

Abstract
Low-dose interleukin-2 (IL-2) represents a new therapeutic approach to regulate immune homeostasis to promote immune tolerance in patients with autoimmune diseases, including type 1 diabetes. We have developed a new IL-2–based biologic, an IL-2/CD25 fusion protein, with greatly improved pharmacokinetics and pharmacodynamics when compared with recombinant IL-2 to enhance this type of immunotherapy. In this study, we show that low-dose mouse IL-2/CD25 (mIL-2/CD25), but not an equivalent amount of IL-2, prevents the onset of diabetes in NOD mice and controls diabetes in hyperglycemic mice. mIL-2/CD25 acts not only to expand regulatory T cells (Tregs) but also to increase their activation and migration into lymphoid tissues and the pancreas. Lower incidence of diabetes is associated with increased serum levels of IL-10, a cytokine readily produced by activated Tregs. These effects likely act in concert to lower islet inflammation while increasing Tregs in the remaining inflamed islets. mIL-2/CD25 treatment is also associated with lower anti-insulin autoantibody levels in part by inhibition of T follicular helper cells. Thus, long-acting mIL-2/CD25 represents an improved IL-2 analog that persistently elevates Tregs to maintain a favorable Treg/effector T cell ratio that limits diabetes by expansion of activated Tregs that readily migrate into lymphoid tissues and the pancreas while inhibiting autoantibodies.
Introduction
Recombinant interleukin-2 (IL-2) is currently under clinical investigation to increase regulatory T cells (Tregs) in patients with autoimmunity to suppress inflammation associated with autoreactive T cells. The rationale for this immunotherapy is: 1) IL-2 is essential for Treg development, homeostasis, and function (1); 2) low IL-2 receptor (IL-2R) signaling effectively supports Tregs, but not effector T (Teff) cells (2,3); and 3) autoimmunity was effectively controlled using low amounts of IL-2 in preclinical studies (4–6). A common theme of completed clinical trials is that this therapy is safe, with no indications of activation of self-reactive T cells, in which Tregs increased in all patients (7,8). Initial studies also demonstrated clinical improvement in patients with chronic graft-versus-host disease, hepatitis C virus–induced vasculitis, alopecia areata, and systemic lupus erythematosus (9–13). More recently, these types of findings have been extended to 11 other autoimmune diseases (14). With respect to type 1 diabetes (T1D), completed phase 1 trials have identified an optimal dose of 500,000 IU/m2 that supports Treg increases without adverse effects on C-peptide levels after a mixed-meal tolerance test (15–17). Currently, a phase 2 study in patients with recently diagnosed T1D, which assesses the efficacy of low-dose IL-2 therapy, is near completion of enrollment (DIABIL-2; NCT02411253).
A major drawback of IL-2 in this type of immunotherapy is that it must be frequently administered due to the short in vivo half-life of IL-2 (<10 min) (18,19), and the Treg increases achievable are limited by the potential for non-Treg activation and expansion. Efforts, therefore, have been underway to develop analogs of IL-2 to improve the selectivity toward Tregs and/or extend the persistence of IL-2R signaling (20–29). In this regard, we have developed a novel IL-2 biologic, a fusion protein in which mouse IL-2 is covalently linked to mouse CD25 (mIL-2/CD25) using a Gly/Ser linker (30). mIL-2/CD25 predominately exists as an inactive noncovalent head-to-tail dimer that slowly dissociates into a biologically active mIL-2/CD25 monomer. At low doses, mIL-2/CD25 persistently and selectively stimulates Tregs due to its long half-life (16 h) and to the dissociation of dimer to monomer that limits the available amount of active fusion protein to stimulate IL-2R signaling. A single administration of low-dose mIL-2/CD25 in mice increased Tregs, but not Teff and NK cells, while an equivalent amount of IL-2 was not effective in stimulating Tregs. Moreover, low-dose mIL-2/CD25 prevented diabetes when administered for 5 weeks to young prediabetic NOD mice as well as when administration was delayed to a time that overlapped with the diabetes onset (30).
In the current study, we have evaluated the outcome of persistent IL-2R signaling induced by mIL-2/CD25 in Tregs. We have also further assessed the effectiveness of mIL-2/CD25 to limit diabetes, including therapy applied to hyperglycemic NOD mice. Our data favor a model in which mIL-2/CD25 persistently elevates Tregs to generate a favorable ratio of Tregs to Teff cells that controls diabetes by expansion and development of activated Tregs that also promotes migration into lymphoid tissues and the pancreas.
Research Design and Methods
Mice
Female NOD-ShiLtJ, NOD.SCID (NOD.CB17-PrkdcSCID/J), and NOD CD45.2 congenic (NOD.B6-Ptprcb/6908MrkJacJ) mice were purchased from The Jackson Laboratory. Mice were housed in a specific pathogen-free animal facility at the University of Miami. All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Miami.
IL-2 and mIL-2/CD25
mIL-2 was purchased from Thermo Fisher Scientific, and human IL-2 (hIL-2) (aldesleukin/Proleukin) was manufactured by Novartis and purchased through the pharmacy. mIL-2/CD25 was purified from culture supernatants from mIL-2/CD25-transfected CHO cells by Ni affinity chromatography columns, as previously described (30). hIL-2 and mIL-2/CD25 were administered to mice by i.p. injections.
Diabetes Assessment
Urine and blood glucose levels were monitored at least twice a week for diabetes. For the prevention study, mice were considered diabetic when two consecutive blood glucose readings were >250 mg/dL. For the experiments assessing the effect of mIL-2/CD25 on hyperglycemic mice (two consecutive daily blood glucose measurements between 150 and 275 mg/dL), mice were considered diabetic when blood glucose levels were >400 mg/dL. These mice were monitored until their blood glucose reached 600 mg/dL to ascertain whether mIL-2/CD25 might reverse diabetes.
Determination of Islet Infiltration
To assess islet infiltration, pancreas sections (5 μm) were fixed in 10% neutral buffered formalin, embedded in paraffin, and stained by hematoxylin and eosin. Islet infiltration of lymphocytes was blindly assessed by light microscopy and scored as described (31): 1) no insulitis, 2) lymphocytic infiltration around periphery of islet (peri/polar-insulitis), 3) <50% lymphocytic infiltration, 4) ≥50% of lymphocytic infiltration, and 5) end-stage insulitis (≥90% of lymphocytic infiltration).
Measurement of Insulin Autoantibodies and Cytokines in the Serum
Insulin autoantibodies (IAA) were assessed using the radiobinding assay developed at the Barbara Davis Center for Childhood Diabetes as previously published (32), and the results are expressed as an index. The sensitivity and specificity were 66% and 99%, respectively, in the 2018 Islet Autoantibody Standardization Program workshop. To measure serum cytokines, mouse-specific Luminex reagents were sourced from Millipore Sigma. Cytokine measurements were performed using manufacturer’s protocols that have been miniaturized to 384-well format (33). Lyophilized cytokine standard cocktails were also purchased from Millipore Sigma and prepared according to the manufacturer’s instruction. Mouse serum samples were kept at −80°C prior to analysis. Samples were thawed on ice and diluted 1:1 in the Luminex kit assay buffer. A total of 10 µL of diluted serum, standard, and kit-provided control samples were transferred to 384-well assay plates (catalog number 781096; Greiner Bio-One) and mixed with 10 µL Luminex bead reagents. Plates were covered with plastic seals and shaken overnight at 4°C in the dark. Subsequently, plates were washed two times using 90 µL of 1× wash buffer using a magnetic plate washer (BioTek). Then, 10 µL of detection antibody solution was added to each well and mixed. Plates were shaken for 1 h at room temperature in the dark. Finally, 10 µL of streptavidin-labeled phycoerythrin (PE) was added to each well. The assay plates were incubated for 30 min at room temperature in the dark before two washing steps using 90 µL of 1× wash buffer. After the last wash, 80 µL of sheath fluid was added to each well to prevent the samples from drying out. Plates were measured using the Bio-Plex 3D system (Bio-Rad Laboratories), and data were analyzed using Bio-Plex Software 3.0. A total of 70 µL and a minimum of 50 bead events/analyte were acquired. The raw data (mean fluorescence intensity) was converted into picograms per milliliter from the standard curve of each analyte (triplicates of 12 individual points based on one to two serial dilutions) using four- or five- parametric logistic fit models.
Antibodies for Flow Cytometry
Antibodies to mouse Foxp3 (eFluor 450; clone FJK-16s), CD62L (Qdot 605; clone MEL-14), CD103 (PE; clone 2E7), Klrg-1 (allophycocyanin [APC]; clone 2F1), and APC-labeled streptavidin were purchased from Thermo Fisher Scientific. Antibodies to mouse CD25 (PE; clone PC61), STAT5 (Alexa Fluor 647; clone pY694), Ki67 (Alexa Fluor 700; clone B56), and CXCR5 (not conjugated; clone 2G8) were purchased from BD Biosciences. Antibodies to mouse CD4 (PE; clone RM4-5), CD8α (PerCP/Cy5.5 and Alexa Fluor 700; clone 56.6.7), CD69 (PE/Cy7; clone H1.2F3), CD45.2 (APC; clone 104), and PD-1 (PE; clone RMPI-14) were purchased from BioLegend. Biotinylated anti-rat IgG was purchased from Jackson ImmunoResearch Laboratories. Antibody to mouse CD4 (FITC; clone GK1.5) was prepared in house.
FACS Analysis
Single-cell suspensions were prepared in Hanks’ balanced salt solution (HBSS) containing BSA (2 mg/mL) and sodium azide (1 mg/mL) and stained with monoclonal antibodies to relevant surface molecules. For assessment of peripheral blood mononuclear cells, red blood cells were first lysed using Tris (0.2%, pH 7.6) ammonium chloride (0.75%). To detect Foxp3, cells were then fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffers Set (Thermo Fisher Scientific) according to the manufacturer’s instruction. Tyrosine phosphorylation of STAT5 (pSTAT5) was determined as previously described (30). In brief, to measure pSTAT5 directly ex vivo, spleen cell suspensions were immediately fixed and permeabilized prior to staining for pSTAT5 and relevant T-cell molecules. For IL-2–induced pSTAT5 in vitro, spleen cells were cultured for 30 min in medium and then stimulated with mIL-2 for 15 min. These cells were then fixed and permeabilized prior to staining for pSTAT5 and relevant T-cell molecules. CXCR5 was detected by three-step staining (i.e., anti-CXCR5 followed by biotinylated anti-rat IgG and then APC streptavidin) (34). Cells were subjected to FACS analysis using BD LSRFortessa HTS and BD LSR II analyzers. Typically, 1 × 106 events/sample were acquired. All FACS data were analyzed using BD FACSDiva software version 8.0.1 or 8.0.2.
Islet Imaging
Heat-induced antigen retrieval was performed on formalin-fixed paraffin-embedded pancreas sections after submersion in 1× Antigen Decloaker (CB910M; Biocare Medical) using a pressure chamber. Sections were washed with PBS and then blocked for nonspecific binding with Triton X-100 (0.2%), 5% normal goat serum (catalog number 005-000-121; Jackson ImmunoResearch Laboratories), and 2% BSA in PBS and then stained overnight with primary antibodies (i.e., rat anti-mouse Foxp3 [clone FJK-16s, catalog number 14-5773-82; Thermo Fisher Scientific], polyclonal guinea pig anti-insulin [catalog number AB7842; Abcam], and rabbit anti-glucagon [catalog number AB92517; Abcam]) in blocking solution. Sections were washed with PBS three times and incubated with secondary antibodies (i.e., goat anti-rabbit IgG Alexa Fluor 488 [catalog number A-11008; Thermo Fisher Scientific], goat anti-rat IgG Alexa Fluor 568 [catalog number A-11077; Thermo Fisher Scientific], and goat anti–guinea pig IgG Alexa Fluor 647 [catalog number A-21450; Thermo Fisher Scientific]) in blocking solution for 1 h. Finally, sections were washed six times with PBS and mounted with Prolong Gold Antifade with DAPI (P36935; Life Technologies). Islets were imaged at ×10 original magnification using a KEYENCE BZ-X710 fluorescence microscope.
Statistical Analysis
Data were analyzed using GraphPad Prism 7 software. All data are represented as ± SEM. The specific statistical analyses used are listed in the figure legends. When exact values are not shown, P values are indicated as: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; and ****P ≤ 0.0001.
Data and Resource Availability
Data supporting the findings of this study are available from the authors upon reasonable request.
Results
The Effect of Acute Administration of mIL-2/CD25 on Tregs and Teff Cells
Our previous study showed that 4 µg of mIL-2/CD25 twice a week was the lowest effective dose to prevent diabetes in female NOD mice (30). To further evaluate the selectivity of Tregs to mIL-2/CD25, NOD mice received a single injection of various amounts of mIL-2/CD25 (1–100 µg), and their T lymphocytes were evaluated 3 days later ex vivo. CD4+Foxp3+ Tregs, but not CD4+Foxp3− conventional T (Tconv) cells, from the spleen showed mIL-2/CD25-dependent pSTAT5 activation over the entire dose response (Fig. 1A).
Figure 1.
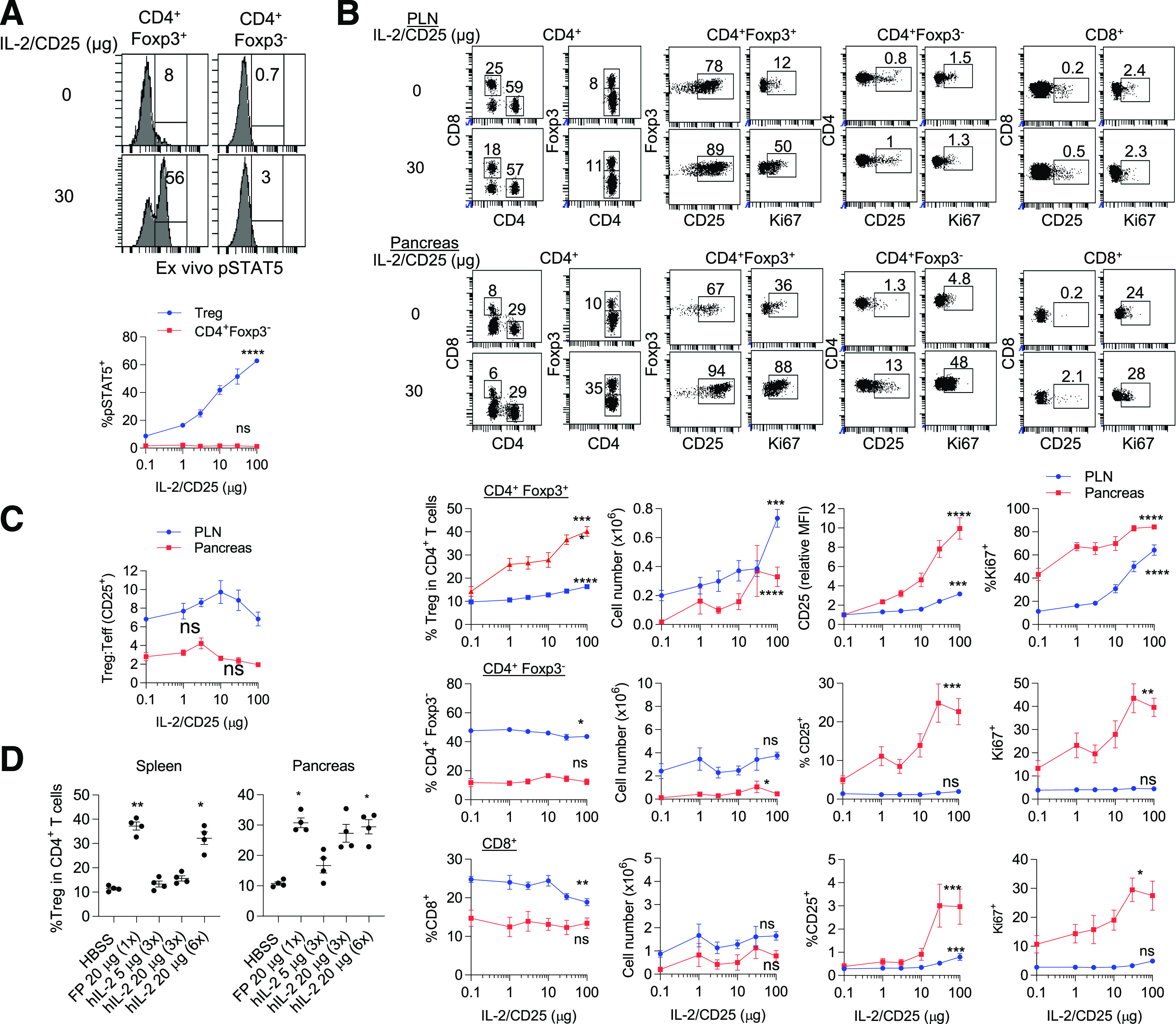
Low-dose mIL-2/CD25 selectively increases Tregs in NOD mice. Six- to 7-week-old female NOD mice received a single dose of mIL-2/CD25 or HBSS, and the indicated cell populations from spleen, PLN, and pancreas were assessed 72 h posttreatment. A: Representative FACS histograms (top) and quantitative data (bottom) for pSTAT5 amounts in splenic Tregs assessed directly ex vivo (n = 5–6/group). B: Representative FACS dot plots (top) and quantitative data (bottom) of the proportions, numbers of CD4+Foxp3+ Treg, CD4+Foxp3− Tconv, and CD8+ T cells, and their expression of CD25 and Ki67 in the PLN and pancreas (n = 7–10/group). C: The ratio of CD4+Foxp3+ Tregs to CD4+CD25+ Teff cells was enumerated for the PLN and pancreas after gating on each cell population (n = 4/group) as represented in Supplementary Fig. 1. D: The expansion of Tregs 3 days after administering a single injection of mIL-2/CD25 or daily (3 times) or every 12 h (6 times) injections of IL-2, as indicated (n = 4/group). B–D: Data are shown as mean ± SEM and were analyzed by unpaired one-way ANOVA Kruskal-Wallis test. Significance of the dose response (B) or the comparison of mIL-2/CD25 vs. IL-2 (D) was assessed relative to control HBSS-treated mice. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. FP, fusion protein; MFI, mean fluorescence intensity.
To determine whether this high selectivity was maintained in autoimmune-related tissues, responses by Treg and CD4+ and CD8+ Tconv cells in the pancreatic lymph node (PLN) and the pancreas were also examined 3 days postadministration. Representative FACS plots for a single relatively high dose (30 µg) of mIL-2/CD25 are shown in the top panel of Fig. 1B. Quantitative results (Fig. 1B, bottom) showed a dose-dependent increase in Treg proportions and numbers that was greater for the pancreas. Tregs also showed a proportional increase in proliferation, as assessed by Ki67 expression, and in the mean fluorescence intensity of CD25, which is upregulated by IL-2–dependent signaling (30,35). In contrast, the proportion and numbers (Fig. 1B) of CD4+Foxp3− and CD8+ T cells were unchanged or declined with increasing dose of mIL-2/CD25. However, at higher doses (30 and 100 µg) of mIL-2/CD25, CD4+ and CD8+ T cells in the pancreas exhibited increased percentage of CD25+ and Ki67+ cells. Thus, high-dose mIL-2/CD25 stimulated some Tconv cells in the autoimmune target tissue.
The proportion of CD4+Foxp3+ Tregs to CD4+CD25+ Tconv cells in the PLN and pancreas was also evaluated. The gating strategy to identify these cell populations is shown in Supplementary Fig. 1. This ratio did not significantly change over this dose response even though CD4+Foxp3−CD25+ T cells, which likely represent Teff cells, markedly increased in the pancreas at high doses of mIL-2/CD25 (Fig. 1C). A trend for higher Treg/Teff ratios was noted at lower doses (3–10 µg) of mIL-2/CD25, which further supports the use of low-dose mIL-2/CD25 in treatment of autoimmunity. Consistent with these observations, a low dose (4 µg) of mIL-2/CD25 was effective in controlling diabetes in NOD mice (30). Collectively, these data indicate that at doses <30 µg, mIL-2/CD25 is largely selective for Tregs in which the Treg/CD4 Tconv ratio was at least maintained at all doses in the PLN and pancreas, target tissue related to autoimmune diabetes in NOD mice.
The Relative Potency of mIL-2/CD25 and IL-2 to Expand Tregs
mIL-2/CD25 substantially expands Tregs when assessed 3 days later (Fig. 1B). To evaluate the potency of mIL-2/CD25 relative to IL-2 in vivo, the expansion of Tregs by a single injection of mIL-2/CD25 was compared with multiple (daily and twice daily) injections of IL-2. mIL-2/CD25 and IL-2 were administered based on their molecular mass rather than bioactivity because the fusion protein exists dominantly as inactive transdimers, which underestimates its IL-2 activity when using IL-2–dependent CTLL cells in vitro (30). As the molecular mass of IL-2 is four times smaller than the fusion protein, an equivalent dose of IL-2 represents four times more molecules than mIL-2/CD25. Similar increased proportions and numbers of Tregs, with an accompanying increased expression of CD25 and Ki67, were found in the spleen and pancreas of NOD mice after administering 20 µg of mIL-2/CD25 once or 20 µg of IL-2 six times (Fig. 1D and Supplementary Fig. 2). Thus, on a per-molecule basis, frequent administration of 24-fold more IL-2 is required to approximate the same effects on Tregs by a single administration of mIL-2/CD25.
mIL-2/CD25, but Not Equivalent IL-2, Controls Diabetes in NOD Mice
The dose-response data (Fig. 1) suggest that mIL-2/CD25 might control T1D in NOD mice over a larger dose range than the twice-weekly 4-µg dose, which is the minimum required to delay the onset of diabetes in NOD mice (30). To assess this point and evaluate more prolonged administration of mIL-2/CD25, we investigated a relatively low (4 μg) and a higher dose (20 μg) of mIL-2/CD25 to prevent the onset of diabetes in NOD mice. Both doses of mIL-2/CD25 significantly delayed the onset of diabetes when compared with control HBSS-treated NOD mice or a dose of hIL-2 (1 µg), which provides equivalent total moles to 4 µg of mIL-2/CD25 (Fig. 2A). The inability of IL-2 to control diabetes in this experiment was expected, as the dose and frequency of IL-2 was less than used in previous studies that established that recombinant IL-2 limits diabetes in NOD mice (4,5). Although mIL-2/CD25 has a prolonged half-life compared with IL-2, with a half-life of 16 h versus <10 min, the concentration of mIL-2/CD25 in circulation would be minimal in less than a week after dosing cessation. Despite this, onset of diabetes was delayed by >5 weeks after cessation of dosing. Collectively, these data indicate that an identical twice-a-week treatment for 10 weeks using mIL-2/CD25, but not IL-2, leads to significant reduction of diabetes even in the off-drug period after dosing and demonstrate the pharmacologic and therapeutic benefit of long-acting mIL-2/CD25-mediated IL-2R agonism.
Figure 2.
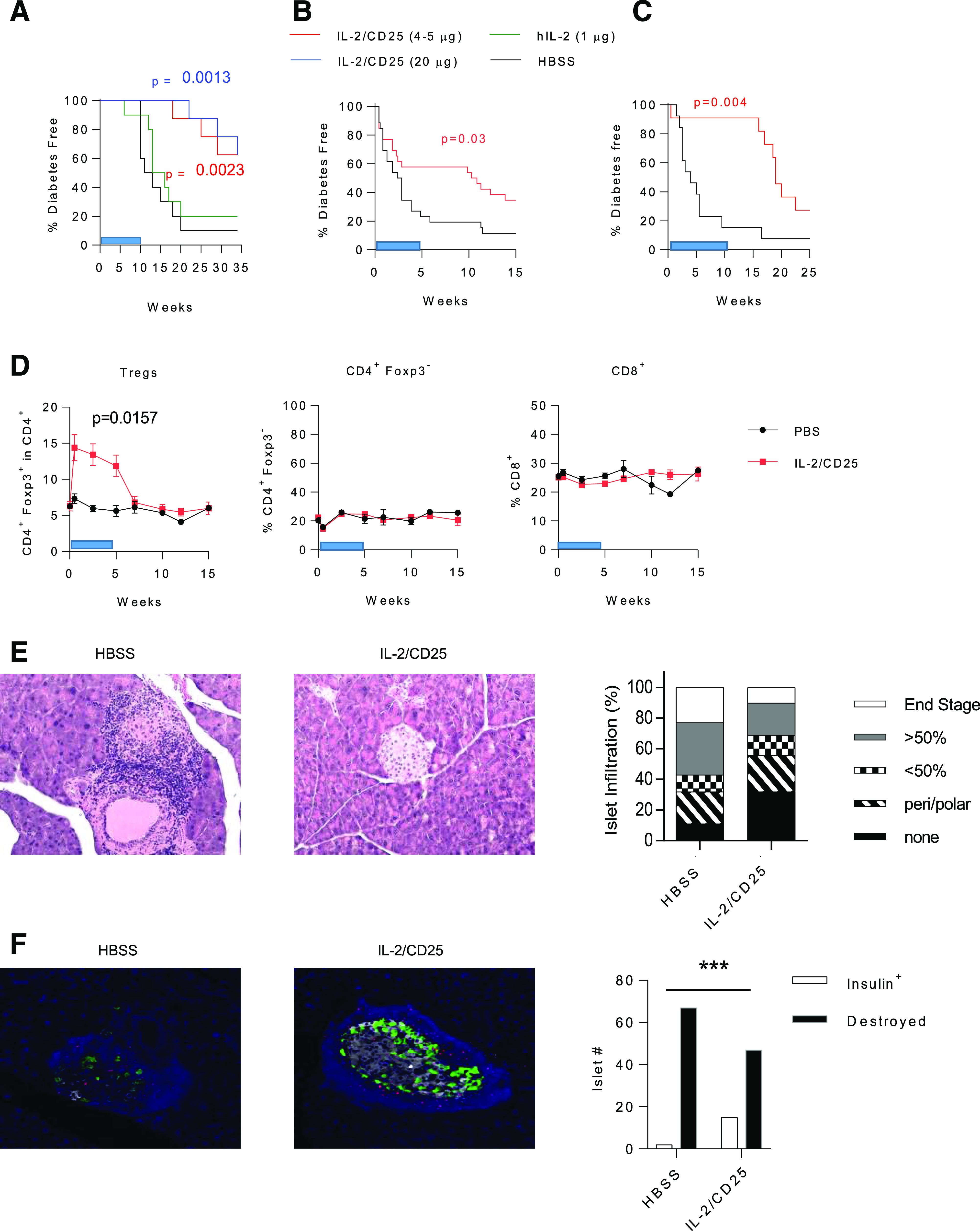
mIL-2/CD25 controls diabetes in NOD mice. A: Six- to 7-week-old female NOD mice received the indicated dose of mIL-2/CD25, hIL-2, or HBSS twice a week for 10 weeks. Diabetes-free survival (n = 8–10/group) was analyzed by Mantel-Cox log-rank test. Hyperglycemic female NOD mice (two consecutive blood glucose readings between 150 and 275 mg/dL glucose) were treated twice a week with HBSS or mIL-2/CD25 (5 µg) for 5 (B) or 10 (C) weeks. Diabetes-free survival data (B, n = 26 mice/group; C, n = 13–14/group) were analyzed by Mantel-Cox log-rank test. D: The representation of the indicated T-cell subpopulations in the blood from hyperglycemic NOD mice treated with HBSS or mIL-2/CD25 for 5 weeks. The 0-week time point represents a bleed prior to the start of mIL-2/CD25 treatment. Data (n = 13/group) were analyzed by unpaired two-sided t test after determining the area under each curve. The blue bars (A–D) indicate the treatment period with mIL-2/CD25 or HBSS. E: Representative hematoxylin and eosin staining (left) (×12.5 original magnification) of diabetic HBSS control-treated and a diabetes-free mIL-2/CD25-treated (5 weeks) hyperglycemic NOD mouse; inflammation scoring (right) of pancreas sections of control HBSS- (209 islets scored from 25 mice) and mIL-2/CD25-treated mice (200 islets scored from 26 mice). F: Representative staining (left) and quantitative data (right) of insulin-producing islets and their inflammatory status of pancreas sections (10 times) of diabetic HBSS control-treated (n = 8/group) and diabetes-free mIL-2/CD25-treated (5 weeks) hyperglycemic (n = 9/group) NOD mouse. Pancreas sections were stained for insulin (white), glucagon (green), and Foxp3 (red) and counterstained with DAPI. Insulin-expressing islets and destroyed islets were counted. Data were analyzed by χ2 test. ***P ≤ 0.001.
mIL-2/CD25 Controls Diabetes in Hyperglycemic Mice
We also investigated the efficacy of mIL-2/CD25 to reverse hyperglycemia in recent-onset diabetic NOD mice using a model of late prediabetes that is representative of stage 2 T1D. When mice exhibited two consecutive daily blood glucose readings of ≥150 mg/dL but ≤275 mg/dL, they received twice-weekly injections of low-dose (5 µg) mIL-2/CD25. When mice were treated for 5 weeks with the mIL-2/CD25, diabetes progression was delayed in ∼60% of the mice (Fig. 2B). Another cohort of mice was treated with 5 µg of mIL-2/CD25 for 10 weeks. This longer treatment with mIL-2/CD25 controlled diabetes in ∼90% of the mice (Fig. 2C). Blood glucose levels of individual mice in each experiment were evaluated (Supplementary Fig. 3), which revealed several points. First, for those mice that responded to mIL-2/CD25 therapy, some mice eventually progressed to diabetes when treatment with mIL-2/CD25 was stopped, while others remained diabetes free for 10–15 weeks off therapy. Second, a number of mice, especially in the shorter (5-week) treatment experiment, did not respond to mIL-2/CD25 treatment, as their serum glucose levels continued to rise during the treatment period. When the data from both experiments were combined and stratified based on their age and time to progression to diabetes, mice that became hyperglycemic at a younger age (≤14 weeks of age) were less responsive to mIL-2/CD25 treatment than older mice (Supplementary Fig. 4A). This result is more easily seen when the number of mIL-2/CD25-treated mice with delayed diabetes (>10 weeks) is evaluated based on age of onset of hyperglycemia (Supplementary Fig. 4B). We found that 45% were responsive to mIL-2/CD25 when hyperglycemia was noted at ≤14 weeks of age, but 78% were responsive when hyperglycemia developed at >14 weeks of age. This result suggests that mIL-2/CD25 is more effective in animals when diabetes takes a longer time to develop.
Tregs in the blood increased during mIL-2/CD25 therapy but diminished to baseline after treatment ceased (Fig. 2D). In contrast, CD4+Foxp3− and CD8+ T cells were minimally affected by mIL-2/CD25 (Fig. 2D), pointing to the selectivity of Treg expansion to mIL-2/CD25. Hematoxylin and eosin staining of the pancreas revealed that NOD mice treated with the mIL-2/CD25 exhibited lower inflammation of the islets when compared with diabetic control-treated mice (Fig. 2E). This result was confirmed when pancreas sections were stained for insulin, glucagon, Foxp3, and DAPI; insulin-producing β-cells were more readily found in mIL-2/CD25-treated mice (Fig. 2F). Insulin-producing islets after mIL-2/CD25 therapy, nevertheless, often exhibited inflammation surrounding the islet that contained numerous Foxp3+ Tregs, consistent with Treg immunoregulation of an autoimmune attack. Collectively, these data show that mIL-2/CD25 controls progression of diabetes in hyperglycemic mice. Although long-lasting immunoregulation occurred in some mice (Fig. 2A–C), inflamed insulin-producing islets were noted (Fig. 2F). Thus, low-dose mIL-2/CD25 therapy does not completely eliminate autoaggressive cells. Furthermore, mIL-2/CD25-dependent immunoregulation appears to be less effective in animals that rapidly develop early onset diabetes. Although evidence of off-drug efficacy was noted in animals that responded to treatment, the effect of mIL-2/CD25 treatment diminishes with time in some mice.
Consequences of Persistent mIL-2/CD25 Treatment on T Cells From NOD Mice
To assess the effects of mIL-2/CD25 therapy on T cells in the PLN and pancreas, an independent cohort of NOD mice was administered 4 and 20 µg of mIL-2/CD25 twice a week for 10 weeks, and their T cells were analyzed 3 days following the last administration. A significant dose-dependent increase in Treg frequency and number (Fig. 3A) was detected at both sites, with a more noticeable change in the pancreas. After treatment with mIL-2/CD25, CD25 increased in Tregs in the PLN and pancreas, while the percentage of Ki67+ Tregs increased in the PLN, but not the pancreas (Fig. 3A). This latter response was less striking than observed after a single injection of mIL-2/CD25 (Fig. 1A). The reason for this difference is not clear, but may reflect persistent versus acute responses to mIL-2/CD25 by Tregs or increased inflammation in the pancreas associated with older mice that received a therapeutic regimen of mIL-2/CD25.
Figure 3.
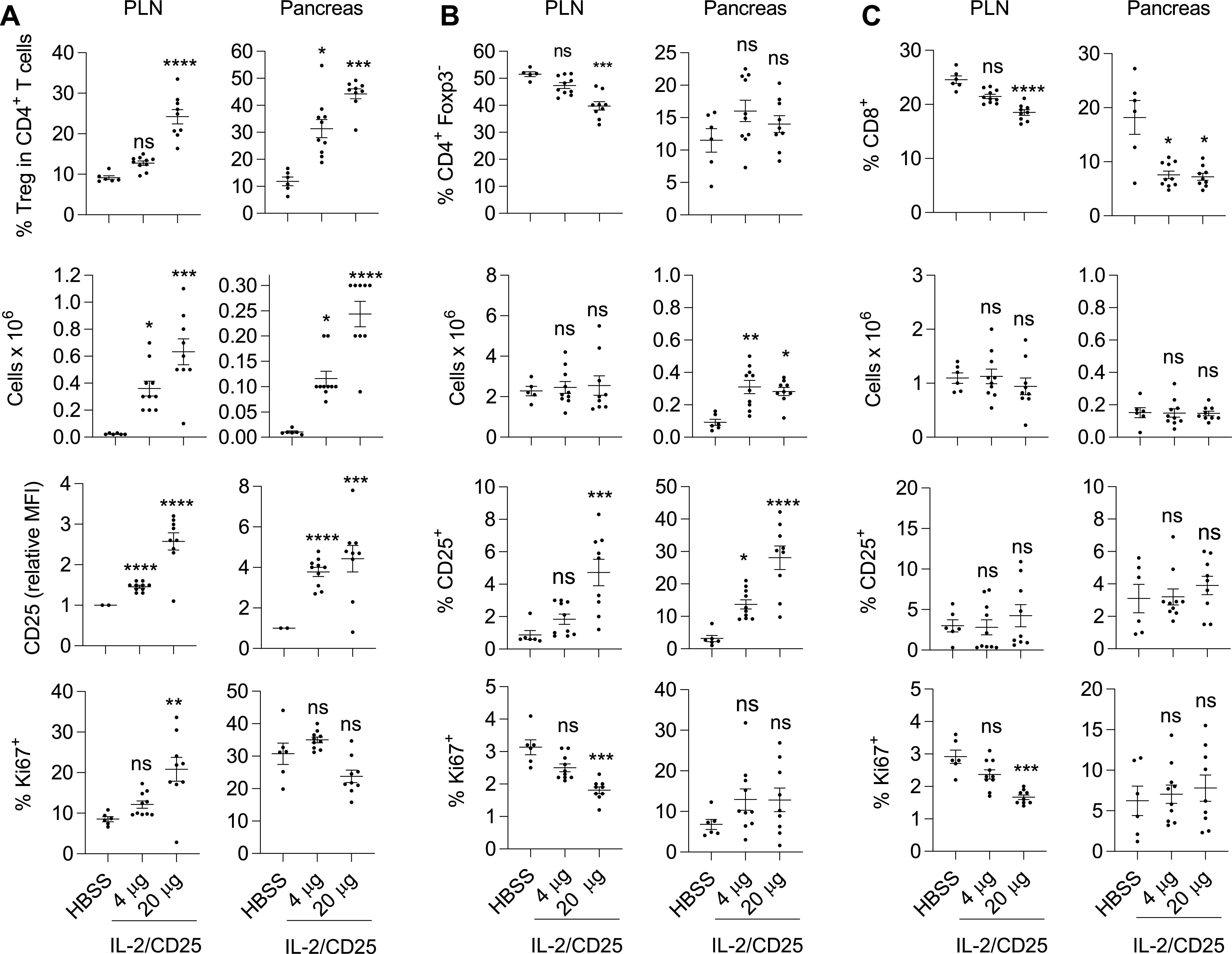
Persistent low-dose mIL-2/CD25 selectively expands Tregs. Seven- to 8-week-old female NOD mice received the indicated dose of mIL-2/CD25 or HBSS twice a week for 10 weeks. At 72 h after the last injection, the proportions and numbers of CD4+Foxp3+ Treg (A), CD4+Foxp3− Tconv (B), and CD8+ T cells (C), including their expression of CD25 and Ki67 (n = 6–10/group), were enumerated in the PLN and pancreas. A–C: Data (n = 6–10/group) were analyzed by one-way ANOVA (Kruskal-Wallis test) and Dunn multiple-comparison test. Significance was assessed in comparison with mice treated with HBSS. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. MFI, mean fluorescence intensity.
The influence of low-dose mIL-2/CD25 on CD4+Foxp3− and CD8+ T cells was minimal. The proportion and number of CD4+Foxp3− T cells (Fig. 3B) remained unchanged, while the proportion of CD8+ T cells decreased, although their numbers did not change (Fig. 3C). mIL-2/CD25 did not increase the proliferation of CD4+Foxp3− and CD8+ T cells, as measured by Ki67 (Fig. 3B and C) at either dose. Rather, proliferation of both were slightly lower at the higher dose (20 µg) of mIL-2/CD25 in the PLN, suggesting suppression by the accompanying increase in Tregs. However, the percentage of CD4+Foxp3− T cells that were CD25+ increased, especially in the pancreas (Fig. 3B). CD25 is one of the most highly upregulated genes upon IL-2R signaling (36). This observation, therefore, is consistent with a potential increase in IL-2R signaling in non-Tregs, which was particularly evident at the high dose of mIL-2/CD25. Thus, persistent mIL-2/CD25 results in a substantial increase in Tregs that appears to control any stimulation of non-Tregs, even at a relatively high dose of 20 µg.
Tregs can be divided into two major subsets, resting or central Tregs (cTregs) and activated or effector Tregs (eTregs). Phenotypic and single-cell RNA-sequencing data indicate that CD62L expression defines these two subsets, in which cTregs are CD62Lhi and eTregs are CD62Llo (37,38). Tregs that express CD69, CD103, Klrg1, and ICOS, among other markers, identify more activated Tregs and are often associated with subsets of eTregs (38–40). mIL-2/CD25 led to a dose-dependent increase in CD62Lhi cTregs with a corresponding decrease in highly activated CD62LloCD69+ eTregs (Fig. 4A). However, these mIL-2/CD25-dependent cTregs did not exhibit the classic resting Treg phenotype, as they expressed increased amounts of Treg activation molecules such as CD103, Klrg1, and ICOS, and they showed increased proliferation based on Ki67 expression (Fig. 4B and Supplementary Fig. 5). The mIL-2/CD25-dependent eTregs were also more activated, as an increased proportion expressed CD103 and Klrg1 (Fig. 4C and Supplementary Fig. 4). These results indicate that mIL-2/CD25 favors expansion of cTregs while simultaneously enhancing their activation, which likely promotes Treg-suppressive function. This conclusion is consistent with past global gene expression of Tregs from C57BL/6 mice that received a single injection of mIL-2/CD25 that increased mRNAs related to Treg growth, survival, metabolism, suppressive function, activation, and tissue homing (30).
Figure 4.

Persistent mIL-2/CD25 promotes cTregs with a more activated phenotype. Mice were treated for 10 weeks as described in the legend to Fig. 3 and assayed 72 h after the last injection. A: Representative FACS contour plots (top) and quantitative results (bottom) from the PLN and pancreas for the distribution of cTregs and highly activated eTregs based on expression of CD62L and CD69. Expression of markers of activation on CD62LhiCD69− cTregs (B) and CD62LloCD69+ eTregs (C). Data (n = 7–10/group) are shown as the mean ± SEM and analyzed by one-way ANOVA (Kruskal-Wallis test). Significance of the dose response was assessed relative to control HBSS-treated mice. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. Rel MFI, relative mean fluorescence intensity.
mIL-2/CD25 Supports Trafficking of Tregs to Lymphoid Tissues and the Pancreas
The above results indicate that mIL-2/CD25 supports a substantial and selective increase of Tregs within the pancreas. This led us to test the extent to which this effect might be due to migration of Tregs into this tissue sites. NOD CD45.2+ mice received a single administration of 10 µg of mIL-2/CD25, and 48 h later, spleen cells from these mice were transferred into NOD/SCID recipient mice. At 24–28 h posttransfer, donor CD45.2+ T cells were enumerated in spleen, mesenteric lymph node (MLN), and pancreas of the recipient mice (Fig. 5A). The PLN was not evaluated as it was too small to be identified. The input donor spleen cells had similar proportions of total CD4+ and CD8+ T cells, including the proportion of Tregs in CD4+ T cells (Fig. 5B). However, the Tregs from mIL-2/CD25-treated mice had increased CD25 and a trend for increased expression of Ki67 (Fig. 5B), properties expected when Tregs respond to the fusion protein.
Figure 5.
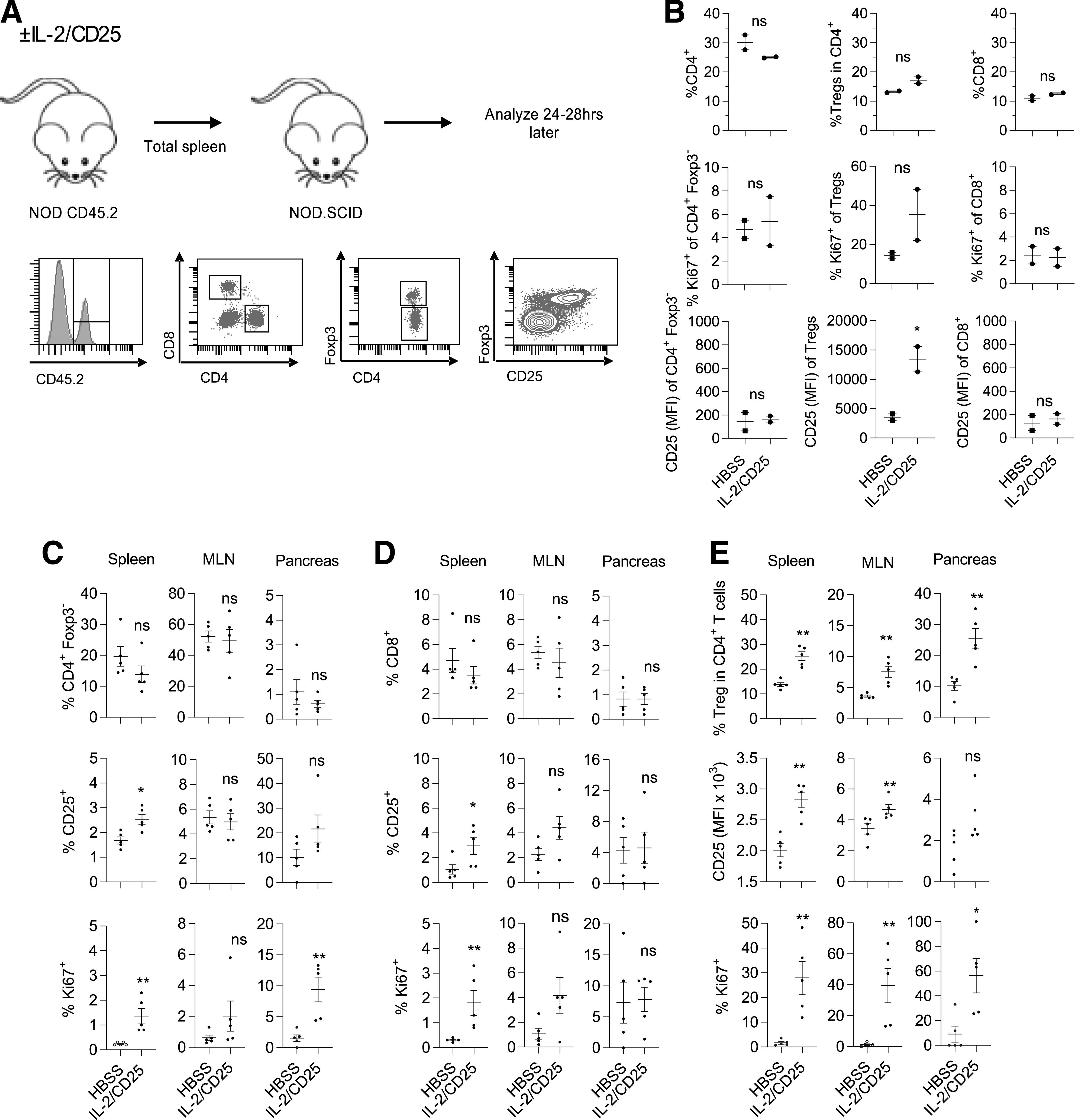
mIL-2/CD25 enhances trafficking of Tregs into lymphoid tissues and the pancreas of NOD.SCID mice. Seven- to 8-week-old female CD45.2+ NOD mice received a single injection of mIL-2/CD25 (10 μg) or HBSS. Forty-eight hours later, unfractionated spleen cells (3 × 107 cells) were transferred i.v. into 6- to 7-week-old female CD45.1+ NOD.SCID mice, and their spleen, MLN, and pancreas were analyzed 24–28 h posttransfer for donor T cells. A: Experimental flowchart and representative FACS gating. B: Distribution of T cells in the donor inoculum. Enumerated are total CD4+ T cells, the fraction of Tregs in CD4+ T cells, and CD8+ T cells, including their expression of CD25 and Ki67. Data represent two pools of donor cells, are shown as mean ± range, and were analyzed by an unpaired two-sided t test. C–E: Distribution of CD45.2-donor–derived T cells in recipient NOD.SCID mice. Enumerated are CD4+Foxp3− T cells (C), CD8+ T cells (D), and CD4+Foxp3+ Tregs (E), including their expression of CD25 and Ki67, from the indicated tissues. C–E: Data (n = 5/group) are combined from two independent experiments, are shown as mean ± SEM, and were analyzed by an unpaired two-sided t test. *P ≤ 0.05; **P ≤ 0.01. hrs, hours; MFI, mean fluorescence intensity.
Similar proportions of CD45.2+ lymphoid cells were detected in each tissue from the recipients that received donor-derived spleen cells from HBSS- or mIL-2/CD25-treated mice (Supplementary Fig. 6). Donor T cells were found in all three tissues, but they made up a higher proportion of the donor cells in the spleen and MLN (Fig. 5C and D, top panels). In addition, a higher frequency of donor Tregs was detected in all three tissues, with the highest frequency in the spleen and pancreas, when Tregs were derived from mIL-2/CD25-treated, but not HBSS-treated, donor spleen cells (Fig. 5E). In contrast, donor CD4+ Foxp3− (Fig. 5C) and CD8+ (Fig. 5D) T cells, when enumerated in relationship to the total fraction of donor CD45.2+ cells, were detected at a similar frequency in all tissues regardless of whether they were derived from mIL-2/CD25- or control-treated donor spleen cells. The preferential accumulation of Tregs from mIL-2/CD25-treated donor spleen cells was accompanied by increased proliferation, measured by Ki67 expression and upregulation of CD25 (Fig. 5E), whereas small changes in these parameters were observed for CD4+ Foxp3− (Fig. 5C) and CD8+ (Fig. 5D) T cells. These results, therefore, support the hypothesis that mIL-2/CD25 promotes the trafficking and accumulation of proliferating Tregs in tissues, including the pancreas, spleen, and lymph nodes. The increased accumulation of Tregs in these tissues when the donor cells were derived from the spleen of mIL-2/CD25-treated mice may reflect that the fusion protein has programmed the proliferation of these cells, where trafficking of Tregs is favored to these lymphoid tissue sites.
Low-Dose mIL-2/CD25 Minimally Changes Circulating Levels of Inflammatory Cytokines
Serum was also collected from NOD mice after 10 twice-weekly treatments with mIL-2/CD25. The levels of 33 inflammatory cytokines were measured by multiplex analysis. Of the measured cytokines (Supplementary Fig. 7), an increase in only three cytokines was observed and only at the highest dose tested. Increases in IL-10, CXCL9 (MIG), and CXCL10 (IP10) were seen for mice treated with 20 µg of the mIL-2/CD25 (Fig. 6), with a trend for higher IL-10 at the lower (4 µg) dose. The cellular origin of these cytokines is not known, but the increase in activated Tregs by mIL-2/CD25 may account for increased IL-10, which would favor dampening autoreactive T cells.
Figure 6.

Persistent mIL-2/CD25 increases serum cytokines in NOD mice. Mice were treated for 10 weeks as described in the legend to Fig. 3. Serum was collected 3 days after the last injection. The serum was evaluated by Luminex assay for 33 cytokines. Data for the only three cytokines differentially detected are shown. Data (n = 9–10/group) are shown as the mean ± SEM and analyzed by one-way ANOVA (Kruskal-Wallis test) and Dunn multiple-comparison test. Significance was assessed in comparison with mice treated with HBSS. *P ≤ 0.05; ***P ≤ 0.001.
Effect of Persistent mIL-2/CD25 on IL-2R Signaling by Tregs
When Treg levels were assessed in the blood over time, the greatest increase induced by the mIL-2/CD25 was noted at the initial bleed (Fig. 2D), with slightly lower responses upon repeated administration. We evaluated the extent that persistent IL-2R–dependent activation by mIL-2/CD25 might affect IL-2R signaling. Tregs were obtained from the spleen of NOD mice treated twice a week with 4 and 20 µg of mIL-2/CD25 for 10 weeks. Three days after the last administration of mIL-2/CD25, spleen cells were stimulated with IL-2 in vitro, and pSTAT5 amounts were determined as a measure of proximal IL-2R signaling. Shown are representative FACS plots (Fig. 7A) and dose-response curves by Tregs (Fig. 7B). These data revealed that IL-2R signaling is not desensitized after persistent stimulation in vivo with mIL-2/CD25, but rather appears to be modestly enhanced. Nonlinear regression analysis of these data (Fig. 7B, bottom) indicates the Tregs from the mIL-2/CD25-treated mice are 2.4–3.3 times more sensitive to IL-2 when compared with Tregs from control-treated mice. This effect is likely due at least in part to the upregulation of CD25 by mIL-2/CD25. Thus, the slight attenuation in circulating Tregs after persistent application of mIL-2/CD25 must be accounted for by a mechanism independent of initial activation of STAT5.
Figure 7.
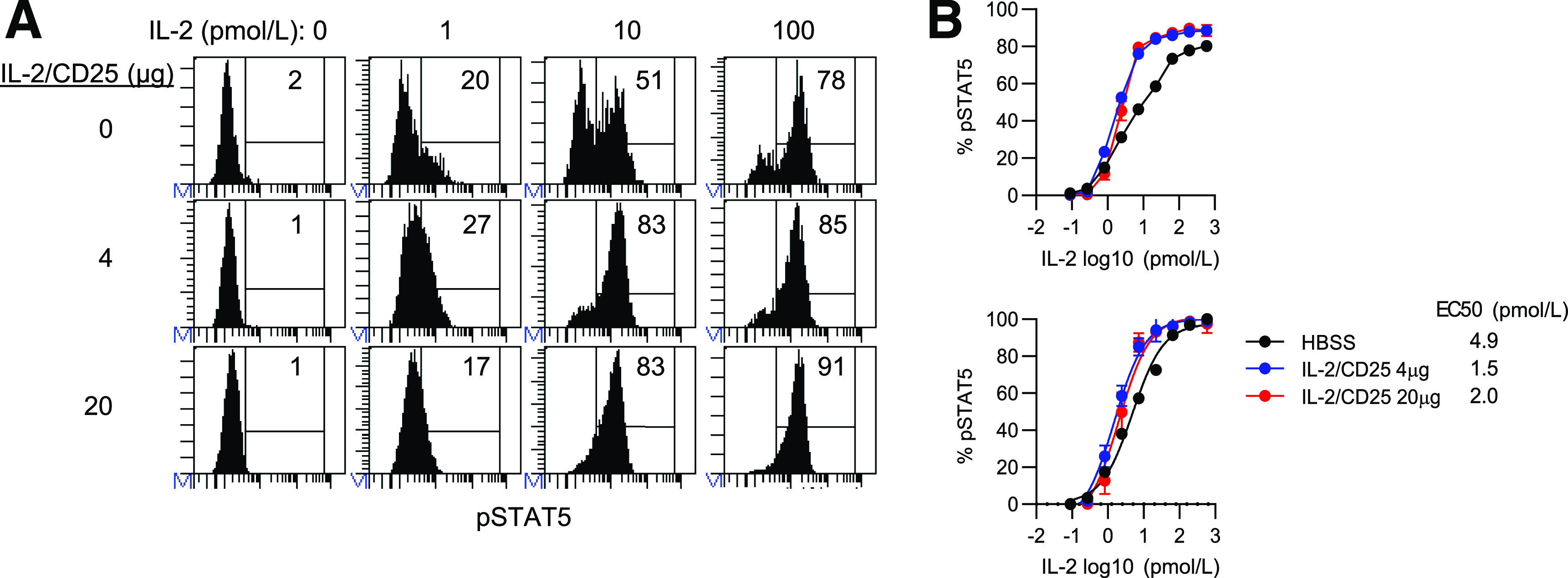
Tregs are more responsive to IL-2 after persistent stimulation with mIL-2/CD25. Mice were treated for 10 weeks as described in the legend to Fig. 3. At 72 h after the last injection, spleen cells from indicated mice were stimulated with mIL-2 for 15 min, and the amounts of pSTAT5 were determine by FACS analysis. Representative FACS histograms (A) and resulting dose-response curves (B), in which the top panel represents the binding data and the bottom panel represents nonlinear regression analysis of those data. Data (n = 5/group) are shown as the mean ± SEM.
mIL-2/CD25 Lowers Anti-IAA and T Follicular Helper Cells
IL-2R signaling opposes T follicular helper (Tfh) cell development (41). Therefore, we also tested whether mIL-2/CD25 affected IAA and Tfh cells. Serum collected after treatment with mIL-2/CD25 for 10 weeks (Fig. 2A) revealed that IAA titers in mIL-2/CD25-treated mice were below the level of detection in all but one mouse. In contrast, a higher frequency of mice with detectable IAA titers occurred in HBSS control-treated mice (Fig. 8A). Tfh cells were identified by CD4+Foxp3− T cells that coexpressed CXCR5 and PD-1. Representative dot plots (Fig. 8B) and quantitative data (Fig. 8C) revealed a mIL-2/CD25 dose-dependent reduction in Tfh cells for the spleen and PLN. Thus, besides promoting Tregs, mIL-2/CD25 lowers Tfh cells, which likely contributed to minimal IAA titers, and represents another beneficial feature of low-dose mIL-2/CD25 in the treatment of autoimmunity.
Figure 8.

Persistent mIL-2/CD25 lowers anti-IAA titers and inhibits Tfh cells. Mice were treated for 10 weeks with HBSS or mIL-2/CD25 (4 and 20 µg) and assayed 72 h after the last injection. A: Assessment of IAA levels after the 10-week treatment period. Data points above the horizontal line in the graph are considered IAA+. Data (n = 9/group) were analyzed by χ2 test as a function of IAA+ or IAA−. Representative contour plots (B) and quantitative data (C) of Tfh cells within the indicated tissues after gating on CD4+Foxp3− T cells. Data are expressed as the number of T follicular helper (Tfh) in 1 × 106 of CD4+Foxp3− T cells. Data (n = 6–10/group) are shown as the mean ± SEM and analyzed by one-way ANOVA (Kruskal-Wallis test) and Dunn multiple-comparison test. Significance was assessed in comparison with mice treated with HBSS. *P ≤ 0.05; ****P ≤ 0.0001.
Discussion
Low-dose IL-2 is a promising new therapy for patients with autoimmune disorders, including those with T1D. The short half-life of IL-2 and potential stimulation of non-Tregs have led to the development of new IL-2–based biologics with increased persistence and selectivity. mIL-2/CD25 is a novel fusion protein that provides active IL-2R agonism in which mIL-2/CD25 transdimers slowly dissociate into biologically active monomers (30). This mechanism leads to a steady, long-lived, and relatively low amount of active IL-2R agonism that favors capture by CD25-expressing cells and stimulation of cells bearing the high-affinity IL-2R. At low doses, mIL-2/CD25 is selective for Tregs due to their high surface expression of CD25 and other cell-intrinsic mechanisms, including increased activity of the serine/threonine phosphatase PP2A (3,42). In the current study, the superiority of mIL-2/CD25 over IL-2 was illustrated by similar expansion of Tregs using 24-fold lower mIL-2/CD25 than IL-2 and by the ability of only the mIL-2/CD25 to prevent diabetes in NOD mice after administering both forms of IL-2 using equal molar amounts at the same frequency.
We defined four important properties by which mIL-2/CD25 favorably controls diabetes in NOD mice. First, mIL-2/CD25 causes preferential expansion of Tregs. This finding is consistent with past preclinical studies in NOD mice (4,5). Second, mIL-2/CD25 activates Tregs to express markers (e.g., CD103, Klrg1, and ICOS) associated with highly suppressive Tregs. We also found that serum from mIL-2/CD25-treated mice contained elevated IL-10. Previous gene expression profiling experiments of Tregs demonstrated that mIL-2/CD25 increased RNAs for: Gzmb, Fgl2, Il10, and Ctla4, molecules implicated in Treg-suppressive function in vivo; Myb, Txb21, and Prdm1, transcriptional regulators associated with activated Tregs; and Klrg1, Tigit, Icos, and Itgae (CD103), surface markers associated with activated Tregs (30). Third, mIL-2/CD25 supports migration of activated proliferating Tregs into the inflamed pancreas and other tissues. Correspondingly, mIL-2/CD25 upregulated mRNAs for Ccr2, Ccr4, Ccr5, Ccr10, and Cxcr3, which favor trafficking of cells to inflamed tissues, and downregulated Ccr7, which promotes egress of activated lymphocytes from lymph nodes (30). Although mIL-2/CD25 supported migration of Tregs into the pancreas of NOD.SCID mice, the overall representation of T cells was higher in the spleen and MLN. Thus, the high numbers of Tregs in the pancreas of mIL-2/CD25-treated wild-type NOD mice may reflect chemokines released by pancreatic Teff cells that promote Treg trafficking and/or proliferation by resident Tregs to the fusion protein. Fourth, mIL-2/CD25 also inhibits Tfh cell numbers, which was associated with decreased IAA titers. IL-2R signaling opposes development of Tfh and Th17 cells (41). Thus, inhibition of Tfh and potentially Th17 cells likely represents another therapeutic benefit of low-dose mIL-2/CD25 therapy.
The acute and chronic responses of Tregs to mIL-2/CD25 were generally similar. However, Treg expansion in the blood was slightly lower when measured after chronic application of the mIL-2/CD25. This observation is not accounted for by desensitization of proximal IL-2R signaling. Alternatively, this might reflect other downstream aspect of IL-2R signaling or a difference in the numbers of Tregs in circulation upon prolonged application of mIL-2/CD25, perhaps due to migration out of the circulation. With respect to the latter point, the increased proportion and number of Tregs in the pancreas were similar after a single and 10 twice-weekly injections of mIL-2/CD25, consistent with some variability in the circulation of Tregs during chronic administration of the mIL-2/CD25. Thus, the overall accumulation of Tregs in the pancreas after long-term persistent IL-2R signaling may be complex and reflect factors related to proliferation, survival, and tissue retention, the latter of which may be due to activation of CD103, which is associated with tissue-resident memory cells (43,44).
Over a wide range of concentrations, mIL-2/CD25 supported much more extensive expansion of Tregs than Tconv cells. However, at high doses of mIL-2/CD25 (30 µg and 100 µg), CD25+ cells, which may represent Teff cells, were increased in the CD4+Foxp3− T-cell population. The overall relevance of these T cells to disease progression is not fully apparent, and they may not be autoreactive. Nevertheless, the ratio of Tregs/CD4+CD25+ Teff cells was generally maintained over the dose response (1–100 µg) of mIL-2/CD25, with a slightly higher ratio at lower doses of the fusion protein, consistent with Treg selectivity at the lower doses. Even at high doses of mIL-2/CD25, the response to mIL-2/CD25 by Tregs was sufficiently large to keep up with expansion of CD25+ Teff cells. In this regard, we showed that longer-term treatment with mIL-2/CD25 not only prevented T1D in NOD mice at a low dose (4–5 µg), but also at a substantially higher dose (20 µg), at which the potential to activate Tconv cells maybe greater. Protection at the high dose of mIL-2/CD25 was similar to the low dose. Thus, persistent IL-2R signaling over a range of amounts of mIL-2/CD25 leads to a favorable Treg/Teff ratio that controls diabetes. However, Tconv cells are not blind to mIL-2/CD25, as some increase in CD4+CD25+ T cells was noted, particularly at the higher doses and with long-term treatment. Moreover, increased serum levels of CXCL9 (MIG) and CXCL10 (IP10) were found in the serum of NOD mice chronically treated with mIL-2/CD25. CXCL9 and CXCL10 are associated with activated T cells and are chemoattractants that are induced by interferon-γ that would recruit T cells, including Tregs, to inflammatory sites.
Three therapeutic outcomes of low-dose mIL-2/CD25 (4–5 µg) therapy were noted after using a model of late prediabetes that is representative of stage 2 T1D. Some NOD mice were not responsive to mIL-2/CD25 therapy, which more frequently was associated with hyperglycemia at a young age, indicative of more rapid onset of diabetes. This result implies that the therapeutic response to mIL-2/CD25 therapy may depend upon the aggressiveness with which diabetes develops. Nevertheless, the majority of NOD mice responded to the mIL-2/CD25 therapy, in which diabetes in these animals did not progress even upon cessation of therapy and after circulating levels of mIL-2/CD25 waned. However, some of these mice eventually became diabetic while others remained diabetes free long-term. Past work has characterized two categories of diabetes onset in NOD mice (i.e., acute, with a rapid raise in blood glucose levels that is often associated with younger NOD mice, and progressive, with a more gradual increase in blood glucose associated with older NOD mice) (45). Similar to our experience with mIL-2/CD25, NOD mice with progressive disease were more likely to respond to antithymocyte globulin plus granulocyte colony-stimulating factor immunotherapy. What leads to acute diabetes in NOD mice is not well defined, but some studies have linked it to a preponderance of high-affinity autoreactive T cells and the level of cytotoxicity expressed by autoreactive cytotoxic T lymphocytes (46,47).
Our study in NOD mice may provide some key insights for translating low-dose mIL-2/CD25-based therapy to patients with T1D and other autoimmune diseases. Compared with IL-2, mIL-2/CD25 requires less frequent administration to increase Tregs due to its more favorable pharmacokinetics and pharmacodynamics. However, only some NOD mice were fully responsive to this monotherapy, which may be related to the rapidity of disease progression or other factors that might affect responsiveness to a Treg-targeted therapy; by analogy, depending upon disease characteristics or patient-related variability in Treg expansion, mIL-2/CD25 may benefit only a subgroup of patients with T1D. Our results also point to the potential for off-drug efficacy but also for a need for continual administration of mIL-2/CD25 for optimal benefit in the widest number of patients. Indeed, continual administration of low-dose IL-2 is necessary to optimally control unwanted autoaggressive T cells in patients with chronic graft-versus-host disease (48). Some NOD mice remained nondiabetic long term. Thus, in some cases, extended use of mIL-2/CD25 may effectively regulate the immune system to re-establish immune tolerance, which would permit this immunotherapy to be discontinued. A critical issue will be to determine what dictates distinctive responses to mIL-2/CD25 therapy and identify biomarkers that are predictive. Limited data examining normal individuals suggest variability in Treg responsiveness to IL-2 in vitro (3), raising the possibility that distinctive responses to mIL-2/CD25 are complex and not solely a function of disease severity and rapidity.
Article Information
Acknowledgments. The authors thank the following individuals at the University of Miami: Michael Dee for technical assistance; Patricia Guevara, Shannon Saigh, and Jay Enten from the Flow Cytometry Core of the Sylvester Comprehensive Cancer Center (supported by National Institutes of Health grant P30CA240139) and Oliver Umland from the Flow Cytometry Core Center of the Diabetes Research Institute for help with FACS analyses; Kevin Johnson for help with preparation of slides for histology; and Carmen Fotino for help with islet imaging. The authors also thank the following individuals at Bristol-Myers Squibb: James Young and Priyanka Madia for helpful discussions and Nathan Cheadle, Leads Discovery & Optimization, for help with the mouse cytokine data.
Funding. This research was supported by funding to T.R.M. from the National Institute of Diabetes and Digestive and Kidney Diseases (RO1DK093866) and a sponsored research agreement from Bristol-Myers Squibb.
Duality of Interest. The University of Miami, R.H., and T.R.M. have a patent pending (Wo2016022671A1) on mIL-2/CD25 fusion proteins and their use (PCT/US20/13152; R.H., T.R.M.) that has been licensed exclusively to Bristol-Myers Squibb, and this research in part has been supported by a collaboration and sponsored research and licensing agreement with Bristol-Myers Squibb. M.S. and J.X. are employees of Bristol-Myers Squibb. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. N.C.W., J.B.L., M.S., J.X., and T.R.M. were responsible for conception and design of the study. N.C.W., J.B.L., R.H., L.Y., C.J.D., S.H., and A.Y. performed experiments. A.S.S. prepared and validated the fusion protein. N.C.W., J.B.L., R.H., M.S., A.Y., and T.R.M. analyzed and interpreted data. N.C.W. and T.R.M. wrote the manuscript. All authors edited and approved the manuscript. T.R.M. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented at the “Promise of Interleukin-2 Therapy” sponsored by the International Union of Immunological Sciences, Paris, France, 13–16 November 2019.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.12841220.
N.C.W. and J.B.L. contributed equally to this work.
References
- 1.Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity 2010;33:153–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 2009;30:204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu A, Snowhite I, Vendrame F, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes 2015;64:2172–2183 [DOI] [PubMed] [Google Scholar]
- 4.Grinberg-Bleyer Y, Baeyens A, You S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 2010;207:1871–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang Q, Adams JY, Penaranda C, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 2008;28:687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster KE, Walters S, Kohler RE, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med 2009;206:751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 2015;15:283–294 [DOI] [PubMed] [Google Scholar]
- 8.Sharabi A, Tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC. Regulatory T cells in the treatment of disease. Nat Rev Drug Discov 2018;17:823–844 [DOI] [PubMed] [Google Scholar]
- 9.Castela E, Le Duff F, Butori C, et al. Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Dermatol 2014;150:748–751 [DOI] [PubMed] [Google Scholar]
- 10.Koreth J, Matsuoka K, Kim HT, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med 2011;365:2055–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saadoun D, Rosenzwajg M, Joly F, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 2011;365:2067–2077 [DOI] [PubMed] [Google Scholar]
- 12.von Spee-Mayer C, Siegert E, Abdirama D, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis 2016;75:1407–1415 [DOI] [PubMed] [Google Scholar]
- 13.He J, Zhang X, Wei Y, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med 2016;22:991–993 [DOI] [PubMed] [Google Scholar]
- 14.Rosenzwajg M, Lorenzon R, Cacoub P, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis 2019;78:209–217 [DOI] [PubMed] [Google Scholar]
- 15.Hartemann A, Bensimon G, Payan CA, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2013;1:295–305 [DOI] [PubMed] [Google Scholar]
- 16.Rosenzwajg M, Churlaud G, Mallone R, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun 2015;58:48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Todd JA, Evangelou M, Cutler AJ, et al. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PLoS Med 2016;13:e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lotze MT, Frana LW, Sharrow SO, Robb RJ, Rosenberg SA. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J Immunol 1985;134:157–166 [PubMed] [Google Scholar]
- 19.Konrad MW, Hemstreet G, Hersh EM, et al. Pharmacokinetics of recombinant interleukin 2 in humans. Cancer Res 1990;50:2009–2017 [PubMed] [Google Scholar]
- 20.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006;311:1924–1927 [DOI] [PubMed] [Google Scholar]
- 21.Yao Z, Dai W, Perry J, Brechbiel MW, Sung C. Effect of albumin fusion on the biodistribution of interleukin-2. Cancer Immunol Immunother 2004;53:404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell CJ, Sun Y, Nowak UM, et al. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J Autoimmun 2015;56:66–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng XX, Steele AW, Hancock WW, et al. IL-2 receptor-targeted cytolytic IL-2/Fc fusion protein treatment blocks diabetogenic autoimmunity in nonobese diabetic mice. J Immunol 1999;163:4041–4048 [PubMed] [Google Scholar]
- 24.Levin AM, Bates DL, Ring AM, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 2012;484:529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao BM, Driver I, Lauffenburger DA, Wittrup KD. Interleukin 2 (IL-2) variants engineered for increased IL-2 receptor alpha-subunit affinity exhibit increased potency arising from a cell surface ligand reservoir effect. Mol Pharmacol 2004;66:864–869 [DOI] [PubMed] [Google Scholar]
- 26.Shanafelt AB, Lin Y, Shanafelt MC, et al. A T-cell-selective interleukin 2 mutein exhibits potent antitumor activity and is well tolerated in vivo. Nat Biotechnol 2000;18:1197–1202 [DOI] [PubMed] [Google Scholar]
- 27.Peterson LB, Bell CJM, Howlett SK, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun 2018;95:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spangler JB, Trotta E, Tomala J, et al. Engineering a single-agent cytokine/antibody fusion that selectively expands regulatory T cells for autoimmune disease therapy. J Immunol 2018;201:2094–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sockolosky JT, Trotta E, Parisi G, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018;359:1037–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ward NC, Yu A, Moro A, et al. IL-2/CD25: a long-acting fusion protein that promotes immune tolerance by selectively targeting the IL-2 receptor on regulatory T cells. J Immunol 2018;201:2579–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faleo G, Fotino C, Bocca N, et al. Prevention of autoimmune diabetes and induction of β-cell proliferation in NOD mice by hyperbaric oxygen therapy. Diabetes 2012;61:1769–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu L, Robles DT, Abiru N, et al. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A 2000;97:1701–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang H, Panemangalore R, Yarde M, Zhang L, Cvijic ME. 384-Well multiplexed luminex cytokine assays for lead optimization. J Biomol Screen 2016;21:548–555 [DOI] [PubMed] [Google Scholar]
- 34.Choi YS, Kageyama R, Eto D, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011;34:932–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malek TR, Ashwell JD. Interleukin 2 upregulates expression of its receptor on a T cell clone. J Exp Med 1985;161:1575–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malek TR The biology of interleukin-2. Annu Rev Immunol 2008;26:453–479 [DOI] [PubMed] [Google Scholar]
- 37.Smigiel KS, Richards E, Srivastava S, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med 2014;211:121–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol 2018;19:291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol 2013;34:74–80 [DOI] [PubMed] [Google Scholar]
- 40.Toomer KH, Yuan X, Yang J, Dee MJ, Yu A, Malek TR. Developmental progression and interrelationship of central and effector regulatory T cell subsets. J Immunol 2016;196:3665–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013;38:13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding Y, Yu A, Tsokos GC, Malek TR. CD25 and protein phosphatase 2A cooperate to enhance IL-2R signaling in human regulatory T cells. J Immunol 2019;203:93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szabo PA, Miron M, Farber DL. Location, location, location: tissue resident memory T cells in mice and humans. Sci Immunol 2019;4:eaas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol 2005;174:5444–5455 [DOI] [PubMed] [Google Scholar]
- 45.Mathews CE, Xue S, Posgai A, et al. Acute versus progressive onset of diabetes in NOD mice: potential implications for therapeutic interventions in type 1 diabetes. Diabetes 2015;64:3885–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han B, Serra P, Yamanouchi J, et al. Developmental control of CD8 T cell-avidity maturation in autoimmune diabetes. J Clin Invest 2005;115:1879–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin H, Trudeau JD, Reid GS, et al. Progression of spontaneous autoimmune diabetes is associated with a switch in the killing mechanism used by autoreactive CTL. Int Immunol 2004;16:1657–1662 [DOI] [PubMed] [Google Scholar]
- 48.Koreth J, Kim HT, Jones KT, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood 2016;128:130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]