

Abstract
Introduction
Caesarean delivery is steadily becoming one of the more common surgical procedures in Australia with over 100 000 caesarean sections performed each year. Over the last 10 years in Australia, the caesarean section rate has increased from 28% in 2003 to 33% in 2013. On the international stage, the Australian caesarean delivery rates are higher than the average for the Organisation for Economic Co-operation and Development, Australia ranked as 8 out of 33 and is second to the USA. Postoperative surgical site infections (SSIs) and wound complications are the most common and costly event following a caesarean section. Globally, complication rates following a caesarean delivery vary from 4.9% to 9.8%. Complications such as infection and wound breakdown affect the postpartum mother’s health and well-being, and contribute to healthcare costs for clinical management that often spans the acute, community and primary healthcare settings. Published level one studies using advanced wound dressings in the identified ‘at-risk’ population prior to surgery for prophylactic intervention are yet to be forthcoming.
Methods and analysis
A parallel group randomised control trial of 448 patients will be conducted across two metropolitan hospitals in Perth, Western Australia, which provide obstetric and midwifery services. We will recruit pregnant women in the last trimester, prior to their admission into the healthcare facility for delivery of their child. We will use a computer-generated block sequence to randomise the 448 participants to either the interventional (negative pressure wound therapy (NPWT) dressing, n=224) or comparator arm (non-NPWT dressing, n=224). The primary outcome measure is the occurrence of surgical wound dehiscence (SSWD) or SSI. The Centres for Disease Control reporting definition of either superficial or deep infection at 30 days will be used as the outcome measure definition. SWD will be classified as per the World Union of Wound Healing Societies grading system (grade I–IV). We will assess recruitment rate, and adherence to intervention and follow-up. We will assess the potential effectiveness of NPWT in the prevention of postpartum surgical wound complications at three time points during the study; postoperative days 5, 14 and 30, after which the participant will be closed out of the trial. We will use statistical methods to determine efficacy, and risk stratification will be conducted to determine the SWD risk profile of the participant. Follow-up at day 30 will assess superficial and deep infection, and wound dehiscence (grade I–IV) and the core outcome data set for wound complications. This study will collect health-related quality of life (European Quality of Life 5-Dimensions 5-Level Scale), mortality and late complications such as further surgery with a cost analysis conducted. The primary analysis will be by intention-to-treat. This clinical trial protocol follows the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) and the Consolidated Standards of Reporting Trials guidelines.
Ethics and dissemination
Ethics approval was obtained through St John of God Health Care (HREC1409), Western Australia Department of Health King Edward Memorial Hospital (HREC3111). Study findings will be published in peer-reviewed journals and presented at international conferences. We used the SPIRIT checklist when writing our study protocol.
Trial registration number
Australian and New Zealand Clinical Trials Registry (ACTRN12618002006224p).
Keywords: wound management, surgery, obstetrics, clinical trials
Strengths and limitations of this study.
The EffeCtiveness of neGative pressure wound therapY in the preventioN of surgical woUnd complicationS in the cesarean section at risk population: a randomised multi-centre trial, the CYGNUS trial (CYGNUS) trial is a large multicentre trial that will provide important evidence on the effectiveness of a therapy for prevention of a wound complication after caesarean section.
This study will address an important gap in the current evidence for early identification of those at risk prior to surgery.
Pragmatically designed and reviewed by clinicians, and consumer groups to allow for integration into routine clinical practice.
Due to the nature of the intervention, blinding of participants and providers is not possible.
Trial is available only to women who have proficiency in English language.
This study will recruit from scheduled elective admissions at a tertiary women’s hospital with a probable and coincidental inclusion of urgent or emergency cases.
Introduction
Currently, in Western Australia, the caesarean section delivery rate is reportedly 37% of births compared with the Organisation for Economic Co-operation and Development average of 25%.1 Surgical site infection (SSI) or wound complications such as dehiscence is a common cause of morbidity with reported rates of 4.9%–9.8% in some acute care settings.2–4 The potential risk of surgical wound complications following the caesarean procedure is considerable, and there is still a gap in the literature as to best practice in prevention of wound complications following this type of procedure.2 3 Several studies in the field have retrospectively investigated the effect of negative pressure wound therapy (NPWT) in the reduction of postsurgical wound complications in the obstetric population.5 6 However, there is a lack of published level one studies investigating the prophylactic use of in the prevention of surgical complications in high-risk patients with multiple risk factors such as obesity, diabetes and previous caesarean-section.7–9 While a recent study from Odense University Hospital, Denmark, has identified an effect of the use on patients with one risk factor (body mass index (BMI) of 30 kg/m2+),10 there remain considerable gaps in the evidence addressing patients with multiple risk factors (ie, diabetes, smoking and previous caesarean section) which may be more generalisable to an average caesarean section population. The utility of risk stratification and the impact of prophylactic initiated for use in high-risk cohorts for the prevention of wound dehiscence following a caesarean section remains to be determined. Furthermore, and more important, is the measure of patient perception of their wound healing after the procedure. An evidence-based approach is important to inform the development of clinical pathways and protocols in patient management, and this study will form the basis of the development of a postoperative wound management pathway for caesarean section surgery patients both at the participating trial sites, and more broadly in both national and international clinical settings.
Study design
CYGNUS is a parallel group randomised control trial (RCT) testing the effectiveness of an intervention (NPWT dressing) to a control (non-NPWT dressing) with respect to wound healing, complications, and cost and health-related quality of life. Figure 1 summarises the design of the trial and each of the trial aspects described below as per Consolidated Standards of Reporting Trials (CONSORT).11 This study was designed using the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) checklist.12
Figure 1.
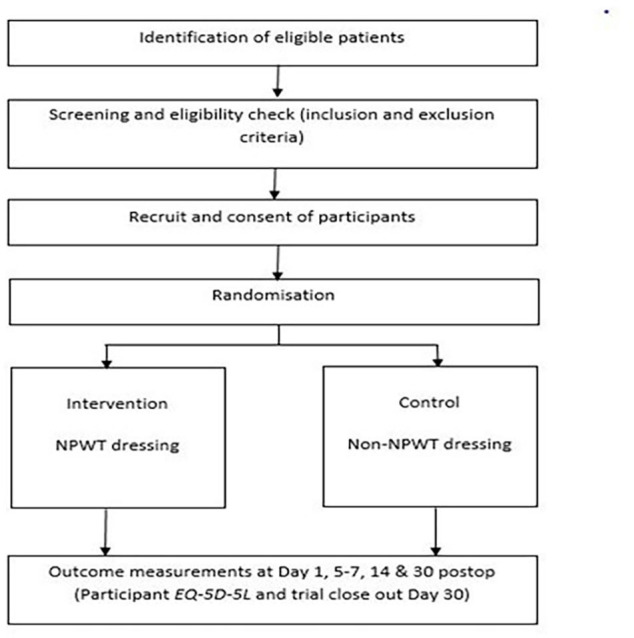
CYGNUS Consolidated Standards of Reporting Trials participant. EQ-5D-5L, European Quality of Life 5-Dimensions 5-Level Scale; NPWT, negative pressure wound therapy.
Study aim and objectives
The aim of the CYGNUS trial is to determine the effectiveness of NPWT (intervention) in the prevention of postcaesarean section complications in the postpartum mother compared with non-NPWT dressing (control). Our primary objectives are to conduct an RCT to determine the effectiveness of NPWT in prevention of postoperative complications such as surgical wound dehiscence (SWD) and SSI following a caesarean section in low-to-high risk cohorts and determine the clinical utility of the intervention. Our secondary objectives are to conduct a health-related quality of life European Quality of Life 5-Dimensions 5-Level Scale (EQ-5D-5L) assessment, and to evaluate patient perceptions of wound healing and pain following the surgical procedure in the intervention and control arms. Finally, we aim to determine the cost-effectiveness of the intervention relative to the control using a stepped health economic analysis, concluding with a cost-utility analysis reporting a cost per quality-adjusted life year (QALY).
Study setting
The CYGNUS trial will be conducted in two metropolitan Perth maternity hospitals over a period of 2 years (October 2019 to October 2021). All eligible pregnant women at participating sites will be considered for enrolment.
Study participants and eligibility criteria
Pregnant women eligible for recruitment to the CYGNUS trial are those with a viable pregnancy, able to provide written informed consent in English and have no known allergies to hydrocolloid, polysiloxanes or silicone resin. Women who are not fluent in English are ineligible to participate.
Recruitment and randomisation
All pregnant women booked for antenatal care will be screened against the eligibility criteria. Participants will be screened according to their risk level using the validated Perth Surgical Wound Dehiscence Risk Assessment Tool (PSWDRAT), which has a number of risk factors embedded into the tool. Any participant who has a score above 2 will be deemed at risk. Eligible participants will receive the CYGNUS patient information sheet (PIS) at least 24 hours prior to their admission booking. This will allow time to consider participation in the trial. The PIS will be accompanied by a letter from the principal investigator informing patients about the trial and inviting them to participate. The PIS will be discussed with eligible women by a member of the research team and potential participants will have the opportunity to discuss the study and ask questions. If a patient wishes to participate, they will be provided with a CYGNUS trial consent form where written consent to participate will be sought. The person who interviews the potential study participants and obtains their consent to participate will be blinded to the treatment allocation schedule. Once the consent has been obtained, the staff member who is recruiting the patient will contact the senior research fellow at their site to request the next study identifier and treatment allocation. Baseline participant information, demographic and related medical history will be collected. A participant risk profile for SWD will be obtained using the PSWDRAT.13 This will determine the SWD risk profile of the trial participant. The EQ-5D-5L,14 a validated questionnaire, will be administered at baseline and at the end of the trial to allow estimation of QALYs of all participants. The study statistician will generate the allocation sequence for each site before the study commences, using a random number generator on a computer. There will be a separate sequence for each site, and each sequence will be generated using a permuted random block strategy to ensure that recruitment to the two arms of the study occurs at approximately equal rates within each site. The allocation list for each site will be provided to the senior research fellow at each site, and will be kept private from all other personnel at the site. Due the nature of the study device, blinding of participants or study personnel after treatment allocation is not possible. However, the study statistician will be blinded to group allocation.
Patient and public involvement
No patient was involved in the study design.
Postrandomisation withdrawals/exclusions
Participants may choose not to participate in the CYGNUS trial or withdraw from the study at any time without prejudice. Choosing either of these options will not affect the standard of care the patient receives.
Study dressings
Participants who elect to have a caesarean section as the chosen method of delivery usually have a scheduled time and day for the procedure. Some patients may be required to undergo an urgent or an emergency caesarean delivery due to a number of uncontrolled factors. Routine antibiotic prophylaxis will be given to patients immediately before surgery and all intraoperative procedures will adhere to the WHO15 surgical site safety checklist and local infection prevention policies in accordance with Australian Commission on Safety and Quality in Healthcare.16
Control dressing
The standard dressing for a surgical wound consists of a non-adherent film dressing applied directly over the incision to cover the incision site (3M Tegaderm film dressing). The control dressing does not use negative pressure over the incision site. The standard wear time of the control dressing is up to, and including, 7 days. Details of the dressing and wear time will be recorded in the case report forms (CRFs).
Intervention
The NPWT system (Avelle; ConvaTec), consists of a non-adherent dressing pad with an adhesive cover that adheres to the surrounding skin near the incision site. The dressing pad is attached to a small silent pump via a soft tube that creates a partial vacuum over the wound. The pump delivers a continual negative pressure of 80 mm Hg and is a battery-operated device. The NPWT pump can operate for up to 30 days, and the dressing can be worn for 7 days as per the manufacturer’s recommendation. In this trial, the participants randomised to the intervention group will receive the portable negative pressure device on day 0. The dressing will be worn for a standardised period of 5 days (intervention and controls arms). Any further wound dressings after the initial dressing application will be recorded in the CRFs, and following the allocated treatment, unless otherwise clinically indicated.
Outcome measures
Primary outcome
The primary outcome for this study is SWD as defined by the World Union of Wound Healing Societies SWD grading system,17 and the Centres for Disease Control definitions of SSI18 will be used as the primary outcome measure for confirmed wound infection. The primary outcome measure definitions include a wound complication that occurs within 30 days of surgery. The treating clinical team will determine the diagnosis of SWD or infection as per routine clinical wound assessment protocol if there is a confirmed SSI or SWD. Rapid diagnosis and treatment of wound infection is central to patient’s standard care and the attending clinician will document any changes in the patient’s medical notes and adhere to local wound management protocols.
Secondary outcomes
Health-related quality of life assessment
A qualitative assessment of the participant’s perceptions of wound healing will be conducted.
EuroQol EQ-5D-5L
The EuroQol EQ-5D-5L is a validated measure of health-related quality of life, comprising a five-dimension health status classification system and a separate visual analogue scale.19 The EQ-5D-5L consists of five dimensions (mobility, self-care, usual activities, pain/discomfort and anxiety/depression), each with either three or five levels. We will use the five-level version of the instrument, which is likely to be more sensitive to small but important changes in health-related quality of life.14 The responses will be converted into an index score, and total QALYs for each woman will be obtained by estimating the area under the curve defined by their baseline and final EQ-5D-5L responses.20
Economic analysis
All resources used in the study will be recorded to help inform the economic analysis. Cost data will be derived from the hospital finance departments and any related community nursing service or primary healthcare centre where the participant was attended. Cost consequences following discharge, including out-of-pocket expenses (if any), will be recorded in the CRFs at day 30 following the procedure. The incremental cost of the intervention relative to the control will be estimated, and divided by incremental outcomes reported in the study. Each resultant incremental cost-effectiveness ratio will be reported separately, with the last step being the cost per QALY. We will conduct univariate and probabilistic sensitivity analyses around the cost per QALY to assess the robustness of the result, with the threshold for cost-effectiveness determined by recent work by Edney et al.21
Adverse event management
Adverse device effect is an adverse event (AE) related to the use of an investigational medical device (IMD).22 AEs related to an IMD are defined as any untoward medical occurrence, unintended disease or injury, or untoward clinical signs (including abnormal laboratory findings) in participants, users or other persons, whether or not related to the IMD.22 Definitions of AEs, serious adverse device effect (SADE), serious AE, significant safety issue, unanticipated SADE (USADE) or urgent safety measure are as per the National Health and Medical Research Council (2016) Guidance on Safety monitoring and reporting in clinical trials involving therapeutic goods.22 During the treatment protocol, any USADE will be reported directly to the data safety monitoring board (DSMB) and within 7 days to the Australian Government’s Therapeutic Goods Administration via the electronic medical device incident reporting system. Reports will also be sent to the human research ethics committees of the local study sites. The safety aspects of the study will be closely monitored by the DSMB, which will receive unblinded data for review. In the case of a device-related AE, the manufacturer will be notified.
Follow-up
Each participant will be followed up during the trial as close as practically possible to the specific time points: days 5, 14 and 30 following surgery. The close-out time point of the trial participant is day 30 postoperatively. In the event that a participant has an unresolved complication beyond 30 days, follow-up will continue until complete wound healing, and participants will have the opportunity to opt out of the extended data collection beyond day 30. All participants will be followed up by the visiting midwifery service and a scripted phone call at the trial close-out time point. Various forms of communication will be used in engaging the participation, email, phone call and face-to-face consultation, to reduce loss to follow-up. All data recorded during the follow-up time points will be recorded on the CRFs and clinical assessment will follow standard postoperative wound care management and clinical procedures.
Sample size
Participants will be randomised in a 1:1 ratio of trial intervention to control. The sample size calculation is based on the following estimates: a complication rate of 20% in caesarean section patients was observed following a retrospective medical note audit at the major tertiary women’s hospital in Perth. If the intervention can reduce this figure to 10% (a reduction of 50% from the current figure), then a sample of size n=199 in each arm would be required to detect this difference with power=80% and α=0.05. To allow for an attrition rate of 11%, we plan to recruit 224 patients to each arm of the study. The total sample size for the study is 448 participants. Loss to follow-up and non-adherence will be reviewed as the trial progresses and numbers will be revised as required.
Data management
The CRFs have been designed by the senior research fellow in consultation with the trial co-investigators. All hard and electronic-based patient identifiable information will be stored on a secure password-protected database purpose built for the trial. All CRFs will be stored in a locked filing cabinet in a locked office at the participating site. Participants will be identified by a code number only on the database, but a file linking the code number to the participant name and contact details will be kept separately and securely. This will allow re-identification of the patient for follow-up purposes. Direct access to source data and/or documents may be granted for trial-related monitoring/audit by the regulatory authorities on written request only. All paper and electronic data will be retained for 5 years after completion of the trial.
Statistical analysis
Simple descriptive statistics (means, SDs, medians, IQR for continuous variables and frequencies, and percentages for categorical variables) will be used to summarise the profile of the study participants. Comparison of participants in the two groups (control vs intervention) will be performed using the χ2 test or t-test as appropriate. These tests will be used to identify any differences in baseline characteristics between groups. Recruitment and retention rates will be reported as per the CONSORT11 statement. Reasons for ineligibility, protocol deviation or participant withdrawal will be stated, and any trends reported. A generalised estimating equation (GEE) will be used to analyse each of the primary outcomes over all the time points of the study. This analysis is similar to a logistic regression for each of the binary outcome variables, but takes into account the correlation between the repeated measurements made on the same participant. The results of the GEE will be expressed as ORs, their 95% CIs and p values. The GEE model will include a term for the time point, so that changes over time can be assessed, as well as a term for the treatment group allocation (on which the main conclusions of the study will be based). In addition, the GEE model will be extended to include anthropometric measurements (eg, BMI), presence of health conditions (eg, diabetes, hypertension), the recruitment site and other variables collected at baseline. In this way, variables which are identified as being associated with the outcomes may be used to form a ‘risk score’ for each outcome. Analysis of the pain scores (which are measured on a continuous scale at each time point through the study) will be performed using a mixed regression model where the random effect will be the patient identifier, and the time point and treatment allocation group will be fixed effects. The distribution of the pain scores will be assessed for normality and transformed to improve normality (if necessary) prior to analysis.
All statistical analyses will be performed using the SAS V.9.4 software and, following convention, a p value <0.05 will be taken to indicate a significant association in all tests.
Trial oversight
A trial committee (TC) and DSMB will be set up. The DSMB advocates for the ethical and safety interests of the participants while the trial progresses by making non-binding recommendations to the TC. A DSMB will be formed to monitor the study at interim periods: first third participants closed out (n=148) and last quarter closed out. The DSMB consists of three independent reviewers; a statistician, a surgeon and a nurse. The DSMB will be bound by the DSMB terms of reference and will provide a written report to the TC. The TC consists of the principal and site investigators, the study biostatistician and health economist. This trial will use the Haybittle-Peto23 24 boundary as the designated trial statistic for stopping the trial.
Ethics and dissemination
Ethics approval was obtained through St John of God Health Care (HREC1409), Western Australia Department of Health King Edward Memorial Hospital (HREC3111). Study findings will be published in peer-reviewed journals and presented at local and international conferences. We used the SPIRIT checklist when writing our study protocol.
Discussion on strengths and limitations of the CYGNUS trial
The CYGNUS trial is a designed as a multicentre randomised control trial that is powered to determine treatment effectiveness. This robust study design has been engaged to ensure that any differences between the two arms of the study are attributable to the intervention. Hyldig et al 2019 have yielded positive findings with the application of NPWT in the form of potential reduction in the occurrence of surgical wound complications in postpartum mothers who have a BMI of 35+.10 The CYGNUS trial will contribute further research to this particular issue, and is the first to use a prescreening risk assessment tool designed to identify patients at risk with multiple risk factors. Another strength of this study is a within trial health economic evaluation comparing the NPWT to standard care from multiple healthcare perspectives. This will include the acute, community and primary healthcare setting. In light of the increased use of NPWT in patients with high BMI of 35+, there remains a considerable gap in the evidence base for clinical or cost-effectiveness.
This study will challenge the current rationale for initiating NPWT based on a single risk factor (BMI of 35+), by using a validated risk assessment tool with multiple predictors, which is more reflective of a real world setting.
Due to the nature of the intervention, blinding of participants and providers is not possible. However, statistical analysis will be blinded.
The exclusion of emergency cases may result in sample bias and exclude an already ‘at-risk’ cohort.
Participants will be followed up via face-to-face meetings or telephone call to ensure participant well-being and data capture. This may potentially halt any loss to follow-up.
Supplementary Material
Footnotes
Contributors: KSH and RP designed the study. KSH wrote the background and methodology, and developed the research question with input from RP and SWW. RP wrote the statistical design section and RN wrote the health economic sections of the protocol. Reviews were conducted by MWF and FMW. All authors reviewed and agreed with the final manuscript.
Funding: This work is funded by an unrestricted educational grant from ConvaTec. Grant ID UWA52001100.
Competing interests: None declared.
Patient and public involvement: Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer-reviewed.
References
- 1.AIHW Mothers and babies. 'Australia’s mothers and babies 2013—in brief. Perinatal statistics series no. 31'. Canberra: AIHW, 2017. [Google Scholar]
- 2.Wilson J, Wloch C, Saei A, et al. Inter-Hospital comparison of rates of surgical site infection following caesarean section delivery: evaluation of a multicentre surveillance study. J Hosp Infect 2013;84:44–51. 10.1016/j.jhin.2013.01.009 [DOI] [PubMed] [Google Scholar]
- 3.Wloch C, Wilson J, Lamagni T, et al. Risk factors for surgical site infection following caesarean section in England: results from a multicentre cohort study. BJOG 2012;119:1324–33. 10.1111/j.1471-0528.2012.03452.x [DOI] [PubMed] [Google Scholar]
- 4.Ward VP, Charlett A, Fagan J, et al. Enhanced surgical site infection surveillance following caesarean section: experience of a multicentre collaborative post-discharge system. J Hosp Infect 2008;70:166–73. 10.1016/j.jhin.2008.06.002 [DOI] [PubMed] [Google Scholar]
- 5.Orth TA, Gerkovich MM, Heitmann E, et al. Cesarean delivery with external negative pressure dressing system: a retrospective cohort study. Surg J 2016;2:e59–65. 10.1055/s-0036-1585470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smid MC, Dotters-Katz SK, Grace M, et al. Prophylactic negative pressure wound therapy for obese women after cesarean delivery: a systematic review and meta-analysis. Obstet Gynecol 2017;130:969–78. 10.1097/AOG.0000000000002259 [DOI] [PubMed] [Google Scholar]
- 7.Shree R, Park SY, Beigi RH, et al. Surgical site infection following cesarean delivery: patient, provider, and Procedure-Specific risk factors. Am J Perinatol 2016;33:157–64. 10.1055/s-0035-1563548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneid-Kofman N, Sheiner E, Levy A, et al. Risk factors for wound infection following cesarean deliveries. Int J Gynaecol Obstet 2005;90:10–15. 10.1016/j.ijgo.2005.03.020 [DOI] [PubMed] [Google Scholar]
- 9.Krieger Y, Walfisch A, Sheiner E. Surgical site infection following cesarean deliveries: trends and risk factors. J Matern Fetal Neonatal Med 2017;30:8–12. 10.3109/14767058.2016.1163540 [DOI] [PubMed] [Google Scholar]
- 10.Hyldig N, Vinter CA, Kruse M, et al. Prophylactic incisional negative pressure wound therapy reduces the risk of surgical site infection after caesarean section in obese women: a pragmatic randomised clinical trial. BJOG 2019;126:628–35. 10.1111/1471-0528.15413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulz KF, Altman DG, Moher D, et al. Consort 2010 statement: updated guidelines for reporting parallel group randomized trials. Ann Intern Med 2010;152:726–32. 10.7326/0003-4819-152-11-201006010-00232 [DOI] [PubMed] [Google Scholar]
- 12.Chan A-W, Tetzlaff JM, Altman DG, et al. Spirit 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med 2013;158:200–7. 10.7326/0003-4819-158-3-201302050-00583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandy-Hodgetts K, Carville K, Santamaria N, et al. The Perth surgical wound dehiscence risk assessment tool (PSWDRAT): development and prospective validation in the clinical setting. J Wound Care 2019;28:332–44. 10.12968/jowc.2019.28.6.332 [DOI] [PubMed] [Google Scholar]
- 14.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727–36. 10.1007/s11136-011-9903-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.WHO Implementation manual surgical safety checklist. 1st ed Geneva, Switzerland: World Health Organisation, 2008. [Google Scholar]
- 16.ACSQH National safety and quality health service standards. 2nd ed Sydney: ACSQHC, 2017. [Google Scholar]
- 17.Societies WUoWH Consensus Document. Surgical wound dehiscence: improving prevention and outcomes Wounds International - a division of Omnia-med Ltd, 2018. [Google Scholar]
- 18.Horan TC, Gaynes RP, Martone WJ, et al. Cdc definitions of nosocomial surgical site infections, 1992: a modification of CDC definitions of surgical wound infections. Infect Control Hosp Epidemiol 1992;13:606–8. 10.2307/30148464 [DOI] [PubMed] [Google Scholar]
- 19.Rabin R, de Charro F. EQ-5D: a measure of health status from the EuroQol group. Ann Med 2001;33:337–43. 10.3109/07853890109002087 [DOI] [PubMed] [Google Scholar]
- 20.Norman R, Cronin P, Viney R. A pilot discrete choice experiment to explore preferences for EQ-5D-5L health states. Appl Health Econ Health Policy 2013;11:287–98. 10.1007/s40258-013-0035-z [DOI] [PubMed] [Google Scholar]
- 21.Edney LC, Haji Ali Afzali H, Cheng TC, Karnon J, Tc C, et al. Estimating the reference incremental cost-effectiveness ratio for the Australian health system. Pharmacoeconomics 2018;36:239–52. 10.1007/s40273-017-0585-2 [DOI] [PubMed] [Google Scholar]
- 22.NHMRC Guidance: safety monitoring and reporting in clinical trials involving therapuetic goods. Canberra: National Health and Medical Research Council, 2016. [Google Scholar]
- 23.Haybittle JL. Repeated assessment of results in clinical trials of cancer treatment. Br J Radiol 1971;44:793–7. 10.1259/0007-1285-44-526-793 [DOI] [PubMed] [Google Scholar]
- 24.Peto R, Pike MC, Armitage P, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. I. Introduction and design. Br J Cancer 1976;34:585–612. 10.1038/bjc.1976.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.