

Abstract
Synthetic lethality between poly(ADP-ribose) polymerase (PARP) inhibition and BRCA deficiency is exploited to treat breast and ovarian tumors. However, resistance to PARP inhibitors (PARPi) is common. To identify potential resistance mechanisms, we performed a genome-wide RNAi screen in BRCA2-deficient mouse embryonic stem cells and validation in KB2P1.21 mouse mammary tumor cells. We found that resistance to multiple PARPi emerged with reduced expression of TET2 (ten eleven translocation), which promotes DNA demethylation by oxidizing 5-methylcytosine (5mC) to 5-hydroxymethycytosine (5hmC) and other products. TET2 knockdown in BRCA2-deficient cells protected stalled replication forks (RFs) due to reduced 5hmC amounts at sites of double-stranded breaks (DSBs). Increasing 5hmC abundance induced the degradation of stalled RFs in KB2P1.21 and human cancer cells by recruiting the base excision repair-associated apurinic/apyrimidinic endonuclease APE1, independent of the BRCA2 status. TET2 loss did not affect the recruitment of the repair protein RAD51 to sites of DSBs or the abundance of proteins associated with RF integrity, or BRCA2 status. The loss of TET2, of its product 5hmc, and of APE1 recruitment to stalled RFs promoted resistance to the chemotherapeutic cisplatin. Our findings reveal a previously unknown role for the epigenetic mark 5hmC in maintaining the integrity of stalled RFs and a potential resistance mechanism to PARPi and cisplatin.
INTRODUCTION
Breast cancer is the most common malignancy in women and one of the leading causes of death worldwide. Germline mutations in BRCA1 and BRCA2 genes accounts for 20–25% of all hereditary breast cancer cases (1). Functional loss of BRCA proteins leads to a defect in homologous recombination (HR)-dependent repair of DNA double strand breaks (DSBs). This renders BRCA-deficient cells sensitive to poly(ADP)-ribose (PARP) inhibitors (PARPi) (2–5). PARPi-induced synthetic lethality is being successfully used in the clinic for the targeted, non-invasive treatment of BRCA-deficient breast and ovarian tumors (6, 7). However, tumors frequently acquire resistance to PARPi. Secondary mutations in the BRCA1 or BRCA2 that restore their HR function cause resistance not only to PARPi but also to platinum compounds (8). Mutation in other genes have also been reported to restore BRCA1-mediated HR. Inactivation of 53BP1 promotes end resection of DSBs and restores HR in BRCA1-deficient cells (9, 10). Similarly, loss of the shieldin complex components REV7 (DNA polymerase zeta processivity subunit), C20ORF196 (an uncharacterized protein), and FAM35A (shieldin complex subunit 2) also promotes end resection, thus restoring HR and PARPi resistance (11, 12). PARPi resistance can occur due to increased expression of ATP-dependent drug efflux pump ABCB1 (13). Resistance to PARPi can also be caused by reduced expression of PARP proteins, because PARP inhibitors trap PARP-DNA complexes and contribute to synthetic lethality (14). Loss of dynein light chain 1 (DYNLL1) has also been shown to contribute to HR restoration based on its direct binding to meiotic recombination 11 (MRE11) and restricting end resection (15).
An HR-independent mechanism of PARPi resistance is the protection of stalled replication forks (RFs) in BRCA-deficient cells (16, 17). Loss of PTIP (PAX transactivation domain interacting protein) prevents MRE11 recruitment to stalled RFs and protects fork integrity in BRCA-deficient cells. Similarly, loss of CHD4 (chromodomain helicase DNA binding protein 4) in BRCA2-deficient cells also prevented MRE11 recruitment and protected stalled forks, which contributed to PARPi resistance (17, 18). Epigenetic modification of histones by EZH2 (enhancer of zeste homolog 2) can regulate the stability of replication forks and confer PARPi resistance in BRCA2 deficient cells. Loss of EZH2 reduces H3K27me3 modification of histones, which prevents recruitment of crossover junction endonuclease MUS81 to RFs and suppresses fork degradation (19).
In this study, we investigated novel regulators of resistance to PARPi. We performed a genome-wide siRNA screen in PARP-sensitive mESCs expressing a hypomorphic allele of BRCA2 to identify proteins whose loss conferred olaparib resistance. We found loss of TET2 (Ten Eleven Translocation 2) contributes to PARPi resistance. Tet2 a tumor suppressor that, notably, is frequently mutated in hematopoietic malignancies (20). TET family proteins are responsible for catalyzing the demethylation of 5mC by its sequential oxidation to 5hmC and then to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (21). Among these oxidation products of 5mC, 5hmC is the most stable form. 5hmC levels are 10 to 100-fold higher than 5fC and 5caC (21–23). Differential prevalence of these cytosine modifications is due to differential affinity of TET family members for these cytosines. TET1 and TET2 have greater affinity for 5mC and 5fC than for 5hmC and 5caC, whereas TET3 has affinity for 5caC (24, 25). Because TET family members have the least affinity for 5hmC, its catalytic conversion to 5fC is low, making 5hmC the most stable cytosine modification. These cytosine intermediates are converted to apurinic/apyrimidinic (AP) or abasic sites by thymine DNA glycosylases (TDG) and repaired by the base excision repair (BER) pathway (26, 27). Given that TET proteins are involved in DNA demethylation, they regulate various cellular processes such as gene expression and cell differentiation (28). In response to DNA damage, checkpoint kinases Ataxia telangiectasia-mutated (ATM) and Ataxia telangiectasia- and Rad3-related (ATR) phosphorylate TET enzymes and stimulate 5hmC production (29, 30). However, the precise role of 5hmC in DNA damage response (DDR) pathways is not currently well understood.
Our findings reveal that loss of TET2 protects stalled RFs in BRCA2-deficient cells. Unexpectedly, increasing 5hmC levels caused by TET2 overexpression reduced fork stability and contributed to genomic instability, even in the presence of functional BRCA2. Additionally, 5hmC, 5fC and 5caC at stalled forks contributed to fork degradation by apurinic/apyrimidinic endonuclease 1 (APE1), a BER-associated enzyme that initiates the repair of abasic sites.
RESULTS
Loss of TET2 results in olaparib resistance in BRCA2-deficient cells
To identify genes that contribute to PARPi resistance in BRCA2-deficient cells, we performed a genome-wide siRNA screen targeting 17,575 genes in mouse embryonic stem cells (mESCs) expressing the R2336H BRCA2 variant (PL2F7-Brca2ko/ko;BRCA2(R2336H)] (fig. S1, A and B). We have previously reported that the BRCA2R2336H supports mESC viability, but the cells are defective in homologous recombination and are hypersensitive to olaparib (31), confirmed here (fig. S1C). We identified Tet2 as the top gene with the highest mean Z score of 4.52 (data file S1). The TCGA database revealed that survival was lower in breast cancer patients whose tumors had low TET2 expression than that in patients whose tumors had high TET2 expression (fig. S1D).
We validated the effect of TET2 knockdown on olaparib resistance by stably knocking down TET2 in PL2F7-Brca2ko/ko;BRCA2(R2336H) mESCs (Fig. 1A, and fig. S1, E and F). PL2F7-Brca2cko/ko mESCs with one floxed wildtype allele (cko) and one knockout allele (ko) of Brca2 were used as olaparib-resistant control cell line, whereas PL2F7-Brca2ko/ko;BRCA2(R2336H)-shControl cells expressing hypomorphic mutant of BRCA2 were used as olaparib-sensitive control cell line. Because of functional similarity between TET family members, we further validated the effect of stable knockdown of TET2 and TET3 on olaparib resistance using two shRNAs for each in the BRCA2- and TP53-deficient mouse mammary tumor cell line KB2P1.21 (32). As a BRCA2-proficient isogenic control, we used a derivative line, KB2P1.21R2, which was reconstituted with human BRCA2. We found that either TET2 or TET3 loss significantly increased the survival of KB2P1.21 cells after olaparib treatment (Fig. 1, B and C; and fig. S1, G and H). Loss of TET2 or TET3 conferred resistance to other PARP inhibitors as well, namely veliparib and talazoparib (Fig. 1C and fig. S1, I and J), and also to cisplatin (Fig. 1, C and D). The role of TET1 was not examined, because its expression was not detected in KB2P1.21 cells by RT-PCR (fig. S1K).
Figure 1. Loss of TET confers drug resistance to PARPi and cisplatin and restores RF stability in BRCA2-deficient cells.
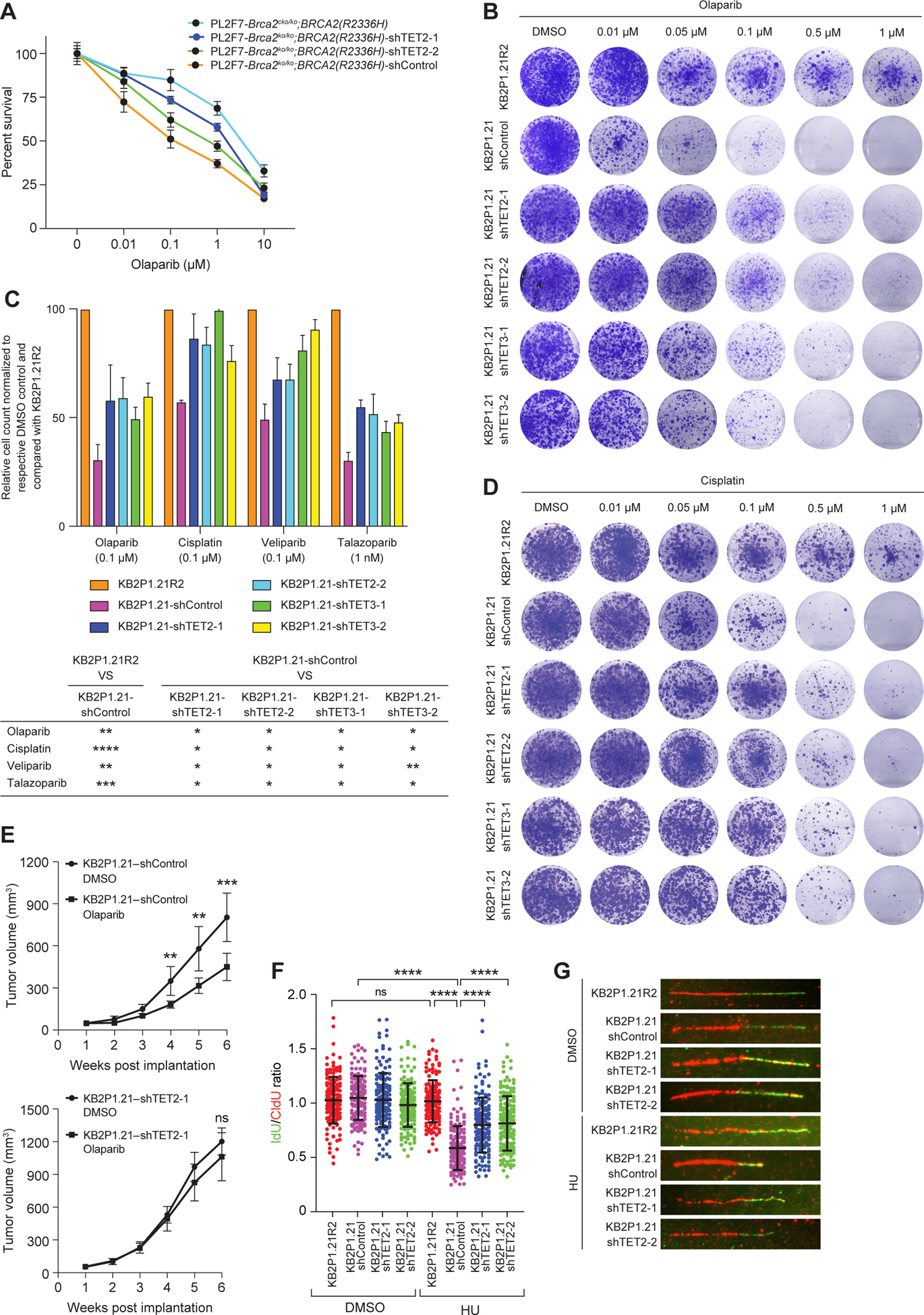
(A) Viability of the indicated control or TET2-knockdown PL2F7-Brca2ko/ko;BRCA2(R2336H) and PL2F7-Brca2cko/ko;BRCA2(R2336H) mES cells to increasing concentrations of olaparib (DMSO, 0.01, 0.1, 1, 10μM), as measured by the XTT assay for 96 hours. Values were normalized to a respective DMSO-treated control. Data are mean ± SD of technical triplicates from 2 independent experiments. (B) Clonogenic survival assay was performed for 14 days in KB2P1.21-shControl, KB2P1.21-shTET2 and KB2P1.21-shTET3 cells (both with each of two shRNAs) treated with increasing concentrations of olaparib (DMSO, 0.01, 0.5, 0.1, 0.5, or 1 μM). Images are representative of 3 independent experiments. (C) Quantification of viability was assessed by hemocytometer in KB2P1.21-shTET2 and KB2P1.21-shTET3 cells treated with olaparib (0.1 μM), cisplatin (0.1 μM), veliparib (0.1 μM) and talazoparib (1 nM) for 14 days. Values are normalized to a respective DMSO-treated control, and calculated relative to KB2P1.21R2 cells treated with the respective drug. Below, table lists P-value thresholds for the indicated comparisons, by paired t-test: * P ≤0.05, ** P ≤ 0.01, *** P≤0.001, and **** P≤0.0001. Graph data are mean ± SD of technical triplicate from 2 independent experiments. (D) As described in (B), in cells treated with increasing concentrations of cisplatin. (E) Growth of allografted KB2P1.21-shControl (top; n = 7 and 8 mice) and KB2P1.21-shTET2 (bottom; n = 10 mice) cells in athymic nude mice treated with DMSO control or olaparib. (F and G) Scatter plot of the ratio of IdU:CldU DNA fibers [green:red in representative images, (G)] upon HU treatment (4 mM for 3 hours) in the indicated cell lines. N = 4 experiments, ~150 DNA fibers. Data in (E and F) are mean ± SD; ns P≥0.05, ** P ≤ 0.01, *** P≤0.001, and **** P≤0.0001, by ANOVA (E) or Mann-Whitney test (F).
To examine the effect of Tet2 knockdown on olaparib resistance in vivo, we injected mouse mammary tumor cell lines KB2P1.21-shControl and KB2P1.21-shTET2 cells into 6- to 8-week-old male athymic nude mice. One week after implantation, mice were treated with olaparib (60mg/kg) or the vehicle control, DMSO. Olaparib-treated KB2P1.21-shControl tumors regressed significantly compared to those treated with DMSO; in contrast, there was no statistically significant difference in the growth of KB2P1.21-shTET2 tumors treated with olaparib or DMSO (Fig. 1E), which supported our in vitro findings. Notably, the KB2P1.21-shTET2 tumors grew faster than KB2P1.21-shControl tumors, although they did not exhibit any significant difference in proliferation in vitro (fig. S1L).
Loss of TET2 restores fork protection but not RAD51 recruitment in BRCA2 deficient cells
Increased expression of p-glycoprotein drug transporters is frequently associated with PARPi resistance in human tumors. This enhances drug efflux and renders cells resistant to chemotherapy (13). We examined the effect of TET loss on the expression of Abcb1α and Abcb1β that encode drug transporter subunits that are frequently upregulated (13). Quantitative RT-PCR did not reveal any significant difference in their expression in KB2P1.21 cells with TET2/TET3 knockdown. (fig. S2A). Furthermore, because TET loss also conferred resistance to cisplatin, which is not substrate of the drug transporter ABCB1 (33), this chemoresistance mechanism was ruled out. PARPi resistance can also be acquired due to partial or complete restoration of BRCA2 function. Therefore, we examined the recruitment of RAD51 to the sites of double-strand breaks (DSBs) to ascertain the effect of TET loss on homologous recombination. Whereas BRCA2-proficient KB2P1.21R2 cells formed distinct RAD51 foci in response to 10Gy γ-irradiation, no foci were observed in KB2P1.21-shControl or KB2P1.21-shTET2 cells (fig. S2, B and C). Because RF stability in BRCA-deficient cells confers chemoresistance, we examined the effect of TET2 loss on the integrity of stalled RFs using the DNA fiber assay (34). This assay involves labeling replication forks sequentially with CldU (red) and then with IdU (green) for 15 to 30 min each, followed by application of hydroxyurea (HU) to stall RFs. The RF is considered to be stable if the ratio of green:red tracks is close to 1 and the fork is degraded if the ratio is significantly reduced. While BRCA2 proficient KB2P1.21R2 cells exhibited stable RFs, BRCA2-deficient KB2P1.21-shControl cells exhibited degradation of RFs upon replicative stress (Fig. 1, F and G). Notably, we observed a significant increase in RF protection in KB2P1.21-shTET2 and KB2P1.21-shTET3 cells compared to KB2P1.21-shControl cells (Fig. 1, F and G; fig. S2D). These findings suggested that TET loss contributes to the stability of stalled RF in BRCA2-deficient cells and contribute to PARPi resistance. Notably, we also observed a significant increase in the stability of stalled forks in BRCA1-deficient human MDA-MB-436 breast cancer cells in response to TET2 knockdown (fig. S2, E and F).
We have previously reported that protection of stalled RFs can contribute to the viability of Brca2ko/ko mESC (16, 17, 35). Therefore, we tested whether TET loss can also rescue the lethality of Brca2ko/ko mESC. We used PL2F7 cells carrying one null allele (ko) and one floxed allele (cko) of Brca2 and stably knocked down Tet2 or Tet1 using each of two shRNAs. We genotyped viable clones obtained after CRE expression and identified multiple Brca2ko/ko clones with Tet2 and Tet1 knockdown but none from PL2F7-shControl cells (fig. S3, A, B, E, and F).
We then examined the effect of Tet1 and Tet2 loss on RF integrity in the rescued PL2F7-shTET2-Brca2ko/ko mESC clones. Expression of a truncated BRCA2 (Y3308X) allele in mESCs resulted in RF degradation in response to HU-induced replicative stress. Two independent rescued Brca2ko/ko ESC clones generated in PL2F7-shTET2 (fig. S3, C and D) and PL2F7-shTET1 (fig. S3G) cells exhibited a significant protection of stalled forks. As observed in KB2P1.21-shTET2 cells, HR was not restored in PL2F7-shTET2-Brca2ko/ko cells based on the failure to recruit RAD51 after IR (fig. S3, H and I).
Global increase in 5hmC levels causes degradation of stalled replication forks
CpG islands in the mammalian genome can undergo TET-mediated demethylation and affect gene expression (28). Published RNA-seq data from mESC lacking TET1 and TET2 did not reveal any significant change in the expression of genes known to protect stalled forks in BRCA2-deficient cells (36). We confirmed the expression of some of these proteins by Western blotting and found no change in the expression of PARP1, MRE11, PTIP, CHD4 and RAD51 (fig. S4A), suggesting that TET loss-mediated RF protection may not be due to altered expression of these proteins.
Given the role of TET proteins in catalyzing the oxidation of 5mC to 5hmC, which is one of the stable intermediates of 5mC modification, we explored the possibility that change in 5hmC levels on the DNA may have a direct impact on the RF stability. We examined the effect of enhancing 5hmC levels in KB2P1.21R2 (BRCA2-proficient) and KB2P1.21 (BRCA2-deficient) cells by treating them with vitamin C (L-ascorbic acid, or LAA). Vitamin C is a known cofactor of TET proteins and has been shown to increase global 5hmC levels in a TET-dependent manner (37). Mass spectrometric analysis revealed increase in global 5hmC levels upon LAA treatment (Fig. 2A). Notably, KB2P1.21 cells had higher 5hmC levels than KB2P1.21R2 after all drug treatments. We also observed a marked increase in 5hmC levels in response to HU treatment in KB2P1.21 cells alone (Fig. 2A); however, when HU treatment was preceded by LAA, both cells showed significant increase in 5hmC levels (Fig. 2A). There was no significant change in global 5mC levels observed under these conditions (fig. S4B). This increase in 5hmC in cells lacking functional BRCA2 is consistent with the reports showing an increase in global 5hmC in response to DNA damage (29, 30). Similar increase in global 5hmC and no change in 5mC levels in response to HU alone and in combination with LAA was observed upon transient knockdown of BRCA2 in human osteosarcoma U2OS cells (Fig. 2B and fig. S4, C and D). We do not precisely understand why 5hmC levels are high in BRCA2-deficient cells; however, we have observed an increase in Tet2 mRNA levels in BRCA2-deficient KB2P1.21 cells compared to BRCA2-proficient KB2P1.21R2 cells (fig. S4E). Similarly, we observed an increase in TET2 mRNA in response to BRCA2 knockdown in U2OS cells (fig. S4F). Because some of the 5hmC is converted into abasic sites by the base excision repair (BER) pathway, we examined whether there is an increase in abasic sites in response to LAA treatment in BRCA2 deficient cells KB2P1.21 cells. We quantitated global abasic sites by using an aldehyde reactive probe-based colorimetric assay and observed increase in abasic sites in the genomic DNA in response to LAA and HU in these cells (fig. S4G).
Figure 2. Increase in 5hmC at RFs is correlated with fork degradation.
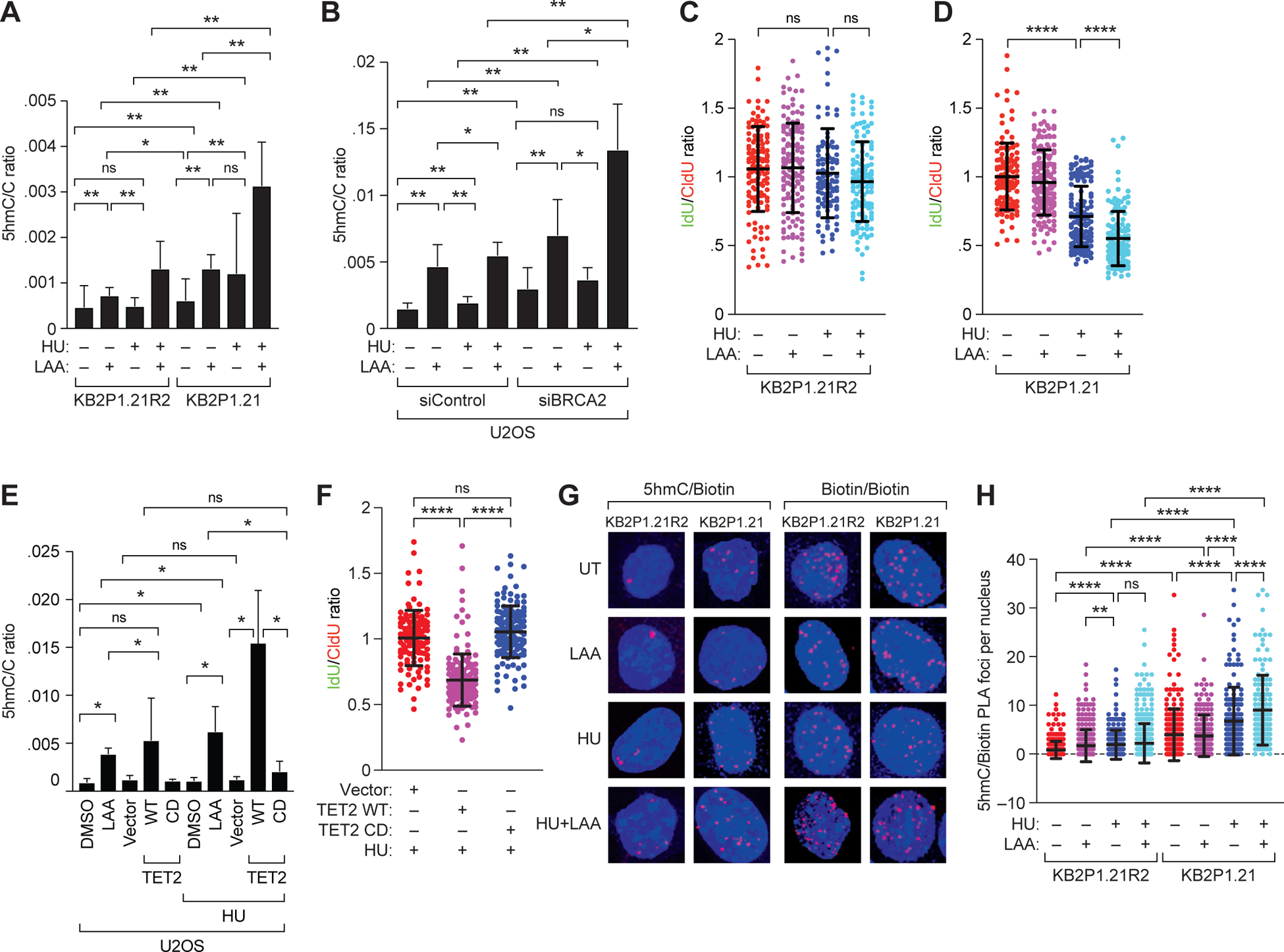
(A and B) Mass spectrometry-based quantitation of 5hmC levels, as a ratio to that of cytosine, in (A) mouse mammary tumor cells KB2P1.21R2 (BRCA2-proficient) and KB2P1.21 (BRCA2-deficient) and (B) U2OS cells after transient knockdown of BRCA2, each upon treatment with L-ascorbic acid (LAA; 250 μM, 72 hours), HU (4mM, 3 hours), or both. (C and D) Scatter plot showing ratio of IdU:CldU DNA fibers in KB2P1.21R2 (C) and KB2P1.21 (D) cells upon treatment with HU, LAA or both. (E) Mass spectrometry-based quantitation of 5hmC levels, as a ratio to that of cytosine, in U2OS cells treated with DMSO or LAA, each with or without HU, and in U2OS cells after transient overexpression of wild-type (WT) or catalytically deficient (CD) TET2 (or empty vector), either untreated or treated with HU. (F) Scatter plot showing ratio of IdU:CldU DNA fibers in U2OS cells after transient expression of vector or wild-type (WT) or catalytically deficient (CD) TET2 followed by treatment with HU. (G and H) Representative images (G) and quantiation (H) of proximity ligation assays in EdU-biotin-labeled KB2P1.21R2 and KB2P1.21 cells using 5hmC and biotin antibodies in DMSO-treated cells or those treated with HU, LAA, or both. Scale bar, 5 μm. Biotin/biotin PLA using two distinct biotin antibodies served as an internal control for EdU incorporation. Data are mean ± SD from n = 2 (C, D, and H) or 3 (F) experiments, or from 2 experiments each in triplicate (A, B, and E); in each, N = ~150 DNA fibers. ns P ≥ 0.05, * P ≤ 0.05, ** P ≤ 0.01, and **** P ≤ 0.0001 by Mann-Whitney test.
After assessing global 5hmC increase on chromatin, we performed DNA fiber assays in KB2P1.21R2 and KB2P1.21 cells treated with LAA. RFs remained relatively protected in KB2P1.21R2 cells after HU treatment as well as after combined treatment with HU and LAA (Fig. 2C); whereas, as expected, the fork was degraded in KB2P1.21 cells upon HU treatment (Fig. 2D). However, unexpectedly, the extent of degradation in KB2P1.21 cells was significantly enhanced when HU treatment was combined with LAA (Fig. 2D). A similar effect on stalled RF degradation was observed in BRCA2-knockdown U2OS cells treated with HU or LAA (fig. S4, H and I). To rule out TET-independent effects of vitamin C on RF stability, we assessed the effect of combined treatment with HU and LAA in KB2P1.21-shTET2 and KB2P1.21-shTET3 cells. We found that the enhanced degradation seen in KB2P1.21-shControl cells was abrogated in KB2P1.21-shTET2 and KB2P1.21-shTET3 cells (fig. S4J). This suggested that the enhanced RF degradation in response to LAA treatment was TET-dependent.
In contrast to KB2P1.21 cells (Fig. 2D), combined treatment of HU and LAA did not affect the stability of RF in BRCA2 proficient KB2P1.21R2 cells (Fig. 2C) as well as U2OS cells (fig. S5A). We hypothesized that the levels of 5hmC may not be high enough to cause fork degradation. We therefore enhanced 5hmC levels by TET2 overexpression. We transfected U2OS cells with WT TET2 or a catalytically deficient mutant of TET2 (TET2H1304Y/D1306A) and quantitated global levels of 5mC and 5hmC (Fig. 2E and fig. S5, B and C). Overexpression WT TET2 resulted in a marked increase in 5hmC levels that was 1.5- to 3-fold higher than that seen in LAA-treated U2OS cells (Fig. 2E). In contrast, U2OS cells expressing either vector (Fig. 2E) or TET2H1304Y/D1306A revealed no change in 5hmC levels (Fig. 2E). Unexpectedly, we found that WT TET2 overexpression significantly reduced stalled RF integrity even in the presence of functional BRCA2 (Fig. 2F and Fig. S5D). fExpression of vector and the catalytically deficient TET2 mutant had no effect on fork stability, consistent with lack of a significant change in 5hmC levels (Fig. 2, E and F; fig. S5, B and C).
Level of 5hmC increases at stalled replication forks
Our findings suggest that an increase in global 5hmC impacts stability of stalled forks. Next, we investigated whether fork instability correlated with 5hmC levels at the fork. To quantify 5hmC levels on active replication forks, we performed a proximity ligation assay (PLA) (38). PLA is a technique used to detect the presence of two targets that are in close proximity (<40nm) using primary antibodies specific to each target. Secondary antibodies conjugated with short DNA oligonucleotides probes are used to generate long DNA products by rolling circle amplification that can be visualized using fluorescent probes under a microscope. We labeled the RF in KB2P1.21R2 and KB2P1.21 cells with a short pulse of EdU and treated the cells with LAA and/or HU. Biotin was conjugated to EdU and antibodies against biotin and 5hmC were used to quantify 5hmC at the fork. Biotin/biotin PLA using two biotin antibodies was used as a control to assess EdU incorporation under different conditions. As expected, the number of biotin/biotin PLA foci remained constant even after LAA, HU or both treatments (Fig. 2G and fig. S5E). However, the increase in the number of 5hmC/biotin PLA foci both after HU treatment and HU in combination with LAA indicates the presence of 5hmC at stalled replication forks. It is evident that KB2P1.21 cells have significantly higher 5hmC/biotin foci than KB2P1.21R2 cells (Fig. 2, G and H). These results clearly indicate that along with the global 5hmC increase, there is a significant increase in 5hmC at RF, which can cause degradation of stalled RF. Similar increase in 5hmC at RF after HU and LAA treatment was also observed in U2OS cells with transient BRCA2 knockdown (fig. S5, F and H). Notably, global increase in 5hmC levels did not correlate with an increase in 5hmC levels at the fork after LAA or HU treatments in KB2P1.21R2, KB2P1.21 and U2OS cells. Global increase in 5hmC in response to LAA treatment was higher than that in response to HU (Fig. 2, A and B), whereas 5hmC increase at RF after HU treatment is higher than LAA treatment (Fig. 2H and fig. S5G). This suggests that TET2 might get recruited to RFs in response to HU treatment.
APE1 endonuclease causes 5hmC-mediated degradation of stalled replication forks
Our results demonstrate that increased levels of 5hmC causes degradation of stalled replication forks and it is independent of the BRCA2 status. MRE11 endonuclease is responsible for fork degradation in BRCA2-deficient cells. However, MRE11 knockdown did not rescue fork degradation caused by TET2 overexpression (Fig. 3, A and B), suggesting that some other endonuclease is responsible for the 5hmC-mediated fork degradation. To identify the nuclease responsible for 5hmC-mediated RF degradation, we knocked down the nucleases APE1 (apurinic/apyrimidinic endonuclease), APE2, CTIP, DNA2, EXO1, FAN1, MRE11 and MUS81 in U2OS cells. Only APE1 knockdown rescued TET2 overexpression-mediated RF degradation (Fig. 3A and fig. S6A). This result corroborated well with previous findings that APE1, which plays a key role in the BER pathway, is responsible for removal of 5hmC and conversion back to cytosine. We knocked down APE1 protein abundance using siRNA or inhibited its activity by using a competitive active site inhibitor (APE1 Inhibitor III, Millipore) in TET2-overexpressing U2OS cells and observed that each resulted in a significant protection of stalled RF (Fig. 3B and fig. S6B). Because APE1 is an essential component of the BER pathway, we tested whether the RF instability in cells overexpressing TET2 can be rescued by blocking the abasic sites. It has been reported that the chemical drug methoxyamine binds to abasic sites and masks it from processing by APE1 (39, 40). Indeed, methoxyamine treatment abrogated TET2 overexpression-mediated fork degradation, similar to the protection observed after APE1 inhibition/knockdown (Fig. 3B and fig. S6B). The protein HMCES (5-hydroxymethylcytosine binding, ES cell-specific) has been implicated in the protection of abasic sites on single-stranded DNA (41). Notably, HMCES knockdown in U2OS cells also degraded stalled forks, which was also rescued by APE1 knockdown or inhibition (Fig. 3C and fig. S6C).
Figure 3. The endonuclease APE1 promotes 5hmC mediated degradation of stalled RFs.
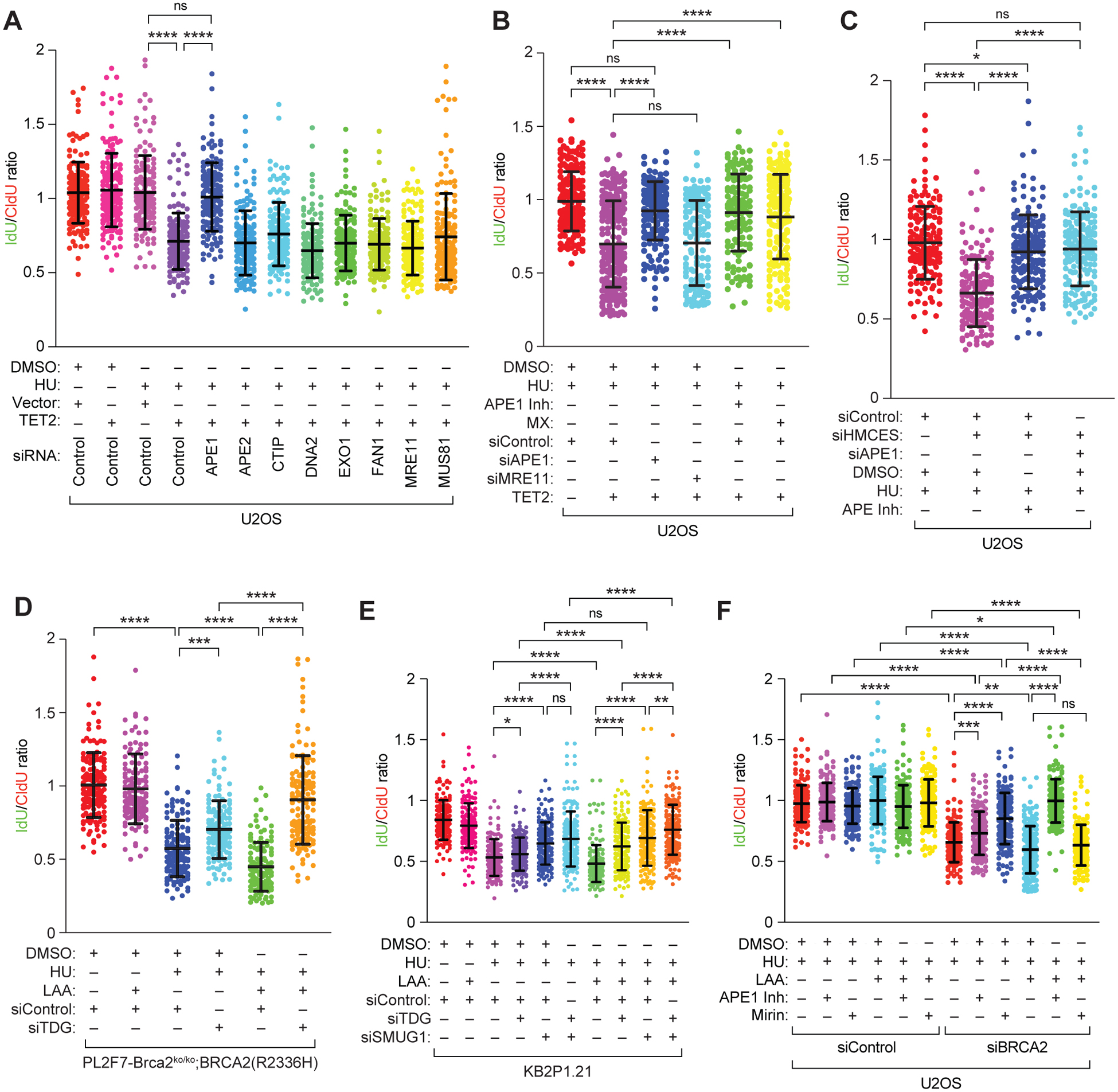
(A to F) Scatter plots showing ratio of IdU:CldU DNA fibers in U2OS cells (A to C, and F), PL2F7-Brca2ko/ko;BRCA2(R2336H) cells (D), and KB2P1.21 cells (E) after the indicated treatment with drugs and/or inhibitor (Inh), after transfection with the indicated expression vector and/or siRNA (si-). MX, methoxyamine (50 μM). Data are mean ± SD from n = 2 (A, D to F) or 3 (B and C) experiments, each N = ~150 DNA fibers. ns P≥0.05, * P ≤0.05, ** P ≤ 0.01, ***P≤0.001, and **** P≤0.0001 by Mann-Whitney test.
Abasic sites are generated by DNA glycosylases by targeting primarily modified cytosine residues such as 5fC and 4caC (42). Notably, thymine DNA glycosylase (TDG) knockdown was reported previously to contribute to PARPi resistance in mESCs (43). Therefore, to understand the role of glycosylases in RF stability, we knocked down TDG in mESCs expressing the hypomorphic mutant of BRCA2 (BRCA2R2336H), cellsthat were used in our siRNA screen. We observed partial rescue of RF stability in HU-treated, TDG-knockdown cells compared to the treated control-transfected cells (Fig. 3D and fig. S6D). However, TDG knockdown completely rescued stalled RF degradation in cells treated with HU preceded by LAA treatment (Fig. 3D). Unexpectedly, TDG knockdown resulted in marginal RF protection in KB2P1.21 cells treated with HU subsequent to LAA (Fig. 3E and fig. S6E). We attributed this to functional redundancy of glycosylases. We examined the effect of single-strand-selective monofunctional uracil-DNA glycosylase 1 (SMUG1) glycosylase, which is also responsible for conversion of 5fC and 5caC to abasic sites (42). SMUG1 knockdown resulted in significant protection of RF degradation after HU treatment (Fig. 3E). Fork protection by combined knockdown of TDG and SMUG1 was not better than SMUG1 knockdown alone in the absence of LAA treatment (Fig. 3E). Notably, in response to LAA treatment, combined knockdown of the two glycosylases completely rescued RF degradation (Fig. 3E). We conclude from these findings that the presence of abasic sites can contribute to fork instability.
Our observation that TET-loss protects the forks raises the question of whether APE1 plays a physiological role in fork instability in BRCA2-deficent cells. To address this, we chemically inhibited APE1 in U2OS cells with transient knockdown of BRCA2 (Fig. 3F and fig. S6F). As expected, Mirin, which inhibits the exonuclease activity of MRE11, rescued RF degradation in BRCA2-knockdown cells after HU treatment (Fig 3F). We observed partial rescue of RF degradation when compared to untreated control with BRCA2 knockdown (Fig. 3F). APE1 inhibition was able to rescue RF degradation completely in HU treated cells preceded by LAA. However, under similar conditions, MRE11 inhibition did not protect the forks suggesting that APE1 is solely responsible for RF degradation when 5hmC levels are high at the forks (Fig. 3F). A similar level of RF stability was observed in KB2P1.21 (BRCA2-deficient) cells after inhibition of APE1 and MRE11 after treatment with HU and LAA (fig. S6G).
It was unclear whether the 5hmC on the parental strand or the nascent strand is responsible for fork degradation by APE1. Therefore, we tested the impact of blocking methylation of the nascent strand by transiently inhibiting DNMT1 with 2’ deoxy-5’azacytidine (DAC) only during the DNA fiber analysis(44) (Fig. 4A). This was based on our observation that DNMT1 knockdown mediated reduction in DNA methylation protects stalled RFs in TET2-overexpressing U2OS cells (Fig. 4B). Unexpectedly, DAC-mediated inhibition of DNMT prevented the degradation of stalled RFs in TET2-overexpressing cells (Fig. 4B and fig. S7A), suggesting that nascent strand methylation contributes to fork degradation. To demonstrate that 5hmC, 5fC or 5caC on the nascent strand can degrade stalled forks, we treated BRCA2-proficient U2OS and KB2P1.21R2 cells with 2-deoxy-cytidine (cytidine), 2-deoxy-5-methyl-cytidine (5mC), 2-deoxy-5-hydroxymethyl-cytidine (5hmC), 2-deoxy-5-formyl-cytidine (5fC) or 2-deoxy-5-carboxy-cytidine (5caC) along with CldU and IdU (each for 30 mins) during fiber assays. Incorporation of cytidine did not affect RF stability, but incorporation of modified cytidine residues resulted in RF degradation after HU treatment (Fig. 4C and fig. S7B). Furthermore, APE1 inhibition prevented RF degradation due to incorporation of these modified cytidine residues (Fig. 4C and fig. S7B). Together, these results clearly demonstrate that an increase in 5hmC, 5fC or 5caC in the nascent DNA strand can contribute to the degradation of stalled forks by APE1.
Figure 4. 5hmC modification of nascent strand causes degradation of stalled replication forks.
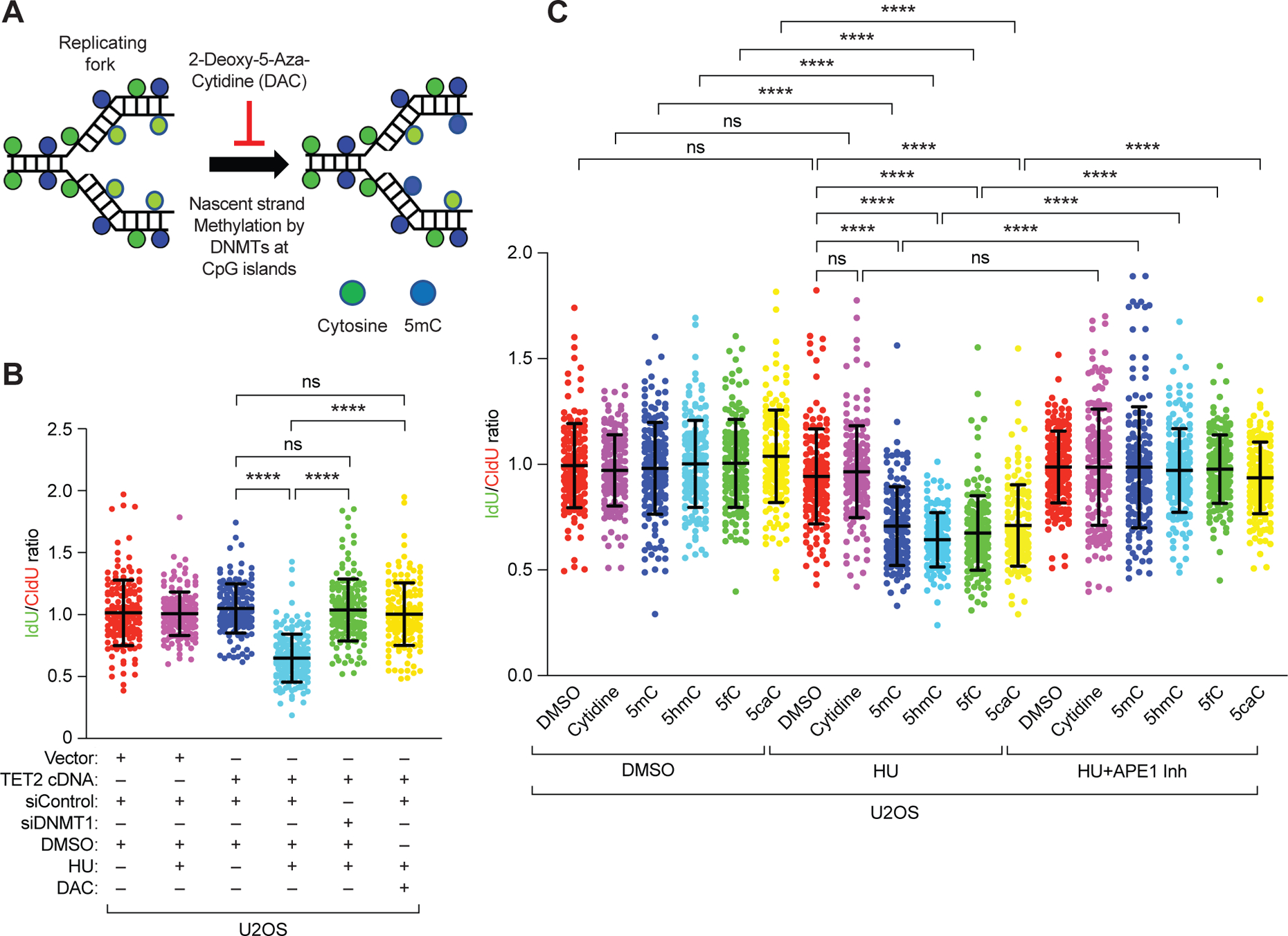
(A) Schematic of the hypothesis that inhibiting DNMT1-mediated methylation of DNA in replication forks with 2-deoxy-5-aza-cytidine (DAC) during DNA fiber analysis will inhibit methylation of nascent DNA strand. (B) Scatter plot of the ratio of IdU:CldU (green:red) in DNA fibers of U2OS cells after TET2 overexpression and DNMT1 knockdown or its inhibition by DAC and in response to either DMSO or HU. (C) Scatter plots showing ratio of IdU:CldU DNA fibers in U2OS cells after treatment with cytidine or modified cytidine residues (1 μM), in the presence of either DMSO, HU, or HU and APE1 inhibitor (600 nM). Data are mean ±SD from n = 2, N = ~150 DNA fibers. ns P≥0.05, and **** P≤0.0001 by Mann-Whitney test.
Finally, we examined whether an increase in 5hmC can contribute to genomic instability. We overexpressed TET2 in U2OS cells and examined the effect of inducing replicative stress. We observed a two-fold increase in chromosomal aberrations and radial structures as well as broken radials in HU-treated, TET2-overexpressing cells compared to HU-treated control cells (Fig. 5, A and B). Our findings demonstrate that an increase in TET2 levels results in genomic instability, suggesting that a reduction in TET2 levels or activity can contribute to the genomic integrity of cells under conditions of replicative stress.
Figure 5. Mechanism of 5hmC-mediated RF degradation.
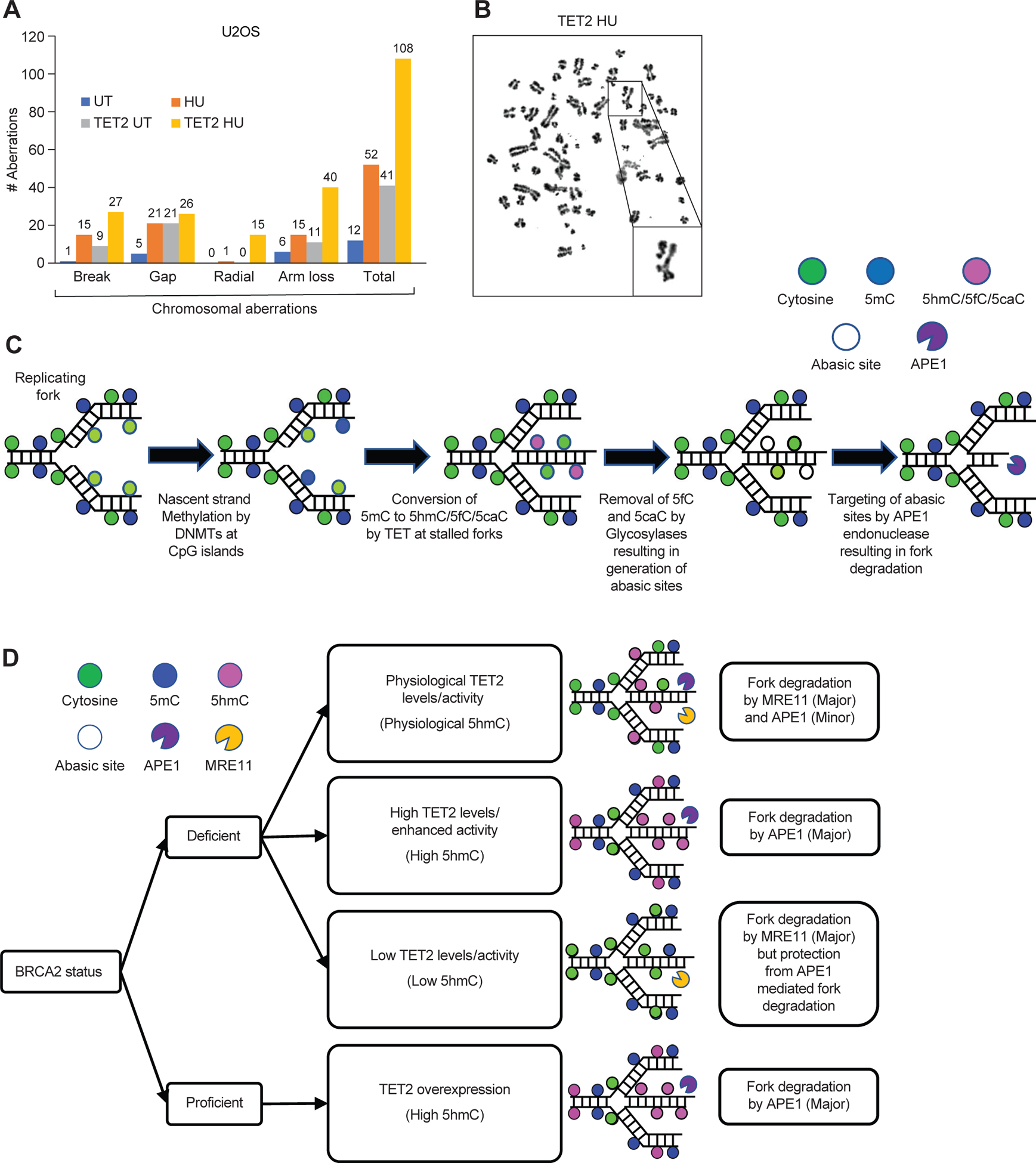
(A and B) Quantification of chromosomal aberrations (A), such as radial structures (B), in 30 randomly selected U2OS cells with and without TET2 overexpression and either untreated (UT) or treated with HU. Data are from a representative of 2 independent experiments. (C and D) Schematic representation of the proposed mechanism of 5hmC-mediated, APE1-dependent degradation of stalled RFs (C) and the effects thereon of the presence or absence of BRCA2, the levels of TET2 expression or activity, and the level of 5hmC (D).
DISCUSSION
DNA methylation is a well-known epigenetic mechanism of regulation of gene expression. Consequently, precise control of DNA methylation patterns is essential for normal growth and development. Whereas DNA methyltransferases (DNMTs) are essential for DNA methylation, the TET family of methylcytosine dioxygenases are considered to play a vital role in maintaining the fidelity of DNA methylation patterns by catalyzing DNA demethylation. TET proteins are known to play an important role in normal hematopoiesis and immune regulation (45, 46). Mutations in TET genes are frequently associated with hematopoietic malignancies (47). In this study, starting with a genome-wide siRNA-based screen, we identified that loss of TET proteins results in resistance to multiple PARP inhibitors as well as cisplatin in BRCA2-deficient cells. We uncovered their role in the stability of stalled replication forks by affecting the levels of 5hmC at the fork. Although fork degradation in BRCA2-deficient cells is primarily mediated by MRE11, APE1 may also have a physiological role in RF stability. This is supported by our observation that APE1 knockdown/inhibition contributes to the partial fork protection in BRCA2-deficient cells. Given the impact of TET proteins on the global gene expression, we cannot rule out the indirect effects of their loss on replication fork protection. However, based on our studies and published reports, we did not observe any significant change in the expression of any of the known key regulators of RF integrity (36). In contrast, we uncovered a direct correlation between 5hmC levels and fork stability. We have shown that increase in 5hmC/5fC/5caC renders stalled replication forks vulnerable to degradation by base excision repair associated APE1 endonuclease.
We propose that the nascent strands are methylated after replication by DNMTs. This is followed by sequential conversion of 5mC to 5hmC/5fC/5caC by TET proteins at stalled forks. These modified cytosine intermediates (5fC and 5caC) are processed by glycosylases resulting in generation of abasic sites (Fig. 5C). These abasic sites are excised by APE1 endonuclease. It is known that in addition to its endonuclease activity, APE1 also possess 3’ to 5’ exonuclease activity (48). Although the exonuclease activity has been reported to cleave a few bases in vitro after excising the abasic sites, it is possible that in vivo, APE1 may have higher processivity at stalled forks (48, 49). We cannot rule out the possibility of an APE1-dependent recruitment of other exonucleases to the stalled RF that may also contribute to fork degradation. However, none of the other nucleases we tested had any effect on TET2-mediated instability of stalled forks. Excessive 5hmC-mediated fork degradation due to replicative stress induces genomic instability. We observed an increase in radial and radial break structures in metaphase spreads of TET2 overexpressing cells in response to HU treatment.
Because BER-mediated repair of 5hmC to cytosine does not require BRCA2, RAD51 or MRE11, an increase in 5hmC at stalled RF can cause fork instability even in BRCA2-proficient cells. This also suggests that RF degradation by APE1 is independent of the classical model of fork protection, wherein stalled forks undergo a process called fork reversal and the recessed arms of reversed forks are loaded with RAD51, thereby conferring protection from nucleases, such as MRE11 (36). Our findings suggest that the 5hmC-mediated degradation is an additional source of RF instability, which may not be significant under normal physiological conditions. However, when there is an increase in 5hmC at stalled forks (such as in response to DNA damage), APE1 has a substantial impact on fork integrity. This is supported by our observation that TET loss provides fork protection in BRCA2-deficient cells and causes PARPi and cisplatin resistance (diagrammed in Fig. 5D).
Given the overall low levels of 5hmC in the genome, it is remarkable that it has a significant effect on the fork integrity. To reconcile these observations, we postulate that along with a global increase in 5hmC in response to DNA damage or increase in TET activity, there must be a targeted increase at the fork as well. This is supported by our PLA-based experiments, which revealed a significant increase in 5hmC localized at RFs after HU treatment. How TET2 is recruited to the stalled forks is unknown. In spite of a clear increase in 5hmC at the fork, we were not successful in detecting TET2 at stalled RFs by PLA. Mass-spectrometric analysis of proteins at stalled forks have also not detected TET proteins at the fork (35). It is possible that this is due to the transient nature of its recruitment to the fork.
The effect of 5hmC on fork instability is dependent on the generation of abasic sites, the target of APE1 endonuclease. 5hmC is not the only source of generation of abasic sites. In fact, up to 10,000 abasic sites are generated in mammalian cells per day, making them the most common DNA lesions (50). Some of the most common sources of abasic site are spontaneous hydrolysis of the N-glycoside bond and DNA glycosylase-mediated removal of DNA bases damaged due to oxidative stress or exposure to X-ray and UV irradiation. HMCES (also known as SRAP1) has been reported to bind to abasic sites and suppress genomic instability in cells (41). We have demonstrated here that if spontaneously generated abasic sites are left unprotected by HMCES knockdown, stalled forks are targeted for degradation by APE1. Although HMCES was reported to protect the abasic sites on ssDNA, because it interacts with PCNA and is localized to the replication forks it may protect abasic sites also at the fork (41). In the absence of HMCES, we predict that the abasic sites are targeted by APE1 and contribute to fork degradation just like they do when 5hmC, 5fC or 5caC levels are increased at the fork. Instability of stalled forks in HMCES-deficient cells can be suppressed by APE1 knockdown, demonstrating a broader physiological role of BER in fork stability. Interestingly, a previous study reported TDG loss to contribute to resistance to olaparib resistance (43). Because PARP1 associates with abasic sites, it is possible that PARP1 trapping may be affected and that may contribute to PARPi resistance (51). The study ruled out that TDG loss affected PARP1 trapping in response to PARPi treatment, but the precise mechanism remained unknown (43). We believe that TDG loss may suppress formation of abasic sites, which may contribute to fork protection and olaparib resistance.
Our findings on the impact of TET proteins on PARPi resistance may have broader clinical implications. It was recently reported that hypoxia reduces TET activity and 5hmC levels in tumor microenvironment (52). Tumor-associated hypoxia contributes to resistance to both chemo- and radiation-therapy because of its effect on a number of biological processes (53, 54). Future studies will focus on exploring the possibility of enhancing TET activity or 5hmC levels as a means to restore tumor sensitivity to chemotherapy.
MATERIALS AND METHODS
Plasmids, Antibodies and Reagents.
pCDNA3-Flag-mTET2 plasmid expressing wildtype mouse TET2 was purchased from Addgene (#89735). pCDNA3-Flag-mTET2 catalytic domain-catalytic dead mutant expressing TET2 with H1304Y-D1306A mutations was purchased from Addgene (#72220). The full-length TET2 catalytic-dead mutant construct was generated by cloning missing N-terminal region from wildtype plasmid (#89735) into (#72220) plasmid using Fse1 and NotI restriction sites. The pCDNA3.1+ (Invitrogen) vector was used as vector control. Immunofluorescence (IF), Western blotting (WB), DNA fiber assays, and proximity ligation assays (PLA) used the following antibodies: BRCA2 (A303–434A; Bethyl Laboratories, 1:2000 dilution for WB), TET1 (09–872; EMD-Millipore, 1:1000 dilution for WB), TET2 (45010S; Cell Signaling Technology, 1:1000 dilution for WB), actin (I-19: sc-1616; Santa Cruz Biotechnology, 1:10000 dilution for WB), MRE11 (a kind gift from Andre Nussenzweig, NCI-Bethesda, NIH; 1:20000 dilution for WB), HMCES (PA5–60876; Invitrogen, 1:5000 dilution for WB), APE1 (ab92744;Abcam, 1:5000 dilution for WB), PARP1 (9542S; Cell Signaling Technology, 1:2000 dilution for WB), CHD4 (ab74603; Abcam, 1:1000 dilution for WB), PTIP (a kind gift from Andre Nussenzweig, NCI-Bethesda, NIH; 1:2500 dilution for WB), GAPDH (sc-32233, Santa Cruz Biotechnology, 1:2000 dilution for WB), FLAG (F3165; Sigma, 1:5000 dilution for WB), RAD51 (PC130; Calbiochem, 1:2000 dilution for WB; 1:500 dilution for IF), γH2AX (07–164; Upstate, 1:500 dilution for IF), 5hmC (39769; Active Motif, 1:500 dilution for PLA), biotin (200-;002-211, mouse; BD Biosciences, 1:500 dilution for PLA; A150–109A, rabbit, 1:500 dilution for PLA), CldU (ab6326; Abcam, 1:500 dilution for DNA fiber assays), IdU (347580; BD Biosciences, 1:500 dilution for DNA fiber assays). The following chemicals were purchased: Olaparib (AZD2281, S1060; Selleckchem), hydroxyurea (HU; H8627; Sigma), L-ascorbate (LAA, A7631; Sigma), APE1 Inhibitor (APE1 Inhibitor III, 262017; Calbiochem), methoxyamine (MX, 226904; Sigma), Mirin (M9948; Sigma), 2-deoxy-5-aza-cytidine (A3656; Sigma), 5-(hydroxymethyl)-2’-deoxycytidine (H946630; Toronto Research Chemicals), 5-methyl-2’-deoxy cytidine (M295900; Toronto Research Chemicals), 2’-deoxycytidine-5-carboxylic acid, sodium salt (PY7593; Berry and Associates), 5-formyl-2’-deoxycytidine (PY7589; Berry and Associates), 2′-deoxycytidine (D3897; Sigma). veliparib (S1004; Selleck Chemicals LLC), talazoparib (S7048; Selleck Chemicals LLC).
Genome-wide siRNA library screening.
Transfections were performed in 384 well plates (Corning 3707). Plates were coated with 0.1% gelatin in H2O overnight at ambient temperature. Gelatin solution was aspirated (BioTek EL406) and siRNAs were printed using a Bravo VPrep liquid handler integrated within a BioCel automation platform (Agilent). Cell viability was measured using CellTiter Glo (Promega). For transfections, 20 μL of serum free media containing Lipofectamine RNAiMax (0.1 μL) was added to wells containing siRNA (0.8pmol). Lipid and siRNA were allowed to complex for 45 min at ambient temperature before addition of 1000 cells in DMEM, 30% FBS to yield final transfection mixtures containing 20 nM siRNA in DMEM, 15% FBS. The screen was conducted using an Ambion (Thermo) Silencer Mouse Genome siRNA collection. The library targets 17,575 mouse genes with 3 siRNAs per gene. Olaparib was added 48 h post-transfection (500 nM) and viability (CellTiter Glo, Promega) was assayed 72 h later on a PerkinElmer Envision 2104 Multilabel plate reader. Ambion Silencer Select Negative Control #2 was incorporated on all screening plates (16 wells) and the median negative control value on each plate was used for normalization. An siRNA targeting Trp53 was used as a positive control (Ambion, catalog# 69843, target sequence AAGGAAAUUUGUAUCCCGAGU). This control yielded a median protective effect of ~1.9 fold versus negative control throughout the screen. The median absolute deviation (MAD) based z-score was calculated for each siRNA (55). Genes were then ranked by taking the median value for all siRNAs tested against a given gene. Scores were also adjusted for seed-based off-target effects to help deprioritize likely false positives (56).
Stable and transient knockdown of proteins.
Lentiviral shRNA vector against mouse TET1 (shRNA1-TRCN0000341847, shRNA2-TRCN0000341848), mouse TET2 (for mouse mammary tumor cells knockdown; shRNA1-TRCN0000250896, shRNA2-TRCN0000250894; for mESC knockdown; shRNA1-TRCN0000250892, shRNA2-TRCN0000250895), mouse TET3 (shRNA1-TRCN0000376843, shRNA2-TRCN0000375394), mouse BRCA2 (shRNA1-TRCN0000071009, shRNA2-TRCN0000071010, shRNA3-TRCN0000071011), human TET2 (shRNA1-TRCN0000144344, shRNA2-TRCN0000418976) and control shRNA (SHC202) were purchased from Sigma (MISSION shRNA). HEK293T cells were used for viral packaging. HEK293T cells were co-transfected with shRNA vector, pRSV-Rev, pMDLg-pRRE, and pHCMVG using FuGENE transfection reagent (E2311) from Promega. Packaging vectors were kind gifts from Dr. Steven Hou (NCI-Frederick, NIH). After 72 h transfection, the supernatant was collected and filtered 0.45 μm filter before being used for infecting either mESCs or BRCA2 deficient mouse mammary tumor cells KB2P1.21. After 48 h infection, cells were subjected to 3 μg/ml puromycin selection for 5 days and puromycin-resistant cells were pooled and used further. Transient knockdown of mentioned proteins was carried out by siRNA transfection using Lipofectamine RNAiMAX transfection reagent (Life Technologies). All siRNA was purchased as SMARTpool siGENOME category from Dharmacon. Mouse TET1 (L-062861-01-0005), Mouse TET2 (L-058965-00-0005), mouse TET3 (L-054156-01-0005), Mouse TDG (M-040666-01-0005), Mouse SMUG1 (M-041546-01-0005), Human HMCES1 (L-020333-02-0005), Human APE1 (L-010237-00-0005), Human MRE11 (L-009271-00-0005), Human DNMT1 (M-004605-01-0005), Human APE2 (M-013730-00-0005), Human CTIP (M-011376-00-0005), Human DNA2 (M-026431-01-0005), Human EXO1 (M-013120-00-0005), Human FAN1 (M-020327-01-0005), Human MUS81 (M-016143-01-0005). siRNA transfections were performed using Lipofectamine RNAiMAX (Life Technologies) according to manufacturer’s protocol.
Cell culture, transfection and drug treatments.
All mESCs were cultured on top of mitotically inactive SNL feeder cells in M15 media (knockout DMEM media (Life Technologies) supplemented with 15% fetal bovine serum (FBS; Life Technologies), 0.00072% beta-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.292 mg ml-1 L-glutamine) at 37 °C, 5% CO2. PL2F7 cells are derivatives from AB2.2 mouse embryonic stem cell line (57). PL2F7 cells expressing BRCA2 R2336H and Y3308X variants were generated by electroporating respective BACs in PL2F7 cells(31, 57). PL2F7-Brca2cko/ko;BRCA2(R2336H) cells express a conditional allele (cko) of Brca2 along with BRCA2(R2336H) hypomorphic allele but PL2F7-Brca2ko/ko;BRCA2(R2336H) cells express only the hypomorphic allele. Mouse mammary tumor cell lines (KB2P1.21R2 and KB2P1.21) were cultured at 37 °C, 5% CO2, 3% O2 in DMEM/F-12 (Life Technologies) supplemented with 10% FBS, antibiotics. The human osteosarcoma cell line U2OS (obtained from American Type Culture Collection (ATCC), Rockville, MD) was cultured at 37 °C, 5% CO2 in DMEM/F-12 (Life Technologies) supplemented with 10% FBS, antibiotics. U2OS cells were transfected with indicated plasmids using Lipofectamine 2000 (Life Technologies) according to manufacturer’s protocol. MDA-MB-436 and MDA-MB-436-BRCA1ko cells were kind gift from Neil Johnson. These cells were cultured in RPMI1640 media (Life Technologies) supplemented with 10% FBS, antibiotics. Combined treatments for LAA and HU treatments were given sequentially. Cells were treated with 250 μM of LAA for 72 hrs. followed by 4mM HU treatment for 3 hrs. APE1 Inhibitor (600 nM) and MX (50 μM) treatments were given in combination with HU. EdU labelling for proximity ligation assay was performed at 125 μM for 8 mins. 2-deoxy-5-aza-cytidine (DAC) treatment (1μM) was given along with CldU-IdU-HU during fiber assay. 5-Hydroxymethyl-2’-deoxycytidine (5hmC), 5-Methyl-2’-deoxy Cytidine (5mC), 2’-Deoxycytidine-5-carboxylic acid, sodium salt (5aC), 5-Formyl-2’-deoxycytidine, 2′-Deoxycytidine (Cytidine) treatments were given in combination with CldU and IdU treatments for 30 mins during DNA fiber assay.
XTT-based cell survival.
Sensitivity of mESC to olaparib was performed by plating 10,000 ES cells per well in gelatinized 96-well plates in three replicas. The next day, cells were treated with M15 medium containing different concentrations of olaparib . After 48 hours, cell viability was measure by XTT (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide) assay as described previously (58).
Clonogenic survival assay.
Cells were seeded at 5,000 per well into 6-well plates and continuously treated with the PARP inhibitor at mentioned concentrations. Colony forming units were stained 14 days after treatment using 0.5% (w/v) crystal violet in methanol. Cell count was performed for 0.1 μM concentration of olaparib, where cells were at 10000 per well in 12 well plate in triplicate. After 14 days on olaparib treatment, cells were trypsinized, harvested, and counted using Beckman coulter counter. Cell survival was estimated by normalizing all values to values from respective DMSO treated wells. Each well was counted thrice, and the average was taken. For each time point, an average of three independent wells was plotted.
DNA fiber assay.
Cells were sequentially treated with 8 μg/ml CldU for 30 min followed by 90 μg/ml IdU for 30 min followed by 4 mM HU for 3 hr. Post drug treatments cells were trypsinized and resuspended at approximate concentration of 3 × 105 cells in PBS. 5 μl of cells were mixed with 15 μl lysis buffer (200 mM Tris-HCl (pH 7.4), 50 mM EDTA, 0.5% SDS) on glass slides and incubated at room temperature for 8 min before DNA fiber was spread. Fibers were then fixed in methanol and acetic acid (3:1) at 4 °C overnight, rehydrated by PBS followed by denaturation in 2.5 M HCl for 1 h. After rinsing away HCl by PBS, slides were then blocked in PBS with 5% bovine serum albumin (BSA) for 40 min and incubated with anti-BrdU antibody (mouse, #347580, Becton Dickinson, 1:250 dilution) and anti-BrdU antibody (rat, ab6326, Abcam, 1:500 dilution) for 2 hr at room temperature. Slides were rinsed with PBS with 0.1% Tween-20 (PBST) and incubated with AlexaFluor488-conjugated anti-mouse IgG secondary antibody and AlexaFluor594-conjugated anti-rat IgG secondary antibody for 1 h at room temperature. After rinsing with PBST, the slides were mounted by mounting media (Prolong Gold, Invitrogen). the images were captured in Zeiss Axio Imager Z1 microscope and the fiber length was measured by ImageJ software (NIH). All DNA fiber analysis were performed blindly. All siRNA mediated knockdown of proteins associated with DNA fiber assay were carried out for 48 hrs followed by drug treatments, except LAA treatment. LAA treatment (250 μM, 72 hrs) were initiated 24 hrs after siRNA transfection. APE1 Inhibitor (600 nM), mirin (100 μM), methoxyamine (50 μM) treatments were given along with HU treatment (4mM, 3 hrs). 2-deoxy-5-aza-cytidine (DAC) 5-(hydroxymethyl)-2’-deoxycytidine (5hmC), 5-methyl-2’-deoxy cytidine (5mC), 2’-deoxycytidine-5-carboxylic acid (5caC), 5-formyl-2’-deoxycytidine (5fC), 2′-deoxycytidine (Cytidine) treatment was given at 1μM concentrations along with CldU (30 mins) and IdU (30 mins) treatments. All experiments were performed at least twice and in several cases 3 times to verify the results.
Immunofluorescence and Proximity Ligation Assay (PLA).
For immunofluorescence, cells were fixed with 4% paraformaldehyde for 10 mins, permeabilized by 0.25% Triton X-100 for 10 mins and blocked with 3% BSA for 1 hr. Cells were incubated with the indicated primary antibodies at 4 °C overnight. After washing four times with PBST, cells were incubated with AlexaFluo488-conjugated anti-rabbit IgG antibody and AlexaFluo568-conjugated anti-mouse IgG antibody (Life Technologies) at room temperature for 2 h. Nucleus was counterstained by 4,6-diamidino-2-phenylindole. Images were taken on Zeiss LSM 510 confocal microscope.
Proximity Ligation Assay.
Cells were treated with EdU for 20 min at 125 μM, followed by either 4 mM HU treatment for 4 hrs. or were left untreated. Then cells were fixed with 4% paraformaldehyde for 10 mins, followed by DNA denaturation with 2mM HCl for 10 mins, then K2BrO7 treatment at 0.1 mM for 10 mins. Cells were washed with PBS and then permeabilized with 0.25% Triton X-100 for 20 mins. Replication fork in cells labelled with EdU were tagged with Biotin using Click reaction buffer prepared in 1X PBS (10mM sodium-L-ascorbate, 20 mM biotin azide (Life Technologies) and 2mM CuSO4) at room temperature for 30 mins. Then cells were washed with PBS and blocked with Duolink blocking buffer at room temperature for 1 hr. Cells were then stained with either 5hmC (rabbit)-biotin (mouse) or biotin (mouse)-biotin (rabbit) antibody pair at 4°C overnight. Afterwards, in situ proximity ligation assay was performed using the Duolink Detection Kit (Sigma Aldrich Duolink). Briefly, anti-Mouse PLUS and anti-Rabbit MINUS oligonucleotide labelled secondary antibodies (PLA probes) were bound to primary antibodies during a 60 min reaction at 37 °C; then PLA probe-coupled oligonucleotides were ligated during a 30 min reaction performed at 37 °C. An amplification reaction was performed for 140 min at 37 °C to allow generation of a fluorescent signal (PLA signal) at sites where two oligonucleotide probes were ligated. Nuclear PLA foci was imaged by using a Zeiss LSM 510 confocal microscope.
Southern blot.
Electrophoresis was performed on EcoRV-digested DNA on 1% agarose gel in 1X TBE (0.1M Tris, 0.1M boric acid and 2mM EDTA, pH 8.0) and transferred to nylon membrane. [α−32P]-dCTP labelled radioactive DNA probe that distinguishes conditional Brca2 allele (cko-Brca2, 4.8 kb) and Brca2 knockout allele (ko-Brca2, 2.2 kb)20 was hybridized with Hybond-N+ nylon membrane (GE Healthcare) at 65 °C overnight. Membrane was washed twice with saline sodium citrate phosphate (SSCP) buffer containing 0.1% SDS and exposed in phosphor image screen overnight and subsequently developed in Typhoon image scanner.
Growth analysis of TET2 knockdown cells.
A total of 10,000 cells were seeded in triplicate for each cell line in a 12 well plate. At indicated time points, cells were trypsinized, harvested, and counted using Beckman coulter counter. Fold growth was estimated by dividing the cell number at a particular time point by the cell number at the time of seeding (Day 0). Each well was counted thrice, and the average was taken. For each time point, an average of three independent wells was plotted.
Karyotyping of U2OS cells.
U2OS cells post transient expression of TET2 and hydroxyurea treatment were arrested by incubation with Colcemid (10 mg/ml) (KaryoMax Colcemid Solution, Invitrogen, Carlsbad, Calif., USA) 3 hr before harvest to prepare metaphase spread. Cells were collected and treated with hypotonic solution (KCl 0.075 M) for 15 min at 37 °C and fixed with methanol:acetic acid 3:1. Slides were prepared, and chromosomal aberrations were analyzed.
Quantitative PCR.
qPCR to determine TET2, TET3, ABCB1α, ABCB1β, DNMT1 and BRCA2 mRNA levels in mentioned cell line was performed by using iTaq Universal SYBR Green Supermix (Bio-Rad). qPCR reaction was run on Mx3000P qPCR system (Agilent Technologies).
TET2 mRNA expression levels in human breast cancer data sets.
Kaplan-Meier graph was plotted using breast cancer data set from The Cancer Genome Atlas. Prognosis levels of breast cancer patients was classified on basis of high or low expression of TET2 mRNA.
Mass spectrometry-based quantitation of cytosine, 5mC and 5hmC.
Genomic DNA was isolated by Phenol:chloroform method. 5 μg of genomic DNA was digested to nucleoside level by using Nucleoside digestion mix (M0649S; NEB). Mass spectrometry-based quantitation of cytosine, 5mC and 5hmC was performed as described (59, 60). Briefly, Cytosine, 5mC, 5hmC were quantitated using a Thermo Vanquish UHPLC coupled to a Thermo TSQ-Altis tandem mass spectrometer through an electrospray ion source operating in positive ion mode at 3.5kV. Stock standard solutions were prepared in deionized water at a concentration of 1 mmol/L each. Calibration standard mixtures were prepared at concentrations between 1.0–250umol/L for dC, 0.04–10 umol/L for mdC, and 0.002–0.5 umol/L for hmdC. Linear calibration plots were prepared using concentration versus peak area integration (zero intercept) with a R2 greater than 0.999. By comparing the internal standard normalized peak areas in the digest sample to the corresponding retention times from the calibration standards, the micromolar concentrations of the nucleosides were determined against the standard curve. The molar ratio as a percent was then calculated as follows:
Growth analysis of TET2-knockdown cells.
A total of 10,000 cells were seeded in triplicate for each cell line in a 12-well plate. At indicated time points, cells were trypsinized, harvested, and counted using Beckman coulter counter. Fold growth was estimated by dividing the cell number at a particular time point by the cell number at the time of seeding (day 0). Each well was counted thrice, and the average was taken. For each time point, an average of three independent wells was plotted.
Allograft assay for olaparib resistance.
BRCA2-deficient mouse mammary tumor cell lines (KB2P1.21)-transfected with a TET2-targeted shRNA or control shRNA were injected into 6- to 8-week-old male athymic nude mice at a concentration of 2×106 cells /200μl L15 media (KB2P1.21-shControl DMSO n=7 mice; KB2P1.21 shControl olaparib, n=8; KB2P1.21 shTET2 DMSO, n=10; KB2P1.21 shTET2 olaparib, n=10). No selection criteria were used for selecting the mice for each group. One week after implantation, mice were intraperitoneally injected either with olaparib (60mg/kg) in 20μl of DMSO or equal volume of DMSO alone as vehicle control, daily for 4 weeks. Tumor growth (volume, mm3) was measured once every week for the subsequent 6 weeks, calculated as a product of (length × width2)/2. Mice were maintained according to the procedures outlined in the Guide for the Care and Use of Laboratory Animals, under an approved Animal Care and Use Committee (ACUC) protocol.
Abasic site quantitation.
Genomic DNA was purified by phenol:chloroform extraction method and resuspended in H2O. Abasic site quantitation was carried using DNA Damage Quantification Kit-AP Site Counting Kit (DK02–12, Dojindo Molecular Technologies, Inc.). Abasic site quantitation was carried as per manufacturer’s protocol.
Statistics.
Statistics was performed by two-tailed t-test, Mann-Whitney test. All error bars represent standard deviation. P<0.05 was considered statistically significant: ns P≥0.05, * P ≤0.05, ** P ≤ 0.01, *** P≤0.001, and **** P≤0.0001. No statistical methods or criteria were used to estimate sample size or to include or exclude samples. The investigators were not blinded to the group allocation during the experiments.
Supplementary Material
Figure S1: Loss of TET confers drug resistance to PARPi and cisplatin.
Figure S2: Loss of TET confers RF stability in BRCA2-deficient cells.
Figure S3: Loss of TET protects stalled RFs in BRCA2-deficient cells and rescues BRCA2-loss-induced lethality in PL2F7 mES cells.
Figure S4: Effect of global 5hmC change on RF stability.
Figure S5: Determination of threshold 5hmC levels that promote degradation of RFs.
Figure S6: APE1 endonuclease mediates the degradation of 5hmC-marked stalled RFs.
Figure S7: 5hmC modification of nascent strands induces RF degradation.
Data file S1. Genome-wide siRNA screen data.
ACKNOWLEDGEMENTS
We thank members of our laboratory for helpful discussions and suggestions. We thank Drs. Jairaj Acharya, Kajal Biswas, Arnab Ray Chaudhury, Ira Daar, Ken Miller, Andre Nussenzweig and Sounak Sahu for their comments and critical review of the manuscript. We thank Dr. David Cortez for his suggestion to examine the role of APE1 in 5hmC-mediated fork degradation, and Drs. Jairaj Acharya and Joseph Barchi for help with 5hmC MS analysis. We thank Drs. Jos Jonkers and Peter Bouwman (Netherland Cancer Institute) for mouse mammary tumor cell lines, Dr. Neil Johnson (Fox Chase Cancer Center, Philadelphia) for MDA-MB-436-BRCA1 WT/KO cells and Drs. Shalini Oberdoerffer (National Cancer Institute) and Xiaochun Yu (Beckman Research Institute) for WT and catalytically dead mutant TET2 cDNA. We also thank Allen Kane (SAIC-Frederick, Inc., Scientific Publications, Graphics & Media Department) for help with the illustrations. We also thank Dr. Ken Cheng and Lu Chen of the National Center for Advancing Translational Sciences for help with RNAi data submission to the PubChem database.
Funding: X.D. received Department of Defense, Breast Cancer Research Program, Postdoctoral Fellowship (W81XWH-13-1-0362). This research was sponsored by the Intramural Research Program, Center for Cancer Research, National Cancer Institute, US National Institutes of Health.
Footnotes
Competing interests: S.E.M. is currently a full-time Genentech employee. E.C.B. is an AstraZeneca employee with unvested stock options The other authors declare that they have no competing interests.
Data and materials availability: The data from the siRNA screen have been deposited to PubChem database, AID: 1347423 (https://pubchem.ncbi.nlm.nih.gov/bioassay/1347423). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Turner N, Tutt A, Ashworth A, Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 4, 814–819 (2004); published online EpubOct (10.1038/nrc1457). [DOI] [PubMed] [Google Scholar]
- 2.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T, Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005); published online EpubApr 14 (10.1038/nature03443). [DOI] [PubMed] [Google Scholar]
- 3.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A, Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005); published online EpubApr 14 (10.1038/nature03445). [DOI] [PubMed] [Google Scholar]
- 4.Scully R, Livingston DM, In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 408, 429–432 (2000); published online EpubNov 23 (10.1038/35044000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welcsh PL, King MC, BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet 10, 705–713 (2001); published online EpubApr ( [DOI] [PubMed] [Google Scholar]
- 6.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS, Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361, 123–134 (2009); published online EpubJul 9 (10.1056/NEJMoa0900212). [DOI] [PubMed] [Google Scholar]
- 7.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, Bowen K, Fielding A, Domchek SM, Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33, 244–250 (2015); published online EpubJan 20 (10.1200/JCO.2014.56.2728). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhillon KK, Swisher EM, Taniguchi T, Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci 102, 663–669 (2011); published online EpubApr (10.1111/j.1349–7006.2010.01840.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A, 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (2010); published online EpubApr 16 (10.1016/j.cell.2010.03.012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, Doroshow JH, Cranston A, Martin NM, Lau A, O’Connor MJ, Ganesan S, Borst P, Jonkers J, Rottenberg S, Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov 3, 68–81 (2013); published online EpubJan (10.1158/2159–8290.CD-12–0049). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, Herzog M, Lam J, Shea A, Demir M, Wu Q, Yang F, Fu B, Lai Z, Balmus G, Belotserkovskaya R, Serra V, O’Connor MJ, Bruna A, Beli P, Pellegrini L, Caldas C, Deriano L, Jacobs JJL, Galanty Y, Jackson SP, Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20, 954–965 (2018); published online EpubAug (10.1038/s41556-018-0140-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, Jaspers JE, Watanabe K, Pieterse M, Kersbergen A, Sol W, Celie PHN, Schouten PC, van den Broek B, Salman A, Nieuwland M, de Rink I, de Ronde J, Jalink K, Boulton SJ, Chen J, van Gent DC, Bartek J, Jonkers J, Borst P, Rottenberg S, REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 (2015); published online EpubMay 28 (10.1038/nature14328). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, Ferguson MJ, Smith G, ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer 115, 431–441 (2016); published online EpubAug 9 (10.1038/bjc.2016.203). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, Pommier Y, Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther 349, 408–416 (2014); published online EpubJun (10.1124/jpet.113.210146). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He YJ, Meghani K, Caron MC, Yang C, Ronato DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A, Doench JG, Legube G, Root DE, D’Andrea AD, Drane P, De S, Konstantinopoulos PA, Masson JY, Chowdhury D, DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 563, 522–526 (2018); published online EpubNov (10.1038/s41586-018-0670-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding X, Ray Chaudhuri A, Callen E, Pang Y, Biswas K, Klarmann KD, Martin BK, Burkett S, Cleveland L, Stauffer S, Sullivan T, Dewan A, Marks H, Tubbs AT, Wong N, Buehler E, Akagi K, Martin SE, Keller JR, Nussenzweig A, Sharan SK, Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat Commun 7, 12425 (2016); published online EpubAug 8 (10.1038/ncomms12425). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A, Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016); published online EpubJul 21 (10.1038/nature18325). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, Cantor SB, Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev 29, 489–494 (2015); published online EpubMar 1 (10.1101/gad.256214.114). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, D’Andrea AD, EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 19, 1371–1378 (2017); published online EpubNov (10.1038/ncb3626). [DOI] [PubMed] [Google Scholar]
- 20.Pastor WA, Aravind L, Rao A, TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14, 341–356 (2013); published online EpubJun (10.1038/nrm3589). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y, Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011); published online EpubSep 2 (10.1126/science.1210597). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A, Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009); published online EpubMay 15 (10.1126/science.1170116). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C, Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 29, 68–72 (2011); published online EpubJan (10.1038/nbt.1732). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin SG, Zhang ZM, Dunwell TL, Harter MR, Wu X, Johnson J, Li Z, Liu J, Szabo PE, Lu Q, Xu GL, Song J, Pfeifer GP, Tet3 Reads 5-Carboxylcytosine through Its CXXC Domain and Is a Potential Guardian against Neurodegeneration. Cell Rep 14, 493–505 (2016); published online EpubJan 26 (10.1016/j.celrep.2015.12.044). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu L, Lu J, Cheng J, Rao Q, Li Z, Hou H, Lou Z, Zhang L, Li W, Gong W, Liu M, Sun C, Yin X, Li J, Tan X, Wang P, Wang Y, Fang D, Cui Q, Yang P, He C, Jiang H, Luo C, Xu Y, Structural insight into substrate preference for TET-mediated oxidation. Nature 527, 118–122 (2015); published online EpubNov 5 (10.1038/nature15713). [DOI] [PubMed] [Google Scholar]
- 26.Melamed P, Yosefzon Y, David C, Tsukerman A, Pnueli L, Tet Enzymes, Variants, and Differential Effects on Function. Front Cell Dev Biol 6, 22 (2018) 10.3389/fcell.2018.00022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi DQ, Ali I, Tang J, Yang WC, New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front Genet 8, 100 (2017) 10.3389/fgene.2017.00100). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen KD, Helin K, Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30, 733–750 (2016); published online EpubApr 1 (10.1101/gad.276568.115). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang D, Zhang Y, Hart RP, Chen J, Herrup K, Li J, Alteration in 5-hydroxymethylcytosine-mediated epigenetic regulation leads to Purkinje cell vulnerability in ATM deficiency. Brain 138, 3520–3536 (2015); published online EpubDec (10.1093/brain/awv284). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang D, Wei S, Chen F, Zhang Y, Li J, TET3-mediated DNA oxidation promotes ATR-dependent DNA damage response. EMBO Rep 18, 781–796 (2017); published online EpubMay (10.15252/embr.201643179). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biswas K, Das R, Alter BP, Kuznetsov SG, Stauffer S, North SL, Burkett S, Brody LC, Meyer S, Byrd RA, Sharan SK, A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. Blood 118, 2430–2442 (2011); published online EpubSep 1 (10.1182/blood-2010-12-324541). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PW, Holstege H, Liu X, van Drunen E, Beverloo HB, Smith GC, Martin NM, Lau A, O’Connor MJ, Jonkers J, Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res 14, 3916–3925 (2008); [DOI] [PubMed] [Google Scholar]
- 33.Breier A, Gibalova L, Seres M, Barancik M, Sulova Z, New insight into p-glycoprotein as a drug target. Anticancer Agents Med Chem 13, 159–170 (2013); published online EpubJan ( [PubMed] [Google Scholar]
- 34.Merrick CJ, Jackson D, Diffley JF, Visualization of altered replication dynamics after DNA damage in human cells. J Biol Chem 279, 20067–20075 (2004); published online EpubMay 7 (10.1074/jbc.M400022200). [DOI] [PubMed] [Google Scholar]
- 35.Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, Cortez D, RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol Cell 67, 374–386 e375 (2017); published online EpubAug 3 (10.1016/j.molcel.2017.06.023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, Zepeda-Martinez JA, Pape UJ, Jacobsen SE, Peters B, Rao A, Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci U S A 111, 1361–1366 (2014); published online EpubJan 28 (10.1073/pnas.1322921111). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, Grillo I, Bakogianni S, Ndiaye-Lobry D, Martin MT, Guillamot M, Banh RS, Xu M, Figueroa ME, Dickins RA, Abdel-Wahab O, Park CY, Tsirigos A, Neel BG, Aifantis I, Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095 e1020 (2017); published online EpubSep 7 (10.1016/j.cell.2017.07.032). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U, Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000 (2006); published online EpubDec (10.1038/nmeth947). [DOI] [PubMed] [Google Scholar]
- 39.Rosa S, Fortini P, Karran P, Bignami M, Dogliotti E, Processing in vitro of an abasic site reacted with methoxyamine: a new assay for the detection of abasic sites formed in vivo. Nucleic Acids Res 19, 5569–5574 (1991); published online EpubOct 25 ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fortini P, Rosa S, Zijno A, Calcagnile A, Bignami M, Dogliotti E, Methoxyamine modification of abasic sites protects CHO cells from the cytotoxic and mutagenic effects of oxygen alkylation. Carcinogenesis 13, 87–93 (1992); published online EpubJan ( [DOI] [PubMed] [Google Scholar]
- 41.Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, Cortez D, HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 176, 144–153 e113 (2019); published online EpubJan 10 (10.1016/j.cell.2018.10.055). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drohat AC, Coey CT, Role of Base Excision “Repair” Enzymes in Erasing Epigenetic Marks from DNA. Chem Rev 116, 12711–12729 (2016); published online EpubOct 26 (10.1021/acs.chemrev.6b00191). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, Frankum J, Ranes M, Pemberton HN, Rafiq R, Fenwick K, Swain A, Guettler S, Lee JM, Swisher EM, Stoynov S, Yusa K, Ashworth A, Lord CJ, Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun 9, 1849 (2018); published online EpubMay 10 (10.1038/s41467-018-03917-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christman JK, 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495 (2002); published online EpubAug 12 (10.1038/sj.onc.1205699). [DOI] [PubMed] [Google Scholar]
- 45.Mercher T, Quivoron C, Couronne L, Bastard C, Vainchenker W, Bernard OA, TET2, a tumor suppressor in hematological disorders. Biochim Biophys Acta 1825, 173–177 (2012); published online EpubApr (10.1016/j.bbcan.2011.12.002). [DOI] [PubMed] [Google Scholar]
- 46.Tsagaratou A, Rao A, TET proteins and 5-methylcytosine oxidation in the immune system. Cold Spring Harb Symp Quant Biol 78, 1–10 (2013) 10.1101/sqb.2013.78.020248). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, Godley L, Opolon P, Tilly H, Solary E, Duffourd Y, Dessen P, Merle-Beral H, Nguyen-Khac F, Fontenay M, Vainchenker W, Bastard C, Mercher T, Bernard OA, TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38 (2011); [DOI] [PubMed] [Google Scholar]
- 48.Chou KM, Cheng YC, The exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1). Biochemical properties and inhibition by the natural dinucleotide Gp4G. J Biol Chem 278, 18289–18296 (2003); published online EpubMay 16 (10.1074/jbc.M212143200). [DOI] [PubMed] [Google Scholar]
- 49.Chou KM, Cheng YC, An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3’ mispaired DNA. Nature 415, 655–659 (2002); published online EpubFeb 7 (10.1038/415655a). [DOI] [PubMed] [Google Scholar]
- 50.Dyrkheeva NS, Lebedeva NA, Lavrik OI, AP Endonuclease 1 as a Key Enzyme in Repair of Apurinic/Apyrimidinic Sites. Biochemistry (Mosc) 81, 951–967 (2016); published online EpubSep (10.1134/S0006297916090042). [DOI] [PubMed] [Google Scholar]
- 51.Prasad R, Horton JK, Chastain PD 2nd, Gassman NR, Freudenthal BD, Hou EW, Wilson SH, Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair. Nucleic Acids Res 42, 6337–6351 (2014); published online EpubJun (10.1093/nar/gku288). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, Ghesquiere B, Van Dyck L, Boeckx B, Schoonjans L, Hermans E, Amant F, Kristensen VN, Peng Koh K, Mazzone M, Coleman M, Carell T, Carmeliet P, Lambrechts D, Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537, 63–68 (2016); published online EpubSep 1 (10.1038/nature19081). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeng W, Liu P, Pan W, Singh SR, Wei Y, Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett 356, 263–267 (2015); published online EpubJan 28 (10.1016/j.canlet.2014.01.032). [DOI] [PubMed] [Google Scholar]
- 54.Graham K, Unger E, Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int J Nanomedicine 13, 6049–6058 (2018) 10.2147/IJN.S140462). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chung N, Zhang XD, Kreamer A, Locco L, Kuan PF, Bartz S, Linsley PS, Ferrer M, Strulovici B, Median absolute deviation to improve hit selection for genome-scale RNAi screens. J Biomol Screen 13, 149–158 (2008); published online EpubFeb (10.1177/1087057107312035). [DOI] [PubMed] [Google Scholar]
- 56.Marine S, Bahl A, Ferrer M, Buehler E, Common seed analysis to identify off-target effects in siRNA screens. J Biomol Screen 17, 370–378 (2012); published online EpubMar (10.1177/1087057111427348). [DOI] [PubMed] [Google Scholar]
- 57.Kuznetsov SG, Liu P, Sharan SK, Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med 14, 875–881 (2008); published online EpubAug (10.1038/nm.1719). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR, Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 48, 4827–4833 (1988); published online EpubSep 1 ( [PubMed] [Google Scholar]
- 59.Le T, Kim KP, Fan G, Faull KF, A sensitive mass spectrometry method for simultaneous quantification of DNA methylation and hydroxymethylation levels in biological samples. Anal Biochem 412, 203–209 (2011); published online EpubMay 15 (10.1016/j.ab.2011.01.026). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, Balasubramanian S, 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem 6, 1049–1055 (2014); published online EpubDec (10.1038/nchem.2064). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Loss of TET confers drug resistance to PARPi and cisplatin.
Figure S2: Loss of TET confers RF stability in BRCA2-deficient cells.
Figure S3: Loss of TET protects stalled RFs in BRCA2-deficient cells and rescues BRCA2-loss-induced lethality in PL2F7 mES cells.
Figure S4: Effect of global 5hmC change on RF stability.
Figure S5: Determination of threshold 5hmC levels that promote degradation of RFs.
Figure S6: APE1 endonuclease mediates the degradation of 5hmC-marked stalled RFs.
Figure S7: 5hmC modification of nascent strands induces RF degradation.
Data file S1. Genome-wide siRNA screen data.