

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a major cause of chronic liver disease and is associated with significant morbidity and mortality worldwide, with a high incidence in Western countries and non‐Western countries that have adopted a Western diet. NAFLD is commonly associated with components of the metabolic syndrome, type 2 diabetes mellitus and cardiovascular disease, suggesting a common mechanistic basis. An inability to metabolically handle free fatty acid overload–metabolic inflexibility–constitutes a core node for NAFLD pathogenesis, with resulting lipotoxicity, mitochondrial dysfunction and cellular stress leading to inflammation, apoptosis and fibrogenesis. These responses can lead to the histological phenotype of nonalcoholic steatohepatitis (NASH) with varying degrees of fibrosis, which can progress to cirrhosis. This perspective review describes the key cellular and molecular mechanisms of NAFLD and NASH, namely an excessive burden of carbohydrates and fatty acids that contribute to lipotoxicity resulting in hepatocellular injury and fibrogenesis. Understanding the extrahepatic dysmetabolic contributors to NASH is crucial for the development of safe, effective and durable treatment approaches for this increasingly common disease.
Keywords: insulin resistance, lipotoxic stress, metabolic inflexibility, mitochondrial dysfunction
Nonalcoholic fatty liver disease (NAFLD) commonly leads to chronic liver disease and is associated with morbidity and mortality, as well as components of metabolic syndrome, type 2 diabetes mellitus and cardiovascular disease. The underlying state of metabolic inflexibility with NAFLD may contribute to the development and progression to nonalcoholic steatohepatitis (NASH), which can evolve into cirrhosis and hepatocellular carcinoma. This review describes the key cellular and molecular mechanisms of NAFLD and NASH, namely an excessive burden of carbohydrates and free fatty acids that contributes to lipotoxicity resulting in hepatocellular injury and fibrogenesis.
1. INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is an increasing cause of liver disease that is associated with significant morbidity and mortality, with a global prevalence of 25%.1 Among those patients with NAFLD, the global estimate of nonalcoholic steatohepatitis (NASH), a more advanced form of liver disease, is 59% in those who had liver biopsies for further evaluation.1 NAFLD is the most common liver disease in children, with a prevalence of 13% in one retrospective review of histological data from 742 children who had an autopsy performed from 1993 to 2003; the highest rate (38%) was seen in obese children.2
Despite the significant disease burden of NASH worldwide, there are currently no approved pharmacological treatments, though multiple drug candidates are in clinical development.3, 4 Current interventions largely focus on lifestyle modification—healthy eating habits, weight loss and increased physical activity—to help manage the patient's metabolic syndrome.5, 6, 7 Owing to the increasing burden of obesity and metabolic disease, cirrhosis caused by NASH has become a leading indication for liver transplantation.8, 9, 10 This perspective review summarizes the pathogenesis of NASH in the context of obesity, type 2 diabetes mellitus (T2D), and cardiovascular disease (CVD) and provides a perspective into the shared mechanisms, largely driven by insulin resistance and underlying lipotoxic stress, contributing to the cluster of these metabolic diseases.
2. NAFLD AND NASH
2.1. Definitions
NAFLD is defined as the accumulation of excess triglyceride droplets in the liver (>5% of hepatocytes with droplets detected histologically or >5% proton density fat fraction by magnetic resonance imaging [MRI]) in people who consume little or no alcohol.7, 11 The same pathophysiology can be present in people who drink excessively; in fact, obesity is a recognized risk factor for more advanced alcoholic liver disease. Unfortunately, there are currently no tests to reliably identify the relative contributions of alcohol and metabolic disturbances to liver disease in patients who drink excessively and have metabolic disease comorbidities. NASH is a subset of NAFLD characterized by biopsy evidence of hepatocellular injury and death, inflammation and varying degrees of fibrosis.12, 13, 14 Nonalcoholic fatty liver (NAFL) is the term currently used for the subset of NAFLD that has minimal or no evidence of inflammation or cellular injury in the setting of steatosis.7, 11 Not all patients with NAFL progress to NASH, and considerable efforts have been undertaken to better understand disease progression and pathogenesis.
2.2. Pathogenesis
High‐calorie diets, excessive consumption of sucrose or fructose and sedentary lifestyle have been linked to the development of NAFLD, its progression to NASH and the presence or development of other components of the metabolic syndrome.15, 16, 17 Genetics also plays a key role across all aspects of NAFLD pathogenesis, as it does in metabolic disease,18, 19 with genetic heterogeneity involved in eating behaviours, modulating energy balance, regulating lipotoxic stress, controlling the inflammatory and regenerative responses to that metabolic stress and regulating extracellular matrix production and turnover. The major genes that have been identified with NAFLD pathogenesis are briefly summarized:
Patatin‐like phospholipase domain‐containing protein 3 (PNPLA3, also called adiponutrin): an enzyme found in hepatocytes and adipocytes that plays a role in the remodelling of triglyceride (TG) lipid droplets in the liver;20, 21, 22, 23 loss of function of PNPLA3 is associated with higher serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels and increased liver steatosis and fibrosis.24 Increased expression of the I148M variant of PNPLA3 leads to impaired proteasomal degradation of PNPLA3, and the accumulated PNPLA3 indirectly impedes lipolysis.23, 25
Trans‐membrane 6 super family 2 (TM6SF2): involved in very low‐density lipoprotein (VLDL) production; loss of function of TM6SF2 is associated with reduced secretion of TG‐rich lipoproteins and increased cellular TG concentration and lipid droplet content;20 a TM6SF2 variant has been shown to be associated with steatosis but not fibrosis.24
17‐beta hydroxysteroid dehydrogenase‐13 (HSD17B13): has retinol dehydrogenase activity and regulates retinoic acid signalling with its enzymatic activity dependent on lipid droplet targeting and cofactor binding;26 loss of function polymorphisms in HSD17B13 result in an unstable and truncated protein with reduced enzymatic activity, which was found to lower the risk of alcoholic liver disease by 53% and of nonalcoholic liver disease by 30%, including a reduced risk of NASH associated with the PNPLA3 I148M allele.27
Membrane bound O‐acyltransferase domain‐containing 7 (MBOAT7): involved in the phospholipid remodelling pathway; loss of function of MBOAT7 is associated with a decrease in phosphatidylinositol‐containing arachidonic acid, which increases liver fibrosis.20, 24
Glucokinase regulatory protein (GCKR): controls de novo lipogenesis by regulating glucokinase function, which controls the flux of glucose into hepatocytes21; loss of function of GCKR protein leads to greater TG accumulation in the liver.
2.3. Disease progression
The hepatocellular inflammation and injury in NASH often promote fibrogenesis, and the resulting accumulation of fibrotic tissue can evolve to cirrhosis. Patients with NAFLD with advanced fibrosis have increased mortality within 8 years compared to those without NAFLD (35% vs 5.5%)28 as fibrosis stage is a key predictor of mortality and time to development of severe liver disease.29, 30, 31 Cirrhosis associated with NASH is commonly unrecognized clinically and can be associated with hepatocellular carcinoma.32 There is increasing recognition of hepatocellular carcinoma developing in patients with NASH who have not progressed to cirrhosis.33
2.4. Diagnosis and prognostic markers
Although liver biopsy remains the gold standard for diagnosing NASH, liver biopsy is associated with significant risks including bleeding, infection, pain and even death. Additionally, sampling differences may occur because the biopsy uses a small sliver of tissue (at best 1/50 000th of the liver).34 Thus, several noninvasive disease monitoring techniques are being utilized and possibly qualified in clinical trials with the expectation that one (or more likely, a combination) of these may eventually replace liver biopsy. These include blood markers to assess fibrosis (eg fibrosis‐4 [FIB‐4], NAFLD fibrosis score [NFS], AST to platelet ratio index [APRI], Enhanced Liver Fibrosis [ELF] score, ProC3 and composite panels); imaging techniques to reliably quantify steatosis and possibly fibroinflammation (MRI‐PDFF, MRI‐corrected T1 and transient elastography); and quantitative functional assessments (methacetin breath test and cholate clearance).35 Liver fibrosis is currently considered the strongest predictor for liver‐related complications and disease‐specific mortality,29, 36 and thus, assessment of fibrosis has become an important end‐point in treatment trials for NASH. Hepatic venous pressure gradient, Model for End‐Stage Liver Disease (MELD) score and albumin levels independently predict clinical decompensation in patients with compensated cirrhosis.37
3. NAFLD IS PART OF A METABOLIC DISEASE CLUSTER
The metabolic basis of NAFLD is supported by its strong links to obesity, T2D, insulin resistance, hyperlipidemia and CVD, all components of the “metabolic” cluster.38 A recent analysis found that patients with NAFLD have 1.6‐2.6 times greater relative risk of developing first, second and third metabolic comorbidities (eg T2D, hypertension or dyslipidemia).39 Patients with NAFLD have a shorter life expectancy than their healthy counterparts (79 vs 83 years for men; 82 vs 86 years for women) and a higher risk of developing one or more metabolic comorbidities after age 50 (76% vs 55% for men; 75% vs 53% for women).39
3.1. NAFLD is a systemic disease with heterogenous clinical presentation
NAFLD pathogenesis is heterogeneous; genetic, epigenetic and dietary contributions play a role to differing degrees in individual patients as they do in patients with other manifestations of metabolic disease.19 NAFLD has been shown to develop in those who are not obese,40 with a prevalence ranging from 4.2% to 27.4%.41 Some patients with risk factors for NASH have little or no liver disease, whereas some patients, such as nonobese patients without T2D, present with NASH and progress to cirrhosis. This disparity suggests that patients vary in terms of their susceptibility to developing NASH, with most patients existing between these two ends of the spectrum with respect to the relative roles of the dysmetabolic state and the genetic predisposition determining their phenotype and rate of progression. Thus, NAFLD development and progression involves a complex interplay between metabolic, genetic and other disease‐modifying (eg iron, gut microbiome) factors that potentially act in concert, further highlighting the heterogeneity of the disease spectrum (Figure 1).
Figure 1.
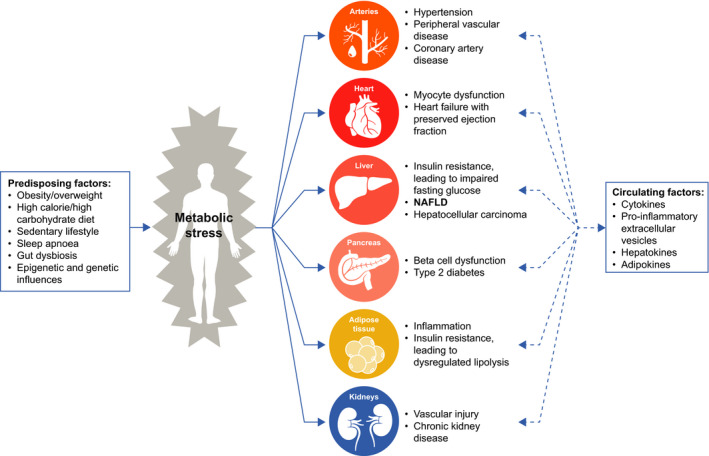
NAFLD is part of a systemic disease with strong associations with components of the metabolic syndrome. Multifactorial metabolic stress has direct effects on multiple tissues and cell types, which then release cytokines, adipokines, hepatokines and pro‐inflammatory extracellular vesicles. These circulating mediators can amplify or reduce the pathologic changes in various target tissues to create a feed‐forward mechanism of disease progression in the case of amplification. NAFLD, nonalcoholic fatty liver disease
There is also substantial heterogeneity in the metabolic phenotype of patients with NAFLD as demonstrated by body composition data from the UK Biobank (UKBB).42 Whole‐body MRI data from nearly 10 000 individuals in the UKBB were analysed for body composition, showing that in both patients with T2D and those with CVD, liver fat was increased (defined in the UKBB population as proton density fat fraction >5%). However, body composition profiles were distinct from each other in that patients with T2D tended to have greater propensity for liver fat accumulation, whereas patients with CVD had more propensity for visceral fat accumulation.42
As shown in Figure 1, there is a complex relationship among the manifestations of metabolic stress in various organs and tissues, which can potentially amplify or dampen the response to the stress in other organs. The liver secretes multiple signalling factors in response to metabolic stress, as does adipose tissue.43, 44, 45, 46 Lipid accumulation in the form of TG is thought to be a sensitive index or measure of a tissue's exposure to fatty acids; this exposure to fatty acids and their non‐TG derivatives leads to cellular dysfunction and cell death in multiple tissues.47 Accumulation of fat in the liver is a sensitive marker of whole‐body metabolic stress and may appear before abnormalities are identified in other organs.48, 49 The temporal sequence of the liver being affected first has been sometimes interpreted to imply that fat in the liver is then the cause of the other abnormalities, but the temporal sequence does not prove causality.50
3.2. Relationship between NAFLD and insulin resistance, T2D and metabolic syndrome
3.2.1. NAFLD and insulin resistance
NAFLD is associated with insulin resistance in liver, muscle and adipose tissue, with hyperinsulinemia demonstrated even in nonobese individuals with normal glucose tolerance.51, 52, 53 Therefore, it is not surprising that the homeostasis model of insulin resistance index (HOMA‐IR) has been shown to be independently correlated with steatosis, ballooning and advanced fibrosis in patients with NAFLD.54 In a study of 154 obese patients divided into four groups (control with no T2D or NAFLD; T2D without NAFLD; T2D with isolated steatosis; and T2D with NASH), insulin secretion and resistance were measured via an oral glucose tolerance test and euglycaemic‐hyperinsulinaemic clamp with glucose turnover measurements, respectively. Only those individuals with NAFLD or NASH (despite underlying T2D) had both hepatic and adipose tissue insulin resistance (increased endogenous glucose production and free fatty acid [FFA] release, respectively) even during hyperinsulinaemic conditions.55
3.2.2. NAFLD and T2D
A recent meta‐analysis estimated that the global prevalence of NAFLD among patients with T2D is 55.5%, and the prevalence of NASH among patients with T2D is 37.3%.56 Regardless of the noninvasive technique used to diagnose NAFLD (plasma ALT, 1H‐MRS, computed tomography, ultrasonography and controlled attenuation parameter) or fibrosis (FibroTest, NFS and vibration controlled transient elastography), the prevalence of NAFLD and advanced fibrosis has been found to be consistently higher in those with T2D compared with the general population.57, 58
The link between diabetes and liver‐related complications is complex and multifaceted. Here, we briefly mention a few of the pathophysiological mechanisms. Firstly, insulin resistance is proposed to be a mechanism for liver damage in NAFLD.59 A study of nondiabetic patients with NAFLD found that increased peripheral insulin resistance was strongly associated with liver fibrosis (but not steatosis or obesity), suggesting that insulin resistance is the key mechanism, over and above its phenotypic expression (diabetes).59 Secondly, increased adiposity observed in T2D is associated with adipocyte insulin resistance and dysfunction. The resulting effect is an increase in FFAs and thereby excess lipid uptake by the liver, eventually leading to progression to NASH and cirrhosis.60 Glucotoxicity, which refers to alterations caused by chronically elevated glucose concentrations present in T2D, is closely linked to lipotoxicity and therefore plays a role in NASH disease progression.60 Finally, it is well established that amino acids play an important role in NASH pathogenesis and that these are impacted in T2D.61 For example, it has been observed that branched chain amino acid (BCAA) metabolism is reduced in patients with NAFLD.61 BCAAs are important regulators of mTOR signalling and may promote insulin resistance and glucose dysregulation through activation of this pathway.62, 63
The presence of NAFLD is also associated with the subsequent development of new‐onset T2D. A study in 13,218 South Korean nondiabetic individuals who were followed over 5 years as a function of their liver fat status (as measured by ultrasound) showed the following: (a) development of new fatty liver was associated with a 2.5‐fold increased risk of incident diabetes; (b) in those individuals in whom severity of fatty liver worsened from mild to moderate/severe over 5 years, there was a 6‐fold increase in new‐onset diabetes; and (c) improvement or resolution of NAFLD was associated with a reduction in T2D incidence similar to that in individuals without steatosis.64 The latter finding was also replicated in a separate cohort.65 Although it is tempting to conclude that NAFLD contributes mechanistically to the development of T2D via altered hepatokines and cytokines or other signalling mechanisms, it is more likely that fat accumulation in the liver is a sensitive barometer of the metabolic abnormalities that also lead to T2D.49, 66, 67
3.2.3. NAFLD, obesity and dyslipidemia
NAFLD prevalence was estimated at 80%‐90% in obese adults and up to 90% in patients with hyperlipidemia.68 The role of central adiposity seems crucial in NAFLD as visceral fat is an important source of TGs leading to steatosis.38, 69, 70 Among patients with T2D and NAFLD, body mass index (BMI), anthropometric measures (waist circumference, waist‐to‐hip ratio) and insulin were common factors in the model for predicting both NASH and advanced fibrosis.71 Moreover, in patients with NASH, weight loss induced by lifestyle changes72 or bariatric surgery73 is associated with histological improvements in NAFLD activity score, NASH resolution and regression of fibrosis.
Taken together, the emerging high‐quality evidence suggests that the interaction between metabolic syndrome components and NAFLD is complex and that both NAFLD and T2D/insulin resistance could be effect modifiers of each other. These findings strongly support the notion that patients with T2D, particularly those with obesity and insulin resistance, should be routinely screened for NAFLD/NASH, which currently remains largely undiagnosed.57
3.3. Relationship between NAFLD and CVD
A growing body of evidence suggests that the presence of NAFLD is associated with a greater risk of CVD and that the presence of CVD dictates outcomes in patients with NAFLD more frequently and to a greater extent than does the progression of liver disease.74 NAFLD is characterized by markers of subclinical CVD,75 higher odds of fatal and/or nonfatal CVD events,76 atherogenic dyslipidemia including elevation of TG‐enriched VLDLs, increased small‐dense LDL and low/dysfunctional HDL.77 The association of NAFLD with subclinical atherosclerosis and CVD events is independent of other risk factors such as obesity and metabolic syndrome.39, 78
Taken together, the findings from multiple studies suggest that on the one hand, individuals with fatty livers are at greater risk of incident hypertension and T2D. On the other hand, individuals with hypertension, hypertriglyceridemia, impaired fasting glucose or T2D have a greater risk of incident fatty liver compared with individuals without those conditions. Thus, T2D, metabolic syndrome, CVD and NAFLD might impact the course of each disease independently, squarely placing NAFLD as a metabolic disease, suggesting a complex disease continuum with common mechanistic underpinnings (Figure 1). Consequently, a better understanding of the pathophysiological links between NAFLD, T2D and CVD could provide insight into the most effective interventional nodes to manage these inter‐related diseases.
4. METABOLIC INFLEXIBILITY—A POTENTIAL CONTRIBUTOR TO NAFLD PATHOGENESIS AND DISEASE PROGRESSION
Faced with an excess of metabolic energy substrates, the body must decide where and how to metabolize or store these substrates. Energy homeostasis is achieved when there is a balance between energy input (calories consumed), energy output (calories burned) and energy stored (primarily in adipose tissue). Maintaining this balance requires the body to have the ability to adequately handle fuels and use appropriate substrates at the appropriate times (eg oxidize fat during periods of starvation), a concept often referred to as metabolic flexibility (Figure 2A).79 When the system is unable to handle fuels and substrates appropriately, the balance tips towards excess fuel storage with or without excess intake, referred to as metabolic inflexibility (Figure 2B), eventually leading to systemic lipotoxic cell stress (see below).
Figure 2.
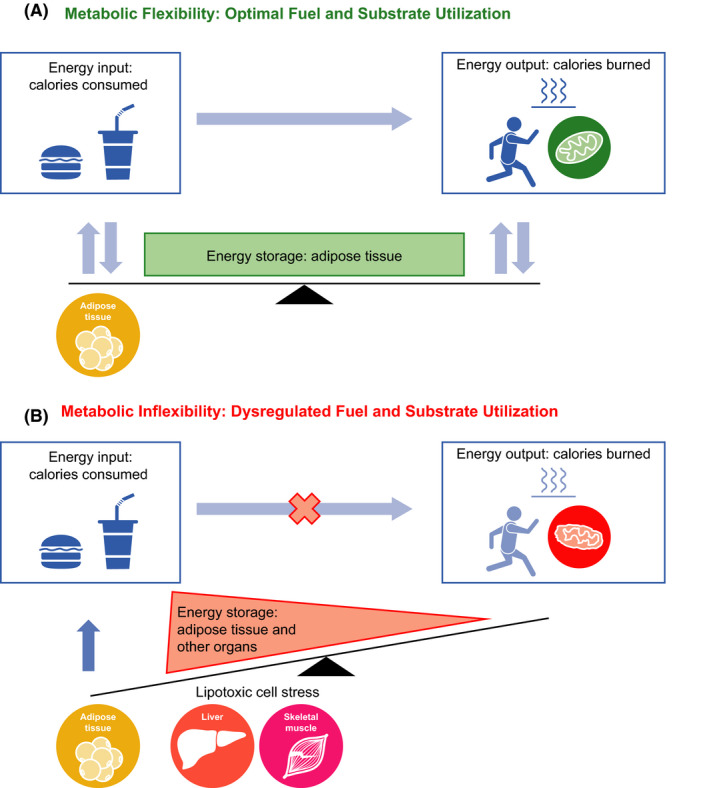
Metabolic flexibility requires the body to have the ability to adequately handle fuels and use appropriate substrates at the appropriate times (A). Inability to maintain homeostatic control to handle fuel needs and appropriately use substrates tips the energy balance scale towards higher intake and storage, ultimately leading to lipotoxic cell stress (B)
NAFLD/NASH is typically associated with higher plasma TGs, FFAs and insulin, reflecting the inability to adequately handle fuel substrates. Thus, it has emerged that NAFLD/NASH is typically associated with dysregulation of energy homeostasis, likely as a result of metabolic inflexibility. A consequence of metabolic inflexibility is dysregulated glucose and lipid metabolism resulting in insulin resistance and dyslipidemia, which are the hallmarks of T2D and metabolic syndrome. Insulin resistance and dyslipidemia, in turn, may contribute to systemic lipotoxicity and mitochondrial dysfunction.80 Mitochondrial perturbations form a core mechanistic node that can lead to inflammation and fibrosis via endoplasmic reticulum (ER) stress and, together with potential underlying genetic and other disease‐modifying factors, eventually contribute to the development and progression of NAFL to NASH. These mechanisms are further described below.
4.1. Hepatic substrate load and mechanisms of fat accumulation in liver
NAFLD is a systemic disease as illustrated by the fact that multiple organ systems (eg adipose, skeletal muscles, gut, liver) are involved in its pathogenesis (Figure 1). In healthy individuals consuming a healthy balanced diet, postprandial glycemia triggers insulin release by the pancreas, which results in increased glucose oxidation in muscles and storage of glucose as muscle glycogen (glucose clearance), whereas FFAs are esterified into TGs and primarily stored in adipose tissue (Figure 3). In times of caloric need, such as between meals and during exercise, TGs in lipid droplets are hydrolysed back to FFAs, which are used in various tissues for energy (via fatty acid β‐oxidation in the mitochondria), and glycogen is broken down into glucose or glucose is generated in the liver via gluconeogenesis, which is used by muscles and the brain (glucose oxidation) as a source of energy to generate adenosine triphosphate.
Figure 3.
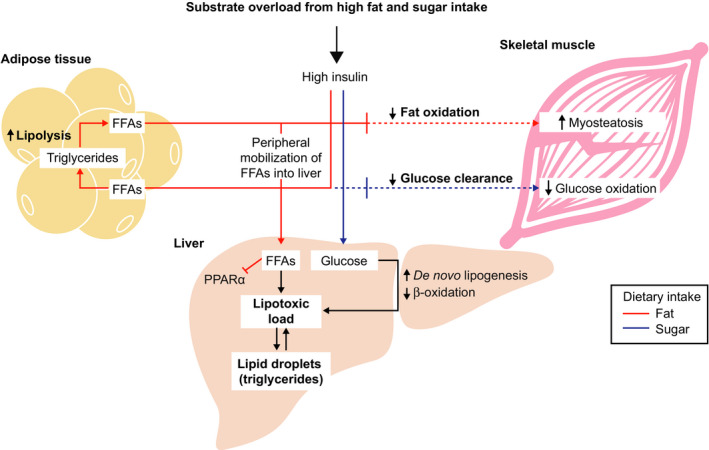
Metabolic inflexibility contributes to dysregulated glucose and lipid metabolism in nonalcoholic fatty liver disease. Chronically high sugar and fat intake in the context of obesity and insulin resistance results in a multiorgan dysregulation, resulting in an inability to appropriately dispose of those fuels. Lipolysis is increased in adipose, fat and glucose oxidation is reduced in muscle, and de novo lipogenesis is increased with a concomitant decrease in fat oxidation in the liver. These disruptions ultimately culminate in multiorgan metabolic stress and inflexibility to adapt to a nutrient overload state. Such a state can subsequently impair signalling through canonical fuel‐sensing master regulators such as PPARα and several others. FFA, free fatty acid; PPARα, peroxisome proliferator‐activated receptor α
4.1.1. Insulin resistance
Chronically high sugar and fat intake in the context of obesity and insulin resistance results in: (a) the inability to store FFAs in the adipose tissue combined with an increase in lipolysis of stored TGs into FFAs; (b) a reduction in fat oxidation by the muscle (leading to accumulation of fat, myosteatosis) and decreased muscle glucose oxidation; and (c) lipotoxic stress induced by increased delivery of FFA to the liver combined with increased conversion of carbohydrates into fat (de novo lipogenesis) (Figure 3). Peripheral mobilization of FFAs from adipocytes and their esterification to TG (“old” fat) within hepatocytes inhibits peroxisome proliferator‐activated receptor‐α (PPARα) signalling81 perpetuating the increased intrahepatic FFA abundance. Other key signalling pathways mediated by JNK,82 TLR483 and novel protein kinase C isoform PKCε84 exacerbate hepatic insulin resistance, increase FFA, leading to more TG accumulation and formation of toxic lipid species such as diacylglycerols, lysophospholipids and ceramides85, 86, 87 to cause cellular stress (see below).
Cumulatively, these processes contribute to high circulating insulin levels and FFA and ectopic fat deposition (ie outside the adipose tissue such as in liver and muscle). The high insulin, FFA and lipotoxic stress serve as central triggers for the development of NAFLD. These factors of insulin resistance and lipotoxicity are further accentuated in the context of T2D, metabolic syndrome and other contributing factors (dysbiosis, epigenetic and genetic). One consequence of chronic insulin resistance and lipotoxicity is an impaired repair response of the liver, further contributing to progressive scarring.87, 88, 89
4.1.2. Lipotoxicity
Circulating FFAs are mostly derived from adipocyte TG lipolysis and are transported to the liver where they typically have three major fates: (a) oxidation for energy production or ketone body synthesis in the mitochondria, (b) esterification to TGs and combination of the TGs with apolipoprotein B for secretion as very low‐density lipoprotein particles or (c) esterification to TGs for storage in lipid droplets. The last point suggests that liver steatosis observed in NAFLD could be considered an epiphenomenon of an excessive supply of FFA attributed to the liver's attempt to store FFA in a less toxic form, that is, in TG lipid droplets.47, 90, 91 Accumulation of liver fat is strongly associated with adipose tissue insulin resistance, further supporting the concept of adipose tissue‐derived fatty acids as a driver of hepatic lipotoxicity with the accumulation of TG serving as a marker for these processes.48, 49
Several studies have systematically profiled patients across the NAFLD spectrum and have revealed distinct lipidomic signatures in the blood.92, 93, 94, 95, 96 Compared with control participants, patients with NAFLD/NASH show more TG‐rich species and accumulate lipid metabolites (eg diacylglycerol and ceramides, sphingomyelin), which constitute a highly toxic mixture to hepatocytes and, consequently, are responsible for inflammation (see below) and hepatocellular damage.87, 97, 98, 99 Comprehensive lipidomic analysis on human biopsy samples from healthy, NAFL and NASH livers revealed decreased activity of fatty acid desaturase‐1 as a key bottleneck resulting in the accumulation of toxic lipids during NASH progression.93 Differences in the lipidomic profile appear to be dependent on the patient's BMI, suggesting that the NAFLD pathogenesis mechanism may be different depending on an individual's level of obesity.92
Taken together, these findings underscore the notion that the consequences of a dysregulated lipid metabolism and resultant lipotoxicity, not the amount of fat in the liver, lead to inflammation and subsequent fibrosis.100 Thus, it follows that interventional approaches to treat NASH should account for the significant complexity of the lipotoxic milieu and the consequent fibroinflammatory changes, rather than acting only to lower liver fat.101
4.2. Consequences of metabolic overload on mitochondrial function
Emerging evidence indicates that the pathogenesis and progression of NAFLD is multifactorial and involves the effects of several fundamental biochemical and immunomodulatory processes, a perspective which differs from the commonly ascribed sequential two‐hit hypothesis.87, 88, 101, 102 Insulin resistance and lipotoxicity in critical organs such as the liver, cardiac myocytes and skeletal muscle develop when the adaptive mechanisms that mitigate the deleterious effects of excess FFA are overwhelmed, which initiates a cascade of lipotoxicity, ER stress, oxidative stress, autophagy and mitochondrial dysfunction.103 In the liver, these processes trigger necrotic and apoptotic cell death pathways, immune‐mediated hepatocellular injury and stellate cell activation and fibrogenesis, initially as an adaptive repair response to the injury and eventually leading to extracellular matrix deposition that exceeds turnover (fibrosis) (Figure 4).98 One potential unifying mechanism that may integrate the multifactorial etiopathogenesis of NAFLD and its progression to NASH in some patients could be mitochondrial dysfunction.104, 105
Figure 4.
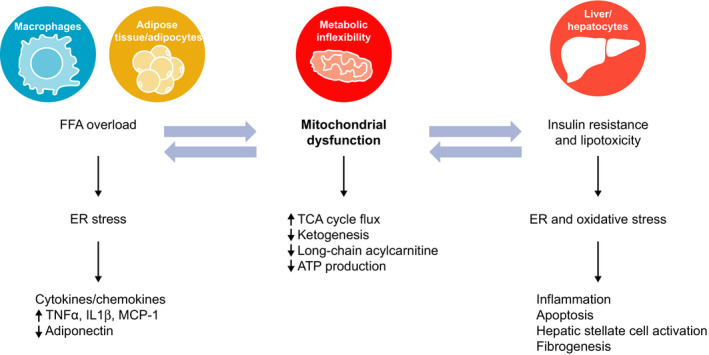
Insulin resistance and lipotoxicity within the liver are associated with mitochondrial dysfunction, oxidative stress/reactive oxygen species production and ER stress in multiple tissues. These processes contribute to hepatocellular injury, release of inflammatory cytokines, apoptosis and liver fibrogenesis that can progress to cirrhosis. ER, endoplasmic reticulum; FFA, free fatty acid; IL‐1β, interleukin‐1 β; MCP‐1, monocyte chemoattractant protein‐1; TCA, tricarboxylic acid; TNFα, tumour necrosis factor α
4.2.1. Disruption of the TCA cycle
Mitochondrial dysfunction has been noted as the earliest manifestation of the NAFL to NASH transition. While mitochondria in NAFL patients maintain a normal morphology of well‐defined cristae, mitochondria in NASH patients are swollen and rounded, with a loss of cristae and presence of multilamellar membranes.52 Early structural changes within the mitochondria have been associated with functional defects as well. A core pathway impacted within mitochondria is the tricarboxylic acid (TCA) cycle, which is at the crossroads of substrate oxidation, respiration and free radical generation. A 50% increase in mitochondrial anaplerosis (the nonoxidative flux of intermediates into the TCA cycle) was observed in those with high intrahepatic TG (defined as liver fat content >6%) compared with the low (≤6%) liver fat group, indicating increased TCA cycle flux through the combined pathways of mitochondrial pyruvate carboxylase and phosphoenolpyruvate carboxykinase (ie increased rate of gluconeogenesis) in patients with NAFLD.106 Interestingly, although oxidative metabolism in the mitochondrial TCA cycle was not impaired, ketone production assessed by tracer dilution of β‐hydroxybutyrate failed to correspondingly increase in patients with NAFLD, indicating that excess acetyl coenzyme A was selectively partitioned to oxidation in the TCA cycle rather than ketogenesis.106
4.2.2. Ketogenic insufficiency leads to abnormal hepatic glucose and lipid metabolism
In the setting of high‐fat feeding, knocking out the rate‐limiting enzyme of ketogenesis, 3‐hydroxy‐3‐methylglutaryl‐coenzyme A synthase, resulted in extensive hepatocyte injury and inflammation, dysglycemia, deranged concentration of hepatic TCA cycle intermediates and impaired hepatic gluconeogenesis due to sequestration of free coenzyme A in mice.107 Hyperinsulinemia itself due to underlying insulin resistance and impaired insulin clearance further suppresses hepatic ketogenesis, impairing hepatic TCA cycle flux and increasing hepatic gluconeogenesis and lipogenesis.107, 108 PPARα deficiency was also shown to decrease HMG‐coenzyme A synthase with an inability to augment ketone body synthesis in the face of prolonged fasting in mice.109 Thus, hepatic maladaptation to ketogenic insufficiency may be a key mechanism underlying NAFLD pathogenesis as ketogenesis plays a significant role in disposing as much as two‐thirds of the fat entering the liver.110
Impairment of ketogenesis and consequently the dysregulated TCA cycle appears to be mediated by BCAA metabolism. BCAAs are essential to mediate efficient channelling of carbon substrates for oxidation through the mitochondrial TCA cycle. In patients with NAFLD undergoing a hyperinsulinaemic‐euglycaemic clamp, high levels of BCAAs and, in particular, a strong positive correlation of insulin‐stimulated levels of plasma leucine and plasma isovaleryl carnitine were observed.111 As isovaleryl carnitine is a degradation product of leucine, this positive correlation suggests that attenuation of complete oxidative BCAA catabolism is a potential mechanism that contributes to elevated plasma BCAA in patients with NAFLD. Further dissection of the biochemical pathways revealed a co‐ordinated inability to dampen TCA cycle flux, induce long‐chain acylcarnitines (which are fuels for mitochondrial fat oxidation and the TCA cycle) and upregulate ketogenesis in response to an acute 4‐hour BCAA challenge in a diet‐induced (high fructose, high trans‐fat) mouse model of NAFLD.111 Together, these data indicate inflexibility within the core bioenergetic nodes impairs the molecular cross‐talk between BCAA and the hepatic TCA cycle, consequently contributing to the mitochondrial dysfunction in NAFLD.
4.3. Consequences of metabolic overload on cellular stress and NASH progression
4.3.1. Effects of mitochondrial dysfunction on oxidative stress
Mitochondrial dysfunction decreases the production of adiponectin by adipocytes.112 Because adiponectin can induce hepatic β‐oxidation, reduced adiponectin secretion contributes to the impaired FFA disposal within hepatocytes, worsening lipotoxicity, exacerbating insulin resistance and inducing oxidative stress.113, 114 TCA cycle dysregulation also leads to the activation of cytochrome P450 2E1 that induces lipid peroxidation, leading to the production of reactive oxygen species (ROS) and creating an environment of oxidative stress.115, 116 Disease‐modifying factors, such as hepatic iron content, can also exacerbate hepatic oxidative stress.117 Oxidative stress results in oxidation of various biomolecules thereby creating a cycle that further degrades mitochondrial structure and function, promotes hepatocellular damage and insulin resistance and triggers inflammation. Mitochondrial damage and dysfunction results in a reduction in respiratory chain efficiency, decrease in adenosine triphosphate production and accumulation of protein oxidation products.118 These processes, combined with excess cholesterol, contribute to the ER stress in hepatocytes (Figure 4).
4.3.2. Effects of ER stress on apoptosis, inflammation and fibrogenesis
In hepatocytes, the ER constitutes a pivotal node controlling inflammation, cell death and lipid metabolism. Excess cholesterol from dietary intake and hepatic cholesterol synthesis can trigger ER stress not only in hepatocytes, but also in macrophages and adipocytes.119, 120 Consequences of systemic chronic ER stress include the following: (a) disruption of hepatic lipid and cholesterol synthesis perpetuating a cycle that further aggravates mitochondrial dysfunction and ER homeostasis121; (b) induction of insulin resistance that drives the cell death response to lipotoxic stress believed to be a major contributor for NASH progression122, 123, 124; and (c) release of pro‐inflammatory cytokines and chemokines, including tumour necrosis factor α, interleukin (IL)‐1β, IL‐6, IL‐8 and monocyte chemoattractant protein‐1 (Figure 4).125 Together, these processes contribute to hepatocellular injury and death, promoting liver fibrogenesis and proliferation of hepatocyte progenitors to compensate for hepatocyte loss. The latter is considered a hallmark of the progression from NAFL to NASH and is also believed to contribute to the development of cirrhosis and hepatocellular carcinoma.88, 126
ER stress induces apoptosis in hepatocytes, another characteristic feature of NASH progression.127, 128, 129 Apoptosis induced by ER stress within the arterial wall (smooth muscle cells, endothelial cells, macrophages), combined with hypercholesterolaemia, induces atherosclerosis progression and CVD,120 the most common cause of death in patients with NASH.
5. CONCLUSION
NAFLD is a systemic and heterogenous disease with variable presentations and disease courses and is integrally linked to T2D and CVD. Progressive disease is determined by the complex interplay of a number of factors including environmental (diet, exercise), microbiome and epigenetic and genetic factors transposed upon multiple pathways that drive metabolic, inflammatory and fibrotic changes. Heterogenous disease presentation provides an opportunity to identify patient subsets via body composition profiling, other methods of phenotyping and genotyping. The identification of patient subsets should allow for greater understanding of disease processes, opening the possibility to tailor therapies for specific patient and/or disease drivers.
The inability to biochemically handle FFA overload–metabolic inflexibility–constitutes a core node for NAFLD pathogenesis and its sequelae (Figure 5). The resulting lipotoxicity, mitochondrial dysfunction and cellular stress in multiple organs and tissues lead to inflammation, apoptosis, fibrogenesis and, ultimately, progression from NAFL to NASH. Interventions to re‐establish or reprogramme metabolic flexibility may be a beneficial treatment approach to NAFLD, as well as T2D and CVD.
Figure 5.
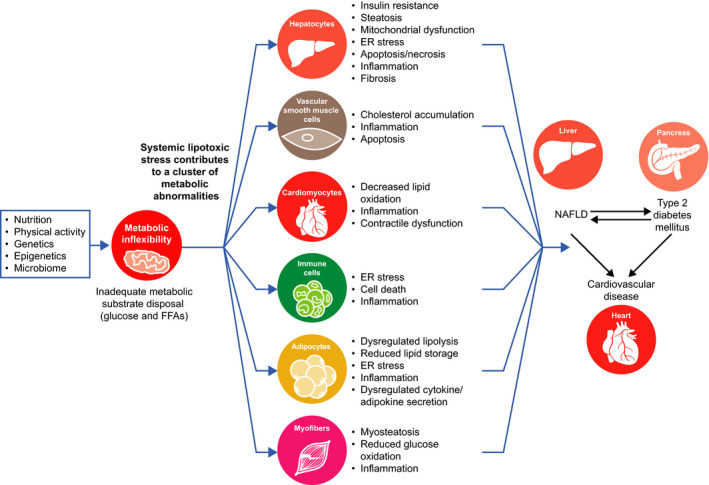
Metabolic inflexibility may constitute a core node for NAFLD pathogenesis and its sequelae. Inability to handle fuel substrates results in insulin resistance and an increased flux of FFA and other toxic lipids that lead to lipotoxicity. Consequences of systemic lipotoxicity include dysregulation of fundamental biological pathways including mitochondrial dysfunction, ER stress, inflammation and apoptosis, leading to a cluster of related metabolic diseases. NAFLD has been associated with increased incident T2D by increasing hepatic gluconeogenesis and exacerbating lipid metabolism and release of pro‐inflammatory cytokines with diabetogenic properties. Similarly, T2D may exacerbate progression of NAFLD from NAFL to NASH and cirrhosis. CVD remains the most common cause of death for both NAFLD and T2D. CVD, cardiovascular disease; ER, endoplasmic reticulum; FFA, free fatty acid; NAFL, nonalcoholic fatty liver; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; T2D, type 2 diabetes mellitus
Thus, effective and durable treatments will likely require addressing the core metabolic drivers of NAFLD (insulin resistance, inability to process excess FFAs, mitochondrial dysfunction) and the ensuing lipotoxic stress and oxidative damage, while simultaneously addressing other more downstream features such as cell injury and fibrosis, to restore the adaptive repair responses to chronic lipotoxicity.3, 88, 130
CONFLICT OF INTEREST
MVC is an employee of Axcella Health, Inc and owns stock options in the company. BAN‐T is a consultant for Allergan, Allysta, Arrowhead, ARTham, Axcella, Blade, Boehringer Ingelheim, BMS, Coherus, Consynance, Durect, Enanta, Fortress, Gelesis, Gilead, HistoIndex, Intercept, Lipocine, Madrigal, Medimmune, Merck, Metacrine, Mundipharma, NGM, pH‐Pharma, Prometheus and Siemens. He has received institutional research grants from Allergan, BMS, Cirius, Cymabay, Enanta, Galectin, Genfit, Gilead, Intercept, Madrigal, NGM and Prometheus.
AUTHORS' CONTRIBUTIONS
MVC reviewed literature, created content, drafted manuscript and created figures. BAN‐T revised manuscript and figures for important intellectual content and identified additional supporting references.
ETHICAL APPROVAL
Not applicable to this review article.
ACKNOWLEDGEMENTS
Editing assistance was provided by Caryne Craige, PhD, of Fishawack Communications Inc; funding for this assistance was provided by Axcella Health, Inc
Chakravarthy MV, Neuschwander‐Tetri BA. The metabolic basis of nonalcoholic steatohepatitis. Endocrinol Diab Metab. 2020;3:e00112 10.1002/edm2.112
DATA AVAILABILITY STATEMENT
Not applicable to this review article; all information is available in the literature cited.
REFERENCES
- 1. Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73‐84. [DOI] [PubMed] [Google Scholar]
- 2. Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118(4):1388‐1393. [DOI] [PubMed] [Google Scholar]
- 3. Neuschwander‐Tetri BA. Non‐alcoholic fatty liver disease. BMC Med. 2017;15(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Konerman MA, Jones JC, Harrison SA. Pharmacotherapy for NASH: current and emerging. J Hepatol. 2018;68(2):362‐375. [DOI] [PubMed] [Google Scholar]
- 5. Ahmed A, Wong RJ, Harrison SA. Nonalcoholic fatty liver disease review: diagnosis, treatment, and outcomes. Clin Gastroenterol Hepatol. 2015;13(12):2062‐2070. [DOI] [PubMed] [Google Scholar]
- 6. Romero‐Gomez M, Zelber‐Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67(4):829‐846. [DOI] [PubMed] [Google Scholar]
- 7. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328‐357. [DOI] [PubMed] [Google Scholar]
- 8. Goldberg D, Ditah IC, Saeian K, et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152(5):1090‐1099.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noureddin M, Vipani A, Bresee C, et al. NASH Leading cause of liver transplant in women: updated analysis of indications for liver transplant and ethnic and gender variances. Am J Gastroenterol. 2018;113(11):1649‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wong RJ, Aguilar M, Cheung R, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148(3):547‐555. [DOI] [PubMed] [Google Scholar]
- 11. European Association for the Study of the Liver (EASL) , European Association for the Study of Diabetes (EASD) , European Association for the Study of Obesity (EASO) . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388‐1402. [DOI] [PubMed] [Google Scholar]
- 12. Bedossa P. Histological assessment of NAFLD. Dig Dis Sci. 2016;61(5):1348‐1355. [DOI] [PubMed] [Google Scholar]
- 13. Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7(4):195‐203. [DOI] [PubMed] [Google Scholar]
- 14. Lonardo A, Nascimbeni F, Maurantonio M, Marrazzo A, Rinaldi L, Adinolfi LE. Nonalcoholic fatty liver disease: evolving paradigms. World J Gastroenterol. 2017;23(36):6571‐6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gerber L, Otgonsuren M, Mishra A, et al. Non‐alcoholic fatty liver disease (NAFLD) is associated with low level of physical activity: a population‐based study. Aliment Pharmacol Ther. 2012;36(8):772‐781. [DOI] [PubMed] [Google Scholar]
- 16. Jensen T, Abdelmalek MF, Sullivan S, et al. Fructose and sugar: a major mediator of non‐alcoholic fatty liver disease. J Hepatol. 2018;68(5):1063‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kechagias S, Ernersson A, Dahlqvist O, et al. Fast‐food‐based hyper‐alimentation can induce rapid and profound elevation of serum alanine aminotransferase in healthy subjects. Gut. 2008;57(5):649‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Severson TJ, Besur S, Bonkovsky HL. Genetic factors that affect nonalcoholic fatty liver disease: a systematic clinical review. World J Gastroenterology. 2016;22(29):6742‐6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sookoian S, Pirola CJ. Nonalcoholic fatty liver disease and metabolic syndrome: shared genetic basis of pathogenesis. Hepatology. 2016;64(5):1417‐1420. [DOI] [PubMed] [Google Scholar]
- 20. Barbara M, Scott A, Alkhouri N. New insights into genetic predisposition and novel therapeutic targets for nonalcoholic fatty liver disease. Hepatobiliary Surg Nutr. 2018;7:372‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin Y‐C, Chang P‐F, Chang M‐H, Ni Y‐H. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am J Clin Nutr. 2014;99(4):869‐874. [DOI] [PubMed] [Google Scholar]
- 22. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH. PNPLA3, CGI‐58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology. 2019;69(6):2427‐2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krawczyk M, Rau M, Schattenberg JM, et al. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: a multicenter biopsy‐based study. J Lipid Res. 2017;58(1):247‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66(4):1111‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma Y, Belyaeva OV, Brown PM, et al. 17‐beta hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology. 2019;69(4):1504‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abul‐Husn NS, Cheng X, Li AH, et al. A protein‐truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. 2018;378(12):1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Le MH, Devaki P, Ha NB, et al. Prevalence of non‐alcoholic fatty liver disease and risk factors for advanced fibrosis and mortality in the United States. PLoS ONE. 2017;12(3):e0173499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Angulo P, Kleiner DE, Dam‐Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149(2):389‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dulai PS, Singh S, Patel J, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology. 2017;65(5):1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hagström H, Nasr P, Ekstedt M, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy‐proven NAFLD. J Hepatol. 2017;67(6):1265‐1273. [DOI] [PubMed] [Google Scholar]
- 32. Bertot LC, Jeffrey GP, Wallace M, et al. Nonalcoholic fatty liver disease‐related cirrhosis is commonly unrecognized and associated with hepatocellular carcinoma. Hepatol Commun. 2017;1(1):53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mittal S, El‐Serag HB, Sada YH, et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2016;14(1):124‐131.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ratziu V, Charlotte F, Heurtier A, et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128(7):1898‐1906. [DOI] [PubMed] [Google Scholar]
- 35. Castera L, Friedrich‐Rust M, Loomba R. Noninvasive assessment of liver disease in patients with nonalcoholic fatty liver disease. Gastroenterology. 2019;156(5):1264‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ekstedt M, Hagstrom H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology. 2015;61(5):1547‐1554. [DOI] [PubMed] [Google Scholar]
- 37. Ripoll C, Groszmann R, Garcia‐Tsao G, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133(2):481‐488. [DOI] [PubMed] [Google Scholar]
- 38. Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia. 2009;13:9‐19. [PMC free article] [PubMed] [Google Scholar]
- 39. Allen AM, Therneau TM, Larson JJ, Coward A, Somers VK, Kamath PS. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: a 20 year‐community study. Hepatology. 2018;67(5):1726‐1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sookoian S, Pirola CJ. Systematic review with meta‐analysis: the significance of histological disease severity in lean patients with nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2018;47(1):16‐25. [DOI] [PubMed] [Google Scholar]
- 41. Kim D, Kim WR. Nonobese fatty liver disease. Clin Gastroenterol Hepatol. 2017;15(4):474‐485. [DOI] [PubMed] [Google Scholar]
- 42. Linge J, Whitcher B, Borga M, Dahlqvist LO. Sub‐phenotyping metabolic disorders using body composition: an individualized, nonparametric approach utilizing large data sets. Obesity. 2019;27(7):1190‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ennequin G, Sirvent P, Whitham M. Role of exercise‐induced hepatokines in metabolic disorders. Am J Physiol Endocrinol Metab. 2019;317(1):E11‐E24. [DOI] [PubMed] [Google Scholar]
- 44. Lonardo A, Lugari S, Ballestri S, Nascimbeni F, Baldelli E, Maurantonio M. A round trip from nonalcoholic fatty liver disease to diabetes: molecular targets to the rescue? Acta Diabetol. 2019;56(4):385‐396. [DOI] [PubMed] [Google Scholar]
- 45. Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957‐969. [DOI] [PubMed] [Google Scholar]
- 46. Meex RCR, Watt MJ. Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol. 2017;13(9):509‐520. [DOI] [PubMed] [Google Scholar]
- 47. Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta. 2010;1801(3):209‐214. [DOI] [PubMed] [Google Scholar]
- 48. Bril F, Barb D, Portillo‐Sanchez P, et al. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology. 2017;65(4):1132‐1144. [DOI] [PubMed] [Google Scholar]
- 49. Rotman Y, Neuschwander‐Tetri BA. Liver fat accumulation as a barometer of insulin responsiveness again points to adipose tissue as the culprit. Hepatology. 2017;65(4):1088‐1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Farese RV Jr, Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012;15(5):570‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marchesini G, Brizi M, Morselli‐Labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107(5):450‐455. [DOI] [PubMed] [Google Scholar]
- 52. Sanyal AJ, Campbell‐Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183‐1192. [DOI] [PubMed] [Google Scholar]
- 53. Portillo‐Sanchez P, Bril F, Maximos M, et al. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J Clin Endocrinol Metab. 2015;100(6):2231‐2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ballestri S, Nascimbeni F, Romagnoli D, Lonardo A. The independent predictors of non‐alcoholic steatohepatitis and its individual histological features. Hepatol Res. 2016;46:1074‐1087. [DOI] [PubMed] [Google Scholar]
- 55. Lomonaco R, Bril F, Portillo‐Sanchez P, et al. Metabolic impact of nonalcoholic steatohepatitis in obese patients with type 2 diabetes. Diabetes Care. 2016;39(4):632‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta‐analysis. J Hepatol. 2019;71(4):793‐801. [DOI] [PubMed] [Google Scholar]
- 57. Bril F, Cusi K. Management of nonalcoholic fatty liver disease in patients with type 2 diabetes: a call to action. Diabetes Care. 2017;40(3):419‐430. [DOI] [PubMed] [Google Scholar]
- 58. Cusi K, Sanyal AJ, Zhang S, et al. Non‐alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes Metab. 2017;19(11):1630‐1634. [DOI] [PubMed] [Google Scholar]
- 59. Rosso C, Mezzabotta L, Gaggini M, et al. Peripheral insulin resistance predicts liver damage in nondiabetic subjects with nonalcoholic fatty liver disease. Hepatology. 2016;63(1):107‐116. [DOI] [PubMed] [Google Scholar]
- 60. Gastaldelli A, Cusi K. From NASH to diabetes and from diabetes to NASH: mechanisms and treatment options. JHEP Rep. 2019;1:312‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gaggini M, Carli F, Rosso C, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. 2018;67(1):145‐158. [DOI] [PubMed] [Google Scholar]
- 62. Dodd KM, Tee AR. Leucine and mTORC1: a complex relationship. Am J Physiol Endocrinol Metab. 2012;302(11):E1329‐E1342. [DOI] [PubMed] [Google Scholar]
- 63. Newgard CB, An J, Bain JR, et al. A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9(4):311‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sung KC, Wild SH, Byrne CD. Resolution of fatty liver and risk of incident diabetes. J Clin Endocrinol Metab. 2013;98(9):3637‐3643. [DOI] [PubMed] [Google Scholar]
- 65. Yamazaki H, Tsuboya T, Tsuji K, Dohke M, Maguchi H. Independent association between improvement of nonalcoholic fatty liver disease and reduced incidence of type 2 diabetes. Diabetes Care. 2015;38(9):1673‐1679. [DOI] [PubMed] [Google Scholar]
- 66. Chitturi S, Farrell GC. Fatty liver now, diabetes and heart attack later? The liver as a barometer of metabolic health. J Gastroenterol Hepatol. 2007;22(7):967‐969. [DOI] [PubMed] [Google Scholar]
- 67. Dhir G, Cusi K. Glucagon like peptide‐1 receptor agonists for the management of obesity and non‐alcoholic fatty liver disease: a novel therapeutic option. J Investig Med. 2018;66(1):7‐10. [DOI] [PubMed] [Google Scholar]
- 68. Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non‐alcoholic fatty liver disease. Dig Dis. 2010;28(1):155‐161. [DOI] [PubMed] [Google Scholar]
- 69. Gastaldelli A, Cusi K, Pettiti M, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133(2):496‐506. [DOI] [PubMed] [Google Scholar]
- 70. Guerrero R, Vega GL, Grundy SM, Browning JD. Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology. 2009;49(3):791‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bazick J, Donithan M, Neuschwander‐Tetri BA, et al. Clinical model for NASH and advanced fibrosis in adult patients with diabetes and NAFLD: guidelines for referral in NAFLD. Diabetes Care. 2015;38(7):1347‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vilar‐Gomez E, Martinez‐Perez Y, Calzadilla‐Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149(2):367‐378. [DOI] [PubMed] [Google Scholar]
- 73. Lassailly G, Caiazzo R, Buob D, et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology. 2015;149(2):379‐388. [DOI] [PubMed] [Google Scholar]
- 74. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J Hepatol. 2018;68(2):335‐352. [DOI] [PubMed] [Google Scholar]
- 75. Oni ET, Agatston AS, Blaha MJ, et al. A systematic review: burden and severity of subclinical cardiovascular disease among those with nonalcoholic fatty liver; should we care? Atherosclerosis. 2013;230(2):258‐267. [DOI] [PubMed] [Google Scholar]
- 76. Targher G, Byrne C, Lonardo A, Zoppini G, Barbui C. Non‐alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta‐analysis. J Hepatol. 2016;65:589‐600. [DOI] [PubMed] [Google Scholar]
- 77. DeFilippis AP, Blaha MJ, Martin SS, et al. Nonalcoholic fatty liver disease and serum lipoproteins: the Multi‐Ethnic Study of Atherosclerosis. Atherosclerosis. 2013;227(2):429‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Thakur ML, Sharma S, Kumar A, et al. Nonalcoholic fatty liver disease is associated with subclinical atherosclerosis independent of obesity and metabolic syndrome in Asian Indians. Atherosclerosis. 2012;223(2):507‐511. [DOI] [PubMed] [Google Scholar]
- 79. Galgani J, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab. 2008;295:E1009‐E1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gonzalez‐Franquesa A, Patti ME. Insulin resistance and mitochondrial dysfunction. Adv Exp Med Biol. 2017;982:465‐520. [DOI] [PubMed] [Google Scholar]
- 81. Chakravarthy MV, Pan Z, Zhu Y, et al. "New" hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005;1(5):309‐322. [DOI] [PubMed] [Google Scholar]
- 82. Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Holland WL, Bikman BT, Wang LP, et al. Lipid‐induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid‐induced ceramide biosynthesis in mice. J Clin Invest. 2011;121(5):1858‐1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jornayvaza FR, Shulman GI. Diacylglycerol activation of protein kinase C ε and hepatic insulin resistance. Cell Metab. 2012;15(5):574‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2011;108(39):16381‐16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Magkos F, Su X, Bradley D, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 2012;142:1444‐1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1049‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology. 2016;150(8):1769‐1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mogler C, Wieland M, Konig C, et al. Hepatic stellate cell‐expressed endosialin balances fibrogenesis and hepatocyte proliferation during liver damage. EMBO Mol Med. 2015;7(3):332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28(4):360‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yilmaz Y. Review article: is non‐alcoholic fatty liver disease a spectrum, or are steatosis and non‐alcoholic steatohepatitis distinct conditions? Aliment Pharmacol Ther. 2012;36(9):815‐823. [DOI] [PubMed] [Google Scholar]
- 92. Barr J, Caballería J, Martínez‐Arranz I, et al. Obesity‐dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J Proteome Res. 2012;11(4):2521‐2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chiappini F, Coilly A, Kadar H, et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci Rep. 2017;7:46658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Marukian S, Afeyan R, Tramontin T, et al. AXA1125, a novel composition of amino acids reprograms the multifactorial pathophysiology in NAFLD (abstract). Hepatology. 2018;68:67A. [Google Scholar]
- 95. Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46(4):1081‐1090. [DOI] [PubMed] [Google Scholar]
- 96. Loomba R, Quehenberger O, Armando A, Dennis EA. Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis. J Lipid Res. 2015;56(1):185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chaurasia B, Summers SA. Ceramides ‐ lipotoxic inducers of metabolic disorders. Trends Endocrinol Metab. 2015;26(10):538‐550. [DOI] [PubMed] [Google Scholar]
- 98. Hirsova P, Ibrahim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. 2016;57(10):1758‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kasumov T, Li L, Li M, et al. Ceramide as a mediator of non‐alcoholic fatty liver disease and associated atherosclerosis. PLoS ONE. 2015;10(5):e0126910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142(4):711‐725. [DOI] [PubMed] [Google Scholar]
- 102. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774‐788. [DOI] [PubMed] [Google Scholar]
- 103. Bozaykut P, Sahin A, Karademir B, Ozer NK. Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic steatohepatitis. Mech Ageing Dev. 2016;157:17‐29. [DOI] [PubMed] [Google Scholar]
- 104. Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6(1):1‐28. [DOI] [PubMed] [Google Scholar]
- 105. Pessayre D. Role of mitochondria in non‐alcoholic fatty liver disease. J Gastroenterol Hepatol. 2007;22(suppl 1):S20‐S27. [DOI] [PubMed] [Google Scholar]
- 106. Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14(6):804‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cotter DG, Ercal B, Huang X, et al. Ketogenesis prevents diet‐induced fatty liver injury and hyperglycemia. J Clin Invest. 2014;124(12):5175‐5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Puchalska P, Crawford PA. Multi‐dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25(2):262‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kersten S, Seydoux J, Peters JM, et al. Peroxisome proliferator–activated receptor α mediates the adaptive response to fasting. J Clin Invest. 1999;103(11):1489‐1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Williamson J, Scholz R, Browning E. Interactions between fatty acid oxidation and the citric acid cycle in perfused rat liver. J Biol Chem. 1969;244:4617‐4627. [PubMed] [Google Scholar]
- 111. Sunny NE, Kalavalapalli S, Bril F, et al. Cross‐talk between branched‐chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab. 2015;309(4):E311‐E319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bugianesi E, Pagotto U, Manini R, et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J Clin Endocrinol Metab. 2005;90(6):3498‐3504. [DOI] [PubMed] [Google Scholar]
- 113. Ghadge AA, Khaire AA, Kuvalekar AA. Adiponectin: a potential therapeutic target for metabolic syndrome. Cytokine Growth Factor Rev. 2018;39:151‐158. [DOI] [PubMed] [Google Scholar]
- 114. Polyzos SA, Toulis KA, Goulis DG, Zavos C, Kountouras J. Serum total adiponectin in nonalcoholic fatty liver disease: a systematic review and meta‐analysis. Metabolism. 2011;60(3):313‐326. [DOI] [PubMed] [Google Scholar]
- 115. Aubert J, Begriche K, Knockaert L, Robin MA, Fromenty B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin Res Hepatol Gastroenterol. 2011;35(10):630‐637. [DOI] [PubMed] [Google Scholar]
- 116. Chalasani N, Gorski JC, Asghar MS, et al. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology. 2003;37(3):544‐550. [DOI] [PubMed] [Google Scholar]
- 117. Fujita N, Miyachi H, Tanaka H, et al. Iron overload is associated with hepatic oxidative damage to DNA in nonalcoholic steatohepatitis. Cancer Epidemiol Biomarkers Prev. 2009;18(2):424‐432. [DOI] [PubMed] [Google Scholar]
- 118. Teodoro JS, Rolo AP, Duarte FV, Simões AM, Palmeira CM. Differential alterations in mitochondrial function induced by a choline‐deficient diet: understanding fatty liver disease progression. Mitochondrion. 2008;8(5–6):367‐376. [DOI] [PubMed] [Google Scholar]
- 119. Ioannou G. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab. 2016;27:84‐95. [DOI] [PubMed] [Google Scholar]
- 120. Sozen E, Ozer NK. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: An updated mini‐review. Redox Biol. 2017;12:456‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15(5):623‐634. [DOI] [PubMed] [Google Scholar]
- 122. Han J, Kaufman RJ. The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. 2016;57(8):1329‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kim JY, Garcia‐Carbonell R, Yamachika S, et al. ER stress drives lipogenesis and steatohepatitis via caspase‐2 activation of S1P. Cell. 2018;175(1):133‐45.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front Genet. 2014;5:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. 2019;76(1):99‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Richardson MM, Jonsson JR, Powell EE, et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology. 2007;133(1):80‐90. [DOI] [PubMed] [Google Scholar]
- 127. Kanda T, Matsuoka S, Yamazaki M, et al. Apoptosis and non‐alcoholic fatty liver diseases. World J Gastroenterol. 2018;24(25):2661‐2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lebeaupin C, Vallée D, Hazari Y, et al. Endoplasmic reticulum stress signalling and the pathogenesis of non‐alcoholic fatty liver disease. J Hepatol. 2018;69(4):927‐947. [DOI] [PubMed] [Google Scholar]
- 129. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Goedeke L, Perry RJ, Shulman GI. Emerging pharmacological targets for the treatment of nonalcoholic fatty liver disease, insulin resistance, and type 2 diabetes. Annu Rev Pharmacol Toxicol. 2019;59:65‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable to this review article; all information is available in the literature cited.