

Abstract
HLA-DQA1 and -DQB1 genes have significant and potentially causal associations with autoimmune type 1 diabetes (T1D). To follow up on the earlier analysis on high-risk HLA-DQ2.5 and DQ8.1, the current analysis uncovers seven residues (αa1, α157, α196, β9, β30, β57, and β70) that are resistant to T1D among subjects with DQ4-, 5-, 6-, and 7-resistant DQ haplotypes. These 7 residues form 13 common motifs: 6 motifs are significantly resistant, 6 motifs have modest or no associations (P values >0.05), and 1 motif has 7 copies observed among control subjects only. The motifs “DAAFYDG,” “DAAYHDG,” and “DAAYYDR” have significant resistance to T1D (odds ratios [ORs] 0.03, 0.25, and 0.18; P = 6.11 × 10−24, 3.54 × 10−15, and 1.03 × 10−21, respectively). Remarkably, a change of a single residue from the motif “DAAYHDG” to “DAAYHSG” (D to S at β57) alters the resistance potential, from resistant motif (OR 0.15; P = 3.54 × 10−15) to a neutral motif (P = 0.183), the change of which was significant (Fisher P value = 0.0065). The extended set of linked residues associated with T1D resistance and unique to each cluster of HLA-DQ haplotypes represents facets of all known features and functions of these molecules: antigenic peptide binding, peptide–MHC class II complex stability, β167-169 RGD loop, T-cell receptor binding, formation of homodimer of α-β heterodimers, and cholesterol binding in the cell membrane rafts. Identification of these residues is a novel understanding of resistant DQ associations with T1D. Our analyses endow potential molecular approaches to identify immunological mechanisms that control disease susceptibility or resistance to provide novel targets for immunotherapeutic strategies.
Introduction
Autoimmune type 1 diabetes (T1D) affects children and young adults worldwide (1,2) and has been shown to involve the host immune system in attacking the pancreatic islet β-cells (3). During the initial attack, the host immune system produces biomarker autoantibodies against specific β-cell autoantigens, leading to seroconversion and a pathogenesis that eventually results in the clinical onset of diabetes (4). Despite decades of research on T1D, the etiology is largely unknown (5), although viruses are suspected (6,7). The genetic etiology is strongly associated with class II HLA genes emanating from classical HLA studies to recent genome-wide association studies (8–18). Like most genetic association studies, efforts are made to identify “causal variants” that increase risk of first-appearing islet autoantibody (19,20), clinical onset, or both. Indeed, we recently identified three residues in HLA-DQ heterodimers, α44, β57, and β135, together with those residues in linkage disequilibrium (LD) with α44 and β135, that appear to be responsible for T1D risks among subjects with DQ2 and DQ8, including the two well-known risk haplotypes HLA-DQ2.5 and DQ8.1 (21).
HLA-DQ linkage to T1D has a dual association, being risk or resistant, in the sense that some HLA-DQ haplotypes are known to be negatively associated with T1D, also when coexisting with high-risk HLA-DQ haplotypes (16,22,23). In our previous study, risk residues were identified (21); consequently, our primary interest in this study was to discover HLA-DQ residues that are negatively associated with T1D risks. A unique feature in HLA-DQ associations with T1D is that multiple DQ haplotypes are resistant to T1D, most prominently DQA1*01:02-DQB1*06:02 (DQ6.2) subjects who are at much lower risk for T1D than noncarriers (22), although the protection is attenuated with age at onset (16). Presence of such multiple resistant HLA-DQ haplotypes begs the question of which unique genetic characteristics are associated with the resistance.
In this follow-up work, our goal was to identify polymorphic residues that reduce the risk or protect subjects with HLA-DQ4, 5, 6, and 7 against T1D while addressing two analytic challenges (i.e., high LD and high-order interactions among residues). To meet these challenges, we pursue our earlier work using the hierarchically organized haplotype (HOH) approach (21). First, clusters of residue sequences are identified based on their sequence similarities and hence similarities of protein structures. Second, individuals with neutral or resistant DQ haplotypes are next searched for specific T1D residues. Among 962 patients and 636 control subjects, high-resolution sequencing-based HLA-DQA1 and -DQB1 genotypes were obtained along with their haplotypes as the basic analytic units (24,25). After filtering out monomorphic residues, all observed 45 unique HLA-DQ haplotypes were hierarchically organized to reveal the five clusters that include either neutral or resistant HLA-DQ haplotypes. Variable T1D associations afford us the opportunity to uncover residues that contribute to observed resistances. Further, among all patients with T1D, we finally analyzed the association between resistant residues as well as their motifs and autoantibodies against GAD65 (GAD antibody [GADA]), insulin (insulin autoantibody [IAA]), IA-2 (IA-2 antigen [IA-2A]), and the three variants (residue R, W, or Q on position 325) of ZnT8 (26–28) to evaluate immunological pathways for those patients with T1D with relatively lower genetic risks.
Research Design and Methods
The study population, HLA genotypes, autoantibodies, and statistical methods are described in detail in recent publications (21,24,25). Briefly, this is a case-control study with 636 population-based control subjects and 962 patients with newly diagnosed T1D 1–18 years of age from Sweden. Using next-generation targeted sequencing technology, we genotyped HLA class II genes and allelic sequences (24). Then we used various statistical methods to perform allelic and haplotypic associations with phylogenic analysis. The two high-risk clusters (DQ2 and DQ8/9) were published (21). For convenience, we also included details in the Supplementary Materials.
Data and Resource Availability
Genotype data with case/control status are available from the corresponding author (L.P.Z.), after submitting a brief application to the senior author (A.L.) and obtaining an approval from the Steering Committee of the Better Diabetes Diagnosis (BDD) study.
Results
Identifying Associated Residues Among Individuals With T1D-Resistant DQ Molecules
This study was focused on residues that reduced T1D risk among individuals with T1D-resistant HLA-DQ molecules, based on the clustering results by applying HOH. There were five T1D-resistant and neutral HLA-DQ haplotype clusters (Fig. 1). Among all resistant DQ clusters, DQ6 was of interest, because it included DQ*01:02-*06:02 (DQ6.2), the most common and most resistant DQ haplotype to T1D. The cluster had four resistant haplotypes (odds ratios [ORs] 0.03, 0.13, 0.28, and 0.07; P = 2.25 × 10−60, 0.0291, 3.37 × 10−13, and 1.14 × 10−3, respectively), two neutral haplotypes (P = 0.0391 and 0.922), and two rare haplotypes. Excluding the “nearly monomorphic residue,” α81, left a total of 28 residues, including 3 complete LD blocks. Specifically, the first LD block included five residues (α22F/Y, α38K/R, α126H/Q, α127A/S, and α204V/M), in which the residue α22 took one of two amino acids (F and Y) and represented two possible polymorphic sequences (FKHAV and YRQSM) for these residues in complete LD block. Similarly, residue β3, with two amino acids (P and S) captured polymorphic sequences (β3, β[-21], β[-18], β[-17], β[-6], β[-5], β[-4], β13, β26, β37, β38, β66, β67, β167, β197, and β203). Lastly, the polymorphic amino acids (V and D) for the residue β57 captured the polymorphic sequence of four residues (β57, β86, β87, and β130) (Table 1). Together with those singleton residues, six residues (α22, β3, β9, β30, β57, and β70) and their motifs captured all haplotypic polymorphisms in the DQ6 cluster D.
Figure 1.

Hierarchically organized DQA1-B1 haplotypes by similarities of their residues observed in the BDD case-control study of T1D. Haplotypes were colored red, black, gray, and green, corresponding to protective, neutral, rare, and risk haplotypes, respectively.
Table 1.
Direct comparisons of allelic sequences within DQ6 cluster of haplotypes that differentiate between protective and neutral alleles
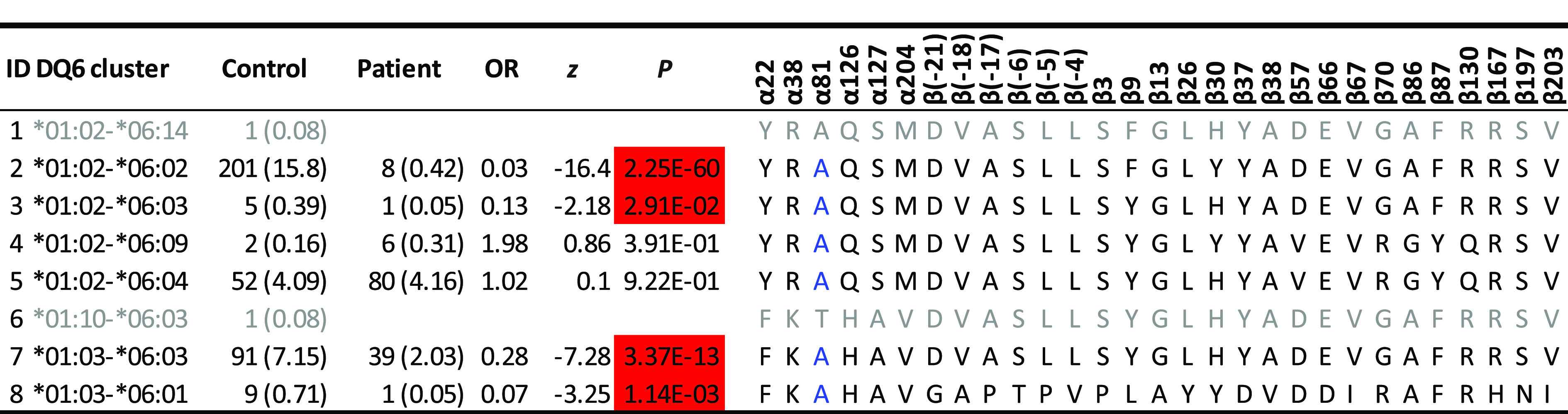 |
Estimated haplotype frequencies (percentages) among control subjects, those among patients, estimated ORs, z scores, and P values are listed. Results on allelic sequences on all resistant clusters are listed in Supplementary Table 1. Protective haplotypes (highlighted in red) have ORs <1 and P values <0.05, and neutral haplotypes have P values >0.05. Uncommon haplotypes with fewer than three copies are shown in gray font. Residue α81, in blue font, is monomorphic among those relatively common DQ haplotypes.
Repeating the same process of organizing residues, we could identify three residues (α135, β9, and β13) in the DQ4 cluster, five residues (αa1, α31, α196, β57, and β126) in DQ5, three residues (α[-6], α157, and β57) in DQ7a, and two residues (α[-13] and α157) in DQ7b. For details, please consult Supplementary Table 1.
Four Residues Account for the T1D Resistance in Individuals Within the DQ6 Cluster
Four protective haplotypes, two neutral and two rare ones in the DQ6 cluster (Table 1), were uniquely characterized by 5 motifs of 30 residues represented by 6 residues (α22, β3, β9, β30, β57, and β70). Specifically, residue α22 was either F (for phenylalanine) or Y (tyrosine) (Table 2). Both had significant resistance to T1D (ORs 0.26 and 0.24; P = 1.02 × 10−15 and 4.83 × 10−40, respectively). However, these two estimated ORs were not significantly different from each other (Fisher P = 0.738), implying that α22 was not responsible for the differential T1D associations among DQ6 carriers. Similar association patterns were observed for the residue β3. In contrast, residue β9 could be F, L (leucine), or V (valine). While all had significant resistance (ORs 0.03, 0.07, and 0.55; P = 8.98 × 10−61, 1.14 × 10−3, and 8.16 × 10−8, respectively), the magnitudes of the resistance were significantly different among them (Fisher P = 4.15 × 10−28). Similarly, residues β30, β57, and β70 had variable T1D associations that were responsible for variability observed across haplotypes within this DQ cluster (Fisher P = 4.88 × 10−23, 2.38 × 10−25, and 5.36 × 10−23). Hence, these four residues (β9, β30, β57, and β70) and their motifs were responsible for variable T1D associations across all DQ haplotypes among DQ6 carriers.
Table 2.
T1D associations with individual residues within DQ6 haplotype cluster
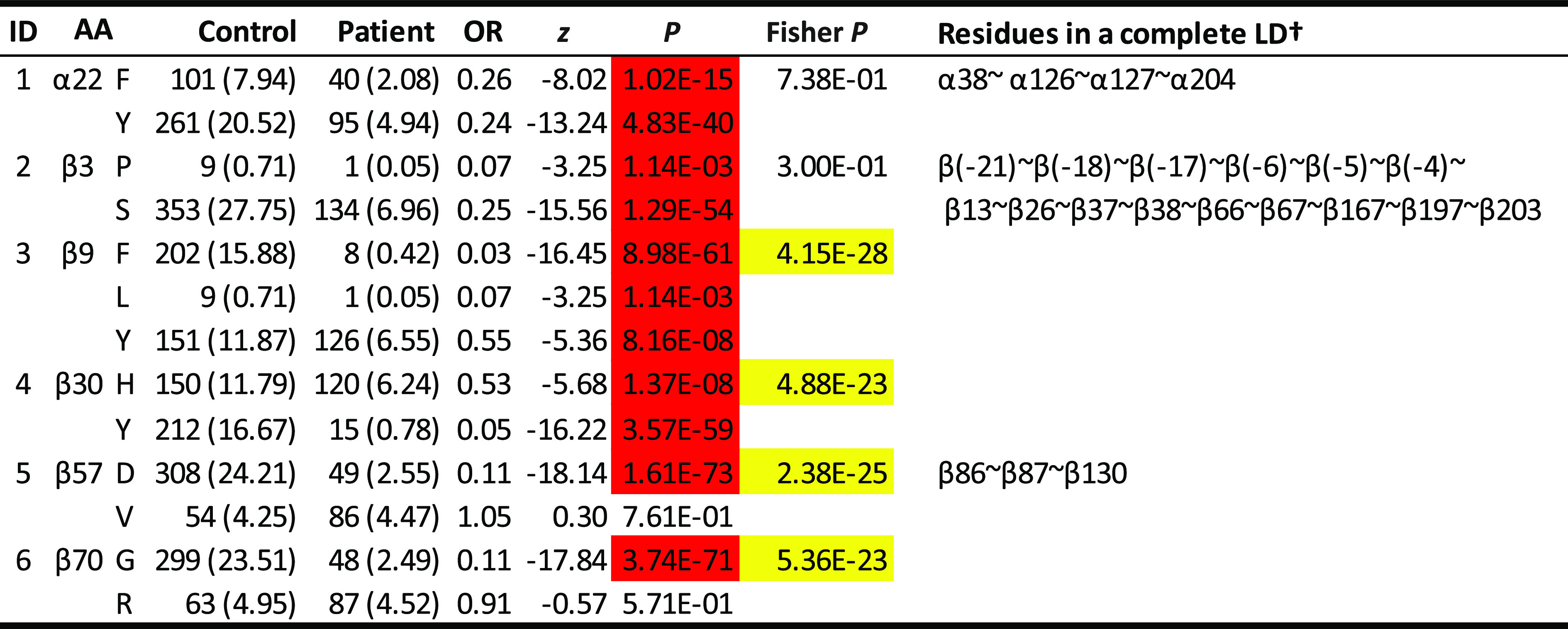 |
Thirty polymorphic residues are represented by six residues (α22, β3, β9, β30, β57, and β70), some of which are in complete LD with one or more residues (listed in the far right column). Residue associations in other DQ clusters are shown in Supplementary Table 2. P values are highlighted in red if they are <0.05 with ORs <1. P values highlighted in yellow have a significance level of 5% and indicate that associations of residues differ significantly. †All residues that are in complete LD with the residue in discussion.
Repeating the same analytic process for all selected residues within respective DQ clusters, we identified a single residue, β9, in the DQ4 cluster, three residues (αa1, α196, and β57) in the DQ5 cluster, and a single residue, α157, in the DQ7a and DQ7b clusters (Supplementary Materials and Supplementary Table 2).
Seven Residues (αa1, α157, α196, β9, β30, β57, and β70) Capture T1D Resistance in Subjects With Resistant DQ Haplotypes
Integrating residues identified in DQ6 with those in DQ4, 5, and 7 clusters resulted in a set of 7 residues that, together with those residues in LD blocks, formed 13 motifs, m1 to m13 (Table 3). To assess the motif associations with T1D among carriers of resistant DQ haplotypes, we computed their association statistics, excluding three rare motifs. Collectively, six motifs (m1–m4, m6, and m13) were T1D resistant, and the relatively uncommon motif m12 was potentially resistant, while six motifs (m1, m5, m7, and m9–m11) had neutral associations with T1D (Table 3). These motifs were highly correlated to those in individual DQ clusters.
Table 3.
Motif association results with selected residues from both α-chain and β-chain (αa1,α157,α196, β9, β30, β57, and β70) with at least five copies of haplotypes
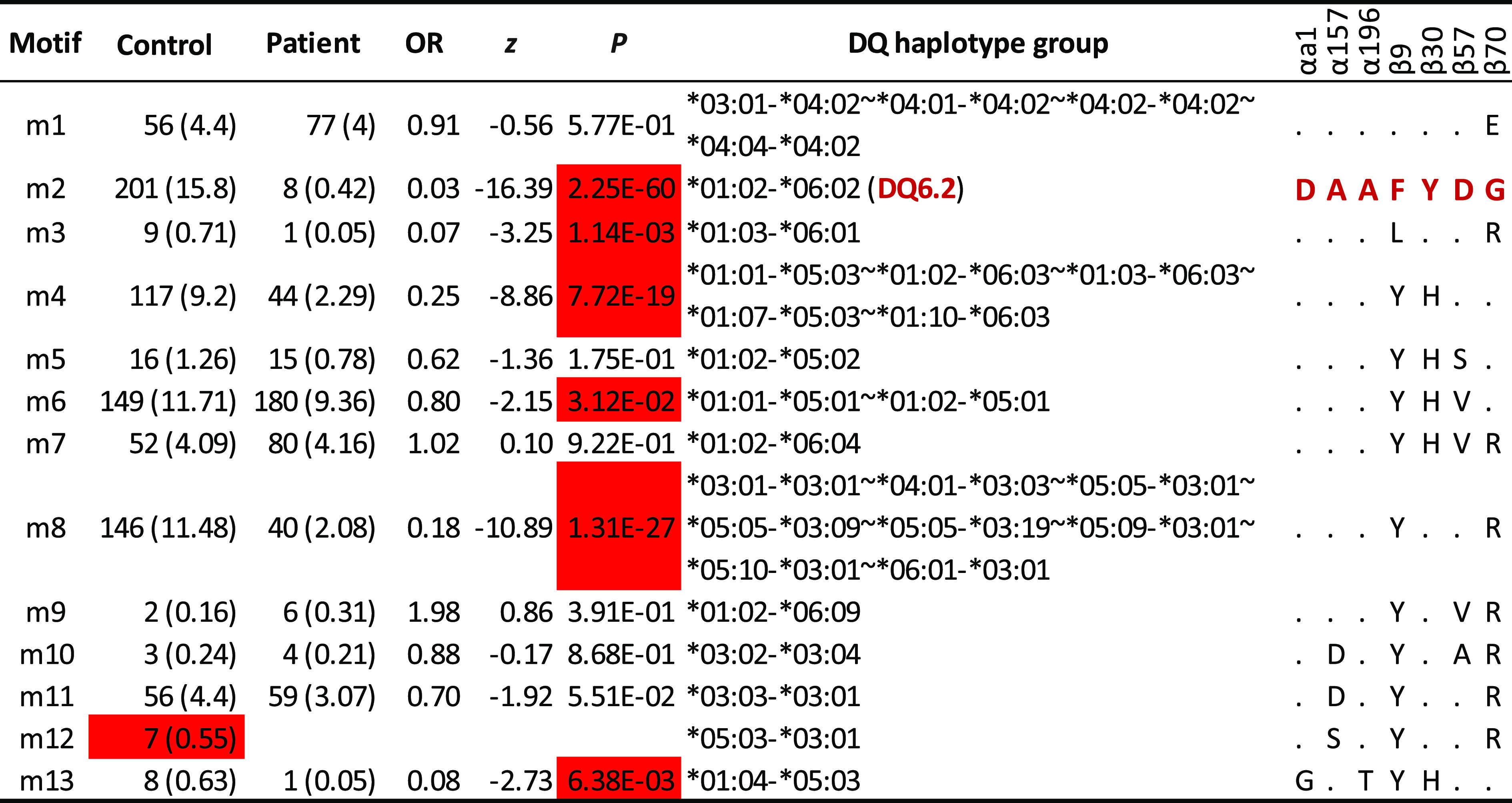 |
Estimated haplotype frequencies (percentages) (excluding those motifs with fewer than 3 copies) among control subjects, those among patients, estimated ORs, z scores, and P values (from association analysis with T1D in case-control study) are listed. For given haplotypes, one or more DQA1-DQB1 haplotypes may be merged into haplotype groups. Motif association results for individual DQ clusters are listed in Supplementary Table 3. The haplotype (DQ6.2) is highlighted in red, to indicate that its motif (also highlighted in red, “DAAFYDG”) is treated as the reference. For other motifs, the same amino acids to those in the reference motif are denoted by “.”
Across these 13 motifs, it was informative to compare motifs and to correlate them with subtle variations in their resistant associations. For example, motif m1 differed from m2 by a single residue, β70, E to G. However, their associations (OR 0.91 compared with 0.03; P = 0.577 vs. 2.25 × 10−60, respectively) had been substantially altered (Fisher P value <10−5). Similarly, motifs m4 and m5 differed by a single residue, β57D, compared with β57S, respectively, but their associations (OR 0.25 vs. 0.62; P = 7.72 × 10−19 vs. 0.175, respectively) were different (Fisher exact test; P = 0.032). Similar comparisons could be made for motif m5 and m6, m8 and m9, and m11 and m12.
Identified T1D-Resistant Haplotypes Had Limited Associations With Six Different Islet Autoantibodies in Subjects With Resistant DQ Haplotypes
The islet autoantibodies were measured only in the patients, and we therefore evaluated their associations with DQ motifs, if they had at least eight observations, netting eight DQ motifs (Table 4). Noticeably, the motif m2 (“DAAFYDG”), corresponding to DQ6.2, had eight copies. Due to the limited small sample size, association statistics were not meaningful, but it was of interest to note that GADA and IA-2A tended to be elevated among 6 and 7, respectively, out of the eight patients.
Table 4.
Associations of seven selected residues (α1b, α157, α196, β9, β30, β57, and β70) among carriers of DQ4, 5, 6, 7a, and 7b, with six autoantibody levels
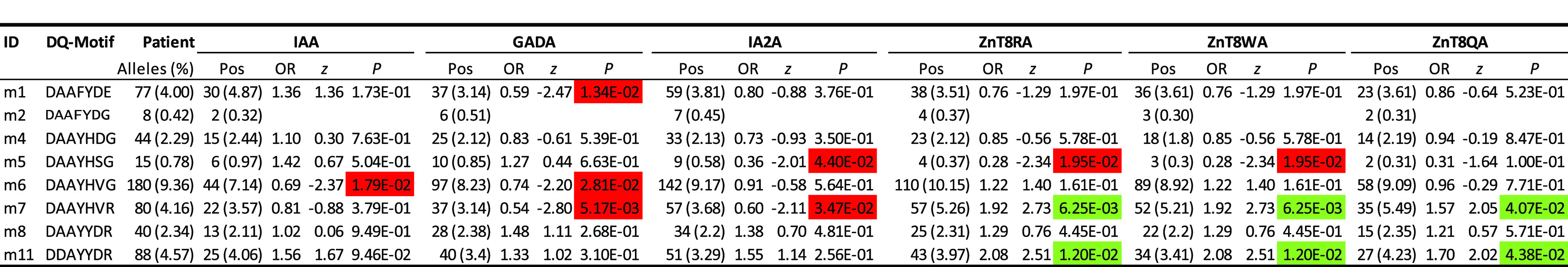 |
With respect to each autoantibody, presented results include estimated frequencies (percentages) among all patients with positive (Pos) autoantibody levels, estimated ORs, z scores, and P values. P values are highlighted green or red if they are significantly positive or negative, respectively, at the significance level of 5%.
Among other motifs with relatively greater frequencies, motif 6 was negatively associated with IAA (OR 0.69; P = 0.0179). Associations with GADA tended to be negative (ORs 0.59, 0.74, and 0.54 and P = 0.0134, 0.0281, and 0.0052, for motifs m1, m6, and m7, respectively). A negative association with IA-2A was observed for motifs m5 and m7 (ORs 0.36 and 0.60; P = 0.044 and 0.0347, respectively). In contrast, autoantibodies to ZnT8-arginine (ZnT8RA), ZnT8-tryptophan (ZnT8WA), and ZnT8-glutamine (ZnT8QA) were associated with both motif m7 (ORs 1.92, 1.92, and 1.57; P = 0.00625, 0.00625, and 0.041, respectively) and m11 (ORs 2.08, 2.08, and 1.70; P = 0.0120, 0.0120, and 0.0439, respectively).
Structural Analysis
In order to gain a better understanding of the three-dimensional structure of the HLA-DQ molecules involved in T1D resistance, the structural correlates of the resistant residues were examined in available crystal structures and models thereof. As there is no crystal structure of any HLA-DQ resistance haplotype bound to a T1D-related epitope, available models were used in cases in which such epitopes had been identified by virtue of binding to respective HLA-DQ molecules. The complex of HLA-DQ6.2–InsB5-15 has been modeled based on the crystal structure of DQ6.2–hypocretin 4-13 peptide (29). The former complex had been identified as a strongly binding epitope, both to DQ6.2 as well as to DQ6.4 (30,31). Only residues in the identified crystal structure can be shown (Fig. 2), so residues from the signal sequences, intramembranous, and cytoplasmic sequences are excluded. Of the depicted residues, we noted those lining the anchoring pockets in the antigen-binding groove (β9, β13, β26, β30, β37, β38, β57, β67, β70, and β86) and immediately neighboring residues that may affect binding (α22 and β87), potential T-cell receptor (TCR) contact residues (α38, α81, β66, and β70), potential CD4 contact residues (α127) or their neighbors (α126 and β130), and one member of the β167-169RGD loop (β167) (32,33) (Fig. 2A). The pockets of DQ6.2 are eminently suited for binding peptides bearing anchor residues fulfilling the derived motif, and the particular insulin β-chain peptide is proof of this motif (31). As the TCR orientation in T cells linked to susceptibility to autoimmune disease seems different from the orientation adopted when recognizing microbial peptide–MHC class II (pMHCII) complexes, no precise statements can be made regarding the role of the putative TCR contact residues (34–37).
Figure 2.

A: Depiction of the modeled structure of the HLA-DQ6.2–insulin B5-15 complex, based on the structure of the HLA-DQ6.2–hypocretin 5-17 peptide (from 1uvq.pdb) highlighting the amino acid (AA) residues shown by HOH analysis to be involved in T1D protection via this cluster of MHCII alleles (DQ6 cluster). This modeled structure was generated and reported in Ettinger et al. (30). The orientation of the molecule is with its long axis nearly perpendicular to the plane of the paper/screen. The top-right quadrant contains the same figure with its long axis parallel to the plane of the paper/screen in order to better reveal the position of certain depicted residues. The cognate TCR would bind on top of the main depiction of the molecule, albeit in an unknown orientation, recognizing simultaneously peptide residues as well as HLA-DQ6 residues from the α1β1 antigen-binding domain. The HLA-DQ6–insulin peptide complex is depicted in its secondary structure formation (α-helix in red, β-sheet in turquoise, and β-turn, random coil, or any other form, such as polyproline II helix of the antigenic peptide backbone, in gray). The AA residues in question are depicted in stick form (atom color convention: carbon, orange; oxygen, red; nitrogen, blue; sulfur, yellow; and hydrogen, white). Four anchor residues of the antigenic peptide (p1Leu [p1L], p4Ala [p4A], p6Leu [p6L], and p9Ala [p9A]) are shown in space-filling form and the same atomic color conventions (except for carbon that is in green), only for orientation purposes in order to appreciate the position and role of the identified residues from the HLA-DQ6.2–insulin peptide complex. Residues β13Gly and β86Ala are, respectively, below p4Ala and the overlying β1-helix, thus barely seen. B: Depiction of the crystal structure of the HLA-DQ5 (A1*01:02-B1*05:01)–myelin basic protein 85-99 complex (from 3pl6.pdb) highlighting the residues shown by HOH analysis to be involved in T1D resistance via the HLA-DQ5 cluster (the cognate TCR is not shown at all). A similar orientation and the same color and depiction conventions for secondary structures and individual AA renderings hold as in A (except that β-turns are in green and carbon atoms are always shown in gray). Four anchor residues of the antigenic peptide (p1Val [p1V], p4Phe [p4F], p6Asn [p6N], and p9Thr [p9T]) are again shown, only for orientation purposes. The sequence β105-β113 did not diffract satisfactorily and was not depicted in the originally reported structure. We label in this figure residues β104Ser (β104S) and β114Leu (β114Q) in order to identify the break in the β-chain. We also depict the N- and COOH-terminal residues of the two DQ chains: αa1Asp (αa1D), β3Ser (β3S), α180Pro (α180P), and β191Gln (β191Q). C: Depiction of the crystal structure of the HLA-DQ8trans (A1*05:01-B1*03:02)–γ1-gliadin complex (from 5ksa.pdb) (39) without the cognate TCR, highlighting the residues shown by HOH analysis to be involved in T1D resistance via the HLA-DQ7a/7b cluster and the DQ4 cluster (the DQ4 cluster α alleles all have the α52 deletion, as DQA1*05:01). The long axis of the molecule is almost parallel to the plane of the paper/screen in order to render visible as many as possible of the implicated residues. The same color and depiction conventions for secondary structures and individual AA renderings hold as in B. The five anchor residues of the antigenic peptide (p1Pro [p1P], p4Ser [p4S], p6Pro [p6P], p7Glu [p7E], and p9Glu [p9E]) are again shown, only for orientation purposes. As noted in our previous publication, celiac disease epitopes are rich in proline residues (21). We are not aware of any T1D epitope, associated either with a susceptibility or a resistance allele, plentiful in prolines. We depict the N- and COOH-terminal residues of the two crystallographically determined DQ chains in blue text: αa1Asp (αa1D), β2Asp (β2D), α181Glu (α181E), and β191Gln (β191Q). Residues involved only in the DQ7a/b clusters are in italics, residues involved in both the DQ7a/b and DQ4 clusters are shown in roman text, and residues involved only in the DQ4 cluster are underlined.
Examination of the structure of HLA-DQ5 complexes is based on the crystal structure of HLA-DQA1*01:02-B1*05:02–myelin basic protein 85-99 and its complex with the Hy.1B11 TCR isolated from CD4+ T cells of a patient with multiple sclerosis (34). The TCR contact in this complex is akin to other such complexes from persons suffering from autoimmune disease (36,37). Again, the set of linked residues includes residues lining the anchoring pockets (α31, β30, β57, β67, β70, and β71), one potential TCR contact residue (β66), one residue in the α2β2 domain (β126), and two intramembranous residues (α196 and α204) (Fig. 2B).
There is no available crystal structure of any DQ4 molecule and no identification of any T1D-related epitope. We merely note that the set of residues contains again those aligning anchoring pockets (β9, β13, β26, β67, β70, β71, and β74) or close to them (α22 and β75), potential TCR contact residues (β66, β70, β71, and β74), members of the β167-169RGD loop (β167), residues in the α2β2 domain of unknown function (α135, α150, and β185), and residue β45 that is a crucial determinant of the electrostatic surface potential of the α1β1-peptide domain and thus a key component of cognate TCR docking (38).
The crystal structures of two molecules very closely related to the clusters of DQ7a and DQ7b alleles are known, and they concern HLA-DQ8cis (A1*03:01-B1*03:02) and DQ8trans (A1*05:01-B1*03:02), which are both susceptible to T1D (12,14,15,17,18). As the residues implicated in DQ7a associations with T1D resistance are also included in the respective DQ7b list, these residues depicted the structure of DQ8trans in complex with a gluten epitope and bound to a cognate TCR (39) (Fig. 2C). Again, the residues implicated were found either in anchoring pockets (β13, β26, and β57), or proximal to them (α74), participating in the β167-169RGD loop, and right next to it (β170), and participating in the formation of the putative homodimer of heterodimers (α157) and in the membrane-proximal extracellular region (β185).
Seven HLA-DQ Residues Account for Resistance to T1D in the General Population
As noted earlier, residues coded for by HLA-DQ genes were in high LD with each other, and their penetrance to T1D was also subject to high-order interactions. Hence, it was of interest to assess how these seven resistance-linked residues associate with T1D risks in the entire case-control population (Supplementary Table 4). The association analysis of these seven residues in the case-control study resulted in 20 unique DQ motifs. Adjusting for motifs of these 7 residues, T1D associations were assessed with each of the 104 individual residues in HLA-DQA1 and B1 (Fig. 3A). Logarithmic P values (i.e., 0, 1, 2, and 10), corresponding to P values of 1.00, 0.10, 0.01, and 10−10, respectively (y-axis), are shown over individual residues (x-axis). By the threshold of 0.05, seven residues explained most of the T1D associations with all residues on the β-chain except β135 (P = 2.27 × 10−7) (Fig. 3A).
Figure 3.

Results from adjusted and unadjusted association analyses with the base 10 logarithmically transformed P values: transformed P values from the conditional analysis of individual residues within HLA-DQA1 and -DQB1, after adjusting for seven residues (α1b, α157, α196, β9, β30, β57, and β70) (A), comparison of transformed P values from the conditional analysis with those from the unconditional analysis of the HLA-DQA1 and -DQB1 residues (B), and comparison of transformed P values from the conditional analysis on α44, β57, and β135, selected residues from high-risk DQ haplotypes, with those from the conditional analysis on α1b, α157, α196, β9, β30, β57, and β70 (C).
Because of adjusting for the seven DQB1 residues, they were referred to as conditional P values and were plotted on the y-axis against the marginal P values (x-axis) obtained without any adjustment (Fig. 3B). Note that the x-axis scale was from 0 to 120, because some of the marginal P values were on the order of 10−100. By the threshold of 0.05, there were four quadrants. In the bottom left quadrant, 12 residues were deemed as insignificant by both marginal and conditional analyses. There were 67 residues in the bottom right quadrant, in which 7 residues effectively adjusted out those marginal associations. In the top right corner, 24 residues remained significant even after the adjustment. Finally, it was interesting to note a single residue that was insignificant in the marginal association analysis and became highly significant in the conditional analysis, indicating that the conditional analysis reduced confounding effects of other residues and improved the power of identifying such a residue that was hidden in the marginal association analysis.
To assess if these seven residues were complementary to three risk residues identified from subjects with either DQ2 or DQ8 (21), we compared two conditional P values from adjusting for the three risk residues (conditional P values for three residues on the y-axis) against the conditional P values from adjusting for the seven risk residues on the x-axis (Fig. 3C). In the first quadrant in the bottom left corner, 63 residues had insignificant P values from both conditional analyses, indicating that both sets of identified residues explained their marginal associations. Similarly, there were 14 residues in the top right corner that remain significant in both conditional analyses, indicating that other factors may still play a role in T1D associations with HLA-DQ. Lastly, 11 and 16 residues in off-diagonal quadrants represented those that were adjusted by one conditional analysis but not by the other, hence suggesting that these sets of risk and resistant residues were complementary to each other.
Discussion
In this manuscript, the aim was to identify T1D-resistant residues among individuals with HLA-DQ4–7 haplotypes. Our discovery analysis resulted in three residues on the α-chain (αa1, α157, and α196) and four β-chain residues (β9, β30, β57, and β70), demonstrating that these residues and motifs explained T1D resistance among individuals with HLA-DQ4, -DQ5, -DQ6, -DQ7a, and -DQ7b (Fig. 1). Collectively, these 7 residues formed 13 motifs (m1, m2, ..., and m13) among individuals with 36 different HLA-DQ4–7 haplotypes, in addition to 3 rare motifs, and captured 6 resistant motifs (m2, m3, m4, m6, m8, and m13) and 1 potentially resistant motif (m12) (Table 3). The remaining six motifs (m1, m5, m7, m9, m10, and m11) were associated with variable ORs, but none of them were significantly different from 1.00. In all, these identified motifs captured all known resistant associations between HLA-DQ haplotypes and T1D. With respect to motif association with autoantibody elevations, resistant motif m6 had modest reduction in levels of autoantibodies (IAA and GADA) and m2, m4, and m8 had no obvious associations with any autoantibodies (Table 4), unlike those risk motifs found earlier that were significantly associated with GADA and IA-2A (21). It was also interesting to note that the change at a single residue could alter the motif from resistance to neutral association. For example, the change of β57S→D between motif m5 and m4 changed a neutral to a resistant motif. The change in OR was statistically significant (Table 3). These data would seem to explain the well-publicized observation that position 57 of the HLA-DQ β-chain protects against T1D (40), as the negative association of this single residue is secondary to the primary negative association of the motif to which the β57 belongs. It was also of interest to note the existence of LD blocks within respective HLA-DQ clusters. Empirically, one or more residues within the LD block could be responsible for the observed associations, and differentiating their functional roles would have to depend on functional data.
The LD blocks are important in that they recapture all putative residues for resistance to T1D. The possible roles that many of these residues may play in protection may include accessory functions of the MHCII molecules that have yet to be investigated. Previous genetic analyses focused both on the HLA-DQA1*01:02-B1*06:02 haplotype (16,22) and simplified to the β57 single residue position (40). The possible association with other negatively associated DQ haplotypes has been largely ignored. As there exist HLA-DQβ57Asp− molecules not as resistant to T1D as in DQB1*06:02 (e.g., m1 and m11), this residue is of paramount importance, yet it alone and with residues in LD does not protect from T1D. In fact, β57Asp is crucial in many respects: 1) It forms a salt bridge with invariant α76-Arginine, leading to several hydrogen bonds with amide and carbonyl groups of the bound antigenic peptide (30,31). Such an arrangement renders pMHCII complexes stable (41,42). 2) β57Asp+ generally excludes acidic residues from pocket 9, favoring instead aliphatic (size dependent on occupancy at β37/β38) and small polar residue (29–31,43), unless there is an excellent fit in the other four pockets (44,45). By contrast, in β57Asp− MHCII molecules, acidic p9 anchors are favored (46). In the case of HLA-DQ2, conferring susceptibility to T1D and celiac disease, aromatic residues are also accommodated due to a spacious pocket (47–49). 3) It influences greatly the cognate TCR selection in the thymus and corresponding responsiveness in the periphery (50).
In DQ6.2, the possible contribution of a single residue in an epitope may be identified as epitope stealing of the InsB6-23 peptide by DQ6.2 over DQ8.1, as has been demonstrated (51,52). DQ6.2 binds this peptide in the InsB6-14 nonamer register (31), while DQ8.1 binds in the InsB13-21 register with a lower affinity (46,51). The T1D neutral-risk DQ6.4 binds the InsB6-14 core in the same register as DQ6.2 but with lower affinity (30). The modeled structures of DQ6.2-InsB6-14 and DQ6.4-InsB6-14 show the largest variability around pockets 1, 4, 6, and 9, justifying the divergence in published peptide-binding motifs (29,30,53). All T1D autoantigenic epitopes that bind well to DQ6.4 bind even better to DQ6.2, except GAD65 339-349 (30). Thus, residue replacements from DQ6.4 to DQ6.2 yield stronger binding by the latter molecule. Likewise, in three comparisons of the binding of human preproinsulin, proinsulin, and insulin peptides to HLA-DR-DQ molecules susceptible and resistant to T1D, peptides containing the InsB6-14 core nonamer have the highest affinity for DQ6.2 (30,31,54) (Supplementary Fig. 1). The remarkable ability of DQ6.2 to retain bound peptides plus the high-affinity binding of T1D epitopes lead to epitope stealing and probably thymic selection of regulatory T cells (Tregs) when such complexes are presented (51,52). This latter hypothesis was verified with T1D-resistant HLA-DRB1*15:01 (55). In contrast, a DQ6.4–InsB6-14 complex would have different potential TCR contact residues (e.g., β70Arginine instead of Glycine), as well as lesser overall stability due to β57Val (30,41,56). Such analyses cannot be extended to any alleles of the DQ4, DQ5, DQ7a, and DQ7b clusters because of lack of precise data regarding respective T1D autoantigenic epitopes linked to resistance from T1D.
The overall architecture of TCR-pMHCII complexes in T1D of protective HLA-DR-DQ has been documented once (35). It concerns the binding to a DRB1*04:01–insulin C-peptide complex by a cognate TCR from a Tr1 regulatory clone in a reversed polarity; no generalizations can be made before more such complexes are available. As pathogenic CD4+ T-cell clones from patients with multiple sclerosis and T1D demonstrate several irregularities in the formation of the respective immunological synapses with antigen-presenting cells, it is well worth exploring the basis of this irregularity as well as the meaning of the reversed polarity in the reported Tr1 clone (34–37,57). By contrast, there are several complexes of gluten peptides with DQ2, DQ8.1, and DQ8trans (DQA1*05:01-B1*03:02), all associated with celiac disease (and T1D), with the cognate TCRs bound in the canonical diagonal orientation relative to the pMHCII axis. Gluten epitopes bound to the these disease-associated HLA-DQ molecules compared with the nonassociated DQ2.2 (DQA1*02:01-B1*02:02) showed: 1) the wide spectrum of gluten-derived and transglutaminase-transformed peptides stably bound to the three susceptible molecules compared with the nonsusceptible one; and 2) a restrictive TCR spectrum that could bind the trimolecular complexes in the susceptible DQ2 compared with a wide spectrum of TCR in DQ2.2 (39,48,56,58,59). Remarkably, the first point centers on the presence of α22 phenylalanine in DQ2.2 instead of the hydrogen bond forming tyrosine in three other alleles and a wider selection of peptides. Stable binding to DQ2.2 was related to few p3 serine-bearing gluten peptides. Hence, the precise TCR docking to particular pMHCII complexes is the key determinant of thymic selection and consequent T-cell fate and the subsequent immune response (36,37,60).
Examination of nonpocket-related residues reveals a set of different residues, some specific to certain motifs and some common for all motifs connected with resistance to T1D. This could all be related to the specific autoantigenic peptides that differentially bind to these DQ molecules and the subsequent path to T1D pathogenesis or resistance. The role of nonpocket residues could be investigated by comparison of the properties, concerning T1D pathogenesis, of very closely related HLA-DQ molecules differing one at a time in such residues. The extensive LD that connects the seven residues linked to T1D resistance includes residues in the signal peptides of the α- and the β-chains, potential TCR contact residues, certain residues in the α2β2 domains participating in CD4 binding and in the formation of the β167-169RGD loop, residues in the proximal membrane stalk, and residues within the intramembrane stretch. The signal peptides of the α- and β-chains have not been examined for any immunological roles, such as antigenicity, in contrast to the signal peptide for preproinsulin (52,60). Residue β45 has been shown as a major determinant of the surface electrostatic potential of the pMHCII complex; thus, it is a factor in the selection of cognate TCRs even though it is at an appreciable distance away from such contact (35–37).
Of the α2β2 domain residues, we single out α127 and α157: the latter participates in the formation of the putative homodimer of DQα-β heterodimers (32,33,42,61), while the former is a putative CD4 contact residue, once such a homodimer is formed (42). Residues β167-170 form the β167-169RGD loop that has the shape of a typical RGD loop involved in cell adhesion, yet still unknown function, while present in the standard shape in five DQcis and two DQtrans alleles with crystallographically determined structures (29,33,39,46,48,53,58,59). Residues α126, α135, α150, β126, and β130 have not been identified in any interactions with third molecules. They may be involved in noncovalent interactions with neighboring residues, adding to the stability of the HLA-DQ heterodimer. Residues β185 and β197 precede the transmembrane segment (α193-α214 and β199-β219), and in a single report, polymorphisms in the β179-191 region of HLA-DRB3*/4*/5* appeared involved in the binding to CD4, but not extending to HLA-DQ (62). Last, the intramembranous residues α196, α204, and β203 are presumed positioned with the first and the last nearly opposed to each other within the lipid bilayer (Fig. 4). Certain intramembrane and intracytoplasmic α-β residues were identified in cholesterol binding within signal-transducing lipid rafts (63).
Figure 4.

Proximal membrane, transmembrane, and intracytoplasmic amino acid sequences of select HLA-DQA-DQB alleles, showing the positions of linked residues associated with resistance to T1D. The two intramembrane sequences have been aligned in nearly identical topographical positions, despite different numberings of the sequences, for ease of comparison. For complete listing of associated alleles, see Table 1. The alleles are color coded in order to show which HLA-DQA molecules can pair with their structural matches from HLA-DQB molecules (purple color with purple color and olive green color with olive green color). The gray-shaded sequence concerns the intramembranous stretch.
The structures of several HLA-DQ molecules linked to T1D susceptibility or resistance have been determined, but only one of the former in complex with a T1D-linked epitope, InsB11-23 (46). The number of peptide epitopes derived from T1D autoantigens are increasing, but their relative order of importance in the progression from environmental exposure to the appearance of β-cell autoimmunity and the subsequent pathogenesis resulting in clinical onset remains to be clarified (64–66). Several studies show that IAA are the first to appear in young (<5 years of age) children, with a subsequent decreased incidence with increasing age, while GADA as the first autoantibody appears at 2–3 years and remains elevated (19,64). It was observed that the HLA genotype was the best predictor for β-cell autoimmunity (i.e., the first appearing autoantibody but not for the pathogenesis and progression to clinical onset). Our analysis shows that knowledge of the specific autoantigenic peptides bound to particular HLA-DR-DQ molecules will be instrumental in understanding both the trigger of β-cell autoimmunity and progression to disease. The motifs of protection should also prove useful when devising means to block the appearance of the pathogenesis or reverse its consequences once it is manifested.
Conditional analysis, used to assess residual associations with residues after adjusting for seven selected residues, was informative to indicate if adjusted associations with residues remained to be significant. If so, it was reasonable to infer that selected residues had not accounted for such residue-specific associations. However, it was important to caution if some residues became insignificant in the conditional analysis. These residues should not be automatically deemed as if their associations were completely accounted for by these seven residues. One interesting autoimmune disease is the Goodpasture syndrome, in which autoimmunity to collagen type IVα leads to kidney disease and the susceptibility is associated with HLA-DR15, while dominant resistance to it is associated with HLA-DR1 (67). Recently, the HLA-DR1–protective core nonamer epitope has been found to be collagen IV α135-143, while the respective HLA-DR15–susceptible epitope is α136-144. It is remarkable that the respective autoantigen-DR tetramers reactive to CD4+ T cells in peripheral blood from patients with Goodpasture syndrome showed a predominance of inflammatory Th1 cells over regulatory FoxP3+CD25highCD127− T cells; by contrast, in the blood of DR1 homozygous control subjects, there was a predominance of tetramer-positive Tregs (67). In a humanized mouse, it was shown that in the HLA-DR1/DR15 heterozygous state, Tregs would protect the mice from disease. It will be interesting to investigate the way antigen-specific Tregs can be induced in susceptible individuals in order to ameliorate or completely eradicate the symptoms of the syndrome. Along similar lines, a just-published study (55) has shown that GAD65- and IGRP-specific effector T cells and Tregs were at higher frequency in individuals carrying the DR1501-DQ6 T1D-resistant haplotype than susceptible or neutral haplotypes. Moreover, such Tregs suppressed DR1501-restricted, GAD65- and IGRP-specific effector T cells and DQB1*03:02-restricted GAD65-specific effector T cells, demonstrating another possible mechanism of T1D resistance by such alleles. Last, our analyses do not consider the upstream regions of HLA-DR-DQ, which appear rich in regulatory sequences and, when linked to certain T1D-resistant alleles, result in higher protein expression in antigen-presenting cells, compared with certain T1D-susceptible alleles (68–70). Deep sequencing of these allele-specific HLA-DR-DQ upstream regions should reveal possible allele-specific additional factors for susceptibility and resistance to T1D.
Being major histocompatibility genes, HLA-DQA and -DQB have complex roles in the human immunity. With respect to their association with T1D, HLA-DQ genes are mostly known as susceptibility genes that increase risks of T1D among carriers of DQ2 and DQ8 (21). However, their protective role against T1D has been observed, but is much less elaborated. In this study, we focus on the resistant DQ haplotypes, and our investigation uncovers an array of specific residues that explain their resistance to T1D. Identification of these residues provides novel insight into the mechanisms underlying the resistance and may provide an impetus to develop immunotherapeutic strategies for treating T1D.
Article Information
Acknowledgments. The authors thank Dr. Gun Forsander (Department of Pediatrics, Queen Silvia Children’s Hospital, Sahlgrenska University Hospital, Gothenburg, Sweden), Dr. Sten A. Ivarsson (Department of Clinical Sciences, Lund University/Clinical Research Centre, Skåne University Hospital, Malmö, Sweden), and Dr. Martina Persson (Department of Clinical Science and Education and Clinical Epidemiology Division, Department of Medicine, Karolinska Institutet, Stockholm, Sweden) for the clinical expertise in the BDD study. The authors also thank Anita Ramelius, Linda Faxius, Karl Moritz, Petra Moritz, Ingrid Wigheden, Ida Jönsson, and Rasmus Bennet, all at the Department of Clinical Sciences, Lund University/Clinical Research Centre, Malmö, Sweden, for expert technical assistance. Finally, the authors also thank the many other clinicians, research nurses, and parents/guardians of participants in the BDD study.
Funding. The study was supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grants 1R56 DK117276, DK63861, and DK26190, a grant from the European Foundation for the Study of Diabetes Clinical Research Grants Programme 2013 and in part the Swedish Child Diabetes Foundation (Barndiabetesfonden), the Swedish Research Council including a Linné grant to Lund University Diabetes Centre, the Skåne County Council for Research and Development, and the Swedish Association of Local Authorities and Regions.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. L.P.Z., G.K.P., and Å.L., researched and analyzed the data and wrote the manuscript. W.W.K. reviewed and commented on the manuscript with respect to structural implications and discussions. D.E.G. led the team of R.W., C.-W.P., and W.C.N. in the next-generation sequencing and researched data. A.K.M. and G.P.B. carried out graphical representations of select HLA-DQ molecules, contributed to discussion, and reviewed the manuscript. J.L. critically reviewed statistical results and improved presentation of results. A.C., H.E.L., J.L., C.M., U.S., and Å.L. designed the BDD study, researched data, contributed to discussion, and reviewed the manuscript. Å.L. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
G.K.P. has been retired from Technological Educational Institute (TEI) of Epirus, Arta, Greece since 1 September 2018. The affiliation is given for identification purposes only. As of 1 October 2018, the TEI of Epirus has been absorbed by the University of Ioannina. The respective department is now called Department of Agriculture.
This article contains supplementary material online at https://doi.org/10.2337/figshare.12860438.
References
- 1.Patterson CC, Karuranga S, Salpea P, et al. Worldwide estimates of incidence, prevalence and mortality of type 1 diabetes in children and adolescents: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract 2019;157:107842. [DOI] [PubMed] [Google Scholar]
- 2.DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet 2018;391:2449–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan NG, Richardson SJ. Fifty years of pancreatic islet pathology in human type 1 diabetes: insights gained and progress made. Diabetologia 2018;61:2499–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katsarou A, Gudbjörnsdottir S, Rawshani A, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers 2017;3:17016. [DOI] [PubMed] [Google Scholar]
- 5.Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet 2016;387:2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sioofy-Khojine AB, Lehtonen J, Nurminen N, et al. Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018;61:1193–1202 [DOI] [PubMed] [Google Scholar]
- 7.Vehik K, Lynch KF, Wong MC, et al.; TEDDY Study Group . Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat Med 2019;25:1865–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rich SS, Concannon P, Erlich H, et al. The type 1 diabetes genetics consortium. Ann N Y Acad Sci 2006;1079:1–8 [DOI] [PubMed] [Google Scholar]
- 9.Cooper JD, Howson JM, Smyth D, et al.; Type 1 Diabetes Genetics Consortium . Confirmation of novel type 1 diabetes risk loci in families. Diabetologia 2012;55:996–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia X, Han B, Onengut-Gumuscu S, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PloS One 2013;8:e64683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med 2009;360:1646–1654 [DOI] [PubMed] [Google Scholar]
- 12.Concannon P, Erlich HA, Julier C, et al.; Type 1 Diabetes Genetics Consortium . Type 1 diabetes: evidence for susceptibility loci from four genome-wide linkage scans in 1,435 multiplex families. Diabetes 2005;54:2995–3001 [DOI] [PubMed] [Google Scholar]
- 13.Barrett JC, Clayton DG, Concannon P, et al.; Type 1 Diabetes Genetics Consortium . Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009;41:703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson G, Valdes AM, Noble JA, et al. Relative predispositional effects of HLA class II DRB1-DQB1 haplotypes and genotypes on type 1 diabetes: a meta-analysis. Tissue Antigens 2007;70:110–127 [DOI] [PubMed] [Google Scholar]
- 15.Kockum I, Sanjeevi CB, Eastman S, et al. Complex interaction between HLA DR and DQ in conferring risk for childhood type 1 diabetes. Eur J Immunogenet 1999;26:361–372 [DOI] [PubMed] [Google Scholar]
- 16.Graham J, Kockum I, Sanjeevi CB, et al. Negative association between type 1 diabetes and HLA DQB1*0602-DQA1*0102 is attenuated with age at onset. Swedish Childhood Diabetes Study Group. Eur J Immunogenet 1999;26:117–127 [PubMed] [Google Scholar]
- 17.Erlich H, Valdes AM, Noble J, et al.; Type 1 Diabetes Genetics Consortium . HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 2008;57:1084–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valdes AM, Thomson G, Erlich HA, Noble JA. Association between type 1 diabetes age of onset and HLA among sibling pairs. Diabetes 1999;48:1658–1661 [DOI] [PubMed] [Google Scholar]
- 19.Krischer JP, Lynch KF, Schatz DA, et al.; TEDDY Study Group . The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: the TEDDY study. Diabetologia 2015;58:980–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilonen J, Hammais A, Laine AP, et al. Patterns of β-cell autoantibody appearance and genetic associations during the first years of life. Diabetes 2013;62:3636–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao LP, Papadopoulos GK, Kwok WW, et al. Motifs of three HLA-DQ amino acid residues (α44, β57, β135) capture full association with the risk of type 1 diabetes in DQ2 and DQ8 children. Diabetes 2020;69:1573–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 1987;329:599–604 [DOI] [PubMed] [Google Scholar]
- 23.Simmons KM, Mitchell AM, Alkanani AA, et al. Failed genetic protection: type 1 diabetes in the presence of HLA-DQB1*06:02. Diabetes 2020;69:1763–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao LP, Alshiekh S, Zhao M, et al.; Better Diabetes Diagnosis (BDD) Study Group . Next-generation sequencing reveals that HLA-DRB3, -DRB4, and –DRB5 may Be associated with islet autoantibodies and risk for childhood type 1 diabetes. Diabetes 2016;65:710–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao LP, Papadopoulos GK, Kwok WW, et al. Eleven amino acids of HLA-DRB1 and fifteen amino acids of HLA-DRB3, 4, and 5 include potentially causal residues responsible for the risk of childhood type 1 diabetes. Diabetes 2019;68:1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlsson A, Kockum I, Lindblad B, et al.; Swedish Better Diabetes Diagnosis Study Group . Low risk HLA-DQ and increased body mass index in newly diagnosed type 1 diabetes children in the Better Diabetes Diagnosis study in Sweden. Int J Obes 2012;36:718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delli AJ, Vaziri-Sani F, Lindblad B, et al.; Better Diabetes Diagnosis Study Group . Zinc transporter 8 autoantibodies and their association with SLC30A8 and HLA-DQ genes differ between immigrant and Swedish patients with newly diagnosed type 1 diabetes in the Better Diabetes Diagnosis study. Diabetes 2012;61:2556–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson C, Larsson K, Vaziri-Sani F, et al. The three ZNT8 autoantibody variants together improve the diagnostic sensitivity of childhood and adolescent type 1 diabetes. Autoimmunity 2011;44:394–405 [DOI] [PubMed] [Google Scholar]
- 29.Siebold C, Hansen BE, Wyer JR, et al. Crystal structure of HLA-DQ0602 that protects against type 1 diabetes and confers strong susceptibility to narcolepsy. Proc Natl Acad Sci U S A 2004;101:1999–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ettinger RA, Papadopoulos GK, Moustakas AK, Nepom GT, Kwok WW. Allelic variation in key peptide-binding pockets discriminates between closely related diabetes-protective and diabetes-susceptible HLA-DQB1*06 alleles. J Immunol 2006;176:1988–1998 [DOI] [PubMed] [Google Scholar]
- 31.Ettinger RA, Kwok WW. A peptide binding motif for HLA-DQA1*0102/DQB1*0602, the class II MHC molecule associated with dominant protection in insulin-dependent diabetes mellitus. J Immunol 1998;160:2365–2373 [PubMed] [Google Scholar]
- 32.Routsias J, Papadopoulos GK. Polymorphic structural features of modelled HLA-DQ molecules segregate according to susceptibility or resistance to IDDM. Diabetologia 1995;38:1251–1261 [DOI] [PubMed] [Google Scholar]
- 33.Paliakasis K, Routsias J, Petratos K, Ouzounis C, Kokkinidis M, Papadopoulos GK. Novel structural features of the human histocompatibility molecules HLA-DQ as revealed by modeling based on the published structure of the related molecule HLA-DR. J Struct Biol 1996;117:145–163 [DOI] [PubMed] [Google Scholar]
- 34.Sethi DK, Schubert DA, Anders AK, et al. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med 2011;208:91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beringer DX, Kleijwegt FS, Wiede F, et al. T cell receptor reversed polarity recognition of a self-antigen major histocompatibility complex. Nat Immunol 2015;16:1153–1161 [DOI] [PubMed] [Google Scholar]
- 36.Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol 2015;33:169–200 [DOI] [PubMed] [Google Scholar]
- 37.La Gruta NL, Gras S, Daley SR, Thomas PG, Rossjohn J. Understanding the drivers of MHC restriction of T cell receptors. Nat Rev Immunol 2018;18:467–478 [DOI] [PubMed] [Google Scholar]
- 38.Sanjeevi CB, Lybrand TP, DeWeese C, et al. Polymorphic amino acid variations in HLA-DQ are associated with systematic physical property changes and occurrence of IDDM. Members of the Swedish Childhood Diabetes Study. Diabetes 1995;44:125–131 [DOI] [PubMed] [Google Scholar]
- 39.Petersen J, Kooy-Winkelaar Y, Loh KL, et al. Diverse T cell receptor gene usage in HLA-DQ8-associated celiac disease converges into a consensus binding solution. Structure 2016;24:1643–1657 [DOI] [PubMed] [Google Scholar]
- 40.Morel PA, Dorman JS, Todd JA, McDevitt HO, Trucco M. Aspartic acid at position 57 of the HLA-DQ beta chain protects against type I diabetes: a family study. Proc Natl Acad Sci U S A 1988;85:8111–8115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ettinger RA, Liu AW, Nepom GT, Kwok WW. Exceptional stability of the HLA-DQA1*0102/DQB1*0602 alpha beta protein dimer, the class II MHC molecule associated with protection from insulin-dependent diabetes mellitus. J Immunol 1998;161:6439–6445 [PubMed] [Google Scholar]
- 42.Brown JH, Jardetzky TS, Gorga JC, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 1993;364:33–39 [DOI] [PubMed] [Google Scholar]
- 43.Sanjeevi CB, Lybrand TP, Stevanovic S, Rammensee HG. Molecular modeling of eluted peptides from DQ6 molecules (DQB1*0602 and DQB1*0604) negatively and positively associated with type 1 diabetes. Ann N Y Acad Sci 2002;958:317–320 [DOI] [PubMed] [Google Scholar]
- 44.Moustakas AK, van de Wal Y, Routsias J, et al. Structure of celiac disease-associated HLA-DQ8 and non-associated HLA-DQ9 alleles in complex with two disease-specific epitopes. Int Immunol 2000;12:1157–1166 [DOI] [PubMed] [Google Scholar]
- 45.Kwok WW, Domeier ME, Johnson ML, Nepom GT, Koelle DM. HLA-DQB1 codon 57 is critical for peptide binding and recognition. J Exp Med 1996;183:1253–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol 2001;2:501–507 [DOI] [PubMed] [Google Scholar]
- 47.Van de Wal Y, Kooy YM, Drijfhout JW, Amons R, Papadopoulos GK, Koning F. Unique peptide binding characteristics of the disease-associated DQ(alpha 1*0501, beta 1*0201) vs the non-disease-associated DQ(alpha 1*0201, beta 1*0202) molecule. Immunogenetics 1997;46:484–492 [DOI] [PubMed] [Google Scholar]
- 48.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A 2004;101:4175–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koelle DM, Johnson ML, Ekstrom AN, Byers P, Kwok WW. Preferential presentation of herpes simplex virus T-cell antigen by HLA DQA1*0501/DQB1*0201 in comparison to HLA DQA1*0201/DQB1*0201. Hum Immunol 1997;53:195–205 [DOI] [PubMed] [Google Scholar]
- 50.Yoshida K, Corper AL, Herro R, Jabri B, Wilson IA, Teyton L. The diabetogenic mouse MHC class II molecule I-Ag7 is endowed with a switch that modulates TCR affinity. J Clin Invest 2010;120:1578–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eerligh P, van Lummel M, Zaldumbide A, et al. Functional consequences of HLA-DQ8 homozygosity versus heterozygosity for islet autoimmunity in type 1 diabetes. Genes Immun 2011;12:415–427 [DOI] [PubMed] [Google Scholar]
- 52.Van Lummel M, Buis DTP, Ringeling C, et al. Epitope stealing as a mechanism of dominant protection by HLA-DQ6 in type 1 diabetes. Diabetes 2019;68:787–795 [DOI] [PubMed] [Google Scholar]
- 53.Schinkelshoek MS, Fronczek R, Kooy-Winkelaar EMC, et al. H1N1 hemagglutinin-specific HLA-DQ6-restricted CD4+ T cells can be readily detected in narcolepsy type 1 patients and healthy controls. J Neuroimmunol 2019;332:167–175 [DOI] [PubMed] [Google Scholar]
- 54.Astill TP, Ellis RJ, Arif S, Tree TI, Peakman M. Promiscuous binding of proinsulin peptides to Type 1 diabetes-permissive and –protective HLA class II molecules. Diabetologia 2003;46:496–503 [DOI] [PubMed] [Google Scholar]
- 55.Wen X, Yang J, James E, Chow IT, Reijonen H, Kwok WW. Increased islet antigen-specific regulatory and effector CD4+ T cells in healthy individuals with the type 1 diabetes-protective haplotype. Sci Immunol 2020;5:eaax8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Z, Reyes-Vargas E, Escobar H, et al. Peptidomic analysis of type 1 diabetes associated HLA-DQ molecules and the impact of HLA-DM on peptide repertoire editing. Eur J Immunol 2017;47:314–326 [DOI] [PubMed] [Google Scholar]
- 57.Schubert DA, Gordo S, Sabatino JJ Jr., et al. Self-reactive human CD4 T cell clones form unusual immunological synapses. J Exp Med 2012;209:335–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ting YT, Dahal-Koirala S, Kim HSK, et al. A molecular basis for the T cell response in HLA-DQ2.2 mediated celiac disease. Proc Natl Acad Sci U S A 2020;117:3063–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Broughton SE, Petersen J, Theodossis A, et al. Biased T cell receptor usage directed against human leukocyte antigen DQ8-restricted gliadin peptides is associated with celiac disease. Immunity 2012;37:611–621 [DOI] [PubMed] [Google Scholar]
- 60.James EA, Gillette L, Durinovic-Bello I, et al. DRB4*01:01 has a distinct motif and presents a proinsulin epitope that is recognized in subjects with type 1 diabetes. J Immunol 2018;201:3524–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stern LJ, Brown JH, Jardetzky TS, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 1994;368:215–221 [DOI] [PubMed] [Google Scholar]
- 62.Fleury S, Thibodeau J, Croteau G, et al. HLA-DR polymorphism affects the interaction with CD4. J Exp Med 1995;182:733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dixon AM, Roy S. Role of membrane environment and membrane-spanning protein regions in assembly and function of the Class II Major Histocompatibility complex. Hum Immunol 2019;80:5–14 [DOI] [PubMed] [Google Scholar]
- 64.Krischer JP, Liu X, Vehik K, et al.; TEDDY Study Group . Predicting islet cell autoimmunity and type 1 diabetes: an 8-year TEDDY Study progress report. Diabetes Care 2019;42:1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Babad J, Mukherjee G, Follenzi A, et al. Generation of β cell-specific human cytotoxic T cells by lentiviral transduction and their survival in immunodeficient human leucocyte antigen-transgenic mice. Clin Exp Immunol 2015;179:398–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.James EA, Mallone R, Kent SC, DiLorenzo TP. T-cell epitopes and neo-epitopes in type 1 diabetes: a comprehensive update and reappraisal. Diabetes 2020;69:1311–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ooi JD, Petersen J, Tan YH, et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature 2017;545:243–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beaty JS, Sukiennicki TL, Nepom GT. Allelic variation in transcription modulates MHC class II expression and function. Microbes Infect 1999;1:919–927 [DOI] [PubMed] [Google Scholar]
- 69.Aydemir Ö, Noble JA, Bailey JA, et al.; Better Diabetes Diagnosis (BDD) Study Group . Genetic variation within the HLA-DRA1 gene modulates susceptibility to type 1 diabetes in HLA-DR3 homozygotes. Diabetes 2019;68:1523–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Majumder P, Lee JT, Rahmberg AR, et al. A super enhancer controls expression and chromatin architecture within the MHC class II locus. J Exp Med 2020;217:e20190668. [DOI] [PMC free article] [PubMed] [Google Scholar]