

Key Points
Question
What is the effect of praliciguat, a soluble guanylate cyclase stimulator, on functional capacity in patients with heart failure with preserved ejection fraction?
Findings
In this randomized clinical trial that included 196 patients, there was no significant difference in the change in peak rate of oxygen consumption from baseline to week 12 in the placebo group compared with the 40-mg praliciguat group (0.04 vs −0.26 mL/kg/min).
Meaning
The findings do not support the use of praliciguat in patients with heart failure with preserved ejection fraction.
Abstract
Importance
Heart failure with preserved ejection fraction (HFpEF) is often characterized by nitric oxide deficiency.
Objective
To evaluate the efficacy and adverse effects of praliciguat, an oral soluble guanylate cyclase stimulator, in patients with HFpEF.
Design, Setting, and Participants
CAPACITY HFpEF was a randomized, double-blind, placebo-controlled, phase 2 trial. Fifty-nine sites enrolled 196 patients with heart failure and an ejection fraction of at least 40%, impaired peak rate of oxygen consumption (peak V̇o2), and at least 2 conditions associated with nitric oxide deficiency (diabetes, hypertension, obesity, or advanced age). The trial randomized patients to 1 of 3 praliciguat dose groups or a placebo group, but was refocused early to a comparison of the 40-mg praliciguat dose vs placebo. Participants were enrolled from November 15, 2017, to April 30, 2019, with final follow-up on August 19, 2019.
Interventions
Patients were randomized to receive 12 weeks of treatment with 40 mg of praliciguat daily (n = 91) or placebo (n = 90).
Main Outcomes and Measures
The primary efficacy end point was the change from baseline in peak V̇o2 in patients who completed at least 8 weeks of assigned dosing. Secondary end points included the change from baseline in 6-minute walk test distance and in ventilatory efficiency (ventilation/carbon dioxide production slope). The primary adverse event end point was the incidence of treatment-emergent adverse events (TEAEs).
Results
Among 181 patients (mean [SD] age, 70 [9] years; 75 [41%] women), 155 (86%) completed the trial. In the placebo (n = 78) and praliciguat (n = 65) groups, changes in peak V̇o2 were 0.04 mL/kg/min (95% CI, –0.49 to 0.56) and −0.26 mL/kg/min (95% CI, −0.83 to 0.31), respectively; the placebo-adjusted least-squares between-group difference in mean change from baseline was −0.30 mL/kg/min ([95% CI, −0.95 to 0.35]; P = .37). None of the 3 prespecified secondary end points were statistically significant. In the placebo and praliciguat groups, changes in 6-minute walk test distance were 58.1 m (95% CI, 26.1-90.1) and 41.4 m (95% CI, 8.2-74.5), respectively; the placebo-adjusted least-squares between-group difference in mean change from baseline was –16.7 m (95% CI, −47.4 to 13.9). In the placebo and praliciguat groups, the placebo-adjusted least-squares between-group difference in mean change in ventilation/carbon dioxide production slope was −0.3 (95% CI, −1.6 to 1.0). There were more dizziness (9.9% vs 1.1%), hypotension (8.8% vs 0%), and headache (11% vs 6.7%) TEAEs with praliciguat compared with placebo. The frequency of serious TEAEs was similar between the groups (10% in the praliciguat group and 11% in the placebo group).
Conclusions and Relevance
Among patients with HFpEF, the soluble guanylate cyclase stimulator praliciguat, compared with placebo, did not significantly improve peak V̇o2 from baseline to week 12. These findings do not support the use of praliciguat in patients with HFpEF.
Trial Registration
ClinicalTrials.gov Identifier: NCT03254485
This randomized trial compares the safety and effect of the oral soluble guanylate cyclase stimulator praliciguat on functional capacity (peak V̇o2) at 8 weeks among patients with HFpEF.
Introduction
Heart failure with preserved ejection fraction (HFpEF) is associated with increased risk of hospitalization and mortality as well as poor quality of life.1,2,3,4 Augmentation of the nitric oxide (NO)–soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP) signaling pathway is a potential therapeutic target for HFpEF.5,6 It is hypothesized that a systemic inflammatory state induced by comorbidities results in endothelial inflammation, which reduces NO bioavailability and results in the decreased production of cGMP by sGC.7 This could lead to decreased protection against myocardial injury, vascular and ventricular stiffening, fibrosis, hypertrophy, and cardiorenal syndrome. Direct stimulation of sGC represents a potential pharmacologic strategy for addressing the impaired cGMP signaling.8,9,10
Praliciguat is a selective sGC stimulator with extensive distribution to tissues (including myocardial, vascular, kidney, adipose, and skeletal muscle tissue) and nonkidney clearance.6,11,12,13,14 In vitro, praliciguat demonstrated concentration-dependent stimulation of cGMP production.11 In vivo, praliciguat dose-dependently reduced blood pressure, attenuated cardiac and kidney damage, and reduced expression of fibrotic markers in Dahl salt-sensitive rats.11,12 In healthy volunteers and patients with diabetes and hypertension, praliciguat resulted in dose-related increases in plasma cGMP and reductions in blood pressure sustained for 24 hours, indicating target engagement.13,14 The results from these studies support investigation of praliciguat as a potential therapy for individuals with HFpEF. The objective of this randomized clinical trial was to evaluate the efficacy and adverse effect profile of praliciguat in patients with HFpEF.
Methods
Study Design and Patient Population
This was a randomized, double-blind, placebo-controlled, phase 2 trial that enrolled patients at 59 sites in the United States and Canada and evaluated the effects of praliciguat vs placebo over 12 weeks in patients with HFpEF. This study was conducted in compliance with International Conference on Harmonisation Good Clinical Practice guidelines. Approval of the protocol was obtained from all institutional review boards. All patients provided written informed consent prior to participation. Details of the trial design have been described previously,15 and the study protocol and statistical analysis plan are provided in Supplement 1 and Supplement 2.
The trial enrolled patients aged at least 45 years with a left ventricular ejection fraction of at least 40% (encompassing patients with midrange ejection fraction) and at least 1 of the following to show objective evidence of heart failure: well-documented hospitalization for heart failure within 12 months; N-terminal fragment of brain natriuretic peptide (NT-proBNP) greater than 300 pg/mL or BNP of at least 100 pg/mL within 6 months; echocardiographic evidence (≥2 of the following findings: left ventricular hypertrophy, left atrial enlargement, or diastolic dysfunction [medial E/e prime ratio ≥15]) within 12 months; or elevated pulmonary capillary wedge pressure at rest (≥15 mm Hg) or with exercise (>25 mm Hg) or left ventricular end-diastolic pressure of at least 15 mm Hg within 12 months. All patients were required to have New York Heart Association (NYHA) class II to IV symptoms and limited exercise capacity (peak rate of oxygen consumption [V̇o2] <80% of age- and sex-adjusted normal values with a respiratory exchange ratio ≥1.0 [indicating adequate effort] determined by a cardiopulmonary exercise test [CPET]).16 The patient population was enriched for potentially impaired NO-sGC-cGMP signaling by requiring participants to have at least 2 of the following 4 conditions associated with NO deficiency: diabetes, hypertension, obesity, or advanced age (≥70 y). The percentage of patients with permanent or persistent atrial fibrillation was limited to less than 20% because atrial fibrillation may affect peak V̇o2 and, if not limited in the sample population, may mask a drug effect. Because it is important to understand the diversity of the population sample in a trial, information on race and ethnicity was collected. Participants self-reported race and ethnicity based on an open-ended question.
Initially, patients were randomized in a 1:1:1:1 ratio to receive 12 weeks of treatment with 1 of 3 praliciguat doses (40, 20, or 10 mg daily) or placebo. The 40-mg dose was selected because drug levels in healthy participants were comparable to exposures in the Dahl salt-sensitive rat model.11 Emerging data from other clinical trials13,14 suggested that the 40-mg dose would be adequately tolerated in patients with HFpEF. Therefore, the academic leadership of the trial in conjunction with the sponsor decided that the objectives of this study could be most efficiently met by discontinuing randomization to the 2 lower dose levels. This change in the protocol was codified by an amendment dated June 27, 2018, under which patients were to be randomized in a 1:1 ratio to receive 40 mg of praliciguat or placebo. Implementation of the change in protocol followed review and approval by each site’s institutional review board.
Patients were stratified by atrial fibrillation status (yes or no) and by baseline peak V̇o2 (<60% or ≥60% of age- and sex-adjusted normal values) and randomized in a 1:1 ratio to receive 40 mg of praliciguat or placebo through a centralized interactive web response system. An upper limit of approximately 36 patients with permanent or persistent atrial fibrillation was set in the interactive web response system. The randomization schedule was prepared by an independent statistician using a block size of 4. The lowest randomization number within a stratum was assigned to the first patient that qualified for randomization and subsequent assignments proceeded in increasing sequential order within a block as patients were qualified for the study.
During weeks 1 and 2, the study drug (praliciguat or placebo) was taken twice daily. For weeks 3 to 12, the study drug was taken once daily. Praliciguat and placebo were supplied as oral tablets that matched in appearance.
Study End Points
The primary efficacy end point was the change from baseline in peak V̇o2 (evaluated by a core laboratory following site training and certification) after 12 weeks of treatment. Secondary end points were the change from baseline to 12 weeks in 6-minute walk test (6-MWT) distance, change in ventilatory efficiency (defined by ventilation/carbon dioxide production relationship on CPET), and number of CPET responders (defined as patients who showed a peak V̇o2 improvement of at least 1.5 mL/kg/min from baseline).
Exploratory efficacy end points included change from baseline in additional CPET parameters (including power output at ventilatory anaerobic threshold and peak workload), echocardiography parameters (left ventricular ejection fraction, left atrial volume index, mitral E/e prime ratio, left ventricular end-diastolic volume, left ventricular end-systolic volume, and tricuspid annular place systolic excursion, analyzed blindly in a core laboratory), percent change in peak V̇o2, and NYHA functional classification. Patient-reported outcomes were assessed using the Kansas City Cardiomyopathy Questionnaire (KCCQ) physical limitation score, total symptom score, clinical summary score, and overall summary score (range, 0-100; higher scores indicate better health; minimal clinically important change, ≥5). Other exploratory end points included 6-MWT responders (defined as having improvement ≥15 m) and KCCQ responders (defined as having ≥5-point improvement from baseline) at week 12.
Biomarkers of myocardial stress and injury, inflammation, fibrosis, and endothelial function (NT-proBNP, troponin T, soluble suppression of tumorigenicity 2, growth differentiation factor 15, l-arginine, asymmetric dimethylarginine, symmetrical dimethylarginine, and l-arginine/asymmetric dimethylarginine ratio) were also evaluated as exploratory end points. Urine albumin creatinine ratio was determined from first-void urine specimens and estimated glomerular filtration rate was calculated using the Chronic Kidney Disease Epidemiology Collaboration creatinine equation. Post hoc evaluations of change from baseline to week 12 were performed for glycated hemoglobin A1c in the subset of patients with diabetes and for homeostatic model assessments to estimate insulin resistance, determined from fasting plasma glucose and insulin levels in the subset of patients with diabetes not using concomitant insulin.
Subgroup analyses of the primary efficacy end point, change from baseline in peak V̇o2, were prospectively defined for sex, baseline peak V̇o2, atrial fibrillation status, presence of diabetes, left ventricular ejection fraction, and NT-proBNP. Post hoc subgroup analyses by the age enrichment criterion (<70 and ≥70 y) and post hoc assessment of treatment by subgroup interactions were also performed.
The primary adverse event end point was the incidence of treatment-emergent adverse events (TEAEs) and study drug–related TEAEs. Additional safety parameters included blood pressure, heart rate, cholesterol, triglycerides, creatinine, and body weight.
Statistical Analysis
The planned sample size of approximately 184 randomized patients was chosen to provide at least 90% power to detect a difference of 1.3 mL/kg/min in the change from baseline to week 12 in peak V̇o2 (based on the observed favorable differences reported in a trial of diet and exercise in patients with HFpEF)17 at a 2-sided significance level of .05 between the 40-mg praliciguat group and the placebo group, assuming a common SD of 2.5 mL/kg/min, 10% attrition from the study, and 10% of patients excluded from analyses due to randomization to the 2 discontinued treatment groups.
The primary efficacy end point was analyzed using an analysis of covariance model with treatment group and stratification factors as the main effects and the corresponding baseline measurement as the covariate. Hypothesis testing of the primary efficacy end point was 2-sided with a 5% significance level. Sensitivity analyses of the primary end point were performed adjusting for geographic region in the primary model by using a last observation carried forward approach to impute missing week 12 assessments and by using a mixed-effects model repeated-measures analysis with change from baseline in peak V̇o2 as the response variable; treatment, visit, treatment × visit interaction, and baseline atrial fibrillation status as fixed effects; baseline value as covariate; and unstructured as the variance-covariance structure. For responder events, the percentage of responders in the 40-mg praliciguat and placebo groups were compared using a Cochran-Mantel-Haenszel test controlling for baseline stratification factors. In the CPET responder analysis, patients who were hospitalized or died due to heart failure during the study treatment period were considered nonresponders. All adverse event parameters were analyzed using descriptive statistics. Due to the exploratory nature of this study, the analyses focused primarily on estimation rather than inferential testing and no multiplicity adjustments were performed. Because of the potential for type I error due to multiple comparisons, findings for analyses of secondary end points should be interpreted as exploratory.
The adverse event analysis set included all patients who received at least 1 dose of the study drug. The primary analytic population included all patients who received the assigned dose of the study drug for at least 8 weeks, had at least 1 evaluable postbaseline assessment, and did not have a major protocol deviation that might have a potential effect on efficacy evaluations. The primary efficacy end point and all secondary and exploratory analyses were assessed in this population (target of 147 participants) as well as in the subgroup excluding patients with permanent or persistent atrial fibrillation at baseline. An analysis of the primary end point was also performed in all patients who received at least 1 dose of study drug and had at least 1 evaluable baseline measurement. Data for patients randomized to receive 10 mg or 20 mg of praliciguat under earlier protocol versions were only included in analyses of adverse events, because the numbers in those groups were judged to be too small for meaningful analysis. Data are summarized by mean (SD) or median (interquartile range) for continuous data and by frequencies and percentages for categorical data. SAS version 9.4 (SAS Institute) was used for all analyses.
Results
Between November 7, 2017, and April 30, 2019, a total of 196 patients were enrolled at 59 centers in the United States and Canada. The final date of follow-up was August 19, 2019. The flow of patients is shown in Figure 1.
Figure 1. Flow of Participants in a Study of the Effect of Praliciguat on Peak Rate of Oxygen Consumption in Patients With Heart Failure With Preserved Ejection Fraction.
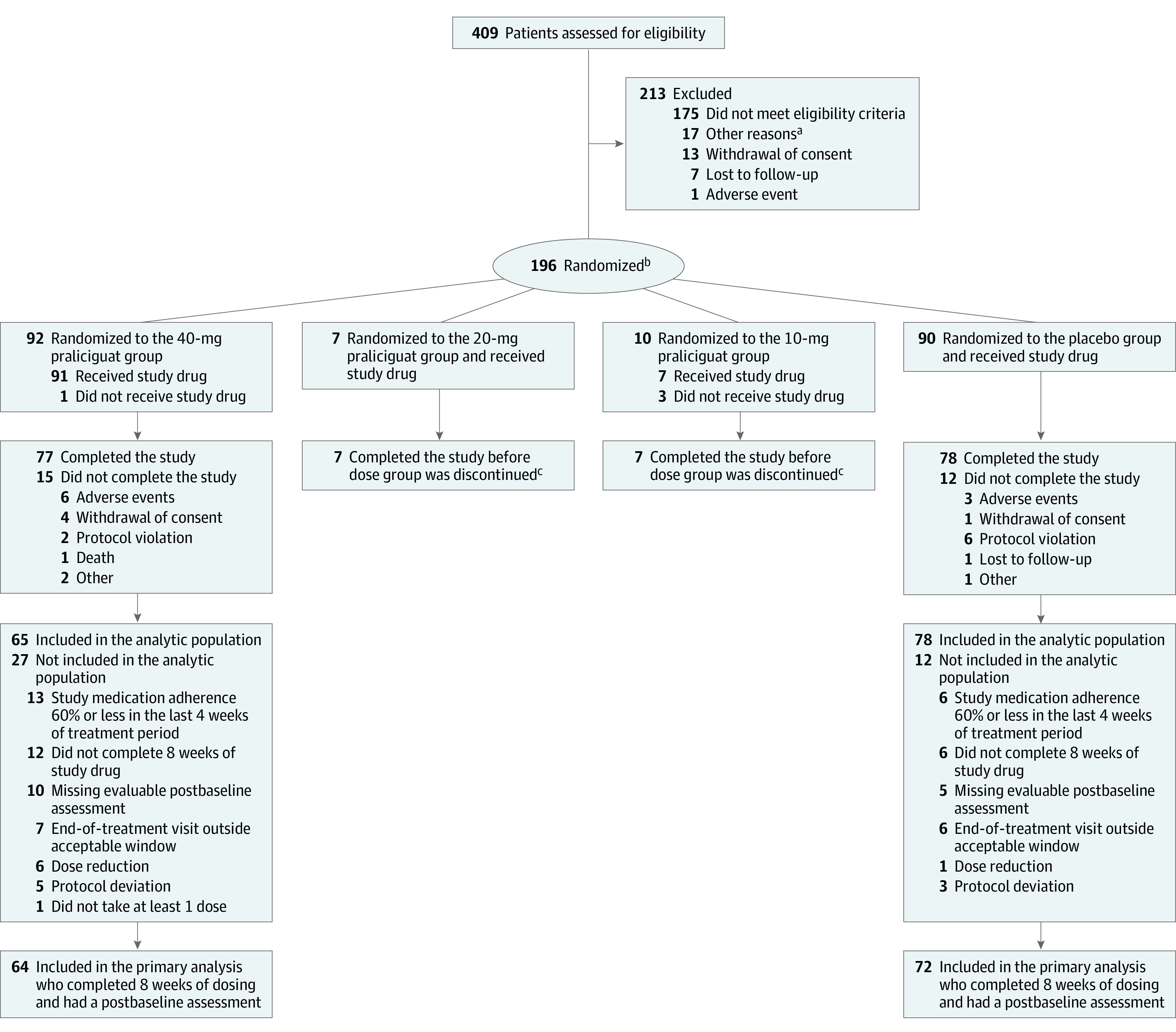
aOther reasons for exclusion were primarily administrative (eg, informed consent form updates, outside of window, end of recruitment).
bPatients were initially randomized in a 1:1:1:1 ratio to 1 of 3 praliciguat dose groups or a placebo group. The study protocol was revised on June 27, 2018, and patients were subsequently randomized in a 1:1 ratio to either the 40-mg praliciguat group or the placebo group.
cData for these patients were not included in the efficacy data analysis.
dPatients may have had more than 1 reason for exclusion.
Baseline Characteristics
Baseline characteristics for patients in the 40-mg praliciguat and placebo groups are shown in Table 1 (mean [SD] age, 70.4 years, 75 [41%] women; mean body mass index, 34; ejection fraction ≤50% in 23 patients [13%]; 129 patients [71%] were in NYHA class II; 59 [33%] had a history of heart failure hospitalization; and 31 [17%] had atrial fibrillation). Among 181 patients in those groups who received the study drug, 155 (86%) completed the trial. By design, conditions associated with HFpEF were common, with 98% of patients having a history of hypertension, 53% having a history of diabetes, 75% fulfilling criteria of obesity (body mass index ≥30), and 57% being aged at least 70 years. Overall, 32% of the population met 2 of the comorbidity clustering criteria, 45% met 3 of the criteria, and 22% met 4 of the criteria. By design, an elevated NT-proBNP was not required for eligibility, and 56% of the enrolled population had NT-proBNP less than or equal to 300 pg/mL.
Table 1. Baseline Characteristics of Participants in a Study of the Effect of Praliciguat on Peak Rate of Oxygen Consumption in Patients With Heart Failure With Preserved Ejection Fraction.
| Characteristic | Mean (SD) | |
|---|---|---|
| 40-mg praliciguat group (n = 91)a | Placebo group (n = 90) | |
| Age, y | 70.7 (9.2) | 70.1 (9.0) |
| Sex, No. (%) | ||
| Women | 35 (38.5) | 40 (44.4) |
| Men | 56 (61.5) | 50 (55.6) |
| Race, No. (%)b | ||
| White | 71 (78.0) | 65 (72.2) |
| Black or African American | 17 (18.7) | 19 (21.1) |
| Asian | 0 | 1 (1.1) |
| Systolic blood pressure, mm Hgc | 133.4 (18.0) | 131.2 (15.5) |
| Diastolic blood pressure, mm Hgc | 74.7 (10.4) | 74.3 (10.7) |
| Heart rate/minc | 67.2 (9) | 67.4 (10.6) |
| BMI | 34.1 (6.1) | 34.7 (7.3) |
| NT-proBNP, median (range), ng/L | 260 (7-4283) | 228.5 (9-5138) |
| Left ventricular ejection fraction, No. (%)d | ||
| ≤50% | 8 (8.8) | 15 (16.7) |
| >50% | 80 (87.9) | 71 (78.9) |
| Atrial fibrillation, No. (%)e | 14 (15.4) | 17 (18.9) |
| New York Heart Association class, No. (%)f | ||
| II | 63 (69.2) | 66 (73.3) |
| III | 28 (30.8) | 24 (26.7) |
| Evidence of heart failure, No. (%)g | ||
| Echocardiographic evidence | 65 (71.4) | 66 (73.3) |
| Elevated BNP or NT-proBNP level | 34 (37.4) | 33 (36.7) |
| Heart failure hospitalization | 28 (30.8) | 31 (34.4) |
| Elevated filling pressures | 18 (19.8) | 17 (18.9) |
| Enrichment eligibility criteria, No. (%)h | ||
| Hypertension | 90 (98.9) | 87 (96.7) |
| Obesity (BMI ≥30) | 71 (78.0) | 64 (71.1) |
| Age ≥70 y | 58 (63.7) | 46 (51.1) |
| Type 2 diabetes/prediabetes | 46 (50.5) | 50 (55.6) |
| 3 of the criteria | 39 (42.8) | 42 (46.7) |
| 2 of the criteria | 29 (31.9) | 29 (32.2) |
| 4 of the criteria | 23 (25.3) | 16 (17.8) |
| NT-proBNP categories, No. (%)i | ||
| ≤300 pg/mL | 52 (57.1) | 50 (55.6) |
| >300 pg/mL | 39 (42.9) | 40 (44.4) |
| Medical history, No. (%)j | ||
| Coronary artery disease | 36 (39.6) | 35 (38.9) |
| Chronic kidney disease | 24 (26.4) | 14 (15.6) |
| Concomitant medication, No. (%) | ||
| Lipid-lowering agent | 72 (79.1) | 66 (73.3) |
| Antiplatelet agent | 63 (69.2) | 60 (66.7) |
| Beta-adrenergic blocker | 42 (46.2) | 47 (52.2) |
| β-Blocker | 39 (42.9) | 24 (26.7) |
| Calcium channel blocker | 28 (30.8) | 26 (28.9) |
| Angiotensin-converting enzyme inhibitor | 27 (29.7) | 36 (40.0) |
| Anticoagulant agent | 21 (23.1) | 17 (18.9) |
| Metformin | 19 (20.9) | 24 (26.7) |
| Diuretic | 17 (18.7) | 17 (18.9) |
| Insulin | 17 (18.7) | 13 (14.4) |
| Glucagon-like peptide 1 receptor agonist | 4 (4.4) | 2 (2.2) |
| Sacubitril/valsartan | 2 (2.2) | 3 (3.3) |
| Sodium glucose co-transporter 2 inhibitor | 2 (2.2) | 1 (1.1) |
Abbreviation: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared).
Data were not available for 1 patient who was randomized but did not receive the study drug.
Patient race was recorded and determined by self-report.
Systolic and diastolic blood pressure and heart rate were assessed at randomization visit.
Left ventricular ejection fraction was assessed by echocardiography; the data presented here reflect the last assessment prior to the start of dosing at the randomization visit.
Atrial fibrillation was defined as history of persistent or permanent atrial fibrillation.
New York Heart Association functional class quantifies clinician-estimated severity of functional limitation. Class I indicates no limiting symptoms; class II, slight limitation; class III, marked symptoms develop with even ordinary activity; and class IV, symptomatic at rest or during minimal activity.
Patient has evidence supporting clinical heart failure syndrome consisting of at least 1 of the following: well-documented hospitalization for heart failure within 12 months; N-terminal fragment of brain natriuretic peptide (NT-proBNP) greater than 300 pg/mL or brain natriuretic peptide of at least 100 pg/mL within 6 months; echocardiographic evidence (≥2 of the following findings: left ventricular hypertrophy, left atrial enlargement, or diastolic dysfunction [medial E/e prime ratio ≥15]) within 12 months; or elevated pulmonary capillary wedge pressure at rest (≥15 mm Hg) or with exercise (>25 mm Hg) or left ventricular end-diastolic pressure of at least 15 mm Hg within 12 months. Of note, hospitalization or emergency department visit were collected as 1 criteria, and numbers of heart failure hospitalizations or emergency department visits individually cannot be assessed.
Included patient population was enriched by at least 2 of the listed 4 conditions with potentially impaired nitric oxide–soluble guanylate cyclase-cyclic guanosine monophosphate signaling.
NT-proBNP levels higher than 300 pg/mL were considered elevated.
Medical history data were determined by study investigators based on medical record review.
Primary and Secondary Efficacy End Points
Results of the primary and secondary end points in patients who received the assigned dose of the study drug for 8 weeks, had at least 1 evaluable postbaseline assessment, and did not have a major protocol deviation (prespecified as the primary analytic population; n = 143) are shown in Table 2. At baseline, patients had impaired functional capacity with mean peak V̇o2 of 13 mL/kg/min. There were no statistically significant between-group differences in the change in peak V̇o2 from baseline to week 12: the change in peak V̇o2 was 0.04 mL/kg/min (95% CI, −0.49 to 0.56) in the placebo group and −0.26 mL/kg/min (95% CI, –0.83 to 0.31) in the praliciguat-treated group (between-group difference, −0.30 mL/kg/min [95% CI, −0.95 to 0.35]; P = .37). As shown in Table 2, there were also no significant between-group differences in the secondary end points, including 6-MWT distance (least-squares mean change from baseline to week 12, −6.74 m [95% CI, −47.38 to 13.90]; nominal P = .28), ventilation/carbon dioxide production slope (between-group difference, −0.30 [95% CI, −1.59 to 1.00]; nominal P = .65), or CPET responders (21.8% of participants in the placebo group and 20.0% in the praliciguat group; odds ratio, 0.91 [95% CI, 0.40-2.06]; nominal P = .82). In the analysis of peak V̇o2 response among patients without atrial fibrillation, there was also no significant between-group difference (least-squares mean change from baseline to week 12, −0.40 mL/kg/min [95% CI, −1.14 to 0.33]; nominal P = .28). Results of post hoc analyses that included all patients who received at least 1 dose of the study drug and had at least 1 evaluable baseline measurement were consistent with the results of the primary analysis (eTable 1 in Supplement 3).
Table 2. Primary and Secondary Functional Efficacy End Points in a Study of the Effect of Praliciguat on Peak Rate of Oxygen Consumption (V̇o2) in Patients With Heart Failure With Preserved Ejection Fractiona.
| End point | 40-mg praliciguat group (n = 65) | Placebo group (n = 78) | Adjusted difference in difference (95% CI)b | P value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean (SD) | Absolute difference (SD) | Adjusted difference (95% CI)b | Mean (SD) | Absolute difference (SD) | Adjusted difference (95% CI)b | |||||
| Baseline | Week 12 | Baseline | Week 12 | |||||||
| Primary | (n = 64) | (n = 72) | ||||||||
| Peak V̇o2, mL/kg/minc | 13.2 (4.1) | 12.9 (3.9) | −0.3 (2.1) | −0.3 (−0.8 to 0.3) | 12.8 (3.5) | 12.9 (3.8) | 0.1 (1.8) | 0.04 (−0.5 to 0.6) | −0.3 (−1.0 to 0.4) | .37 |
| Secondary | Nominal P value | |||||||||
| 6-min walk test distance, md | (n = 63) | (n = 72) | ||||||||
| 333.5 (128.2) | 339.9 (144.4) | 6.4 (82.2) | 41.4 (8.3 to 74.5) | 314.5 (128.5) | 341.4 (125.4) | 27.0 (105.2) | 58.1 (26.1 to 90.1) | −16.7 (−47.4 to 13.9) | .28 | |
| Ventilatory efficiency, VE/VCo2 slopee | (n = 64) | (n = 72)c | ||||||||
| 31.6 (6.9) | 31.4 (6.4) | −0.2 (3.8) | 0.3 (−1.1 to 1.7) | 30.8 (7.2) | 31.1 (7.8) | 0.3 (3.9) | 0.6 (−0.8 to 1.9) | −0.3 (−1.6 to 1.0) | .65 | |
| CPET responders, No. (%)f | (n = 65) | (n = 78) | ||||||||
| 13 (20.0) | 17 (21.8) | Odds ratio, 0.9 (0.4 to 2.1) | .82 | |||||||
| Planned subgroup analysis | (n = 56) | (n = 62) | Nominal P value | |||||||
| Peak V̇o2 in patients without atrial fibrillation, mL/kg/min | (n = 55) | (n = 59) | ||||||||
| 13.4 (4.0) | 13.0 (4.0) | −0.4 (2.1) | −0.3 (−0.9 to 0.2) | 12.8 (3.6) | 12.9 (4.0) | 0.1 (1.8) | 0.1 (−0.4 to 0.6) | −0.4 (−1.1 to 0.3) | .28 | |
Abbreviation: VE/VCo2, ventilation/carbon dioxide production.
Data are for the primary analytic population (all patients who completed 8 weeks of dosing, had at least 1 evaluable postbaseline assessment, and did not have a dose reduction or major protocol deviations). Change from baseline analysis for an end point included patients with measurements at both baseline and week 12 for that specific end point. Cardiopulmonary exercise test (CPET) responder analyses considered patients who were hospitalized or died due to heart failure or had missing assessments performed after the participant received their first dose of the study drug as nonresponders.
Adjusted treatment differences are estimates from an analysis of covariance model with treatment group and stratification factors as categorical variable terms and baseline value as a covariate. CPET responders were analyzed using a Cochran-Mantel-Haenszel test controlling for treatment group and stratification factors.
Normal values vary by age and sex, with higher values representing better functional capacity.
An increase (positive value) denotes improvement and a decrease (negative value) denotes worsening.
Ventilatory efficiency is a unitless index that is defined by minute ventilation (in L/min) relative to carbon dioxide production (in L/min) throughout exercise. Higher values represent worse efficiency.
Patients who improved by at least 1.5 mL/kg/min in peak V̇o2 from baseline to week 12.
Results of post hoc subgroup analyses of the primary end point (Figure 2) suggested significant treatment interactions with age and sex; no favorable effect on peak V̇o2 was seen.
Figure 2. Subgroup Analysis of the Primary Efficacy End Point in a Study of the Effect of Praliciguat on Peak Rate of Oxygen Consumption (V̇o2) in Patients With Heart Failure With Preserved Ejection Fraction.
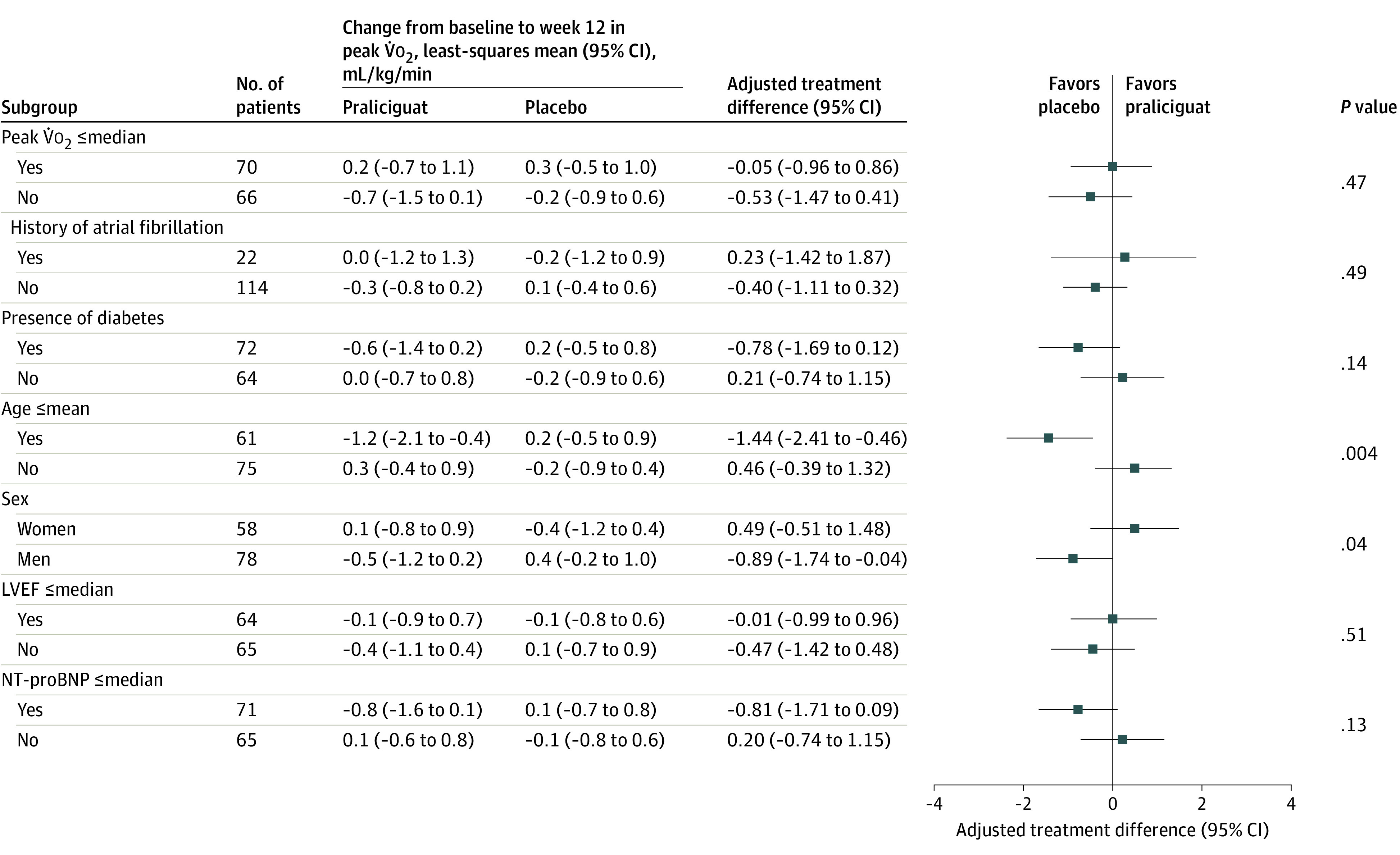
The median peak V̇o2 was 12.6 mL/kg/min; mean age, 70.3 years; median left ventricular ejection fraction (LVEF), 61.5%; and median N-terminal fragment of brain natriuretic peptide (NT-proBNP), 232 ng/L. The analysis models were adjusted for the stratification factors and the baseline assessments.
Exploratory End Points
Exploratory end point results are shown in eTables 2 and 3 in Supplement 3. There were significantly fewer 6-MWT responders (defined as patients who improved by at least 15 m in 6-MWT distance from baseline) in the praliciguat group than in the placebo group (37% vs 59%; odds ratio, 0.39 [95% CI, 0.19-0.77]; nominal P = .007). There were no significant differences between the 40-mg praliciguat group and placebo group in least-squares mean change from baseline to week 12 for NT-proBNP (6.9 ng/L [95% CI, −12.0 to 29.9]; nominal P = .50), troponin T (2.32 pg/mL [95% CI, −1.25 to 5.89]; nominal P = .20), or estimated glomerular filtration rate (−0.1 mL/min/1.73 m2 [95% CI, −3.7 to 3.5]; nominal P = .94). Compared with placebo, the 40-mg praliciguat group showed no significant difference in the homeostatic model assessments to estimate insulin resistance among patients with type 2 diabetes not receiving concomitant insulin therapy (least-squares mean change, −3.1 [95% CI, −6.7 to 0.4]; nominal P = .08). Based on a post hoc analysis, the change in glycated hemoglobin A1c among patients with type 2 diabetes in the 40-mg praliciguat group compared with the placebo group was −0.25% (95% CI −0.65% to 0.15%).
Echocardiographic end points and KCCQ results are shown in eTable 3 in Supplement 3. There were no significant differences between the 40-mg praliciguat and placebo groups in any structural or functional echocardiographic measures. There were also no significant improvements seen in any KCCQ parameter in the 40-mg praliciguat group compared with the placebo group. Change from baseline in the KCCQ overall summary score was significantly greater in the placebo group than in the 40-mg praliciguat group (least-squares mean change, −7.2 [95% CI, −12.3 to −2.0]; nominal P = .007), with a significantly greater percentage of responders (patients whose score change from baseline was ≥5; 63% vs 41%; odds ratio, 0.43 [95% CI, 0.22-0.84]; nominal P = .01).
Adverse Events
Vitals signs and laboratory measures are shown in eTable 4 in Supplement 3. The change in mean systolic blood pressure at week 12 was −6.3 mm Hg (95% CI, −10.4 to −2.3) in the 40-mg praliciguat group compared with −1.1 mm Hg (95% CI, −4.4 to 2.2) in the placebo group. Mean change in body weight from baseline was not different between the 40-mg praliciguat and placebo groups (0.27 kg [95% CI, −0.22 to 0.75] vs −0.41 kg [95% CI, −1.04 to 0.22]).
Rates of TEAEs and serious adverse events are shown in Table 3 and eTable 5 in Supplement 3. There was 1 death in the 40-mg praliciguat group due to cardiovascular complications, which was considered unrelated to the study drug. The incidence of serious adverse events was 11% in the 40-mg praliciguat group and 10% in the placebo group. Eight participants (5 [5.5%] in the 40-mg praliciguat group vs 3 [3.3%] in the placebo group) discontinued treatment due to TEAEs. There was a higher incidence of TEAEs related to the study drug in the 40-mg praliciguat group compared with the placebo group, most commonly headache (11% vs 7%), dizziness (10% vs 1%), and hypotension (9% vs 0%).
Table 3. Adverse Events in a Study of the Effect of Praliciguat on Peak Rate of Oxygen Consumption in Patients With Heart Failure With Preserved Ejection Fractiona.
| Adverse event | No. (%) | |
|---|---|---|
| 40-mg praliciguat group (n = 91) | Placebo group (n = 90) | |
| Any treatment-emergent adverse eventb | 72 (79.1) | 61 (67.8) |
| Any treatment-emergent adverse events related to study drugb | 22 (24.2) | 9 (10.0) |
| Most frequent treatment-emergent adverse eventsb | ||
| Headache | 10 (11.0) | 6 (6.7) |
| Dizziness | 9 (9.9) | 1 (1.1) |
| Hypotension | 8 (8.8) | 0 |
| Any treatment-emergent adverse event leading to discontinuation of study drugb | 5 (5.5) | 3 (3.3) |
| Any serious adverse eventc | 10 (11.0) | 9 (10.0) |
| Deathd | 1 (1.1) | 0 |
Data are for the adverse event analysis set that included all patients who received at least 1 dose of the study drug (lower-dose groups are shown in eTable 5 in Supplement).
Treatment-emergent adverse events were defined as adverse events that started or worsened in severity after the first dose of the study drug.
Serious adverse events were considered any adverse event occurring at any dose that resulted in any of the following outcomes: death or life-threatening event, hospitalization or prolongation of existing hospitalization, persistent or significant disability/incapacity, congenital anomaly/birth defect, or important medical events (events that may not result in death, be life threatening, or require hospitalization that may be considered serious when, based on appropriate medical judgment, it may jeopardize the patient and may require medical or surgical intervention to prevent 1 of the outcomes listed in this definition).
One death in the 40-mg praliciguat group due to cardiovascular complications was considered unrelated to the study drug.
Discussion
The results of this phase 2 trial suggest that in patients with HFpEF and impairment of functional capacity, stimulation of sGC using praliciguat at 40 mg daily for 12 weeks did not significantly improve peak V̇o2, the primary trial end point. Moreover, there were no favorable changes in secondary or exploratory end points that assessed other measures of functional capacity or symptoms or changes in biomarkers or echocardiographic parameters.
Given the reasonably strong support for the concept that the NO-sGC-cGMP pathway may be impaired in patients with HFpEF7 and the strong preclinical data for sGC stimulation with praliciguat,11,12 the neutral results in this trial prompt speculation as to why no favorable clinical effect was seen. Possibilities include suboptimal dosing, patient selection did not identify patients with HFpEF with impaired NO-sGC-cGMP signaling, impaired NO-sGC-cGMP signaling is not of major pathophysiological relevance in patients with HFpEF, or the drug might not be effective in this population. The findings do not exclude the possibility that correcting such impairment, if present, may slow or reverse pathologic myocyte hypertrophy and interstitial fibrosis in the longer term.
Regarding dosing, there was a detectable effect of 40 mg of praliciguat on the vasculature, in that systolic blood pressure was lower by 6.3 mm Hg and diastolic blood pressure was lower by 1.4 mm Hg in the praliciguat group compared with the placebo group at 12 weeks, a slightly greater reduction than seen in a trial of isosorbide mononitrate.18 Moreover, there was a higher prevalence of dizziness and hypotension in the praliciguat group reported as TEAEs. Thus, it is unlikely that the 40-mg daily dose was too low to affect the pathway.
Several lines of evidence link inflammation and abnormalities in metabolic pathways with impaired NO-sGC-cGMP signaling in HFpEF.7,10 The inclusion criteria for this trial were designed with the assumption that clinical factors would identify patients who had impaired NO-sGC-cGMP signaling. Whether these patients had impaired NO-sGC-cGMP signaling at the tissue level cannot be definitively established.
It is also possible that abnormalities in NO-sGC-cGMP signaling are present in patients with HFpEF, but are more of a marker of the comorbidities than a mediator of the complex pathophysiology that results in functional impairment. Previous trials that addressed this pathway18,19,20,21,22 failed to show improvements in exercise tolerance or other end points. The only strategies that have improved functional status have been exercise and caloric restriction,17,23 interventions that would have multifaceted effects across many pathophysiologic pathways.
Elevated natriuretic peptide levels were not required for eligibility in this trial, allowing exploration of the possibility that patients who have NT-proBNP levels below thresholds typically used to define heart failure may have cardiopulmonary reserve that would allow response to an intervention.24,25 Elevated levels of natriuretic peptides are associated with markers of myocardial fibrosis in patients with HFpEF.26 Although a substantial number of patients in this trial had normal or minimally elevated BNP levels, no differential effect on the primary end point was observed.
Limitations
This study has several limitations. First, a trial using CPET end points enrolls patients capable of performing symptom-limited exercise test to peak V̇o2 with an adequate effort. This may not be representative of frailer patients with HFpEF. Second, a higher attrition of patients was observed in the study than planned in the sample size calculations, but because the study also enrolled more patients than planned, it is unlikely that the study conclusion is underpowered. Third, there was differential loss to follow-up in the praliciguat group compared with the placebo group. Fourth, there was a relatively short duration of follow-up. Fifth, although all patients enrolled in the trial had a prior diagnosis of symptomatic HFpEF and objective functional limitation, the use of loop diuretics was low (19%), NT-proBNP values on average were not very elevated, and left atrial volume was not enlarged in some patients. By design, this trial aimed to enroll symptomatic, functionally limited patients with HFpEF in whom augmenting sGC to increase cGMP was hypothesized to be beneficial, and the results demonstrated that praliciguat was not beneficial. Given the type of patients with HFpEF enrolled in this trial (which previous studies have shown may truly have the HFpEF syndrome based on invasive hemodynamic criteria27,28,29), the ability to determine whether praliciguat would have resulted in improved exercise capacity in patients with HFpEF who had overt volume overload and congestion is limited.
Conclusions
Among patients with HFpEF, the soluble guanylate cyclase stimulator praliciguat, compared with placebo, did not significantly improve peak V̇o2 from baseline to week 12. The findings do not support the use of praliciguat in patients with HFpEF.
Trial protocol
Statistical analysis plan
eTables
Data sharing statement
References
- 1.Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2017;14(10):591-602. doi: 10.1038/nrcardio.2017.65 [DOI] [PubMed] [Google Scholar]
- 2.Oktay AA, Rich JD, Shah SJ. The emerging epidemic of heart failure with preserved ejection fraction. Curr Heart Fail Rep. 2013;10(4):401-410. doi: 10.1007/s11897-013-0155-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah KS, Xu H, Matsouaka RA, et al. . Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J Am Coll Cardiol. 2017;70(20):2476-2486. doi: 10.1016/j.jacc.2017.08.074 [DOI] [PubMed] [Google Scholar]
- 4.Butler J, Fonarow GC, Zile MR, et al. . Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail. 2014;2(2):97-112. doi: 10.1016/j.jchf.2013.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greene SJ, Gheorghiade M, Borlaug BA, et al. . The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J Am Heart Assoc. 2013;2(6):e000536. doi: 10.1161/JAHA.113.000536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buys ES, Zimmer DP, Chickering J, et al. . Discovery and development of next generation sGC stimulators with diverse multidimensional pharmacology and broad therapeutic potential. Nitric Oxide. 2018;78:72-80. doi: 10.1016/j.niox.2018.05.009 [DOI] [PubMed] [Google Scholar]
- 7.Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263-271. doi: 10.1016/j.jacc.2013.02.092 [DOI] [PubMed] [Google Scholar]
- 8.Gheorghiade M, Marti CN, Sabbah HN, et al. ; Academic Research Team in Heart Failure (ART-HF) . Soluble guanylate cyclase: a potential therapeutic target for heart failure. Heart Fail Rev. 2013;18(2):123-134. doi: 10.1007/s10741-012-9323-1 [DOI] [PubMed] [Google Scholar]
- 9.Breitenstein S, Roessig L, Sandner P, Lewis KS. Novel sGC stimulators and sGC activators for the treatment of heart failure. Handb Exp Pharmacol. 2017;243:225-247. doi: 10.1007/164_2016_100 [DOI] [PubMed] [Google Scholar]
- 10.Evgenov OV, Pacher P, Schmidt PM, Haskó G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5(9):755-768. doi: 10.1038/nrd2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tobin JV, Zimmer DP, Shea C, et al. . Pharmacological characterization of IW-1973, a novel soluble guanylate cyclase stimulator with extensive tissue distribution, antihypertensive, anti-inflammatory, and antifibrotic effects in preclinical models of disease. J Pharmacol Exp Ther. 2018;365(3):664-675. doi: 10.1124/jpet.117.247429 [DOI] [PubMed] [Google Scholar]
- 12.Shea CM, Price GM, Liu G, et al. . Soluble guanylate cyclase stimulator praliciguat attenuates inflammation, fibrosis, and end-organ damage in the Dahl model of cardiorenal failure. Am J Physiol Renal Physiol. 2020;318(1):F148-F159. doi: 10.1152/ajprenal.00247.2019 [DOI] [PubMed] [Google Scholar]
- 13.Hanrahan JP, Wakefield JD, Wilson PJ, et al. . A randomized, placebo-controlled, multiple-ascending-dose study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of the soluble guanylate cyclase stimulator praliciguat in healthy subjects. Clin Pharmacol Drug Dev. 2019;8(5):564-575. doi: 10.1002/cpdd.627 [DOI] [PubMed] [Google Scholar]
- 14.Hanrahan JP, Seferovic JP, Wakefield JD, et al. . An exploratory, randomised, placebo-controlled, 14 day trial of the soluble guanylate cyclase stimulator praliciguat in participants with type 2 diabetes and hypertension. Diabetologia. 2020;63(4):733-743. doi: 10.1007/s00125-019-05062-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Udelson JE, Lewis GD, Shah SJ, et al. . Rationale and design for a multicenter, randomized, double-blind, placebo-controlled, phase 2 study evaluating the safety and efficacy of the soluble guanylate cyclase stimulator praliciguat over 12 weeks in patients with heart failure with preserved ejection fraction (CAPACITY HFpEF). Am Heart J. 2020;222:183-190. doi: 10.1016/j.ahj.2020.01.009 [DOI] [PubMed] [Google Scholar]
- 16.Fletcher GF, Balady G, Froelicher VF, Hartley LH, Haskell WL, Pollock ML. Exercise standards: a statement for healthcare professionals from the American Heart Association. Circulation. 1995;91(2):580-615. doi: 10.1161/01.CIR.91.2.580 [DOI] [PubMed] [Google Scholar]
- 17.Kitzman DW, Brubaker P, Morgan T, et al. . Effect of caloric restriction or aerobic exercise training on peak oxygen consumption and quality of life in obese older patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2016;315(1):36-46. doi: 10.1001/jama.2015.17346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redfield MM, Anstrom KJ, Levine JA, et al. ; NHLBI Heart Failure Clinical Research Network . Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med. 2015;373(24):2314-2324. doi: 10.1056/NEJMoa1510774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borlaug BA, Koepp KE, Melenovsky V. Sodium nitrite improves exercise hemodynamics and ventricular performance in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2015;66(15):1672-1682. doi: 10.1016/j.jacc.2015.07.067 [DOI] [PubMed] [Google Scholar]
- 20.Zamani P, Rawat D, Shiva-Kumar P, et al. . Effect of inorganic nitrate on exercise capacity in heart failure with preserved ejection fraction. Circulation. 2015;131(4):371-380. doi: 10.1161/CIRCULATIONAHA.114.012957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redfield MM, Chen HH, Borlaug BA, et al. ; RELAX Trial . Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309(12):1268-1277. doi: 10.1001/jama.2013.2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borlaug BA, Anstrom KJ, Lewis GD, et al. ; National Heart, Lung, and Blood Institute Heart Failure Clinical Research Network . Effect of inorganic nitrite vs placebo on exercise capacity among patients with heart failure with preserved ejection fraction: the INDIE-HFpEF randomized clinical trial. JAMA. 2018;320(17):1764-1773. doi: 10.1001/jama.2018.14852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitzman DW, Brubaker PH, Morgan TM, Stewart KP, Little WC. Exercise training in older patients with heart failure and preserved ejection fraction: a randomized, controlled, single-blind trial. Circ Heart Fail. 2010;3(6):659-667. doi: 10.1161/CIRCHEARTFAILURE.110.958785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anand IS, Rector TS, Cleland JG, et al. . Prognostic value of baseline plasma amino-terminal pro-brain natriuretic peptide and its interactions with irbesartan treatment effects in patients with heart failure and preserved ejection fraction: findings from the I-PRESERVE trial. Circ Heart Fail. 2011;4(5):569-577. doi: 10.1161/CIRCHEARTFAILURE.111.962654 [DOI] [PubMed] [Google Scholar]
- 25.Anand IS, Claggett B, Liu J, et al. . Interaction between spironolactone and natriuretic peptides in patients with heart failure and preserved ejection fraction: from the TOPCAT trial. JACC Heart Fail. 2017;5(4):241-252. doi: 10.1016/j.jchf.2016.11.015 [DOI] [PubMed] [Google Scholar]
- 26.Schelbert EB, Fridman Y, Wong TC, et al. . Temporal relation between myocardial fibrosis and heart failure with preserved ejection fraction: association with baseline disease severity and subsequent outcome. JAMA Cardiol. 2017;2(9):995-1006. doi: 10.1001/jamacardio.2017.2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borlaug BA, Nishimura RA, Sorajja P, Lam CS, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail. 2010;3(5):588-595. doi: 10.1161/CIRCHEARTFAILURE.109.930701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anjan VY, Loftus TM, Burke MA, et al. . Prevalence, clinical phenotype, and outcomes associated with normal B-type natriuretic peptide levels in heart failure with preserved ejection fraction. Am J Cardiol. 2012;110(6):870-876. doi: 10.1016/j.amjcard.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. 2017;136(1):6-19. doi: 10.1161/CIRCULATIONAHA.116.026807 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial protocol
Statistical analysis plan
eTables
Data sharing statement