

Abstract
Ocean metaproteomics is an emerging field enabling discoveries about marine microbial communities and their impact on global biogeochemical processes. Recent ocean metaproteomic studies have provided insight into microbial nutrient transport, colimitation of carbon fixation, the metabolism of microbial biofilms, and dynamics of carbon flux in marine ecosystems. Future methodological developments could provide new capabilities such as characterizing long-term ecosystem changes, biogeochemical reaction rates, and in situ stoichiometries. Yet challenges remain for ocean metaproteomics due to the great biological diversity that produces highly complex mass spectra, as well as the difficulty in obtaining and working with environmental samples. This review summarizes the progress and challenges facing ocean metaproteomic scientists and proposes best practices for data sharing of ocean metaproteomic data sets, including the data types and metadata needed to enable intercomparisons of protein distributions and annotations that could foster global ocean metaproteomic capabilities.
Keywords: Metaproteomics, ocean, biogeochemistry, data sharing, best practices
INTRODUCTION
The measurement of many proteins within environmental microbial communities, known as metaproteomics, is of increasing interest to oceanographers and protein scientists. The capacity to directly examine a multitude of functional attributes of microbial communities and their linkages to both ecology and biogeochemistry was once aspirational, but now appears achievable with recent improvements in genomic sequencing and mass spectrometry technology. Emerging metaproteomic methodologies, in concert with other traditional and new approaches, could be particularly powerful in the study of how complex environmental systems operate, as well as how they respond to environmental changes.
Since the development of mass spectrometry based proteomic technologies, there has been an increasing number of metaproteomic or community-based analyses (Table S1), including those of marine/ocean biota (Table 1). Metaproteomics of complex environmental samples such as seawater, sediments, sinking particles, and biofilms have great potential for revealing insight into biogeochemical cycling and microbial response to environmental change in marine systems. For example, recent ocean metaproteomic studies have provided new insights into microbial nutrient transport,1,2 colimitation of carbon fixation processes,3 biogeochemical processes within oxygen minimum zones,4 the composition of microbial biofilms,5 dynamics of carbon flux in marine ecosystems,6–8 and seasonal shifts in microbial metabolic diversity.9 Future methodological developments should lead to new capabilities such as characterizing large scale ecosystem changes, estimating biogeochemical reaction rates from enzyme concentrations and conducting in situ stoichiometric measurements. In the short time since the emergence of these metaproteomic methods, they have been applied to environments around the world: including coastal and open ocean pelagic environments from the Atlantic and Pacific Oceans, and even to the rapidly changing polar environments of the Arctic and Antarctic regions. Diverse biological communities have been sampled including free-living microbial and algal communities (including microbiomes), sinking particles, marine sediments, and even biofilms attached to human built environments.1–3,5,7,8,10–15 Also critical to the development and deployment of metaproteomic approaches in natural environments are controlled laboratory experiments on cultivated microbes from the environment,10,11,16–23 which can enable the identification and verification of protein biomarkers that characterize environmental processes.
Table 1.
Examples of Ocean Metaproteomic Studies
| North Atlantic Ocean, Bermuda Atlantic Time Series Station | Sowell et al., 2008; Bridoux et al., 2015; Saito et al., 2017 |
| Ocean scale metaproteomics in the Atlantic Ocean | Morris et al., 2010; Bergauer et al., 2018 |
| Antarctic Peninsula, Southern Ocean | Williams et al., 2012 |
| Bering Sea Algae | Moore et al., 2012, 2014 |
| Targeted metaproteomics of Central Pacific Ocean | Saito et al., 2014; Saito et al., 2015 |
| Marine biofilms, shiphull environments | Leary et al., 2014 |
| Metaproteomics of the Saniitch Inlet Oxygen Minimum Zone, Coastal Pacific Ocean | Hawley et al., 2014 |
| Metaproteomics of aquatic estuary microbial communities | Colatriano et al., 2015 |
| Marine sediments | Moore et al., 2012, 2012, 2014 |
| Phaeocystis and diatom blooms in the Ross Sea of Antarctica | Bertrand et al., 2013; Bender et al., 2018 |
CONFRONTING CHALLENGES IN METAPROTEOMIC RESEARCH
Despite this progress, key challenges remain in the application of proteomic methods in environmental contexts.24 These challenges can be organized into four broad categories: (1) environmental sample acquisition and protein extraction, (2) chromatographic separation and mass spectrometry analysis, (3) informatic data processing, and (4) data archiving and sharing (Figure 1). A defining feature that affects all of these categories is that the ocean and other natural environments contain a multitude of organisms that are not easily separated, and hence are typically studied together in this “meta” community context. For example, in a typical ocean seawater sample, the microbial diversity includes prominent taxa from each of the three major domains of life as well as from viruses. This natural biological diversity manifests itself in a tremendous chemical complexity for proteomics analysis, where proteins from many organisms are digested into peptides and analyzed together, resulting in peptides that have the potential to be shared across multiple species or ecotypes, or whose sequences are not within available DNA databases. New generations of fast scanning high resolution mass spectrometry instrumentation, such as orbitrap and time-of-flight instruments, now allow deep interrogation of these complex samples and the many low abundance or chimeric peaks within them, thereby improving and elevating the confidence of identification. However, shared chemical similarities across this biologically diverse environment creates a number of challenges throughout the metaproteomic workflow. In this document we identify and describe the status of these challenges in order to enable researchers from environmental fields and beyond to focus efforts on resolving them. In addition, we propose a set of best practices for current and future data sharing for ocean metaproteomic data sets in order for researchers to make maximal use of current and incoming data sets. This effort is necessary to enable interoperability and accessibility as this exciting new data type becomes more widely adopted and to allow critical temporal comparisons as the field evolves.
Figure 1.
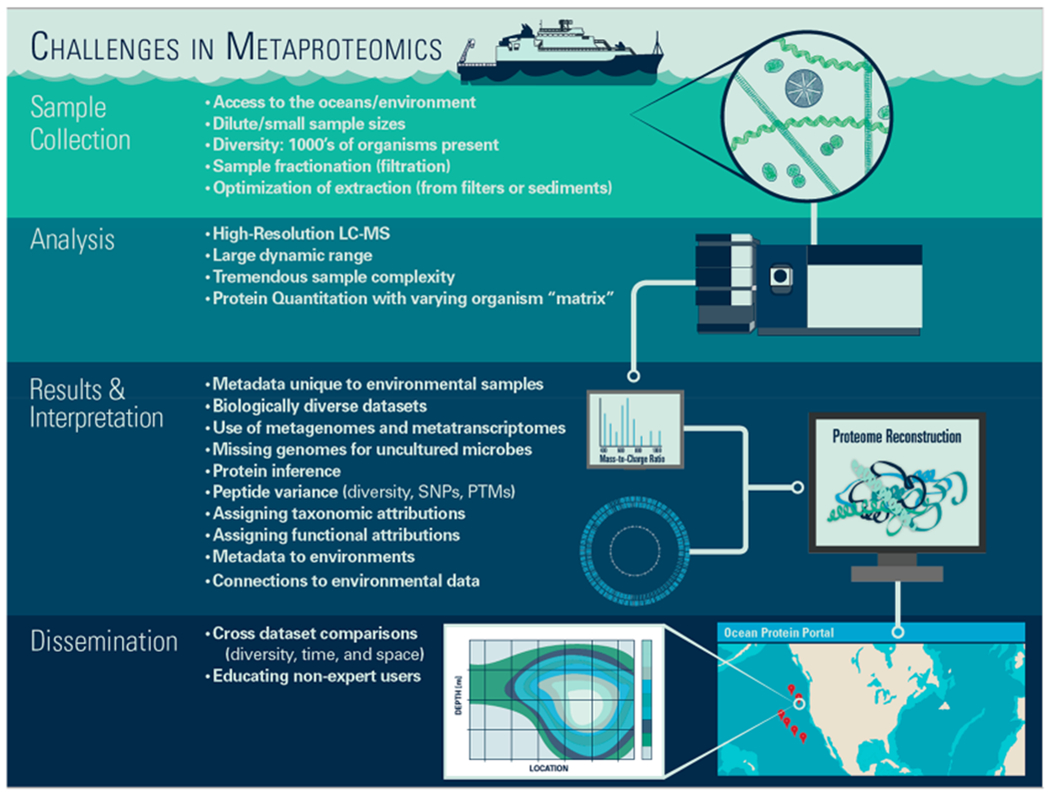
Analysis of proteins within natural environments presents unique challenges that can be improved upon to allow this new type data to inform ecosystem function and change. These challenges span sample collection and extraction, mass spectrometry analysis, informatic approaches, and data management and dissemination.
Sample Acquisition from Natural Environments and Protein Extraction
The study of natural marine communities presents significant challenges in sample collection far beyond that involved in laboratory-based studies. First, accessing the vast oceans that cover 70% of the Earth’s surface can require expeditions on research vessels to reach remote oceanic locations. Second, in seawater environments microbes are often 3–4 orders of magnitude more dilute than model organism laboratory cultures. For example, marine microbial populations can range from 1000 to 100 000 cells per milliliter in seawater compared to model microorganism cultures, such as Escherichia coli that exceed a billion cells per milliliter. This dilute cellular abundance in freshwater and marine environments requires filtration oftens to hundreds of liters of seawater by combining multiple sampling bottles or using specialized in situ underwater pumping systems to yield useful quantities of protein for mass spectrometry analyses (Figure 2a). Similarly, collection of sinking particles and sedimentary samples can require specialized sediment traps and coring devices. There is considerable room for improvement in engineering of sample collection as well as methodological verification of sample handling processes, due to the combined challenges posed by large geographical and depth space to be sampled and the need to concentrate dilute biological material without altering the proteomic signal within those samples. Preservation of proteins at ambient temperatures appears to be possible for some marine microbes using high salt RNA preservatives, allowing in situ environmental samplers to be designed and built, and time series to be taken. For example, a commercially available RNA preservative was shown to preserve proteins within cyanobacteria biomass at room temperature for a month with no reduction in the number of protein identifications, although periplasmic and extracellular protein alkaline phosphatase was observed to be variable, implying loss during filtration.25 This study of dissolved proteins and their role in biogeochemical cycling is also of interest but will likely require separate sampling procedures to concentrate them from seawater. There are new robotic autonomous underwater vehicles (AUVs) being developed that are specifically designed for proteomic sampling in natural environments. For example, the Clio AUV incorporates recent developments in in situ pumping systems26 to collect a suite of discrete protein and other biogeochemical samples by vertically moving and holding position at 16 depths over 6 km of a vertical ocean water column. Integrated over typical ocean expeditions, improvements in sampling efficiency allowed by AUVs such as Clio will enable greatly increased sampling depth resolution and geographic coverage of the vast ocean basins.27
Figure 2.
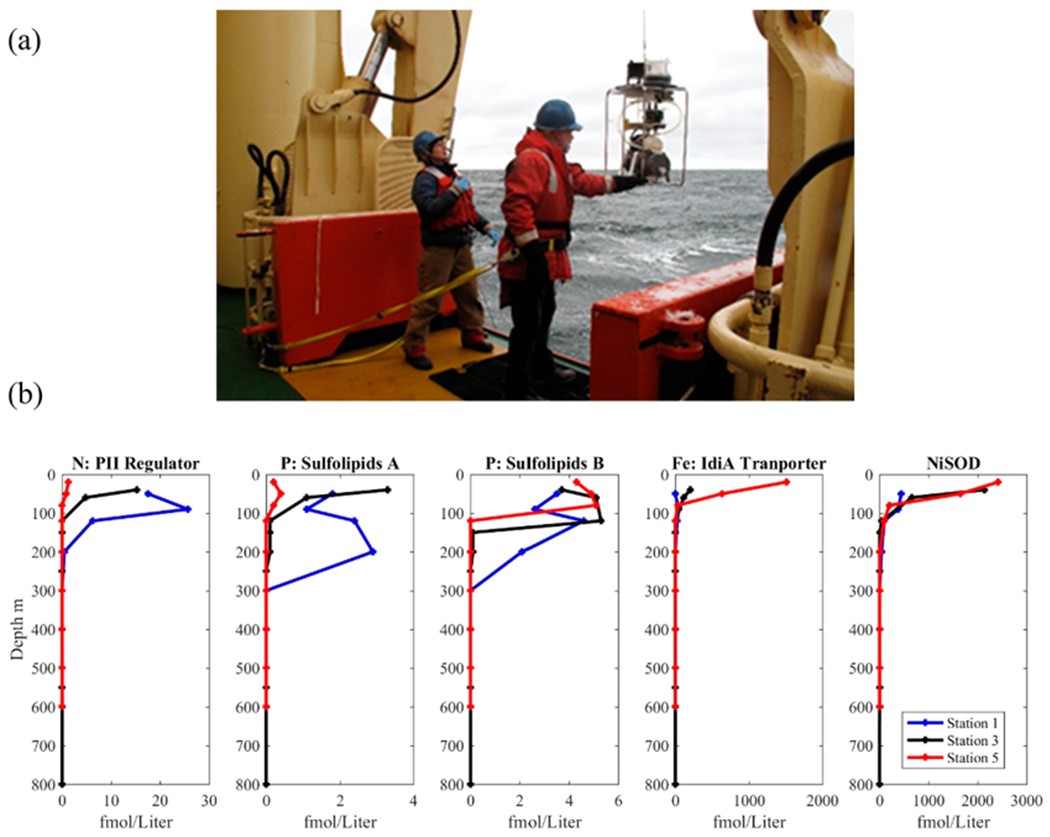
(a) Collection of ocean metaproteomic samples by in situ underwater McLane pump sampler as deployed in Terra Nova Bay of the Ross Sea in Antarctica aboard the icebreaker R/V Palmer to capture the microbial and algal communities as well as larger sinking particles by filtration of several hundreds of liters. (b) Example vertical distributions of three microbial proteins in the Equatorial Pacific Ocean using targeted metaproteomics that are biomarkers of nitrogen (N), phosphorus (P), iron (Fe) nutrient stress, and nickel (Ni) biogeochemical cycling (data from Saito et al., 2014, https://www.bco-dmo.org/dataset/646115). Proteins shown include the nitrogen PII regulator protein from Prochlorococcus (sequence VNSVIDAIAEAAK), the sulfolipid biosynthesis protein from Prochlorococcus (NEAVENDLIVDNK), UDP sulfolipid biosynthesis protein from multiple taxa (FDYDGDYGTVLNR), the IdiA iron transporter from Prochlorococcus (SPYNQSLVANQIVNK), and the nickel superoxide dismutase enzyme from Prochlorococcus and Synechococcus (VAAEAVLSMTK). Taxonomic assignments determined using METATRYP.14
When laboratory and environmental scientists interact, confusion can arise from differing definitions/expectations of biological replication. The scientific approach and objectives of environmental sampling are distinct from laboratory experiments. There are clear differences between laboratory experiments that can be easily replicated and sampling the constantly changing natural environment. The challenge in sample acquisition in marine metaproteomics described above can preclude the collection of replicates; for example, commonly used in situ pumps are tethered to a single wire and deployed at predetermined depths and take several hours to filter large volumes. Since the ocean is a fluid environment, a second sampling deployment would collect a slightly different water mass in space or time, depending on if the sampler was placed adjacent on the vertical hydrowire or as a successive sampling deployment after completion of the first sampling. As a result, real variations (albeit small) in biological communities and chemical properties could be captured in attempts at sampling replication, and true biological duplicates are aspirational, if not impossible. In place of replication, oceanographers often look for “oceanographic consistency” in trends across vertical depth structure (or horizontal structure in the case of ocean basin sections) as a useful means to validate results.28 Single samples have demonstrated this oceanographic consistency in capturing large scale oceanographic and metabolic processes across chemical and biological gradients.2,3,29
The comprehensive extraction of proteinaceous material from biomass is another challenge in metaproteomic studies. Environmental samples can be extraordinarily complex due to being composites of significant biological diversity, as well as having additional biogenic and nonbiogenic materials within them. Moreover, the biological composition of metaproteomic samples can be largely unknown prior to extraction. Hence, the ability to tailor and optimize extraction protocols to the environmental sample type presents unique difficulties. In water column environments, depending on the environment and collection strategy, an environmental microbial sample will contain dozens of major biological species and hundreds to thousands of trace species.30,31 Sediment and sinking particle samples contain not only a mixture of organisms, but partially degraded peptides created by a phalanx of microbial proteases produced by heterotrophic bacteria consuming those particles. There are also numerous complex symbiotic communities such as corals, hydrothermal vent tube worms, and other symbiotic systems where the proteins of the microbial assemblage will be present within the extensive proteome of a eukaryotic host. Studies have examined the recovery efficiency of different extraction buffers on sedimentary and microbial biomass.25,32 Moreover, the presence of biogenic soft and hard parts, including mucilage, calcium carbonate, and siliceous components, as well as mineral phases, can complicate chemical separation of proteins and impair protein extraction efficiencies and require development of matrix-specific extraction protocols.6,12,17,22,33
Mass Spectrometry Analyses
To date, the mass spectrometry measurement component of metaproteomics has utilized three types of approaches: data-dependent acquisition (DDA) for discovery proteomics,10,34,35 data-independent acquisition (DIA2,36), and targeted metaproteomics for quantitative analysis using multiple or parallel reaction monitoring approaches (MRM/PRM3,11,14).
Briefly, these approaches differ in how they select ions for fragmentation: DDA approaches continually select abundant features within ms1 spectra for further ms2 fragmentation analysis (isolating the most abundant peaks within each parent ms1 spectra for fragmentation, with various user parameters such as excluding recently fragmented precursors for a short period),37 while DIA methods conduct ms2 fragmentation on small sequential mass windows across the entire mass range of interest,38 thereby potentially fragmenting spectra of all ions, assuming sufficient intensity. In contrast, targeted methods focus their fragmentation analyses only on precursor ions found on the target list, thereby increasing sensitivity by focusing mass spectrometry time on target ions.39–41 DDA approaches continue to be most prevalent in metaproteomics, but targeted and DIA approaches are increasingly being explored for their ability to provide absolute and relative quantitation, respectively. An example DDA workflow is shown in Figure 3, and examples of vertical profiles of targeted peptides from MRM/PRM experiments are shown in Figure 2b.
Figure 3.
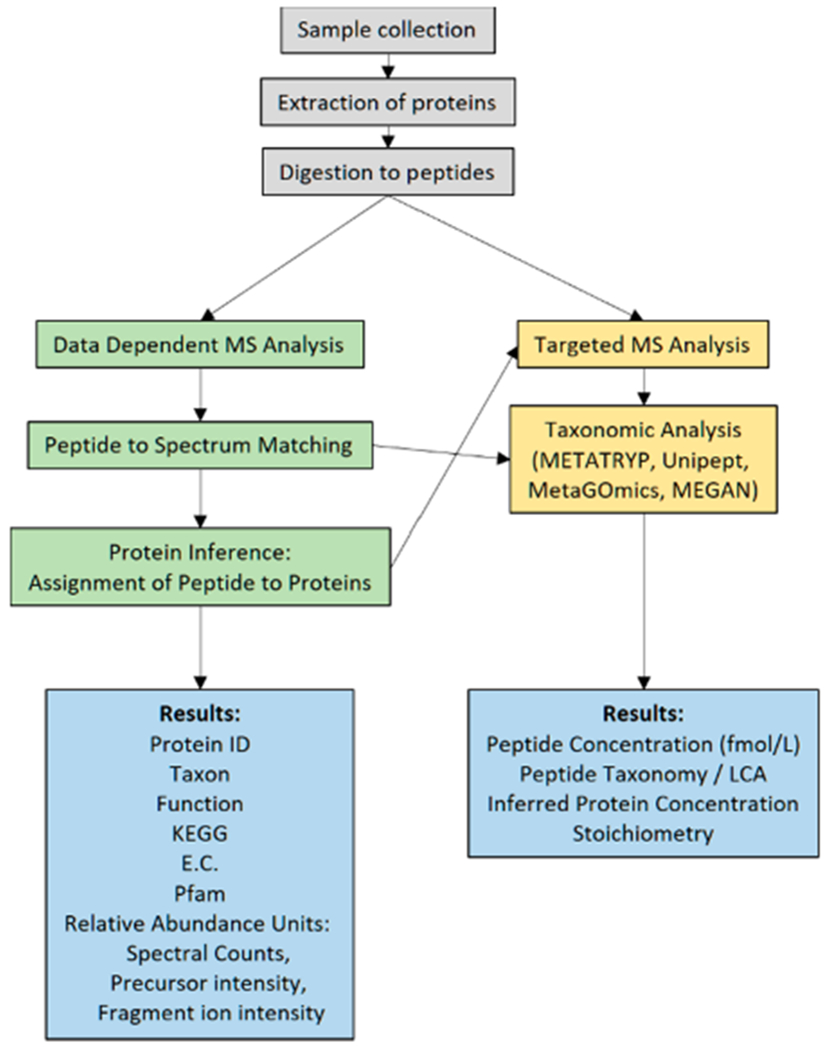
An example environmental metaproteomic workflow where environmental samples are collected and extracted (gray), discovery proteomics are conducted (green), and peptide targets from selected proteins of interest can be assayed using isotopically labeled peptide standards whose taxonomic assignment can be queried against databases of genomes and metagenomes (yellow). The results can provide relative and absolute abundance measurements of the protein from the microbial and algal community, including functional and taxonomic information (blue).
While these proteomic methods have become common in proteomic analyses on single organisms, the complexity of metaproteome samples presents challenges for each method with regard to both the chromatographic separation and mass spectrometry components. For comparison, the complexity of ocean seawater metaproteome samples appears to be significantly greater than the human proteome, despite the latter typically being considered to be one of the more complex proteome sample types. This is illustrated in Figure 4a where a three-dimensional (3D) visualization of the mass spectra acquired from a surface sample in the central Pacific Ocean is shown (filtered by 0.2–3.0 μm size fraction range), and in Figure 4b–c with spectra from a small mass range examined at equivalent chromatographic elution times (575–578 m/z ms1 window and 140–141 min) revealing more observable mass peaks events in an ocean sample (Figure 4c) when compared to a human cell line (HeLa) sample (Figure 4b). These observations of metaproteome complexity were also quantitatively confirmed across entire samples by analysis of ms1 peaks within triplicate HeLa injections and five metaproteome samples from the Pacific Ocean at varying depths in Figure 5. These HeLa-ocean comparisons used identical chromatographic and mass spectrometry settings and were run within 1 week of each other using the same nanospray column, with 0.5 μg of HeLa analyzed per injection, while 1.0 μg ocean sample was analyzed per sample injection. In this example, the number of peaks was higher in the metaproteomes compared to HeLa (Figure 5a–c), while the total ion current (TIC) was considerably lower across all metaproteome samples (Figure 5a–b,d), implying more peaks of lower intensity in the metaproteome samples. This high complexity of metaproteomics samples presents significant challenges to current chromatographic and mass spectrometry workflows. For example, real-time feature identification (peak picking) software on mass spectrometers has not been optimized to process chimeric peak features that appear to be ubiquitous in metaproteomic samples, where chimeric features are peaks so close in mass to other peaks preventing a successful charge state estimate that is needed to trigger ms2 fragmentation in bottom up DDA experiments. Moreover, the low abundance of many ions in metaproteomic samples (as observed in Figure 4c) poses an additional challenge, where the numerous low abundance peaks among more abundant ones remain uncharacterized due to physical limits on the number of ions entering the mass spectrometer at any time, a problem that can challenge both DDA and DIA methods.
Figure 4.
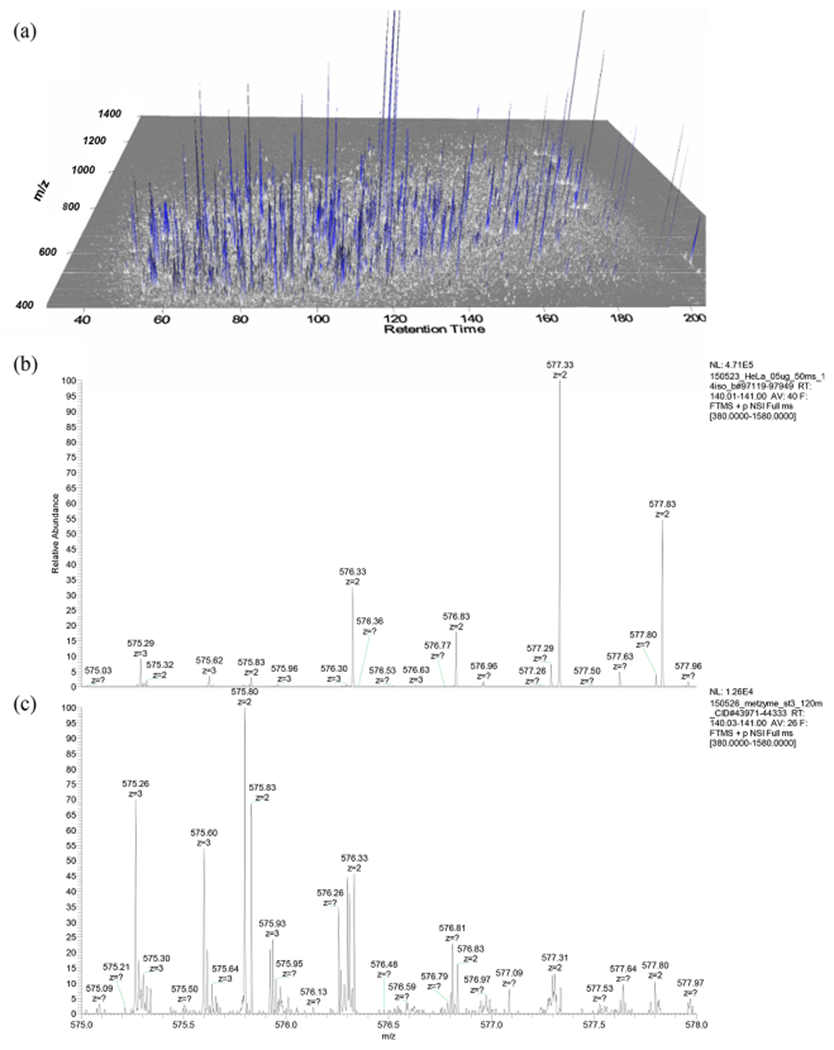
(a) Three-dimensional representation (axes of retention time (min), m/z, and intensity) of complex spectra associated with an environmental ocean sample from the Equatorial Pacific from the METZYME expedition (200 m depth, produced in MzMine242). Comparison of a 3 m/z ms1 mass window (575–578 m/z, 140–141 min) from (b) human proteome spectra (HeLa cell line) and (c) ocean metaproteome (120 m depth) provides an example of the high complexity of environmental samples due to the biological diversity.
Figure 5.
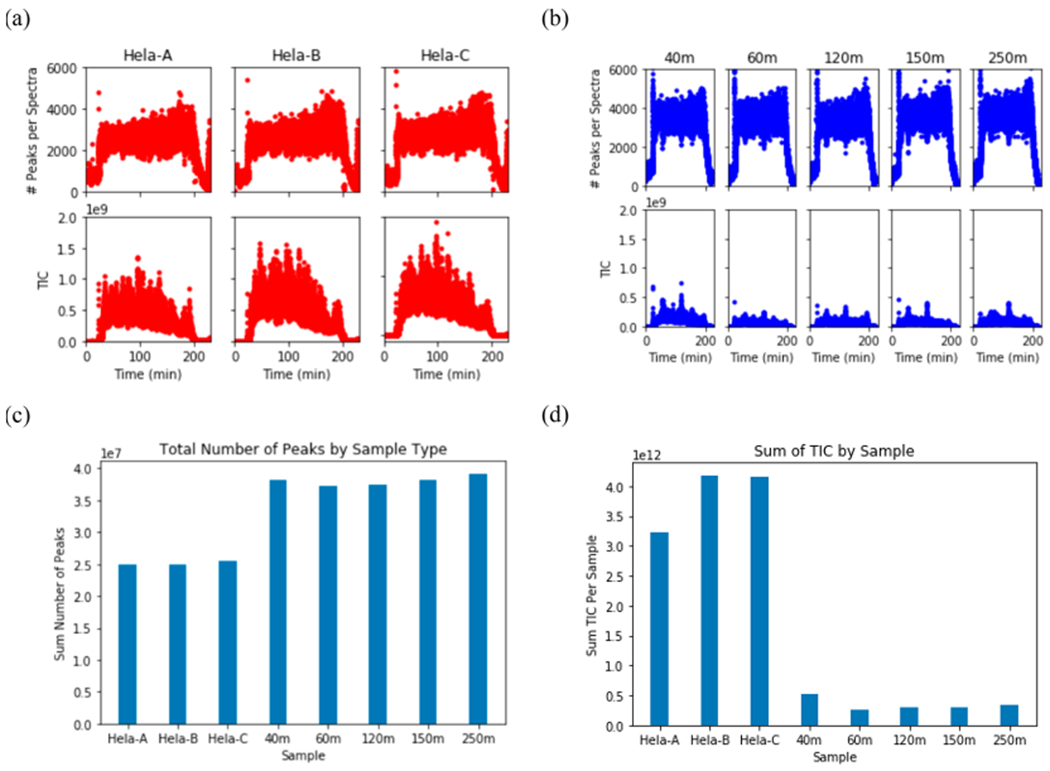
Peak analysis of human cell line and ocean metaproteome samples by identical chromatographic and mass spectrometry conditions. (a–b) Number of peaks identified in replicates (top rows) and the total ion current (TIC, bottom rows) of the sample in Hela (Panel A, replicates Hela-A, Hela-B, and Hela-C) and an ocean metaproteome sample (Panel B, depth 40, 60, 120, 150, and 250 m, Metzyme Expedition Station 3). Samples were run during the same week on the same nanospray column (see methods) with similar amounts of protein injected (0.5 μg for Hela per injection, 1 μg for ocean metaproteome). (c) Total number of peaks by sample type showed a higher number peaks in ocean metaproteome samples consistent with Figure 4, while (d) TIC by sample showed much lower summed peak intensity within the metaproteome samples.
Metaproteomic approaches have made progress in addressing the challenges of this sample complexity. For example, chromatographic approaches have been improved by applying two-dimensional chromatography10,34 or gas phase fractionation2,7,36,43 to distribute the sample complexity for mass spectrometry analysis across subsamples or temporal chromatographic separation as a means to obtain deeper metaproteomes. Moreover, DIA approaches have been utilized to address the crowded and complex nature of ion chromatograms that are specific to metaproteomics,2,36 although bioinformatic pipelines for mixed community DIA data sets are still being developed. Finally, the application of targeted methods offers improved sensitivity and absolute quantitation of biomarkers for environmental stress by targeting representative peptides (Figure 2).3,11,14
Future collaboration with hardware and software developers could also greatly improve metaproteomic research efforts. For example, effort could be expended to capture greater information about the numerous low intensity ions that are missed by real-time and postprocessing algorithms due to several factors including insufficient ions trapped for high-quality ms2 fragmentation spectra, ions being chimeric with other nearby peaks, and lack of charge state assignments. Recent efforts in improving detection of chimeric peaks maybe useful in this regard when applied to metaproteomic applications.44
Finally, there is an important need for intercomparison and intercalibration efforts with regard to protein extraction efficiency and mass spectrometry accuracy and precision. Chemical oceanographers have a legacy of successful intercalibration efforts that have enabled global scale studies of ocean chemistry, such as the recent GEOTRACES (an international study of the marine biogeochemical cycles of trace elements and their isotopes) trace elements and isotope global section program.45 For ocean metaproteomics, uniform preparation of large batches of intercalibration samples may be challenging given that samples can vary in biological composition and sampling methodologies, and likely multiple smaller initial intercomparison studies might first be needed. Alternatively, simpler “synthetic” metaproteome samples could be created by mixing of laboratory microbial isolates that could be made in large batches and distributed, although these may not reproduce the depth of biological diversity nor a realistic environmental chemical matrix. Intercalibrations could be applied to the two current major approaches to ocean metaproteomic mass spectrometry analysis: global discovery data sets and targeted metaproteomics, with studies providing metagenomic databases and isotopically labeled peptide standard materials to facilitate analyses, respectively. Moreover, intercalibration exercises could be conducted on consensus standard sample sets of some example biological communities initially, such as seawater microbial communities that are well-characterized with respect to metagenomic data, although eventually many types of biological materials could be selected for intercalibration (sediments, biofilms, etc.). Finally, future additional types of metaproteomic analyses could be added for intercomparison such as data independent analysis and post-translational modifications within metaproteomic environmental samples.
Metaproteomic Data Analysis
Data analysis of mass spectra from metaproteomics experiments presents many challenges compared to single organism proteomics. In particular, each metaproteome mass spectral data set can contain tremendous biological diversity whose composition is often largely unknown. Furthermore, established proteomic workflows that conduct peptide-to-spectrum matching (PSM) by comparing peptide precursor and fragment ion masses to corresponding predicted masses using genomic reference databases were never designed to handle the inherent complexity and multiple biological entities within metaproteomic data sets, and hence approaches thus far have been improvised adaptations. The expanse of unknown biological diversity often results in metaproteomic protein database searches that are typically large and of redundant nature. This has an effect on database selection, data-search algorithm utilized, subsequent FDR statistics,46,47 and protein inference.48,49 Additionally, metaproteomics shares the challenge of functional and taxonomic assignments with metagenomics, relying on a comparative approach with model organisms, resulting in many proteins of unknown function or taxon. Finally, metaproteomic workflows typically involve the integration of multiple software tools, making documentation and reproducibility difficult as tools evolve. Despite these challenges, multiple approaches have been developed over the last 13 years (Table S1). The analytical workflows that have been developed to date are mainly comprised of (a) database generation, (b) database search, and (c) taxonomy and functional analysis.
Database Type (Genome, Metagenome, Metatranscriptome, Custom).
In order for PSM algorithms to assign peptide sequences to spectra from MS experiments, the observed tandem mass spectra are cross-correlated and scored against theoretical spectra generated in silico from the provided protein sequences. The collection of protein sequences is generated from available genomic, metagenomic, or metatranscriptomic sequence information and commonly referred to as the genomic or protein database. High scoring peptide spectral matches (PSMs) are then reported with their corresponding protein sequence and annotation from the original database. Importantly for metaproteomics, the peptides and proteins reported are dependent on the coherence of the original genomic sequence information relative to the organism(s) present in the sample. More often than not in metaproteomics, each sample’s composition of biological diversity is unknown or its characterization is limited by the depth of DNA sequencing and final assembly. As a result, if a peptide sequence is not in the database, the peptide will not be identified nor will its contribution to a protein identification be included in the experiment. Furthermore, quality of gene prediction algorithms can affect protein detection: if protein-encoding genes are missed during the initial gene prediction phase, then they will not be included in the protein search database. While gene prediction from prokaryotic genomes is relatively straightforward, it becomes challenging for more complex microbial eukaryotic genomes, owing to the complexity and diversity of eukaryotic gene structure (e.g., predicting introns and exons). However, eukaryotic gene prediction algorithms are continually advancing, and indeed proteomics plays a large role in the accurate identification of protein encoding regions of eukaryotic genomes through proteogenomic efforts.50–52 Additionally, the incomplete nature of peptide fragmentation yields high variability in final peptide interpretations, making database choice and construction pivotal.53 Finally, the occurrence of similar but not identical protein sequences (homologues) in closely related organisms adds significant complexity to metaproteomic search databases.
There are three main approaches for creating metaproteomic databases: (1) sequence and assemble a metagenome, (2) assemble a database from the public environmental genomic repositories, and (3) create a pseudo-metagenome by including desired taxonomic classes or species. The composition of the protein search database used to search the mass spectra from a metaproteomic sample has a profound effect on biological conclusions.54 Timmins-Schiffman et al. recommended a best practice for environmental proteomics of corresponding site and time specific metagenomes in order to generate accurate proteomic databases to assign peptide sequences and protein annotations.3,4,10,55,56 While this avenue represents the ideal scenario, at some point sufficient metagenomic coverage of specific environments should allow decoupling between genomic and proteomic analyses as a large inventory accrues of deeply sequenced data sets from diverse environments.57 However, as evolution is a dynamic process, resequencing of these environments will be required to capture continued community adaptation to changing environments and evolutionary forces which are already evident in repeated marine sequencing efforts over seasonal time scales.58
There are a variety of publicly available metagenomics data sets that marine metaproteomics researchers have used, for example, the J.C. Venter Institute’s Global Ocean Sampling (GOS) database.1,59–61 In addition, there are environmental metagenomics databases available at major repositories and portals such as EBI, JGI, and iMicrobe (https://www.ebi.ac.uk/metagenomics, https://img.jgi.doe.gov, http://www.imicrobe.us). For eukaryotic phytoplankton and protists, genomic, transcriptomic, and metagenomic resources are considerably more scarce, though recent availability of the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP) has begun to address this challenge.62 The application of large databases (either public or metagenomics) still suffers from limitations with respect to sensitivity of identifications. One approach to alleviating this problem applied a “metapeptide database” from shotgun metagenomics sequencing and demonstrated a significant increase in the number of identifications presumably due to a more accurate and compact database as compared to an assembled predicted metaproteome and NCBInr.63
Finally, the selection and compilation use of individual microbial genomes for a metaproteomic database can also be useful in metaproteome analysis. Given that many of the major microbial taxa in the oceans were discovered in recent decades and in many cases there are few laboratory isolates and accompanying genomes, there is significant need to amend large public databases with new representative microbial genomes.64–66 In contrast to metagenomic data sets, these genomes of cultivated isolates also provide clarity with regard to taxonomic attribution that can be obfuscated by limitations of metagenomics assembly and annotation. Increasing availability of single cell genomic data (single amplified genome; SAGs) can also contribute significantly to databases for metaproteomic analysis. Notably however, the SAG technology does not produce complete genomic sequences (unclosed genomes), and hence care must be taken when trying to interpret the absence of a protein using metagenomics or SAG databases to avoid false negatives. For eukaryotic metaproteome analysis, transcriptome data can also serve as a useful source of sequence for the protein database generation since full genomic information of marine eukaryotes is relatively rare and the DNA architecture is more complex due to the prevalence of noncoding intron regions intertwined with the protein-coding exons. A recent study from the Ross Sea of Antarctica found that for a diverse bloom community with abundant eukaryotic phytoplankton, a combined database of transcriptomes from cultured isolates and field metatranscriptome provided a richer metaproteome result than either database alone.10
Search Engine.
In common DDA proteomic workflows, the search engine that conducts the PSM analysis is central to protein discovery and identification. Application of these PSM algorithms (e.g., SEQUEST, X!Tandem)67–69 have been successfully applied to metaproteomic analyses, despite the fact that they were never designed to deal with the complexity of metaproteomic data sets. Search algorithms are chosen based on the following factors such as the ability to search large databases, speed, and the ability to generate outputs that are compatible with downstream processing steps such as peptide or PSM output with robust FDR threshold calculations. Most of the suggested database generation strategies generate large databases, which in turn affect the sensitivity of identifications. Multiple strategies have been suggested to increase peptide identifications. This includes the two-step method for searching large databases;70–74 and a cascaded search method.75 Muth et al. have recommended using a database sectioning approach so that searches against subsets of a large database may increase the number of identifications.53 They have also suggested using multiple search algorithms in order to increase the percentage of peptide spectral matches in a data set. For example, SearchGUI/PeptideShaker,76 which uses at least eight open-source search algorithms, can facilitate this multipronged approach and can be used to search against large databases.53 Irrespective of the choice of search algorithms, the goal is to generate outputs with maximal coverage of mass spectra that are compatible with the next steps of taxonomic analysis, functional analysis, and subsequent targeted validation.
Despite these initial successes, it is apparent that these workflows and algorithms could be improved upon to confront significant challenges of spectral complexity, metaproteomic protein inference, and taxonomic attribution within environmental samples. Specifically, the presence of numerous low abundance peak features, discerning chimeric peaks (Figure 2), and assignment of corresponding peptide charge states are difficult for current PSM algorithms and likely result in significant underestimation of peptide identifications within metaproteomic spectra. Application of de novo search algorithms and spectral libraries could also improve identification of peptides from metaproteomics samples.77
Taxonomic and Functional Annotation.
Metaproteomics has a distinct utility in determining the protein functional expression by a microbial community. However, the functional interpretation of a metaproteomics data set is inherently reliant upon the underlying annotation of the protein search database, including the prediction of protein-encoding genes from genomic data and the subsequent functional annotation of the predicted proteins. While much of the taxonomic and functional attribution of metaproteomic results can leverage metagenomic annotation pipelines, there are aspects that are unique to metaproteomics. In particular, the basal unit of proteomic identification is generally the tryptic peptide (due to the effectiveness of trypsin in proteolytic digestions), resulting in amino acid sequence coverage without overlaps, except in cases of missed cleavages. Due to the presence of unknown biological diversity, it is possible to have tryptic peptides that are shared within or across species. As a result, the greatest confidence in metaproteomic discovery occurs on the peptide level, and creates a need in metaproteomic research for investigation of sequence taxonomy on the peptide level. This is also an issue since inference of specific taxonomy tends to be more difficult than function in typical sequence analysis (e.g., BLAST), due to the sharing of biochemical capabilities by many organisms. Two web-based applications are available for this, Unipept and Metatryp, that search DNA sequence data for the presence of user entered tryptic peptide sequences and estimate the lowest common ancestor (LCA; kingdom, phylum, genus or species) for each peptide query14,78,79 (Figure 2b). The applications are distinct in the DNA sequence databases they search, with Unipept searching the UniProt genomic database as well as providing cross-referenced EC numbers and GO terms,80 while Metatryp allows use of custom genomic data and metagenomic data (including single amplified genomes and metagenome assembled genomes) with a focus on marine environments (http://metatryp.whoi.edu).14 The choice of database can affect results; for example, currently Unipept maps 51% of the peptides from the Morris et al. South Atlantic data set2 to sequences within Uniprot, implying that genomic data availability still hinders interpretation of ocean metaproteomic data sets (https://unipept.ugent.be/mpa). There are additional bioinformatic tools for taxonomic analyses that may be useful for metaproteomic research such as MEGAN microbiome software that computes taxonomic profile by assigning PSMs from a metaproteomics experiment to an appropriate taxonomic unit within the NCBI taxonomy. In addition, the recently developed MetaProteomeAnalyzer that uses outputs from SearchGUI/PeptideShaker81 has taxonomic analysis capability.
Connecting protein functions with metaproteomic data sets is a key goal that can be accomplished in variety of ways. BLAST analyses of the metagenomics contigs being used for PSMs provide high-quality searches by using longer sequences, but require availability of well-annotated metagenomics databases. Additional software is available for downstream metaproteomic functional analysis including peptide-level MetaGomics82 or Unipept,83 protein-level (for example, MEGAN84), protein orthologs (for example, EggNOG mapper85 or metaprotein/protein-group level (for example, MetaProteomeAnalyzer). Each of these methods uses distinct annotation databases, such as UniProt (for example software tools such as MetaGomics, Unipept or MetaProteomeAnalyzer53) or NCBInr (MEGAN) or EggNOG database (EggNOG mapper) to assign functional categories. Functional analysis tools generate functional ontologies such as Gene Ontology (GO; for example, MetaGOmics, MEGAN, Unipept and EggNOG mapper), KEGG orthology groups (for example, EggNOG mapper, MEGAN and MetaProteomeAnalyzer), EC numbers (for example, Unipept and MetaProteomeAnalyzer), and EggNOG orthologous groups (EggNOG mapper) are used for deciphering the functional state of a microbiome.
While these annotation approaches described above are useful, it is worth acknowledging that there is a continuing challenge in interpretation of metaproteomics data that is inherited from metagenomic research regarding the process of annotating protein function from gene sequence data. The vast majority of protein annotations are assigned not from direct experimental evidence, but rather from sequence similarity to a previously annotated protein or protein family in a metabolic/ortholog database (e.g., KEGG, COG, METACYC, PFAM). This leads to several issues. The first is that annotation transfer based upon sequence similarity has resulted in the propagation of misannotations in large genomic databases over time, with the most common form of misannotation resulting from “over annotation”—annotation of a gene to a deeper level of functional characterization than the supporting evidence provides.86 Even minor errors or discrepancies in annotation transfer can result in massive error propagation.87 Common irregularities in gene annotation can cause serious issues for the metaproteomics researcher who is reliant upon these annotations to give biological context to proteomic data. Some of these annotation irregularities include gene annotations in which one gene name is assigned very different functional descriptions, instances where the gene name for a particular function has changed over time, or cases where only one function of a multifunctional enzyme is provided.87 Similarly, novel functions can be discovered for previously unannotated hypothetical proteins.16,88 These issues can be compounded if a custom search database comprised of genomes annotated by different means is used for peptide and protein identification, as is common in oceanographic studies. Moreover, as genomic data are updated with improved annotation information, an ability to pass this new information onto deposited processed metaproteomic results will be needed, and could be accomplished with versioning of deposited data sets.
A barrier to managing the spread of misannotations is that for some databases, such as GenBank NR, there are currently no means for the community to submit additional manually curated annotations and corrections. Fortunately, newer techniques for genome annotation which rely on methods beyond simple pairwise sequence similarity—most notably, the use of machine learning based algorithms—outperform the former pairwise similarity and BLAST based annotation transfer methods.89 Protein functional prediction from sequence data is a growing field itself, and will likely benefit from coupling powerful predictive algorithms with high-quality systems data to provide deeper, more accurate, and more meaningful characterizations.
Another common issue is where only a single function is reported for a protein family that is comprised of proteins of divergent function. For example, in Colatriano et al.,56 numerous proteins are assigned to the DMSO reductase enzyme superfamily, which is comprised of a number of functionally distinct proteins including nitrate reductases involved in anaerobic respiration as well as nitrite oxidoreductase involved in dissimilatory nitrite reduction. Only through fine-scale phylogenetic analysis of the identified proteins could the true function as nitrite reductases be determined. However, in many cases, the relationship between phylogeny and function within protein families is unknown. In metaproteomics, this is especially problematic for transporter proteins which are often abundant in metaproteomics data sets and are attractive since they may be used to infer substrate utilization patterns that are directly relevant to global biogeochemical cycles. However, transport function is often poorly conserved with these families, and hence sequence analysis is no substitution for the critical biochemical and genetic studies capable of characterizing protein function directly.16,90 In conclusion, there are significant challenges and room for improvement in the assignment of annotation information to metaproteomic data sets, and cooperation with genomics researchers and organizations, as well as incorporating an ability to reanalyze data sets and submit updated versions will be important components of future metaproteomic data management.
Challenges in Ocean Metaproteomic Data Sharing
There is potentially great value in sharing raw and processed environmental metaproteomics data within ocean sciences and beyond. As with most ‘omics sciences, each data set contains far more information than the data-generator’s laboratory can interpret. Proof that a gene is synthesized into protein form, as well as its variation in spatial or temporal distribution, can provide valuable biological and chemical information about the environment. Yet due to the complexity and newness of this data type, there are challenges unique to metaproteomics in reporting and disseminating this information. In a workshop in 2017, we discussed these challenges, and organized the following information in an attempt to provide a first set of best practices to Ocean Metaproteomics Data Sharing.
Interoperability between ocean metaproteomic observations and their related environmental data requires that the relationships between these data are explicitly known. Defining these relationships helps to communicate proper use when integrating disparate data within a shared domain.91 While defining these relationships helps humans properly integrate data, software and tools need something more. In the past, this meant developing software to a specific set of data types that forced data to follow certain conventions for variables names and structure. Yet current standards for integrating data on the web enable software to infer how disparate data can be integrated when they are described using semantic web schemas and rulesets. In doing so, these disparate data do not need to be transformed to conform to the software. Instead, the data are described using semantic web technologies for proper integration. The semantic web provides a framework for classification of data and its relationships in what are called ontologies, or vocabularies. These ontologies are logical groupings of terms and the axioms that define the how data from that domain are to be described. These terms can define cardinality rules or other logical expressions that enable humans and machines to make inferences over the data. Instead of transforming the original data to force it to conform to software, software can be written to conform to an ontology. As a result, this leaves the data intact and moves the work of integrating data to describing how it maps to the ontology.92
CONNECTIONS TO KEY ENVIRONMENTAL METADATA AND DATA
Environmental research requires unique metadata to provide context for comparisons across space and time. These metadata include numerous attributes associated with sampling from the oceans or other natural environments that are most often not included in the data model in biomedically focused proteomics repositories. For example, there is geospatial and environmental contextual information that is critical to interpreting results such as latitude and longitude, depth, and sampling environment (e.g., pelagic water column or benthic sedimentary location). For the pelagic environments, there is critical methodological information regarding sample collection parameters including filtration pore size range(s) or sediment trap deployment conditions for sinking particles. For benthic environments, key sedimentary environmental details, organism (coral, whale, plankton, zooplankton, etc.), or human built environments (ship hull surface) need to be described. In addition, local time of sampling can be important to detect short-term (diurnal) changes or long-term (seasonal or environmental change) processes. In addition to these metadata, there is also important contextual information derived from co-occurring chemical, biological, or physical measurements, such as temperature, macronutrient and micronutrient abundances, salinity, light intensity, and biological diversity, to name a few parameters. Carefully defining the data and metadata model will also facilitate connections to environmental data management holdings such as those at the Biological and Chemical Data Management Office for Ocean Science in the US (www.bco-dmo.org), and various national data repositories will facilitate access to this contextual information. Table 2 provides a list of recommended metadata for best practices of ocean metaproteomic samples data management to be provided at the time of deposition.
Table 2.
Recommended Metadata for Best Practices in Ocean Metaproteomics Data Sharing
| reporting metric | notes/units | available |
|---|---|---|
| Project Metadata | ||
| expedition identifier | ||
| lead PI, contact info, ORCID identifier | ||
| Co PI, contact info, ORCID identifier | √ | |
| Contextual Metadata | ||
| latitude | degrees N | |
| longitude | degrees W | |
| sampling depth | ||
| habitat type | Pelagic, benthic, reef, ship-hull, host-associated, other | |
| temperature | degrees Celsius | √ |
| salinity | √ | |
| chlorophyll-a concentration | √ | |
| oxygen concentration | √ | |
| other analytes measured | links to environmental data repositories | |
| Sample Acquisition Metadata | ||
| sampling method | filtration, sediment trap, coring, other | |
| volume of water sample represents | liters | √ |
| filter type | membrane (PC, PS), glass fiber, quartz, other | √ |
| filter size | micron pore size | √ |
| prefilter(s) used | if applicable: micron pore size, filter type | √ |
| Sample Extraction Methods | see Table S2 | |
| Mass Spectrometry Methods | see Table S2 | |
| Data Analysis Methods | see Table S2 | |
| Metaproteomics Data Analysis Metadata | ||
| database used for PSM or targeted method development | metagenomic, metatranscriptomic, genomic sequences used; link to sequence repository | |
| taxonomy analysis method | software/algorithm used | |
| functional analysis method | software/algorithm used |
Making connections between metaomics data sets and environmental data is a widely sought goal that is difficult to achieve. Enabling interoperability between ocean metaproteomic observations and their related environmental data requires that the relationships between these data are explicitly known. Defining these relationships helps to communicate proper use when integrating disparate data within a shared domain.91 While defining these relationships helps humans properly integrate data, software and tools need something more. In the past, this meant developing a certain piece of software to a specific set of data types. Yet current technologies, such as the semantic web, enable software to understand how data can be integrated through well-defined schemas and rulesets. Using ontologies, a semantic web technology, data, and their necessary relationships can be described in ways that machines can enforce cardinality constraints and make inferences that are helpful for ensuring a proper integration.92
Due to this fundamental importance of metadata associated with metaproteomics results, deposition of raw data into existing proteomic repositories designed primarily for laboratory studies (e.g., Pride, Massive, and Chorus) could create challenges for researchers in locating and manipulating collections of environmental data sets.93 While these raw data repositories are already valuable in hosting environmental proteomic data, the proper data management of large amounts of metadata can be viewed as a burden beyond what those entities are funded to provide, as has been observed in metagenomics data management spheres. As a result, intermediary environmental metaproteomics portals that host processed data sets and full metadata archives can serve a valuable function as a link to raw data repositories and cocollected or colocated environmental data sets. Hopefully either raw data repositories will work with environmental communities in collecting environmental metadata as well as cooperating to enable web-based connections between raw data repositories and processed data portals. In order to foster a high level of data sharing, reanalysis, and intercomparison, it is important for data generators to preserve a number of data facets and metadata, at the time of publication and archiving.
DATA TYPES USEFUL FOR METAPROTEOMIC DATA SHARING
In addition to the unique and critical metadata and environmental data that need to be provided, the metaproteomic data sets also require several distinct types of raw and processed data (see Table 3) in order to allow reproducibility and to enable a deep interrogation of their attributes in environmental data portals such as the future Ocean Protein Portal. Within processed data sets these include protein identifications with associated functional and taxonomic information (if known), full amino acid sequences, corresponding peptide sequences of discovered peptides, quantitative information for both proteins and peptides (e.g., spectral counts, precursor or fragment intensities), and associated statistical threshold used for generating these data (e.g., protein and peptide FDRs). For raw data, these include the raw mass spectrometry files converted to a platform-independent format as well as the sequence databases used to generate them. An important distinction from model organism studies is the fundamental importance of the peptide-level data to metaproteomics: the ability to have access to detailed peptide-level information with corresponding geospatial and temporal information will be critical to enabling users to directly interrogate the peptides that were actually measured in the oceans, as opposed to relying on protein inference that may be incorrect due to insufficient metagenomic coverage.
Table 3.
Key Datatypes Needed for Ocean Metaproteomic Data Sharing
| data type | attributes |
|---|---|
| raw mass spectra files | open data format, parameters, corresponding environmental and mass spectrometry metadata (see Tables 2 and S2) |
| protein identifications | MS/MS sample name |
| sequence identifier (e.g., metagenome locus or genome ORF) | |
| product name, taxon, taxon ID, KEGG, E.C., PFam | |
| quantitative value(s) (spectral count) | |
| peptide identifications | MS/MS sample name, sequence identifier (e.g., metagenome locus or genome ORF), peptide sequence, peptide start index, peptide stop index, precursor mass, retention time, statistical score of peptide, quantitative value (spectral count, MS1 peak area, fragment ion peak areas from SRM/MRM/PRM analyses, calibrated SI unit concentrations) |
| FASTA of amino acid sequences of all identified proteins | full sequence of DNA or RNA used for peptide-to-spectrum mapping |
| corresponding sequence identifier (e.g., metagenome locus or genome ORF) |
QUANTIFICATION: UNITS, INTERCALIBRATION, INTEROPERABILITY, AND NORMALIZATION
The ability to make comparisons of results across global scales of time and space in the oceans is a key appeal for embarking on ocean metaproteomic research science. Indeed, the ability of proteins to record the functional attributes of each population of marine microbes could allow a “personalized medicine” of the oceans,27 where long-term metaproteomic records would track changes in environmental stresses experienced by major taxa, and their resultant influence on global biogeochemical cycles and implications for sustainability. In ocean sciences, the few long-term time series available allow studies of the impacts of global change on the oceans. However, achieving the ambitious goal of integrating metaproteome studies into global change science will require a sufficient level of confidence regarding the accuracy and precision of analyses to allow detection of changes between samples sets. Metaproteomic data sets have reported quantitative results in a variety of units thus far, including total or normalized spectral counts, precursor intensities, or calibrated absolute concentrations (fmol L−1 seawater). Because the biological “matrix” of an environmental location can change with time, there is a particular value in absolute measurements that record peptide and protein abundances in SI units per liter that can be unequivocally compared across time. As a result, focusing on attributes that enable interoperability between samples, even as technologies (including chromatography, mass spectrometry, and informatics) and reference databases improve, is an important aspect of ocean metaproteomic data sharing. Efforts to harmonize across analytical platforms to improve intercomparability may be possible even in relative measurements (nonabsolute) through the calibration of signal intensity using a common reference material.94 As described earlier, efforts toward intercalibration of targeted metaproteomic analyses, as well as intercomparison of global relative abundance studies are critical to validating current measurements and enabling future comparisons. Similarly, allowing versioning of data sets will enable reanalysis of and reinterpretation of historical data sets that can then be used for temporal comparisons as both reference metagenomic databases and PSM algorithms improve.
Another consideration in the use of metaproteomic data is the choice of whether to normalize protein data to another protein or parameter. This choice may reflect scientific culture to some degree: in biological spheres normalization is routine in order to provide organismal or ecological context, while in chemical oceanography normalization is rarer due to an appreciation for the importance of relaying absolute quantities of molecules or elements per volume of seawater and the fundamental interoperability of absolute units. Notably, normalization approaches developed for single-organism proteomics are not always applicable or appropriate for metaproteomics. For example, assumptions of a constant background proteome (in terms of uniformity of biological organism(s) present) are not valid in many environments depending on spatial or temporal scales being studied. Moreover, the influence of differences in species abundance across samples should be considered when considering normalization of metaproteomic data sets;95 for example, when biological community composition changes across the sampling regime normalization to a particular organism may not be appropriate. While there have been advances in data processing approaches that address aspects of this issue,82 significant challenges remain.
PEPTIDE LEVEL REPORTING
Most publication guidelines for proteomics experiments recommend at least two peptides be identified to confidently report the identification of a specific protein. In the case of metaproteomics, however, it is understood that SNPs, amino acid variations, and substitutions associated with natural biological diversity within species or strain-level populations are common. As a result, it is possible to generate numerous high-quality PSMs in metaproteomics that are “one-hit-wonders”, likely due to a combination of challenges described above, including limitations associated with the application of mass spectrometry and PSM algorithms to highly complex samples, availability of a suitable database due to limited metagenomic coverage and quality, as well as the inherent biological diversity of a given protein (and its host organism) present in each nonclonal population found in the natural environment. In most cases, the natural biological diversity within species or strain-level populations is of great interest. It is not uncommon for peptide sequences (typically tryptic peptides) to be shared between closely related organisms. In marine microbiology, it is becoming common for multiple strains from a single species to have their genomes sequenced and physiology studied. These strains are described as being ecotypes that inhabit distinct environmental niches and can have overlapping distributions allowing co-occurrence within individual environmental samples.96 As a result, the assignment of multiple high-quality PSMs to a single protein sequence derived from a single isolate genome sequence is not as straightforward as with clonal populations of model laboratory organisms. If multiple ecotypes are present with slight variations in peptide sequence, the assignment of peptides to protein sequences could need reconsideration. Indeed, this subject intersects with the larger question regarding the appropriate definition of microbial species itself. Nevertheless, it has been demonstrated that this complexity can be taken advantage of in order to design targeted metaproteomic workflows that can interpret peptide biomarker abundances on a large ocean biome scale and that multiple peptide biomarkers provide consistent results.3
As a result, the two-peptide rule may not be appropriate for metaproteomics. There is precedence for not using the two-peptide rule in splice variant analysis and detection of post-translationally modified amino acid sites. With the subsequent arrival of high resolution mass spectrometry and stringent FDR-based analyses, high-quality PSM that do not map to a single protein sequence can have considerable value, and their inability to have multiple peptides mapping to a protein are likely related to numerous other challenges associated with metaproteomic diversity and dynamic range as described above, rather than necessarily being false positive identifications. The ability to report multiple peptides to a specific protein and its resultant percent peptide coverage itself becomes associated with uncertainty if there are multiple species with similar but not identical protein sequences. The adoption of identification of protein families may become a useful approach in metaproteomics where detection of peptides with small variation in sequence diversity can aggregate to a high confidence detection of a protein family belonging to multiple ecotypes of a species, or a defined higher taxonomic level particularly when interested in the biogeochemical impact of an enzyme.
Because of these challenges, targeted metaproteomic and informatics efforts have focused on the tryptic peptide level, using suites of tryptic peptide biomarkers as proxies for proteins and processes of interest. Informatic tools such as Unipept and Metatryp focus on tryptic peptides by conducting analysis of shared tryptic peptides between different genomes or metagenomes in order to maximize taxonomic interpretation of peptide identification. While the consensus on what should be considered the best practice for a high-quality peptide identification is beyond the scope of this review, it is clear that combining high-resolution mass spectrometry capabilities, low false discovery rate, observance of peptides in multiple spectra, visual inspection when possible, and other factors can contribute to high-quality peptide identifications.
ENCOURAGING PROPER DATA USE
As metaproteomics is a relatively young data type, there is potential for misunderstanding or misuse of results leading to incorrect interpretations. These could have an inadvertent detrimental effect of resulting in lost time chasing false leads or loss of confidence in metaproteomic methods.97 When used by expert data generators, this risk is lessened due to a thorough understanding of the limitations and methodology behind the data. However, in the effort to share metaproteomic results with a broader community of nonexpert users, there is considerable risk that researchers will incorrectly attempt to merge data units inappropriately and/or apply inappropriate data transformations that could result in incorrect interpretation. For example, spectral counts are a popular quantitative unit in proteomics that is powerful in assessing changes in each individual protein’s relative abundance across a range of samples. However, efforts to compare abundances between different proteins using relative abundance measurements such as these should be minimized or replaced by calibrated targeted measurements due to the variable influence of protein size (and resultant number of tryptic peptides) and the ionization efficiency of those peptides on each protein’s spectral count amplitude range. Nonexpert users may be tempted to conduct meta-analyses of spectral count results that could lead to faulty conclusions. As a result, efforts to educate and encourage dialogue among data generators and nonexpert users are important in fostering proper use of shared data sets.
Providing effective means for attribution of effort for those involved in data generation is also important. Ideally, this could include inviting the generator of a data set of interest to collaborate and be a coauthor in studies. In addition, acknowledging the use of a data set by citing original data release manuscripts and DOI identifiers assigned to the data set will be important in enabling data use to have metrics. The attribution component is important in the sustainability of data sharing projects, as this will incentivize the use of data sharing portals and repositories by generators. If data generators feel they are not being properly attributed, they may be reluctant to share data and/or may seek more obscure avenues for meeting data sharing requirements. Learning about data use policy experience of prior metagenomics and large ocean programs such as GEOTRACES will be valuable in this regard.
CONCLUSIONS
Metaproteomics data sets have the potential to become a valuable data type to the ocean science community in that they represent a metabolic record of the status of the key microbial components within specific geographic environments through time. With significant regional and global ecosystem changes now occurring,98 having access to detailed metabolic records through proteomic analyses of key environments could be particularly useful in providing an understanding of anthropogenic impacts on natural ecosystems. Given that marine ecosystems are important to human society in a variety of ways, including maintaining Earth’s habitability through microbial biogeochemical cycling, economic activities such as fisheries and aquaculture, and strategic and security importance to naval operations, the development and sharing of marine metaproteomic data sets will likely contribute to the long-term goal of developing a sustainable human society. This creates a distinct set of use cases for environmental proteomic data sets compared to those of laboratory cultivated organism or clinical proteomes, where planetary scale geospatial and temporal information are critically important metadata, and corresponding environmental data are fundamental to contextualizing environmental metaproteomic results. Moreover, the amount of research funding going into biomedical proteomic research vastly outweighs comparable resources in the ocean environment, making scarce ocean data sets of considerable value. These underlying differences in data usage and investment between environmental and biomedical proteomic data sets demonstrate a need for distinct data sharing strategies, and we have proposed some best practices with regard to metadata needs for ocean metaproteome data sharing, as well as summarized challenges associated with conducting metaproteomic research in hopes of inspiring innovation and collaboration.
EXPERIMENTAL METHODS
While the data presented in this review manuscript were largely previously published,3 some novel interpretations of the data have been included to demonstrate the complexity of metaproteome samples. The methods for these comparisons are briefly described below. A human cell line (HeLa) and ocean metaproteome samples (METZYME KM1128, Station 5 0°N 158°W 40, 60,120,150, and 200 m depth, 0.2 μm filter pore size, prefiltered with 3.0 μm pore size) were analyzed under identical chromatographic and mass spectrometry conditions to provide examples of sample complexity run within 5 days of each other. Protein extraction for metaproteomics was conducted using SDS detergent and tube gel purification as previously described.3 Protein extracts were analyzed by liquid chromatography–mass spectrometry (LC– MS) (Michrom Advance HPLC coupled to a Thermo Scientific Fusion Orbitrap mass spectrometer with a Thermo Flex source). A total of 0.5 μg (HeLa) or 1 μg (ocean) of each sample (measured before trypsin digestion) was concentrated onto a trap column (0.2 × 10 mm ID, 5 μm particle size, 120 Å pore size, C18 Reprosil-Gold, Dr. Maisch GmbH) and rinsed with 100 μL of 0.1% formic acid, 2% acetonitrile (ACN), 97.9% water before gradient elution through a reverse phase C18 nanospray column (0.1 × 400 mm ID, 3 μm particle size, 120 Å pore size, C18 Reprosil-Gold, Dr. Maisch GmbH) at a flow rate of 300 nL/min. The chromatography consisted of a nonlinear 200 min gradient from 5% to 95% buffer B, where A was 0.1% formic acid in water and B was 0.1% formic acid in ACN (all solvents were Fisher Optima grade). The mass spectrometer was set to perform MS scans on the Orbitrap (240 000 resolution at 200 m/z) with a scan range of 380 m/z to 1580 m/z. MS/MS was performed on the ion trap using data-dependent settings (top speed, dynamic exclusion 15 s, excluding unassigned and singly charged ions, precursor mass tolerance of ±3 ppm, with a maximum injection time of 150 ms).
Quantitive Peak Comparisons
Comparisons of the relative complexity of ocean metaproteomic samples (METZYME expedition, Station 3, depths 40, 60, 120, 150, and 250 m) with a human cell line sample run in triplicate was conducted. A total of 0.5 μg of Hela was injected per replicate, while 1 μg of ocean metaproteomic sample was injected per sample, as described above. Precursor peak data were extracted from raw files using ProteoWizard’s MSCon-vertGUI to text file using the vendor (Thermo) peak picking algorithm, and applying two filters: the peak picking algorithm (set for MS Levels 1 only) followed by MS level filter MS level 1 only. The number of precursor peaks per MS1, total ion count (TIC), and chromatographic time information were then extracted from the output files using a custom Python script, summed, and visualized (Figure 5).
Supplementary Material
ACKNOWLEDGMENTS
The workshop that led to this manuscript was funded by an NSF EarthCube Grant No. 1639714, the Gordon and Betty Moor Foundation, and an anonymous donor to M.A.S. and D.K. We thank Mary Zawoysky for assistance with workshop planning and manuscript editing, two anonymous reviewers for their useful comments and suggestions, and Natalie Renier for graphics assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00761.
Table S1. Examples of environmental and biomedical metaproteomics studies. Table S2. Additional metadata associated with sample processing and analysis for, but not exclusive to, metaproteomics data (PDF)
The authors declare no competing financial interest.
The data in this manuscript was previously published3,14 and is available at the Biological and Chemical Oceanography Data Management Office repository (https://www.bco-dmo.org/dataset/646115). The raw mass spectra files are available at PRIDE under project name Pacific Ocean metaproteomics METZYME KM1128, Project accession: PXD009712; Project DOI: 10.6019/PXD009712.
REFERENCES
- (1).Sowell SM; Wilhelm LJ; Norbeck AD; Lipton MS; Nicora CD; Barofsky DF; Carlson CA; Smith RD; Giovanonni SJ Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 2009, 3, 93–105. [DOI] [PubMed] [Google Scholar]
- (2).Morris RM; Nunn BL; Frazar C; Goodlett DR; Ting YS; Rocap G Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 2010, 4, 673–685. [DOI] [PubMed] [Google Scholar]
- (3).Saito MA; McIlvin MR; Moran DM; Goepfert TJ; DiTullio GR; Post AF; Lamborg CH Multiple nutrient stresses at intersecting Pacific Ocean biomes detected by protein biomarkers. Science 2014, 345 (6201), 1173–1177. [DOI] [PubMed] [Google Scholar]
- (4).Hawley AK; Brewer HM; Norbeck AD; Paša-Tolić L; Hallam SJ Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc. Natl. Acad. Sci. U. S.A 2014, 111 (31), 11395–11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Leary DH; Li RW; Hamdan LJ; Hervey WJ IV; Lebedev N; Wang Z; Deschamps JR; Kusterbeck AW; Vora GJ Integrated metagenomic and metaproteomic analyses of marine biofilm communities. Biofouling 2014, 30 (10), 1211–1223. [DOI] [PubMed] [Google Scholar]
- (6).Moore EK; Harvey HR; Faux JF; Goodlett DR; Nunn BL Protein recycling in Bering Sea algal incubations. Mar. Ecol.: Prog. Ser 2014, 515, 45–59. [Google Scholar]
- (7).Moore EK; Nunn BL; Goodlett DR; Harvey HR Identifying and tracking proteins through the marine water column: Insights into the inputs and preservation mechanisms of protein in sediments. Geochim. Cosmochim. Acta 2012, 83, 324–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bridoux MC; Neibauer J; Ingalls AE; Nunn BL; Keil RG Suspended marine particulate proteins in coastal and oligotrophic waters. Journal of Marine Systems 2015, 143, 39–48. [Google Scholar]
- (9).Georges AA; El-Swais H; Craig SE; Li WK; Walsh DA Metaproteomic analysis of a winter to spring succession in coastal northwest Atlantic Ocean microbial plankton. ISME J. 2014, 8 (6), 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bender SJ; Moran DM; McIlvin MR; Zheng H; McCrow JP; Badger J; DiTullio GR; Allen AE; Saito MA Colony formation in Phaeocystis antarctica connecting molecular mechanisms with iron biogeochemistry. Biogeosciences 2018, 15 (16), 4923–4942. [Google Scholar]
- (11).Bertrand EM; Moran DM; McIlvin MR; Hoffman JM; Allen AE; Saito MA Methionine synthase interreplacement in diatom cultures and communities: Implications for the persistence of B12 use by eukaryotic phytoplankton. Limnol Oceanogr. 2013, 58 (4), 1431–1450. [Google Scholar]
- (12).Moore EK; Nunn BL; Faux JF; Goodlett DR; Harvey HR Evaluation of electrophoretic protein extraction and database-driven protein identification from marine sediments. Limnol. Oceanogr.: Methods 2012, 10 (5), 353–366. [Google Scholar]
- (13).Kan J; Hanson T; Ginter J; Wang K; Chen F Metaproteomic analysis of Chesapeake Bay microbial communities. Saline Syst. 2005, 1 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Saito MA; Dorsk A; Post AF; McIlvin M; Rappé MS; DiTullio G; Moran D Needles in the Blue Sea: Sub Species Specificity in Targeted Protein Biomarker Analyses Within the Vast Oceanic Microbial Metaproteome. Proteomics 2015, 15 (20), 3521–3531. [DOI] [PubMed] [Google Scholar]
- (15).Williams TJ; Long E; Evans F; DeMaere MZ; Lauro FM; Raftery MJ; Ducklow H; Grzymski JJ; Murray AE; Cavicchioli R A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J. 2012, 6 (10), 1883–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bertrand EM; Allen AE; Dupont CL; Norden-Krichmar T ; Bai J; Saito MA; Valas RE Influence of Cobalamin Starvation on Diatom Molecular Physiology and the Identification of a Cobalamin Acquisition Protein. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (26), E1762–E1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dyhrman ST; Jenkins BD; Rynearson TA; Saito MA; Mercier ML; Alexander H; Whitney LP; Drzewianowski A; Bulygin VV; Bertrand EM; Wu Z; Benitez-Nelson C; Heithoff A The transcriptome and proteome of the diatom Thalassiosira pseudonana reveal a diverse phosphorus stress response. PLoS One 2012, 7 (3), No. e33768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cox AD; Saito MA Proteomic responses of oceanic Synechococcus WH8102 to phosphate and zinc scarcity and cadmium additions. Front. Microbiol 2013, 4, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mackey KR; Post AF; McIlvin MR; Cutter GA; John SG; Saito MA Divergent responses of Atlantic coastal and oceanic Synechococcus to iron limitation. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (32), 9944–9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wurch LL; Bertrand EM; Saito MA; Van Mooy BAS; Dyhrman ST Proteome Changes Driven by Phosphorus Deficiency and Recovery in the Brown Tide-Forming Alga Aureococcus anophagefferens. PLoS One 2011, 6 (12), No. e28949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Swanner ED; Wu W; Hao L; Wüstner ML; Obst M; Moran DM; McIlvin MR; Saito MA; Kappler A Physiology, Fe (II) oxidation, and Fe mineral formation by a marine planktonic cyanobacterium grown under ferruginous conditions. Frontiers in Earth Science 2015, 3, 60. [Google Scholar]
- (22).Nunn B; Aker J; Shaffer S; Tsai Y; Strzepek R; Boyd P; Freeman T; Brittnacher M; Malmstrom L; Goodlett D Deciphering diatom biochemical pathways via whole-cell proteomics. Aquat. Microb. Ecol 2009, 55 (3), 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nunn BL; Slattery KV; Cameron KA; Timmins-Schiffman E; Junge K Proteomics of Colwellia psychrerythraea at subzero temperatures–a life with limited movement, flexible membranes and vital DNA repair. Environ. Microbiol 2015, 17 (7), 2319–2335. [DOI] [PubMed] [Google Scholar]
- (24).Heyer R; Schallert K; Zoun R; Becher B; Saake G; Benndorf D Challenges and perspectives of metaproteomic data analysis. J. Biotechnol 2017, 261, 24–36. [DOI] [PubMed] [Google Scholar]
- (25).Saito MA; Bulygin VV; Moran DM; Taylor C; Scholin C Examination of Microbial Proteome Preservation Techniques Applicable to Autonomous Environmental Sample Collection. Front. Microbiol 2011, 2, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Breier J; Gomez-Ibanez D; Reddington E; Huber J; Emerson D A precision multi-sampler for deep-sea hydrothermal microbial mat studies. Deep Sea Res. Part I 2012, 70, 83–90. [Google Scholar]
- (27).Saito MA; Breier C; Jakuba M; McIlvin M; Moran D Envisioning a Chemical Metaproteomics Capability for Biochemical Research and Diagnosis of global Ocean Microbiomes In The Chemistry of Microbiomes: Proceedings of a Seminar Series; National Academies Press: National Academies of Sciences, Engineering, Medicine, 2017; pp 29–36. [Google Scholar]
- (28).Boyle EA; Bergquist BA; Kayser RA; Mahowald N Iron, manganese, and lead at Hawaii Ocean Time-series station ALOHA: Temporal variability and an intermediate water hydrothermal plume. Geochim. Cosmochim. Acta 2005, 69 (4), 933–952. [Google Scholar]
- (29).Bergauer K; Fernandez-Guerra A; Garcia JAL; Sprenger RR; Stepanauskas R; Pachiadaki MG; Jensen ON; Herndl GJ Organic matter processing by microbial communities throughout the Atlantic water column as revealed by metaproteomics. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (3), E400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Venter JC; Remington K; Heidelberg JF; Halpern AL; Rusch D; Eisen JA; Wu D; Paulsen I; Nelson KE; Nelson W; Fouts DE; Levy S; Knap AH; Lomas MW; Nealson K; White O; Peterson J; Hoffman J; Parsons R; Baden-Tillson H; Pfannkoch C; Rogers YH; Smith HO Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304 (5667), 66–74. [DOI] [PubMed] [Google Scholar]
- (31).DeLong EF; Preston CM; Mincer T; Rich V; Hallam SJ; Frigaard N-U; Martinez A; Sullivan MB; Edwards R; Brito BR; Chisholm SW; Karl DM Community Genomics Among Stratified Microbial Assemblages in the Ocean’s Interior. Science 2006, 311 (5760), 496–503. [DOI] [PubMed] [Google Scholar]
- (32).Nunn BL; Keil RG A comparison of non-hydrolytic methods for extracting amino acids and proteins from coastal marine sediments. Mar. Chem 2006, 98 (1), 31–42. [Google Scholar]
- (33).Keil RG; Tsamakis E; Fuh CB; Giddings JC; Hedges JI Mineralogical and textural controls on the organic composition of coastal marine sediments: hydrodynamic separation using SPLITT-fractionation. Geochim. Cosmochim. Acta 1994, 58 (2), 879–893. [Google Scholar]
- (34).Ram RJ; VerBerkmoes NC; Thelen MP; Tyson GW; Baker BJ; Blake RC II; Shah M; Hettich RL; Banfield JF Community Proteomics of a Natural Microbial Biofilm. Science 2005, 308 (5730), 1915–1920. [PubMed] [Google Scholar]
- (35).VerBerkmoes NC; Hervey WJ; Shah M; Land M; Hauser L ; Larimer FW; Van Berkel GJ; Goeringer DE Evaluation of “shotgun” proteomics for identification of biological threat agents in complex environmental matrixes: experimental simulations. Anal. Chem 2005, 77 (3), 923–932. [DOI] [PubMed] [Google Scholar]
- (36).Mattes TE; Nunn BL; Marshall KT; Proskurowski G; Kelley DS; Kawka OE; Goodlett DR; Hansell DA; Morris RM Sulfur oxidizers dominate carbon fixation at a biogeochemical hot spot in the dark ocean. ISME J. 2013, 7 (12), 2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sinitcyn P; Rudolph JD; Cox J Computational Methods for Understanding Mass Spectrometry-Based Shotgun Proteomics Data. Annual Review of Biomedical Data Science 2018, 1 (1), 207–234. [Google Scholar]
- (38).Gillet LC; Navarro P; Tate S; Rost H; Selevsek N; Reiter L ; Bonner R; Aebersold R Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 2012, 11 (6), O111-O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gallien S; Bourmaud A; Kim SY; Domon B Technical considerations for large-scale parallel reaction monitoring analysis. J. Proteomics 2014, 100, 147–159. [DOI] [PubMed] [Google Scholar]
- (40).Gallien S; Duriez E; Crone C; Kellmann M; Moehring T; Domon B Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 2012, 11 (12), 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gallien S; Duriez E; Domon B Selected reaction monitoring applied to proteomics. J. Mass Spectrom 2011, 46 (3), 298–312. [DOI] [PubMed] [Google Scholar]
- (42).Pluskal T; Castillo S; Villar-Briones A; Orešič M MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 2010, 11 (1), 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nunn BL; Faux JF; Hippmann AA; Maldonado MT; Harvey HR; Goodlett DR; Boyd PW; Strzepek RF Diatom proteomics reveals unique acclimation strategies to mitigate Fe limitation. PLoS One 2013, 8 (10), No. e75653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Dorfer V; Maltsev S; Winkler S; Mechtler K CharmeRT: Boosting peptide identifications by chimeric spectra identification and retention time prediction. J. Proteome Res 2018, 17 (8), 2581–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cutter GA Intercalibration in chemical oceanography—getting the right number. Limnol Oceanogr.: Methods 2013, 11 (7), 418–424. [Google Scholar]
- (46).The M; Tasnim A; Käll L How to talk about protein-level false discovery rates in shotgun proteomics. Proteomics 2016, 16 (18), 2461–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Noble WS Mass spectrometrists should search only for peptides they care about. Nat. Methods 2015, 12 (7), 605–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Huang T; Wang JJ; Yu WC; He Z Protein inference: a review. Briefings Bioinf. 2012, 13 (5), 586–614. [DOI] [PubMed] [Google Scholar]
- (49).Nesvizhskii AI; Aebersold R Interpretation of shotgun proteomic data - The protein inference problem. Mol. Cell. Proteomics 2005, 4 (10), 1419–1440. [DOI] [PubMed] [Google Scholar]
- (50).Brosch M; Saunders GI; Frankish A; Collins MO; et al. Shotgun proteomics aids discovery of novel protein coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome. Genome Res. 2011, 21 (5), 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Volders PJ; Verheggen K; Menschaert G; Vandepoele K; et al. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43 (8), 4363–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Slavoff SA; Mitchell AJ; Schwaid AG; Cabili MN; Ma J; Levin JZ; Karger AD; Budnik BA; Rinn JL; Saghatelian A Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol 2013, 9, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Muth T; Kolmeder CA; Salojarvi J; Keskitalo S; Varjosalo M ; Verdam FJ; Rensen SS; Reichl U; de Vos WM; Rapp E; Martens L Navigating through metaproteomics data: A logbook of database searching. Proteomics 2015, 15 (20), 3439–3453. [DOI] [PubMed] [Google Scholar]
- (54).Timmins-Schiffman E; May DH; Mikan M; Riffle M; Frazar C; Harvey H; Noble WS; Nunn BL Critical decisions in metaproteomics: achieving high confidence protein annotations in a sea of unknowns. ISMEJ. 2017, 11 (2), 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Teeling H; Fuchs BM; Becher D; Klockow C; Gardebrecht A; Bennke CM; Kassabgy M; Huang S; Mann AJ; Waldmann J; Weber M; Klindworth; Otto A; Lange J; Bernhardt J; Reinsch C; Hecker M; Peplies J; Bockelmann FD; Callies U; Gerdts G; Wichels A; Wiltshire KH; Glockner FO; Schweder T; Amann R. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336 (6081), 608–611. [DOI] [PubMed] [Google Scholar]
- (56).Colatriano D; Ramachandran A; Yergeau E; Maranger R; Gelinas Y; Walsh DA Metaproteomics of aquatic microbial communities in a deep and stratified estuary. Proteomics 2015, 15 (20), 3566–3579. [DOI] [PubMed] [Google Scholar]
- (57).Burton JN; Liachko I; Dunham MJ; Shendure J Species-level deconvolution of metagenome assemblies with Hi-C-based contact probability maps. G3: Genes, Genomes, Genet. 2014, 4, 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kashtan N; Roggensack SE; Rodrigue S; Thompson JW; Biller SJ; Coe A; Ding H; Marttinen P; Malmstrom RR; Stocker R ; Follows MJ; Stepanauskas R; Chisholm SW Single-Cell Genomics Reveals Hundreds of Coexisting Subpopulations in Wild Prochlorococcus. Science 2014, 344 (6182), 416–420. [DOI] [PubMed] [Google Scholar]
- (59).Rusch DB; Martiny AC; Dupont CL; Halpern AL; Venter JC Characterization of Prochlorococcus clades from iron-depleted oceanic regions. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (37), 16184–16189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Yooseph S; Sutton G; Rusch DB; Halpern AL; Williamson SJ; Remington K; Eisen JA; Heidelberg KB; Manning G; Li W; Jaroszewski L; Cieplak P; Miller CS; Li H; Mashiyama ST; Joachimiak MP; van Belle C; Chandonia J-M; Soergel DA; Zhai Y; Natarajan K; Lee S; Raphael BJ; Bafna V; Friedman R; Brenner SE; Godzik A; Eisenberg D; Dixon JE; Taylor SS; Strausberg RL; Frazier M; Venter JC The Sorcerer II Global Ocean Sampling Expedition: Expanding the Universe of Protein Families. PLoS Biol. 2007, 5 (3), No. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Williamson AJ; Smith DL; Blinco D; Unwin RD; Pearson S; Wilson C; Miller C; Lancashire L; Lacaud G; Kouskoff V; Whetton AD Quantitative proteomics analysis demonstrates post-transcriptional regulation of embryonic stem cell differentiation to hematopoiesis. Mol. Cell. Proteomics 2008, 7 (3), 459–472. [DOI] [PubMed] [Google Scholar]
- (62).Keeling PJ; Burki F; Wilcox HM; Allam B; Allen EE; Amaral-Zettler LA; Armbrust EV; Archibald JM; Bharti AK; Bell CJ; Beszteri B The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryoticlife in the oceans through transcriptome sequencing. PLoS Biology 2014, 12 (6), No. e1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).May DH; Timmins-Schiffman E; Mikan MP; Harvey HR; Borenstein E; Nunn BL; Noble WS An alignment-free “metapeptide” strategy for metaproteomic characterization of microbiome samples using shotgun metagenomic sequencing. J. Proteome Res 2016, 15 (8), 2697–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Biller SJ; Berube PM; Berta-Thompson JW; Kelly L; Roggensack SE; Awad L; Roache-Johnson KH; Ding H; Giovannoni SJ; Rocap G; Moore LR; Chisholm SW Genomes of diverse isolates of the marine cyanobacterium Prochlorococcus. Sci. Data 2014, 1, 140034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Grote J; Thrash JC; Huggett MJ; Landry ZC; Carini P; Giovannoni SJ; Rappe MS Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 2012, 3 (5), e00252–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Santoro AE; Dupont CL; Richter RA; Craig MT; Carini P; McIlvin MR; Yang Y; Orsi WD; Moran DM; Saito MA Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (4), 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Eng J; McCormack A; Yates J An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–989. [DOI] [PubMed] [Google Scholar]
- (68).Eng JK; Jahan TA; Hoopmann MR Comet: an open source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–24. [DOI] [PubMed] [Google Scholar]
- (69).Craig R; Beavis RC A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom 2003, 17 (20), 2310–2316. [DOI] [PubMed] [Google Scholar]
- (70).Jagtap P; McGowan T; Bandhakavi S; Tu ZJ; Seymour S; Griffin TJ; Rudney JD Deep metaproteomic analysis of human salivary supernatant. Proteomics 2012, 12 (7), 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Jagtap P; Goslinga J; Kooren JA; McGowan T; Wroblewski MS; Seymour SL; Griffin TJ A two step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics 2013, 13 (8), 1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Jagtap PD; Johnson JE; Onsongo G; Sadler FW; Murray K; Wang Y; Shenykman GM; Bandhakavi S; Smith LM; Griffin TJ Flexible and accessible workflows for improved proteogenomic analysis using the Galaxy framework. J. Proteome Res 2014, 13 (12), 5898–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Jagtap PD; Blakely A; Murray K; Stewart S; Kooren J; Johnson JE; Rhodus NL; Rudney J; Griffin TJ Metaproteomic analysis using the Galaxy framework. Proteomics 2015, 15 (20), 3553–3565. [DOI] [PubMed] [Google Scholar]
- (74).Rudney JD; Jagtap PD; Reilly CS; Chen R; Markowski TW; Higgins L; Johnson JE; Griffin TJ Protein relative abundance patterns associated with sucrose-induced dysbiosis are conserved across taxonomically diverse oral microcosm biofilm models of dental caries. Microbiome 2015, 3 (1), 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kertesz-Farkas A; Keich U; Noble WS Tandem mass spectrum identification via cascaded search. J. Proteome Res 2015, 14 (8), 3027–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Vaudel M; Burkhart JM; Zahedi RP; Oveland E; Berven FS; Sickmann A; Martens L; Barsnes H PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol 2015, 33, 22. [DOI] [PubMed] [Google Scholar]
- (77).Muth T; Hartkopf F; Vaudel M; Renard BY A Potential Golden Age to Come—Current Tools, Recent Use Cases, and Future Avenues for De Novo Sequencing in Proteomics. Proteomics 2018, 18 (18), 1700150. [DOI] [PubMed] [Google Scholar]
- (78).Mesuere B; van der Jeugt F; Devreese B; Vandamme P; Dawyndt P The unique peptidome: Taxon-specific tryptic peptides as biomarkers for targeted metaproteomics. Proteomics 2016, 16 (17), 2313–2318. [DOI] [PubMed] [Google Scholar]
- (79).Mesuere B; Devreese B; Debyser G; Aerts M; Vandamme P; Dawyndt P Unipept: Tryptic Peptide-Based Biodiversity Analysis of Metaproteome Samples. J. Proteome Res 2012,11 (12), 5773–5780. [DOI] [PubMed] [Google Scholar]
- (80).Mesuere B; Willems T; van der Jeugt F; Devreese B; Vandamme P; Dawyndt P Unipept web services for metaproteomics analysis. Bioinformatics 2016, 32 (11), 1746–1748. [DOI] [PubMed] [Google Scholar]
- (81).Muth T; Kohrs F; Heyer R; Benndorf D; Rapp E; Reichl U ; Martens L; Renard BY MPA Portable: A Stand-Alone Software Package for Analyzing Metaproteome Samples on the Go. Anal. Chem 2018, 90 (1), 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Riffle M; May D; Timmins-Schiffman E; Mikan MP; Jaschob D; Noble WS; Nunn BL MetaGOmics: A Web-Based Tool for Peptide Centric Functional and Taxonomic Analysis of Metaproteomic Data. Proteomes 2018, 6 (1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Gurdeep Singh R; Tanca A; Palomba A; van der Jeugt F; Verschaffelt P; Uzzau S; Martens L; Dawyndt P; Mesuere B Unipept 4.0: Functional Analysis of Metaproteome Data. J. Proteome Res 2019, 18, 606–615. [DOI] [PubMed] [Google Scholar]
- (84).Huson DH; Auch AF; Qi J; Schuster SC MEGAN analysis of metagenomic data. Genome Res. 2007, 17 (3), 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Huerta-Cepas J; Forslund K; Coelho LP; Szklarczyk D; Jensen LJ; von Mering C; Bork P Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol 2017, 34 (8), 2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Schnoes AM; Brown SD; et al. Annotation error in public databases: misannotation of molecular function in enzyme super-families. PLoS Comput. Biol 2009, 5 (12), No. e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Brenner SE Errors in genome annotation. Trends Genet. 1999, 15 (4), 132–133. [DOI] [PubMed] [Google Scholar]
- (88).McQuaid JB; Kustka AB; Oborník M; Horák A; McCrow JP; Karas BJ; Zheng H; Kindeberg T; Andersson AJ; Barbeau KA; Allen AE Carbonate-sensitive phytotransferrin controls high-affinity iron uptake in diatoms. Nature 2018, 555, 534. [DOI] [PubMed] [Google Scholar]
- (89).Radivojac P; Clark WT; et al. A large-scale evaluation of computational protein function prediction. Nat. Methods 2013, 10 (3), 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Webb EA; Moffett JW; Waterbury JB Iron Stress in Open Ocean Cyanobacteria (Synechococcus Trichodesmium and Crocosphaera): Identification of the IdiA protein. Appl Environ. Microbiol 2001, 67, 5444–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Hitzler P; Janowicz K Semantic Web. 3rd ed.; J. Chapman and Hall/CRC: 2014; Vol. 50, p 50–10–13. [Google Scholar]
- (92).Patton EW; Seyed P; Wang P; Fu L; Dein FJ; Bristol RS; McGuinness DL SemantEco: A semantically powered modular architecture for integrating distributed environmental and ecological data. Future Generation Computer Systems 2014, 36, 430–440. [Google Scholar]
- (93).Martens L; Hermjakob H; Jones P; Adamski M; Taylor C; States D; Gevaert K; Vandekerckhove J; Apweiler R PRIDE: The proteomics identifications database. Proteomics 2005, 5 (13), 3537–3545. [DOI] [PubMed] [Google Scholar]
- (94).Pino LK; Searle BC; Huang EL; Noble WS; Hoofnagle AN; MacCoss MJ Calibration Using a Single-Point External Reference Material Harmonizes Quantitative Mass Spectrometry Proteomics Data between Platforms and Laboratories. Anal. Chem 2018, 90 (21), 13112–13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Kleiner M, Normalization of metatranscriptomic and metaproteomic data for differential gene expression analyses: The importance of accounting for organism abundance. PeerJ. Preprints 2017, 5, e2846v1 [Google Scholar]
- (96).Johnson ZI; Zinser ER; Coe A; McNulty NP; Woodward EMS; Chisholm SW Niche Partitioning Among Prochlorococcus Ecotypes Along Ocean-Scale Environmental Gradients. Science 2006, 311 (5768), 1737–1740. [DOI] [PubMed] [Google Scholar]
- (97).White FM The potential cost of high-throughput proteomics. Sci. Signaling 2011, 4 (160), pe8. [DOI] [PubMed] [Google Scholar]
- (98).Doney SC The Growing Human Footprint on Coastal and Open-Ocean Biogeochemistry. Science 2010, 328 (5985), 1512–1516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.