

Abstract
The first hypotheses about how the immune system affects cancers were proposed in the early 20th century. These early concepts about cancer immunosurveillance were further developed in the decades that followed, but a detailed understanding of cancer immunity remained elusive. It was only recently, through the advent of high-throughput technologies, that scientists gained the ability to profile tumors with a resolution that allowed for granular assessment of both tumor cells and the tumor microenvironment. The advent of immune checkpoint inhibitors (ICIs), which have proven to be effective cancer therapies in many malignancies, has spawned great interest in developing biomarkers for efficacy, an endeavor that highlighted the value of dissecting tumor immunity using large-scale methods. Response to ICI therapy has been shown to be a highly complex process, where the dynamics of tumor and immune cells is key to success. The need to understand the biologic mechanisms at the tumor–immune interface has given rise to the field of cancer immunogenomics, a discipline that aims to bridge the gap between cancer genomics and classical immunology. We provide a broad overview of this emerging branch of translational science, summarizing common platforms used and recent discoveries in the field, which are having direct clinical implications. Our discussion will be centered around the genetic foundations governing tumor immunity and molecular determinants associated with clinical benefit from ICI therapy. We emphasize the importance of molecular diversity as a driver of anti-tumor immunity and discuss how these factors can be probed using genomic approaches.
Introduction
Cancer has affected humans throughout history. Although its first descriptions go back to ancient Egypt (1), it was not until the 18th century, with the advent of microscopy, that tumors were recognized as collections of individual cells (2). The technological revolution that ensued in the following decades led to a growing number of realizations about cancer biology, and by the early 1900s, the first hypotheses about cancer evolution (3) and immune surveillance (4) were being proposed. These ideas served as the basis for the study of cancer immunology, eventually leading to the modern understanding that tumors represent dynamic cell populations able to evolve and diversify in response to environmental pressures such as immune surveillance (5).
The significant degree of heterogeneity that exists within tumors continues to pose a significant challenge to cancer research (6). The pervasiveness of intratumoral heterogeneity (ITH) as well as its association with poor prognosis has been extensively reported (7,8). ITH has been described at many levels—from mutations and chromosomal aberrations in the genome (9–13) to the transcriptome (14) and the epigenome (15,16). However, somatic genetic heterogeneity is the most well documented in the literature, and investigators have been able to demonstrate that genomic ITH arises as a result of ongoing tumor evolution and clonal selection (17,18). Genomic instability has been proposed as one of the primary mechanisms driving these evolutionary forces (19,20), characterized by a process where cell transition from a normal stable genome to one that is plastic and ever changing, characterized by the ability to adapt to new environments and grow indiscriminately (5,6). Opposing this tendency for tumor cells to grow are microenvironmental factors, which limit tumor cell growth or keep growth in check. It has been shown that tumor cell accumulation is greatly dependent on interactions between tumors and host cells that populate the tumor microenvironment (TME) (21). Some of the key mechanisms that regulate tumor growth include neovascularization (22), intercellular growth signaling (23) and immune evasion (24). Notably, TMEs often contain large amount of immune cells, which are known to cooperate and perform activities of immune surveillance (25). It has been shown that the continuous predation of tumors by the immune system puts a strong evolutionary pressure on tumor cells to adapt in order to avoid destruction, leading to phenomena such as immunoediting (19,26,27).
The degree to which tumors are able to avoid recognition and destruction by immune cells is only beginning to be more clearly understood, and initial discoveries in this field have led to the development of a new class of drugs known as immune checkpoint inhibitors (ICIs), drugs capable of reinvigorating immune cells to facilitate tumor clearance (28,29). ICIs have begun to challenge the paradigms of cancer therapy, while at the same time renewing interest in the interplay between cancer cells and immune microenvironment. Indeed, it is becoming clear that tumors are composed of a heterogeneous population of aberrant cells and how these tumor cells behave is, in large part, dictated by the interaction of these cells with distinct immune cell populations (30–32). This realization has led to growing efforts to develop tools that allow for ever more detailed assessment of the TME. Through the development of high-throughput next-generation sequencing (NGS), we have acquired the ability to interrogate these genetic features of the evolving TME at useful resolution (33). The use of these platforms in cancer research has led to an exponential expansion in our knowledge of cancer evolution and anti-tumor immune responses, as well as changed the way we think about and treat cancer (34). Both genotypic and phenotypic features of tumor and immune cells have been shown to have predictive ability for ICI therapy response (28). We will discuss some of the most common platforms used in the field of immunogenomics as well as some of the molecular features that can be explored with these tools to evaluate the tumor–immune interface. Our goal is to provide a general summary of the field of immunogenomics—touching on the main technologies used, the biologic processes that can be interrogated and some of the key findings that have been made in recent years. A review on the established and emerging biomarkers of response to ICI therapy has been presented elsewhere (28).
High-throughput sequencing approaches to dissecting immune phenotypes
As mentioned previously, NGS has exponentially increased the amount and depth of the molecular data obtained from tumor profiling, bringing both new opportunities and challenges. These technologies now allow scientists to interrogate the highly variable tumor genome and look for characteristics that might help affect disease course and response to therapy (34); by enabling a high-resolution and reproducible assessment of the genome or transcriptome, these approaches allow us to take ‘snapshots’ of dynamic processes (6). Implementation of these technologies has significantly increased our understanding of clonal dynamics and immune evasion (27). However, there is still a pressing need to continue to develop and improve more accurate bioinformatic tools (33). Bulk sequencing platforms, such as exome-wide and whole-genome sequencing, allow for a comprehensive assessment of somatic DNA alterations, while at the same time enabling the assessment of certain germline features that are known to influence tumor dynamics (28). Our increased ability to target and manipulate DNA and RNA sequences has led to the development of even more sophisticated technologies, specifically geared to answer questions about tumor and immune heterogeneity. Examples include approaches such as T-cell receptor (TCR) and B-cell receptor repertoire sequencing, which allows characterization of the immune repertoire (35), and single-cell sequencing (36).
Exome-wide and targeted DNA sequencing: examining genomic features and tumor evolution associated with immunosurveillance
Exome-wide sequencing is a relatively common platform that involves capture-based sequencing of all the exons of the genome (37). Given that it provides a good balance between coverage and depth of sequencing (6), this approach allows investigators to evaluate a wide array of somatic aberrations, such as mutations, copy number alterations (CNAs) and translocations, as well as genomic signatures such as presence of microsatellite instability (MSI) (38,39). Exome sequencing can be used to measure tumor mutational burden (TMB), which has been shown to be a strong predictor of clinical benefit from immune checkpoint therapy and adoptive cell therapy with tumor infiltrating lymphocyte treatment. Other DNA sequencing platforms that target a smaller subset of genes have also been used in the field of cancer genomics, and their utility in the detection of somatic mutations and CNAs has been widely demonstrated (40,41). These assays often involve a higher sequencing depth (typically >500×), improving the detection of subclonal mutations in target genes (6). Because targeted sequencing has been easier to implement and can be done at lower cost, companies such as Foundation Medicine or Tempest that provide clinical sequencing services have adopted this approach first. The utility of targeted panels in the assessment of genome-wide features such as the TMB or MSI is possible, and the results have been largely consistent with those from exome sequencing as long as the panels sample adequate amount of the genome (42). For example, the concordance of TMB as measured by targeted panels and whole-exome sequencing is good as long as the panels sample at least 1.1–1.3 megabases (MB). Recently, the Food and Drug Administration (FDA) has approved pembrolizumab for the treatment of TMB-hi tumors in patients who have not responded to previous therapies, regardless of tumor histology. This approval was based on the use of the FoundationOne NGS panel. Although this is a first step towards utilizing TMB in the clinic, the extensive variability in probe design that exists between different platforms can make cross platform comparisons difficult. As such, there is still a need to harmonize methods to calculate TMB. Exome sequencing, on the other hand, is a rather less biased alternative to targeted panel sequencing and may, in many ways, provide a reasonable alternative, especially as sequencing prices continue to drop over time.
Although there is a lot of flexibility in the design of these DNA sequencing assays, those that encompass sequencing of tumor and normal specimens have unique advantages over tumor-only sequencing platforms (40). For example, the detection of somatic mutations is simplified by comparing the tumor and normal sequences with the reference genome, while simultaneously allowing for the distinction between somatic and germline variants (43). Without matched normal sequencing, upwards of 10–20% of potential germline variants may be mistaken for somatic alterations, even with the use of the latest germline reference databases. Germline sequencing also allows for the assessment of features such as MSI (39) and germline HLA diversity (44), which have been implicated in response to ICI therapy (28). Furthermore, by examining allelic frequencies of single-nucleotide polymorphisms across large regions of the genome, matched tumor/normal sequencing can infer tumor purity and ploidy more effectively (45,46), which in combination with conventional mutation calling algorithms can be used to determine mutation cancer cell fraction (i.e. the percent of cancer cells bearing a specific variant) and tumor clonality (12). Although multi-region tumor sequencing constitutes the preferred approach to assess ITH (5,6), a decent approximation of tumor heterogeneity can be obtained through implementation of computational approaches to model clonality such as PyClone, QuantumClone, PhyloWGS and others.
Whole transcriptome RNA sequencing: expression programs of the TME
Whole transcriptome sequencing (WTS) involves sequencing of all the messenger RNA transcripts present in a given tissue and has multiple advantages over older RNA assessment technologies, such as the potential for a more unbiased assessment of all genes and a more accurate tally of relative transcript number (47). WTS has already changed the way we study cancer, even leading to new tools such as single-cell and spatial transcriptomics that promise an even more detailed picture into the transcriptional states in cancer (48). However, the fact that samples are composed of a heterogeneous mix of tumor and normal cells poses a significant challenge when trying to study complex cellular populations. Given the inherent variability in the expression profiles of different cell types, RNA analyses can have vastly different interpretations depending on the analysis approach used. Although bioinformatic algorithms for the deconvolution of the cellular composition of the sample have been developed (49–51), these approaches only estimate the relative frequencies of different cell populations in a given sample. Deconvolution can at best suggest cell type frequency based on known gene expression signatures. It cannot identify novel cell types easily nor can it segregate the cell type-specific gene expression patterns that are unique to individual populations of cells. Single-cell sequencing has recently been able to address that need. Empowering investigators with the ability to examine the discrete transcriptomes of individual cells, single-cell sequencing is able to compartmentalize the gene expression patterns of individual cells so that differences between transcriptional states of cells can be clearly observed. Single-cell sequencing has enabled the detection and characterization of immune cell subsets at a level of resolution that was previously impossible.
TCR sequencing: T-cell repertoire and heterogeneity
DNA and RNA sequencing technologies have also been adapted to allow for the evaluation of the TCR repertoire (35). By targeting the VDJ segments of the TCR genes, which determine the antigen specificity of CD8+ T-lymphocytes, researchers are able to assess the composition of the immune repertoire (52). However, one of the challenges when trying to do so is the high degree of diversity in this sequence. The VDJ recombination that produces functional TCR genes can result in the production of up to 1015–1020 different sequences. However, humans display a significantly lower number (53), suggesting that VDJ recombination is not completely random and that constraints and preferences exist in this process. Despite this, the majority of TCRs are rare, leading to a high degree of inter-individual variability (54,55). By identifying the individual clonotypes (i.e. TCR sequences), investigators can evaluate features such as antigen specificity and clonal heterogeneity (52). Population metrics, such as entropy, evenness, richness and other measures, can be calculated to provide hints about the diversity of TCR repertoires and the behaviors of repertoires after specific challenges. Given that TCRs have been shown to be a source of great diversity between individuals and that CD8+ T cell dynamics have been directly implicated in ICI response (30,56–58), they represent obvious targets of study in the field of immunogenomics. Several TCR sequencing platforms have been developed, and a detailed review on the methods of sequencing and analysis has been presented elsewhere (35). Other platforms targeting additional immune populations such as B-cells have also been described (59), but their relationship to ICI response is far from clear.
Single-cell sequencing platforms: maximum resolution into cell population genetics
The last few years have seen the advent of single-cell sequencing technologies, which are providing an even bigger lens into tumor and microenvironmental heterogeneity (36,60,61). These platforms are still in relatively early phases of development and refinement, but this has not prevented investigators from using it to make important insights about cancer and other biological processes (36,60,62–65). Both single-cell RNA sequencing (scRNAseq) (36) and single-cell genome sequencing (9) have been used in the study of cancer biology. Even single-cell TCR technologies have been reported in the literature (66), effectively providing the maximum resolution that could be achieved with this platform (i.e. one TCR per cell). Single-cell technologies represent a new and promising research frontier, and their increasing adoption will continue to deepen our understanding of the dynamics at the tumor–immune interface.
Computational assessment of antigen presentation machinery function
Many cellular processes have been interrogated in the search for predictors of ICI response (28). The antigen presentation machinery, and specifically, the major histocompatibility complex (MHC) pathway, has been of particular interest due to its well-established role in the presentation of antigens that are targeted by the adaptive immune system (67,68). As discussed previously, NGS technologies have provided unparalleled scale and resolution for tumor DNA assessment, and scientists are now able to take a ‘snapshot’ of the genomes of cancer cells in order to make predictions on the immunogenicity of the mutations they bear. We will discuss the molecular features of tumor and immune cells that have been implicated in response to ICI therapy, with emphasis on properties that can be evaluated using genomics (Figure 1). A detailed description of the mechanisms behind antigen presentation through the MHC class I pathway, as well as the alterations to this machinery that have been described in cancer, has been presented elsewhere (69).
Figure 1.
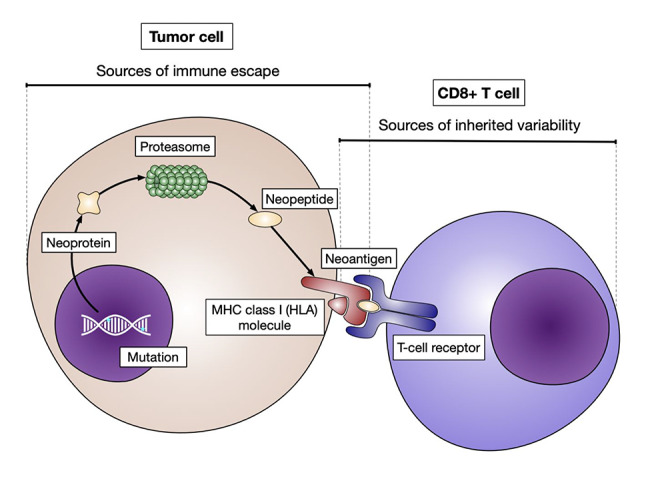
Sources of inter-individual variability in the neoantigen presentation process. A summary of the MHC-I antigen presentation pathway is shown. The integrity of these processes can be interrogated using sequencing, which also provides an unbiased assessment of many features leading to inter-individual variability in immune responses. The tumor mutational repertoire, the breadth of the immunopeptidome and the composition of the immune repertoire are sources of inter-individual variability that can be interrogated. Refinement of these platforms is already paving the way towards the individualization of cancer care.
HLA diversity and the immunopeptidome
Virtually all nucleated cells express some type of cell defense mechanism against pathogens such as viruses, which reproduce by using cellular machinery for reproduction. The MHC-I pathway is a line of defense against viruses. MHC-I presents both pathogen peptides and self-antigens on the cell surface and, hence, plays critical roles in dictating immune tolerance and pathogen recognition (67). The genes that encode the molecules directly responsible for antigen presentation [i.e. the human leukocyte antigen (HLA) genes] are the most polymorphic across the human species (70–72), leading to a high degree of inter-individual immunopeptidome variability (i.e. the total collection of peptides that a cell is potentially able to present as antigens). This diversity appears to be a result of different microbial pressure exerted upon human populations during their evolution (70,71,73). Notably, some of the polymorphisms described in the HLA genes (i.e. HLA-A, HLA-B, HLA-C) have been implicated in differences in effectiveness of antiviral immune responses (74,75) as well as in a wide array of immunologic-related diseases (76). However, their importance in anti-tumor immune responses is only beginning to be understood. Although polymorphisms in other immune-related genes have been described in association with response to ICI therapy (77), the HLA genotype imparts the strongest effects by far.
The ability of tumor cells to present antigens depends first and foremost on the genetic background from which they originate. It has been shown that individuals with a more diverse array of HLA-I alleles are more likely to experience clinical benefit after ICI therapy (58,78). Additionally, the presence of a specific HLA supertype (i.e. clusters of HLA alleles with similar anchor specificities (79)), such as HLA-B44, has also been associated with an increased likelihood of response to ICI therapy in specific tumor types (58). Consistent with these initial reports, investigators have demonstrated that the degree of evolutionary divergence exhibited by an individual’s HLA alleles (80) is in direct association with improved response and survival after ICI therapy (81). HLA diversity could represent a potential biomarker in the quest to personalize immunotherapy, and further research is needed to establish the utility of these features in the clinical setting.
TMB and neoantigen immunogenicity
Tumor cells can present peptides to the immune system through the MHC-I pathway in very much the same way that normal cells do. Briefly, expressed intracellular proteins are processed in the proteasome and converted into short peptides (of 8–11 amino acids in length) that fit the HLA-I binding pocket (82,83). These peptides are then transported to the endoplasmic reticulum where they are bound to MHC-I molecules and are transported by the secretory pathway through the Golgi apparatus, until they finally migrate to the cell membrane for presentation (84–86). Somatic alterations in the coding regions of the exome can result in the production of a neopeptides that can be presented as neoantigens (56,87–91). The degree to which somatic mutations are able to produce an immunogenic neoantigen is a topic under active study (57), and several features have been shown to affect the likelihood that a mutation is presented. First, the degree of change in the amino acid sequence can have a great impact on the likelihood of presentation. Mutations leading to frameshifts, such as insertions or deletions, can produce new protein sequence that is drastically different than any sequence in the germline. The amount of small insertions and deletion mutations has been linked to ICI response in several studies (92–94). Missense mutations can change a single amino acid in either anchor locations or more central regions of the epitope that contact the CDR3 domain of TCRs. These types of mutations, while producing more subtle changes in amino acid sequence, can also produce immunogenic neoantigen peptides. Point mutations involving the MHC anchor positions can make a peptide that was previously not presented by MHC molecules to ones that are presented. These types of mutations can produce very immunogenic epitopes (95).
Many bioinformatic methods have been developed to predict whether antigens are presented by MHC molecules or are immunogenic. Neoantigens have been characterized based on their predicted MHC-binding affinity by using the differential agretopicity index (DAI), a measure that expresses binding affinity relative to the wild-type sequence (95). Neoantigen DAI has been shown to be a surrogate of clinical benefit after ICI therapy in melanoma cohorts (96,97). Similarly, sequence homology methods, using previously validated immunogenic microbial epitopes as reference, have been proposed as a means to evaluate neoantigen immunogenicity (98). However, given the complexity of the actual presentation process, and the need to individualize predictions to each tumor context, no single approach has proven sufficient to accurately predict immunogenicity. Challenges still remain and many improvements need to be made to improve the accuracy of prediction methods. Recent attempts have been made using multi-parametric machine learning algorithms (99,100), and these have shown substantial promise.
Despite the complexity of the antigen presentation process, TMB is known to strongly correlate with neoantigen burden (101). In line with this, response to ICI therapy was first described in cancer types with high TMBs such as melanoma (101–103) and NSCLC (104–108). Furthermore, high TMB has been shown to predict for response to ICI therapy in these (101,108,109) and other solid malignancies (42,110,111). Examples include tumors such as HPV-negative head and neck squamous cell carcinoma (112), urothelial carcinoma (113) and small cell lung cancer (114). However, TMB is not the only factor determining response, and notable exceptions to this rule have been described in tumors such as renal cell (115) and certain subtypes of Merkel cell carcinomas (116), which are responsive to ICI therapy despite an overall low TMB. In these tumor types, high TMB does not seem to be sufficient alone to predict benefit after ICI therapy (42,110). The bulk of the evidence, and the consistency of the findings across multiple disease contexts, has led to the FDA approval of pembrolizumab for the treatment of any solid malignancy with a high TMB (≥10 mutations/megabase) (117).
It is important to note, however, that some tumors with high TMB such as melanoma and NSCLC are known to arise in the context of exposure to carcinogenic agents like ultraviolet light and smoke (118). As a consequence, the neoantigens derived from these mutations will be most likely derived from these mutational processes. The type of mutation may play a role in immunogenicity and not just the quantity of mutations. Furthermore, clonality of neoantigens may also be important. The clonality of neoantigens has been proposed as a contributor to the immunogenicity of mutations (30,119).
Antigen presentation disruption and immune escape
Owing to the fact that the MHC-I pathway represents a key factor in the development of cellular anti-tumor immune responses, tumors are under constant selective pressure to ‘lose’ these mechanisms in order to avoid immune recognition (27,120,121). Examples of tumors displaying somatic aberrations that promote immune evasion have been reported in the literature (121–125), and detailed reviews have been presented elsewhere (123,126,127). We will focus on the genomic aberrations described in the MHC-I pathway, which have been linked not only to immune evasion but also to poor prognosis (128,129) and resistance to ICI therapy in different contexts (58,130). Among the proteins that have been found to be somatically inactivated are the HLA molecules themselves (131), the beta-2-microglobulin protein (B2M gene) (131–133) and the transporter associated with antigen processing (TAP1 gene) (125,128,129,132,134). Notably, tumors that show disruption of key components of the MHC-I pathway seem to be able to tolerate higher neoantigen burdens in their genomes, providing evidence of ongoing immunoediting (44,131). Interestingly, the frequency in which these alterations occur is relatively low. Of particular interest are the HLA genes, which seem to be recurrent substrates of immune evasion. Both somatic mutations and CNAs (including unbalanced losses and copy-neutral loss-of-heterozygosity) have been described in these genes in the setting of immune escape (58,121,135), and the polymorphic nature of these loci represents a special challenge to their evaluation using DNA sequencing. Although this has already prompted researchers to develop specific computational algorithms for their assessment (44,135,136), further research is needed both to refine them and to evaluate their clinical utility (137).
Genomic features of tumors associated with immune response
We briefly mentioned some of the mechanisms by which tumors can ‘learn’ how to avoid immune recognition. However, certain intrinsic tumor characteristics have been implicated in ICI response. For example, certain mutational signatures (i.e. patterns of base changes in the DNA that serve as indicators of causal processes of mutations) (118), such as the smoking and the APOBEC signatures in NSCLC specimens, have been shown to be associated with clinical benefit after ICI therapy (107,138). Similarly, the presence of certain broad genomic characteristics, such as MSI (139), and alterations that result in a deficiency of DNA mismatch repair, leading to subsequent mutation accumulation (140), have been associated with response to ICIs (141). Solid tumors with mismatch repair deficiency have been approved by the FDA to be treated with pembrolizumab therapy regardless of tumor histology (142).
Other somatic alterations that have been implicated in the development of anti-tumor immune responses after ICI therapy include somatic aberrations that produce inactivation of genes in the IFNγ-JAK/STAT signaling pathway (143–145) and copy number events such as PD-L1 amplifications (146), which have been implicated in ICI resistance and sensitivity, respectively. Additionally, somatic mutations commonly used to subclassify tumors can have important clinical implications in the immunotherapy context. For example, patients with mutant BRAF melanomas have been reported to respond better to ICI combination therapy (147) as a group; and the KRAS mutant STK11/LKB1 mutant subtype of NSCLC has been shown to be rather unresponsive to PD-1 inhibitors (148). Other examples of tumor subtypes with differential ICI sensitivity include the higher response rates to PD-L1 inhibitors in the luminal cluster II subtype of urothelial carcinoma (113) and the relative resistance to checkpoint inhibition reported in tumors with PTEN inactivation (149,150). Mutations in chromatin remodeling genes, such as PBRM1, have also been proposed as potential biomarkers of response to ICI therapy in tumors like renal cell carcinoma (151), but conflicting findings have been reported (115) and initial results reporting the association of PBRM1 inactivation with sensitivity to immunotherapy were likely confounded by previous treatment with anti-angiogenic agents.
Finally, another pervasive feature of cancer that has been implicated in anti-tumor immune response is the degree of genomic instability displayed by tumor cells, a feature commonly represented by the overall burden of CNAs observed in the tumor genome (152). Somatic CNA profiles can be effectively assessed using DNA sequencing technologies (45,46), and the burden of these chromosomal anomalies has been associated with poor prognosis, immune evasion and ICI resistance (121,152–155). However, this metric is not yet used in the clinic and additional research is needed to standardize copy number algorithms, validate these findings and explore their practical utility.
Microenvironment composition: gene expression and immune cell activity
As discussed briefly before, the microenvironmental composition of the tumor plays an important role in tumor adaptation, survival and therapeutic response (156). For the purposes of this discussion, we will consider the tumor as having three compartments: the tumor compartment, whose genomic alterations have already been discussed; the immune compartment of the TME, composed of several immune subpopulations; and the non-immune compartment of the TME, which encompasses the rest of the stromal cells. Because the individual cells composing each of these groups are deeply intermixed with those from the other groups, expression assays such as bulk WTS can only measure a combined expression signal from the entire specimen (47). RNA expression signatures (i.e. phenotypic profiles based on the expression of groups of genes) have been widely studied in the setting of ICI therapy. Notably, some of the expression profiles that have been associated with response to ICI include T-cell inflamed (157,158), T-cell dysfunction (159) and T-cell effector/IFN-γ signatures (115). Similarly, TGF-β phenotypes (160,161) and an innate anti-PD-1 resistance signature (162) have been proposed to be enriched in non-responders. These findings make clear that microenvironmental signals determining response to ICI therapy are partly driven by the immune microenvironment (163). Interestingly, CD8+ TIL populations that show high PD-1 expression have been implicated in both response (164,165) and resistance to checkpoint inhibition (164,166), and questions remain about implications of these findings.
Although several transcriptomic features have been implicated in ICI response, none of them have been adopted into clinical practice. The only exception is the expression of PD-L1 in tumor specimens (which is actually evaluated as an immunohistochemical readout), which has been widely described in the literature in association to clinical benefit after checkpoint blockade. High PD-L1 expression levels have been shown to serve as a surrogate for T-cell dysfunction (167) as well as a predictor of response to ICIs (106,113,168–170). However, conflicting reports are also widespread in the literature (112,171,172), and the potential reasons behind these discrepancies have been extensively discussed before (28,173,174). Besides the biases associated with single-sample assessment, most inconsistencies are thought to arise from the lack of standardization among different immunohistochemistry (IHC) assays and differences in interpretation of the staining (175,176). This has prompted some researchers to argue that a better way to standardize the assessment of PD-L1 expression levels is using RNA sequencing approaches (177). However, due to the increased costs associated with this technology and the relative instability of the RNA molecule, it is unlikely that an RNA-based assay would be adopted solely for this purpose, and furthermore, bulk RNA sequencing imposes inherent restrictions on IHC with regard to spatial resolution, a factor that has also been implicated in ICI response (160,178). Nevertheless, one could envision a future where comprehensive and contextualized assessment of tumor expression profiles could aid clinicians in individualizing cancer therapeutics.
Finally, the clonal architecture of the T-cell repertoire is also a topic under active study that is amenable to immunogenomics approaches, and changes in the diversity of the TCR repertoire during ICI therapy have been described (30). Notably, clonal T-cell populations have been shown to arise in ICI responders, in proportion to the number of neoantigens depleted (30). However, the ability of TCR repertoire characteristics to predict ICI response is far from clear, with different studies reporting mixed findings (104,154,165,179–182). Importantly, prior exposure to ICI therapy can drastically alter T-cell dynamics (30), suggesting that the immune repertoire is highly dependent on disease and therapeutic contexts. Even though T-cell identity and dynamics are a key factor in the development of anti-tumor responses, other cell populations have also been implicated. For example, a cluster of CD4 + FOXP3-PD1hi T cells, which confers a T follicular helper phenotype, has been associated with anti-tumor immunity and prolonged survival after ICI therapy (183,184), further emphasizing the subtlety of these findings and the need for new technologies that provide increased resolution of cellular states. Tumor microenvironmental composition assessment is an area of research where scRNAseq technologies promise significant improvements over current approaches, and studies using these platforms have already led to important realizations about this process. For example, a memory-like phenotype of CD8+ T cells, defined by the expression of TCF7, and certain CD4+ populations have been linked to ICI response (185).
Conclusion
Tumors represent dynamic cell populations that are constantly subject to a wide array of environmental pressures. Their multiplicative nature allows them to respond by evolving and adapting to their environment, often leading to the modification of intrinsic biologic mechanisms to increase their fitness. Immune evasion has now been recognized as one of the core hallmarks of cancer, and many biologic mechanisms that underlie this process have been reported. Realization about the complexity of the tumor–immune interface dynamics has pushed investigators to develop assays that can probe cancer and immune cell populations with increasing resolution.
The field of immunogenomics has thus arisen as a bridge between classical immunology and cancer genomics, leading to a new and exciting frontier in translational medicine that promises to convert a once phenomenological discipline into one steeped in large-scale data. An example of the utility of NGS approaches has been the dissection of the complex facets of how MHC-I presentation plays a role in tumor immunity. Although several processes have been associated with sensitivity and resistance to ICI therapy, the MHC-I antigen presentation pathway is of particular interest to the field due to its direct relationship with immune evasion as well as the high degree of inter-individual variability that it exhibits. These features make it an ideal target for assessment using NGS platforms such as exome-wide sequencing, which can now be scaled for use with each individual patient.
Acknowledgements
We thank the Chan lab members for helpful discussions. We thank the staff and physicians of the MSK Department of Medicine Kidney Program and the Urology Service for helpful suggestions. We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Confict of Interest. T.A.C. is a co-founder of Gritstone Oncology and holds equity. T.A.C. holds equity in An2H. T.A.C. acknowledges grant funding from Bristol-Myers Squibb, AstraZeneca, Illumina, Pfizer, An2H and Eisai. T.A.C. has served as an advisor for Bristol-Myers, MedImmune, Squibb, Illumina, Eisai, AstraZeneca and An2H. T.A.C. holds ownership of intellectual property on using tumor mutation burden to predict immunotherapy response, with pending patent, which has been licensed to PGDx.
Contributor Information
Renzo G DiNatale, Immunogenomics and Precision Oncology Platform, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA; Urology Department, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
A Ari Hakimi, Urology Department, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Timothy A Chan, Immunogenomics and Precision Oncology Platform, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA; Department of Radiation Oncology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA; Center for Immunotherapy and Precision Immuno-Oncology, Cleveland Clinic, Cleveland, OH 44195, USA; Lerner Research Institute and Taussig Cancer Center, Cleveland Clinic, Cleveland, OH 44195, USA.
Funding
National Institutes of Health (R35 CA232097, DOD KC180165 to T.A.C.); Mellnikoff Fund (T.A.C.); Weiss Family Fund (A.A.H.).
References
- 1.. Patterson J.T. and Olson J.S. (1991) The history of cancer: an annotated bibliography. J. Am. Hist., 77, 1475. [Google Scholar]
- 2.. Morgagni (1761) De Sedibus, et Causis Morborum per anatomen Indagatis libri quinque. In Dissectiones, et Animadversiones, nunc primum editas, complectuntur propemodum innumeras, medicis, chirurgis, anatomicis profuturas. Remondini, Venice, 1761.
- 3.. Boveri T. (1914) Zur frage der entstehung maligner tumoren. Fischer, 1914.
- 4.. Ehrlich P. (1909) Über den jetzigen Stand der Karzinomforschung. In Beiträge zur experimentellen Pathologie und Chemotherapie, 117–164.
- 5.. McGranahan N. and Swanton C. (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell, 168, 613–628. [DOI] [PubMed] [Google Scholar]
- 6.. Turajlic S., Sottoriva A., Graham T. et al. (2019) Resolving genetic heterogeneity in cancer. Nat. Rev. Genet., 20, 404–416. [DOI] [PubMed] [Google Scholar]
- 7.. Caswell D.R. and Swanton C. (2017) The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med., 15, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.. Morris L.G.T., Riaz N., Desrichard A. et al. (2016) Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget, 7, 10051–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.. Baslan T., Kendall J., Volyanskyy K. et al. (2020) Novel insights into breast cancer copy number genetic heterogeneity revealed by single-cell genome sequencing. elife, 9, e51480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.. Hashimoto T., Osoegawa A., Takumi Y. et al. (2018) Intratumoral heterogeneity of copy number variation in lung cancer harboring L858R via immunohistochemical heterogeneous staining. Lung Cancer, 124, 241–247. [DOI] [PubMed] [Google Scholar]
- 11.. Gerlinger M., Rowan A.J., Horswell S. et al. (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med., 366, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.. McGranahan N., Favero F., Bruin E.C. et al. (2015) Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med., 7, 283ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.. Mamlouk S., Childs L.H., Aust D. et al. (2017) DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun., 8, 14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.. Sharma A., Merritt E., Hu X. et al. (2019) Non-genetic intra-tumor heterogeneity is a major predictor of phenotypic heterogeneity and ongoing evolutionary dynamics in lung Tumors. Cell Rep., 29, 2164–2174.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.. Easwaran H., Tsai H.-C. and Baylin S.B. (2014) Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell, 54, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.. Mazor T., Pankov A., Song J.S. et al. (2016) Intratumoral heterogeneity of the epigenome. Cancer Cell, 29, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.. Turajlic S., Xu H., Litchfield K. et al. (2018) Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell, 173, 581–594.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.. Jiang Y., Qiu Y., Minn A.J. et al. (2016) Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A., 113, E5528–E5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.. Hanahan D. and Weinberg R.A. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 20.. Bakhoum S.F. and Landau D.A. (2017) Chromosomal instability as a driver of tumor heterogeneity and evolution. Cold Spring Harb. Perspect. Med., 7(6), a029611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.. Quail D.F. and Joyce J.A. (2013) Microenvironmental regulation of tumor progression and metastasis. Nat. Med., 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.. De Palma M., Biziato D. and Petrova T.V. (2017) Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer, 17, 457–474. [DOI] [PubMed] [Google Scholar]
- 23.. Di Virgilio F., Sarti A.C., Falzoni S. et al. (2018) Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer, 18, 601–618. [DOI] [PubMed] [Google Scholar]
- 24.. Ferrone S. and Whiteside T.L. (2007) Tumor microenvironment and immune escape. Surg. Oncol. Clin. N. Am., 16, 755–774. [DOI] [PubMed] [Google Scholar]
- 25.. Binnewies M., Roberts E.W., Kersten K. et al. (2018) Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med., 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.. Vinay D.S., Ryan E.P., Pawelec G. et al. (2015) Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin. Cancer Biol., 35, S185–S198. [DOI] [PubMed] [Google Scholar]
- 27.. O’Donnell J.S., Teng M.W.L. and Smyth M.J. (2019) Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol., 16, 151–167. [DOI] [PubMed] [Google Scholar]
- 28.. Havel J.J., Chowell D. and Chan T.A. (2019) The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer, 19, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.. Darvin P., Toor S.M., Sasidharan Nair V. et al. (2018) Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med., 50, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.. Riaz N., Havel J.J., Makarov V. et al. (2017) Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell, 171, 934–949.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.. Anagnostou V., Forde P.M., White J.R. et al. (2019) Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Res., 79, 1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.. Gide T.N., Quek C., Menzies A.M. et al. (2019) Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell, 35, 238–255.e6. [DOI] [PubMed] [Google Scholar]
- 33.. Koboldt D.C., Steinberg K.M., Larson D.E. et al. (2013) The next-generation sequencing revolution and its impact on genomics. Cell, 155, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.. Gagan J. and Van Allen E.M. (2015) Next-generation sequencing to guide cancer therapy. Genome Med, 7(80), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.. Rosati E., Dowds C.M., Liaskou E. et al. (2017) Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol., 17, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.. Hwang B., Lee J.H. and Bang D. (2018) Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med., 50, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.. Bao R., Huang L., Andrade J. et al. (2014) Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform., 13, 67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.. Nakagawa H. and Fujita M. (2018) Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci., 109, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.. Niu B., Ye K., Zhang Q. et al. (2014) MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics, 30, 1015–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.. Cheng D.T., Mitchell T.N., Zehir A. et al. (2015) Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn., 17, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.. Weiss G.J., Hoff B.R., Whitehead R.P. et al. (2015) Evaluation and comparison of two commercially available targeted next-generation sequencing platforms to assist oncology decision making. Onco Targets Ther., 8, 959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.. Samstein R.M., Lee C.-H., Shoushtari A.N. et al. (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet., 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.. Schrader K.A., Cheng D.T., Joseph V. et al. (2016) Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol, 2, 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.. Shukla S.A., Rooney M.S., Rajasagi M. et al. (2015) Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol., 33, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.. Carter S.L., Cibulskis K., Helman E. et al. (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol., 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.. Shen R. and Seshan V.E. (2016) FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res., 44, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.. Wang Z., Gerstein M. and Snyder M. (2009) RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet., 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.. Stark R., Grzelak M. and Hadfield J. (2019) RNA sequencing: the teenage years. Nat. Rev. Genet., 20, 631–656. [DOI] [PubMed] [Google Scholar]
- 49.. Wang X., Park J., Susztak K. et al. (2019) Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun., 10, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.. Monaco G., Lee B., Xu W. et al. (2019) RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep., 26, 1627–1640.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.. Chen B., Khodadoust M.S., Liu C.L. et al. (2018) Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol., 1711, 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.. Schatz D.G. and Ji Y. (2011) Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol., 11, 251–263. [DOI] [PubMed] [Google Scholar]
- 53.. Laydon D.J., Bangham C.R.M. and Asquith B. (2015) Estimating T-cell repertoire diversity: limitations of classical estimators and a new approach. Philos. Trans. R. Soc. B Biol. Sci., 370, 20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.. Freeman J.D., Warren R.L., Webb J.R. et al. (2009) Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res., 19, 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.. Warren R.L., Freeman J.D., Zeng T. et al. (2011) Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res., 21, 790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.. Yang W., Lee K.-W., Srivastava R.M. et al. (2019) Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med., 25, 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.. Jiang T., Shi T., Zhang H. et al. (2019) Tumor neoantigens: from basic research to clinical applications. J. Hematol. Oncol., 12, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.. Chowell D., Morris L.G.T., Grigg C.M. et al. (2018) Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science, 359, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.. Yaari G. and Kleinstein S.H. (2015) Practical guidelines for B-cell receptor repertoire sequencing analysis. Genome Med., 7, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.. Tang X., Huang Y., Lei J. et al. (2019) The single-cell sequencing: new developments and medical applications. Cell Biosci., 9, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.. González-Silva L., Quevedo L. and Varela I. (2020) Tumor functional heterogeneity unraveled by scRNA-seq technologies. Trends Cancer Res., 6, 13–19. [DOI] [PubMed] [Google Scholar]
- 62.. Farrell J.A., Wang Y., Riesenfeld S.J. et al. (2018) Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science, 360(6392), eaar3131. doi: 10.1126/science.aar3131. Epub 2018 Apr 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.. Zhang L., Dong X., Lee M. et al. (2019) Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. U. S. A., 116, 9014–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.. Pellegrino M., Sciambi A., Treusch S. et al. (2018) High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res., 28, 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.. Macaulay I.C., Ponting C.P. and Voet T. (2017) Single-cell multiomics: multiple measurements from single cells. Trends Genet., 33, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.. De Simone M., Rossetti G. and Pagani M. (2018) Single cell T cell receptor sequencing: techniques and future challenges. Front. Immunol., 9, 1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.. Rock K.L., Reits E. and Neefjes J. (2016) Present yourself! By MHC class I and MHC class II molecules. Trends Immunol., 37, 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.. Hewitt E.W. (2003) The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology, 110, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.. Leone P., Shin E.-C., Perosa F. et al. (2013) MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J. Natl. Cancer Inst., 105, 1172–1187. [DOI] [PubMed] [Google Scholar]
- 70.. Trowsdale J. and Knight J.C. (2013) Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics Hum. Genet., 14, 301–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.. Prugnolle F., Manica A., Charpentier M. et al. (2005) Pathogen-driven selection and worldwide HLA class I diversity. Curr. Biol., 15, 1022–1027. [DOI] [PubMed] [Google Scholar]
- 72.. Parham P. and Ohta T. (1996) Population biology of antigen presentation by MHC class I molecules. Science, 272, 67–74. [DOI] [PubMed] [Google Scholar]
- 73.. Barreiro L.B. and Quintana-Murci L. (2010) From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat. Rev. Genet., 11, 17–30. [DOI] [PubMed] [Google Scholar]
- 74.. Frías M., Rodríguez-Cano D., Cuenca-López F. et al. (2017) HLA-B18 as a risk factor of short-term progression to severe liver fibrosis in HIV/HCV co-infected patients with absent or minimal fibrosis: implications for timing of therapy. Pharmacogenomics J., 17, 551–555. [DOI] [PubMed] [Google Scholar]
- 75.. Migueles S.A., Sabbaghian M.S., Shupert W.L. et al. (2000) HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci., 97, 2709–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.. Gough S.C.L. and Simmonds M.J. (2007) The HLA region and autoimmune disease: associations and mechanisms of action. Curr. Genomics, 8, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.. Arce Vargas F., Furness A.J.S., Litchfield K. et al. (2018) Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell, 33, 649–663.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.. Goodman A.M., Castro A., Pyke R.M. et al. (2020) MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med., 12, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.. Sidney J., Peters B., Frahm N. et al. (2008) HLA class I supertypes: a revised and updated classification. BMC Immunol., 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.. Pierini F. and Lenz T.L. (2018) Divergent allele advantage at human MHC genes: signatures of past and ongoing selection. Mol. Biol. Evol., 35, 2145–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.. Chowell D., Krishna C., Pierini F. et al. (2019) Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat. Med., 25, 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.. Parham P., Lomen C.E., Lawlor D.A. et al. (1988) Nature of polymorphism in HLA-A, -B, and -C molecules. Proc. Natl. Acad. Sci. U. S. A., 85, 4005–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.. Reche P.A. and Reinherz E.L. (2003) Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J. Mol. Biol., 331, 623–641. [DOI] [PubMed] [Google Scholar]
- 84.. Klein J. and Sato A. (2000) The HLA system. First of two parts. N. Engl. J. Med., 343, 702–709. [DOI] [PubMed] [Google Scholar]
- 85.. Carrington M., Nelson G.W., Martin M.P. et al. (1999) HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science, 283, 1748–1752. [DOI] [PubMed] [Google Scholar]
- 86.. Zinkernagel R.M. and Doherty P.C. (1974) Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature, 248, 701–702. [DOI] [PubMed] [Google Scholar]
- 87.. Tran E., Ahmadzadeh M., Lu Y.-C. et al. (2015) Immunogenicity of somatic mutations in human gastrointestinal cancers. Science, 350, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.. Tran E., Turcotte S., Gros A. et al. (2014) Cancer immunotherapy based on mutation-specific CD4 T cells in a patient with epithelial cancer. Science, 344, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.. Monach P.A., Meredith S.C., Siegel C.T. et al. (1995) A unique tumor antigen produced by a single amino acid substitution. Immunity, 2, 45–59. [DOI] [PubMed] [Google Scholar]
- 90.. Robbins P.F., El-Gamil M., Li Y.F. et al. (1996) A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med., 183, 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.. Lennerz V., Fatho M., Gentilini C. et al. (2005) The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. U. S. A., 102, 16013–16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.. Linnebacher M., Gebert J., Rudy W. et al. (2001) Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int. J. Cancer, 93, 6–11. [DOI] [PubMed] [Google Scholar]
- 93.. Saeterdal I., Bjorheim J., Lislerud K. et al. (2001) Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci., 98, 13255–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.. Saeterdal I., Gjertsen M.K., Straten P. et al. (2001) A TGF betaRII frameshift-mutation-derived CTL epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol. Immunother., 50, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.. Duan F., Duitama J., Al Seesi S. et al. (2014) Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med., 211, 2231–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.. Ghorani E., Rosenthal R., McGranahan N. et al. (2018) Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann. Oncol., 29, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.. Rech A.J., Balli D., Mantero A. et al. (2018) Tumor immunity and survival as a function of alternative neopeptides in human cancer. Cancer Immunol. Res., 6, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.. Łuksza M., Riaz N., Makarov V. et al. (2017) A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature, 551, 517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.. Sarkizova S. and Hacohen N. (2017) How T cells spot tumour cells. Nature, 551, 444–446. [DOI] [PubMed] [Google Scholar]
- 100.. Kim S., Kim H.S., Kim E. et al. (2018) Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol., 29, 1030–1036. [DOI] [PubMed] [Google Scholar]
- 101.. Snyder A., Makarov V., Merghoub T. et al. (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med., 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.. Hodi F.S., O’Day S.J., McDermott D.F. et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med., 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.. Hodi F.S., O’Day S.J., McDermott D.F. et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med., 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.. Lommatzsch M., Bratke K. and Stoll P. (2018) Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med., 379, e14. [DOI] [PubMed] [Google Scholar]
- 105.. Brahmer J.R., Tykodi S.S., Chow L.Q.M. et al. (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med., 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.. Topalian S.L., Hodi F.S., Brahmer J.R. et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med., 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.. Rizvi N.A., Hellmann M.D., Snyder A. et al. (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science, 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.. Hellmann M.D., Nathanson T., Rizvi H. et al. (2018) Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell, 33, 843–852.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.. Van Allen E.M., Miao D., Schilling B. et al. (2015) Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science, 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.. Yarchoan M., Hopkins A. and Jaffee E.M. (2017) Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med., 377, 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.. Legrand F.A., Gandara D.R., Mariathasan S. et al. (2018) Association of high tissue TMB and atezolizumab efficacy across multiple tumor types. J. Clin. Oncol., 36, 12000–12000. [Google Scholar]
- 112.. Hanna G.J., Lizotte P., Cavanaugh M. et al. (2018) Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight, 3(4), e98811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.. Rosenberg J.E., Hoffman-Censits J., Powles T. et al. (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet, 387, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.. Hellmann M.D., Callahan M.K., Awad M.M. et al. (2019) Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell, 35, 329. [DOI] [PubMed] [Google Scholar]
- 115.. McDermott D.F., Huseni M.A., Atkins M.B. et al. (2018) Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med., 24, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.. Knepper T.C., Montesion M., Russell J.S. et al. (2019) The genomic landscape of Merkel cell carcinoma and clinicogenomic biomarkers of response to immune checkpoint inhibitor therapy. Clin. Cancer Res., 25, 5961–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.. Subbiah V., Solit D.B., Chan T.A. et al. (2020) The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) ≥10: a decision centered on empowering patients and their physicians. Ann. Oncol, 31(9), P1115–1118. [DOI] [PubMed] [Google Scholar]
- 118.. Alexandrov L.B., Nik-Zainal S., Wedge D.C. et al. (2013) Signatures of mutational processes in human cancer. Nature, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.. McGranahan N., Furness A.J.S., Rosenthal R. et al. (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science, 351, 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.. Tripathi R., Modur V., Senovilla L. et al. (2019) Suppression of tumor antigen presentation during aneuploid tumor evolution contributes to immune evasion. Oncoimmunology, 8, 1657374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.. Rosenthal R., Cadieux E.L., Salgado R. et al. (2019) Neoantigen-directed immune escape in lung cancer evolution. Nature, 567, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.. Foukas P.G., Tsilivakos V., Zacharatos P. et al. (2001) Expression of HLA-DR is reduced in tumor infiltrating immune cells (TIICs) and regional lymph nodes of non-small-cell lung carcinomas. A putative mechanism of tumor-induced immunosuppression? Anticancer Res., 21, 2609–2615. [PubMed] [Google Scholar]
- 123.. Qin A., Coffey D.G., Warren E.H. et al. (2016) Mechanisms of immune evasion and current status of checkpoint inhibitors in non-small cell lung cancer. Cancer Med., 5, 2567–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.. Korkolopoulou P., Kaklamanis L., Pezzella F. et al. (1996) Loss of antigen-presenting molecules (MHC class I and TAP-1) in lung cancer. Br. J. Cancer, 73, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.. Lou Y., Vitalis T.Z., Basha G. et al. (2005) Restoration of the expression of transporters associated with antigen processing in lung carcinoma increases tumor-specific immune responses and survival. Cancer Res., 65, 7926–7933. [DOI] [PubMed] [Google Scholar]
- 126.. Menter T. and Tzankov A. (2018) Mechanisms of immune evasion and immune modulation by lymphoma cells. Front. Oncol., 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.. Bates J.P., Derakhshandeh R., Jones L. et al. (2018) Mechanisms of immune evasion in breast cancer. BMC Cancer, 556, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.. Kamarashev J., Ferrone S., Seifert B. et al. (2001) TAP1 down-regulation in primary melanoma lesions: an independent marker of poor prognosis. Int. J. Cancer, 95, 23–28. [DOI] [PubMed] [Google Scholar]
- 129.. Ling A., Löfgren-Burström A., Larsson P. et al. (2017) TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology, 6, e1356143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.. Rodig S.J., Gusenleitner D., Jackson D.G. et al. (2018) MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med., 10(450), eaar3342. [DOI] [PubMed] [Google Scholar]
- 131.. Castro A., Ozturk K., Pyke R.M. et al. (2019) Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. BMC Med. Genet., 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.. Campo A.B., Campo A.B., Kyte J.A. et al. (2014) Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int. J. Cancer, 134, 102–113. [DOI] [PubMed] [Google Scholar]
- 133.. Ozcan M., Janikovits J., Knebel Doeberitz M. et al. (2018) Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology, 7, e1445453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.. Tao J., Li Y., Liu Y.-Q. et al. (2008) Restoration of the expression of transports associated with antigen processing in human malignant melanoma increases tumor-specific immunity. J. Investig. Dermatol., 128, 1991–1996. [DOI] [PubMed] [Google Scholar]
- 135.. McGranahan N., Rosenthal R., Hiley C.T. et al. (2017) Allele-specific HLA loss and immune escape in lung cancer evolution. Cell, 171, 1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.. Matey-Hernandez M.L., Danish Pan Genome Consortium, Brunak S. et al. (2018) Benchmarking the HLA typing performance of Polysolver and Optitype in 50 Danish parental trios. BMC Bioinformatics, 19, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.. Kiyotani K., Mai T.H. and Nakamura Y. (2017) Comparison of exome-based HLA class I genotyping tools: identification of platform-specific genotyping errors. J. Hum. Genet., 62, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.. Wang S., Jia M., He Z. et al. (2018) APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene, 37, 3924–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.. Le D.T., Durham J.N., Smith K.N. et al. (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science, 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.. Meier B., Volkova N.V., Hong Y. et al. (2018) Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res., 28, 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.. Germano G., Lamba S., Rospo G. et al. (2017) Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature, 552, 116–120. [DOI] [PubMed] [Google Scholar]
- 142.. Food and Drug Administration FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. [Updated 2017 May 30; cited 2020 April 15]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication. [Google Scholar]
- 143.. Gao J., Shi L.Z., Zhao H. et al. (2016) Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell, 167, 397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.. Shin D.S., Zaretsky J.M., Escuin-Ordinas H. et al. (2017) Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov., 7, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.. Zaretsky J.M., Garcia-Diaz A., Shin D.S. et al. (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med., 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.. Goodman A.M., Piccioni D., Kato S. et al. (2018) Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid Tumors. JAMA Oncol, 4, 1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.. Wolchok J.D., Chiarion-Sileni V., Gonzalez R. et al. (2017) Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med., 377, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.. Skoulidis F., Goldberg M.E., Greenawalt D.M. et al. (2018) Mutations and PD-1 inhibitor resistance in -mutant lung adenocarcinoma. Cancer Discov., 8, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.. Peng W., Chen J.Q., Liu C. et al. (2016) Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov., 6, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.. George S., Miao D., Demetri G.D. et al. (2017) Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity, 46, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.. Miao D., Margolis C.A., Gao W. et al. (2018) Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science, 359, 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.. Taylor A.M., Shih J., Ha G. et al. (2018) Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell, 33, 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.. Davoli T., Uno H., Wooten E.C. et al. (2017) Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science, 355(6322), eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.. Roh W., Chen P.-L., Reuben A. et al. (2017) Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med., 9(379), eaah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.. McGrail D.J., Federico L., Li Y. et al. (2018) Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat. Commun., 9, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.. Whiteside T.L. (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene, 27, 5904–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.. Cristescu R., Mogg R., Ayers M. et al. (2018) Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science, 362(6411), eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.. Charoentong P., Finotello F., Angelova M. et al. (2017) Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep., 18, 248–262. [DOI] [PubMed] [Google Scholar]
- 159.. Jiang P., Gu S., Pan D. et al. (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med., 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.. Mariathasan S., Turley S.J., Nickles D. et al. (2018) TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature, 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.. Chakravarthy A., Khan L., Bensler N.P. et al. (2018) TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun., 9, 4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.. Hugo W., Zaretsky J.M., Sun L. et al. (2017) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell, 168, 542. [DOI] [PubMed] [Google Scholar]
- 163.. Fridman W.H., Pagès F., Sautès-Fridman C. et al. (2012) The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer, 12, 298–306. [DOI] [PubMed] [Google Scholar]
- 164.. Thommen D.S., Koelzer V.H., Herzig P. et al. (2018) A transcriptionally and functionally distinct PD-1 CD8 T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med., 24, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.. Tumeh P.C., Harview C.L., Yearley J.H. et al. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.. Kansy B.A., Concha-Benavente F., Srivastava R.M. et al. (2017) PD-1 status in CD8 T cells associates with survival and anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res., 77, 6353–6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.. Zhang Y., Huang S., Gong D. et al. (2010) Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell. Mol. Immunol., 7, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.. Motzer R.J., Rini B.I., McDermott D.F. et al. (2015) Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J. Clin. Oncol., 33, 1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.. Garon E.B., Rizvi N.A., Hui R. et al. (2015) Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med., 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- 170.. Ferris R.L., Blumenschein G. Jr., Fayette J. et al. (2016) Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med., 375, 1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.. Brahmer J., Reckamp K.L., Baas P. et al. (2015) Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med., 373, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.. Motzer R.J., Escudier B., McDermott D.F. et al. (2015) Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med., 373, 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.. Topalian S.L., Taube J.M., Anders R.A. et al. (2016) Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer, 16, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.. Cogdill A.P., Andrews M.C. and Wargo J.A. (2017) Hallmarks of response to immune checkpoint blockade. Br. J. Cancer, 117, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.. Ribas A. and Hu-Lieskovan S. (2016) What does PD-L1 positive or negative mean? J. Exp. Med., 213, 2835–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.. Hendry S., Byrne D.J., Wright G.M. et al. (2018) Comparison of four PD-L1 immunohistochemical assays in lung cancer. J. Thorac. Oncol., 13, 367–376. [DOI] [PubMed] [Google Scholar]
- 177.. Conroy J.M., Pabla S., Nesline M.K. et al. (2019) Next generation sequencing of PD-L1 for predicting response to immune checkpoint inhibitors. J Immunother Cancer, 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.. Herbst R.S., Soria J.-C., Kowanetz M. et al. (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature, 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.. Inoue H., Park J.-H., Kiyotani K. et al. (2016) Intratumoral expression levels of, and along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma. Oncoimmunology, 5, e1204507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.. Reuben A., Spencer C.N., Prieto P.A. et al. (2017) Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med., 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.. Snyder A., Nathanson T., Funt S.A. et al. (2017) Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: an exploratory multi-omic analysis. PLoS Med., 14, e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.. Hopkins A.C., Yarchoan M., Durham J.N. et al. (2018) T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight, 3(13), e122092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.. Zappasodi R., Budhu S., Hellmann M.D. et al. (2018) Non-conventional inhibitory CD4Foxp3PD-1 T cells as a biomarker of immune checkpoint blockade activity. Cancer Cell, 33, 1017–1032.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.. Bindea G., Mlecnik B., Tosolini M. et al. (2013) Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity, 39, 782–795. [DOI] [PubMed] [Google Scholar]
- 185.. Sade-Feldman M., Yizhak K., Bjorgaard S.L. et al. (2018) Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell, 175, 998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]