

Abstract
Background -
Arrhythmia syndromes associated with KCNJ2 mutations have been described clinically, however little is known of the underling arrhythmia mechanism. We create the first patient inspired KCNJ2 transgenic mouse and study effects of this mutation on cardiac function, IK1, and Ca2+ handling, to determine the underlying cellular arrhythmic pathogenesis.
Methods -
A cardiac-specific KCNJ2-R67Q mouse was generated and bred for heterozygosity (R67Q+/−). Echocardiography was performed at rest, under anesthesia. In vivo electrocardiogram (ECG) recording and whole heart optical mapping of intact hearts was performed before and after adrenergic stimulation in wild-type (WT) littermate controls and R67Q+/− mice. IK1 measurements, action potential (AP) characterization, and intracellular Ca2+ imaging from isolated ventricular myocytes at baseline and after adrenergic stimulation were performed in WT and R67Q+/− mice.
Results -
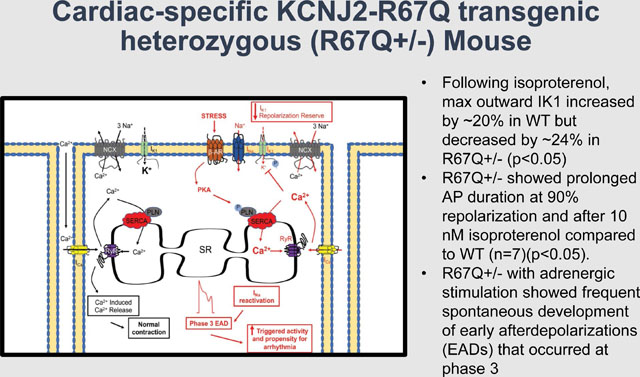
R67Q+/− mice (n=17) showed normal cardiac function, structure, and baseline electrical activity compared to WT (n=10). Following epinephrine and caffeine, only the R67Q+/− mice had bidirectional ventricular tachycardia (BiVT), ventricular tachycardia, frequent ventricular ectopy and/or bigeminy and optical mapping demonstrated high prevalence of spontaneous and sustained ventricular arrhythmia. Both R67Q+/− (n=8) and WT myocytes (n=9) demonstrated typical n-shaped IK1 IV relationship; however, following isoproterenol, max outward IK1 increased by ~20% in WT but decreased by ~24% in R67Q+/− (p<0.01). R67Q+/− myocytes (n=5) demonstrated prolonged AP duration at 90% repolarization and after 10 nM isoproterenol compared to WT (n=7)(p<0.05). Ca2+ transient amplitude, 50% decay rate and SR Ca2+ content were not different between WT (n=18) and R67Q+/− (n=16) myocytes. R67Q+/− myocytes (n=10) under adrenergic stimulation showed frequent spontaneous development of early afterdepolarizations (EADs) that occurred at phase 3 of AP repolarization.
Conclusions -
KCNJ2 mutation R67Q+/− causes adrenergic-dependent loss of IK1 during terminal repolarization and vulnerability to phase 3 EADs. This model clarifies a heretofore unknown arrhythmia mechanism and extends our understanding of treatment implications for patients with KCNJ2 mutation.
Keywords: potassium channels, catecholaminergic polymorphic ventricular tachycardia, ventricular tachycardia, ventricular arrhythmia, Kir2.1, KCNJ2
Journal Subject Terms: Arrhythmias, Animal Models of Human Disease, Basic Science Research, Ion Channels/Membrane Transport, Translational Studies
Graphical Abstract

Introduction
Human cardiac myocyte electrical stability depends on normal polarization of the resting potential achieved by the inward-rectifier K+ current (IK1). KCNJ2 encodes for the pore-forming subunit Kir2.1 and creates the dominant component for IK1 that functions to maintain the resting membrane potential, completes phase 3 repolarization, and indirectly determines cellular excitability1,2. Four different arrhythmia syndromes based on clinical phenotype have been associated with KCNJ2 mutations: Anderson-Tawil Syndrome (ATS1)3, Short QT Syndrome 3 (SQT3)4, Familial Atrial Fibrillation (FAF)5, and Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)6. The cellular correlate to Kir2.1 dysfunction for ATS1 is a “loss of function”, while SQT3 and FAF are thought to be due to a “gain of function”5,7. Moreover, some KCNJ2 mutations have been clinically described in patient cohorts presenting with a CPVT-like phenotype yet the cellular correlates have not been clarified. Unraveling the arrhythmia mechanism related to KCNJ2 mutations has been hindered by the lack of transgenic in vivo models. Prior investigation of KCNJ2 mutation syndromes has been limited to heterologous expression of mutations or employed the use of Kir2.1 blockers to mimic the cellular effects8,9. Such approaches underestimate more complex channel regulation and important subcellular interactions may be misunderstood by reducing the mutation effect to a “gain” or “loss” of function. For example, we found that certain KCNJ2 mutations can influence the rectification index with a preferential effect of loss of outward (physiologic) IK110.
We previously reported a patient who harbors KCNJ2 mutation, R67Q, and presented with exertion-induced polymorphic ventricular tachycardia (PMVT), bidirectional ventricular tachycardia (BiVT) and syncope11, a similar phenotype to CPVT12. Arrhythmia in CPVT is related to altered intracellular calcium [Ca2+]i handling leading to a propensity to develop delayed afterdepolarizations (DADs) and genetically linked to mutations in the cardiac ryanodine receptor (RYR2)13, and calsequestrin-2 (CASQ2)14. Additionally, patients presenting with a CPVT-like phenotype have been noted in association with mutations in triadin (TRDN), ankyrin-B (ANK2), calmodulin (CALM1)15 and Kir2.1 (KCNJ2)6, 16–18. It remains unclear if CPVT-mimicking phenotypes share the same arrhythmia mechanism as RYR2 or CASQ2 mutations19,20. In a pharmacologic induced loss of IK1 in a heart failure rabbit model, loss of Kir2.1 function has been linked to Ca2+ overload arrhythmia related to Vm/Cai imbalance causing DADs21. In contrast to this in a drug induced LQT model, loss of IK1 was shown to underlie the mechanism for phase 3 early afterdepolarizations (EADs)22. These studies highlight that the clinical presentation may not mirror the arrhythmia mechanism; discerning these subtleties is important for effective arrhythmia suppression as the treatment approach toward DAD vs. EAD suppression is divergent. Unfortunately, the current strategy of extrapolating from diseases with a similar phenotype may result in inaccurate treatment and exposing patients to an increased risk of SCD23.
It is with this premise that we undertook the creation and characterization of the arrhythmia mechanism in a cardiac-specific knock-in (KI) in vivo model inspired by our patient presenting with a CPVT-like phenotype and a KCNJ2 mutation11. Together, these data not only link the clinical arrhythmia presentation with the KCNJ2 mutation R67Q but may help inform clinical treatment approaches.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files. Mouselines are available from The Jackson Laboratory Model Repository.
Human subjects and Animal use assurances
The University of Wisconsin Institutional Review Board approved this research in accordance with the National Institutes of Health guidelines for human research. All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH) and approved by the University of Wisconsin-Madison Institutional Animal Care and Use Committee.
Cardiac-specific knock-in mice
Cardiac specific KI mice were generated in collaboration with University of Wisconsin-Madison Biotechnology Center using the Cre-loxP system24. Due to the extra-cardiac effects of Kir2.1 in development25, a cardiac specific KI was designed to avoid effects of mal-development on the whole animal and to focus on the patient’s arrhythmic phenotype which occurs in absence of extra-cardiac effects. For gene targeting, HindIII-linearized PL253 targeting vector DNA containing R67Q-KCNJ2 was electroporated into embryonic stem (ES) cells26. The target vector contained R67Q-KCNJ2 with a neocassette inserted into the primary sequence, flanked with loxP sites. This results in silencing of R67Q-KCNJ2 in tissues that do not express Cre recombinase. To generate the cardiac-specific knock-in, resultant targeted mice were crossed with mice expressing Cre recombinase under the control of the myosin heavy chain 6 promoter (MYHC6) to excise the neocassette (B6.FVB-Tg(Myh6-cre)2182Mds/J, Jax stock: #011038(4)). ES cells were plated in 100-mm dishes and were cultured for 48 hours. Positive and negative selections were performed using Geneticin (G418) and neomycin respectively. Positive clones were isolated and confirmed by Southern blot using an external probe to the target sequence and microinjected into the pronucleus of C57BL/6J one-cell mouse embryos. Following microinjections, embryos were transferred into pseudo-pregnant recipients and pups born (University of Wisconsin-Madison Biotechnology Center). Chimeric mice were bred to C57BL/6 mice to establish a hybrid line for 10 generations. Germ-line transmission has generated KCNJ2+/KCNJ2R67Q-neo mice with genetic background 129SV/J from embryonic stem cells and C57BL/6 from blastocysts. Genotypes from F1 and F2 generations were determined by PCR on DNA from ear and tail biopsy specimens. Cardiac-specific transgenic mice were generated by crossing KCNJ2+/KCNJ2R67Q-neo mice with mice obtained from The Jackson Laboratory expressing Cre cDNA under the control of the mouse α-MHC promoter (Tg(Myh6-cre)1Jmk/J, JAX stock #009074). The genotypes from the F1 generation without the neocassette were determined by PCR on DNA from ear, tail biopsy as well as cardiac specimens to ensure the neocassette had been excised. Age-matched male and female littermates (8–12 weeks) wild-type (Cre+/R67Q−/− mice are designated WT for the purposes of this study) and heterozygous R67Q (Cre+/R67Q+/−) were used in whole animal physiological studies (N=8), biochemistry (N=4), whole-cell patch clamp electrophysiology (IK1; N=5, n=9 WT, N=5, n=8 for R67Q+/−; Action potentials; WT – N=4, n=7; WT + ISO – N=4, n=6; R67Q+/− and R67Q+/− + ISO - N=3, n=5) and calcium transient studies (Line scan calcium transients - WT – N4, n=18; R67Q+/− - N=4, n=16; Ratiometric calcium transients – WT – N=6, n=7; R67Q+/− - N=4, n=10).
Echocardiography
Transthoracic echocardiography was performed on 8–12-week-old age mice under 1% isoflurane gas anesthesia using a Visual Sonics 770 ultrasonograph with a 30-MHz transducer (RMV 707B) (Visual Sonics, Toronto)27. Two-dimensionally guided M-mode images were acquired in the long axis. LV mass-to-body weight ratio (LV/BW), LV volume during diastole and systole, heart rate, LV dimension in diastole (LVIDd), thickness of the posterior walls in diastole were recorded. Parameters were measured over at least three consecutive cycles. Following measurements, mice were sacrificed, and hearts snap frozen for biochemical analyses. LV mass was calculated using the following equation; LV mass = [1.05 × ((Posterior Walldiastole+Anterior Walldiastole+LV diameterdiastole)3 – (LV diameterdiastole)3)].
Electrocardiography
Adult (8–12 weeks) WT and R67Q+/− mice were anesthetized by 2% isoflurane inhalation and maintained at 1–2% for the duration of the measurements. Mice were placed on the monitor (Indus Rodent Surgical Monitor, Indus Instruments, Portland, OR) in the dorsal position and baseline ECGs (Notocord) recorded for 5 minutes. A single induction for arrhythmia initiation was performed with an I.P. injection of caffeine (120mg/kg) and epinephrine (2mg/kg). ECG examples were taken 10 minutes following injection. Following cessation of ECG recordings, mice were euthanized, and hearts excised for cardiac genotyping28. ECGs were analyzed blinded to animal genotype in accordance with standard protocols. Arrhythmic events were calculated as follows; ECGs were examined for each animal following injection of caffeine and epinephrine and recorded if arrhythmia phenomenon were observed. The number of mice with arrhythmic events for each group was divided by the total number of mice investigated to give an overall percentage of arrhythmic events for each group.
Isolated right ventricular myocytes
Calcium tolerant myocytes were isolated from the right ventricular (RV) free wall of adult WT and R67Q+/− male and female mice of 8–12 weeks in age by perfusion of collagenase II (Worthington Labs) or Liberase™ (Roche) in the Langendorff model29. Mice were anesthetized prior with 5% isoflurane prior to cardiac excision. Anesthesia was confirmed by absence of response to stimuli (firm toe pinch). Due to know regional variation in IK1 within the left ventricular chamber including transmural differences, the RV free wall was selected due to the homogeneity of IK1 known to occur in the chamber to create a robust and reproducible dataset. These myocytes were used for cellular electrophysiology and for calcium transient measurements.
Cellular electrophysiology
Electrophysiology experiments were performed using an Axopatch 200B amplifier and pCLAMP10 (Molecular Devices, Sunnyvale, CA). All electrophysiology recordings were performed blinded to the animal genotype.
Voltage clamp data was recorded at room temperature from isolated calcium tolerant adult myocytes from WT and R67Q+/− mice using whole cell technique30. Borosilicate glass pipettes were to pulled to resistances of 2–3 MΩ when filled (Model P-97, Sutter Instruments, Novato, CA). Bath solution for IK1 measurements contained (mM): NaCl 148, KCl 5.4, CaCl2 1.8, MgCl2 1, HEPES 15, NaHPO4 0.4, D-glucose 5.5 and pH adjusted to 7.4 (NaOH). Calcium currents and calcium-sensitive chloride currents were blocked with nifedipine (10μM) in the bath solution. Pipette filling solution contained (mM): K-gluconate 150, EGTA 5, MgATP 5, HEPES 10 and pH adjusted to 7.2 (KOH). IK1 was recorded from a holding potential of −50 mV sequential 10 mV steps from −120 mV to +50 mV in 100 ms steps. This was repeated in the presence of isoproterenol (10 nM). Finally, cells were perfused with bath solution containing 0.5mM barium chloride and I-V protocol repeated (Supplemental Figure 3). This was then subtracted from previously recorded I-V protocols in addition to measuring I-V relationships at 500 ms of protocol to eliminate possible contaminating currents. Cell stability was ensured by recording using the aforementioned protocol twice at baseline, twice following ISO and twice following barium to ensure consistency. If the cell is unstable or a seal of 250MΩ is not achieved, then the cell is discarded and another selected and the process repeated. Rectification index was calculated as follows: This index was defined as the ratio of the outward current at −50 mV divided by the absolute value of the inward current at −100 mV and then multiplied by 10011 11.
Action potentials (AP) were recorded under current clamp mode at 34 ± 2°C. Myocytes were paced 2–6 Hz with a brief depolarizing square pulse using an analog programmable stimulator (Multichannel Systems, Reutlingen, Germany). AP amplitude and upstroke velocity (dV/dtmax), resting membrane potential (RMP) and AP duration at 10% (APD10), 50% (APD50), 70% (APD70), and 90% (APD90) of repolarization were analyzed (pCLAMP 10). Each myocyte was paced for >100 beats before analysis. The terminal five consecutive APs at each frequency (2–6 Hz) were used for analysis. APs were measured at baseline and following incubation with isoproterenol (10 nM). Bath solution for AP was the same as for IK1 minus nifedipine and pipette solution contained (mM): KCl 150, NaCl 5, CaCl2 2, EGTA 5, MgATP 5, HEPES 10 and pH adjusted to 7.2 (KOH).
Optical Mapping
Male and female mice age 8–12 weeks were anesthetized using Isoflurane. Heparin (100 units) was used to treat and prevent blood clots in the veins and arteries. After a midsternal incision, the heart was removed, cannulated and washed in oxygenated (95% O2-5% CO2) constant-temperature (37 ± 1°C) modified Tyrode solution [in mM: 128.2 NaCl, 4.7 KCl, 1.19 NaH2PO4, 1.05 MgCl2, 1.8 CaCl2, 20.0 NaHCO3, and 11.1 glucose (pH 7.35 ± 0.05)]. Perfusion was performed using a peristaltic pump (Peri-Star, WPI, Sarasota, FL) under constant aortic pressure of 60–80 mmHg as measured by a pressure amplifier. Coronary perfused hearts were stained by perfusion with voltage-sensitive dye [RH-237, Invitrogen (Carlsbad, CA), 5 μl of 1 mg/ml DMSO in Tyrode solution] for 5–7 min. The excitation-contraction uncoupler blebbistatin (10 μM, Tocris Bioscience) was used to prevent the effect of motion artifacts. The fluorescent signals emitted from the preparation was long pass (>700 nm) filtered using an edge pass filter (Thorlabs) before reaching the camera. Emitted signals were directed toward a MiCAM Ultima-L CMOS camera (SciMedia) with high spatial (100 × 100 pixels, 230 ± 20 μm/pixel) and temporal (500–1,000 frames/s) resolution. The acquired fluorescent signal was digitized, amplified, and visualized by custom software (SciMedia).
Confocal Ca2+ Imaging
Following Ca2+ re-introduction, myocytes were incubated in 10 μM fluo-4 AM (ThermoFisher) and 0.4% Pluronic F-127 (ThermoFisher) for 5 min at 37 °C. Then, they were plated on glass-bottom dishes (MatTek) coated with laminin (ThermoFisher) in normal Tyrode’s solution, containing (mM) 135 NaCl, 4 KCl, 1 MgCl2 10 HEPES, 1.2 NaH2PO4, 10 Glucose and 1.8 CaCl2, pH 7.40 with NaOH. Confocal line-scans were collected with a Zeiss LSM800 microscope using a 40x/1.2 water-immersion objective, at 488 nm excitation and >505 nm emission wavelengths. A 512-pixel line was drawn across the long axis of the cell and unidirectional line-scans were obtained at 2.47 ms/line. Ca2+ transients were elicited through field stimulation using a Grass stimulator (2ms pulses, 60–70 V) controlled with a model 3800 Stimulator (A-M Systems). The protocol consisted of 5 s of rest, 30 s at 1 Hz, 15 s at 2 Hz and 15 s at 3 Hz, followed by a pulse of 10 mM caffeine to measure SR content. Recordings were made in basal conditions and in the presence of 300 nM (−)-isoproterenol. Images were analyzed using a custom-made Matlab 2019b script to calculate the transient amplitude, 50% decay time and maximum Ca2+ release velocity (dF/dtMAX). F0 was determined as the basal fluorescence during the resting period.
Ratiometric calcium transients
Calcium tolerant right ventricular myocytes were loaded with fura2-AM ratiometric dye (Sigma, St Louis, MO, USA) at room temperature. Calcium transients were recorded at 34 ± 2°C using IonOptix Calcium and Contractility System equipped with a Hyperswitch and MyoCam-S (IonOptix, Westwood, MA, USA). Myocytes were paced 2–6 Hz with field pacing using Myopacer (IonOptix, Westwood, MA, USA). Bath solution contained (mM): NaCl 148, KCl 5.4, CaCl2 1.8, MgCl2 1, HEPES 15, NaHPO4 0.4, D-glucose 5.5 and pH adjusted to 7.4 (NaOH). Diastolic calcium ratio was analyzed (IonWizard, IonOptix; Origin 6). Each myocyte was paced for 1 minute before recording 10 consecutive calcium transients for analysis. Cells from WT and R67Q+/− mice were used and calcium transients measured at baseline and following incubation with isoproterenol (10 nM). In a separate set of experiments, calcium tolerant right ventricular myocytes were paced for 30 s at 2 Hz then treated with caffeine (10 mM) and sarcoplasmic reticulum (SR) calcium content and Na+/Ca2+-exchanger (NCX) activity were analyzed.
Spontaneous Event Quantification
Occurrence of triggered activity of both early after (EADs) and delayed (DADs) afterdepolarizations observed during action potential recordings were quantified using event detection in pClamp 10. Cells were paced at each frequency for 1 minute and each 1-minute period was analyzed, per frequency per cell during pacing for quantification of EADs. Rejection threshold was set at 20 mV for EAD detection, as EADs rarely passed this threshold, thus excluding action potentials that did pass 20 mV from being included in analyses. DAD quantification occurred in the 1-minute following cessation of pacing per frequency per cell and were observed as membrane oscillations, and oscillations that reached threshold elicited an action potential. Data was analyzed using pClamp 10 and Origin 6 and shown as mean events/cell.
Spontaneous calcium release events during ratiometric calcium transient measurements were quantified by manually counting increases in 340/380 ratio following cessation of pacing over a 1-minute recording time for each frequency for each cell. Data was analyzed using IonWizard and Origin 6 and shown as mean events/cell.
Western blot
Mouse hearts were homogenized under liquid nitrogen to a fine powder and lysed in lysis buffer containing (mM) Tris 50, NaCl 150, Triton X-100 1%, Sodium deoxycholate 0.5% supplemented with 2 mM phenylmethane sulfonyl fluoride (PMSF) and protease inhibitors. Lysates were incubated on ice followed by sonication and insoluble material removed by centrifugation. Approximately 100 μg of whole heart lysates were analyzed by SDS-PAGE and western blotting31. Membranes were incubated with anti-Kir2.1 (Santa Cruz, Dallas, TX, USA), anti-GAPDH (BD Bioscience, San Jose, CA, USA), anti-SERCA2A (ThermoFisher, MA, USA), anti-NCX1 (Swant, Switzerland) and anti-RyR2 (ThermoFisher, MA, USA) then incubated with HRP-conjugated secondary antibodies. Western blots were imaged using a GE Amersham Imager 600. Densitometry data were analyzed using ImageJ and Origin 6.
Hematoxylin and Eosin Histology
Age and sex matched mouse hearts of WT and R67Q+/− were excised and perfused with 4% paraformaldehyde. Hearts were cut in 4 chambered view and embedded in paraffin, then processed for histological analysis at the UW Department of Surgery Histology Core. 5 to 6 μm sections of the paraffin-embedded hearts were stained with Hematoxylin and Eosin (H&E). Briefly, dry paraffin slides were deparaffinized and rehydrated by incubating at 60°C for 20 mins followed by incubation in xylene and then ethanol. Sections were rinsed in water and stained with Harris Hematoxylin and Eosin sequentially, with sections washed with water and ethanol in between each stain. Tissue sections were then dehydrated by rinsing in ethanol and xylene before mounting with cytoseal using coverslips. Images were acquired using an EVOS cell imaging system (ThermoFisher Scientific, MA, USA) with bright field using a 10X objective. Area scanning and tile stitching feature was used to generate the images shown.
Statistics
All data are presented as mean ± SE. Statistical comparisons were carried out using Student’s unpaired t-test (see Table 1, Figures 3B, 3D), Two-way repeated measures ANOVA with post-hoc Bonferroni correction was used for electrophysiological and calcium transient analyses (see Table 2, Figures 3C, 4B, 5B–F, 6B), or non-linear regression analysis (see Figure 6C–D) using OriginLab (Northampton, MA, USA). Differences were significant at p<0.05. Repeated measures for Table 2 were treatment (column factor) and duration (row factor). For duration, p<0.0001 and for treatment, p<0.05. Multiple comparisons were carried out using Prism 8 by GraphPad, to allow comparison of different APDs and treatments within the same dataset. Bonferroni correction is applied to correct for multiple comparisons.
Table 1.
Electrocardiographic assessment of R67Q+/− following epinephrine and caffeine administration
| Parameter | WT Baseline | WT Post Epi and Caffeine | R67Q+/− Baseline | R67Q+/− Post Epi and Caffeine |
|---|---|---|---|---|
| RR (ms) | 122.5 ± 5.01 | 95.9 ± 4.4*** | 118.5 ± 3.7 | 95.9 ± 3.6** |
| PR (ms) | 37.7 ± 0.6 | 43.6 ± 1.8 | 38.9 ± 0.9 | 44.9 ± 2.7 |
| QRS (ms) | 15.6 ± 0.5 | 16.4 ± 0.3 | 15.7 ± 0.4 | 17.4 ± 0.8 |
| QT (ms) | 39.3 ± 1.02 | 39.0 ± 0.5 | 40.1 ± 0.7 | 39.6 ± 1.1 |
Values are means ± SE. Shown is the quantification of electrocardiography parameters in R67Q+/− and WT mice at 8–12 weeks of age, at baseline and following intraperitoneal injection of epinephrine and caffeine. ECGs were quantified 5 minutes’ post injection of adrenergic stimulus.
P < 0.05,
P < 0.01,
and
P < 0.001 compared with those at baseline.
N=8 per group. Students t-test was used to determine significance difference between groups.
Figure 3:
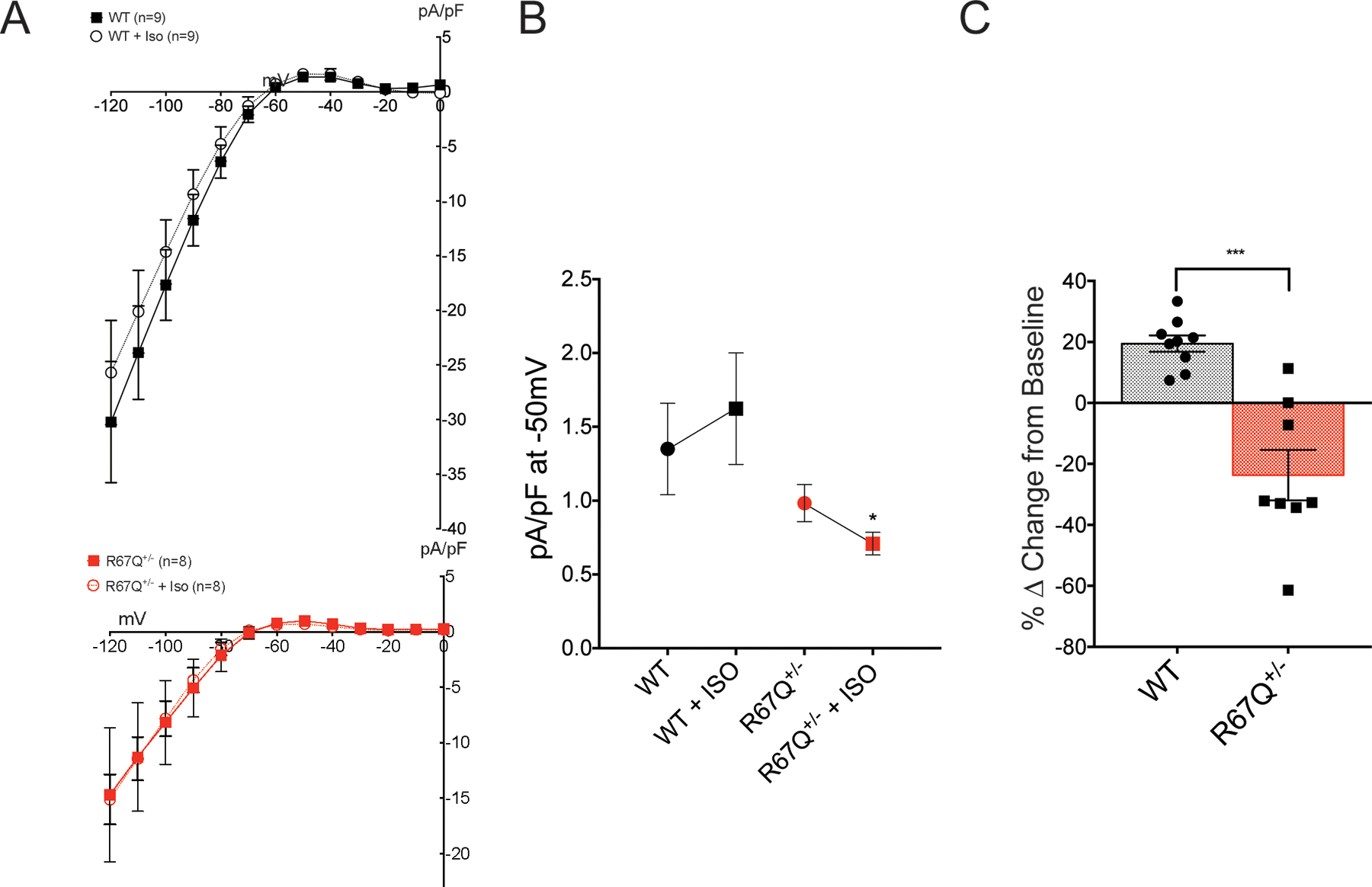
IK1 fails to increase following isoproterenol. A. Baseline current voltage relationship for WT (black squares, N=5, n=9), R67Q+/− (red squares, N=5, n=8). Following isoproterenol (ISO) perfusion for 5 minutes, WT outward current (black open circles, N=5, n=9) increased, however R67Q+/− outward current (red open circles, N=5, n=8) decreased in response to adrenergic stress. Currents shown are calculated following barium subtraction. Barium is perfused for 2 minutes following final ISO measurement. B. Absolute current values at −50mV following ISO treatment: R67Q+/− outward IK1 (0.71 ± 0.08 pA/pF) is significantly decreased compared to WT (1.62 ± 0.38 pA/pF) following isoproterenol treatment (p<0.05). C. Peak outward current was determined at −50 mV and the percentage change for each cell from baseline was calculated. The delta from baseline showed a 19.5 ± 2.7% increase in outward current at −50mV in WT cells (black, N=5, n=9), whereas R67Q+/− cells (red, N=5, n=8) showed a 23.7 ± 8.3% decrease in outward current at −50 mV. *** p<0.001. Students t-test was used to determine significance difference between groups (3B), or Two-way repeated measures ANOVA with post-hoc Bonferroni correction was used (3C).
Table 2:
Action potential characteristics in WT and R67Q+/− myocytes at baseline and following isoproterenol.
| Condition | RMP (mV) |
dV/dTmax (V/sec) |
APA (mV) |
APD10 (msec) |
APD50 (msec) |
APD70 (msec) |
APD90 (msec) |
|---|---|---|---|---|---|---|---|
| WT | |||||||
| 2Hz | −70.91 ± 0.71 | 232.40 ± 15.57 | 117.60 ± 1.88 | 0.37 ± 0.03 | 3.36 ± 0.58 | 7.76 ± 1.22 | 35.97 ± 3.40 |
| 4Hz | −71.27 ± 1.19 | 244.40 ± 6.13 | 115.40 ± 2.06 | 0.40 ± 0.03 | 3.44 ± 0.64 | 8.31 ± 1.35 | 38.88 ± 3.86 |
| 6Hz | −71.85 ± 1.55 | 245.00 ± 8.55 | 112.30 ± 2.10 | 0.42 ± 0.04 | 3.79 ± 0.72 | 9.65 ± 1.62 | 40.66 ± 3.44 |
| WT + ISO | |||||||
| 2Hz | −74.66 ± 0.75* | 256.10 ± 9.51 | 118.20 ± 1.43 | 0.40 ± 0.04 | 4.76 ± 0.77 | 8.94 ± 1.29 | 32.33 ± 2.13 |
| 4Hz | −76.51 ± 1.13* | 257.90 ± 9.03 | 118.20 ± 1.49 | 0.45 ± 0.04 | 5.81 ± 0.77 | 11.46 ± 1.53 | 39.63 ± 1.61 |
| 6Hz | −76.49 ± 1.41 | 256.10 ± 10.05 | 115.80 ± 1.51 | 0.45 ± 0.05 | 6.21 ± 0.89 | 13.42 ± 1.97 | 42.61 ± 3.28 |
| R67Q+/− | |||||||
| 2Hz | −71.10 ± 0.62 | 268.90 ± 6.54 | 120.90 ± 2.30 | 0.54 ± 0.13 | 7.65 ± 3.26 | 18.79 ± 7.50 | 54.34 ± 13.92* |
| 4Hz | −70.76 ± 0.93 | 268.90 ± 7.32 | 118.50 ± 2.20 | 0.55 ± 0.13 | 7.07 ± 2.61 | 17.87 ± 5.67 | 55.70 ± 10.02* |
| 6Hz | −70.10 ± 1.34 | 261.50 ± 8.16 | 114.70 ± 2.03 | 0.59 ± 0.16 | 6.69 ± 2.34 | 19.90 ± 5.19 | 67.76 ± 14.12*** |
| R67Q+/− + ISO | |||||||
| 2Hz | −73.90 ± 1.29 | 264.60 ± 8.31 | 118.20 ± 2.92 | 0.74 ± 0.30 | 9.94 ± 3.12 | 18.44 ± 5.10 | 59.10 ± 11.76** |
| 4Hz | −74.25 ± 1.13 | 264.00 ±10.59 | 115.90 ± 3.33 | 0.81 ± 0.32 | 10.81 ± 2.91 | 20.89 ± 5.01 | 62.81 ± 10.30*** |
| 6Hz | −72.97 ± 1.06 | 267.00 ± 11.35 | 110.50 ± 4.18 | 0.86 ± 0.33 | 11.15 ± 2.18 | 23.00 ± 3.26 | 64.38 ± 9.38*** |
Values are means ± SE. Resting membrane potential (RMP), dV/dTmax, action potential amplitude (APA) and action potential duration (APD) at 2, 4 and 6Hz at baseline and following isoproterenol.
P < 0.05,
P < 0.01,
and
P < 0.001 compared to WT.
(WT - N=4, n=7; WT + ISO – N=4, n=6; R67Q+/− +/− ISO - N=3, n=5). Two-way repeated measures (Treatment (+/− ISO) or duration of action potential) ANOVA with post-hoc Bonferroni correction was used. Significance levels indicated are following Bonferroni correction.
Figure 4:
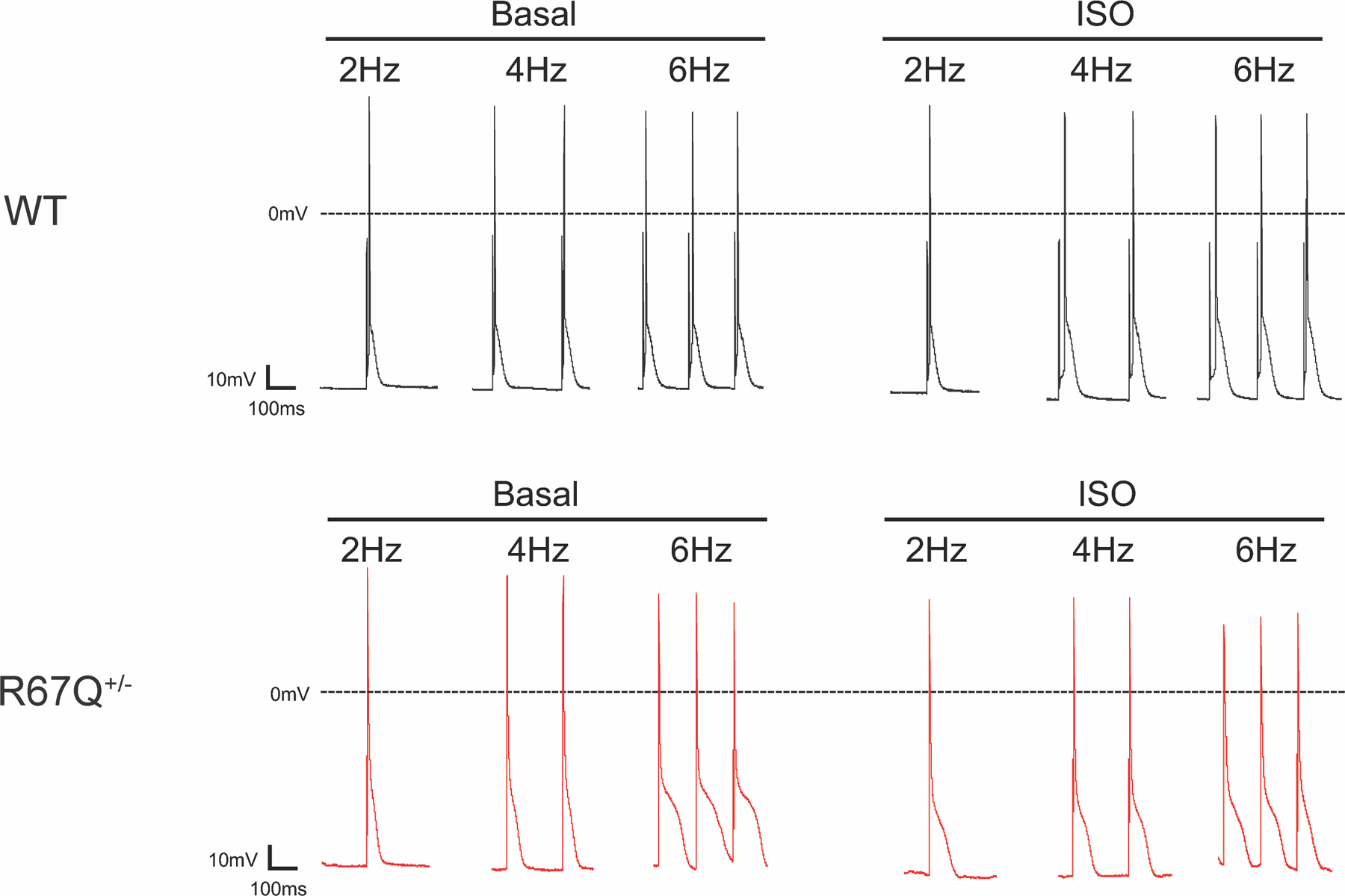
R67Q+/− myocytes have prolonged APD90 at baseline and following adrenergic stress. A. Representative action potentials from WT (left, upper), WT + isoproterenol (ISO) (right, upper), R67Q+/− (left, lower), and R67Q+/− + ISO (right, lower) at 2, 4 and 6Hz. Scale bar represents 100 ms and 10 mV.
Figure 5:
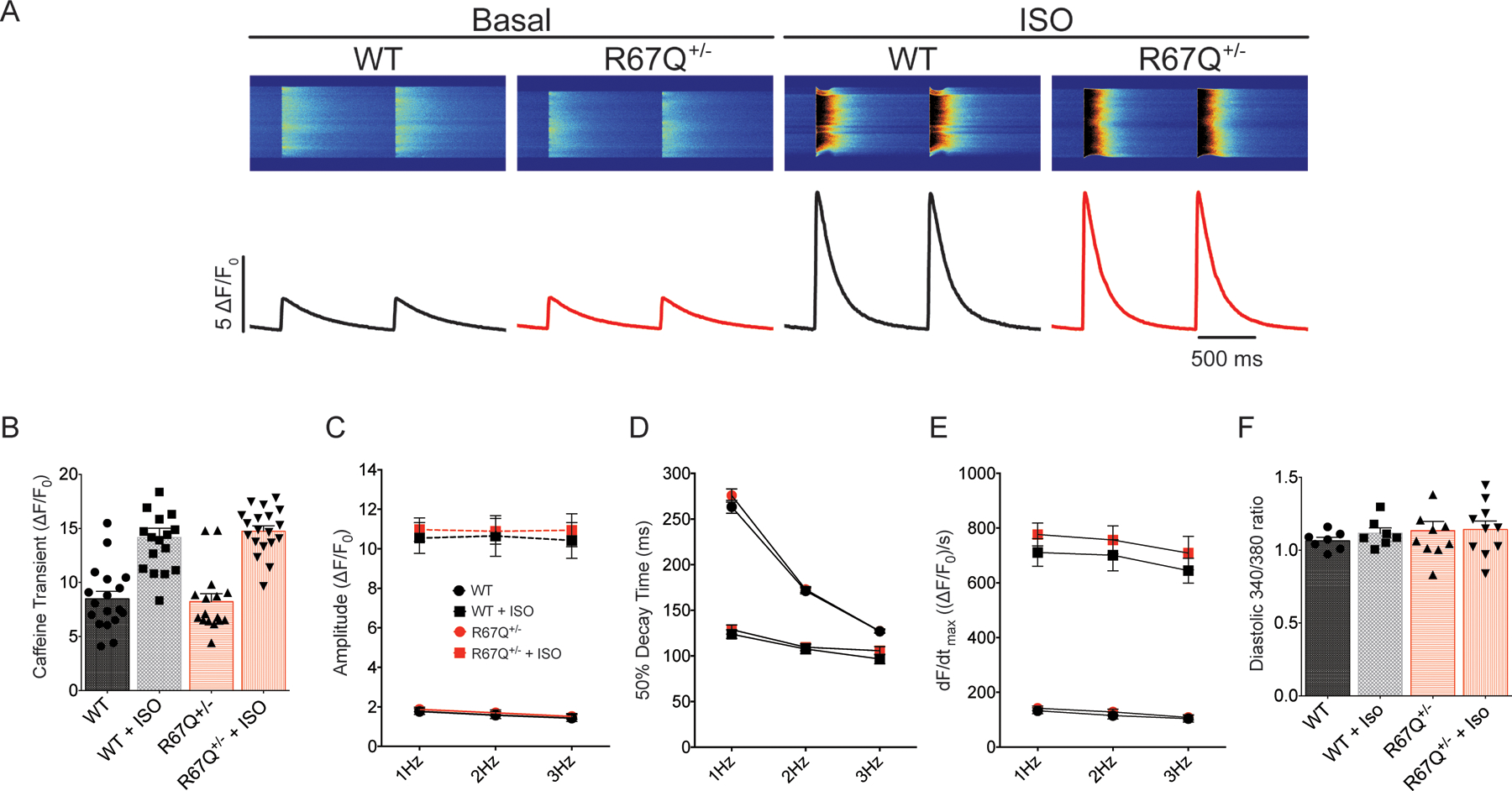
R67Q+/− does not impact Ca2+ handling. A. Representative calcium transient line-scans from WT and R67Q+/− myocytes. Cells were paced for 30s at 1Hz, 15s at 2Hz and 15s at 3Hz, followed by a pulse of 10 mM caffeine to measure SR content. B. WT and R67Q+/− myocytes had comparable SR calcium loads at baseline (8.48 ± 0.70 ΔF/F0 vs. 8.23 ± 0.73 ΔF/F0, respectively, p>0.1) and following treatment with ISO (14.17 ± 0.84 ΔF/F0 vs. 14.75 ± 0.49 ΔF/F0). (WT - N=4, n=18; R67Q+/− - N=4, n=16). C. No significant difference was observed in calcium transient amplitude between WT (black circles) and R67Q+/− (red circles) at baseline or following ISO (WT, black squares, R67Q+/−, red squares) (WT - N=4, n=18; R67Q+/− - N=4, n=16). D. 50% decay time was not significantly different between WT and R67Q+/− at baseline or following ISO. (WT - N=4, n=18; R67Q+/− - N=4, n=16). E. Velocity of the transient (dF/dtmax) is not significantly different between WT and R67Q+/− at baseline or following ISO. (WT - N=4, n=18; R67Q+/− - N=4, n=16). F. Ratiometric calcium transient measurements show no significant difference in diastolic calcium between WT and R67Q+/− VMs (p>0.05). (WT – N=6, n=7; R67Q+/− - N=4, n=10). Two-way repeated measures ANOVA with post-hoc Bonferroni correction was used (5B-F).
Figure 6:
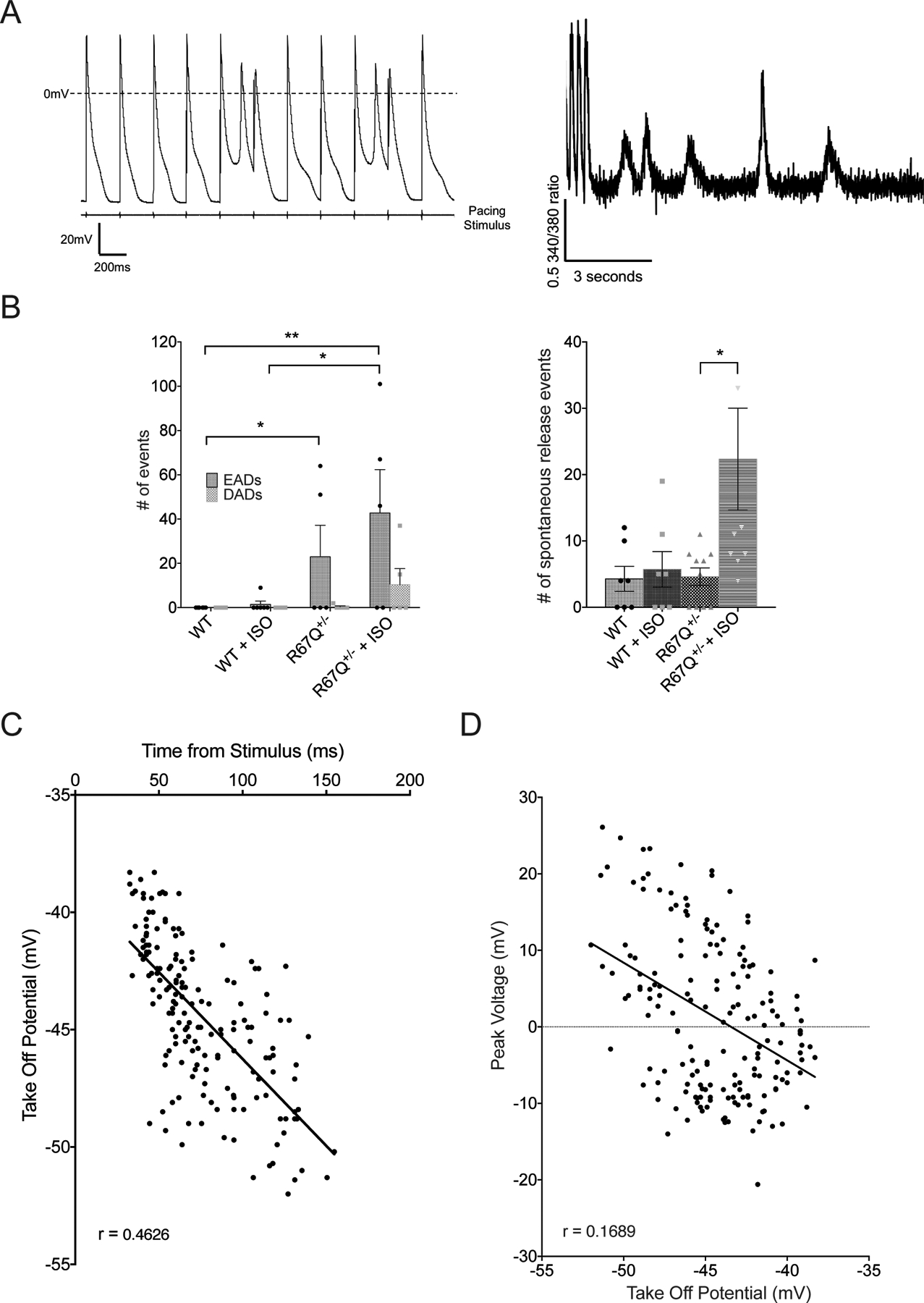
Adrenergic-dependent phase 3 EADs and spontaneous Ca2+ oscillations in R67Q+/− mice. A. Representative traces from action potential recordings (left) and calcium transient measurements (right) showing phase 3 EADs and spontaneous Ca2+ release events, respectively, from isolated R67Q+/− ventricular myocytes. B. R67Q+/− myocytes had more EADs at baseline (23 ± 14.2 events) and following ISO (42.8 ± 19.6 events) compared to WT (0 events at baseline, 1.5 ± 1.5 events following ISO) (left panel) during action potential measurements and increased spontaneous release events following ISO during calcium transient measurements (right panel). C. Analysis of take off potential revealed depolarized potential compared to classical phase 2 EADs, with delayed onset from stimulus. D. Linear regression of take off potential vs peak voltage reached by phase 3 EADs in R67Q+/− myocytes. (Animals/cells for EAD analysis: WT – N=3, n=7; R67Q+/− - N=2, n=5. Animals/cells for spontaneous event analysis - WT – N=6, n=7; R67Q+/− - N=4, n=10). Two-way repeated measures ANOVA with post-hoc Bonferroni correction was used (6B), or non-linear regression (6C, 6D).
Results
Human Cardiac Phenotype is Recapitulate by R67Q+/− Mice: Adrenergic-Dependent BiVT and Structurally Normal Hearts
The index patient who presented to the UW Inherited Arrhythmia Clinic at age 33 with symptoms of teen-age onset of exertional syncope and dyspnea11. She has a structurally normal heart by echocardiogram, no coronary artery disease and is heterozygous for the KCNJ2 R67Q mutation. The patient underwent Holter monitor testing prior to initiating any medications. Quantification of Holter data revealed that 28% of all QRS complexes were ventricular in origin, no atrial arrhythmia nor atrial ectopy occurred, and there were 1616 runs of PMVT and BiVT. Figure 1A includes one of the Holter recorded ventricular runs that occurred during exercise demonstrating 4-beats of BiVT followed by a sinus complex and then 22 beats of PMVT before the arrhythmia spontaneously terminates. Other Holter recorded runs of PMVT occurred with activity but were shorter and none occurred during sleep.
Figure 1:
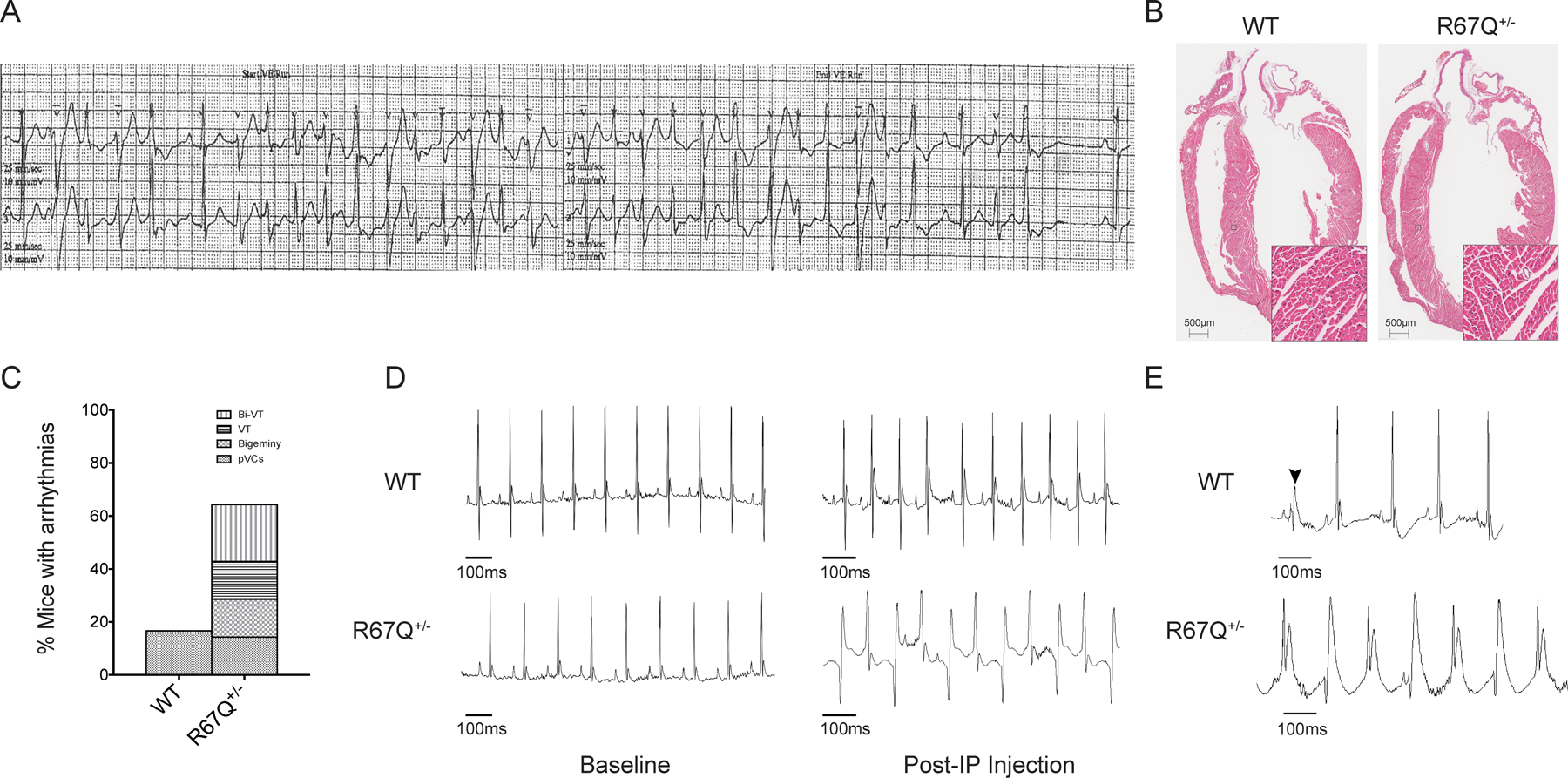
R67Q+/− mice develop paroxysmal bi-directional VT following administration of caffeine and epinephrine. A. Tracing from a Holter monitor of the patient with R67Q mutation showing BiVT and PMVT. B. Hematoxylin and Eosin staining of whole heart from WT and R67Q+/−. Gross histology revealed no significant difference between WT and R67Q+/− in heart structure and size. Scale bar is 500μm. Insert scale is 50μm C. Quantification of arrhythmic events observed in WT and R67Q+/− during ECG analysis. D. ECG recorded from anesthetized WT (top panel) and R67Q+/− (bottom panel) mice at baseline for 5 minutes. Normal sinus rhythm was observed in both groups. Epinephrine (2mg/kg) and caffeine (120mg/kg) were administered by IP injection and ECG recorded for up to 30 minutes. WT showed faster sinus rhythm (top right panel), whereas R67Q+/− mice developed paroxysm of bi-directional ventricular tachycardia (bottom right panel taken at 10 minutes post-injection). E. Upper trace shows a representative PVC observed in WT mice following injection of caffeine and epinephrine. PVC is highlight by the arrow. Lower trace shows representative polymorphic ventricular tachycardia observed in R67Q+/− following injection. N=8 animals per group. Bi-VT – bi-directional ventricular tachycardia; VT – ventricular tachycardia; pVCs – premature ventricular contractions; IP – intraperitoneal injection.
To study the whole animal and arrhythmia phenotype WT and R67Q+/− mice underwent echocardiogram, cardiac morphologic characterization, and ECG analysis. Echocardiographic comparison at 8–12 weeks old (summarized in Supplemental Table 1) revealed all parameters including ejection fraction, ventricular size in systole and diastole, and stroke volume were comparable between WT and R67Q+/−. Shown in Figure 1B is a H&E stained 4-chamber display of WT and R67Q+/− demonstrating normal myocyte architecture and centrally placed nuclei for both genotypes. Together, these results establish that R67Q+/− mice are not functionally distinct from WT littermates and do not have cardiac structural abnormalities.
WT and R67Q+/− mice ECG recordings were analyzed blinded to genotypes and are summarized in Table 1. Baseline parameters in WT and R67Q+/− mice showed sinus rhythm with normal intervals (Table 1 and Figure 1) and without differences between groups. In humans, there is a strong correlation between shorter RR intervals and QT shortening, however, the relationship between QT and RR in mice is weak, and correction formulae, routinely used in human ECG interpretation, have been shown to be misleading32,33. Mice received an intraperitoneal (IP) injection of caffeine (120 mg/kg) and epinephrine (2 mg/kg) and ECGs recorded for up to 30 minutes. As shown in Table 1, RR interval decreased in WT and R67Q+/− following epinephrine and caffeine (p<0.05), but QRS and QT intervals were unaffected (p>0.05). After adrenergic stimulation, R67Q+/− mice developed paroxysms of bi-directional ventricular tachycardia (BiVT), frequent premature ventricular contractions (PVCs), ventricular bigeminy, and ventricular tachycardia (VT) (Figure 1C, example of Bi-VT shown in 1D following adrenergic challenge, VT shown in 1E). WT mice showed sinus tachycardia and 26% of WT also had rare, isolated PVCs (example shown in 1E), but none developed BiVT, VT or sustained arrhythmia (Figure 1C). Frequency of arrhythmia types recorded is shown in Figure 1C and development of arrhythmic phenotype was not dependent upon animal sex (Supplemental Figure 2A).
Spontaneous Sustained Ventricular Arrhythmia in R67Q+/− hearts
We performed optical mapping of the anterior ventricular epicardium of R67Q+/− vs. WT Langendorff-perfused hearts. Baseline rhythm was sinus and pseudo-ECG (pECG, black trace), optical action potential (OAP, blue trace) and activation maps demonstrate reproducible and similar activation patterns between WT (Figure 2A and 2B) and R67Q+/−(Figure 2C and 2E). Figure 2B and Figure 2E are the representative activation map for sinus rhythm for WT and R67Q+/−, respectively, showing impulse generation exiting from the conduction system normally and spreading throughout the heart. Under basal conditions, there were no spontaneous arrhythmias in either group. Based on previous murine models for VT induction34, we treated the excised hearts with caffeine (2.5 mM) plus epinephrine (0.8 μM) with extracellular [Ca2+] 1.8 mM. Following treatment, 100% of the R67Q+/− hearts had induction of spontaneous sustained ventricular tachycardia (VT) within 14–19 min from the time of caffeine plus epinephrine perfusion. Figure 2C shows an example pseudo-ECG from a R67Q+/− heart in sinus rhythm that transitions to wide complex tachycardia as shown in Figure 2D. Focal activity, as demonstrated by the highlighted individual beats (VT1-VT4) in Figure 2F, initiates polymorphic ventricular tachycardia from the ventricular apex region. This then progresses to focal reentry, shown in the activation map (Figure 2F, VT5 and VT6). In contrast, with the same protocol WT hearts failed to demonstrate sustained arrhythmia. Nearly 40 minutes after the infusion of caffeine and epinephrine, 1 of 3 WT hearts had spontaneous arrhythmia that lasted only 2 min and then spontaneously terminated. Using a low dose of caffeine (1 mM) plus epinephrine (0.8 μM) with either 1.2 to 1.8 mM extracellular [Ca2+] did not result in any spontaneous arrhythmias (N=2/group) nor did perfusion of 100 nM isoproterenol (without caffeine or epinephrine) (N=2/group).
Figure 2:
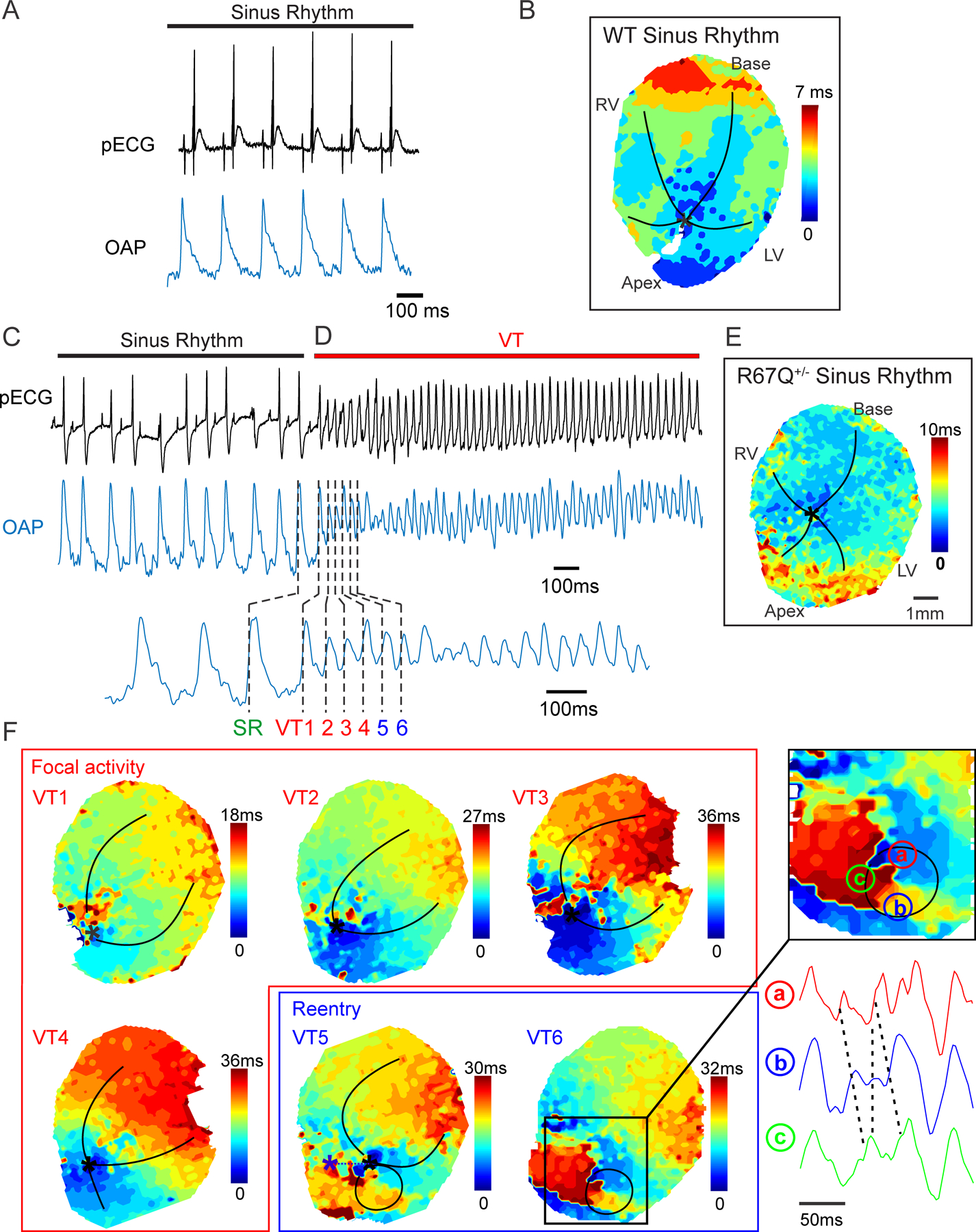
Epinephrine and caffeine administration results in ventricular arrhythmia in Langendorff-perfused R67Q+/− heart. A. Sinus rhythm pseudo ECG (pECG) from intact WT heart (black trace) and optical action potential (OAP, blue trace). B. Activation map for sinus rhythm from intact WT heart. C. Sinus rhythm pseudo ECG (pECG) from intact heart (black trace) and optical action potential (OAP, blue trace). D. Spontaneous ventricular tachycardia following adrenergic stimulation captured on pECG and OAP. E. Activation map for sinus rhythm from intact heart. F. Focal activity activation maps from OAP beats VT1–4. Initiation of focal activity is indicated with a black asterisk. Lower right panel shows progression to focal reentry in beats VT5 and VT6. Insert shows reentry activation pattern and OAP delay between a, b and c. N=3/group.
Paradoxical Response of R67Q+/− to Adrenergic Stimulation Compared to WT
IK1 was measured from isolated cardiomyocytes sequentially at baseline and after 10 nM isoproterenol (representative figures in Supplemental Figure 3 and Supplemental Figure 4A). Myocytes isolated from the right ventricular free wall were used as Kir2.1 expression is variable throughout the ventricles, and expression of Kir2.1 is more consistent cell to cell in the right ventricle35. WT and R67Q+/− myocytes demonstrated a typical inward-rectifier N-shaped current-voltage (IV) relationship with maximal outward current at −50 mV (Figure 3A, rectification index summary data in Supplemental Figure 4B). For comparisons, we focused on the most physiologic component of IK1 which is the outward current as cardiomyocytes are not polarized below −90mV. Outward current amplitude at baseline was not significantly different in R67Q+/− cells compared to WT cells, Figure 3B. With isoproterenol infusion, WT myocytes demonstrated an increase at −50 mV in absolute outward current values as well as percent increase from baseline (Figure 3B and 3C). In contrast, R67Q+/− myocytes showed decreased at −50mV in absolute outward current and percent decreased from baseline (p<0.001) (Figure 3B and 3C). Thus, the difference in IK1 repolarization drive between R67Q+/− and WT is ~43%. Absolute current values showed a similar effect at −40mV with WT IK1 increased, whereas R67Q+/− IK1 decreased to (p<0.05) (Supplemental Figure 5). Importantly, densitometry analysis of Kir2.1 protein expression showed no significant difference between mouse groups (Supplemental Figure 6) suggesting that in R67Q+/− mice adrenergic stimulation results in loss of IK1 decreasing the repolarization reserve despite normal Kir2.1 protein expression.
R67Q+/− mice show action potential prolongation at baseline and following isoproterenol
Example AP traces for WT and R67Q+/− at baseline and after incubation with isoproterenol are shown in Figure 4. Analysis of AP morphology showed a longer APD90 in R67Q+/− myocytes compared to WT at 2, 4 and 6Hz, both at baseline (p<0.05 at 2 and 4Hz, p<0.001 at 6Hz) and following isoproterenol (p<0.01 at 2Hz, p<0.001 at 4 and 6Hz) (Table 2). The dV/dtmax did not differ between WT and R67Q+/− myocytes both at baseline and following isoproterenol (Table 2) at any of the frequencies measured. This is indicative of both normal cellular excitability and normal activation of cardiac sodium channels in R67Q+/− myocytes. Resting membrane potential (RMP) at baseline did not differ between mouse groups; however, following isoproterenol, RMP hyperpolarized in WT cells (p<0.05) at 2 and 4Hz (normal physiologic response), but not in R67Q+/− cells (p>0.1), consistent with lack of IK1 augmentation with isoproterenol (Table 2).
R67Q+/− and WT have similar calcium handling
Calcium-tolerant isolated right ventricular myocytes were loaded with fluo-4 AM and paced at 1Hz for 30s, 2Hz for 15s and 3Hz for 15s followed by a pulse of 10mM caffeine, in the presence and absence of isoproterenol. Representative line scans and transients are shown in Figure 5A. Calcium transient amplitude was not significantly different between WT and R67Q+/− myocytes under basal conditions. Both WT and R67Q+/− calcium transients increased in amplitude in response to isoproterenol perfusion, at all frequencies measured. Additionally, the 50% decay time was not significantly different between WT and R67Q+/− at baseline or following isoproterenol, suggesting that Ca2+ reuptake/extrusion mechanisms are unaffected by the presence of the mutation. The velocity of the transient was comparable between WT and R67Q+/− at baseline and following isoproterenol, Figure 5E. In a separate set of experiments to determine diastolic Ca2+, ratiometric imaging of intracellular Ca2+ cycling in isolated ventricular myocytes revealed a similar baseline Ca2+ ratio in R67Q+/− vs. WT (Figure 5F) that was not significantly different following isoproterenol (p>0.05).
SR Ca2+ content was estimated by the caffeine transient (Figure 5B) and is similar in R67Q+/− vs WT ventricular myocytes. Following isoproterenol, both WT and R67Q+/− SR Ca2+ increase to similar levels. Therefore, R67Q+/− myocytes have no significant differences in SR Ca2+ load or Ca2+ release and removal compared to WT at baseline. Accordingly, Western blot analysis from lysates of WT or R67Q+/− mouse hearts did not show any difference in the expression of primary Ca2+ handling proteins, RyR2, NCX1 and SERCA2a, between groups (Supplemental Figure 7).
Adrenergic stimulation causes phase 3 EADs in R67Q+/− myocytes
An increase in the number of triggered APs during current clamp experiments and spontaneous Ca2+ release events during CaT measurements was observed in cells from R67Q+/− mice following isoproterenol compared to WT (Figure 6A at 4Hz pacing and Supplemental Figure 8 for 6 Hz pacing). In current clamp experiments, R67Q+/− cells had a dramatic increase in the frequency of EADs following isoproterenol while WT had no EADs at baseline and few EADs following isoproterenol. At baseline, R67Q+/− cells had a similar number of DADs as WT cells. A slight increase in DADs was observed in R67Q+/− cells following isoproterenol but did not reach significance (p>0.05). Figure 6B is a summary of the number and type of triggered activity events per cell during AP measurements (left panel). Analysis of the EADs are summarized in Figure 6C and 6D. Figure 6C plots the AP voltage that the EAD take-off potential occurs vs. the time from the pacing stimulus. Notably, EADs were observed to have a take-off potential at −38mV to −52mV along with rapid re-activation. Regression analysis of the EAD take-off potential vs. peak voltage is plotted in Figure 6D. Integrating the stimulation of EADs by isoproterenol, their take-off potential voltage and morphologic characteristics with steep reactivation are consistent with other reports of phase 3 EADs36–38. Unlike phase 2 EADs, the AP is not exceedingly prolonged and some reports of phase 3 EADs not an AP shortening38. The take-off potential is below ICaL window activation range and accordingly may depend on INa at least as the initial inward charge driver38. These are in contrast to phase 2 EADs with a less negative take-off potential and depend on window ICaL activation for the inward charge39,40
Spontaneous Ca2+ release events were noted after rapid pacing and isoproterenol infusion for the R67Q+/− during CaT measurements but not WT. These delayed Ca2+ release events, shown in example traces in Figure 6A, right panel, could be consistent with DADs39,41. Figure 6B right panel quantifies the spontaneous Ca2+ releases during Ca2+ transient measurements. WT cells showed comparable spontaneous release events at baseline and following isoproterenol. At baseline, R67Q+/− cells had comparable events to WT cells. Following isoproterenol, R67Q+/− cells had significantly more events compared to WT cells (see Supplemental Figure 9 for data at 6 Hz pacing). These data parallel the arrhythmia susceptibility demonstrated in R67Q+/− ECG recordings with a dramatic increase in events following adrenergic stimulation similar to the patient harboring R67Q mutation.
Arrhythmia Mechanism
Our collection of in vivo data and analysis in the first transgenic patient-inspired KCNJ2 mutation mouse model points to a cascade of events leading to triggered activity and arrhythmia in R67Q+/− mice. Unifying our observations, we propose a mechanistic concept that is depicted in the graphical abstract. Adrenergic loss of IK1 causes a critical change in membrane voltage that decreases repolarization reserve and drives the development of phase 3 EADs. Our findings are consistent with other models of phase 3 EAD initiation under conditions of decreased repolarization reserve and low extracellular potassium reducing IK122 and have been mechanistically theorized by other groups42. Initially we anticipated that because the clinical phenotype mimics CPVT, the arrhythmia mechanism would be DAD dependent43 and that EADs would not be present due to lack of patient and mouse QT prolongation39, despite that KCNJ2 loss of function mutations were initially categorized as LQTS10. In contrast to our model, QT prolongation and a tendency for phase 2 EADs are described in other LQTS murine models for which the APD of ventricular myocytes can be twice that of the WT controls44,45 and EAD take-off potential and morphology is congruent with phase 2 EADs. Instead, our data supports adrenergic-dependent phase 3 EADs, which are thought to arise from a combination of EADs and DADs mechanistic events46. We propose that understanding the differences in these arrhythmia triggering events, heretofore unknown, will help inform the approach for treatment and arrhythmia suppression strategy.
Discussion
This is the first patient-inspired KCNJ2 mutation in-vivo model and we show that R67Q+/− mice mimic the patient phenotype with adrenergic-induced BiVT with a structurally normal heart. Our data determine that R67Q+/− mice have adrenergic-induced IK1 loss causing decreased repolarization reserve, AP prolongation that together cause phase 3 EADs and the arrhythmogenic milieu for BiVT.
Adrenergic Loss of Function
The critical component of arrhythmogenic vulnerability in the R67Q+/− model lies in the adrenergic loss of IK1. We have shown that WT Kir2.1 outward (physiologic component) current is augmented following adrenergic stimulation and that some KCNJ2 mutations result in either a blunted response or a decrease in outward component of IK1 in heterologous expression systems11,45. In this in vivo model of a KCNJ2 mutation, we demonstrate that adrenergic challenge causes dynamic of loss of function, causing a 40% decrease in repolarization reserve when compared to WT. The underlying molecular mechanism for R67Q-Kir2.1 adrenergic loss of function is not clear at present but could involve PKA channel phosphorylation disruption and/or an increase in sensitivity to the direct inhibitory effect of divalent cation (calcium) on Kir2.1 channels11,47. Kir2.1 channels have at least one known amino acid residue that is phosphorylated by adrenergic activated-PKA. Mutation of the known PKA site, abrogates the effect of adrenergic stimulation on channel function11,45 however, R67Q mutation is physically distant from this PKA site. Alternatively, there may be several PKA targets, yet to be identified, that R67Q interacts with directly or the effect may be related to 3D tetrameric subunit interaction. This and alternative explanations are areas of active investigation in our lab.
Triggered Activity and Phase 3 EAD generation
EADs are secondary membrane voltage depolarizations that occur during AP repolarization as opposed to DADs which occur after repolarization. EADs that occur during the plateau phase are phase 2 EADs and prolonged repolarization and bradycardia creates vulnerable conditions for phase 2 EADs due to a longer window for reactivation of L-type Ca2+ channels39. L-type Ca2+ channels’ time and voltage dependence require phase 2 EADs to occur after a pause or with bradycardia, and phase 2 EADs are suppressed with faster heart rates. In contrast, phase 3 EADs are less well mechanistically understood, but may represent a hybrid of DADs and EADs since some DAD conditions are linked to phase 3 EAD initiation48. It has been proposed that phase 3 EADs result from a combination of adrenergic stimulation and SR Ca2+ release leading to subsequent ITi generation (from NCX) causing sodium channel reactivation with a lesser role from L-type Ca2+ channels49. A defining feature of phase 3 EADs is the negative take-off potential under −35mV and rapid activation. These features are unique from phase 2 EADs and implicate nonequilibrium reactivation of INa (window current between −60mV and −30mV) rather than ICaL (window current peaks at −15mV and spans between −30mV and 0mV)40 as the initial inward current for phase 3 EADs.
As a strong rectifier, IK1 is negligible at AP plateau voltages and therefore loss of IK1 is unlikely to induce phase 2 EADs. However, decreased IK1 will affect phase 3 of repolarization and it has been theorized that this will promote EADs at negative voltages spanning the INa window current42. In murine models, EADs have been described with characteristics of both phase 2 EADs50–52 and those with characteristics of phase 3 EADs, the latter with more negative take-off potential below ICaL activation range and activated by adrenergic stimulation. In our experiments, we predominately note adrenergic dependent triggered activity only in the R67Q+/− mice during action potential repolarization with a take-off potential ~46mV. Thus, the prevailing triggered activity in our model appears to be in the form of phase 3 EADs and fit with the aforementioned theory of loss of IK1 by Qu, A, et al.42. Again, to contextualize our findings with other studies, loss of IK1 during Ca2+ loading can cause DAD-driven arrhythmias due to Ca2+i/Vm coupling gain53 This is important with respect to arrhythmogenesis since BiVT is an arrhythmia associated with Ca2+ overload and DAD activity54. It is logical, then, that a hybrid of EAD and DAD conditions necessary for phase 3 EADs are present in the R67Q+/− mice who demonstrate BiVT under conditions of: 1) adrenergic stimulation, 2) Ca2+ loading (rapid pacing or adrenergic stimulation), 3) adrenergic dependent loss of IK1, and 4) AP90 prolongation.
Arrhythmia Mechanism
The dominant question we attempted to address with this project was why our patient, heterozygous for KCNJ2 mutation R67Q14, as well as the R67Q+/− mouse do not have arrhythmia at baseline but PMVT and BiVT under adrenergic stress. This is in contrast to ATS1 patients who do not require adrenergic stimulation for arrhythmia induction. The added layer of adrenergic stimulation as requisite for arrhythmia induction clinically suggests a CPVT-like phenotype and cellular Ca2+ mishandling. In our case, adrenergic stimulation did not result in Ca2+ mishandling but instead potentiated a loss of IK1 repolarization capacity. The adrenergic loss of IK1, caused critical changes in the membrane potential to drive phase 3 EAD triggered activity and is congruent with both the patient and mouse arrhythmic activity. This is also congruent with other investigators’ hypothesis that an arrhythmia mechanism for IK1 loss is phase 3 EADs42 In contrast to CPVT-associated arrhythmia mechanisms, the primary arrhythmogenic substrate from our in vivo model is the change in membrane voltage due to adrenergic-dependent loss of IK1, resulting lack of repolarizing current of the terminal AP leading to phase 3 EAD triggered activity (see cartoon in Figure 7).
Figure 7:
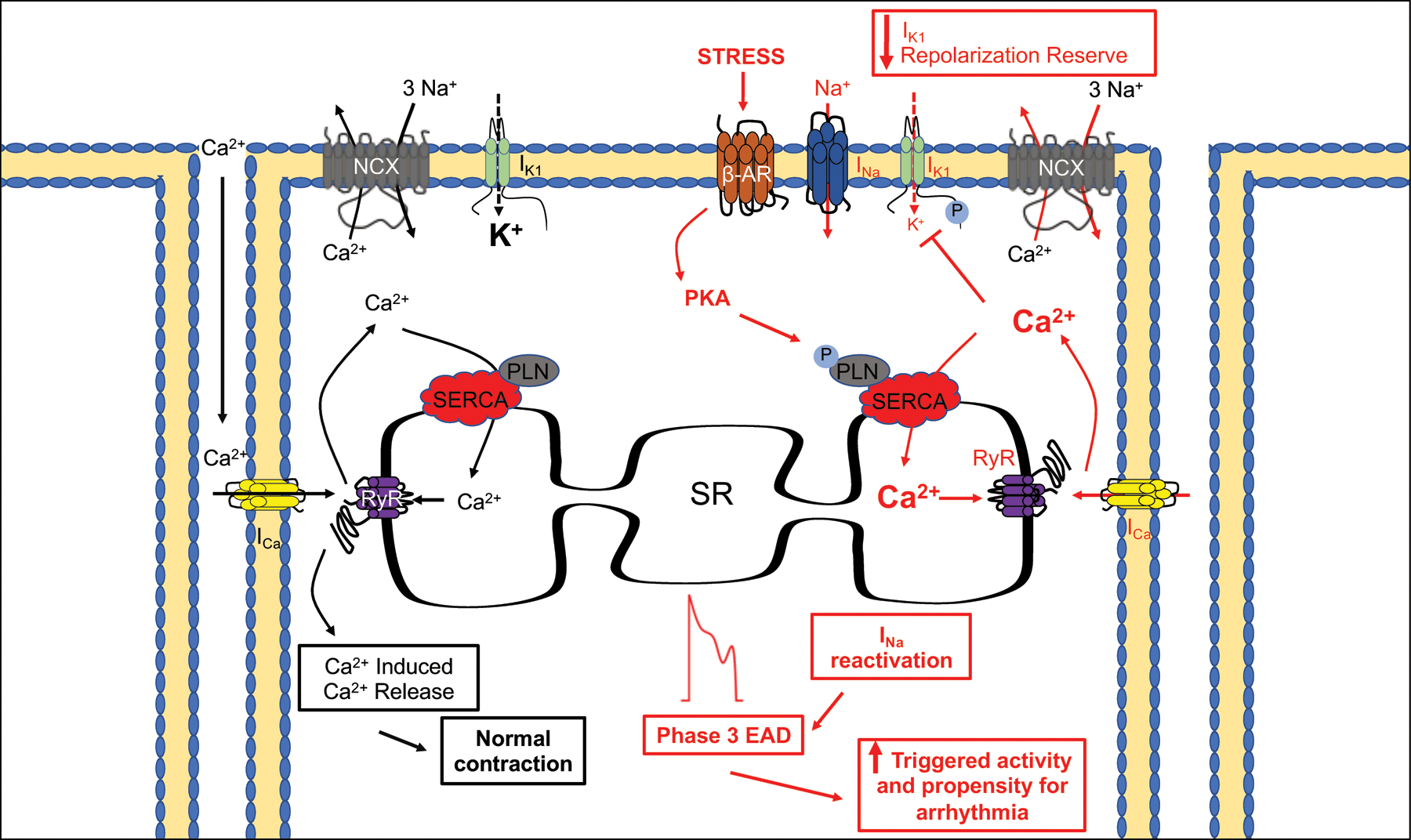
Proposed arrhythmia mechanism for KCNJ2 mutation: Adrenergic loss of IK1 causes a critical change in membrane voltage that decreases repolarization reserve and drives the development of phase 3 EAD.
Study Limitations
The whole animal phenotype recapitulates many of the clinical characteristics observed in patients harboring KCNJ2 mutation. We recognize the limitations and challenges associated with generation of an in vivo model, the differences between human and murine models and translating findings to human clinical phenotypes. However, using in vivo models is necessary to bridge the gaps between mutation to molecular characterization to cellular phenotype and to arrhythmia mechanisms in the whole organ.
Conclusions
R67Q+/− mice demonstrate BiVT associated with IK1 loss causing decreased repolarization reserve and resulting in arrhythmogenic phase 3 EADs. These findings advance our understanding of arrhythmia syndromes related to KCNJ2 mutations and further study of this model will allow us to elucidate more precise treatment approaches based on this arrhythmia mechanism.
Supplementary Material
What Is Known?
Mutations in KCNJ2 can result in a variety of phenotypic presentations but the arrhythmia mechanism(s) remain unknown
What the Study Adds?
We created the first patient-inspired KCNJ2 in vivo model from a patient missense mutation and presented with CPVT-like phenotype.
KCNJ2 in vivo model demonstrates aggressive ventricular arrhythmia including frequent Bi-directional VT similar to the patient.
The arrhythmia mechanism depends not on abnormal calcium handling (as in CPVT) but adrenergic loss of IK1 causing phase 3 EADs
This in vivo model demonstrates a unique arrhythmia mechanism highlights the complexity of KCNJ2 mutation related arrhythmia syndromes as well as phenotypic categorization.
Acknowledgments:
We thank the technical services provided by the University of Wisconsin-Madison Genome Editing and Animal Models (GEAM) Core and Cardiovascular Resource Core at University of Wisconsin-Madison for technical assistance. We thank Dr. Ravi Vaidyanathan for genotyping assistance and Martin Lea for technical advice for myocyte isolation. We thank the members of the Cellular and Molecular Arrhythmia Research Program for constructive discussion. We thank Dr. Elenora Grandi for insightful discussion.
Sources of Funding
This work was supported by National Institutes of Health [NIH R01 HL139738-01 and R01 HL128598-01 to L.L.E., PPG PO1 HL094291 to J.C.M.]; and the American Heart Association [17POST33670358 to L.R, 19CDA34660208 to F.J.A.].
Nonstandard Abbreviations and Acronyms
- AP
action potential
- APD
action potential duration
- ATS
Anderson-Tawil Syndrome
- BiVT
bi-directional VT
- Ca2+
calcium
- CaT
calcium transient
- CPVT3
Catecholaminergic Polymorphic Ventricular Tachycardia 3
- DAD
delayed afterdepolarization
- EAD
early afterdepolarization
- ECG
electrocardiogram
- IP
Intraperitoneal
- LQT7
Long QT Syndrome 7
- NCX1
sodium/calcium exchanger
- PMVT
Polymorphic Ventricular Tachycardia
- PVC
premature ventricular contraction
- SERCA2a
Sarcoplasmic reticulum Ca2+ ATPase 2a
- SQTS3
Short QT Syndrome
- VM
ventricular myocyte
- VT
ventricular tachycardia
- WT
wild-type
Footnotes
Disclosures: None
References:
- 1.Lopatin AN, Nichols CG. Inward Rectifiers in the Heart: An Update on IK1. J Mol Cell Cardiol. 2001;33:625 638. Available from: 10.1006/jmcc.2001.1344 [DOI] [PubMed] [Google Scholar]
- 2.Willis BC, Pandit S, Ponce-Balbuena D, Zarzoso M, Guerrero-Serna G, Limbu B, Deo M, Cammors E, Ramirez RJ, Mironov S, et al. Constitutive Intracellular Na +Excess in Purkinje Cells Promotes Arrhythmogenesis at Lower Levels of Stress than Ventricular Myocytes from Mice with Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation. 2016;133:CIRCULATIONAHA.116.021936 57. Available from: 10.1161/circulationaha.116.021936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donaldson M, Yoon G, Fu Y-H, Ptacek L. Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med. 2009;36:92 97. Available from: 10.1080/17431380410032490 [DOI] [PubMed] [Google Scholar]
- 4.Priori SG. A Novel Form of Short QT Syndrome (SQT3) Is Caused by a Mutation in the KCNJ2 Gene. Circ Res. 2005;96:800 807. Available from: 10.1161/01.res.0000162101.76263.8c [DOI] [PubMed] [Google Scholar]
- 5.Xia M, Jin Q, Bendahhou S, He Y, Larroque M-M, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Bioph Res Co. 2005;332:1012 1019. Available from: 10.1016/j.bbrc.2005.05.054 [DOI] [PubMed] [Google Scholar]
- 6.Tester DJ, Arya P, Will M, Haglund CM, Farley AL, Makielski JC, Ackerman MJ. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm. 2006;3:800 805. Available from: 10.1016/j.hrthm.2006.03.025 [DOI] [PubMed] [Google Scholar]
- 7.Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, Nishio Y, Sasaki K, Itoh H, Yokode M, et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res. 2012;93:666 673. Available from: 10.1093/cvr/cvr329 [DOI] [PubMed] [Google Scholar]
- 8.Maruyama M, Lin SF, Chen PS. Alternans of diastolic intracellular calcium elevation as the mechanism of bidirectional ventricular tachycardia in a rabbit model of Andersen-Tawil syndrome. Heart Rhythm. 2012;9:626 627. Available from: 10.1016/j.hrthm.2010.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morita H, Zipes D, Morita S, Wu J. Mechanism of U wave and polymorphic ventricular tachycardia in a canine tissue model of Andersen–Tawil syndrome. Cardiovasc Res. 2007;75:510 518. Available from: 10.1016/j.cardiores.2007.04.028 [DOI] [PubMed] [Google Scholar]
- 10.Eckhardt LL, Farley AL, Rodriguez E, Ruwaldt K, Hammill D, Tester DJ, Ackerman MJ, Makielski JC. KCNJ2 mutations in arrhythmia patients referred for LQT testing: A mutation T305A with novel effect on rectification properties. Heart Rhythm. 2007;4:323 329. Available from: 10.1016/j.hrthm.2006.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalscheur MM, Vaidyanathan R, Orland KM, Abozeid S, Fabry N, Maginot KR, January CT, Makielski JC, Eckhardt LL. KCNJ2 mutation causes an adrenergic-dependent rectification abnormality with calcium sensitivity and ventricular arrhythmia. Heart Rhythm. 2014;11:885 894. Available from: 10.1016/j.hrthm.2014.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic Polymorphic Ventricular Tachycardia in Children : A 7-Year Follow-up of 21 Patients. Circulation. 1995;91:1512 1519. Available from: 10.1161/01.cir.91.5.1512 [DOI] [PubMed] [Google Scholar]
- 13.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the Cardiac Ryanodine Receptor Gene (hRyR2) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation. 2001;102:196 200. [DOI] [PubMed] [Google Scholar]
- 14.Lahat H, Pras E, Olender T, Avidan N, Ben-Ashder E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, et al. A Missense Mutation in a Highly Conserved Region of CASQ2 Is Associated with Autosomal Recessive Catecholamine-Induced Polymorphic Ventricular Tachycardia in Bedouin Families from Israel. Am J Hum Genet. 2001;69:1378 1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nyegaard M, Overgaard MT, Søndergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G, et al. REPOR T Mutations in Calmodulin Cause Ventricular Tachycardia and Sudden Cardiac Death. Am J Hum Genetics. 2012;91:703 712. Available from: 10.1016/j.ajhg.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff J-M, Costa AD, Sebillon P, Mannens MMAM, Wilde AAM, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21 6. Available from: 10.1161/01.res.0000038886.18992.6b [DOI] [PubMed] [Google Scholar]
- 17.Rooryck C, Kyndt F, Bozon D, Roux-Buisson N, Sacher F, Probst V, Thambo J. New Family With Catecholaminergic Polymorphic Ventricular Tachycardia Linked to the Triadin Gene. J Cardiovasc Electr. 2015;26:1146 1150. Available from: 10.1111/jce.12763 [DOI] [PubMed] [Google Scholar]
- 18.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT, Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137–9142. Available from: 10.1073/pnas.0402546101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barajas-Martinez H, Hu D, Ontiveros G, Caceres G, Desai M, Burashnikov E, Scaglione J, Antzelevitch C. Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry. Circ Cardiovasc Genet. 2011;4:51 57. Available from: 10.1161/circgenetics.110.957696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding W-G, Wu J, Ohno S, Makiyama T, Miyamoto A, et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012;5:344 353. Available from: 10.1161/circgenetics.111.962316 [DOI] [PubMed] [Google Scholar]
- 21.Myles RC, Wang L, Bers DM, Ripplinger CM. Decreased inward rectifying K +current and increased ryanodine receptor sensitivity synergistically contribute to sustained focal arrhythmia in the intact rabbit heart. J Physiology. 2014;593:1479 1493. Available from: 10.1113/jphysiol.2014.279638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maruyama M, Lin S-F, Xie Y, Chua S-K, Joung B, Han S, Shinohara T, Shen MJ, Qu Z, Weiss JN, Chen P-S. Genesis of Phase 3 Early Afterdepolarizations and Triggered Activity in Acquired Long-QT Syndrome. Circ Arrhythm Electrophysiol. 2011;4:103 111. Available from: 10.1161/circep.110.959064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang C-E, Huikuri H, et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia SyndromesExpert Consensus Statement on Inherited Primary Arrhythmia Syndromes. Heart Rhythm. 2013;10:1932 1963. Available from: 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 24.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc National Acad Sci. 1993;90:8424–8428. Available from: 10.1073/pnas.90.18.8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted Disruption of Kir2.1 and Kir2.2 Genes Reveals the Essential Role of the Inwardly Rectifying K+ Current in K+-Mediated Vasodilation. Circ Res. 2000;87:160 166. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, Jenkins NA, Copeland NG. A Highly Efficient Recombineering-Based Method for Generating Conditional Knockout Mutations. Genome Res. 2003;13:476–484. Available from: 10.1101/gr.749203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brody MJ, Feng L, Grimes AC, Hacker TA, Olson TM, Kamp TJ, Balijepalli RC, Lee Y. LRRC10 is required to maintain cardiac function in response to pressure overload. Am J Physiol-heart C. 2016;310:H269 H278. Available from: 10.1152/ajpheart.00717.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salama G, London B. Mouse models of long QT syndrome. J Physiology. 2006;578:43 53. Available from: 10.1113/jphysiol.2006.118745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skrzypiec-Spring M, Grotthus B, Szeląg A, Schulz R. Isolated heart perfusion according to Langendorff—Still viable in the new millennium. J Pharmacol Toxicol. 2007;55:113 126. Available from: 10.1016/j.vascn.2006.05.006 [DOI] [PubMed] [Google Scholar]
- 30.Wischmeyer E, Karschin A. Receptor stimulation causes slow inhibition of IRK1 inwardly rectifying K+ channels by direct protein kinase A-mediated phosphorylation. Proc National Acad Sci. 1996;93:5819–5823. Available from: 10.1073/pnas.93.12.5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reilly L, Howie J, Wypijewski K, Ashford MLJ, Hilgemann DW, Fuller W. Palmitoylation of the Na/Ca exchanger cytoplasmic loop controls its inactivation and internalization during stress signaling. Faseb J. 2015;29:1 12. Available from: 10.1096/fj.15-276493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roussel J, Champeroux P, Roy J, Richard S, Fauconnier J, Guennec J-YL, Thireau J. The Complex QT/RR Relationship in Mice. Sci Rep-uk. 2016;6:1 9. Available from: 10.1038/srep25388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Speerschneider T, Thomsen MB. Physiology and analysis of the electrocardiographic T wave in mice. Acta Physiol. 2013;209:262 271. Available from: 10.1111/apha.12172 [DOI] [PubMed] [Google Scholar]
- 34.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, OConnell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, et al. Arrhythmogenic Mechanisms in a Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Res. 2007;101:1039 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panama BK, McLerie M, Lopatin AN. Heterogeneity of IK1 in the mouse heart. Am J Physiol-heart C. 2007;293:H3558 67. Available from: 10.1152/ajpheart.00419.2007 [DOI] [PubMed] [Google Scholar]
- 36.Bailie DS, Inoue H, Kaseda S, Ben-David J, Zipes DP. Magnesium suppression of early afterdepolarizations and ventricular tachyarrhythmias induced by cesium in dogs. Circulation. 1988;77:1395 1402. [DOI] [PubMed] [Google Scholar]
- 37.Brugada P, Wellens HJJ. Early Afterdepolarizations: Role in Conduction Block, “Prolonged Repolarization-Dependent Reexcitation,” and Tachyarrhythmias in the Human Heart. PACE. 1985;8:889 896. [DOI] [PubMed] [Google Scholar]
- 38.Edwards AG, Grandi E, Hake JE, Patel S, Li P, Miyamoto S, Omens JH, Brown JH, Bers DM, McCulloch AD. Nonequilibrium Reactivation of Na+ Current Drives Early Afterdepolarizations in Mouse Ventricle. Circ Arrhythm Electrophysiol. 2014;7:1205–1213. Available from: 10.1161/circep.113.001666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.January CT, Riddle JM, Salata JJ. A Model for Early Afterdepolarizations: Induction With the Ca2+ Channel Agonist Bay K 8644. Circ Res. 1988;62:563 571. [DOI] [PubMed] [Google Scholar]
- 40.Hirano Y, Moscucci A, January CT. Direct measurement of L-type Ca2+ window current in heart cells. Circ Res. 2018;70:445–455. Available from: 10.1161/01.res.70.3.445 [DOI] [PubMed] [Google Scholar]
- 41.Pogwizd SM, Onufer JR, Kramer JB, Sobel BE, Corr PB. Induction of delayed afterdepolarizations and triggered activity in canine Purkinje fibers by lysophosphoglycerides. Circ Res. 1986;59:416 426. Available from: 10.1161/01.res.59.4.416 [DOI] [PubMed] [Google Scholar]
- 42.Qu Z, Xie L-H, Olcese R, Karagueuzian HS, Chen P-S, Garfinkel A, Weiss JN. Early afterdepolarizations in cardiac myocytes: beyond reduced repolarization reserve. Cardiovasc Res. 2013;99:6 15. Available from: 10.1093/cvr/cvt104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohamed U, Napolitano C, Priori SG. Molecular and Electrophysiological Bases of Catecholaminergic Polymorphic Ventricular Tachycardia. J Cardiovasc Electr. 2007;18:791 797. Available from: 10.1111/j.1540-8167.2007.00766.x [DOI] [PubMed] [Google Scholar]
- 44.Patterson E, Szabo B, Scherlag BJ, Lazzara R. Early and Delayed Afterdepolarizations Associated with Cesium Chloride-Induced Arrhythmias in the Dog. J Cardiovasc Pharm. 1990;15:323–331. Available from: 10.1097/00005344-199002000-00021 [DOI] [PubMed] [Google Scholar]
- 45.Vega AL, Tester DJ, Ackerman MJ, Makielski JC. Protein Kinase A-Dependent Biophysical Phenotype for V227F-KCNJ2 Mutation in Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Arrhythm Electrophysiol. 2009;2:540 547. Available from: 10.1161/circep.109.872309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patterson E, Szabo B, Scherlag BJ, Lazzara R. Early and Delayed Afterdepolarizations Associated with Cesium Chloride-Induced Arrhythmias in the Dog. J Cardiovasc Pharmacol. 1990;15:323 331. [DOI] [PubMed] [Google Scholar]
- 47.Zaza A, Rocchetti M, Brioschi A, Cantadori A, Ferroni A. Dynamic Ca2+-Induced Inward Rectification of K+ current during the ventricular action potential. Circ Res. 1998;82:947 956. [DOI] [PubMed] [Google Scholar]
- 48.Burashnikov A, Antzelevitch C. Late-Phase 3 EAD. A Unique Mechanism Contributing to Initiation of Atrial Fibrillation. PACE. 2006;29:290 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morotti S, McCulloch AD, Bers DM, Edwards AG, Grandi E. Atrial-selective targeting of arrhythmogenic phase-3 early afterdepolarizations in human myocytes. J Mol Cell Cardiol. 2016;96:63 71. Available from: 10.1016/j.yjmcc.2015.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Said M, Becerra R, Valverde CA, Kaetzel MA, Dedman JR, Mundiña-Weilenmann C, Wehrens XH, Vittone L, Mattiazzi A. Calcium-calmodulin dependent protein kinase II (CaMKII): A main signal responsible for early reperfusion arrhythmias. J Mol Cell Cardiol. 2011;51:936–944. Available from: 10.1016/j.yjmcc.2011.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas G, Gurung IS, Killeen MJ, Hakim P, Goddard CA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CL-H. Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in murine hearts containing a modification in the Scn5a gene modelling human long QT syndrome 3. J Physiology. 2007;578:85–97. Available from: 10.1113/jphysiol.2006.121921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Killeen MJ, Gurung IS, Thomas G, Stokoe KS, Grace AA, Huang CL-H. Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol. 2007;191:43–58. Available from: 10.1111/j.1748-1716.2007.01715.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maruyama M, Joung B, Tang L, Shinohara T, On YK, Han S, Choi EK, Kim DH, Shen MJ, Weiss JN, Lin SF, Chen PS. Diastolic Intracellular Calcium-Membrane Voltage Coupling Gain and Postshock Arrhythmias: Role of Purkinje Fibers and Triggered Activity. Circ Res. 2010;106:399 408. Available from: 10.1161/circresaha.109.211292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahlers BA. Identification of an Endogenous Inhibitor of the Cardiac Na+/Ca2+ Exchanger, Phospholemman. J Biol Chem. 2005;280:19875 19882. Available from: 10.1074/jbc.m414703200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.