

Abstract
Objective
To assess whether reshaping of the immune balance by infusion of autologous natural regulatory T cells (nTregs) in patients after kidney transplantation is safe, feasible, and enables the tapering of lifelong high dose immunosuppression, with its limited efficacy, adverse effects, and high direct and indirect costs, along with addressing several key challenges of nTreg treatment, such as easy and robust manufacturing, danger of over immunosuppression, interaction with standard care drugs, and functional stability in an inflammatory environment in a useful proof-of-concept disease model.
Design
Investigator initiated, monocentre, nTreg dose escalation, phase I/IIa clinical trial (ONEnTreg13).
Setting
Charité-University Hospital, Berlin, Germany, within the ONE study consortium (funded by the European Union).
Participants
Recipients of living donor kidney transplant (ONEnTreg13, n=11) and corresponding reference group trial (ONErgt11-CHA, n=9).
Interventions
CD4+ CD25+ FoxP3+ nTreg products were given seven days after kidney transplantation as one intravenous dose of 0.5, 1.0, or 2.5-3.0×106 cells/kg body weight, with subsequent stepwise tapering of triple immunosuppression to low dose tacrolimus monotherapy until week 48.
Main outcome measures
The primary clinical and safety endpoints were assessed by a composite endpoint at week 60 with further three year follow-up. The assessment included incidence of biopsy confirmed acute rejection, assessment of nTreg infusion related adverse effects, and signs of over immunosuppression. Secondary endpoints addressed allograft functions. Accompanying research included a comprehensive exploratory biomarker portfolio.
Results
For all patients, nTreg products with sufficient yield, purity, and functionality could be generated from 40-50 mL of peripheral blood taken two weeks before kidney transplantation. None of the three nTreg dose escalation groups had dose limiting toxicity. The nTreg and reference groups had 100% three year allograft survival and similar clinical and safety profiles. Stable monotherapy immunosuppression was achieved in eight of 11 (73%) patients receiving nTregs, while the reference group remained on standard dual or triple drug immunosuppression (P=0.002). Mechanistically, the activation of conventional T cells was reduced and nTregs shifted in vivo from a polyclonal to an oligoclonal T cell receptor repertoire.
Conclusions
The application of autologous nTregs was safe and feasible even in patients who had a kidney transplant and were immunosuppressed. These results warrant further evaluation of Treg efficacy and serve as the basis for the development of next generation nTreg approaches in transplantation and any immunopathologies.
Trial registration
NCT02371434 (ONEnTreg13) and EudraCT:2011-004301-24 (ONErgt11).
Introduction
Adoptive cellular therapies based on thymus derived natural regulatory T cells (nTregs) are promising candidates for sustainable reshaping of undesired immune reactions in various medical indications.1 2 Conventional immunosuppression targets undesired effector mechanisms, but unfortunately also protective pathways like nTreg control. Consequently, in patients undergoing immunosuppression the immune balance remains disturbed and chronic immunosuppression is often necessary. Reshaping the immune balance by nTreg infusion in immunopathology related diseases might overcome the need for lifelong high dose immunosuppression with its limited efficacy, adverse effects, and high direct and indirect costs. Preclinical studies have already shown the ability of nTregs to delay or prevent graft rejection after solid organ transplantation or graft versus host disease, and to control autoimmunity and undesired immunogenicity of biological drugs and advanced treatment products after adoptive transfer in vivo. However, several open questions and obstacles remain before adoptive cellular treatment with nTregs can be widely implemented.
Transplantation with long term immunosuppression is the current standard of care in solid organ transplantation.3 4 Despite progress in one year patient and allograft survival, long term results have not improved during the past two decades. Chronic rejection and long term complications of chronic immunosuppression such as infections, malignancies, cardiovascular diseases, and pharmacological toxicity associated graft failure (eg, chronic allograft nephropathy) continue to be issues, which have spurred the search for novel treatment strategies.5 6 7 8 9 10 Therefore, minimising or weaning long term immunosuppression after solid organ transplantation remains a major goal.4 Several promising approaches have failed mainly because of the high clonal size of alloreactive naïve and memory or effector T cells and B cells.4 Macrochimerism inducing protocols have shown promising results of tolerance induction, but their complexity limits broad implementation.11
CD4+ CD25+ FoxP3+ nTregs have been identified as key players of immune homoeostasis and are now a major research focus in transplantation.12 13 14 15 16 In a preclinical kidney transplantation model, which mimics the cellular presensitisation frequently found in patients undergoing transplantation, regulatory T cells (Tregs) combined with calcineurin inhibitor and T cell depletion controlled donor reactive memory T cells without the need for permanent calcineurin inhibitor treatment.12 Although clinical experience with nTregs is limited, during the past decade a few phase I clinical trials of Treg treatments have been carried out for different clinical indications. Two small studies have included patients undergoing kidney transplantation and more recently patients with coronavirus disease 2019.1 2 17 18 However, crucial knowledge gaps still exist about the optimal use of Treg treatment, reflected among the large heterogeneity in good manufacturing practice (GMP) protocols, dosing, and timing of Treg applications.
We used a single dose of nTreg treatment after living donor kidney transplantation in an investigator initiated phase I/IIa trial, accompanied by an extensive biomarker programme, as a proof-of-principle study. We addressed several issues relating to the broad implementation of nTreg treatment for various indications, including easy and robust manufacturing, risk of over immunosuppression, interaction with standard care drugs, and biomarkers for monitoring safety and efficacy.
Methods
Trial objectives, study design, safety, and efficacy monitoring
Our trials were independent studies within the European Union funded ONE study consortium, applying different site specific adoptive cellular therapies in patients undergoing living donor kidney transplantation in conjunction with using a consensus immunosuppression protocol and biomarker portfolio in comparison to the reference patients.19 20
The primary objective of the ONEnTreg13 phase I/IIa trial was to assess safety and feasibility of our in-house developed autologous CD4+ CD25+ FoxP3+ nTreg product in patients undergoing living donor kidney transplantation (n=11). The results were compared with the ONErgt11-CHA reference trial (n=9), which was conducted at our centre before the ONEnTreg13 trial, to establish safety margins and biomarker panels (fig S1A). We used a composite primary safety endpoint, consisting of adverse infusion related effects, infections, acute rejection, and graft function or failure (table S1). The secondary objective of the trial was to evaluate whether nTregs allow safe tapering of conventional maintenance immunosuppression from triple drug treatment to monotherapy within 48 weeks of transplantation (fig 1). Figure 1, figure S1, and tables S1 and S2 summarise the primary and secondary trial objectives and endpoints, the assessment of adverse events and efficacy, and patient eligibility criteria. Baseline parameters were similar for the reference group and the nTreg group (table 1 and table S5). Additionally, both groups followed the same approach (fig 1 and table 2) for visits and initial immunosuppression (except the nTreg group did not receive induction treatment with the anti-interleukin 2 receptor basiliximab), and the adjunct biomarker programme.
Fig 1.
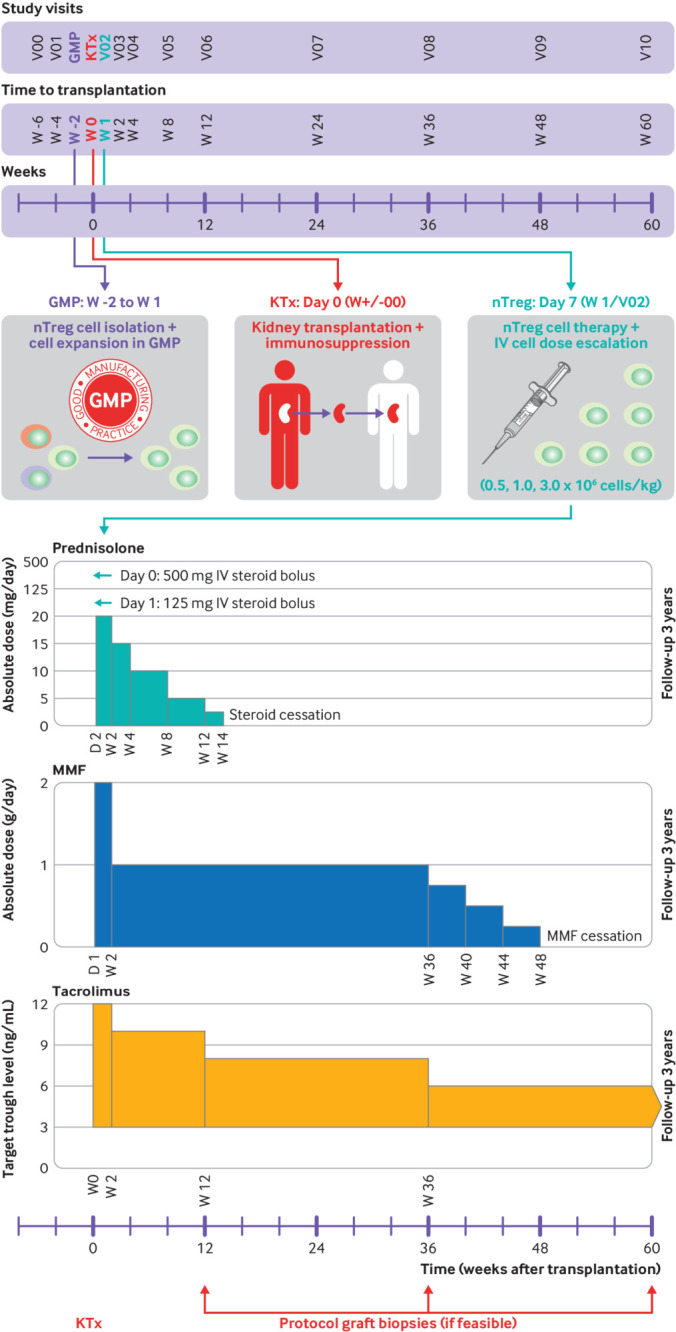
ONE study ONEnTreg13 clinical trial design. Upper panel: time schedule of ONEnTreg13 clinical trial: patient enrolment, cell collection, living donor kidney transplantation, nTreg adoptive cellular therapy (dose escalation of 0.5, 1.0, or 2.5-3.0×106 fresh cells/kg of body weight), and primary 60 week study follow-up. Lower panel: overview of protocol immunosuppressive regimen of ONEnTreg13 clinical trial with doses adjusted to specified levels at indicated time points: first steroid reduction until week 14, followed by MMF reduction at week 36-48, with continuation of tacrolimus monotherapy in nTreg group until study end at week 60 and three year follow-up. GMP=good manufacturing practice; IV=intravenous; KTx=kidney transplantation; MMF=mycophenolate mofetil; nTreg=natural regulatory T cell
Table 1.
Baseline characteristics of patients before transplantation. Data are numbers unless indicated otherwise
| Characteristics | Charité nTreg group ONEnTreg13 (n=11) | Charité reference group ONErgt11-CHA (n=9) |
|---|---|---|
| Median (range) recipient age (years) | 36 (33-58) | 43 (30-63) |
| Recipient female/male | 5/6 | 4/5 |
| Median (range) donor age (years) | 56 (33-63) | 53 (30-67) |
| Donor related/unrelated | 7/4 | 7/2 |
| Cause of end stage renal disease | ||
| Glomerulonephritis or vasculitis | 4 | 5 |
| Diabetic nephropathy | 2 | 2 |
| Polycystic kidney disease | 2 | — |
| Uropathy | 1 | — |
| Other or undetermined | 2 | 2 |
| Median (range) time receiving dialysis (months) | 11 (0-110) | 14 (4-118) |
| Human leukocyte antigen mismatch | ||
| 0 | 2 | 1 |
| 1-3 | 7 | 6 |
| 4-6 | 2 | 2 |
| Panel reactive antibody test | ||
| <10% | 10 | 8 |
| >10% | 1 | 1 |
| Cytomegalovirus serology donor/recipient | ||
| D−/R− | 3 | 3 |
| D-/R+ | 2 | — |
| D+/R− | 2 | 1 |
| D+/R+ | 4 | 5 |
nTreg=natural regulatory T cell.
Table 2.
Clinical outcomes after 60 week study period and three year follow-up. Data are numbers unless indicated otherwise
| Clinical outcome | 60 weeks | 3 years | |||||
|---|---|---|---|---|---|---|---|
| nTreg (n=11) | Reference (n=9) | P value | nTreg (n=11) | Reference (n=9) | P value | ||
| Immunosuppression | |||||||
| Monotherapy (tacrolimus only) | 8 | 0 | 0.002 | 8 | — | 0.001 | |
| Dual therapy (tacrolimus, MMF) | — | 5 | — | 4 | |||
| Triple therapy (tacrolimus, MMF, steroid) | 3 | 4 | 3 | 5 | |||
| Alloreactivity | |||||||
| T cell mediated acute rejection | 0.56 | 1.00 | |||||
| Borderline | — | 1 | — | — | |||
| Ia/Ib | 1 | 1 | — | — | |||
| IIa/IIb/III | 1 | — | — | — | |||
| Mixed T cell and antibody mediated acute rejection | 1 | — | — | — | |||
| De novo donor specific antibodies | 2 | — | 0.18 | 2 | 1 | 0.66 | |
| Infections and malignancies | |||||||
| Cytomegalovirus viraemia | 1 | 1 | 0.51 | — | — | 1.00 | |
| Cytomegalovirus disease | — | — | 1.00 | — | 1 | 0.26 | |
| Polyomavirus viraemia | — | 1 | 0.26 | — | — | 1.00 | |
| Other infections* | 1 | 1 | 0.88 | — | — | 1.00 | |
| Cancer | — | — | 1.00 | — | 1 | 0.26 | |
| Graft function | |||||||
| Delayed graft function | — | 2 | 0.10 | — | — | 1.00 | |
| Median (range) creatinine (mg/dL) | 1.6 (1.3-1.8) | 1.1 (1.0-2.0) | 0.65 | 1.5 (1.2-1.6) | 1.2 (0.9-2.0) | 0.80 | |
| Median (range) estimated glomerular filtration rate (mL/min) | 51 (46-58) | 60 (37-64) | 0.45 | 53 (47-49) | 59 (38-66) | 0.79 | |
| Median (range) proteinuria (mg/g Krea) | 192 (92-336) | 130 (93-430) | 0.89 | 120 (89-221) | 151 (62-278) | 0.89 | |
MMF=mycophenolate mofetil; nTreg=natural regulatory T cell.
nTreg and reference groups were compared after 60 weeks (study endpoint) and at three year follow-up.
Urinary tract infection and pneumonia.
In total, 17 patients were assessed two weeks before kidney transplantation for eligibility to be enrolled in the ONEnTreg13 trial. However, six patients did not receive the nTreg product because of patient related issues occurring before cell infusion which was scheduled seven days after surgery (such as kidney transplantation not performed, surgical complications, withdrawal of consent, early biopsy proven acute rejection; fig S1). The remaining 11 patients were split into cohorts of three or four patients and received nTregs in escalating doses of 0.5, 1.0, or 2.5-3.0×106 fresh cells/kg of body weight seven days after kidney transplantation. Because the targeted dose of 3.0×106 cells/kg could not be obtained in two patients owing to cell loss at bead depletion, two patients in the high dose group received only 2.5×106 cells/kg body weight. The cells were administered through slow intravenous infusion over 30 minutes by Perfusor Space pump (fig 1 upper panel). Paracetamol (acetaminophen) and antihistamine were given prophylactically before cell infusion. With the exception of basiliximab (to prevent targeting of Tregs), nTreg treatment was used in conjunction with the standard immunosuppression regimen (steroids, mycophenolate mofetil, and tacrolimus) as adjunct treatment with the intention to reduce maintenance immunosuppression (fig 1 lower panel).
GMP manufacturing and characterisation of nTregs
The clinical grade nTregs were manufactured at our in-house GMP facility (Berlin Institute of Health Center for Regenerative Therapies, and Berlin Center for Advanced Therapies).21 Table S3 gives an overview of the process and materials. We were able to establish a robust GMP process for isolation and expansion of autologous polyclonal nTregs with good purity and sufficient yield (table 3) from 40-50 mL peripheral blood samples. We studied whether our nTreg manufacturing process is feasible for patients before kidney transplantation in terms of robustness, yield, and product composition. In total, we generated 17 nTreg products from patients before kidney transplantation and 11 were administered to patients in the ONEnTreg13 trial (table S4).
Table 3.
Robust GMP manufacturing of nTregs from small volumes of blood from patients with ESRD and healthy donors. Data are medians (minimum-maximum)
| Quality control analyses | Release criteria | Patients with ESRD (n=17)* | Healthy donors (n=6) |
|---|---|---|---|
| Viability (%) | ≥70 | 96 (87-97.5) | 96.9 (95.27-98.2) |
| Purity | |||
| CD4+ CD25+ FoxP3+ (% of CD4+) | ≥70 | 95.44 (90.4-99.6) | 96.48 (94.55-98.82) |
| CD4+ CD25+ FoxP3+ (% of total) | FIO | 91.9 (80.8-99.6) | 94.0 (90.4-98.8) |
| Impurity | |||
| Interleukin 2 production (% of total) | ≤10 | 2.9 (0.15-6.05) | 0.82 (0-1.5) |
| Interferon γ production (% of total) | ≤10 | 2.2 (0.2-7) | 0.35 (0-1.14) |
| Starting material and cell yield | |||
| Blood collected (mL) | FIO | 45 (38-50) | 50 (50-50) |
| Treg cell No start (E+06) | FIO | 3.9 (2.3-8.02) | 5.5 (2.2-7) |
| Treg cell No end (E+09) | FIO | 4.7 (0.96-37.7) | 3.78 (0.73-13.4) |
ESRD=end stage renal disease; FIO=for information only (no release criteria); GMP=good manufacturing practice; nTreg=natural regulatory T cell.
Quality control analyses of 23 GMP nTreg products from validation and clinical trial runs. Figure S4 shows further details on the process.
Eleven products were infused seven days after kidney transplantation to Treg group.
For validation purposes and to better understand the manufacturing process and to study potential differences in product characteristics, we also generated nTreg products from healthy donors. A sufficient yield of nTregs (>1×109 cells) could be consistently generated from 40-50 mL of heparinised whole blood obtained by venipuncture from the patient’s forearm two weeks before kidney transplantation. All of the donors in the ONE study cohort had end stage renal disease and half were receiving dialysis. The Tregs were enriched by using the CliniMACS Plus system (CD8+ T cell depletion and CD25+ T cell enrichment) with subsequent stimulation and expansion for 23 days under nTreg promoting conditions, followed by magnetic expansion bead depletion (>99.997%) to obtain the final nTreg product (fig S4). We performed several analytical procedures before product release with validated methods (table 3), as described in detail in the supplementary methods (fig S4, tables S3 and S7). The nTreg products from all patients showed high expansion rates from the starting material (mean >2000-fold), high viability (mean 96%) and purity (mean 96% and 91% of CD4+ T cells and total cells, respectively), and low effector cytokine production (<10% interleukin 2 and interferon γ) upon phorbol myristate acetate or ionomycin activation. These data were comparable to those of healthy donors. T cell receptor repertoire analysis of our nTreg GMP products by next generation sequencing confirmed their polyclonal pattern, as described previously.22
Clinical monitoring and exploratory biomarker analysis
More than 100 parameters for determining safety, efficacy, and mechanism of action were monitored in the nTreg and reference groups over the study period of 60 weeks, with follow-up of up to three years, consisting of the consensus biomarker portfolio within the ONE study consortium19 and additional site specific markers. Monitoring included assessing biopsy confirmed acute rejection, clinical analysis and biochemical indices for renal function, and immune monitoring for measures of safety, immune activation or tolerance, and pharmacokinetics or dynamics, as outlined in supplementary table S7. Specifically, patient assessment included monitoring of viral load, monocytic human leukocyte antigen (HLA)-DR expression,23 standardised multiparameter flow cytometry analysis of 60 immune cell subsets according to the ONE study protocol20 24; monitoring of humoral and cellular allosensitisation by screening of antidonor HLA panel reactive antibodies with Luminex and donor reactive T cell frequencies with interferon γ ELISPOT25; cytokine measurements in plasma and urine; T cell receptor repertoire analysis of nTreg GMP products and circulating nTregs in patient blood26 27 28 29; and gene expression of selected tolerance or rejection pattern.30
Statistical analysis
A statistical analysis plan defined the conventions and analysis, and emphasised the exploratory nature of the study; accordingly the proposed statistical examination of clinical and immunological data was in principle descriptive.19 Table S6 summarises all statistical subset definitions, testing methods, and P values. We assessed clinical parameters and biomarkers over the whole time course by using non-parametric analysis of the variance type statistic with F approximation developed by Brunner and colleagues31 in a two way factorial repeated measures design (fig 2, fig 3, fig 4, and fig S2). Testing was conducted for significant interaction; that is, non-parallel response profiles of the two treatment groups and within group changes over time using the nparLD R package.32 Post hoc multiple comparisons between individual time points were performed with P value adjustment by using Holm’s method. A two tailed Wilcoxon matched pairs signed rank test was used when comparing two time points within the nTreg group. A P value less than 0.05 was considered statistically significant for all tests. All analyses were performed with R (version 3.5.1). GraphPad Prism software (version 8, GraphPad, La Jolla, CA) was used to generate graphs. Longitudinal data are presented as medians and interquartile ranges in all line graphs or individual comparisons as boxplots with whiskers indicating the data ranges.
Fig 2.
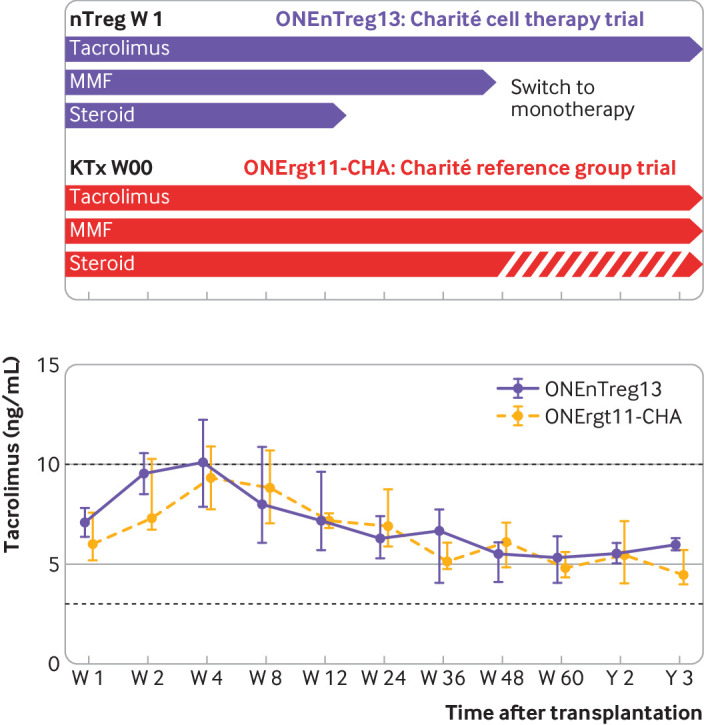
Autologous nTreg infusion might enable minimisation of immunosuppression in patients with kidney transplant. Tapering of immunosuppression in the nTreg trial group (switch to monotherapy) compared with reference group (continuous dual or triple drug regimen) with monitoring of patients’ tacrolimus levels (median±interquartile range, shown on lower panel). KTx=kidney transplantation; MMF=mycophenolate mofetil; nTreg=natural regulatory T cell
Fig 3.

Long term follow-up of renal allograft function (median±interquartile range): glomerular filtration rate (GFR, mL/min), serum creatinine (mg/dL), proteinuria (mg/day), and C reactive protein (CRP, mg/dL)
Fig 4.
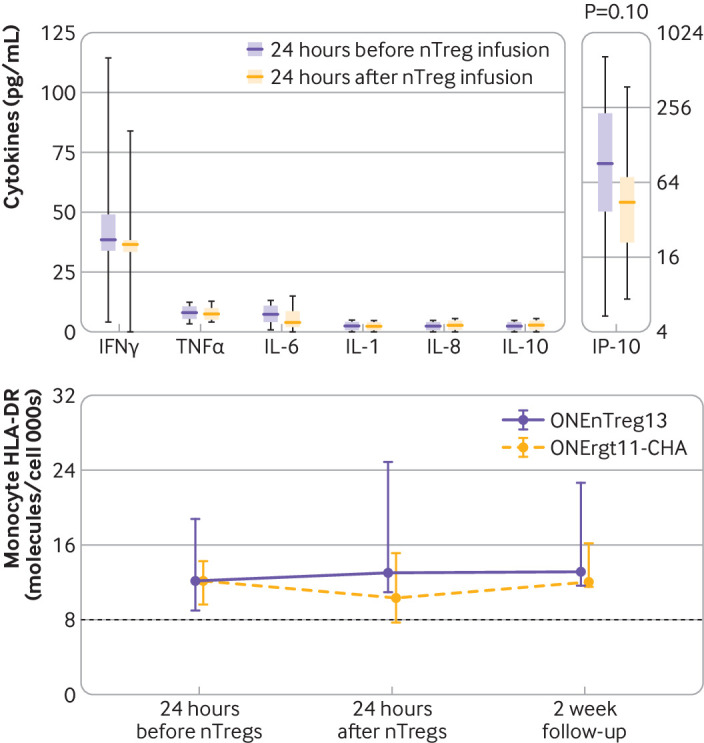
No immune complications after infusion: monitoring of cytokine release after infusion by measuring inflammatory cytokine serum or urinary levels (box plot, minimum-maximum range) with respective typical normal values (serum: TNFα<15.0 pg/mL, IFNγ=10-30 pg/mL, IL-6<5.0 pg/mL, IL-1<5.0 pg/mL, IL-8<10.0 pg/mL, IL-10<5.0 pg/mL, IP-10<200 pg/mL) and monocyte HLA-DR (molecules per cell) either 24 hours before or 24 hours after systemic nTreg infusion, including two week follow-up for HLA-DR. No significant differences between groups for any assessed parameters. HLA=human leukocyte antigen; IFNγ=interferon γ; IL=interleukin; IP-10= urinary interferon induced protein 10; nTreg=natural regulatory T cell; TNFα=tumour necrosis factor α
Patient and public involvement
No patients were involved in setting the research question, nor were they involved in the design and implementation of the study.
Results
Patients and study design
Two weeks before kidney transplantation, patients were assessed for eligibility and blood samples were collected to manufacture the nTreg products. The definitive enrolment of patients for cellular treatment and for the reference group took place seven days after kidney transplantation according to the inclusion and exclusion criteria (fig S1A and table S2). The clinical results of patients treated with Tregs (n=11) were compared with the ONE study reference group patients (n=9), who were enrolled at our centre by using the same criteria and who received the standard of care. Patient characteristics and initial immunosuppression were similar for the two groups (table 1), but the nTreg group did not receive anti-interleukin 2 receptor induction treatment to prevent interaction with the nTreg product. No apparent cell dose-response association was found for any of the parameters investigated.
Safety of nTreg treatment
The primary objective was to assess the safety of cellular treatment. At the time of this analysis after three years of follow-up, all patients in both groups are alive with good graft function. No serious adverse events have been reported; that is, immediate or long term adverse effects arising directly from nTreg cell infusion (fig S1B and table 2). Importantly, no differences were found in the following parameters: tacrolimus blood levels during the three year follow-up (fig 2); renal functional parameters (including estimated glomerular filtration rate, serum creatinine, proteinuria, serum albumin, and serum urea; fig 3 and fig S2A); liver functional parameters (including bilirubin, aspartate aminotransferase, alanine transaminase, and alkaline phosphatase; fig S2B); or systemic inflammation and other safety parameters (such as C reactive protein, urinary interferon induced protein 10 inflammation marker, fibrinogen, and free haemoglobin; fig 4 and fig S2C). In contrast to anticancer T cell treatment, we did not observe any cellular infusion related systemic increase of proinflammatory or anti-inflammatory plasma cytokines, such as interferon γ, tumour necrosis factor α, interleukin 1, interleukin 6, interleukin 8, interleukin 10, and urinary interferon induced protein 10 (fig 4). A trend was observed towards decreased intrarenal inflammation (P=0.1, fig 4). Furthermore, we did not observe any signs of immediate and long term over immunosuppression by nTreg treatment, as shown by monitoring monocytic HLA-DR expression (an immunocompetence marker; fig 4 lower panel), and the rate of infectious complications (eg, cytomegalovirus, Epstein-Barr virus, polyomavirus, urinary tract infections, septic events) and cancer until the three year follow-up (table 2).
First hints for efficacy of nTreg treatment
The secondary objective of the ONEnTreg13 trial was to evaluate whether nTregs allow safe tapering of conventional maintenance immunosuppression from triple drug treatment to monotherapy (fig 1 and fig S3). Weaning of immunosuppression to low dose tacrolimus monotherapy (trough blood levels <6 ng/mL) was achieved within 48 weeks for 10 of 11 patients receiving nTreg treatment (91%). One patient (C5058) was excluded from a switch to monotherapy as he developed a posterior reversible encephalopathy syndrome related to calcineurin inhibitor treatment early after kidney transplantation. Tacrolimus was extremely reduced (trough level 2-3 ng/mL) for more than six months to overcome this complication. When a protocol biopsy revealed acute cellular rejection Banff IB, the patient received antirejection treatment. He was kept on standard triple drug treatment with good three year graft function.
In two of the 10 patients who were weaned of immunosuppression, tacrolimus monotherapy failed: one patient (C5056) developed mixed acute cellular rejection Banff III, antibody mediated acute rejection, and donor specific antibodies shortly before the end of the 60 week study period. Graft function could be rescued by rejection treatment and reversal to triple drug immunosuppression with good three year function. In another patient (C5063) recurrence of the underlying renal disease (immunoglobulin A nephropathy) was observed in the protocol biopsy some weeks later that was associated with proteinuria. For safety reasons, immunosuppression was reversed to the triple drug regimen. Patient C5057 showed mild graft deterioration from uncontrolled type 2 diabetes mellitus and cardiac decompensation early on before protocol switch to monotherapy. He did not show any hints of alloreactivity or inflammation in the biomarkers and recompensation of heart failure and diabetes would probably have been sufficient to normalise kidney function. However, he received a short steroid bolus for safety reasons as the histological picture showed some cellular infiltrations (acute cellular rejection Banff IB). Later he was successfully weaned to tacrolimus monotherapy with excellent three year follow-up. During the three year follow-up, stable good graft function was observed in the eight patients from the nTreg group on low dose tacrolimus monotherapy, and in all three patients from the nTreg group who were kept on standard triple drug treatment. Figure S3 shows the individual patient courses.
The consensus protocol of the ONE study consortium for the reference group was to aim for a steroid free dual immunosuppression after week 12 and to manage patients with standard triple drug regimens in case of impending complications (functional deterioration, proteinuria, de novo donor specific antibodies). At the end of the 60 week study period, five of nine patients were on dual immunosuppression; at the three year follow-up four of nine patients were on dual immunosuppression. The remaining patients were receiving triple drug immunosuppression (P=0.002 and P=0.001; table 2 and fig 2).
Exploratory biomarker analysis of nTreg treatment
Analysis of a broad set of more than 100 exploratory biomarkers revealed only few significant differences between the two groups (table S6). Among the 59 immune cell subsets analysed by multiparameter flow cytometry, we observed significantly enhanced nTreg counts (CD4+ CD25high CD127low or CD4+ CD25high FoxP3+) with a favourable ratio of regulatory to effector T cells in the circulation for up to eight weeks after nTreg infusion with no apparent dose-response association (fig 5 and fig S7). By contrast, the reference group showed decreased nTreg levels compared with baseline for up to 12 weeks after kidney transplantation, partly owing to the basiliximab induction treatment,33 which supports sequestration of nTreg and internalisation of CD25. The nTreg group developed significantly fewer activated HLA-DR+ CD4+ non-Treg conventional T cells (P=0.018) and the counts of activated CD25+ 127high CD8+ conventional T cells did not change over the entire period in the nTreg group. However, in the reference group these cells were initially suppressed by basiliximab, but increased after week 12 in a rebound effect (P=0.012; fig S6 and table S6C-D). Additionally, in the reference group a shift was observed from the CD56dim to the more mature CD56high natural killer cell subset during the study period, while the ratio remained stable in the nTreg group (P=0.08). At the end of the 60 week study period, the nTreg group showed a slightly higher proportion of the marginal zone B cell subset in the circulation (P=0.045). All other immune cell subsets were not significantly affected by nTreg treatment compared with the reference group (fig S6).
Fig 5.

Exploratory biomarker analysis of therapy response to nTreg treatment. More than 100 biomarkers were analysed in patients receiving nTregs, with a more comprehensive summary presented in figures S5-S6. Patients receiving nTregs were evaluated according to cell dose applied and compared with reference group. Transiently enhanced Treg levels and favourable Treg:Teff ratio. Upper panels: multiparameter flow cytometry profiling of nTregs in patient blood either comparing nTreg with reference group or showing the infused cell dose (0.5, 1.0, and 2.5-3.0×106 cells/kg of body weight); table shows significance levels. Lower panels: ratio between CD4+ CD25high CD127low Treg levels relative to either CCR7– CD4+ or CCR7– CD8+Teffs shown as representative result for almost 60 immune cell subsets shown in figure S6; table shows significance levels. nTreg=natural regulatory T cell; Teff= effector T cell; Treg=regulatory T cell
To track nTregs in vivo, we monitored the T cell receptor repertoire of ex vivo sorted Tregs from patients at different time points after kidney transplantation and compared them with the respective infused nTreg product (fig 6 and fig S5). As expected, expanded clones covering more than 0.1% or even more than 1% of the T cell receptor repertoire were rarely detectable or not detectable at all within the polyclonal nTreg product, respectively, and together reached less than 15% of the total repertoire. The peripheral blood nTreg samples from the ONEnTreg13 group showed a significant clonal expansion within a few weeks after infusion, which persisted for up to 60 weeks. Up to 80% of the total nTreg T cell receptor repertoire consisted of in vivo expanded nTreg clones at individual frequencies of more than 0.1%; strongly expanded nTreg clones of more than 1% clonal size covered up to 60% (P<0.01 v baseline). Most of these in vivo expanded clones were detectable at low frequency in the respective nTreg product of the individual patient. Analysis of a tolerance or rejection gene expression panel30 in whole blood did not reveal significant differences, except for the rejection associated gene TMEM176B (TORID), which was downregulated to almost undetectable levels for the first 12 weeks in the nTreg group but not the reference group (group difference P=0.023).
Fig 6.

Decreasing clonal diversity by oligoclonal nTreg expansion. Next generation sequencing based TCR repertoire analysis of individual nTreg GMP products before infusion (GMP columns) and ex vivo Tregs isolated at indicated patient visits (week 4, 12, 36, and 60, according to sample availability) for all 11 patients receiving nTregs, with a more detailed summary shown in figure S5. GMP=good manufacturing practice; nTreg=natural regulatory T cell; TCR=T cell receptor; Treg=regulatory T cell
Discussion
We used kidney transplantation as a proof-of-concept disease model to address several challenges of nTreg cellular treatment for reshaping undesired immune reactions. We were able to show that GMP compliant production of nTreg products from patients with end stage renal disease who had several comorbidities is feasible, robust, and possible at reasonable costs (€17 500/product (£15 800; $20 500))—a prerequisite for trustworthy data from clinical adoptive cell treatment trials.34 35 36 In line with the main trial objective, we also showed that infusion of autologous nTregs at the end of the first week after kidney transplantation was well tolerated. No evidence was found of cell infusion related short term or long term adverse effects, and we observed no signs of over immunosuppression. All patients in the nTreg group and the reference group had good graft function until the three year follow-up despite stepwise minimising immunosuppression within the first 60 weeks in the nTreg group. The data support the feasibility of Treg treatment for many medical indications in solid organ transplantation and many other diseases with immunopathologies.
Strengths and limitations of this study
We were able to establish a robust GMP process for the isolation and expansion of autologous polyclonal nTregs with good purity and yield (table 3) from small peripheral blood samples of 40-50 mL. Starting sources normally use approximately 10 times this amount of blood or leukapheresis material.37 38 39 40 Therefore, the use of nTregs is a more feasible treatment option and its applicability can be widened to different medical indications.
Our attempt to taper the standard triple or dual immunosuppression regimen to less toxic monotherapy with low dose tacrolimus showed stable three year success in 73% of the patients treated with nTregs. In the remaining 27% of patients, in whom tapering to tacrolimus monotherapy was not feasible or had to be reversed, we observed good graft function after three years of follow-up with a standard triple drug immunosuppression regimen. In contrast, reference patients remained on gold standard dual or triple immunosuppression as further tapering of immunosuppression in this group was not justifiable for safety reasons. Despite the limitation of low numbers of patients, the biomarker programme revealed five observations.
Firstly, the absence of any signs of inflammatory reaction after cell infusion, as seen typically after anticancer T cell treatment, underlines the lack of significant effector cell contamination of the nTreg product as shown by the product release test. Secondly, we did not observe any clinical (infections) or laboratory signs of over immunosuppression, previously discussed as a putative safety issue of polyclonal Treg treatment.41 We suggest that without T cell receptor or CD28 stimulation, Tregs lose their activation within a few days or even die. Therefore, only Treg clones that are repeatedly antigen stimulated keep their suppressive capacity and can expand in vivo. Thirdly, we observed that the polyclonal T cell receptor repertoire of infused Treg products shifts in vivo over time to an oligoclonal pattern, suggesting an alloantigen driven selection process. Similarly, the replacement of polyclonal anti-CD3/28 monoclonal antibodies by alloantigen in vitro stimulation induces a biased T cell receptor repertoire within a few days,22 which suggests specific immunoregulation even after administration of polyclonal nTregs.
Fourthly, nTreg infusion induced only a temporary increase in Treg counts. The drop in circulating Tregs after four weeks might be explained by nTregs homing to the inflamed graft or lack of lasting engraftment. Because we hypothesise that only alloantigen triggered Tregs survive as suggested by the oligoclonal expansion, the clonal size might not be sufficient to be visible at total Treg counts. Finally, the nTreg group expressed less conventional T cell activation, natural killer cell maturation, and downregulation of the rejection associated gene TORID, which might give an indication of the mode of action of nTreg in vivo.
The study has some further limitations. As typical for cell treatment phase I/IIa trials, the number of patients is low, which limits the power of statistical analyses. Therefore, interpretation of the data must be done carefully and on a case-by-case basis. However, the consistency of many parameters with the same message makes core statements plausible (eg, no over immunosuppression, stable graft function despite weaning, inhibition of conventional T cell activation). In two patients, tapering of immunosuppression was not successful. The first patient developed mixed acute cellular rejection Banff III, antibody mediated acute rejection, and donor specific antibodies shortly before the study endpoint. This was the only patient in both groups with high levels of pre-existing donor specific T cell immunity. After successful intense rejection treatment, immunosuppression could be reversed to the triple drug regimen. The second patient had recurrence of the underlying kidney disease, immunoglobulin A nephropathy, an event observed in 10-60% of patients after kidney transplantation. Because high immunosuppression by thymoglobuline and weaning of steroids correlates with decreased and enhanced risk of immunoglobulin A nephropathy recurrence, respectively, we decided to change treatment back to the triple drug regimen.42 43 We have seen an improvement in proteinurea and stable graft function for more than three years in this patient. Therefore, in both patients treatment was successfully reversed to the triple drug regimen.
These data might suggest that nTregs together with low dose tacrolimus monotherapy cannot sufficiently control pre-existing pathogenic memory or effector immune cells. This hypothesis should be carefully studied in follow-up studies. The third patient on standard treatment in the nTreg group was not suitable for tapering because he developed tacrolimus related posterior reversible encephalopathy syndrome. We could speculate that the survival of his transplant with good three year function despite subtherapeutically low tacrolimus levels for several months (<2-3 ng/mL) might be partially because of nTreg mediated protection.
Comparison with other studies
Minimising the clinical need for chronic immunosuppression as early and as much as possible is a major task after transplantation because it offers the chance to diminish undesired immunosuppression associated comorbidities.4 Empirical approaches with calcineurin inhibitor sparing regimens that minimise,9 withdraw,44 or avoid calcineurin inhibitors45 have only been partially successful or have even failed to consistently show long term safety.4 46 At first view, Chan and colleagues47 reported promising data after temporary T cell or B cell depletion by alemtuzumab induction in patients who had kidney transplantation (73% on monotherapy after two years). However, several critical points should be noted: firstly, the Chan study reported a high rate of graft loss by graft deterioration or death within two years of follow-up (7.4%), whereas our trial reported 0% graft loss or death within three years of follow-up; secondly, the Chan study had a relatively high number of urinary tract infections (38%), whereas we observed only 10%; and thirdly, the target tacrolimus blood levels at the second half of the first year (and thereafter) were much higher in the Chan study than in our cell treatment group (mean approximately 9 ng/mL v <6 ng/mL tacrolimus). Other studies that used alemtuzumab induction showed higher rates of monotherapy failure, acute rejections, and infections.46 48
The challenge remains to find new approaches that enable sustainable reduction of immunosuppression, thereby diminishing undesirable treatment associated comorbidities.4 This incentive formed the rationale for the ONEnTreg13 clinical trial because adoptive transfer of nTregs is a promising option to reshape undesired alloimmunity. Currently there are more than 50 active and completed early clinical trials that examine the safety and efficacy of Treg treatment for indications such as solid organ transplantation, inflammatory disease, and autoimmune diseases.1 2 17 Recently, two Treg clinical trials have been reported in patients undergoing kidney transplantation. The Leventhal study treated nine patients in a dose escalating phase I/IIa trial with much higher cell doses (up to 15 times higher than our highest tier) at week 4 after kidney transplantation and patients were kept on full rapamycin or mycophenolate mofetil based immunosuppression, allowing only safety analysis. Similar to our observations, this study did not see any major dose dependent effects.22 Chandran and colleagues treated three patients with subclinical rejection months after kidney transplantation without safety issues.49 Our study adds several novel aspects: new, more widely applicable manufacturing processes; application of Tregs early after solid organ transplantation with concomitant tapering immunosuppression to low dose tacrolimus monotherapy; and extensive mechanistic analyses giving insights into the mode of action of nTreg application.
Conclusions
Administration of our nTreg product was safe, well tolerated, and met the primary and secondary objectives. To further improve the efficacy of this treatment and to reach stable low dose immunosuppression or even complete weaning in almost all patients, next generation Treg approaches are required to address more sustainable and functional engraftment of Tregs, redirected specificity, resistance to immunosuppressive drugs, control of pre-existing memory or effector T cell response, as well as randomised trials. We also need to have a better understanding of the optimal dose. Our robust GMP manufacturing process is a key enabling platform technology for the ongoing development of more advanced next generation genetically modified products, such as chimeric antigen receptor nTregs, to improve the efficacy of these highly promising adoptive cellular therapies.34 35 36 50 51
What is already known on this topic
No consensus protocol exists for tapering immunosuppression in patients after solid organ transplantation and pharmacological induction treatment; preclinical data have indicated adoptive transfer of regulatory T cells (Tregs) as a promising option
Although currently published protocols on good manufacturing practice are heterogeneous and inconsistent, several groups have been able to generate Treg products from peripheral blood, cord blood, or thymus with sufficient yield and purity if enough starting material is available; however, manufacturing protocols that are easy to implement and clear defined criteria of product characteristics are lacking
Data from 12 patients who received Treg treatment at later time points after kidney transplantation have been published with limited information on mechanistic side studies to better understand the mode of action and reasons for patients responding or not responding to treatment
What this study adds
Our natural Treg (nTreg) manufacturing process uses small amounts of blood and is easy to use, especially for patients with comorbidities and for those receiving solid organ transplantation from deceased donors, with a short time window for cell collection
These data show stable minimisation of immunosuppression in most patients receiving nTreg treatment after kidney transplantation
Our analyses indicate that inhibition of conventional T cell activation and natural killer cell maturation without signs of over immunosuppression and (allo)antigen driven clonal expansion in the nTreg T cell receptor repertoire might be important mechanisms of nTreg treatment
Acknowledgments
We thank all ONE study participants and staff for the intense discussion in the consortium and the joint development of the consensus reference group protocol and biomarker portfolio used for all site specific ONE study trials. We thank all further members of the BeCAT GMP facility (H Hoffmann, I Lehmann, M Stein, A Forke, C Diezemann), the nurses and staff in the KTx-ward, KTx-outclinic, and KTx-office (A Wilke, H Zobel, SM Fischer, P Hecker), and colleagues from the Department of Transfusion Medicine. Finally, we thank all patients who participated in the study.
Web extra.
Extra material supplied by authors
Web appendix: Supplementary materials
Contributors: AR and NMO contributed equally to this work. Clinical principal investigator of the study: PR; clinical study team: PR, NMO, GB, RO, TS, MC, AS, CG; clinical development and regulatory affairs: PR, MA; GMP manufacturing: PR, AR, DK, CB, SLK; preclinical studies and biomarkers: HDV, NB, BS, MS, SS, GM, DLW, MSH, LA, US; compilation of data and statistical analyses: GM, SS, KJ, NMO, GB, AS; manuscript writing: PR, GM, AR, NMO, HDV. All authors critically revised the paper for important intellectual content and approved the final version to be published. All authors had full access to all of the data in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. HDV and PR supervised the study and are the guarantors. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Funding: The preclinical work, clinical study, and data analysis were supported by the European Union, 7th EU Framework programme and Horizon 2020 programme (grants 260687 HEALTH-F5-2010-260687 (ONE study) and 825392 (ReSHAPE), respectively), the Federal Ministry of Education and Research (BMBF) under grant agreement BCRT and the Berlin Institute of Health (BIH). The funders had no role in considering the study design or in the collection, analysis, interpretation of data, writing of the report, or decision to submit the article for publication.
Competing interests: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf and declare: support from the European Union 7th EU Framework Programme and Horizon 2020 programme and the BMBF under grant agreement BCRT and the BIH for the submitted work; no direct funding or donations from private parties, including the pharmaceutical industry; PR, HDV, and SLK also received unrestricted research funding from the public sources for the project (see funding).
Ethical approval: The trials were conducted in accordance with the ethical principles expressed in the Declaration of Helsinki after written informed consent. The ONE study ONEnTreg13 (EudraCT No 2013-001294-24; NCT02371434) and the ONErgt11 Reference Group Trial (EudraCT No 2011-004301-24) are registered clinical trials and were approved by the local Berlin State Ethics Committee (LAGESO approval: 3 November 2014; file No 14/0168-EK12), the German national regulatory body (PEI approval: 16 April 2014; file No 2118/01), and the institutional review board of the Charité.
Patient confidentiality: Patient data are handled according to the European and German rules of health data protection. Pseudonymised (deidentified) individual participant data were analysed by the data compiling team and the biostatisticians. Participant identities remain known exclusively to the clinical study doctors (PR, GB, NMO, MC, TS) and study nurses (AS, CG). All biomarkers were analysis blinded.
Data sharing: The data from the clinical trial have already been reported to the regulatory body (Paul-Ehrlich Institute, Germany). Most of the data are presented in the main manuscript and the extensive supplementary tables and figures of the paper (open access); any additional data will be made available by the corresponding author on request (petra.reinke@charite.de).
The corresponding author affirms that the manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
Dissemination to participants and related patient and public communities: The dissemination plan for the results allows for the involvement of patients and patient organisations as well as the public, and implementation has already begun: for example, presentation of initial data and introduction of patients in regular doctor-patient seminars at our transplant centre 2017/18, as well as at the RESTORE health by advanced therapies event at the European Parliament in Brussels, annual long night of science event for public in Berlin 2019, production of a film for the public and politics about advanced therapies with an example of nTreg therapy (www.restore-horizon.eu).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Bluestone JA, Tang Q. Treg cells-the next frontier of cell therapy. Science 2018;362:154-5. 10.1126/science.aau2688. [DOI] [PubMed] [Google Scholar]
- 2. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol 2020;20:158-72. 10.1038/s41577-019-0232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med 2004;351:2715-29. 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 4. Ekberg H, Tedesco-Silva H, Demirbas A, et al. ELITE-Symphony Study Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 2007;357:2562-75. 10.1056/NEJMoa067411. [DOI] [PubMed] [Google Scholar]
- 5. Pascual M, Theruvath T, Kawai T, Tolkoff-Rubin N, Cosimi AB. Strategies to improve long-term outcomes after renal transplantation. N Engl J Med 2002;346:580-90. 10.1056/NEJMra011295. [DOI] [PubMed] [Google Scholar]
- 6. Sayegh MH, Carpenter CB. Transplantation 50 years later--progress, challenges, and promises. N Engl J Med 2004;351:2761-6. 10.1056/NEJMon043418. [DOI] [PubMed] [Google Scholar]
- 7. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 2009;4:481-508. 10.2215/CJN.04800908. [DOI] [PubMed] [Google Scholar]
- 8. Rama I, Grinyó JM. Malignancy after renal transplantation: the role of immunosuppression. Nat Rev Nephrol 2010;6:511-9. 10.1038/nrneph.2010.102. [DOI] [PubMed] [Google Scholar]
- 9. Golshayan D, Pascual M. Minimization of calcineurin inhibitors to improve long-term outcomes in kidney transplantation. Transpl Immunol 2008;20:21-8. 10.1016/j.trim.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 10. Ponticelli C, Scolari MP. Calcineurin inhibitors in renal transplantation still needed but in reduced doses: a review. Transplant Proc 2010;42:2205-8. 10.1016/j.transproceed.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 11. Leventhal JR, Mathew JM. Outstanding questions in transplantation: Tolerance. Am J Transplant 2020;20:348-54. 10.1111/ajt.15680. [DOI] [PubMed] [Google Scholar]
- 12. Siepert A, Ahrlich S, Vogt K, et al. Permanent CNI treatment for prevention of renal allograft rejection in sensitized hosts can be replaced by regulatory T cells. Am J Transplant 2012;12:2384-94. 10.1111/j.1600-6143.2012.04143.x. [DOI] [PubMed] [Google Scholar]
- 13. Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol 2012;12:417-30. 10.1038/nri3227. [DOI] [PubMed] [Google Scholar]
- 14. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008;133:775-87. 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 15. Safinia N, Grageda N, Scottà C, et al. Cell therapy in organ transplantation: our experience on the clinical translation of regulatory T cells. Front Immunol 2018;9:354. 10.3389/fimmu.2018.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol 2019;10:43. 10.3389/fimmu.2019.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atif M, Conti F, Gorochov G, Oo YH, Miyara M. Regulatory T cells in solid organ transplantation. Clin Transl Immunology 2020;9:e01099. 10.1002/cti2.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gladstone DE, Kim BS, Mooney K, Karaba AH, D’Alessio FR. Regulatory T cells for treating patients with covid-19 and acute respiratory distress syndrome: two case reports. Ann Intern Med 2020. 10.7326/L20-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sawitzki B, Harden PN, Reinke P, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 2020;395:1627-39. 10.1016/S0140-6736(20)30167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Streitz M, Miloud T, Kapinsky M, et al. Standardization of whole blood immune phenotype monitoring for clinical trials: panels and methods from the ONE study. Transplant Res 2013;2:17. 10.1186/2047-1440-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abou-El-Enein M, Römhild A, Kaiser D, et al. Good Manufacturing Practices (GMP) manufacturing of advanced therapy medicinal products: a novel tailored model for optimizing performance and estimating costs. Cytotherapy 2013;15:362-83. 10.1016/j.jcyt.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 22. Landwehr-Kenzel S, Issa F, Luu SH, et al. Novel GMP-compatible protocol employing an allogeneic B cell bank for clonal expansion of allospecific natural regulatory T cells. Am J Transplant 2014;14:594-606. 10.1111/ajt.12629. [DOI] [PubMed] [Google Scholar]
- 23. Döcke WD, Höflich C, Davis KA, et al. Monitoring temporary immunodepression by flow cytometric measurement of monocytic HLA-DR expression: a multicenter standardized study. Clin Chem 2005;51:2341-7. 10.1373/clinchem.2005.052639. [DOI] [PubMed] [Google Scholar]
- 24. Kverneland AH, Streitz M, Geissler E, et al. Age and gender leucocytes variances and references values generated using the standardized ONE-Study protocol. Cytometry A 2016;89:543-64. 10.1002/cyto.a.22855. [DOI] [PubMed] [Google Scholar]
- 25. Bestard O, Crespo E, Stein M, et al. Cross-validation of IFN-γ Elispot assay for measuring alloreactive memory/effector T cell responses in renal transplant recipients. Am J Transplant 2013;13:1880-90. 10.1111/ajt.12285. [DOI] [PubMed] [Google Scholar]
- 26. Dziubianau M, Hecht J, Kuchenbecker L, et al. TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of T cell-related pathology. Am J Transplant 2013;13:2842-54. 10.1111/ajt.12431. [DOI] [PubMed] [Google Scholar]
- 27. Lei H, Kuchenbecker L, Streitz M, et al. Human CD45RA(-) FoxP3(hi) memory-type regulatory T cells show distinct TCR repertoires with conventional T cells and play an important role in controlling early immune activation. Am J Transplant 2015;15:2625-35. 10.1111/ajt.13315. [DOI] [PubMed] [Google Scholar]
- 28. Bacher P, Heinrich F, Stervbo U, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell 2016;167:1067-78.e16. 10.1016/j.cell.2016.09.050 [DOI] [PubMed] [Google Scholar]
- 29. Bacher P, Hohnstein T, Beerbaum E, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans . Cell 2019;176:1340-55.e15. 10.1016/j.cell.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 30. Sagoo P, Perucha E, Sawitzki B, et al. Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest 2010;120:1848-61. 10.1172/JCI39922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brunner E, Domhof S, Langer F. Nonparametric analysis of longitudinal data in factorial experiments. Wiley, 2002. [Google Scholar]
- 32. Noguchi K, Gel YR, Brunner E, et al. nparLD: an R software package for the nonparametric analysis of longitudinal data in factorial experiments. J Stat Softw 2012;50:23 10.18637/jss.v050.i12 [DOI] [Google Scholar]
- 33. Bluestone JA, Liu W, Yabu JM, et al. The effect of costimulatory and interleukin 2 receptor blockade on regulatory T cells in renal transplantation. Am J Transplant 2008;8:2086-96. 10.1111/j.1600-6143.2008.02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abou-El-Enein M, Elsanhoury A, Reinke P. Overcoming challenges facing advanced therapies in the EU market. Cell Stem Cell 2016;19:293-7. 10.1016/j.stem.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 35. Fritsche E, Volk HD, Reinke P, Abou-El-Enein M. Toward an optimized process for clinical manufacturing of CAR-Treg cell therapy. Trends Biotechnol 2020;38:1099-112. 10.1016/j.tibtech.2019.12.009. [DOI] [PubMed] [Google Scholar]
- 36. Elsallab M, Levine BL, Wayne AS, Abou-El-Enein M. CAR T-cell product performance in haematological malignancies before and after marketing authorisation. Lancet Oncol 2020;21:e104-16. 10.1016/S1470-2045(19)30729-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Safinia N, Vaikunthanathan T, Fraser H, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget 2016;7:7563-77. 10.18632/oncotarget.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fraser H, Safinia N, Grageda N, et al. A rapamycin-based GMP-compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol Ther Methods Clin Dev 2018;8:198-209. 10.1016/j.omtm.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bluestone JA, Buckner JH, Fitch M, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 2015;7:315ra189. 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mathew JM, H-Voss J, LeFever A, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep 2018;8:7428. 10.1038/s41598-018-25574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Q, Lu W, Liang CL, et al. Chimeric antigen receptor (CAR) Treg: a promising approach to inducing immunological tolerance. Front Immunol 2018;9:2359. 10.3389/fimmu.2018.02359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moroni G, Belingheri M, Frontini G, Tamborini F, Messa P. Immunoglobulin A nephropathy. recurrence after renal transplantation. Front Immunol 2019;10:1332. 10.3389/fimmu.2019.01332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wyld ML, Chadban SJ. Recurrent IgA nephropathy after kidney transplantation. Transplantation 2016;100:1827-32. 10.1097/TP.0000000000001093. [DOI] [PubMed] [Google Scholar]
- 44. Oberbauer R. Calcineurin inhibitor withdrawal from sirolimus-based therapy in kidney transplantation: a systematic review of randomized trials. Am J Transplant 2005;5:3023. 10.1111/j.1600-6143.2005.01100.x. [DOI] [PubMed] [Google Scholar]
- 45. Larson TS, Dean PG, Stegall MD, et al. Complete avoidance of calcineurin inhibitors in renal transplantation: a randomized trial comparing sirolimus and tacrolimus. Am J Transplant 2006;6:514-22. 10.1111/j.1600-6143.2005.01177.x. [DOI] [PubMed] [Google Scholar]
- 46. Tan HP, Donaldson J, Basu A, et al. Two hundred living donor kidney transplantations under alemtuzumab induction and tacrolimus monotherapy: 3-year follow-up. Am J Transplant 2009;9:355-66. 10.1111/j.1600-6143.2008.02492.x. [DOI] [PubMed] [Google Scholar]
- 47. Chan K, Taube D, Roufosse C, et al. Kidney transplantation with minimized maintenance: alemtuzumab induction with tacrolimus monotherapy--an open label, randomized trial. Transplantation 2011;92:774-80. 10.1097/TP.0b013e31822ca7ca. [DOI] [PubMed] [Google Scholar]
- 48. van der Zwan M, Baan CC, van Gelder T, Hesselink DA. Review of the clinical pharmacokinetics and pharmacodynamics of alemtuzumab and its use in kidney transplantation. Clin Pharmacokinet 2018;57:191-207. 10.1007/s40262-017-0573-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chandran S, Tang Q, Sarwal M, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant 2017;17:2945-54. 10.1111/ajt.14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cathomen T, Schüle S, Schüßler-Lenz M, Abou-El-Enein M. The human genome editing race: loosening regulatory standards for commercial advantage? Trends Biotechnol 2019;37:120-3. 10.1016/j.tibtech.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 51. Wagner DL, Amini L, Wendering DJ, et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat Med 2019;25:242-8. 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Web appendix: Supplementary materials