

Abstract
Background
Treatment for neuropathic pain as a refractory disease remains unsatisfactory and represents a significant clinical challenge. A highly effective drug is thus urgently needed for neuropathic pain treatment. Dihydromyricetin (DMY) is a flavonoid with a wide range of biological activities. The purpose of this research is to explore the effects of DMY on neuropathic pain and the underlying mechanism of its effect.
Methods
The effect of DMY was investigated in BV-2 cells and lipopolysaccharide (LPS)-induced BV-2 cells. A neuropathic pain model was established via spared nerve injury (SNI) surgery in mice, and the protein expression level was detected via Western blot assay. The percent of M1 and M2 phenotype polarization cells were detected via flow cytometry assay. Immunochemical staining assay was also performed to measure the marker levels of the M1 and M2 phenotype polarization cells and aldehyde dehydrogenase 2 (ALDH2) level, and mechanical pain sensitivity was evaluated via measurement of the mechanical withdrawal threshold.
Results
We found that DMY promoted the transition from M1 to M2 polarization and upregulated the ALDH2 level in vitro and vitro. ALDA-1, an ALDH2 agonist, promoted the switching from M1 to M2 polarization in vivo and vitro. DMY alleviated pain hypersensitivity induced by SNI via enhancing M2 phenotype polarization by elevating ALDH2 activity in mice. After DMY- or ALDA-1-microglia were injected into SNI-induced pain hypersensitive mice, the mechanical withdrawal threshold was increased significantly when compared with the SNI group.
Conclusions
Our data demonstrated that DMY alleviated neuropathic pain via enhancing the polarization transition from the M1 to M2 phenotype by potentially elevating ALDH2 activity in vitro and vivo. DMY- or ALDA-1-microglia may have alleviative effects on neuropathic pain. The findings herein provide a promising avenue for neuropathic pain treatment, suggesting a new target, ALDH2, in the treatment of neuropathic pain.
Keywords: Neuropathic pain, dihydromyricetin (DMY), aldehyde dehydrogenase 2 (ALDH2), polarization, ALDA-1
Introduction
Neuropathic pain is a chronic pain disease which arises from impairment or disease in the somatosensory nervous system (1-3). Neuropathic pain is characterized by hyperalgesia and allodynia and severely worsens patients’ quality of life, placing a heavy burden on society (4,5). The prevalence of neuropathic pain is about 5–10% of the general population (6,7). Neuropathic pain can result from complications in diseases like diabetes, cancer, and stroke (8-10). Its incidence is likely on the rise, which is attributable to the concurrent increase in diabetes. Thus, the mechanisms of neuropathic pain and its related therapeutic approaches urgently need to be elucidated.
The current perspectives of neuropathic pain pathogenesis include central sensitization, inflammatory mediator stimulation, metabolic damage, overactivation of microglia cells, and other factors (11-13). In this regard, convincing evidence has demonstrated that the activation of microglia cells plays a crucial role in neuropathic pain (14,15). Microglia function as the major immune cells in the nervous system. They are activated within 24 h of nerve injury (16,17) via polarization, specifically, M1 phenotype and M2 phenotype polarization. M1 polarization is a pro-inflammatory phenotype that contributes to neuropathic pain (18,19) while, the M2 phenotype, as the alternative path of polarization, exerts an anti-inflammatory effect, thus opposing M1 phenotype polarization (20). The relevant research indicates that switching from M1 phenotype polarization to M2 phenotype polarization is a promising avenue in neuropathic pain treatment (21,22). The process behind the regulation of the M1/M2 polarization process remains unclear, and gaining insight into its mechanism is crucial to developing new drugs for neuropathic pain therapy.
Aldehyde dehydrogenase 2 (ALDH2), the second enzyme of the major oxidative pathway of alcohol metabolism, is highly expressed in the brain (23). ALDH2 is reported to relieve inflammation via regulating autophagy (24) and has been shown to reduce dox-induced toxicity via suppressing oxidative stress and apoptosis (25). In microglial cells, ALDH2 functions downstream of TGR5, reducing oxidative stress and resisting neuronal apoptosis after activation (26) and can exert protective effects in chronic pain (27). However, the relation between ALDH2 and M1/M2 polarization in neuropathic pain remains unknown.
Dihydromyricetin (DMY) is an active extract of many plants including Ampelopsis grossedentata and Hovenia dulcis. Of these, H. dulcis has protective effects against hangover and liver injuries induced by alcohol through elevating ALDH2 activity, which may be a newly discovered strategy of neuroprotection (28,29). We speculated that DMY, as the main active compound of H. dulcis, may have effects on ALDH2 activity and thus has a role in neuropathic pain. DMY is also an extract with a variety biological activity, including anti-inflammatory and anti-oxidative effects. DMY was reported to have protective effects against neuroinflammation and microglial activation APP/PS1 transgenic mice (30), and other recent research has indicated that DMY has alleviative effects on diabetic neuropathic pain (31,32). Despite these findings, the effect and mechanism of DMY in neuropathic pain is not yet fully understood. Thus, this study aimed to explore the effects and potential mechanism of DMY on ALDH2 activity in neuropathic pain.
We present the following article in accordance with the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/atm-20-5838).
Methods
Cell culture and treatment
The BV-2 cells (cat. no. CL-0493, Procell Life science & Technology Co., Ltd., China) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), streptomycin (100 µg/mL), and penicillin (100 U/mL). Cells were preserved in serum-free cell freezing medium (Teyebio, Shanghai, China). The BV-2 cells were seeded in 6-well plates at a density of 2×105, at 37 °C in an incubator containing 5% CO2 and 95% air. The cells were assigned into the following groups: control, 10 µM DMY, 100 µm DMY, 1 mM DMY, lipopolysaccharide (LPS), LPS + 10 µM DMY, LPS + 100 µm DMY, and LPS + 1 mM DMY. In the DMY group, the cells were treated with different concentrations of DMY (10 µM, 100 µm, and 1 mM). For the LPS group, the cells were treated with 100 ng/mg LPS for 24 h. For the LPS + DMY group, the cells were pretreated with LPS and then treated with different concentrations of DMY (10 µM, 100 µm, and 1 mM) for 24 h. For the ALDA-1 group, the cells were incubated with ALDA-1 (20 µm). For the ALDA-1 + LPS group, after LPS treatment, the cells were incubated with ALDA-1 (20 µm). After that, images of the cells were taken under a microscope.
Western blotting
The total proteins in BV-2 cells were extracted with lysis buffer, and then the lysates were centrifuged (15,000 ×g, 10 min). Bicinchoninic acid assay kit (Teyebio, Shanghai, China) was used to detect the concentration of the proteins. Then, 30 µg of proteins were electrophoresed on a 15% SDS-polyacrylamide gels and then blotted on a polyvinylidene fluoride (PVDF) membrane. After blockage with 5% skim milk, the membranes reacted with the primary antibodies against nitric oxide synthase (INOS) (#ab15323, Abcam), arginase 1 (Arg1) (#ab96183, Abcam), cluster of differentiation 206 (CD206) (#ab64693, Abcam), CD86 (#ab242142), and ALDH2 (#ab108306, Abcam). GAPDH (#ab181602, Abcam) was used as the internal reference. Then, IgG-HRP secondary antibody (#ab7090, Abcam) was incubated with the membranes. An enhanced chemiluminescence system (Bio-Rad Laboratories, USA) was used for visualization of the bands.
Flow cytometry
The percentage of M2/ M1 phenotype cells was determined via evaluating the level of their corresponding markers: CD86 and CD206, respectively. The percentage of CD86 and CD206 was determined via flow cytometry assay. After treatment in the study groups, the BV-2 cells were collected and suspended in phosphate-buffered saline (PBS). Subsequently, the BV-2 cells were stained with the fluorescence-labeled CD206 and CD86 antibody for half an hour in the dark at 4 ˚C. For the control group, isotypes of corresponding primary antibodies were incubated with the cells. FACS Calibur (Beckman Coulter, USA) was applied for analyzing the percentage of CD206 and CD86.
Animals
Male CD1 mice (8 weeks old) were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. and kept under the standard conditions (25±2 °C, humidity: 55%±5%, 12:12 h light/dark). The mice had free access to the standard laboratory diet and water. The mice were housed in the experiment environment for 48 h to adapt to the environment before testing began. Animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee at China-Japan Union Hospital of Jilin University (#JU3874).
Establishment of the neuropathic pain model and treatment
The neuropathic pain model was established via spared nerve injury (SNI) surgery. The mice were randomly assigned to the study groups (10 mice per group). The anesthesia was carried out via using (sodium pentobarbital 60 mg/kg i.p.) in mice. After shaving and sterilization, an incision was made on the left hind limb. The sciatic nerve was exposed after the biceps femoris muscle was separated. Then, the common peroneal and tibial nerves at the terminal branches of sciatic nerve were ligated using suture silk 5.0, following the manufacturer instruction. The terminal of the nerve trunk (2–4 mm) was removed. For the sham group, after exposure of sciatic nerve and its branches as described above, no ligation or other operation was performed, apart from direct suture. All procedures were conducted in sterile conditions. After establishment of the model, the mice in the ALDA-1 Ii group were treated with ALDA-1 (10 mg/kg) via intraperitoneal injection, while mice in the ALDA-1 IL group were treated with ALDA-1 (10 µg/10 µL) via local injection as previously described (33). Mice in the SNI + DMY Ii group were administered with DMY (10 mg/kg) once a day via intraperitoneal injection, while mice in the SNI + DMY Li group, were given DMY (10 µg/10 µL) via local injection once a day for 6 days. After modeling, mice in the SNI + DMY microglial group were treated with 103 microglial cells which were pretreated with DMY. Mice in the SNI + ALDA microglial group were treated with 103 microglial cells which were pretreated with ALDA-1. For the microglial groups, 103 microglial cells were injected into the myelin sheath.
Measurement of the mechanical withdrawal threshold
The measurement of the mechanical withdrawal threshold was performed by using a series of von Frey filaments (2, 4, 6, 8, 10, 15 g). All the mice were placed in a cage for 30 min before tests to adapt to the experimental environment. Then, a series Frey filament stimulation (0.6, 1, 2, 4, 6, 8, 10, 15 g) with internal of 15 s treatment was performed on the left hind paw of the mice for 3–4 seconds. It was considered as a positive reaction when the mice withdrew their paw rapidly within 4 s. On the contrary, a negative reaction was characterized by no paw withdrawal. A threshold tracking algorithm method was used for calculation of the paw withdrawal threshold (PWT) (34).
Immunochemistry assay
After 10% fixation, tissue samples of the different study groups were embedded with paraffin. Then, the samples in the different study groups were made into 5 µm sections and subjected to dewaxing and dehydration. Then, the sections were incubated with the primary antibodies against INOS (Cat#15323, Abcam), Arg1 (Cat# ab272887, Abcam), CD206 (Cat#ab64693, Abcam), CD11b (Cat#ab133357, Abcam), and ALDH2 (Cat#ab194587, Abcam) at 4 °C, overnight. Next, the sections were reacted with the secondary antibodies at room temperature for 1 h. Subsequently, hematoxylin and eosin (HE) counterstaining was performed. An RFCA (Olympus, Japan) microscope was used for observation of the slides.
Statistical analysis
The results were analyzed via SPSS 11.0 and GraphPad version 6.0 software. All the data are presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) with Tukey’s multiple test was used for comparison between groups. A P value <0.05 indicated significant difference. We present the following article in accordance with the ARRIVE reporting checklist.
Results
DMY promoted the transition from M1 to M2 polarization and upregulated ALDH2 level in vitro
As M1 phenotype polarization contributes to neuropathic pain and M2 phenotype polarization works in counter fashion, we explored the effects of DMY on M1/M2 polarization. INOS and CD86, which are the common M1 polarization markers, and Arg1 and CD206, which are M2 polarization markers, were subsequently measured. The polarized cells were gradually increased by DMY in a concentration-dependent manner when compared with controls (Figure 1A). Compared with controls, INOS and CD86, were all decreased by DMY, while Arg1 and CD206 were all increased by DMY (Figure 1B,C,D,E,F) in a concentration-dependent manner, suggesting that DMY promotes a polarization switch from M1 to M2. As neuropathic pain is mainly caused by M1 polarization, the M1 phenotype polarization cells served well as a cell model of neuropathic pain. The M1 phenotype polarization cells were induced by LPS. Compared with controls, there was a significant increase in the polarized cells induced by LPS. With the increase of DMY concentration, the polarized cells first decreased and then increased in LPS-induced cells (Figure 1A). Simultaneously, DMY reduced the levels of INOS and CD86 while elevating the levels of Arg1 and CD206 in LPS-induced cells, indicating that DMY promotes the transition from M1 to M2 polarization. With the increase of DMY concentration, there was an initial decrease and a subsequent increase in the number of polarized cells induced by LPS (Figure 1A), which reflected the transition from M1 to M2 under DMY treatment.
Figure 1.

The effects of DMY on cell polarization and ALDH2 level. The cell morphology under microscope, the scale bar is 100 µm (A). The levels of M1 and M2 polarization markers detected by Western blotting (B,C,D,E,F). The ALDH2 level detected by Western blotting (G,H). *, P<0.05 and **, P<0.01 vs. control group; ##, P<0.01 vs. LPS group; &, P<0.05 vs. 100 µM DMY; &&, P<0.01 vs. LPS + 10 µM DMY; $$, P<0.01 vs. LPS + 100 µM DMY; aa, LPS + 10 μM DMY vs. LPS; bb, LPS + 100 μM DMY vs. LPS + 10 μM DMY, and LPS; cc, LPS + 1 mM DMY vs. LPS + 100 μM DMY, LPS+10 μM DMY, and LPS. DMY, dihydromyricetin; LPS, lipopolysaccharide.
As ALDH2 is thought to be a protective enzyme in chronic pain, the effect of DMY on ALDH2 was also examined. The ALDH2 level was elevated significantly by DMY in a concentration-dependent manner in BV-2 cells with or without LPS treatment (Figure 1G,H). The results from flow cytometry showed that the M1 phenotype polarization, as indicated by the percent of CD86, was decreased, while M2 phenotype polarization, as indicated by CD206, was incTreased (Figure 2A,B), further confirming that DMY promotes a transition from M1 to M2 polarization.
Figure 2.

The effects of DMY on the cell percentage of M1 and M2 polarization. The CD86 percentage (A) and CD206 percentage (B) in the study groups. **, P<0.01 vs. control group; ##, P<0.01 vs. LPS group; &&, P<0.01 vs. 100 µM DMY; $$, P<0.01 vs. LPS + 100 µM DMY; aa, LPS + 10 μM DMY vs. LPS; bb, LPS + 100 μM DMY vs. LPS + 10 μM DMY, and LPS; cc, LPS + 1 mM DMY vs. LPS + 100 μM DMY, LPS + 10 μM DMY, and LPS. DMY, dihydromyricetin; LPS, lipopolysaccharide.
DMY promoted polarization from M1 to M2 via upregulating ALDH2 level in vitro
As outlined previously, DMY can elevate ALDH2 level and promote cell polarization from M1 to M2. However, the relation between M1/M2 polarization and ALDH2 remains unknown. The effects of ALDH2 agonist, ALDA-1, was further investigated in BV-2 cells with or without LPS treatment. Compared with controls, the polarized cells were increased in the ALDA-1 group and LPS group (Figure 3A). As seen in the results from Western blotting (Figure 3B,C,D,E,F), the M1 polarization markers, iNOS and CD86, in the ALDA-1 group were decreased, while the M2 polarization makers, Arg1 and CD206, were increased when compared with controls (Figure 3B,C,D,E,F); this indicate that the polarized cells in the ALDA-1 group were mainly M2 polarization cells. Consistent with the results above, the polarized cells in LPS group were mainly M1 polarization cells, which was further supported by the increased levels of cell polarization markers CD86. In the ALDA-1 + LPS group, the M1 polarization markers were decreased and M2 polarization markers were increased when compared with LPS group, suggesting that ALDA-1, being an ALDH2 agonist, can promote the cell polarization switch from M1 to M2. In addition, we found that DYM could elevate the ALDH2 level, with cell polarization from M1 to M2 being simultaneously enhanced by the ALDH2 agonist, ALDA-1. We can therefore conclude that DYM promoted a cell polarization switch from M1 to M2 via elevating the ALDH2 level in vitro.
Figure 3.

The effects of ALDA-1 on cell polarization. The cell morphology under microscope (A). The levels of M1 and M2 polarization markers detected by Western blotting (B,C,D,E,F). **, P<0.01 vs. control group; ##, P<0.01 vs. LPS group; &, P<0.05 vs. LPS; &&, P<0.01 vs. LPS; aa, LPS + 10 μM DMY vs. LPS; bb, LPS + 100 μM DMY vs. LPS + 10 μM DMY, and LPS; cc, LPS + 1 mM DMY vs. LPS + 100 μM DMY, LPS + 10 μM DMY, and LPS. DMY, dihydromyricetin; LPS, lipopolysaccharide.
DMY attenuated pain hypersensitivity induced by SNI by enhancing the transition from M1 to M2 phenotype polarization through elevation of ALDH2 activity in mice
As shown by the immunohistochemistry results, the positive staining of INOS was increased while Arg1, CD206, and CD11b were decreased in the neuropathic pain model induced by SNI when compared with sham group (Figure 4A,B; 10 mice per group), indicating that M1 polarization was the major polarization phenotype in the neuropathic pain model induced by SNI (SNI group). After ALDA-1 or DMY treatment, the INOS staining-positive level via intraperitoneal injection was decreased more obviously than that via local injection when compared with the SNI group, revealing that M1 polarization was inhibited by ALDA-1 or DMY in the neuropathic pain model induced by SNI. Arg1, CD206, and CD11b were decreased in the SNI group when compared with the control group, and appeared elevated by ALDA-1 or DMY via intraperitoneal injection more obviously than via local injection when compared with the SNI group, demonstrating that both ALDA-1 and DMY could promote M2 polarization. Moreover, The ALDH2 positive-staining level was decreased in the SNI group when compared with control, and was more obviously elevated by ALDA-1 or DMY via intraperitoneal injection than via local injection when compared with the SNI group. DMY and ALDA-1 had the same effects on M1/M2 polarization. Moreover, the ALDH2 level could be elevated by DMY. All these results indicate that DMY functions on M1/M2 polarization via elevating ALDH2 activity.
Figure 4.

The effects of DMY/ALDA-1 on the levels of M1 and M2 polarization markers and ALDH2 level. The levels of M1 and M2 polarization markers and ALDH2 (A,B) detected by immunochemical staining assay. The scale bar is 100 μm. DMY, dihydromyricetin.
The effect of DMY on mechanical hyperalgesia was also measured. The PWT in the SNI group was significantly lower than that in the sham group (Figure 5A), confirming that the neuropathic pain model was established successfully. The PWT was more obviously increased by ALDA-1 or DMY treatment via intraperitoneal injection than via local injection when compared with SNI group, confirming that DMY could alleviate neuropathic pain and that this effect is inferior to that of ALDA-1. In addition, DMY could elevate the ALDH2 level, meaning DMY attenuates neuropathic pain via elevating ALDH2 activity. We further explored the therapeutic effects of DMY- and ALDA-1-microglia on neuropathic pain. As shown by the results (Figure 5B), the PWT in the SNI group was significantly lower than that in the sham group, confirming that the neuropathic pain model was consistent. After microglia treatment, there was almost no change in PWT when compared with the SNI group, indicating that microglia has no therapeutic effect on neuropathic pain. Moreover, the DMY- or ALDA-1-microglia treatment elevated the PWT significantly, strongly supporting the notion that DMY- or ALDA-1-microglia can alleviate neuropathic pain, and thus offering a potential new strategy for neuropathic pain treatment.
Figure 5.
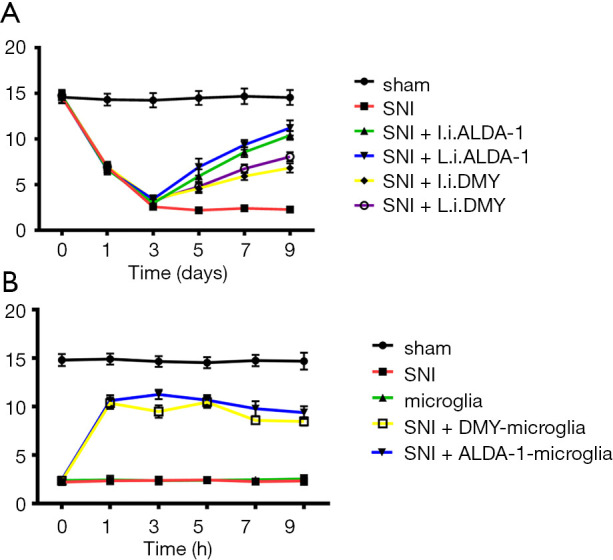
The effects of DMY/ALDA-1 and microglial cells pretreated with DMY/ALDA-1 on the paw withdrawal threshold. The paw withdrawal threshold in the different study groups (A,B). DMY, dihydromyricetin.
Discussion
Neuropathic pain is caused by a lesion or disease of the somatosensory system, including peripheral fibres (Aβ, Aδ and C fibres) and central neurons (35). The mechanism of neuropathic pain including imbalances between excitatory and inhibitory somatosensory signaling (36), alterations in ion channels (37) and second-order nociceptive neuron alterations (38). Neuropathic pain remains a problematic refractory disease. Despite the progress that has been made in the therapy of neuropathic pain, the efficacy of treatment remains unsatisfactory (39). Studies have confirmed that microglial polarization plays a pivotal role in development of neuropathic pain (20,40-42), with the typical activated microglia being M1 phenotype polarization which contributes to progression of neuropathic pain via its pro-inflammatory role. On the other hand, M2 phenotype polarization, as an alternative path of polarization, has an anti-inflammatory effect. Regulation of polarization states towards the M2 phenotype has been considered a promising avenue for neuropathic pain treatment. Substantial evidence has shown that DHM can cross the blood-brain barrier, modulate inflammation, and exert neuroprotective effects (43). It has been reported that DHM can inhibit microglia-mediated neuroinflammation and play a neuroprotective role in Alzheimer’s disease by suppressing inflammation (44). However, the role of DMY in modulating neuropathic pain has not been studied. In the present study, we found that DMY induced a switch from M1 to M2 phenotype polarization in microglia cells, which attenuates neuropathic pain.
As neuropathic pain is mainly caused by M1 phenotype polarization, we constructed M1 phenotype polarization cells using LPS, as it is the common stimulator of M1 phenotype polarization (45,46). We further investigated the effects of DMY on M1/M2 phenotype polarization in the presence of LPS treatment, and found that the polarization of cells was increased with an increase in the DMY concentration in BV-2 cells. Flow cytometer analysis also demonstrated that DMY treatment could promote the M2 microglial phenotype by shifting microglial cells from the M1 to the M2 phenotype in a concentration-dependent manner, which was consistent with the previous study (30).
A growing body of evidence has indicated that oxidative stress is critically involved in the aggravation of neuropathic pain (47-49). However, many studies have focused on the injuries caused by reactive oxygen species (ROS) with fewer studies having examined other products of oxidative stress. One such product, aldehyde (50), is toxic to cells and tissues and is also involved in neuropathic pain. Acrolein, which is a reactive aldehyde, has also been reported to act as a contributor of neuropathic pain (51). Furthermore, acetaldehyde has been confirmed to cause oxidative stress and neuropathic pain (52). Meanwhile, ALDH2 has been shown to be an excellent scavenger of reactive aldehydes (53) and has also been found to suppress oxidative stress and inflammation in many diseases (24,53-56); however, the role of ALDH2 in neuropathic pain remains elusive. Thus, this was the first study to examine the role of ALDH2 in neuropathic pain. We found that the ALDH2 level could be elevated by DMY in a concentration dependent-manner in BV-2 cells with or without LPS treatment, and, as ALDH2 was reported to work as a protective agent in chronic pain (27), ALDH2 may thus be a promising therapeutic target in neuropathic pain. Because DMY promoted M2 polarization, elevated the ALDH2 level, and thus had a protective effect in neuropathic pain, we speculated that there may be a certain relationship between microglia polarization and ALDH2. As expected, our data suggest that ALDA-1, the agonist of ALDH2, elevated M2 phenotype polarization cells and decreased M1 phenotype polarization cells. Furthermore, we found that DMY promoted the transition from M1 to M2 phenotype polarization via the elevation ALDH2.
In order to confirm the validity of our conclusions, the effects of DMY were examined in vivo. The immunochemical staining results showed that DMY could also elevate ALDH2 level in vivo. The M1 phenotype polarization cells were decreased by DMY and ALDA-1, as reflected by the positive staining level of INOS. The M2 phenotype polarization cells were increased by DMY and ALDA-1, as indicated by the markers’ positive staining level. All the observed effects of DMY in vivo were consistent with those observed in vitro. In addition, the mechanical withdrawal threshold was decreased significantly by DMY and ALDA-1, strongly supporting the supposition that DMY attenuates neuropathic pain via promoting M2 polarization by elevating ALDH2 activity. As M2 polarization has therapeutic effects, we further investigated the effects of microglial cells treated by DMY or ALDA-1. The mechanical withdrawal threshold was increased more significantly by the microglial cells treated by DMY /ALDA-1, demonstrating that using DMY- or ALDA-1-induced microglial cells may be a promising strategy for treating neuropathic pain.
Conclusions
In this study, DYM was found to attenuate neuropathic pain by switching from M1 to M2 phenotype polarization via potentially elevating ALDH2 activity. The findings provide a new strategy for neuropathic pain treatment, laying the foundations for future clinical implications.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This work was supported by Jilin Provincial Department of education project (JJKH20190069KJ).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee at China-Japan Union Hospital of Jilin University (#JU3874).
Reporting Checklist: The authors have completed the ARRIVE reporting checklist. Available at http://dx.doi.org/10.21037/atm-20-5838
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-5838
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-5838). The authors have no conflicts of interest to declare.
(English Language Editor: J. Gray)
References
- 1.St John Smith E. Advances in understanding nociception and neuropathic pain. J Neurol 2018;265:231-8. 10.1007/s00415-017-8641-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finnerup NB, Haroutounian S, Kamerman P, et al. Neuropathic pain: an updated grading system for research and clinical practice. Pain 2016;157:1599-606. 10.1097/j.pain.0000000000000492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baad-Hansen L, Benoliel R. Neuropahtic orofacial pain: Facts and fiction. Cephalalgia 2017;37:670-9. 10.1177/0333102417706310 [DOI] [PubMed] [Google Scholar]
- 4.Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol 2014;13:924-35. 10.1016/S1474-4422(14)70102-4 [DOI] [PubMed] [Google Scholar]
- 5.Derry S, Bell RF, Straube S, et al. Pregabalin for neuropathic pain in adults. Cochrane Database Syst Rev 2019;1:CD007076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colloca L, Ludman T, Bouhassira D, et al. Neuropathic pain. Nat Rev Dis Primers 2017;3:17002. 10.1038/nrdp.2017.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sewell RD. Neuropathic pain models and outcome measures: a dual translational challenge. Ann Transl Med 2018;6:S42. 10.21037/atm.2018.09.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shillo P, Sloan G, Greig M, et al. Painful and Painless Diabetic Neuropathies: What Is the Difference? Curr Diab Rep 2019;19:32. 10.1007/s11892-019-1150-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fallon MT. Neuropathic pain in cancer. Br J Anaesth 2013;111:105-11. 10.1093/bja/aet208 [DOI] [PubMed] [Google Scholar]
- 10.Watson JC, Sandroni P. Central Neuropathic Pain Syndromes. Mayo Clin Proc 2016;91:372-85. 10.1016/j.mayocp.2016.01.017 [DOI] [PubMed] [Google Scholar]
- 11.Meacham K, Shepherd A, Mohapatra DP, et al. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr Pain Headache Rep 2017;21:28. 10.1007/s11916-017-0629-5 [DOI] [PubMed] [Google Scholar]
- 12.Gilron I, Baron R, Jensen T. Neuropathic pain: principles of diagnosis and treatment. Mayo Clin Proc 2015;90:532-45. 10.1016/j.mayocp.2015.01.018 [DOI] [PubMed] [Google Scholar]
- 13.Chen G, Zhang YQ, Qadri YJ, et al. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 2018;100:1292-311. 10.1016/j.neuron.2018.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong X, Chen Y, Fu B, et al. Infant nerve injury induces delayed microglial polarization to the M1 phenotype, and exercise reduces delayed neuropathic pain by modulating microglial activity. Neuroscience 2017;349:76-86. 10.1016/j.neuroscience.2017.02.051 [DOI] [PubMed] [Google Scholar]
- 15.Jin J, Guo J, Cai H, et al. M2-Like Microglia Polarization Attenuates Neuropathic Pain Associated with Alzheimer's Disease. J Alzheimers Dis 2020;76:1255-65. 10.3233/JAD-200099 [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, Trapp BD. Microglia and neuroprotection. J Neurochem 2016;136 Suppl 1:10-7. 10.1111/jnc.13062 [DOI] [PubMed] [Google Scholar]
- 17.Cohen SP, Mao J. Neuropathic pain: mechanisms and their clinical implications. BMJ 2014;348:f7656. 10.1136/bmj.f7656 [DOI] [PubMed] [Google Scholar]
- 18.Wang D, Liu F, Zhu L, et al. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J Neuroinflammation 2020;17:257. 10.1186/s12974-020-01921-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin GL, He SD, Lin SM, et al. Koumine Attenuates Neuroglia Activation and Inflammatory Response to Neuropathic Pain. Neural Plast 2018;2018:9347696. 10.1155/2018/9347696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, Wang J, Li H, et al. IL-35 promotes microglial M2 polarization in a rat model of diabetic neuropathic pain. Arch Biochem Biophys 2020;685:108330. 10.1016/j.abb.2020.108330 [DOI] [PubMed] [Google Scholar]
- 21.Piotrowska A, Kwiatkowski K, Rojewska E, et al. Maraviroc reduces neuropathic pain through polarization of microglia and astroglia - Evidence from in vivo and in vitro studies. Neuropharmacology 2016;108:207-19. 10.1016/j.neuropharm.2016.04.024 [DOI] [PubMed] [Google Scholar]
- 22.Jiang Y, Wang J, Li H, Xia L. IL-35 promotes microglial M2 polarization in a rat model of diabetic neuropathic pain. Arch Biochem Biophys 2020;685:108330. 10.1016/j.abb.2020.108330 [DOI] [PubMed] [Google Scholar]
- 23.Stewart MJ, Malek K, Crabb DW. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J Investig Med 1996;44:42-6. [PubMed] [Google Scholar]
- 24.Guo R, Xu X, Babcock SA, et al. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. J Hepatol 2015;62:647-56. 10.1016/j.jhep.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Gao Y, Xu Y, Hua S, et al. ALDH2 attenuates Dox-induced cardiotoxicity by inhibiting cardiac apoptosis and oxidative stress. Int J Clin Exp Med 2015;8:6794-803. [PMC free article] [PubMed] [Google Scholar]
- 26.Zuo G, Zhang T, Huang L, et al. Activation of TGR5 with INT-777 attenuates oxidative stress and neuronal apoptosis via cAMP/PKCε/ALDH2 pathway after subarachnoid hemorrhage in rats. Free Radic Biol Med 2019;143:441-53. 10.1016/j.freeradbiomed.2019.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Sun W, Gu C, et al. Targeting ALDH2 for Therapeutic Interventions in Chronic Pain-Related Myocardial Ischemic Susceptibility. Theranostics 2018;8:1027-41. 10.7150/thno.22414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du J, He D, Sun LN, et al. Semen Hoveniae extract protects against acute alcohol-induced liver injury in mice. Pharm Biol 2010;48:953-8. 10.3109/13880200903300196 [DOI] [PubMed] [Google Scholar]
- 29.Shen Y, Lindemeyer AK, Gonzalez C, et al. Dihydromyricetin as a novel anti-alcohol intoxication medication. J Neurosci 2012;32:390-401. 10.1523/JNEUROSCI.4639-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jing N, Li X. Dihydromyricetin Attenuates Inflammation through TLR4/NF-kappaB Pathway. Open Med (Wars) 2019;14:719-25. 10.1515/med-2019-0083 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Ge H, Guan S, Shen Y, et al. Dihydromyricetin affects BDNF levels in the nervous system in rats with comorbid diabetic neuropathic pain and depression. Sci Rep 2019;9:14619. 10.1038/s41598-019-51124-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan S, Shen Y, Ge H, et al. Dihydromyricetin Alleviates Diabetic Neuropathic Pain and Depression Comorbidity Symptoms by Inhibiting P2X7 Receptor. Front Psychiatry 2019;10:770. 10.3389/fpsyt.2019.00770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Zhao Q, Ye F, et al. Alda-1, an ALDH2 activator, protects against hepatic ischemia/reperfusion injury in rats via inhibition of oxidative stress. Free Radic Res 2018;52:629-38. 10.1080/10715762.2018.1459042 [DOI] [PubMed] [Google Scholar]
- 34.Chaplan SR, Bach FW, Pogrel JW, et al. Quantitative assessment of tactile allodynia in the rat paw. Journal of Neuroscience Methods 1994;53:55-63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- 35.Colloca L, Ludman T, Bouhassira D, et al. Neuropathic pain. Nat Rev Dis Primers 2017;3:17002. 10.1038/nrdp.2017.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Occhipinti E, Colombini D, Cantoni S, et al. Spinal changes in drivers of heavy vehicles. Med Lav 1986;77:280-92. [PubMed] [Google Scholar]
- 37.Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 2004;41:171-4. 10.1136/jmg.2003.012153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyron R. Functional brain imaging: what has it brought to our understanding of neuropathic pain? A special focus on allodynic pain mechanisms. Pain 2016;157 Suppl 1:S67-71. 10.1097/j.pain.0000000000000387 [DOI] [PubMed] [Google Scholar]
- 39.Popiolek-Barczyk K, Kolosowska N, Piotrowska A, et al. Parthenolide Relieves Pain and Promotes M2 Microglia/Macrophage Polarization in Rat Model of Neuropathy. Neural Plast 2015;2015:676473. 10.1155/2015/676473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gui X, Wang H, Wu L, et al. Botulinum toxin type A promotes microglial M2 polarization and suppresses chronic constriction injury-induced neuropathic pain through the P2X7 receptor. Cell Biosci 2020;10:45. 10.1186/s13578-020-00405-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma YM, Li CD, Zhu YB, et al. The mechanism of RvD1 alleviates type 2 diabetic neuropathic pain by influencing microglia polarization in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2017;33:277-81. [DOI] [PubMed] [Google Scholar]
- 42.Yin Y, Phạm TL, Shin J, et al. Arginase 2 Deficiency Promotes Neuroinflammation and Pain Behaviors Following Nerve Injury in Mice. J Clin Med 2020;9:305. 10.3390/jcm9020305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Youdim KA, Dobbie MS, Kuhnle G, et al. Interaction between flavonoids and the blood-brain barrier: in vitro studies. J Neurochem 2003;85:180-92. 10.1046/j.1471-4159.2003.01652.x [DOI] [PubMed] [Google Scholar]
- 44.Feng J, Wang JX, Du YH, et al. Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci Ther 2018;24:1207-18. 10.1111/cns.12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Xu S, Qian Y, et al. Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun 2017;64:162-72. 10.1016/j.bbi.2017.03.003 [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Chen Q, Nai Y, et al. Suppression of miR-155 attenuates neuropathic pain by inducing an M1 to M2 switch in microglia. Folia Neuropathologica 2020;58:70-82. 10.5114/fn.2020.94008 [DOI] [PubMed] [Google Scholar]
- 47.Ge Y, Jiao Y, Li P, et al. Coregulation of endoplasmic reticulum stress and oxidative stress in neuropathic pain and disinhibition of the spinal nociceptive circuitry. Pain 2018;159:894-906. 10.1097/j.pain.0000000000001161 [DOI] [PubMed] [Google Scholar]
- 48.Shim HS, Bae C, Wang J, et al. Peripheral and central oxidative stress in chemotherapy-induced neuropathic pain. Mol Pain 2019;15:1744806919840098. 10.1177/1744806919840098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.‘, Wang Z, Zhao W, et al. The role of P2Y6 receptors in the maintenance of neuropathic pain and its improvement of oxidative stress in rats. J Cell Biochem 2019;120:17123-30. [DOI] [PubMed]
- 50.Singh S, Brocker C, Koppaka V, et al. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic Biol Med 2013;56:89-101. 10.1016/j.freeradbiomed.2012.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin Y, Chen Z, Tang J, et al. Acrolein Contributes to the Neuropathic Pain and Neuron Damage after Ischemic–Reperfusion Spinal Cord Injury. Neuroscience 2018;384:120-30. 10.1016/j.neuroscience.2018.05.029 [DOI] [PubMed] [Google Scholar]
- 52.De Logu F, Li Puma S, Landini L, et al. Schwann cells expressing nociceptive channel TRPA1 orchestrate ethanol-evoked neuropathic pain in mice. J Clin Invest 2019;129:5424-41. 10.1172/JCI128022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhai X, Zhang Z, Liu W, et al. Protective effect of ALDH2 against cyclophosphamide-induced acute hepatotoxicity via attenuating oxidative stress and reactive aldehydes. Biochem Biophys Res Commun 2018;499:93-8. 10.1016/j.bbrc.2018.03.041 [DOI] [PubMed] [Google Scholar]
- 54.Gu X, Fang T, Kang P, et al. Effect of ALDH2 on High Glucose-Induced Cardiac Fibroblast Oxidative Stress, Apoptosis, and Fibrosis. Oxid Med Cell Longev 2017;2017:9257967. 10.1155/2017/9257967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao R, Fang D, Wang J, et al. ALDH2 Overexpression Alleviates High Glucose-Induced Cardiotoxicity by Inhibiting NLRP3 Inflammasome Activation. J Diabetes Res 2019;2019:4857921. 10.1155/2019/4857921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zambelli VO, Gross ER, Chen CH, et al. Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci Transl Med 2014;6:251ra118. 10.1126/scitranslmed.3009539 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as