

Abstract
Introduction
Young age is an adverse prognostic factor in children with ependymomas. Treatment of these infants is challenging since beneficial therapeutic options are limited. As ependymomas are considered a biologically heterogeneous group, we aimed to characterize infant ependymomas with regard to their histological and genetic features.
Materials and methods
We analyzed 28 ependymomas occurring in children younger than 18 months at diagnosis enrolled into the HIT2000-E protocols with the aim to postpone irradiation until the age of 18 months if possible. All cases underwent neuropathological review, including immunohistochemical characterization. Genome-wide copy number alterations (CNA) were assessed by molecular inversion probe assays, and RELA and YAP1 fusions were detected by RT-PCR and sequencing.
Results
All infant ependymomas were anaplastic (WHO grade III). Twenty-one (75%) cases were located in the posterior fossa. Gross total resection was accomplished in 12 (57%) of these cases. All posterior fossa tumors showed loss of H3-K27me3 characteristic of PFA ependymomas. CNA analysis showed a stable genome in all cases with lack of chromosome 1q gain, an adverse prognostic marker in PFA ependymomas of older children. However, after a median follow-up of 5.4 years, 15 (71%) relapsed, and 9 (43%) died. Seven ependymomas (25%) occurred in the supratentorial region. Gross total resection could be achieved in only two of these cases. Four tumors carried C11orf95-RELA fusions, and two cases had typical YAP1-MAMLD1 fusions (one case was not analyzable). The RELA-fused cases did not display CDKN2A loss as an adverse indicator of prognosis in this disease entity. Although three infants (43%) with supratentorial ependymomas relapsed, all patients survived (median follow-up, 8.0 years).
Conclusion
Infant ependymomas seem to fall into three biological entities, with supratentorial tumors carrying RELA or YAP fusions and PFA posterior fossa ependymomas. The latter showed a poor outcome even though chromosome 1q gain was absent.
Keywords: Ependymoma, Infant, Genetics, Neuropathology, Fusion genes
Introduction
Ependymomas are the third most common brain tumor in children [1]. They can arise along the entire neuroaxis. In children, most tumors are located intracranially and two thirds of them in the posterior fossa (PF) [2]. Despite histological resemblance, ependymomas represent distinct disease entities with different cells of origin, genetic pathways activated in their pathogenesis, epigenetic landscape, and clinical behavior [3, 4]. The usefulness of the WHO grading system [5] for risk stratification is controversial [6–11]. Despite major progress in the understanding of the biology of ependymomas, therapeutic options remain limited and mainly include maximal safe surgery and radiation therapy (RT) [7, 8]. Previous trials proved that chemotherapy administered as monotherapy is ineffective [12], but the possible role of chemotherapy is not fully resolved yet and is currently being tested in the European SIOP EP-II trial [7, 11].
Furthermore, the AIEOP trial as well as recently published data by Merchant et al. showed evidence that some patients may be cured by surgery alone [7, 9]. So far, risk stratification as a basis for risk-adapted therapy mainly relies on clinical parameters including age at diagnosis, residual tumor after surgery, and local versus metastatic disease at presentation. In addition, some protocols also differentiate according to location (supratentorial versus infratentorial) and histological grading [7–9].
Although improved risk models have been proposed in the past dividing tumors according to their cytogenetic patterns into numerical, balanced, or structural subgroups [13], evaluating the presence of individual prognostic factors such as gain of chromosome 1q in PF ependymoma [2, 9, 13, 14], RELA and YAP1 fusions in supratentorial (ST) ependymomas [3, 15–18], or analyzing specific epigenetic patterns [3, 19], none of these have yet been included in stratification of patients enrolled in clinical trials.
Most published risk models include children with ependymomas of all ages, locations, and underlying genetics ignoring the differences in biology and treatment among individual age groups. The group of infants with PF ependymoma apparently faces worst prognosis; at the same time, this group seems to be the one most difficult to treat in terms of surgery and adjuvant treatment [20].
In this study, we analyzed a total of 28 patients under the age of 18 months at the time of diagnosis (retrieved from a cohort of 203 pediatric ependymoma), treated according to the HIT ependymoma trial, with the majority harboring PF ependymomas, focusing on their clinical, histological, and genetic particularities.
Material and methods
Patients
Ependymomas in 28 children under the age of 18 months were retrieved from the archives of the Institute of Neuropathology, University of Bonn Medical Center (Bonn, Germany), and of the DGNN German Brain Tumor Reference Center (Bonn, Germany). All patients were diagnosed with anaplastic ependymoma (WHO grade III) in a period from 2002 to 2011 and enrolled into the prospective non-randomized multicenter HIT ependymoma study (ClinicalTrials.gov NCT00303810). This included risk-adapted HIT-SKK chemotherapy (without intraventricular methotrexate) and irradiation. In infant patients, the aim was to postpone irradiation until the age of 18 months if possible. In case of residual tumor, patients were evaluated for potential second-look surgery after each therapy element. All examinations were carried out on the basis and according to the legal requirements of the revised Declaration of Helsinki of the World Medical Association in 1983 and according to the guidelines of the institutional review boards. Informed consent was given at study inclusion by the parents.
Histopathology and immunohistochemistry
All tumors were centrally reviewed at the DGNN German Brain Tumor Reference Center (Bonn, Germany) and reclassified according to the revised WHO classification of tumors of the CNS 2016 [5]. Mitotic activity was assessed in 4μm-thick HE-stained FFPE slides by counting mitotic figures in 10 high power fields (HPF; area, 2.38 mm2). Presence of more than 10 mitoses/10 HPF was defined as high mitotic activity. Further histological features including necrosis, vascular proliferation, and clear cell morphology were assessed. Immunohistochemical staining—performed on an immunostaining system (BenchMark XT, Ventana-Roche, Mannheim, Germany)—included Ki67 (MAb MIB-1, Dako, Glostrup, Denmark), phosphohistone-H3 (Biocare, Concord, USA), p16 protein (MAb E6H4, Ventana-Roche, Darmstadt, Germany), p65-RelA (rabbit antibody D14E12, Cell Signaling, Danvers, U.S.A.), and trimethylated histone3-Lys27 (H3-K27me3; rabbit MAb C36B11, Cell Signaling). For H3-K37me3, positive nuclei of endothelial cells served as internal control.
DNA extraction and copy number analysis
On hematoxylin and eosin (HE)-stained FFPE sections, representative areas with at least 80% tumor cell content were identified for microdissection and DNA extraction from serial sections using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. In order to identify genome-wide copy number alterations and allelic disbalances, we used molecular inversion probe (MIP) assays (OncoScan FFPE express 330 K arrays, versions 2 and 3; Affymetrix, Santa Clara, CA, US) as previously described [21, 22]. Raw data were further analyzed with the Nexus Copy Number software, version 7.5 (BioDiscovery, El Segundo, CA, USA). DNA from FFPE cerebellar tissue served as normal control. The SNP-FASST-2 segmentation algorithm was used for data processing. All cases were reviewed individually, and if necessary, diploidy correction was performed manually.
RNA extraction and detection of C11orf95-RELA and YAP1-MAMLD1 fusions
FFPE or fresh frozen tissue (if available) of supratentorial ependymomas was further tested for presence of C11orf95-RELA and YAP1-MAMLD1 fusions. RNA was extracted, followed by RT-PCR and sequencing, described previously [15, 16, 18].
Statistical analyses
EFS and OS data was shown in Kaplan-Meier plots applying log-rank (Mantel-Cox) method using IBM SPSS statistics version 23 (IBM, Bonn, Germany). Results with a p value < 0.05 were regarded as significant.
Results
Supratentorial ependymomas
Seven patients of 28 (25%) had an ependymoma in a supratentorial location. These occurred in five girls and two boys. Subtotal resection (STR) was performed in five (71%), and in three (43%), a relapse occurred. However, all patients were alive after a median follow-up of 7.96 years (range 0.28–12 years). High mitotic activity, necrosis, and vascular proliferation each were present in all but one case. All cases had at least one of these criteria. All tumors were classified as anaplastic ependymoma (WHO grade III). Two cases showed clear cell morphology, and balanced genomes were found in four cases (57%) compared to structural (two cases) and numerical genomic group (one case). Chromosome 1q gain was not present at all, while one case showed chromosome 22 loss. Six cases could be studied for fusion transcripts; for the remaining one, material for further analysis was not available. RELA fusions were detected in four cases, which showed nuclear accumulation of p65-RelA protein as indicator of activation of NFkB signaling. Two of the RELA-fused cases displayed clear cell morphology, and three of the four relapsed. YAP1-MAMLD1 fusions were found in two other cases, not carrying RELA fusions. Concerning the latter two patients, none relapsed, although in one, gross total resection (GTR) could not be achieved. One of the two cases harboring a YAP1-MAMLD1 fusion showed monosomy of chromosome 22. CDKN2A loss/LOH or p16 protein loss was not identified in any of these 7 cases. For further information, see Table 1 and Fig. 1.
Table 1.
Clinical, histological, and genetic characteristics
| All locations | Supratentorial | Posterior fosssa | ||||
|---|---|---|---|---|---|---|
| Number of cases | 28 | % | 7 | % | 21 | % |
| Female | 15 | 54 | 5 | 71 | 10 | 48 |
| Male | 13 | 46 | 2 | 29 | 11 | 52 |
| Dead | 9 | 32 | 0 | 0 | 9 | 43 |
| Alive | 19 | 68 | 7 | 100 | 12 | 57 |
| Age at diagnosis : Mean | 0.93 y | 0.78 y | 0.97 y | |||
| Median | 0.95 y | 0.89 y | 1.05 y | |||
| 5-year EFS | 46 | 57 | 43 | |||
| 5-year OS | 82 | 100 | 71 | |||
| Subtotal resection | 14 | 50 | 5 | 71 | 9 | 43 |
| Initial metastatic disease | 1 | 4 | 1 | 14 | 0 | 0 |
| Relapse | 18 | 64 | 3 | 43 | 15 | 71 |
| WHO grade II | 0 | 0 | 0 | 0 | 0 | 0 |
| WHO grade III | 28 | 100 | 7 | 100 | 21 | 100 |
| Necrosis | 24 | 86 | 6 | 86 | 18 | 86 |
| Mitotic Activity > 10 mitoses/ 10 HPF | 21 | 75 | 6 | 86 | 15 | 71 |
| Vascular proliferation | 25 | 89 | 6 | 86 | 19 | 90 |
| True ependymal rosettes | 0 | 0 | 0 | 0 | 0 | 0 |
| Clear cell morphology | 2 | 7 | 2 | 29 | 0 | 0 |
| Genomic group: Numerical | 1 | 4 | 1 | 14 | 0 | 0 |
| Balanced | 25 | 89 | 4 | 57 | 21 | 100 |
| Structural | 2 | 7 | 2 | 29 | 0 | 0 |
| Chromosome 1q gain | 0 | 0 | 0 | 0 | 0 | 0 |
| Chromosome 22 loss | 1 | 4 | 1 | 14 | n.a. | |
| C11orf95-RELA fusion | 4 | 14 | 4 | 57 | n.a. | |
| CDKN2A loss | 0 | 0 | 0 | 0 | n.a. | |
| YAP1-MAMLD1 fusion | 2 | 7 | 2 | 29 | n.a. | |
Fig. 1.

Histological and genetic features. 1 PFA ependymoma: (A) H&E, (B) H3-K27me3 immunohistochemistry indicating complete loss in tumor cells, and (C) CNV plot showing a balanced genome (MIP). 2 RELA ependymoma: (A) H&E, (B) nuclear p65-RelA accumulation, and (C) cDNA sequencing of C11orf95-RELA fusion transcript. 3 YAP ependymoma: (A) H&E, (B) cytoplasmatic p65-RelA staining, and (C) cDNA sequencing of YAP1-MAMLD1 fusion transcript
Posterior fossa ependymomas
Twenty-one of 28 patients (75%) developed tumors in the posterior fossa. The male to female ratio was even (11 vs. 10). All cases were classified as anaplastic ependymoma (WHO grade III). In terms of histological criteria associated with anaplasia, 19 (91%) revealed vascular proliferation, 18 (86%) necrosis, and 15 (71%) high mitotic activity, while neither true ependymal rosettes nor clear cell morphology were encountered. Most strikingly, all cases showed a balanced genomic profile with consequently no gain of chromosome 1q and loss of the trimethylated lysine 27 of histone 3, typical of pediatric “group A” PF ependymomas [23, 24] (Table 1, Fig. 1).
Nine patients (43%) had postoperative residual tumor, and 15 (71%) suffered a relapse; none showed initial metastasis. After a median follow-up time of 5.44 years, 5-year EFS and OS were 43% and 71%, respectively.
Statistical analysis
Kaplan-Meier analysis revealed no statistically significant difference between ST and PF ependymoma regarding EFS and OS, respectively. Regarding OS, there was nevertheless a clear trend towards a favorable prognosis of ST ependymoma of infancy, since none of the patients died (Fig. 2).
Fig. 2.
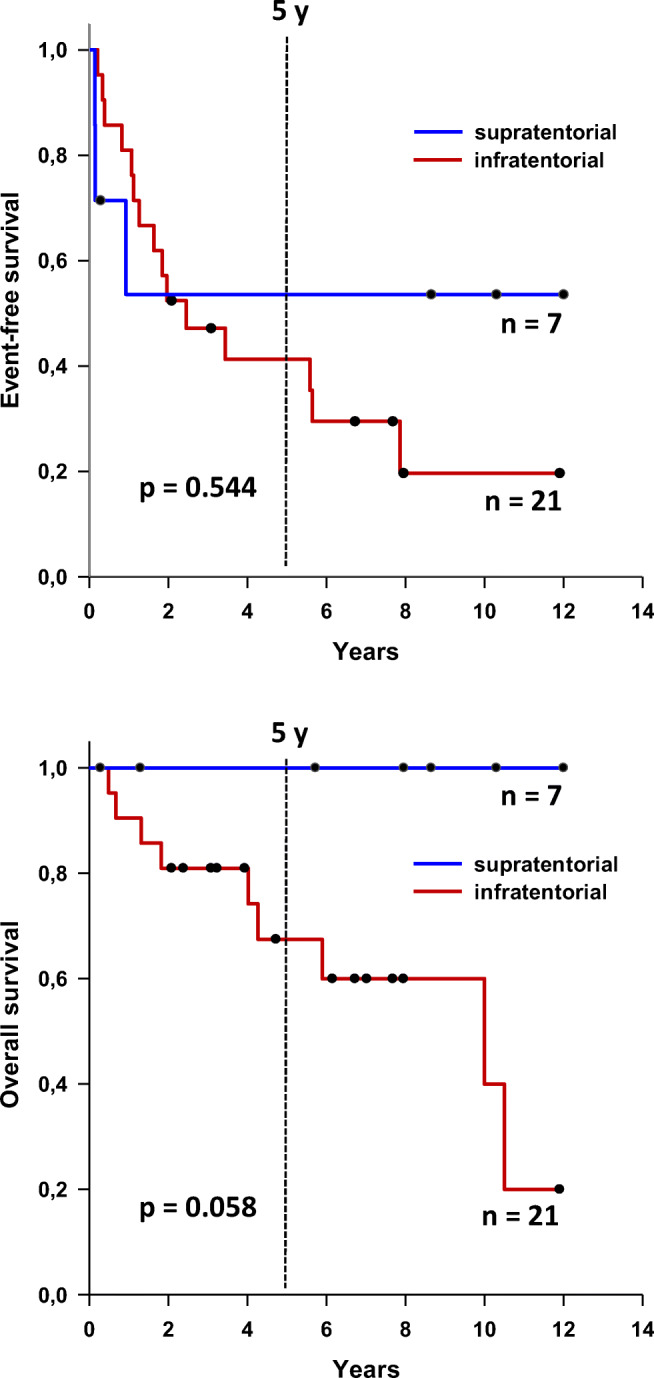
Survival analysis comparing ST and PF ependymoma below 18 months
Discussion
Our results show that ependymomas of infancy (0–18 months) comprise distinct location- and age-associated ependymoma entities in terms of underlying biology and genetics and—probably resulting from the latter—their prognosis. In contrast to previous publications, we considered ST and PF ependymomas as different entities and therefore analyzed the populations separately with regard to clinical, histological, and genetic parameters.
Until now, a separate analysis according to infant age and tumor location was not provided in most published clinical trials. In addition, neuropathological features and genetic characteristics were usually not described in detail. However, Upadhyaya et al. recently published their prospective study for children below the age of 3 years analyzing tumors of different locations individually regarding prognostically relevant factors [11].
In our study, the period of recruitment (10 years) was comparingly short, and patients were enrolled into the risk-adapted HIT trial ruling out major population inhomogeneity. Due to case selection and specific characterization mentioned above, the number of cases—remaining from a large cohort of pediatric ependymoma (n = 203)—unfortunately was limited.
Supratentorial ependymoma in infants show specific fusion transcripts and favorable overall survival
Strikingly, no patient with a supratentorial ependymoma below the age of 18 months at diagnosis died during the period of follow-up although all tumors in this age group were graded as anaplastic (WHO grade III). RELA fusions—representing the most frequent alteration in pediatric supratentorial ependymomas—were present in four cases (57%), which is similar in frequency as previously reported [3, 15, 17]. All three genomic groups are represented in tumors of this location. However, in line with Upadhyaya et al., we cannot confirm the poor prognosis reported by Pajtler et al. in a retrospective case collection [3, 11]. Although some patients relapsed, they were successfully treated, either with second resection, chemotherapy, or radiotherapy. One explanation for a favorable prognosis of this specific subgroup may be the absence of CDKN2A loss/LOH in RELA-positive tumors. CDKN2A loss has been previously reported in ST ependymoma and is known to be an indicator of adverse prognosis in RELA-fused ependymomas [25] and other brain tumors such as IDH-mutated anaplastic astrocytoma [26].
In addition, in this location, tumors harboring a YAP1-MAMLD1 fusion—apparently a parameter of favorable prognosis [3, 11, 15, 17]—accounted for two patients (29%), and neither of them suffered from relapse or died.
Posterior fossa ependymoma in infants show a balanced genome and frequent recurrence
It is generally accepted that patients with PF ependymomas face a worse prognosis, especially at young age; however, a recent publication showed no age-dependent prognostic difference for these tumors [9]. Although radiation therapy can be effective in infants, most neurooncological centers are reluctant to apply irradiation in this age group because of severe long-term secondary effects. Therefore, the treatment of infants imposes difficulties compared to older patients with PF ependymoma; hence, there is a need for identification of specific risk factors and novel treatment options in this subgroup of patients.
The rate of incomplete resection, a parameter of inferior prognosis, occurred in 43% of infratentorial compared to 71% in supratentorial ependymoma of infancy. The shorter overall survival of patients with posterior fossa ependymoma may result from re-resection being more difficult compared to the supratentorial compartment.
All PF ependymomas in this study were classified as anaplastic (WHO grade III). The prognostic impact of WHO grading was demonstrated in some studies but not found in others [9, 11].
Furthermore, genomic analysis revealed that all 21 patients in this group showed balanced genomic profiles and were confirmed to represent PF-A group/CIMP + ependymoma, immunohistochemistry demonstrating a complete loss of H3K27me3, which represents the subgroup of ependymoma with the worst prognosis [3, 27]. Interestingly, chromosome 1q gain and the associated structural genomic profile—both reported indicators of adverse prognosis [2, 5, 13, 14, 28, 29]—were not found in any of the 21 PF tumor samples in our cohort.
Ependymomas of infancy represent distinct tumor entities and impose distinct challenges in terms of clinical management
In summary, we were able to demonstrate that ependymomas of infancy represent distinct location- and age-associated ependymoma entities in terms of underlying biology and genetics. Even though statistical significance regarding survival could not be reached, most probably due to the small cohort sizes, ST ependymomas seem to have a superior prognosis compared to PF ependymoma in terms of OS. The EFS of patients with tumors of the two locations may not be different, but re-resection being more easily achieved in the ST-compartment may lead to the long-term survival benefits noted in the previous studies.
ST ependymomas harbor distinct genetic alterations (such as fusions) which represent possible therapeutic targets; these fusions are absent in PF ependymomas. Besides the optimal timing for radiation therapy, the field of targeted therapies remains to be explored for ependymoma in the future.
Our results show that risk stratification for pediatric ependymoma remains challenging and suggest that separate analysis—regarding risk factors and treatment regimens—is required for tumors in different locations and among different age groups.
Acknowledgments
We thank Dr. Steffen Albrecht, Montreal, for critical reading of the manuscript.
Funding information
Open Access funding provided by Projekt DEAL. The study was supported by grants from the German Children Cancer Foundation DKS 2006.03, 2009.19, 2011.01 and 2014.17 to TP.
Data availability
The data that support the findings of this study are available on reasonable request from the corresponding author.
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncol. 2017;19:v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kilday J-P, Rahman R, Dyer S, Ridley L, Lowe J, Coyle B, Grundy R. Pediatric ependymoma: biological perspectives. Mol Cancer Res. 2009;7:765–786. doi: 10.1158/1541-7786.MCR-08-0584. [DOI] [PubMed] [Google Scholar]
- 3.Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, Reimand J, Warnatz HJ, Ryzhova M, Mack S, Ramaswamy V, Capper D, Schweizer L, Sieber L, Wittmann A, Huang Z, van Sluis P, Volckmann R, Koster J, Versteeg R, Fults D, Toledano H, Avigad S, Hoffman LM, Donson AM, Foreman N, Hewer E, Zitterbart K, Gilbert M, Armstrong TS, Gupta N, Allen JC, Karajannis MA, Zagzag D, Hasselblatt M, Kulozik AE, Witt O, Collins VP, von Hoff K, Rutkowski S, Pietsch T, Bader G, Yaspo ML, von Deimling A, Lichter P, Taylor MD, Gilbertson R, Ellison DW, Aldape K, Korshunov A, Kool M, Pfister SM. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27:728–743. doi: 10.1016/j.ccell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, MacDonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Louis D, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours of the central nervous system. Revised. 4. IARC Press: Lyon; 2016. [Google Scholar]
- 6.Ellison DW, Kocak M, Figarella-Branger D, Felice G, Catherine G, Pietsch T, Frappaz D, Massimino M, Grill J, Boyett JM, Grundy RG. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results BioMed. 2011;10:7. doi: 10.1186/1477-5751-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massimino M, Miceli R, Giangaspero F, Boschetti L, Modena P, Antonelli M, Ferroli P, Bertin D, Pecori E, Valentini L, Biassoni V, Garrè ML, Schiavello E, Sardi I, Cama A, Viscardi E, Scarzello G, Scoccianti S, Mascarin M, Quaglietta L, Cinalli G, Diletto B, Genitori L, Peretta P, Mussano A, Buccoliero A, Calareso G, Barra S, Mastronuzzi A, Giussani C, Marras CE, Balter R, Bertolini P, Giombelli E, la Spina M, Buttarelli FR, Pollo B, Gandola L. Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro-Oncol. 2016;18:1451–1460. doi: 10.1093/neuonc/now108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merchant TE, Li C, Xiong X, Kun LE, Boop FA, Sanford RA. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol. 2009;10:258–266. doi: 10.1016/S1470-2045(08)70342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merchant TE, Bendel AE, Sabin ND, Burger PC, Shaw DW, Chang E, Wu S, Zhou T, Eisenstat DD, Foreman NK, Fuller CE, Anderson ET, Hukin J, Lau CC, Pollack IF, Laningham FH, Lustig RH, Armstrong FD, Handler MH, Williams-Hughes C, Kessel S, Kocak M, Ellison DW, Ramaswamy V. Conformal radiation therapy for pediatric ependymoma, chemotherapy for incompletely resected ependymoma, and observation for completely resected, supratentorial ependymoma. J Clin Oncol. 2019;37:974–983. doi: 10.1200/JCO.18.01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tihan T, Zhou T, Holmes E, Burger PC, Ozuysal S, Rushing EJ. The prognostic value of histological grading of posterior fossa ependymomas in children: a Children’s oncology group study and a review of prognostic factors. Mod Pathol. 2008;21:165–177. doi: 10.1038/modpathol.3800999. [DOI] [PubMed] [Google Scholar]
- 11.Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Billups CA, Bowers DC, Bendel AE, Hassall T, Crawford JR, Partap S, Fisher PG, Tatevossian RG, Seah T, Qaddoumi IA, Vinitsky A, Armstrong GT, Sabin ND, Tinkle CL, Klimo P, Indelicato DJ, Boop FA, Merchant TE, Ellison DW, Gajjar A. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro-Oncol. 2019;21:1319–1330. doi: 10.1093/neuonc/noz069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grill J, Le Deley M-C, Gambarelli D, Raquin M-A, Couanet D, Pierre-Kahn A, et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19:1288–1296. doi: 10.1200/JCO.2001.19.5.1288. [DOI] [PubMed] [Google Scholar]
- 13.Dyer S, Prebble E, Davison V, Davies P, Ramani P, Ellison D, Grundy R. Genomic imbalances in pediatric intracranial ependymomas define clinically relevant groups. Am J Pathol. 2002;161:2133–2141. doi: 10.1016/S0002-9440(10)64491-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilday J-P, Mitra B, Domerg C, Ward J, Andreiuolo F, Osteso-Ibanez T, Mauguen A, Varlet P, le Deley MC, Lowe J, Ellison DW, Gilbertson RJ, Coyle B, Grill J, Grundy RG. Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the Children’s Cancer Leukaemia group (CCLG), Societe Francaise d’Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP) Clin Cancer Res. 2012;18:2001–2011. doi: 10.1158/1078-0432.CCR-11-2489. [DOI] [PubMed] [Google Scholar]
- 15.Andreiuolo F, Varlet P, Tauziède-Espariat A, Jünger ST, Dörner E, Dreschmann V, Kuchelmeister K, Waha A, Haberler C, Slavc I, Corbacioglu S, Riemenschneider MJ, Leipold A, Rüdiger T, Körholz D, Acker T, Russo A, Faber J, Sommer C, Armbrust S, Rose M, Erdlenbruch B, Hans VH, Bernbeck B, Schneider D, Lorenzen J, Ebinger M, Handgretinger R, Neumann M, van Buiren M, Prinz M, Roganovic J, Jakovcevic A, Park SH, Grill J, Puget S, Messing-Jünger M, Reinhard H, Bergmann M, Hattingen E, Pietsch T. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019;29:205–216. doi: 10.1111/bpa.12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figarella-Branger D, Lechapt-Zalcman E, Tabouret E, Jünger S, de Paula AM, Bouvier C, Colin C, Jouvet A, Forest F, Andreiuolo F, Quintin-Roue I, Machet MC, Heitzmann A, Milin S, Sevestre H, Godfraind C, Labrousse F, Metellus P, Scavarda D, Pietsch T. Supratentorial clear cell ependymomas with branching capillaries demonstrate characteristic clinicopathological features and pathological activation of nuclear factor-kappaB signaling. Neuro-Oncol. 2016;18:919–927. doi: 10.1093/neuonc/now025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, Lee R, Tatevossian RG, Phoenix TN, Thiruvenkatam R, White E, Tang B, Orisme W, Gupta K, Rusch M, Chen X, Li Y, Nagahawhatte P, Hedlund E, Finkelstein D, Wu G, Shurtleff S, Easton J, Boggs K, Yergeau D, Vadodaria B, Mulder HL, Becksfort J, Gupta P, Huether R, Ma J, Song G, Gajjar A, Merchant T, Boop F, Smith AA, Ding L, Lu C, Ochoa K, Zhao D, Fulton RS, Fulton LL, Mardis ER, Wilson RK, Downing JR, Green DR, Zhang J, Ellison DW, Gilbertson RJ. C11orf95–RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature. 2014;506:451–455. doi: 10.1038/nature13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietsch T, Wohlers I, Goschzik T, Dreschmann V, Denkhaus D, Dörner E, Rahmann S, Klein-Hitpass L. Supratentorial ependymomas of childhood carry C11orf95–RELA fusions leading to pathological activation of the NF-κB signaling pathway. Acta Neuropathol. 2014;127:609–611. doi: 10.1007/s00401-014-1264-4. [DOI] [PubMed] [Google Scholar]
- 19.Ramaswamy V, Hielscher T, Mack SC, Lassaletta A, Lin T, Pajtler KW, Jones DTW, Luu B, Cavalli FMG, Aldape K, Remke M, Mynarek M, Rutkowski S, Gururangan S, McLendon RE, Lipp ES, Dunham C, Hukin J, Eisenstat DD, Fulton D, van Landeghem FKH, Santi M, van Veelen MLC, van Meir EG, Osuka S, Fan X, Muraszko KM, Tirapelli DPC, Oba-Shinjo SM, Marie SKN, Carlotti CG, Lee JY, Rao AAN, Giannini C, Faria CC, Nunes S, Mora J, Hamilton RL, Hauser P, Jabado N, Petrecca K, Jung S, Massimi L, Zollo M, Cinalli G, Bognár L, Klekner A, Hortobágyi T, Leary S, Ermoian RP, Olson JM, Leonard JR, Gardner C, Grajkowska WA, Chambless LB, Cain J, Eberhart CG, Ahsan S, Massimino M, Giangaspero F, Buttarelli FR, Packer RJ, Emery L, Yong WH, Soto H, Liau LM, Everson R, Grossbach A, Shalaby T, Grotzer M, Karajannis MA, Zagzag D, Wheeler H, von Hoff K, Alonso MM, Tuñon T, Schüller U, Zitterbart K, Sterba J, Chan JA, Guzman M, Elbabaa SK, Colman H, Dhall G, Fisher PG, Fouladi M, Gajjar A, Goldman S, Hwang E, Kool M, Ladha H, Vera-Bolanos E, Wani K, Lieberman F, Mikkelsen T, Omuro AM, Pollack IF, Prados M, Robins HI, Soffietti R, Wu J, Metellus P, Tabori U, Bartels U, Bouffet E, Hawkins CE, Rutka JT, Dirks P, Pfister SM, Merchant TE, Gilbert MR, Armstrong TS, Korshunov A, Ellison DW, Taylor MD. Therapeutic impact of cytoreductive surgery and irradiation of posterior fossa ependymoma in the molecular era: a retrospective multicohort analysis. J Clin Oncol. 2016;34:2468–2477. doi: 10.1200/JCO.2015.65.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merchant TE. Current management of childhood ependymoma. Oncology (Williston Park, NY) 2002;16:629–642. [PubMed] [Google Scholar]
- 21.Japp AS, Gessi M, Messing-Jünger M, Denkhaus D, zur Mühlen A, Wolff JE, Hartung S, Kordes U, Klein-Hitpass L, Pietsch T (2015) High-resolution genomic analysis does not qualify atypical plexus papilloma as a separate entity among choroid plexus tumors. J Neuropathol Exp Neurol 74:110–120 [DOI] [PubMed]
- 22.Wang Y, Cottman M, Schiffman JD. Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet. 2012;205:341–355. doi: 10.1016/j.cancergen.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, Sabari B, Margol AS, Panwalkar P, Parolia A, Pekmezci M, McEachin RC, Cieslik M, Tamrazi B, Garcia BA, La Rocca G, Santi M, Lewis PW, Hawkins C, Melnick A, David Allis C, Thompson CB, Chinnaiyan AM, Judkins AR, Venneti S (2016) Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med 8:366ra161 [DOI] [PMC free article] [PubMed]
- 24.Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, Yip S, Hukin J, Sun Y, Schipper MJ, Chavez L, Margol A, Pekmezci M, Chung C, Banda A, Bayliss JM, Curry SJ, Santi M, Rodriguez FJ, Snuderl M, Karajannis MA, Saratsis AM, Horbinski CM, Carret AS, Wilson B, Johnston D, Lafay-Cousin L, Zelcer S, Eisenstat D, Silva M, Scheinemann K, Jabado N, McNeely PD, Kool M, Pfister SM, Taylor MD, Hawkins C, Korshunov A, Judkins AR, Venneti S. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017;134:705–714. doi: 10.1007/s00401-017-1752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jünger ST, Andreiuolo F, Mynarek M, Wohlers I, Rahmanm S, Klein-Hitpass L, Dörner E, zur Mühlen A, Velez-Char N, von Hoff K, Warmuth-Metz M, Kortmann R, Timmermanm B, von Bueren AO, Rutkowski S, Pietsch T (2020) CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol (in press) [DOI] [PMC free article] [PubMed]
- 26.Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, Koelsche C, Wefers A, Reinhardt A, Huang K, Sievers P, Shimizu H, Nanjo H, Kobayashi Y, Miyake Y, Suzuki T, Adachi JI, Mishima K, Sasaki A, Nishikawa R, Bewerunge-Hudler M, Ryzhova M, Absalyamova O, Golanov A, Sinn P, Platten M, Jungk C, Winkler F, Wick A, Hänggi D, Unterberg A, Pfister SM, Jones DTW, van den Bent M, Hegi M, French P, Baumert BG, Stupp R, Gorlia T, Weller M, Capper D, Korshunov A, Herold-Mende C, Wick W, Louis DN, von Deimling A. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136:153–166. doi: 10.1007/s00401-018-1849-4. [DOI] [PubMed] [Google Scholar]
- 27.Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jäger N, Jones DTW, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJH, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, Hovestadt V, Tzaridis T, Dubuc AM, Northcott PA, Peacock J, Bertrand KC, Agnihotri S, Cavalli FMG, Clarke I, Nethery-Brokx K, Creasy CL, Verma SK, Koster J, Wu X, Yao Y, Milde T, Sin-Chan P, Zuccaro J, Lau L, Pereira S, Castelo-Branco P, Hirst M, Marra MA, Roberts SS, Fults D, Massimi L, Cho YJ, van Meter T, Grajkowska W, Lach B, Kulozik AE, von Deimling A, Witt O, Scherer SW, Fan X, Muraszko KM, Kool M, Pomeroy SL, Gupta N, Phillips J, Huang A, Tabori U, Hawkins C, Malkin D, Kongkham PN, Weiss WA, Jabado N, Rutka JT, Bouffet E, Korbel JO, Lupien M, Aldape KD, Bader GD, Eils R, Lichter P, Dirks PB, Pfister SM, Korshunov A, Taylor MD. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506:445–450. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korshunov A, Witt H, Hielscher T, Benner A, Remke M, Ryzhova M, Milde T, Bender S, Wittmann A, Schöttler A, Kulozik AE, Witt O, von Deimling A, Lichter P, Pfister S. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;28:3182–3190. doi: 10.1200/JCO.2009.27.3359. [DOI] [PubMed] [Google Scholar]
- 29.Jünger ST, Mynarek M, Wohlers I, Dörner E, zur Mühlen A, Velez-Char N, von Hoff K, Rutkowski S, Warmuth-Metz M, Kortmann R, Timmermann B, Rahmann S, Klein-Hitpass L, von Bueren AO, Pietsch T (2019) Improved risk-stratification for posterior fossa ependymoma of childhood considering clinical, histological and genetic features – a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol Comm 7 (1) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on reasonable request from the corresponding author.