4). Using superposition and structural alignment analysis, we predicted the binding pocket of AMP in the SARS-CoV2 N-protein NTD based on the structural configuration of the PDB entry 4LI4. The structure of the N-protein NTD deviated from that of hCoV−OC43 with RMSD of 2.2 nm. The binding conformation of the AMP in its binding pocket was then determined through docking analysis. The structural alignment, superposition and AMP interactions at the binding site have been provided in Fig. 7 . AMP has polar interactions with residues S51, F53, A55, Y109, Y116 and R149 in the SARS-CoV2 N-protein NTD. The corresponding interacting residues in the hCoV N-protein NTD are also shown (see Fig. 7B).
Fig. 7.

Binding site analysis of AMP. (A) Structural superposition of SARS-CoV2 N-protein N-terminal domain model (green) to crystal structure of hCoV N-protein N-terminal domain (red) in complex to AMP (blue). (B) Superposition of active sites showing polar interactions (yellow dashes) between AMP and active site residues in SARS-CoV2 (green) and hCoV (red).
3.2.2. Discovery of potential inhibitors the N-terminal domain
A library of 21 antiviral drugs with varying targets was used in our virtual screening analysis. The nomenclature, molecular weight, chemical structure and autodock score (estimated free energy of binding) are provided in Table 2 . Among the studied ligands, pleconaril (PLE), raltegravir (RAL) and zidovudine triphosphate (ZTP) recorded higher free energy of binding than AMP. The binding energy is referred to as ‘autodock score’ because the value, although efficient at discriminating suitable ligands from non-suitable ones, does not accurately represent the binding affinity.
Table 2.
List of ligands used for the virtual screening. Their binding energies based on autodock analysis are also shown.
| Ligand | IUPAC nomenclature | M.W(g/mol) | Chemical representation | Binding energy (kcal/mol) |
|---|---|---|---|---|
| Adenosine 5′-monophosphate | [(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl dihydrogen phosphate | 347.22 | 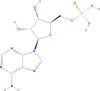 |
−4.94 |
| Amprenavir | [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate | 505.6 | 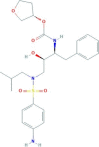 |
−3.24 |
| Entecavir | 2-amino-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylidenecyclopentyl]-1H-purin-6-one | 277.28 | 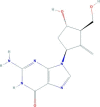 |
−3.28 |
| Indinavir | (2S)-1-[(2S,4R)-4-benzyl-2-hydroxy-5-[[(1S,2R)-2-hydroxy-2,3-dihydro-1H-inden-1-yl]amino]-5-oxopentyl]-N-tert-butyl-4-(pyridin-3-ylmethyl)piperazine-2-carboxamide | 613.8 | 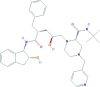 |
−2.85 |
| Pleconaril | 3-[3,5-dimethyl-4-[3-(3-methyl-1,2-oxazol-5-yl)propoxy]phenyl]-5-(trifluoromethyl)-1,2,4-oxadiazole | 381.3 | 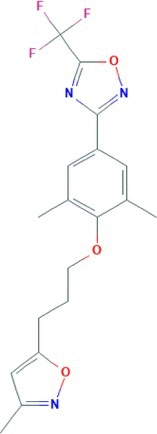 |
−5.44 |
| Raltegravir | N-[2-[4-[(4-fluorophenyl)methylcarbamoyl]-5-hydroxy-1-methyl-6-oxopyrimidin-2-yl]propan-2-yl]-5-methyl-1,3,4-oxadiazole-2-carboxamide | 444.4 | 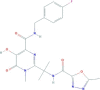 |
−5.5 |
| Trifluridine | 1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-(trifluoromethyl)pyrimidine-2,4-dione | 296.2 | 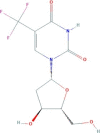 |
−4.26 |
| Umifenovir | ethyl 6-bromo-4-[(dimethylamino)methyl]-5-hydroxy-1-methyl-2-(phenylsulfanylmethyl)indole-3-carboxylate | 477.4 | 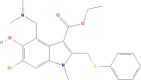 |
−3.62 |
| Vicriviroc | (4,6-dimethylpyrimidin-5-yl)-[4-[(3S)-4-[(1R)-2-methoxy-1-[4-(trifluoromethyl)phenyl]ethyl]-3-methylpiperazin-1-yl]-4-methylpiperidin-1-yl]methanone | 533.6 | 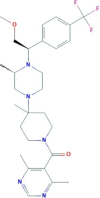 |
−4.3 |
| Zalcitabine | 4-amino-1-[(2R,5S)-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one | 211.22 | 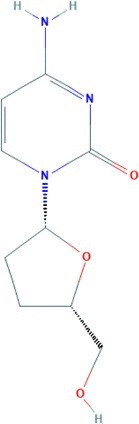 |
−3.48 |
| Penciclovir | 2-amino-9-[4-hydroxy-3-(hydroxymethyl)butyl]-1H-purin-6-one | 253.26 | 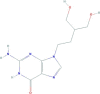 |
−2.87 |
| Brivudine | 5-[(E)-2-bromoethenyl]-1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione | 333.13 | 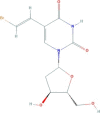 |
−4.87 |
| Famciclovir | [2-(acetyloxymethyl)-4-(2-aminopurin-9-yl)butyl] acetate | 321.33 | 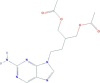 |
−3.61 |
| Cidofovir | [(2S)-1-(4-amino-2-oxopyrimidin-1-yl)-3-hydroxypropan-2-yl]oxymethylphosphonic acid | 279.19 | 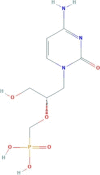 |
−2.83 |
| Ganciclovir | 2-amino-9-(1,3-dihydroxypropan-2-yloxymethyl)-1H-purin-6-one | 255.23 | 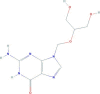 |
−3.58 |
| Valgancyclovir | [2-[(2-amino-6-oxo-1H-purin-9-yl)methoxy]-3-hydroxypropyl] (2S)-2-amino-3-methylbutanoate | 354.36 | 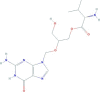 |
−2.18 |
| Acyclovir | 2-amino-9-(2-hydroxyethoxymethyl)-1H-purin-6-one | 225.2 | 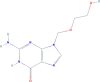 |
−3.17 |
| Valacyclovir | 2-[(2-amino-6-oxo-1H-purin-9-yl)methoxy]ethyl (2S)-2-amino-3-methylbutanoate | 324.34 | 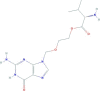 |
−2.26 |
| Ribavirin | 1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazole-3-carboxamide | 244.2 | 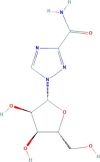 |
−3.02 |
| Remdesivir | 2-ethylbutyl (2S)-2-[[[(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate | 602.6 | 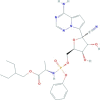 |
−3.02 |
| Zidovudine triphosphate | [[(2S,3S,5R)-3-azido-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate | 469.2 | 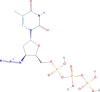 |
−7.83 |
| Lamivudine triphosphate | [[(2R,5S)-5-(4-amino-2-oxopyrimidin-1-yl)-1,3-oxathiolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate | 469.2 | 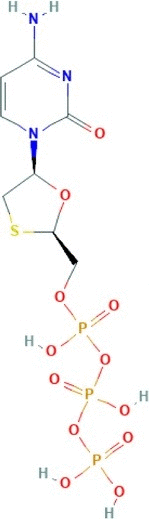 |
−3.09 |
3.2.3. MM/PBSA binding energy analysis
The molecular mechanics with Poisson-Boltzmann and surface area solvation (MM/PBSA) method of binding energy calculation is designed to be more accurate than the docking and scoring approach. For g_mmpbsa binding energy calculations, three energy terms are used namely molecular mechanics, polar solvation and non-polar solvation energies. The molecular mechanics potential energy includes energy of bonded and non-bonded interactions (Van-der Waals and electrostatic energies). The polar solvation term is calculated by solving the Poisson-Boltzmann equation [52]. The non-polar solvation term was calculated using the solvent accessible surface area (SASA) model and indicates repulsion and attraction between solvent and solute [52]. The total binding energy of ZTP (-1312.837 kJ/mol) is significantly higher as compared to AMP (-108.234 kJ/mol) with major contributions from the electrostatic terms in both cases (Table 3 ).
Table 3.
MM/PBSA calculations of energy terms and binding energy governing the protein-ligand interactions.
| Energy terms (kJ/mol) | |||||
|---|---|---|---|---|---|
| Ligand | Van der Waal | Electrostatics | Polar solvation | Non-polar solvation (SASA) | Binding energy (kJ/mol) |
| AMP | −0.001 | −106.428 | −1.855 | 0.051 | −108.234 |
| RAL | −0.002 | 0.175 | −8.177 | 0.068 | −7.936 |
| ZTP | −15.851 | −2012.207 | 721.787 | −6.565 | −1312.837 |
| PLE | −43.228 | −3.938 | 23.653 | −6.002 | −29.514 |
3.2.4. Simulation of protein-ligand complex
The 100 ns simulation revealed that the NTD-ZTP complex seems to be the most stable with RMSD of ≈ 0.3 nm while the NTD-AMP complex recorded RMSD of ≈ 0.35 nm (Fig. 8 A). Evolution of the total hydrogen bonds between the ligands and the NTD are shown in (Fig. 8B) and ranged from 2 to 8 hydrogen bonds for NTD-AMP and NTD-ZTP complexes. The minimum distance between ligands and the protein (Fig. 8C) ranged from 0.15 to 0.25 nm, suggesting close proximity especially for ZTP (≈ 0.15 nm). The compactness of the ZTP-NTD complex seems to be significantly low as compared to AMP-ZTP complex (see Fig. 8D). Root Mean Square Fluctuations (RMSF) values for residues 91–102 (residues of the β-hairpin) were significantly higher upon binding of AMP than ZTP (Fig. 9 A). The flexibility of the β-hairpin residues was similar for the NTD and NTD-AMP complex. ZTP occupied the binding pocket of AMP and interacts with residues T49, S51, F53, R88, Y109, Y111 and R149 (Fig. 9B).
Fig. 8.

Molecular dynamics analysis of selected antiviral drugs complexed with the N-terminal domain (NTD) of SARS-CoV2 N-protein. (A) RMSD of complexes after least square fitting to reference structure. (B) Number of hydrogen bonds formed between the ligands and protein. (C) The minimum distance between the ligands and the protein are shown (D) Radius of gyration (Rg) of the protein-ligand complexes.
Fig. 9.

The interaction between ZTP and the N-terminal domain (NTD). (A) RMSF analysis of the NTD. RMSF of the N-protein is shown alongside the RMSF of the protein in complex with ZTP or AMP. Residual fluctuations for the residues of the β-hairpin of the SARS-CoV2 N-protein NTD and loop regions are demonstrated. (B) The active site of ZTP (red) and the polar interaction (yellow dashes) with active site residues (blue) are shown. The structure of the β-hairpin is also demonstrated (cyan).
4. Discussion
A number of B-cell and T-cell epitopes have been identified using immuno-informatics which were subjected to stringent assessment and evaluation analysis based on desirable immunogenic properties such as allergenicity, toxicity, IFN-γ induction and structural stability. The N-protein epitopes 404SKQLQQSMSSADS416 and 92RRIRGGDGKMKDL104 were selected as potential epitope-based vaccine candidates capable of inducing B-cell immunity against SARS-CoV2 infection. Based on evaluation performance, MHCI-restricted epitope 79SSPDDQIGY87 and MHCII-restricted epitopes 305AQFAPSASAFFGMSR319, 304IAQFAPSASAFFGMS318 and 302PQIAQFAPSASAFFG316 were selected for further evaluations on the basis of cross reactivity across related coronaviruses and population coverage of their response.
A possibility of memory T-cell cross-reactivity specific to the S-protein has been suggested by Braun et al. [53] and a scenario of cross-reactivity in relations to SARS-CoV1 and endemic CoV has also been reported [54]. Based on sequence alignment analysis, our results suggest a possible MHCII-restricted epitope-induced T-cell cross-reactivity to SARS-CoV1, MERS-CoV, hCoV−OC43 and hCoV-NL63. Moreover, the HLA-I restricted epitope is predicted to induce T-cell immunity in SARS-CoV1.
Presentation of antigenic peptides by MHC for recognition by immune cells during COVID-19 infection is not clearly defined, however, extrapolating knowledge from previous coronaviruses suggests presentation by both MHCI and MHCII alleles [55]. The relevance of demonstrating the HLA-epitope complex formation and interactions cannot be disregarded. The epitopes were docked to specific HLA-alleles and the best docked conformations were further characterized by simulation analysis. The findings indicate that the HLA-A*03:01-79SSPDDQIGY87 complex is a more stable structure than HLA-B*35:01-79SSPDDQIGY87 based on RMSD analysis, hydrogen bonds formed and proximity of epitope to HLA protein. Similarly, the HLA-DRB1*09:01-305AQFAPSASAFFGMSR319 complex was the most stable among the MHCII-restricted epitope candidates. Collectively, our results show that the epitopes (79SSPDDQIGY87 and 305AQFAPSASAFFGMSR319) can bind efficiently and form stable structures with their respective HLA proteins.
Although we have an incomplete understanding of the immunological responses during the SARS-CoV2 infection, findings from research on previous coronavirus outbreaks (SARS-CoV1 and MERS-CoV) and the emerged findings on immunologic characteristics of COVID-19 provide substantial answers. Available information suggests the involvement of innate immunity, T- and B-cell immunity as well as specific antiviral neutralizing antibodies in the immunological responses during SARS-CoV2 infection [[56], [57], [58]]. Simulation of the immune responses to the N-protein of SARS-CoV2 yielded results that correlate well with the emerging data on immunological profile of COVID-19. Upon repeated injection of the antigen (N-protein), aspects of innate immunity (Natural killer cells), B- and T-cell immunity and antibody-mediated neutralization were observed accompanied by development of immunological memory that lasted for several months. Both T-helper and T-cytotoxic cell-mediated immunity were observed upon exposure to the N-protein, however, only T-helper cells developed memory for the antigen. Protein specific T-cell responses with induction of IFN-γ, TNF-a, and IL-2 have been detected in COVID-19 patients [59]. Recognition of S and N protein epitopes by virus specific T-helper and T-cytotoxic cells during the COVID-19 infection have also been reported [60]. Immunological characteristics of SARS-CoV1 infection include the development of CD4 and CD8 memory T-cells which persisted for over 4 years [61]. The extent of memory T-cell immunity against COVID-19 is yet to be elucidated, however, our results indicate a different scenario as observed for the SARS-CoV1 infection. The B-cells also elicited a robust response to the SARS-CoV2 N-protein just that the memory B-cell faded significantly over time as compared to the memory T-helper cells. Long term protection courtesy of memory B-cells have been confirmed in SARS-CoV1 infections [62,63] and emerging evidence are supporting a similar situation for COVID-19 [[64], [65], [66]] but the exact life-span is yet to be known.
The B-cell immunity during SARS-CoV2 infection is further confirmed by the detection of virus-specific immunoglobulin titers with particular affinity for S- and N- protein epitopes after about a week of infection [22,24,64]. Current reports suggest IgM, IgG and IgA responses as the main antibody neutralization during the SARS-CoV2 infection with the detection of IgM response at the early stages (7 days of infection) followed by IgG and IgA upon progression to an acute stage [56,58]. Our computational results agree well with these findings. According to the immune simulation analysis, immunocomplex IgG + IgM and IgM are the antibodies responsible for antigen neutralization during the early stages of exposure. IgA was, however not detected but the source of this discrepancy may be attributed to the antigenic protein used as target.
Exposure to the N-protein also elicited certain aspects of both adaptive and innate immune response. According to our computational results, IFN-γ and IL-2 elicited robust responses to the antigen with significant levels of natural killer (NK) cells also being detected. Possible NK-mediated response to the SARS-CoV2 infection has been reported by Pinto et al. [67] and evidence of reduced levels of NK cells in the peripheral blood of infected individuals have also emerged [68,69].
Taking into consideration the novelty of the SARS-CoV2 infection and our limited understanding of its dynamics, other therapeutic options are mandatory for complete containment of the pandemic. If the effects of the vaccine fades over time provided the virus develops means to evade immunity, drugs can be used to eliminate symptoms faster. Moreover, populations or individuals that do not respond well to the vaccine would rely on drugs for managing the pandemic. In view of this, we targeted the N-terminal domain (NTD) of SARS-CoV2 N-protein for potential drug candidates. The NTD, otherwise known as the RNA-binding domain, plays important roles during package of viral genome into a ribonucleoprotein complex [70,71]. Kang et al. [72] solved the structure of the NTD of SARS-CoV2 N-protein (PDB ID: 6M3M) and identified potential pockets for drug targeting. Considering the urgent need to contain the SARS-CoV2 infection, drug repurposing seems to be the best tool in the search for therapeutic options. Existing drugs for different targets such as chloroquine, remdesivir, lopinavir, ribavirin and ritonavir have been re-oriented to target the SARS-CoV2 and several in-vitro and in-vivo studies are providing positive outcomes [[73], [74], [75]]. Based on MM/PBSA binding energy calculations and molecular dynamics simulation analysis, zidovudine triphosphate (ZTP) has higher affinity and interacts better with the NTD of N-protein than AMP. ZTP is an anti-HIV agent that targets the viral reverse transcriptase, thus interfering with the reverse transcription process [76]. Residual fluctuation analysis from the simulation studies revealed that upon binding of ZTP, the flexibility of the NTD β-hairpin (basic palm) residues significantly reduce. The impact could be detrimental for the NTD considering the roles of the flexible, positively charged β-hairpin. The β-hairpin has been proposed to be essential for RNA packaging by interacting with phosphate groups of the viral RNA [[77], [78], [79]]. This observation together with the high binding affinity depicts ZTP as a promising N-protein inhibitor and should be further validated by experimental studies.
5. Conclusion
We have explored the SARS-CoV2 N-protein for potential epitope-based vaccine candidates using immuno-informatics and identified an antiviral drug that binds to the ribonucleoside 5′-monophosphate binding pocket in the N-terminal domain (NTD) of SARS-CoV2 N-protein with much greater affinity and stability. In view of this, we have identified B-cell epitopes (404SKQLQQSMSSADS416 and 92RRIRGGDGKMKDL104), MHCI-restricted epitope (76SSPDDQIGY86) and MHCII-restricted epitope (305AQFAPSASAFFGMSR319) based on performance after stringent evaluation and profiling analysis on the basis of certain desirable vaccine characteristics. We also identified zidovudine triphosphate as a promising inhibitor of the NTD of SARS-CoV2 N-protein. The limitations of our study cannot be disregarded as experimental data are needed to validate our findings. Nonetheless, the integrative bioinformatics methods we used will significantly save time and cost for research communities using wet-laboratory approaches and clinical studies. As the need for COVID-19 therapeutics grow by the day, our findings will assist research scientists to fast-track the discovery of vaccines and drugs to contain the SARS-CoV2 infection.
Authors contribution
AK and EA: conception and design of study, AK, EA, SOP, SAS: Acquisition of data: AK, EA, SOP, SAS: Analysis and interpretation of data, AK, EA, SOP, SAS: Drafting and final revision of manuscript.
Data availability
Data is available upon request
Declaration of Competing Interest
The authors declare they have no competing interest.
Acknowledgement
AK is a grant recipient on EDCTP and African Research Network for Neglected Tropical and Diseases SGP-III. We are also indebted to the staff of the Kwarteng Lab (Kumasi Centre for Collaborative Research in Tropical Medicine) for their unflinching support in the preparation of this manuscript.
References
- 1.Gralinski L.E., Menachery V.D. Return of the coronavirus: 2019-nCoV. Viruses. 2020;12(2):135. doi: 10.3390/v12020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu A., Peng Y., Huang D., Ding X., Wang X., Niu P. Genome composition and divergence of the Novel Coronavirus (2019-nCoV) originating in China. Cell Host Microbe. 2020;27:325–328. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naqvi A.A.T., Fatima K., Mohammad T., Fatima U., Singh I.K., Singh A., Hassan M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochimica et Biophysica Acta (BBA) – Mol. Basis of Dis. 2020;165878 doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan. China, JAMA. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortiz-Prado E., Simbaña-Rivera K., Gómez-Barreno L., Rubio-Neira M., Guaman L.P., Kyriakidis N.C., López-Cortés A. Clinical, molecular and epidemiological characterization of the SARS-CoV2 virus and the Coronavirus disease 2019 (COVID-19), a comprehensive literature review. Diagn. Microbiol. Infect. Dis. 2020;115094 doi: 10.1016/j.diagmicrobio.2020.115094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masters P.S., L.S The molecular biology of coronaviruses. Adv. Virus Res. 2006;64:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshimoto F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020;39(3):198–216. doi: 10.1007/s10930-020-09901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Haan C.A., Rottier P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005;64:165–230. doi: 10.1016/S0065-3527(05)64006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McBride R. M. Van Zyl, B. Fielding, the coronavirus nucleocapsid is a multifunctional protein. Viruses. 2014;6(8):2991–3018. doi: 10.3390/v6082991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Haan C.A., Kuo L., Masters P.S., Vennema H., Rottier P.J. Coronavirus particle assembly: primary structure requirements of the membrane protein. J. Virol. 1998;72:6838–6850. doi: 10.1128/jvi.72.8.6838-6850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Surjit M., Kumar R., Mishra R.N., Reddy M.K., Chow V.T., Lal S.K. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J. Virol. 2005;79(17):11476–11486. doi: 10.1128/JVI.79.17.11476-11486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cavanagh D. Coronaviruses and toroviruses. In: Zuckerman A.J., Banatvala J.E., Griffiths P.D., Pattison J.R., Schoub B.D., editors. Principles and Practice of Clinical Virology. John Wiley & Sons Ltd.; Chichester: 2004. pp. 379–397. [Google Scholar]
- 14.Van der Meer Y., Snijder E.J., Dobbe J.C., Schleich S., Denison M.R., Spaan W.J., Locker J.K. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 1999;73:7641–7657. doi: 10.1128/JVI.73.9.7641-7657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serafin M.B., Bottega A., Foletto V.S., da Rosa T.F., Hörner A., Hörner R. Drug repositioning an alternative for the treatment of coronavirus COVID-19. Int. J. Antimicrob. Agents. 2020;105969 doi: 10.1016/j.ijantimicag.2020.105969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.World Health Organization . 2020. Draft of the Landscape of COVID-19 Candidate Vaccines.https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines [Google Scholar]
- 17.Vartak A., Sucheck S.J. Recent advances in subunit vaccine carriers. Vaccines. 2016;4(2):12. doi: 10.3390/vaccines4020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shang W., Yang Y., Rao Y. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. Npj Vaccines. 2020;5:18. doi: 10.1038/s41541-020-0170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grifoni A., Sidney J., Zhang Y., Scheuermann R.H., Peters B., Sette A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020;27(4):671–680. doi: 10.1016/j.chom.2020.03.002. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li C.K.F., Wu H., Yan H., Ma S., Wang L., Zhang M., Tang X., Temperton N.J., Weiss R.A., Brenchley J.M. T cell responses to whole SARS coronavirus in humans. J. Immunol. 2008;181:5490–5500. doi: 10.4049/jimmunol.181.8.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tay M.Z., Poh C.M., Renia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020 doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amanat F., Stadlbauer D., Strohmeier S. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020;26:1033–1036. doi: 10.1038/s41591-020-0913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ju B., Zhang Q., Ge J. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584:115–119. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 24.To K.K., Tsang O.T., Leung W.S. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect. Dis. 2020;20(5):565–574. doi: 10.1016/S1473-3099(20)30196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jespersen M.C., Peters B., Nielsen M., Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45(W1):W24–W29. doi: 10.1093/nar/gkx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ponomarenko J., Bui H.H., Li W., Fusseder N., Bourne P.E., Sette A., Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9(1):514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta S., Kapoor P., Chaudhary K., Gautam A., Kumar R. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimitrov I., Bangov I., Flower D.R., Doytchinova I. AllerTOP v.2—a server for in silico prediction of allergens. J. Mol. Model. 2014;20(6) doi: 10.1007/s00894-014-2278-5. [DOI] [PubMed] [Google Scholar]
- 29.Dhanda S.K., Vir P., Raghava G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct. 2013;8(1) doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamiable A., Thévenet P., Rey J., Vavrusa M., Derreumaux P., Tufféry P. PEP-FOLD3: fasterde novostructure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016;44(W1):W449–W454. doi: 10.1093/nar/gkw329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozakov D., Hall D.R., Xia B., Porter K.A., Padhorny D., Yueh C., Vajda S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brenke R., Hall D.R., Chuang G.Y., Comeau S.R., Bohnuud T., Beglov D., Kozakov D. Application of asymmetric statistical potentials to antibody-protein docking. Bioinformatics. 2012;28(20):2608–2614. doi: 10.1093/bioinformatics/bts493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozakov D., Brenke R., Comeau S.R., Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 34.Katchalski-Katzir E. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. U.S.A. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks B.R. Charmm - a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 36.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 37.Celada F., Seiden P.E. A computer model of cellular interactions in the immune system. Immunol. Today. 1992;13:56–62. doi: 10.1016/0167-5699(92)90135-T. [DOI] [PubMed] [Google Scholar]
- 38.Seiden P.E., Celada F. A model for simulating cognate recognition and response in the immune system. J. Theor. Biol. 1992;158:329–357. doi: 10.1016/s0022-5193(05)80737-4. [DOI] [PubMed] [Google Scholar]
- 39.Celada F., Seiden P.E. Affinity maturation and hypermutation in a simulation of the humoral immune response. Eur. J. Immunol. 1996;26:1350–1358. doi: 10.1002/eji.1830260626. [DOI] [PubMed] [Google Scholar]
- 40.Haskins K., Kubo R., White J., Pigeon M., Kappler J. The major histocompatibility complex-restricted antigen receptor on T cells. J. Exp. Med. 1983;157:1149–1162. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bere C., Ziegner M. The maturation of the immune response. Immunol. Today. 1993;14:400–404. doi: 10.1016/0167-5699(93)90143-9. [DOI] [PubMed] [Google Scholar]
- 42.Celada F. The cellular basis of immunological memory. Prog. Allergy. 1971;15:223–267. [PubMed] [Google Scholar]
- 43.Sprent J., Lo D., Gao F.K., Ron Y. T cell selection in the thymus. Immunol. Rev. 1988;101:173–190. doi: 10.1111/j.1600-065x.1988.tb00737.x. [DOI] [PubMed] [Google Scholar]
- 44.Allen P.M. Antigen processing at the molecular level. Immunol. Today. 1987;8:270–273. doi: 10.1016/0167-5699(87)90187-3. [DOI] [PubMed] [Google Scholar]
- 45.Berzofski J.A. The nature and role of antigen processing. In: Cruse J.M., Lewis R.E., editors. T Cell Activation, Year in Immunology. 1985. pp. 18–24. [PubMed] [Google Scholar]
- 46.Unanue E.R. Antigen-presenting function of the macrophage. Ann. Rev. Immunol. 1985;2:395–428. doi: 10.1146/annurev.iy.02.040184.002143. [DOI] [PubMed] [Google Scholar]
- 47.Rapin N., Lund O., Bernaschi M., Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One. 2010;5(4):e9862. doi: 10.1371/journal.pone.0009862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan S., Chan H.C.S., Hu Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017;7(2):e1298. doi: 10.1002/wcms.1298. [DOI] [Google Scholar]
- 49.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 50.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A. T.E. Cheatham 3rd, calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 52.Kumari R., Kumar R., Lynn A. g_mmpbsa—a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 53.Braun J., Loyal L., Frentsch M., Wendisch D., Georg P., Kurth F., Hippenstiel S., Dingeldey M., Kruse B., Fauchere F. Presence of SARS-CoV-2 reactive T cells in COVID-19 patients and healthy donors. MedRxiv. 2020 doi: 10.1101/2020.04.17.20061440. [DOI] [PubMed] [Google Scholar]
- 54.Gioia C., Horejsh D., Agrati C., Martini F., Capobianchi M.R., Ippolito G., Poccia F. T-Cell response profiling to biological threat agents including the SARS coronavirus. Int. J. Immunopathol. Pharmacol. 2005;18:525–530. doi: 10.1177/039463200501800312. [DOI] [PubMed] [Google Scholar]
- 55.Liu P., Wu F. Novel immunodominant peptide presentation strategy: a featured HLA-A*2402-restricted cytotoxic T-lymphocyte epitope stabilized by intrachain hydrogen bonds from severe acute respiratory syndrome coronavirus nucleocapsid protein. J. Virol. 2010;84 doi: 10.1128/JVI.01464-10. 11849e11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Azkur A.Z., Akdis M., Azkur D., Sokolowska M., van de Veen W., Brüggen M., Akdis C.A. Immune response to SARS‐CoV‐2 and mechanisms of immunopathological changes in COVID‐19. Allergy. 2020 doi: 10.1111/all.14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X., Geng M., Peng Y., Meng L., Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020;10(2):102–108. doi: 10.1016/j.jpha.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin R., Samstein R.M. 2020. Immunology of COVID-19: Current State of the Science, Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ni L., Ye F., Chen M.L., Feng Y., Deng Y.Q., Zhao H., Wei P., Ge J., Gou M., Li X. 2020. ., Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals, Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu L., Shi Y. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Resp. Med. 2020 doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fan Y.Y., Huang Z.T., Li L. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch. Virol. 2009;154 doi: 10.1007/s00705-009-0409-6. 1093e1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao Z., Liu L., Du L., Zhang C., Jiang S., Li T., He Y. Potent and persistent antibody responses against the receptor-binding domain of SARS-CoV spike protein in recovered patients. Virol. J. 2010;7:299. doi: 10.1186/1743-422X-7-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu J., Li S., Liu J., Liang B., Wang X., Wang H., Li W., Tong Q., Yi J., Zhao L. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. 2020;55:102763. doi: 10.1016/j.ebiom.2020.102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ju B., Zhang Q., Ge X., Wang R., Sun J., Ge X., Yu J., Shan S., Zhou B., Song S. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020 doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 65.Bao L., Deng W., Gao H., Xiao C., Liu J., Xue J., Lv Q., Liu J., Yu P., Xu Y. Reinfection could not occur in SARS-CoV-2 infected rhesus macaques. BioRxiv. 2020 doi: 10.1101/2020.03.13.990226. [DOI] [Google Scholar]
- 66.Adams E.R., Anand R., Andersson M.I., Auckland K., Baillie J.K., Barnes E., Bell J., Berry T., Bibi S., Carroll M. Evaluation of antibody testing for SARS-CoV-2 using ELISA and lateral flow immunoassays. MedRxiv. 2020 doi: 10.1101/2020.04.15.20066407. [DOI] [Google Scholar]
- 67.Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A. Structural and functional analysis of a potent sarbecovirus neutralizing antibody. bioRxiv. 2020 doi: 10.1101/2020.04.07.023903. [DOI] [Google Scholar]
- 68.Song C.Y., Xu J., He J.Q., Lu Y.Q. COVID-19 early warning score: a multi-parameter screening tool to identify highly suspected patient. medRxiv. 2020 doi: 10.1101/2020.03.05.20031906. [DOI] [Google Scholar]
- 69.Zheng M., Gao Y., Wang G. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020;17:533–535. doi: 10.1038/s41423-020-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saikatend K.S., Joseph J.S., Subramanian V., Neuman B.W., Buchmeier M.J., Steven R.C. Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 2007;81:3913e21. doi: 10.1128/JVI.02236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan Y.W., Fang S., Fan H., Lescar J., Liu D.X. Amino acid residues critical for RNA-binding in the N-terminal domain of the nucleocapsid protein are essential determinants for the infectivity of coronavirus in cultured cells. Nucleic Acids Res. 2006;34:4816e25. doi: 10.1093/nar/gkl650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang S., Yang M., Hong Z., Zhang L., Huang Z., Chen X. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta. Pharm. Sin. B. 2020 doi: 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamamoto N., Matsuyama S., Hoshino T., Yamamoto N. Nelfi- navir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv. 2020 doi: 10.1101/2020.04.06.026476. [DOI] [Google Scholar]
- 74.de Wit E., Feldmann F., Cronin J., Jordan R., Okumura A., Thomas T., Scott D., Cihlar T., Feldmann H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. U.S.A. 2020;117:6771–6776. doi: 10.1073/pnas.1922083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Borba M.G.S., de Almeida Val F., Sampaio V.S., Alexandre M.A.A., Melo G.C., Brito M., Moura˜ o M.P.G., Brito Sousa J.D., Baía-da-Silva D.C., Guerra M.V.F. Effect of high vs low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. JAMA Netw. Open. 2020;3:e208857. doi: 10.1001/jamanetworkopen.2020.8857. [DOI] [PubMed] [Google Scholar]
- 76.De Clercq E., Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang Q., Yu L., Petros A.M., Gunasekera A., Liu Z., Xu N., Hajduk P., Mack J., Fesik S.W., Olejniczak E.T. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry. 2004;43:6059–6063. doi: 10.1021/bi036155b. [DOI] [PubMed] [Google Scholar]
- 78.Fan H., Ooi A., Tan Y.W., Wang S., Fang S., Liu D.S., Lescar J. The nucleocapsid protein of coronavirus infectious bronchitis virus: crystal structure of its N-terminal domain and multimerization properties. Structure. 2005;13:1859–1868. doi: 10.1016/j.str.2005.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan Y.W., Fang S., Fan H., Lescar J., Liu D.X. Amino acid residues critical for RNA-binding in the N-terminal domain of the nucleocapsid protein are essential determinants for the infectivity of coronavirus in cultured cells. Nucleic Acids Res. 2006;34:4816–4825. doi: 10.1093/nar/gkl650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data is available upon request