

Abstract
The causal role of abdominal overweight/obesity, insulin resistance and type 2 diabetes (T2D) on the risk of fatty liver disease (FLD) has robustly been proven. A consensus of experts has recently proposed the novel definition of ‘metabolic dysfunction‐associated fatty liver disease, MAFLD’ instead of ‘nonalcoholic fatty liver disease, NAFLD’, emphasizing the central role of dysmetabolism in the disease pathogenesis. Conversely, a direct and independent contribution of FLD per se on risk of developing T2D is still a controversial topic. When dealing with FLD as a potential risk factor for T2D, it is straightforward to think of hepatic insulin resistance as the most relevant underlying mechanism. Emerging evidence supports genetic determinants of FLD (eg PNPLA3, TM6SF2, MBOAT7, GCKR, HSD17B13) as determinants of insulin resistance and T2D. However, recent studies highlighted that the key molecular mechanism of dysmetabolism is not fat accumulation per se but the degree of hepatic fibrosis (excess liver fat content—lipotoxicity), leading to reduced insulin clearance, insulin resistance and T2D. A consequence of these findings is that drugs that will ameliorate liver fat accumulation and fibrosis in principle may also exert a beneficial effect on insulin resistance and risk of T2D in individuals with FLD. Finally, initial findings show that these genetic factors might be directly implicated in modulating pancreatic beta‐cell function, although future studies are needed to fully understand this relationship.
Keywords: diabetes, fatty liver disease, human genetics
Emerging evidence supports genetic determinants of fatty liver disease as determinants of insulin resistance and type 2 diabetes. The key molecular mechanism of dysmetabolism is not fat accumulation per se but the degree of hepatic fibrosis (excess liver fat content—lipotoxicity).
1. INTRODUCTION
Fatty liver disease (FLD) is defined by excessive hepatic fat accumulation mainly due to metabolic derangement and excess in alcohol intake. 1 Abdominal overweight/obesity, insulin resistance and type 2 diabetes (T2D) are among the strongest acquired risk factors for the development of FLD and its progression to advanced fibrosis, cirrhosis and hepatocellular carcinoma. 2 , 3 , 4 The causal role of abdominal overweight/obesity, insulin resistance and T2D on risk of FLD development and progression has robustly been proven. 5 The opposite, namely a direct and independent contribution of FLD per se on risk of developing T2D, is still a controversial topic.
However, it is becoming clear that the link between FLD and T2D is more complex than previously thought. Human genetic variations primarily increasing liver fat content do not have a direct effect on insulin resistance. 6 Indeed, recent evidence suggests that quality of fat, rather than quantity, is more important in causing the increase in insulin resistance. 6 , 7 Furthermore, the effect of gender in the development of FLD should not be dismissed. 8 A growing body of evidence suggests that gender and its related biological components represent strong determinants of FLD development and progression. 9 In agreement, also derangement in glucose metabolism has a sexual dysmorphism. 10 , 11 , 12 , 13 , 14 , 15 , 16 Among the unknown questions, there is also if genetic determinants of FLD interact specifically with sex. Increasing clinical evidence now suggests that FLD may precede and/or promote the development of T2D and other cardiometabolic diseases. 17 Thus, FLD appears to be a good biomarker for predicting risk of incident T2D, irrespective of established risk factors and may be also used to stratify the risk of cardiometabolic diseases and personalize prevention. When dealing with FLD as a new risk factor for T2D, it is straightforward to think of liver fat content contributing directly to hepatic insulin resistance and diabetes as the most likely mechanism. 18 However, as will be discussed in greater detail, emerging data are now challenging this notion.
Very recently, a consensus of experts has proposed to replace the ‘nonalcoholic fatty liver disease, NAFLD’ with a more appropriate term, namely ‘metabolic dysfunction‐associated fatty liver disease, MAFLD’. 19 , 20 This novel term emphasizes that derangement in hepatic lipid and glucose handling, namely metabolic dysfunction, is the key player in the pathogenesis of chronic liver disease. In particular, they propose a set of novel affirmative criteria for diagnosing MAFLD (mainly based on the presence of overweight/obesity, T2D or other metabolic syndrome traits), irrespective of other concomitant liver diseases. However, this term has not been unanimously accepted 21 and therefore, in this review we will use the term FLD.
In this review article, we will focus on the contribution of human genetics to the multifaceted and bidirectional relationship between FLD and T2D, highlighting the potential clinical use of FLD for a better risk stratification of T2D and its related chronic vascular complications (mainly cardiovascular and chronic kidney disease).
2. EPIDEMIOLOGY
2.1. FLD and increased risk of diabetes: epidemiological evidence
A body of evidence shows that FLD, as detected by imaging methods, is an early predictor for the development of incident T2D. 3 , 4 In Table 1, we included the observational studies, published in the last 5 years, investigating the association between FLD and risk of incident T2D. 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 Collectively, all these studies have consistently documented that FLD was strongly associated with an increased risk of incident T2D, independently of age, sex, adiposity measures and other potential confounding factors (Table 1). The increased risk of incident T2D ranged approximately from a 50% 30 to 3.5‐fold increase 36 in individuals with FLD, becoming even higher in sex‐stratified analyses. 35 The significant association between FLD and increased risk of incident T2D was also confirmed among FLD individuals with prediabetes. 39
Table 1.
Observational studies published from 2016 to 2020 that assessed the association between FLD (as detected by imaging or biopsy) and the risk of incident type 2 diabetes
| Author, year | Study characteristics; follow‐up length | Diagnosis of FLD | Diagnosis of diabetes | Covariate adjustment | Main findings |
|---|---|---|---|---|---|
| Chen, 2016 22 | Prospective cohort study of 6,542 (3.2% with FLD) Chinese nondiabetic subjects without known chronic liver diseases; 6 years | Ultrasound | Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5% or drug treatment |
Age, obesity, hypertriglyceridaemia, IFG |
FLD was independently associated with incident diabetes (adjusted HR 2.17, 95% CI 1.6‐3.0) |
| Li, 2017 23 | Prospective cohort study of 18,111 (31.9% with FLD) Chinese nondiabetic subjects without known chronic liver diseases; 4.6 years | Ultrasound | Fasting glucose ≥ 7.0 mmol/L, clinical history or drug treatment |
Age, sex, BMI, waist circumference, alcohol intake, smoking, exercise, family history of diabetes, fasting glucose, triglycerides, total cholesterol |
The adjusted HRs for incident diabetes compared with those without FLD were as follows: 1.88 (95% CI 1.6‐2.2) in the mild FLD group and 2.34 (95% CI 1.9‐3.0) in the moderate‐severe FLD group (P‐trend < 0.001) |
| Ma, 2017 24 | Prospective cohort study of 1,051 (17.8% with FLD) United States nondiabetic subjects without known chronic liver diseases; 6.2 years | Computed tomography |
Fasting glucose ≥ 7.0 mmol/L or drug treatment |
Age, sex, BMI, smoking, exercise, alcohol intake, fasting glucose, changes in BMI and liver phantom ratio during follow‐up |
FLD was independently associated with incident diabetes (adjusted OR 2.85, 95% CI 1.4‐6.0, P = .006) |
| Chen, 2017 25 | Prospective cohort study of 132,377 (32% with FLD, 18.1% with chronic liver diseases) Taiwanese nondiabetic subjects; 5.8 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, clinical history or drug treatment |
Age, obesity, hypertension, dyslipidemia, family history of diabetes, smoking, alcohol intake, exercise, AST, ALT, GGT, ALP | FLD was independently associated with incident diabetes (adjusted HR 2.08, 95% CI 1.9‐2.2, P < .001 in men and adjusted HR 2.65, 95% CI 2.4‐2.9, P < .001 in women). Exclusion of chronic liver diseases did not attenuate the association |
| Liu, 2017 26 | Retrospective cohort study of 18,507 (18.8% with FLD) Chinese elderly nondiabetic males without known chronic liver diseases; 5 years | Ultrasound | Fasting glucose ≥ 7.0 mmol/L, 2‐h plasma glucose ≥ 11.1 mmol/L during 75‐g OGTT, clinical history or drug treatment |
Age, BMI, ALT, smoking, marriage status, alcohol intake, hypertension, dyslipidemia |
FLD was independently associated with incident diabetes (adjusted RR 1.67, 95% CI 1.4‐2.1, P < .001) |
| Björkström, 2017 27 | Retrospective cohort study of 396 (100% with FLD) Swedish nondiabetic subjects without known chronic liver diseases; 18.4 years | Biopsy | Clinical history or drug treatment | Age, sex, BMI, triglycerides | Liver fat content was independently associated with incident diabetes in the fibrosis stages 0‐2 (adjusted HR 1.36, 95% CI 1.0‐1.8; P = .03), not in the fibrosis stages 3‐4 (adjusted HR 1.24, 95% CI 0.4‐3.7, P = .71) |
| Tokita, 2017 28 | Retrospective cohort study of 2,408 (11.2% with FLD) Japanese nondiabetic subjects without known chronic liver diseases; 10 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5% or drug treatment |
Age, sex, HbA1c, HDL, triglycerides, systolic blood pressure | FLD was independently associated with incident diabetes (P = .0001) |
| Mitsuhashi, 2017 29 | Retrospective cohort study of 17,810 (21.6% with FLD) Japanese nondiabetic subjects without known chronic liver diseases; 5.1 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5%, clinical history or drug treatment |
Age, BMI, smoking, exercise, alcohol intake, family history of diabetes, log ALT, fasting glucose | The adjusted HR for incident diabetes compared with those without FLD and MetS were as follows: 2.35 (95% CI 1.9‐2.9, P < .001) in the non‐MetS with FLD group, 1.70 (95% CI 1.3‐2.2, P < .001) in the MetS without FLD group, and 2.33 (95% CI 1.9‐2.9, P < .001) in the MetS with FLD group |
| Bae, 2018 30 | Retrospective cohort study of 7,849 (46.7% with FLD) Korean nondiabetic subjects without known chronic liver diseases; 4 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5%, clinical history or drug treatment |
Age, sex, fasting glucose, HbA1c, BMI, LDL, HDL, triglycerides, systolic blood pressure, HOMA‐IR, smoking | Changes in FLD status were independently associated with incident diabetes. The adjusted HRs compared with those without FLD were as follows: 1.50 (95% CI 1.1‐2.0) in the persistent FLD group and 0.99 (95% CI 0.8‐1.3) in the resolved FLD group |
| Seko, 2018 31 | Retrospective cohort study of 89 (100% with FLD) Japanese nondiabetic subjects (58% with IGT) without known chronic liver diseases; 5.2 years | Biopsy | Fasting glucose ≥ 7.0 mmol/L, 2‐h plasma glucose ≥ 11.1 mmol/L during 75‐g OGTT, HbA1c ≥ 6.5% or drug treatment | Age, sex, BMI, ferritin, fibrosis stage, NAS, AST/ALT ratio, fasting glucose, 30‐min and 1‐h postload plasma glucose, HbA1c, 1‐h postload insulin, HOMA‐B, HOMA‐IR | Insulin resistance was independently associated with incident diabetes (adjusted HR 40.1, 95% CI 1.4‐119.3, P = .033) |
| Kim, 2018 32 | Retrospective cohort study of 2,920 (31.6% with FLD, 3.5% with diabetes) Korean subjects without known chronic liver diseases; 5.1 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5% or drug treatment |
Age, sex, waist circumference, triglycerides, HDL, LDL, uric acid, smoking | FLD was independently associated with incident diabetes. The adjusted HRs compared with the nonobese without FLD group were as follows: 2.69 (95% CI, 1.7‐4.2, P < .001) in the nonobese with FLD group, 1.92 (95% CI, 1.1‐3.4, P = .022) in the obese without FLD group, and 2.89 (95% CI, 1.7‐4.8, P < .001) in the obese with FLD group |
| Shen, 2018 33 | Prospective cohort study of 41,650 (28.4% with FLD) Chinese nondiabetic subjects without known chronic liver diseases; 3.6 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L or drug treatment |
Age, sex, smoking, exercise, education, family incomes, family history of diabetes, waist circumference, ALT, LDL, HDL, triglycerides, fasting glucose, uric acid, C‐reactive protein, hypertension, metabolic syndrome | The severity of FLD was associated with higher risk of incident diabetes. The adjusted HRs compared with those without FLD were as follows: 1.62 (95% CI 1.5‐1.8) in the whole FLD group, 1.46 (95% CI 1.3‐1.6) in the mild FLD group, 1.92 (95% CI 1.7‐2.2) in the moderate FLD group and 2.66 (95% CI 2.2‐3.3) in the severe FLD group (P‐trend < 0.001). Similar associations were observed between FLD and incident IFG |
| Brunner, 2019 34 | Retrospective cohort study of 808 (14% with FLD, 2.5% with diabetes) United States subjects; 6.2 years | Computed tomography | Fasting glucose ≥ 7.0 mmol/L or drug treatment | Age, sex, BMI, smoking, alcohol intake, liver phantom ratio, fasting glucose, changes in BMI during follow‐up | Increasing liver fat content during follow‐up was independently associated with incident diabetes (adjusted OR 1.68, 95% CI 1.2‐2.3, P = .001) |
| Okamura, 2019 35 | Retrospective cohort study of 15,464 (17.7% with FLD) Japanese nondiabetic subjects without known chronic liver diseases; 5.1 years | Ultrasound | Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5% or clinical history | Age, alcohol intake, smoking, exercise, fasting glucose | FLD was independently associated with incident diabetes (adjusted HR 4.74, 95% CI 1.9‐11.7, P = .006 in men and adjusted HR 14.0, 95% CI 7.2‐27.1, P < .001 in women). The clustering of obesity, visceral obesity and FLD markedly increased the risk of developing diabetes (adjusted HR 10.5, 95% CI 8.0‐13.8, P < .001 in men and adjusted HR 30.0, 95% CI 18.0‐50.0, P < .001 in women) |
| Cho, 2019 36 | Retrospective cohort study of 2,726 (30.3% with FLD) Korean nondiabetic subjects without known chronic liver diseases; 62.2 months | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5% or drug treatment |
Age, sex, BMI, fasting glucose, ALT | Changes in FLD status were independently associated with incident diabetes. The adjusted HRs compared with those without FLD were as follows: HRs 3.59 (95% CI 2.1‐6.3, P < .001) in the persistent FLD group, 1.94 (95% CI 1.1‐3.5, P = .026) in the incident FLD group and 1.21, 95% CI, 0.4‐3.6, P = .733) in the resolved FLD group |
| Sung, 2019 37 | Retrospective cohort study of 70,303 (13.1% with FLD) Korean nondiabetic subjects without known chronic liver diseases; 3.7 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5%, clinical history or drug treatment |
Age, education, exercise, smoking, alcohol intake, centre, year, family history of diabetes, waist circumference, BMI, triglycerides, LDL, drugs for hypertension and hyperlipidaemia | FLD was independently associated with incident diabetes (adjusted HR 2.17, 95% CI 1.6‐3.0 in men and adjusted HR 2.86, 95% CI 1.5‐5.5 for women) |
| Wang, 2019 38 | Retrospective cohort study of 10,064 (5.4% with FLD) Japanese nondiabetic subjects without known chronic liver diseases; 6 years | Ultrasound |
Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5% or clinical history |
Age, sex, BMI, alcohol intake, smoking, HbA1c | FLD was independently associated with incident diabetes (adjusted HR 2.52, 95% CI 1.6‐4.0, P < .001) |
| Lee, 2019 39 | Retrospective cohort study of 6,240 (45.4% with FLD) Korean prediabetic subjects without known chronic liver disease; 4.3 years | Ultrasound | Fasting glucose ≥ 7.0 mmol/L, HbA1c ≥ 6.5% or drug treatment | Age, sex, BMI, smoking, alcohol intake, ALT, triglycerides, HDL, systolic blood pressure, HbA1c | FLD was independently associated with incident diabetes (adjusted RR 1.81, 95% CI, 1.5‐2.2, P < .001) |
| Nasr, 2020 40 | Prospective cohort study of 106 (100% with FLD) Swedish nondiabetic subjects without known chronic liver diseases; 23.2 years | Biopsy | Fasting glucose ≥ 7.0 mmol/L with drug treatment or 2‐h plasma glucose ≥ 11.1 mmol/L during 75‐g OGTT | Age, BMI, histologic fibrosis stage | Liver fat content was independently associated with incident diabetes (adjusted HR 1.03 per 1% increase, 95% CI 1.0‐1.1, P = .02) |
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; CI, confidence interval; FLD, fatty liver disease; GGT, gamma‐glutamyl transferase; HbA1c, haemoglobin A1c; HDL, high‐density lipoprotein; HOMA‐B, homeostasis model assessment of beta‐cell function; HOMA‐IR, homeostasis model assessment of insulin resistance; HR, hazard ratio; IFG, impaired fasting glycaemia; IGT, impaired glucose tolerance; LDL, low‐density lipoprotein; MetS, metabolic syndrome; NAS, NAFLD activity score; OGTT, 75‐g oral glucose tolerance test; T2D, type 2 diabetes.
Notably, the increase in the risk of incident T2D was found to be proportional to the severity of liver steatosis assessed by ultrasonography or computed tomography. 23 , 33 , 34 For example, in a large prospective cohort study of 18,111 Chinese nondiabetic subjects, Li et al showed that the incidence rates of T2D at 4.6‐year follow‐up progressively increased with the ultrasonographic severity of FLD at baseline, accounting for 18.1% of incident T2D cases in the moderate‐severe FLD group, 10.6% in the mild FLD group and 4.7% in the normal group, respectively (P < .001). In the multivariable Cox regression analysis, the adjusted hazard ratios (HRs) for incident T2D were, respectively, 2.34 (95% CI 1.9‐3.0) and 1.88 (95% CI 1.6‐2.2) in individuals belonging to the moderate‐severe and mild FLD groups, when compared with those in the non‐FLD group (P‐trend < 0.001). 23
Similarly, in a prospective cohort study of 41,650 Chinese nondiabetic individuals followed for a mean period of 3.6 years, it has been reported that FLD on ultrasonography was independently associated with increased incidence of both T2D (adjusted HR 1.62, 95% CI 1.5‐1.8) and prediabetes (adjusted HR 1.12, 95% CI 1.1‐1.2). In particular, compared with subjects without FLD, the HRs for T2D development were significantly greater in those belonging to the severe FLD group (adjusted HR 2.66, 95% CI 2.2‐3.3), the moderate (adjusted HR 1.92, 95% CI 1.7‐2.2) or mild (adjusted HR 1.46, 95% CI 1.3‐1.6) FLD groups. 33
Interestingly, in a retrospective cohort study of 2,726 South Korean nondiabetic individuals, Cho et al have assessed the risk of incident T2D during 62 months of follow‐up in the following three subgroups of subjects: (1) those with persistent FLD on ultrasonography both at baseline and at follow‐up; (2) those with newly diagnosed FLD at follow‐up; and (3) those with FLD resolution at follow‐up examination. Notably, these authors found that compared with individuals without FLD, the risk of incident T2D was remarkably greater in those with persistent FLD (adjusted HR 3.59, 95% CI 2.1‐6.3, P < .001) and those who developed incident FLD (adjusted HR 1.94, 95% CI 1.1‐3.5, P = .026) over the follow‐up period. Conversely, the risk of incident T2D was not increased in those who resolved FLD at follow‐up (adjusted HR 1.21, 95% CI, 0.4‐3.6, P = .733). 36
Similarly, in a retrospective cohort study of 7,849 South Korean nondiabetic individuals who were followed for a mean period of 4 years, Bae et al reported that the persistence of FLD on ultrasonography was independently associated with an approximately 50% increased risk of incident T2D, whereas the risk of individuals who resolved FLD over the follow‐up was essentially superimposable to that of individuals without FLD. 30
Notably, Mitsuhashi et al have also shown that FLD was a stronger risk factor for incident T2D than the presence of metabolic syndrome (MetS) without fatty liver. Indeed, in a population‐based cohort study of over 17,000 Japanese nondiabetic individuals enrolled in a healthy check‐up programme for more than 5 years, the authors found that the incidence rates of T2D were 1.7% in non‐MetS individuals without FLD, 8.3% in individuals with FLD alone, 12.5% in those with MetS alone and 21.2% in those with both conditions, respectively. Compared with the normal group, the adjusted HRs for incident T2D were 2.35 (95% CI 1.9‐2.9) in non‐MetS individuals with FLD, 1.70 (95% CI 1.3‐2.2) in those with MetS alone and 2.33 (95% CI 1.9‐2.9) in those with both MetS and FLD, respectively. Additionally, patients with FLD (irrespective of coexistence of MetS) had a ~ 38% increased risk of developing T2D compared to those with MetS alone. 29
Using the same population‐based cohort, Okamura et al have subsequently shown that FLD per se had the strongest adverse effect on risk of incident T2D (adjusted HR 4.74, 95% CI 1.9‐11.7, in men and adjusted HR 14.0, 95% CI 7.2‐27.1, in women) compared with either obesity without FLD (adjusted HR 1.85, 95% CI 1.1‐3.3, in men and adjusted HR 1.79, 95% CI 0.2‐13.2, in women) or visceral obesity without FLD (adjusted HR 3.41, 95% CI 2.5‐4.6, in men and adjusted HR 2.30, 95% CI 0.9‐6.1, in women). As expected, the clustering of these three conditions (obesity, visceral obesity and FLD) markedly increased the risk of incident T2D (adjusted HR 10.5, 95% CI 8.0‐13.8, in men and adjusted HR 30.0, 95% CI 18.0‐50.0, in women). 35
In a retrospective cohort study of 396 Swedish nondiabetic adults with biopsy‐confirmed FLD, Björkström et al have reported that the incidence rate of T2D was significantly higher among subjects with fibrosis stages 3‐4 than among those with fibrosis stages 0‐2 (51% vs. 31%) over a mean follow‐up of 18.4 years. 27 Subsequently, in a cohort study of 106 Swedish nondiabetic subjects with biopsy‐proven FLD followed for over 20 years, Nasr et al from the same research group have observed that the severity of hepatic steatosis, quantitatively measured by stereological point counting, was independently associated with increased T2D incidence (adjusted HR 1.03 per 1% increase, 95% CI 1.0‐1.1). 40
In a small retrospective cohort study of 89 Japanese nondiabetic subjects (58% with IGT) with biopsy‐confirmed FLD, Seko et al have shown that HOMA‐estimated insulin resistance was the strongest independent predictor of incident T2D over a 5.2‐year follow‐up (adjusted HR 40.1, 95% CI 1.4‐119.3). 31 Noteworthy, a recent combined meta‐analysis and bias analysis including more than 240,000 middle‐aged individuals (mostly of Asian ethnicity) has provided further strong evidence for a causal relationship between FLD and risk of T2D. 41
Collectively, all these epidemiological studies support the notion that FLD (defined radiologically or histologically) is strongly associated with an increased risk of incident T2D in different ethnic populations and that the magnitude of risk of incident T2D parallels the underlying severity of FLD. However, there are some important limitations to be considered. First, most of the aforementioned observational studies have a retrospective design and are heterogeneous in terms of demographic characteristics, length of follow‐up, covariates included in multivariable regression analyses, as well as severity of FLD. Second, most of the studies included individuals from Asian countries (especially China and South Korea). Third, only few of these studies (Björkström et al, 27 Seko et al 31 and Nasr et al 40 ) have used liver biopsy for diagnosing and staging FLD. Finally, the large majority of studies—except for Liu et al, 26 Seko et al 31 and Nasr et al 40 —did not perform 75‐g oral glucose tolerance test for the diagnosis of diabetes.
Additional larger prospective cohort studies performed on different ethnic groups, considering also the genetic determinants for FLD, are certainly needed to better define the magnitude of risk of incident T2D associated with FLD.
2.2. FLD and risk of T2D chronic complications: epidemiological evidence
The global prevalence of FLD diagnosed by ultrasonography and magnetic resonance spectroscopy among individuals with T2D is currently estimated to be approximately 55%, with the highest rates reported from Europe (68%) and West Asia (67%), followed by South Asia (58%), Latin America (57%), East Asia (52%), the United States (52%) and Africa (30%). 42 These rates for the global FLD prevalence are nearly twice those observed in the general population from the same regions. 42 , 43 Similarly, the global prevalence of histologically proven nonalcoholic steatohepatitis (NASH) and advanced fibrosis among individuals with FLD and T2D is very high, accounting for 37% and 17%, respectively. 42
Additionally, T2D has been adversely related to the onset of FLD long‐term complications, such as cirrhosis, hepatocellular carcinoma, liver‐related mortality and all‐cause mortality. 44 , 45 , 46 , 47 , 48 In this context, T2D seems to be not only a major driven of FLD global burden but also an important risk factor for liver disease progression.
A detailed discussion of the link between FLD and risk of chronic vascular complications of diabetes is beyond the scope of this review article. In brief, the coexistence of FLD and T2D increases not only the risk of developing the more severe forms of FLD (advanced fibrosis, cirrhosis and hepatocellular carcinoma), but also the risk of developing chronic vascular complications of diabetes. Indeed, to date, a number of large population‐based and hospital‐based cohort studies reported an increased incidence of fatal and nonfatal cardiovascular events in individuals with FLD, across a wide range of disease spectra, including T2D. 49 , 50 For instance, a prospective nested case‐control study in 744 T2D outpatient individuals without known cardiovascular and or chronic liver damage at baseline demonstrated that those with ultrasound‐detected FLD had a nearly two‐fold increased risk of major adverse cardiovascular events over a follow‐up period of 5 years. Notably, this association was independent of traditional cardiovascular risk factors, diabetes‐related variables and use of hypoglycaemic, antihypertensive, lipid‐lowering and antiplatelet medications. 51 Similar results were also confirmed in a subsequent larger cohort study of 2,103 outpatients with T2D with a longer follow‐up period (6.5 years). 52 Accumulating evidence also suggests that FLD is associated with valvular heart disease (mainly aortic‐valve sclerosis) and increased risk of cardiac arrhythmias (mainly permanent atrial fibrillation), especially in individuals with T2D. 53 , 54 This supports the notion that the diagnosis of FLD identifies a subset of subjects at higher risk of cardiovascular disease over time. 55
In the last decade, a growing body of epidemiological evidence also suggests that FLD is significantly associated with an increased prevalence and incidence of microvascular complications of diabetes, especially with chronic kidney disease. 56 For instance, in the Valpolicella Heart Diabetes Study cohort involving 1,760 T2D outpatients with normal kidney function at baseline, the presence of ultrasound‐diagnosed FLD was associated with an increased risk of incident chronic kidney disease (CKD stage ≥ 3) over a follow‐up period of 6.5 years, independently of established renal risk factors, diabetes duration, glycaemic control and use of medications. 57 A recent updated meta‐analysis of nine observational studies (including a total of nearly 96,500 adult individuals) confirmed that FLD is associated with a nearly 40% increase in the long‐term risk of incident CKD stage ≥ 3 (ie defined as occurrence of estimated glomerular filtration rate < 60 ml/min/1.73m2, with or without accompanying overt proteinuria). In subgroup analyses, the significant association between FLD and increased risk of CKD was particularly evident among patients with T2D and FLD. 58
However, despite the growing epidemiological evidence that links FLD with the long‐term risk of chronic vascular complications of diabetes, a causal relationship between these two diseases remains to be demonstrated. Additional larger prospective studies in different ethnic populations and translational studies are needed to firmly establish whether FLD (especially in its more advanced forms) actively contributes to the increased risk of macrovascular and microvascular complications observed among patients with T2D and FLD.
3. HUMAN GENETICS
3.1. Common genetic variants associated with risk of FLD
In the last decade, several common genetic variants have been reported to confer increased genetic susceptibility to or protection against FLD. 59 Notably, these common genetic variants had a several fold larger effect if compared to common variants of susceptibility in other complex disease traits, including T2D or obesity. A detailed discussion of the association between rare genetic variants of FLD and risk of insulin resistance and diabetes is beyond the scope of this review article. Briefly, rare mutations in apolipoprotein B (APOB) predispose to familial hypobetalipoproteinaemia and progressive liver disease due to impaired triglycerides assembly into very low‐density lipoproteins and failure to secrete triglycerides from the liver. 60 Consistently with common genetic variations, despite higher liver fat content, the risk of insulin resistance and diabetes seems not to be greatly increased in carriers of APOB variants. 61 , 62 , 63 , 64 , 65 Moreover, although the coexistence of obesity, visceral adiposity and insulin resistance promotes the development of hepatic fat accumulation in these subjects, familial hypobetalipoproteinaemia represents a condition that per se leads to higher degree of FLD. 66 , 67 In this section, we will discuss the evidence of an association between common genetic variants of FLD and T2D or insulin resistance.
3.2. Patatin‐like phospholipase domain‐containing 3
To date, the patatin‐like phospholipase domain‐containing 3 (PNPLA3) rs738409 encoding for an isoleucine to methionine substitution at position 148 (I148M) of the protein is the most robust genetic determinant of FLD. 68 , 69 This genetic variant is associated with insulin resistance or T2D mainly in individuals with obesity but not in those with normal weight. 68 , 70 , 71 , 72 , 73 , 74 , 75 , 76 A possible reason for this association is that obesity uncovers the effect of the PNPLA3 variant, increasing its effect size. 73 , 77 Additionally, quality of intrahepatic lipids, rather than quantity, may exert a major impact on the development of insulin resistance and glucose intolerance. 6 , 7 , 78 , 79 , 80 , 81 , 82 In particular, in metabolically related FLD, but not in PNPLA3‐related FLD, the liver was found to be predominantly enriched with saturated triglycerides and with markers of de novo ceramides synthesis. 6 , 7 Notably, ceramides have been strongly associated with hepatic insulin resistance, thus supporting their key role in the pathogenesis of metabolically related FLD. 6 , 7 , 78 On the other side, in PNPLA3‐associated FLD the quality of triglycerides shifted towards polyunsaturated fatty acids. 7
However, there are also some studies showing a significant relationship between PNPLA3 I148M polymorphism and greater insulin resistance in nonobese individuals from Taiwan and South Korea. 83 , 84 In addition, in a prospective cohort study of 2,189 Chinese middle‐aged and elderly individuals with a follow‐up of 4.2 years, Xia et al showed that the PNPLA3 rs738409 was significantly associated with lower risk of incident T2D. 85 Furthermore, in a study of Brazilian individuals with T2D, Machado et al reported that the PNPLA3 I148M variant was significantly correlated to a better glycaemic control. 86 All these data suggest that in addition to obesity there are also other factors possibly related to ethnicity that can modulate the effect of the PNPLA3 genetic variant on T2D risk. Further studies are needed to establish the magnitude of genetic and environmental risk factors in FLD pathogenesis and to better characterize the different clinical FLD phenotypes resulting from their interactions.
3.3. Transmembrane 6 superfamily member 2
A body of evidence shows that the rs58542926 in transmembrane 6 superfamily member 2 (TM6SF2) (E167K) is a robust genetic determinant of FLD, 87 , 88 , 89 inducing a reduction in APOB100 containing lipoprotein lipidation and secretion. 90 , 91
Furthermore, studies have also investigated the relationship between FLD, insulin sensitivity and T2D among individuals carrying the TM6SF2 E167K. As for the PNPLA3 I148M, lines of evidence have described the TM6SF2 E167K as a potential risk variant for T2D development, 92 , 93 mainly linked to increased hepatic and adipose insulin resistance and impaired pancreatic beta‐cell function. 94 On the other hand, TM6SF2 E167K has been reported to be associated with preserved insulin sensitivity, estimated by HOMA‐IR and adipose insulin resistance or measured by hyperinsulinaemic euglycaemic clamp. 84 , 95
3.4. Membrane bound O‐acyltransferase domain‐containing protein 7
The membrane bound O‐acyltransferase domain‐containing protein 7 (MBOAT7) is a 6‐transmembrane domain protein 96 that promotes the remodelling of membrane phosphatidylinositol with polyunsaturated fatty acids. 96 , 97 , 98 , 99 Depletion of MBOAT7 increases liver fat content by inducing hepatic synthesis of triglycerides fueled by an accelerated turnover of phosphatidylinositol. 100 Hyperinsulinaemia also contributes to liver fat accumulation by enhancing hepatic MBOAT7 down‐regulation, independently of MBOAT7 rs641738 genotype, 99 thus suggesting that MBOAT7 activity might be influenced by insulin signalling pathways.
To date, there are very few studies examining the effect of MBOAT7 rs641738 on T2D‐related metabolic traits. Viitasalo et al did not find any association of MBOAT7 rs641738 with plasma glucose and insulin levels among Caucasian obese children. 101 Similarly, no association was found between the MBOAT7 rs641738 and HOMA‐estimated insulin resistance among Asian adult individuals. 84 However, in a multiethnic cohort of 860 obese youths, Umano et al showed that MBOAT7 rs626283 (ie a genetic variant in strong linkage disequilibrium with the MBOAT7 rs641738) was associated with both hyperisulinaemia and impaired insulin sensitivity in European individuals, but not in Hispanics and African Americans. 102
3.5. Glucokinase regulator
The rs1260326 in glucokinase regulator (GCKR) (P446L) reduces GCKR ability to inhibit glucokinase, resulting in constitutive activation of glucose uptake and increased hepatic de novo lipogenesis. 103 This results in the occurrence of FLD with lower insulin resistance and decreased risk of T2D as shown in several ethnic groups, mostly European and Asian populations. 104 , 105 , 106 , 107 , 108 , 109 , 110 , 111 , 112 , 113 , 114 , 115 , 116
Notably, as for other genetic variants, a GCKR‐related protection against development of T2D was not observed in African American individuals, 113 , 114 , 116 supporting that the impact of GCKR variant on T2D risk and its related clinical traits might differ depending on ethnicity. Moreover, the association of the GCKR variant with fasting glucose, insulin levels and insulin sensitivity seems to be less pronounced in children or adolescents compared to adults, suggesting that the GCKR‐induced hypoglycaemic effect might become more evident with increasing age. 117 , 118 Unexpectedly, the rs1260326 or rs780094 (an intronic variant in high linkage disequilibrium) in GCKR gene variants have been associated with increased 2‐hour postload plasma glucose levels. 106 , 114 , 119 Finally, inconsistent results have been reported regarding the association between GCKR polymorphisms and pancreatic beta‐cell function, as estimated by the HOMA‐B index. 106 , 110 , 114
3.6. Hydroxysteroid 17‐beta dehydrogenase 13
The loss‐of‐function rs72613567:TA in hydroxysteroid 17‐beta dehydrogenase 13 (HSD17B13) was recently found to protect against the development and progression of both alcoholic and nonalcoholic chronic liver disease, while showing no association with simple steatosis. 120 , 121 , 122 It has been hypothesized that the HSD17B13 rs72613567:TA may result in defective HSD17B13 enzymatic activity, leading to impaired synthesis of several proinflammatory lipid species (eg leukotriene B4) into the liver. 120 However, the exact molecular mechanism(s) and the protein function need further investigation.
Similarly, it is still not known whether the HSD17B13 gene locus influences susceptibility to T2D and insulin resistance. A study by Luukkonen et al have recently reported that in European nondiabetic individuals, the HSD17B13 rs72613567:TA was not significantly associated with changes in fasting glucose and insulin levels or insulin sensitivity, as directly quantified by euglycaemic hyperinsulinaemic clamp technique. 123
3.7. Causal relationships between FLD, insulin resistance and diabetes: Mendelian randomization studies
In the last few years, an increasing number of studies have applied a Mendelian randomization approach to establish a possible causal relationship between FLD and its related metabolic traits, that is insulin resistance and T2D. 89 , 124
Interestingly, it has been shown that the presence of genetically determined fatty liver (by using a genetic risk score including PNPLA3, TM6SF2, GCKR and MBOAT7 variants) was causally associated with greater insulin resistance, as estimated by HOMA‐IR, in individuals at risk of progressive liver disease (ie those with suspected NASH or severe obesity), but not in the general population. 89 However, it should be noted that as reported by Stender et al these genetic variants strongly interact with obesity 125 and, therefore, it is not surprising that the deleterious metabolic effect of these genetic variants was observed principally among those at higher risk for FLD. Moreover, this study also suggested that FLD per se does not directly cause insulin resistance, but the risk is mainly mediated by the degree of liver fibrosis, in other words by the duration and severity of liver disease (Figure 1). 89
Figure 1.
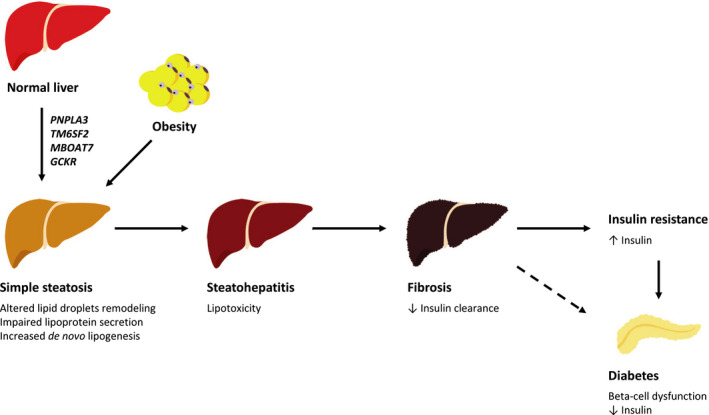
Causal relationship between genetically determined fatty liver disease, insulin resistance and diabetes. A Mendelian randomization study published by Dongiovanni et al 89 showed that: 1) genetically determined fatty liver disease (FLD) is causally associated with insulin resistance in individuals at risk of progressive liver disease (eg those with suspected NASH or severe obesity); 2) impairment of insulin sensitivity is mediated by increased hepatic fibrosis (excess liver fat content—lipotoxicity). Similarly, a Mendelian randomization study by Liu et al 124 confirmed that genetically determined FLD causes the development of type 2 diabetes (T2D), although the underlying molecular mechanism(s) has yet to be entirely elucidated. In accord with the well‐recognized link between cirrhosis and increased T2D onset, 126 the association between genetically determined FLD and enhanced risk of incident T2D might again be largely mediated by increased hepatic fibrosis
Within this context, hyperinsulinaemia might be secondary to intrahepatic accumulation of specific lipotoxic species in addition to fibrosis‐induced defect in hepatic insulin clearance. 59 Similarly, given the well‐recognized association between cirrhosis and increased risk of incident T2D, 126 it would be not surprising if genetically related FLD may cause pancreatic beta‐cell dysfunction through the same underlying mechanism, that is advanced liver fibrosis. However, this issue has yet to be studied in greater detail in future studies. It is worth noting that the accuracy of Mendelian randomization methodology can be compromised by the pleiotropic effects of genetic variants, although this disadvantage is largely minimized by using polygenic risk scores. Another limitation of human‐based studies is partly due to the presence of multiple potential confounding factors (eg comorbidities or use of certain medications) that may weaken or mask the specific genetic associations. For example, the coexistence of severe obesity was found to strongly influence the impact of PNPLA3 I148M on systemic insulin sensitivity. 70 Experimental studies conducted in animal models may help to stem these issues. To support this, a recent experimental study published by Liu et al reported that PNPLA3 I148M was associated with chronic hyperglycaemia and increased visceral adiposity, but not with insulin resistance. Interestingly, the authors proposed that PNPLA3‐induced reduction in glucose tolerance was largely mediated by pancreatic chronic inflammation, leading to impaired pancreatic insulin and glucagon secretion. 124 Taken all this together, it would appear that genetically determined liver steatosis does not carry the same diabetogenic risk associated with the metabolically determined liver steatosis. Moreover, quality of intrahepatic lipids, rather than quantity, decides whether the accumulation of fat in the liver will result in changes in glucose metabolism rather than only a deleterious effect for the hepatocyte.
Based on this evidence, it is likely that the use of drugs that will ameliorate liver steatosis and fibrosis in principle should also exert a beneficial effect on insulin resistance and risk of T2D associated with FLD. Currently, several pharmacological therapies have shown promising results in improving liver fat content and inflammation, such as the peroxisome proliferator‐activated receptor γ (PPAR‐γ) agonist pioglitazone and the glucagon‐like peptide 1 (GLP‐1) receptor agonist liraglutide. 127 , 128 , 129 , 130 In addition to these well‐known antidiabetic drugs, the stearoyl CoA desaturase‐1 (SCD1) modulator aramchol showed improvement in hepatic steatosis and glycaemic control in individuals with prediabetes or T2D and biopsy‐proven NASH (NCT02279524). On the other hand, despite ameliorating hepatic steatosis and fibrosis, the farnesoid X receptor (FXR) agonist obeticholic acid was found to increase a) insulin resistance, estimated by HOMA‐IR and b) circulating levels of low‐density lipoproteins, resulting in a more proatherogenic profile. 131 Similarly, the chemokine receptor (CCR) 2/5 antagonist cenicriviroc, which showed a primary antifibrotic activity, appears to be likely metabolically neutral. 132 , 133 However, larger phase 3 clinical trials are required to further validate these results. Finally, the pleiotropic effects of genetic factors and of drug pathways should be borne in mind when prescribing a drug for individuals with FLD.
3.8. Effect of FLD genetics on T2D chronic complications
To date, emerging evidence supports the existence of a significant relationship between some genetic determinants of FLD and susceptibility to diabetic nephropathy, although the topic needs to be further explored. 134 Notably, the PNPLA3 I148M has been associated with lower estimated glomerular filtration rate and increased risk of chronic kidney disease among European individuals with T2D. 135 , 136 Interestingly, the significant association between the PNPLA3 I148M variant and increased risk of kidney dysfunction was independent of established renal risk factors and severity of FLD, suggesting that the PNPLA3 I148M might be directly involved in the pathophysiology of diabetic nephropathy. In line with this hypothesis, PNPLA3 expression was found to be high in the renal cortex, mainly in podocytes. 136 Conversely, the steatogenic allele in GCKR locus seems to protect against the development of chronic kidney disease among T2D individuals, 137 , 138 consistently with the GCKR‐related hypoglycaemic effect observed in nondiabetic individuals.
Some evidence also suggests that PNPLA3 and TM6SF2 gene variants may protect against cardiovascular risk, whereas variants in GCKR are associated with increased risk of cardiovascular disease, perhaps mediated by a decrease in the atherogenic dyslipidemia in both PNPLA3 and TM6SF2 carriers and an increase in the atherogenic dyslipidemia in GCKR carriers. 139 However, further research is needed to clarify whether ‘genetic‐related FLD’ and ‘metabolic‐related FLD’ exert differential effects on risk of major adverse cardiovascular events. 49 , 140
4. CONCLUSIONS AND FUTURE PERSPECTIVES
New insights by molecular human genetics robustly support that FLD is causally associated with dysmetabolism and T2D. 89 , 124 Recent studies highlighted that the key molecular mechanism of dysmetabolism is not fat accumulation per se but the degree of hepatic fibrosis (excess liver fat content—lipotoxicity), leading to reduced insulin clearance, insulin resistance and T2D. 59 Notably, initial findings show that these genetic factors might be directly implicated in modulating pancreatic beta‐cell function, 124 although future studies are needed to fully understand this relationship. In this context, it is worth noting that a consensus of experts has recently proposed novel criteria for diagnosing MAFLD (mainly based on the presence of overweight/obesity, T2D or other metabolic syndrome traits), irrespective of other concomitant liver diseases. 19 , 20 We believe that this novel definition is the first attempt to define the complexity of FLD and its heterogeneous clinical phenotypes, paving the way for a more fit design of clinical trials that will lead to precision medicine. Finally, it is also reasonable to speculate that the quantitative assessment of liver fat content by novel unconventional methods and the discovery of specific biomarkers of hepatic lipotoxicity will provide a better opportunity to improve the overall risk prediction of incident T2D in all individuals with FLD.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors conceived and designed the review, were involved in drafting and revising the manuscript and approved the final version prior to submission.
ACKNOWLEDGEMENTS
This work was supported by the Swedish Research Council (Vetenskapsrådet (VR), 2016–01527 to SR); by the Swedish state under the Agreement between the Swedish government and the county councils (the ALF‐agreement) (SU 2018–04276 to SR); by the Novonordisk Foundation Grant for Excellence in Endocrinology (Excellence Project, 9321–430 to SR); by Wallenberg Academy Fellows from the Knut and Alice Wallenberg Foundation (KAW 2017.0203 to SR); by the Novonordisk Project grants in Endocrinology and Metabolism (SR); by Astra Zeneca Agreement for Research (SR); by Grant SSF ITM17‐0384, Swedish Foundation for Strategic Research (SR).
Tavaglione F, Targher G, Valenti L, Romeo S. Human and molecular genetics shed lights on fatty liver disease and diabetes conundrum. Endocrinol Diab Metab. 2020;3:e00179 10.1002/edm2.179
Contributor Information
Luca Valenti, Email: luca.valenti@unimi.it.
Stefano Romeo, Email: stefano.romeo@wlab.gu.se.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this study.
REFERENCES
- 1. Valenti L, Pelusi S. Redefining fatty liver disease classification in 2020. Liver Int. 2020;40(5):1016‐1017. [DOI] [PubMed] [Google Scholar]
- 2. European Association for the Study of the Liver, European Association for the Study of Diabetes, European Association for the Study of Obesity . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol 2016;64(6):1388‐1402. [DOI] [PubMed] [Google Scholar]
- 3. Valenti L, Bugianesi E, Pajvani U, Targher G. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int. 2016;36(11):1563‐1579. [DOI] [PubMed] [Google Scholar]
- 4. Mantovani A, Byrne CD, Bonora E, Targher G. Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: a meta‐analysis. Diabetes Care. 2018;41(2):372‐382. [DOI] [PubMed] [Google Scholar]
- 5. Younossi ZM. Non‐alcoholic fatty liver disease – a global public health perspective. J Hepatol. 2019;70(3):531‐544. [DOI] [PubMed] [Google Scholar]
- 6. Yki‐Järvinen H. Ceramides: A cause of insulin resistance in nonalcoholic fatty liver disease in both murine models and humans. Hepatology. 2020;71(4):1499‐1501. [DOI] [PubMed] [Google Scholar]
- 7. Luukkonen PK, Zhou Y, Sädevirta S, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non‐alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167‐1175. [DOI] [PubMed] [Google Scholar]
- 8. Lonardo A, Nascimbeni F, Ballestri S, et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology. 2019;70(4):1457‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balakrishnan M, Patel P, Dunn‐Valadez S, et al. Women have lower risk of nonalcoholic fatty liver disease but higher risk of progression vs men: a systematic review and meta‐analysis. Clin Gastroenterol Hepatol. 2020;S1542‐3565(20):30612–1. 10.1016/j.cgh.2020.04.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gannon M, Kulkarni RN, Tse HM, Mauvais‐Jarvis F. Sex differences underlying pancreatic islet biology and its dysfunction. Mol Metab. 2018;15:82‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mauvais‐Jarvis F, Le May C, Tiano JP, Liu S, Kilic‐Berkmen G, Kim JH. The role of estrogens in pancreatic islet physiopathology. Adv Exp Med Biol. 2017;1043:385‐399. [DOI] [PubMed] [Google Scholar]
- 12. Mauvais‐Jarvis F. Menopause, estrogens, and glucose homeostasis in women. Adv Exp Med Biol. 2017;1043:217‐225. [DOI] [PubMed] [Google Scholar]
- 13. Mauvais‐Jarvis F. Epidemiology of Gender Differences in Diabetes and Obesity. Adv Exp Med Biol. 2017;1043:3‐8. [DOI] [PubMed] [Google Scholar]
- 14. Mauvais‐Jarvis F. Gender differences in glucose homeostasis and diabetes. Physiol Behav. 2018;04(187):20‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mauvais‐Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34(3):309‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tiano JP, Mauvais‐Jarvis F. Importance of oestrogen receptors to preserve functional β‐cell mass in diabetes. Nat Rev Endocrinol. 2012;8(6):342‐351. [DOI] [PubMed] [Google Scholar]
- 17. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J Hepatol. 2018;68(2):335‐352. [DOI] [PubMed] [Google Scholar]
- 18. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014;59(2):713‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eslam M, Sanyal AJ, George J, Panel IC. MAFLD: a consensus‐driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020. 10.1053/j.gastro.2019.11.312 [DOI] [PubMed] [Google Scholar]
- 20. Eslam M, Newsome PN, Anstee QM, et al. A new definition for metabolic associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020. 10.1016/j.jhep.2020.03.039 [DOI] [PubMed] [Google Scholar]
- 21. Younossi ZM, Rinella ME, Sanyal A, et al. From NAFLD to MAFLD: implications of a premature change in terminology. Hepatology. 2020. 10.1002/hep.31420 [DOI] [PubMed] [Google Scholar]
- 22. Chen GY, Cao HX, Li F, et al. New risk‐scoring system including non‐alcoholic fatty liver disease for predicting incident type 2 diabetes in East China: Shanghai Baosteel Cohort. J Diabetes Investig. 2016;7(2):206‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, Wang J, Tang Y, et al. Bidirectional association between nonalcoholic fatty liver disease and type 2 diabetes in Chinese population: Evidence from the Dongfeng‐Tongji cohort study. PLoS One. 2017;12(3):e0174291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma J, Hwang SJ, Pedley A, et al. Bi‐directional analysis between fatty liver and cardiovascular disease risk factors. J Hepatol. 2017;66(2):390‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen SC, Tsai SP, Jhao JY, Jiang WK, Tsao CK, Chang LY. Liver fat, hepatic enzymes, alkaline phosphatase and the risk of incident type 2 diabetes: a prospective study of 132,377 adults. Sci Rep. 2017;7(1):4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu M, Wang J, Zeng J, Cao X, He Y. Association of NAFLD With Diabetes and the Impact of BMI Changes: A 5‐Year Cohort Study Based on 18,507 Elderly. J Clin Endocrinol Metab. 2017;102(4):1309‐1316. [DOI] [PubMed] [Google Scholar]
- 27. Björkström K, Stål P, Hultcrantz R, Hagström H. Histologic scores for fat and fibrosis associate with development of type 2 diabetes in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2017;15(9):1461‐1468. [DOI] [PubMed] [Google Scholar]
- 28. Tokita Y, Maejima Y, Shimomura K, et al. Non‐alcoholic fatty liver disease is a risk factor for type 2 diabetes in middle‐aged Japanese men and women. Intern Med. 2017;56(7):763‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitsuhashi K, Hashimoto Y, Hamaguchi M, et al. Impact of fatty liver disease and metabolic syndrome on incident type 2 diabetes; a population based cohort study. Endocr J. 2017;64(11):1105‐1114. [DOI] [PubMed] [Google Scholar]
- 30. Bae JC, Han JM, Cho JH, et al. The persistence of fatty liver has a differential impact on the development of diabetes: The Kangbuk Samsung Health Study. Diabetes Res Clin Pract. 2018;135:1‐6. [DOI] [PubMed] [Google Scholar]
- 31. Seko Y, Sumida Y, Tanaka S, et al. Insulin resistance increases the risk of incident type 2 diabetes mellitus in patients with non‐alcoholic fatty liver disease. Hepatol Res. 2018;48(3):E42‐E51. [DOI] [PubMed] [Google Scholar]
- 32. Kim SS, Cho HJ, Kim HJ, et al. Nonalcoholic fatty liver disease as a sentinel marker for the development of diabetes mellitus in non‐obese subjects. Dig Liver Dis. 2018;50(4):370‐377. [DOI] [PubMed] [Google Scholar]
- 33. Shen X, Cai J, Gao J, et al. Nonalcoholic fatty liver disease and risk of diabetes: a prospective study in China. Endocr Pract. 2018;24(9):823‐832. [DOI] [PubMed] [Google Scholar]
- 34. Brunner KT, Pedley A, Massaro JM, Hoffmann U, Benjamin EJ, Long MT. Increasing liver fat is associated with incident cardiovascular risk factors. Clin Gastroenterol Hepatol. 2019. 10.1016/j.cgh.2019.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okamura T, Hashimoto Y, Hamaguchi M, Obora A, Kojima T, Fukui M. Ectopic fat obesity presents the greatest risk for incident type 2 diabetes: a population‐based longitudinal study. Int J Obes (Lond). 2019;43(1):139‐148. [DOI] [PubMed] [Google Scholar]
- 36. Cho HJ, Hwang S, Park JI, et al. Improvement of nonalcoholic fatty liver disease reduces the risk of type 2 diabetes mellitus. Gut Liv. 2019;13(4):440‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sung KC, Seo DC, Lee SJ, Lee MY, Wild SH, Byrne CD. Non alcoholic fatty liver disease and risk of incident diabetes in subjects who are not obese. Nutr Metab Cardiovasc Dis. 2019;29(5):489‐495. [DOI] [PubMed] [Google Scholar]
- 38. Wang L. Ultrasound‐diagnosed nonalcoholic fatty liver disease independently predicts a higher risk of developing diabetes mellitus in nonoverweight individuals. Acad Radiol. 2019;26(7):863‐868. [DOI] [PubMed] [Google Scholar]
- 39. Lee J, Cho YK, Kang YM, et al. The impact of NAFLD and waist circumference changes on diabetes development in prediabetes subjects. Sci Rep. 2019;9(1):17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nasr P, Fredrikson M, Ekstedt M, Kechagias S. The amount of liver fat predicts mortality and development of type 2 diabetes in non‐alcoholic fatty liver disease. Liver Int. 2020. 10.1111/liv.14414 [DOI] [PubMed] [Google Scholar]
- 41. Morrison AE, Zaccardi F, Khunti K, Davies MJ. Causality between non‐alcoholic fatty liver disease and risk of cardiovascular disease and type 2 diabetes: a meta‐analysis with bias analysis. Liver Int. 2019;39(3):557‐567. [DOI] [PubMed] [Google Scholar]
- 42. Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta‐analysis. J Hepatol. 2019;71(4):793‐801. [DOI] [PubMed] [Google Scholar]
- 43. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐Meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73‐84. [DOI] [PubMed] [Google Scholar]
- 44. Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non‐alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56(6):1384‐1391. [DOI] [PubMed] [Google Scholar]
- 45. Dongiovanni P, Romeo S, Valenti L. Hepatocellular carcinoma in nonalcoholic fatty liver: role of environmental and genetic factors. World J Gastroenterol. 2014;20(36):12945‐12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stepanova M, Rafiq N, Younossi ZM. Components of metabolic syndrome are independent predictors of mortality in patients with chronic liver disease: a population‐based study. Gut. 2010;59(10):1410‐1415. [DOI] [PubMed] [Google Scholar]
- 47. Zoppini G, Fedeli U, Gennaro N, Saugo M, Targher G, Bonora E. Mortality from chronic liver diseases in diabetes. Am J Gastroenterol. 2014;109(7):1020‐1025. [DOI] [PubMed] [Google Scholar]
- 48. Pang Y, Kartsonaki C, Turnbull I, et al. Diabetes, plasma glucose, and incidence of fatty liver, cirrhosis, and liver cancer: a prospective study of 0.5 million people. Hepatology. 2018;68(4):1308‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lonardo A, Ballestri S, Targher G. "Not all forms of NAFLD were created equal". Do metabolic syndrome‐related NAFLD and PNPLA3‐related NAFLD exert a variable impact on the risk of early carotid atherosclerosis? Atherosclerosis. 2017;257:253‐255. [DOI] [PubMed] [Google Scholar]
- 50. Targher G, Lonardo A, Byrne CD. Nonalcoholic fatty liver disease and chronic vascular complications of diabetes mellitus. Nat Rev Endocrinol. 2018;14(2):99‐114. [DOI] [PubMed] [Google Scholar]
- 51. Targher G, Bertolini L, Poli F, et al. Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes. 2005;54(12):3541‐3546. [DOI] [PubMed] [Google Scholar]
- 52. Targher G, Bertolini L, Rodella S, et al. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care. 2007;30(8):2119‐2121. [DOI] [PubMed] [Google Scholar]
- 53. Anstee QM, Mantovani A, Tilg H, Targher G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2018;15(7):425‐439. [DOI] [PubMed] [Google Scholar]
- 54. Mantovani A, Dauriz M, Sandri D, et al. Association between non‐alcoholic fatty liver disease and risk of atrial fibrillation in adult individuals: An updated meta‐analysis. Liver Int. 2019;39(4):758‐769. [DOI] [PubMed] [Google Scholar]
- 55. Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non‐alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta‐analysis. J Hepatol. 2016;65(3):589‐600. [DOI] [PubMed] [Google Scholar]
- 56. Targher G, Byrne CD. Non‐alcoholic fatty liver disease: an emerging driving force in chronic kidney disease. Nat Rev Nephrol. 2017;13(5):297‐310. [DOI] [PubMed] [Google Scholar]
- 57. Targher G, Chonchol M, Bertolini L, et al. Increased risk of CKD among type 2 diabetics with nonalcoholic fatty liver disease. J Am Soc Nephrol. 2008;19(8):1564‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mantovani A, Zaza G, Byrne CD, et al. Nonalcoholic fatty liver disease increases risk of incident chronic kidney disease: A systematic review and meta‐analysis. Metabolism. 2018;79:64‐76. [DOI] [PubMed] [Google Scholar]
- 59. Romeo S, Sanyal A, Valenti L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab. 2020;31(1):35‐45. [DOI] [PubMed] [Google Scholar]
- 60. Dongiovanni P, Valenti L. Genetics of nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1026‐1037. [DOI] [PubMed] [Google Scholar]
- 61. Della Corte C, Fintini D, Giordano U, et al. Fatty liver and insulin resistance in children with hypobetalipoproteinemia: the importance of aetiology. Clin Endocrinol (Oxf). 2013;79(1):49‐54. [DOI] [PubMed] [Google Scholar]
- 62. Visser ME, Lammers NM, Nederveen AJ, et al. Hepatic steatosis does not cause insulin resistance in people with familial hypobetalipoproteinaemia. Diabetologia. 2011;54(8):2113‐2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Amaro A, Fabbrini E, Kars M, et al. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology. 2010;139(1):149‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lonardo A, Lombardini S, Scaglioni F, et al. Hepatic steatosis and insulin resistance: does etiology make a difference? J Hepatol. 2006;44(1):190‐196. [DOI] [PubMed] [Google Scholar]
- 65. Noto D, Arca M, Tarugi P, Cefalù AB, Barbagallo CM, Averna MR. Association between familial hypobetalipoproteinemia and the risk of diabetes. Is this the other side of the cholesterol‐diabetes connection? A systematic review of literature. Acta Diabetol. 2017;54(2):111‐122. [DOI] [PubMed] [Google Scholar]
- 66. Tanoli T, Yue P, Yablonskiy D, Schonfeld G. Fatty liver in familial hypobetalipoproteinemia: roles of the APOB defects, intra‐abdominal adipose tissue, and insulin sensitivity. J Lipid Res. 2004;45(5):941‐947. [DOI] [PubMed] [Google Scholar]
- 67. Schonfeld G, Patterson BW, Yablonskiy DA, et al. Fatty liver in familial hypobetalipoproteinemia: triglyceride assembly into VLDL particles is affected by the extent of hepatic steatosis. J Lipid Res. 2003;44(3):470‐478. [DOI] [PubMed] [Google Scholar]
- 68. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Valenti L, Al‐Serri A, Daly AK, et al. Homozygosity for the patatin‐like phospholipase‐3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51(4):1209‐1217. [DOI] [PubMed] [Google Scholar]
- 70. Palmer CN, Maglio C, Pirazzi C, et al. Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS One. 2012;7(6):e39362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kotronen A, Johansson LE, Johansson LM, et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia. 2009;52(6):1056‐1060. [DOI] [PubMed] [Google Scholar]
- 72. Sookoian S, Pirola CJ. Meta‐analysis of the influence of I148M variant of patatin‐like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53(6):1883‐1894. [DOI] [PubMed] [Google Scholar]
- 73. Romeo S, Sentinelli F, Dash S, et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obes (Lond). 2010;34(1):190‐194. [DOI] [PubMed] [Google Scholar]
- 74. Kantartzis K, Peter A, Machicao F, et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin‐like phospholipase 3 gene. Diabetes. 2009;58(11):2616‐2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Speliotes EK, Butler JL, Palmer CD, et al. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology. 2010;52(3):904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Park JH, Cho B, Kwon H, et al. I148M variant in PNPLA3 reduces central adiposity and metabolic disease risks while increasing nonalcoholic fatty liver disease. Liver Int. 2015;35(12):2537‐2546. [DOI] [PubMed] [Google Scholar]
- 77. Romeo S, Sentinelli F, Cambuli VM, et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J Hepatol. 2010;53(2):335‐338. [DOI] [PubMed] [Google Scholar]
- 78. Samuel VT, Shulman GI. Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Ceramides. N Engl J Med. 2019;381(19):1866‐1869. [DOI] [PubMed] [Google Scholar]
- 79. Imamura F, Sharp SJ, Koulman A, et al. A combination of plasma phospholipid fatty acids and its association with incidence of type 2 diabetes: The EPIC‐InterAct case‐cohort study. PLoS Medicine. 2017;14(10):e1002409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mannerås‐Holm L, Schönke M, Brozinick JT, et al. Diacylglycerol kinase ε deficiency preserves glucose tolerance and modulates lipid metabolism in obese mice. J Lipid Res. 2017;58(5):907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Montgomery MK, Brown SH, Lim XY, et al. Regulation of glucose homeostasis and insulin action by ceramide acyl‐chain length: A beneficial role for very long‐chain sphingolipid species. Biochim Biophys Acta. 2016;1861(11):1828‐1839. [DOI] [PubMed] [Google Scholar]
- 82. Jordy AB, Kraakman MJ, Gardner T, et al. Analysis of the liver lipidome reveals insights into the protective effect of exercise on high‐fat diet‐induced hepatosteatosis in mice. Am J Physiol Endocrinol Metab. 2015;308(9):E778‐E791. [DOI] [PubMed] [Google Scholar]
- 83. Wang CW, Lin HY, Shin SJ, et al. The PNPLA3 I148M polymorphism is associated with insulin resistance and nonalcoholic fatty liver disease in a normoglycaemic population. Liver Int. 2011;31(9):1326‐1331. [DOI] [PubMed] [Google Scholar]
- 84. Koo BK, Joo SK, Kim D, et al. Additive effects of PNPLA3 and TM6SF2 on the histological severity of non‐alcoholic fatty liver disease. J Gastroenterol Hepatol. 2018;33(6):1277‐1285. [DOI] [PubMed] [Google Scholar]
- 85. Xia MF, Lin HD, Chen LY, et al. The PNPLA3 rs738409 C>G variant interacts with changes in body weight over time to aggravate liver steatosis, but reduces the risk of incident type 2 diabetes. Diabetologia. 2019;62(4):644‐654. [DOI] [PubMed] [Google Scholar]
- 86. Machado CM, Leite NC, França PH, Cardoso CR, Salles GF, Villela‐Nogueira CA. PNPLA3 gene polymorphism in Brazilian patients with type 2 diabetes: A prognostic marker beyond liver disease? Nutr Metab Cardiovasc Dis. 2019;29(9):965‐971. [DOI] [PubMed] [Google Scholar]
- 87. Kozlitina J, Smagris E, Stender S, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dongiovanni P, Petta S, Maglio C, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61(2):506‐514. [DOI] [PubMed] [Google Scholar]
- 89. Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 2018;283(4):356‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem. 2016;291(20):10659‐10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Prill S, Caddeo A, Baselli G, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep. 2019;9(1):11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536(7614):41‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim DS, Jackson AU, Li YK, et al. Novel association of TM6SF2 rs58542926 genotype with increased serum tyrosine levels and decreased apoB‐100 particles in Finns. J Lipid Res. 2017;58(7):1471‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Musso G, Cipolla U, Cassader M, et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J Lipid Res. 2017;58(6):1221‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhou Y, Llauradó G, Orešič M, Hyötyläinen T, Orho‐Melander M, Yki‐Järvinen H. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. J Hepatol. 2015;62(3):657‐663. [DOI] [PubMed] [Google Scholar]
- 96. Caddeo A, Jamialahmadi O, Solinas G, et al. MBOAT7 is anchored to endomembranes by six transmembrane domains. J Struct Biol. 2019;206(3):349‐360. [DOI] [PubMed] [Google Scholar]
- 97. Luukkonen PK, Zhou Y, Hyötyläinen T, et al. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non‐alcoholic fatty liver disease in humans. J Hepatol. 2016;65(6):1263‐1265. [DOI] [PubMed] [Google Scholar]
- 98. Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7‐TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology. 2016;150(5):1219‐1230.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Meroni M, Dongiovanni P, Longo M, et al. Mboat7 down‐regulation by hyper‐insulinemia induces fat accumulation in hepatocytes. EBioMedicine. 2020;52:102658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tanaka Y, Shimanaka Y, Caddeo A, et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut. 2020. 10.1136/gutjnl-2020-320646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Viitasalo A, Eloranta AM, Atalay M, Romeo S, Pihlajamäki J, Lakka TA. Association of MBOAT7 gene variant with plasma ALT levels in children: the PANIC study. Pediatr Res. 2016;80(5):651‐655. [DOI] [PubMed] [Google Scholar]
- 102. Umano GR, Caprio S, Di Sessa A, et al. The rs626283 Variant in the MBOAT7 Gene is Associated with Insulin Resistance and Fatty Liver in Caucasian Obese Youth. Am J Gastroenterol. 2018;113(3):376‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Beer NL, Tribble ND, McCulloch LJ, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18(21):4081‐4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Valenti L, Alisi A, Nobili V. Unraveling the genetics of fatty liver in obese children: additive effect of P446L GCKR and I148M PNPLA3 polymorphisms. Hepatology. 2012;55(3):661‐663. [DOI] [PubMed] [Google Scholar]
- 105. Vaxillaire M, Cavalcanti‐Proença C, Dechaume A, et al. The common P446L polymorphism in GCKR inversely modulates fasting glucose and triglyceride levels and reduces type 2 diabetes risk in the DESIR prospective general French population. Diabetes. 2008;57(8):2253‐2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sparsø T, Andersen G, Nielsen T, et al. The GCKR rs780094 polymorphism is associated with elevated fasting serum triacylglycerol, reduced fasting and OGTT‐related insulinaemia, and reduced risk of type 2 diabetes. Diabetologia. 2008;51(1):70‐75. [DOI] [PubMed] [Google Scholar]
- 108. Saxena R, Voight BF, Lyssenko V, et al. Genome‐wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331‐1336. [DOI] [PubMed] [Google Scholar]
- 109. Speliotes EK, Yerges‐Armstrong LM, Wu J, et al. Genome‐wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3):e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Qi Q, Wu Y, Li H, et al. Association of GCKR rs780094, alone or in combination with GCK rs1799884, with type 2 diabetes and related traits in a Han Chinese population. Diabetologia. 2009;52(5):834‐843. [DOI] [PubMed] [Google Scholar]
- 111. Takeuchi F, Katsuya T, Chakrewarthy S, et al. Common variants at the GCK, GCKR, G6PC2‐ABCB11 and MTNR1B loci are associated with fasting glucose in two Asian populations. Diabetologia. 2010;53(2):299‐308. [DOI] [PubMed] [Google Scholar]
- 112. Kitamoto A, Kitamoto T, Nakamura T, et al. Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits. Endocr J. 2014;61(7):683‐689. [DOI] [PubMed] [Google Scholar]
- 113. Orho‐Melander M, Melander O, Guiducci C, et al. Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C‐reactive protein but lower fasting glucose concentrations. Diabetes. 2008;57(11):3112‐3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bi M, Kao WH, Boerwinkle E, et al. Association of rs780094 in GCKR with metabolic traits and incident diabetes and cardiovascular disease: the ARIC Study. PLoS One. 2010;5(7):e11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li H, Xu R, Peng X, Wang Y, Wang T. Association of glucokinase regulatory protein polymorphism with type 2 diabetes and fasting plasma glucose: a meta‐analysis. Mol Biol Rep. 2013;40(6):3935‐3942. [DOI] [PubMed] [Google Scholar]
- 116. Wang H, Liu L, Zhao J, et al. Large scale meta‐analyses of fasting plasma glucose raising variants in GCK, GCKR, MTNR1B and G6PC2 and their impacts on type 2 diabetes mellitus risk. PLoS One. 2013;8(6):e67665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55(3):781‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Barker A, Sharp SJ, Timpson NJ, et al. Association of genetic Loci with glucose levels in childhood and adolescence: a meta‐analysis of over 6,000 children. Diabetes. 2011;60(6):1805‐1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Saxena R, Hivert MF, Langenberg C, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42(2):142‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Abul‐Husn NS, Cheng X, Li AH, et al. A Protein‐Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med. 2018;378(12):1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pirola CJ, Garaycoechea M, Flichman D, et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J Lipid Res. 2019;60(1):176‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Stender S, Romeo S. HSD17B13 as a promising therapeutic target against chronic liver disease. Liver Int. 2020;40(4):756‐757. [DOI] [PubMed] [Google Scholar]
- 123. Luukkonen PK, Tukiainen T, Juuti A, et al. Hydroxysteroid 17‐β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI . Insight. 2020;5(5): 10.1172/jci.insight.132158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liu Z, Zhang Y, Graham S, et al. Causal relationships between NAFLD, T2D and obesity have implications for disease subphenotyping. J Hepatol. 2020. 10.1016/j.jhep.2020.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Stender S, Kozlitina J, Nordestgaard BG, Tybjærg‐Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49(6):842‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Orsi E, Grancini V, Menini S, Aghemo A, Pugliese G. Hepatogenous diabetes: Is it time to separate it from type 2 diabetes? Liver Int. 2017;37(7):950‐962. [DOI] [PubMed] [Google Scholar]
- 127. Pelusi S, Valenti L. Hepatic fat as clinical outcome and therapeutic target for nonalcoholic fatty liver disease. Liver Int. 2019;39(2):250‐256. [DOI] [PubMed] [Google Scholar]
- 128. Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cusi K, Orsak B, Bril F, et al. Long‐Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med. 2016;165(5):305‐315. [DOI] [PubMed] [Google Scholar]
- 130. Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non‐alcoholic steatohepatitis (LEAN): a multicentre, double‐blind, randomised, placebo‐controlled phase 2 study. Lancet. 2016;387(10019):679‐690. [DOI] [PubMed] [Google Scholar]
- 131. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet. 2015;385(9972):956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Friedman SL, Ratziu V, Harrison SA, et al. A randomized, placebo‐controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67(5):1754‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ratziu V, Sanyal A, Harrison SA, et al. Cenicriviroc Treatment for Adults with Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology. 2020. 10.1002/hep.31108 [DOI] [PubMed] [Google Scholar]
- 134. Byrne CD, Targher G. NAFLD as a driver of chronic kidney disease. J Hepatol. 2020;72(4):785‐801. [DOI] [PubMed] [Google Scholar]
- 135. Mantovani A, Zusi C, Sani E, et al. Association between PNPLA3rs738409 polymorphism decreased kidney function in postmenopausal type 2 diabetic women with or without non‐alcoholic fatty liver disease. Diabetes Metab. 2019;45(5):480‐487. [DOI] [PubMed] [Google Scholar]
- 136. Mantovani A, Taliento A, Zusi C, et al. PNPLA3 I148M gene variant and chronic kidney disease in type 2 diabetic patients with NAFLD: Clinical and experimental findings. Liver Int. 2020;40(5):1130–1141. 10.1111/liv.14419 [DOI] [PubMed] [Google Scholar]
- 137. Bonetti S, Trombetta M, Boselli ML, et al. Variants of GCKR affect both β‐cell and kidney function in patients with newly diagnosed type 2 diabetes: the Verona newly diagnosed type 2 diabetes study 2. Diabetes Care. 2011;34(5):1205‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Deshmukh HA, Palmer CN, Morris AD, Colhoun HM. Investigation of known estimated glomerular filtration rate loci in patients with type 2 diabetes. Diabet Med. 2013;30(10):1230‐1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Brouwers MCGJ, Simons N, Stehouwer CDA, Isaacs A. Non‐alcoholic fatty liver disease and cardiovascular disease: assessing the evidence for causality. Diabetologia. 2020;63(2):253‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Adams LA, Anstee QM, Tilg H, Targher G. Non‐alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. 2017;66(6):1138‐1153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.