

Abstract
Aims
The safety and pharmacokinetics of single and multiple doses of a novel mGlu2/3 receptor agonist prodrug, MGS0274 besylate (TS‐134), were investigated in healthy subjects.
Methods
Phase 1 single‐ascending dose (5–20 mg) and multiple‐ascending dose titration (5–80 mg) studies were conducted in healthy male and female subjects. Both studies were randomized, double‐blinded and placebo‐controlled. In one cohort of single‐ascending dose study (10 mg), concentrations of MGS0008, the active compound, in the cerebrospinal fluid (CSF) were measured for up to 24 hours postdose.
Results
Following single and multiple oral administrations, MGS0274 was rapidly absorbed and extensively converted into MGS0008, which reached a maximum concentration (Cmax) in plasma within 4 hours postdose and declined with a terminal half‐life (t1/2) of around 10 hours. Plasma exposure to MGS0274 was minimal, accounting for approximately 3% of the area under the concentration–time curve (AUC) of MGS0008. Plasma Cmax and AUC of MGS0008 at steady state increased dose proportionally (5–80 mg). MGS0008 penetrated into CSF, with a CSF‐to‐plasma Cmax ratio of 3.66%, and was eliminated with a t1/2 of approximately 16 hours. The most frequent treatment‐emergent adverse events observed following single and multiple oral administration included headache, nausea, somnolence, dizziness and vomiting.
Conclusion
TS‐134 is orally bioavailable in humans and converts rapidly and extensively to MGS0008, which exhibits good CSF penetration. Orally administered TS‐134 was safe and generally well‐tolerated; hence, TS‐134 is a promising candidate for further clinical development for the treatment of disorders in which glutamatergic abnormalities are involved, such as schizophrenia.
Keywords: cerebrospinal fluid, mGlu2/3 agonist, MGS0008, MGS0274, schizophrenia, TS‐134
1.
What is already known about this subject
Metabotropic glutamate (mGlu) receptors, particularly mGlu2 and mGlu3 receptors have been of interest for the development of a novel intervention for schizophrenia due to their distribution within cortical and limbic areas, and their modulatory roles in glutamatergic transmission.
MGS0008, the active form of MGS0274 besylate (TS‐134) is a potent and selective agonist of mGlu2 and mGlu3 receptors, and has been shown to possess antipsychotic‐like activity in some animal models.
What this study adds
These first‐in‐human, single and multiple ascending dose studies assessed the safety, tolerability and pharmacokinetics of MGS0274 besylate (TS‐134), a novel prodrug of mGlu2/3 receptor agonist, administered orally in healthy subjects.
The favourable safety profile of TS‐134, and exposure of its active compound, MGS0008, observed in both plasma and cerebrospinal fluid from current studies, support further evaluation of TS‐134 in a proof‐of‐concept study in patients with schizophrenia.
2. INTRODUCTION
Glutamate is the major excitatory neurotransmitter in the brain, and its dysfunction plays an important role in aetiology and pathophysiology of psychiatric disorders, including schizophrenia. 1 , 2 Increased pyramidal neuron activity in the prefrontal cortex due to hypofunction of N‐methyl‐D‐aspartate (NMDA) receptor on the γ‐aminobutyric acid interneurons was noted in schizophrenic patients, 3 , 4 and the NMDA receptor antagonists cause schizophrenia‐like symptoms in healthy volunteers, and worsen psychosis in schizophrenic patients. 5 , 6 Thus, glutamatergic dysfunction, in particular the hypofunction of the NMDA receptor, has an important role in the pathophysiology of schizophrenia, and the glutamatergic system is an attractive therapeutic target for the treatment of schizophrenia. 7 , 8
Among glutamate receptors, metabotropic glutamate (mGlu) receptors, which consist of 8 subtypes, 9 have emerged as attractive therapeutic targets, and both mGlu2 and mGlu3 receptors are of particular interest for the development of a novel intervention for schizophrenia because of their distribution within cortical and limbic areas, and their modulatory roles in glutamatergic transmission. 8 , 10 , 11 Indeed, mGlu2/3 receptor agonists presynaptically reduce excess glutamate release induced by blockade of NMDA receptor in the medial prefrontal cortex, 12 , 13 and thereby normalize an NMDA receptor antagonist‐induced aberrant activity in the region. 14 Moreover, mGlu2/3 receptor agonists ameliorate abnormal behaviours in animal models which evaluate antipsychotic‐like potential of compounds. 15 , 16 , 17 Despite seemingly promising effects in animal models, clinical results of an mGlu2/3 receptor agonist prodrug, pomaglumetad, after an exciting outcome in the first trial, 18 were not necessarily encouraging in subsequent trials. 19 , 20 , 21 However, exploratory analysis of 5 placebo‐controlled trials with pomaglumetad revealed that patients who are early‐in‐disease or previously treated with dopamine D2 receptor dominant drugs exhibited significantly greater improvement relative to those receiving placebo. 22 Moreover, imaging studies raised the concerns that doses used in the trials may not have been appropriate. 23 Therefore, the potential for mGlu2/3 receptor agonists to be novel therapeutic agents for the treatment of schizophrenia needs to be further explored.
(1S,2S,3S,5R,6S)‐2‐amino‐3‐fluorobicyclo[3.1.0]hexane‐2,6‐dicarboxylic acid (MGS0008) is a potent and selective orthosteric mGlu2/3 receptor agonist which has been demonstrated to exert antipsychotic‐like effects in animal models. 24 , 25 Recently, we synthesized an ester‐based lipophilic prodrug of MGS0008, (1S,2S,3S,5R,6S)‐2‐amino‐3‐ fluoro‐6‐({(1S)‐1‐[({[(1R,2S,5R)‐5‐methyl‐2‐(propan‐2‐yl)cyclohexyl]oxy}carbonyl)oxy]ethoxy}carbonyl)bicyclo[3.1.0]hexane‐2‐carboxylic acid monobenzenesulfonate (MGS0274 besylate), and demonstrated that the prodrug was rapidly converted into MGS0008 in monkeys after oral administration, and that the hydrolytic activity against MGS0274 in the human liver S9 fraction was comparable to that in monkeys, suggesting the possibility of the rapid presystemic hydrolysis of MGS0274 to MGS0008 in humans. 26 Moreover, MGS0008 was detected in the cerebrospinal fluid (CSF) in rats after oral administration of MGS0008. 26 Collectively, MGS0274 besylate is an appropriate tool to be used for the proof‐of‐concept study in human subjects. Here we report safety, tolerability and pharmacokinetics (PK) of single and multiple doses of TS‐134, of which MGS0274 besylate is the active ingredient, in healthy subjects, as well as preclinical data, to show pharmacological profiles of MGS0008.
3. METHODS
3.1. Preclinical pharmacology studies of MGS0008 and nomenclature of mGlu2/3 receptor
We previously reported in vitro and in vivo characteristics of MGS0008. 24 , 25 In the present study, we further investigated the pharmacological profiles of MGS0008, including the effects of MGS0008 on abnormal activity induced by hypofunction of NMDA receptor. Objectives and methods for preclinical pharmacology studies are described in Supporting Information: Materials and Methods. Nomenclature of mGlu2 and mGlu3 receptors was conformed to the IUPHAR/BPS Guide to PHARMACOLOGY nomenclature classification. 27
3.2. Clinical study designs
Two Phase 1 studies, a single‐ascending dose (SAD) study and a multiple‐ascending dose titration (MAD) study, employed randomized, double‐blind (subject, investigator and sponsor), placebo‐controlled, ascending dose designs. Both studies were conducted at a single site in the USA from August 2015 to December 2016. The studies were approved by the Institutional Review Board (IntegReview, Austin, TX, USA) and were conducted in accordance with the Declaration of Helsinki and the International Council on Harmonisation Guideline for Good Clinical Practice. All subjects provided written informed consent before participating in the study.
The SAD study was a first‐in‐human study consisting of 4 cohorts in 3 parts (Parts A [Cohorts 1 and 2], B [Cohort 7] and C [Cohort 8]; Figure 1A). In Parts A and C, subjects were randomized to receive either TS‐134 or placebo in a 3:1 ratio in each cohort (6 active and 2 placebo per cohort). In Part A, Cohort 1 initiated with 5 mg TS‐134. Subjects were dosed orally in a fasted condition except for Cohort 2 (20 mg TS‐134), which was first dosed in a fasted condition and then in a fed condition in a single crossover design. In Cohort 2, treatment‐emergent adverse events (TEAEs) of vomiting and nausea led to early termination of subjects, which limited further dose escalations in Part A, and the planned Cohorts 3–6 were not conducted. Part C was then conducted to examine the tolerability of a dose level of 10 mg TS‐134, and to additionally evaluate the food effect on TS‐134 PK. Part B (CSF cohort) was conducted with a dose level of 10 mg TS‐134, in a fed condition, which was determined following a blinded interim review of safety and PK data from Parts A and C. No subjects received placebo in Part B.
The MAD study evaluated the safety, tolerability and PK of 6 dose levels of TS‐134 (5, 10, 20, 40, 60 and 80 mg), administered orally and daily under a fed condition, according to specific 14‐day multiple dose titration schemes in 6 cohorts (Figure 1B). Each cohort included a titration period (TP) and a maintenance period (MP). TS‐134 dose levels were titrated up during TP until reaching the target dose level and then the respective dose level was maintained during MP. Subjects were randomized to receive either TS‐134 or placebo in a 4:1 ratio in each cohort. Each cohort included 10 subjects (8 active and 2 placebo), except for Cohort 6, which included 9 subjects (7 active and 2 placebo).
FIGURE 1.
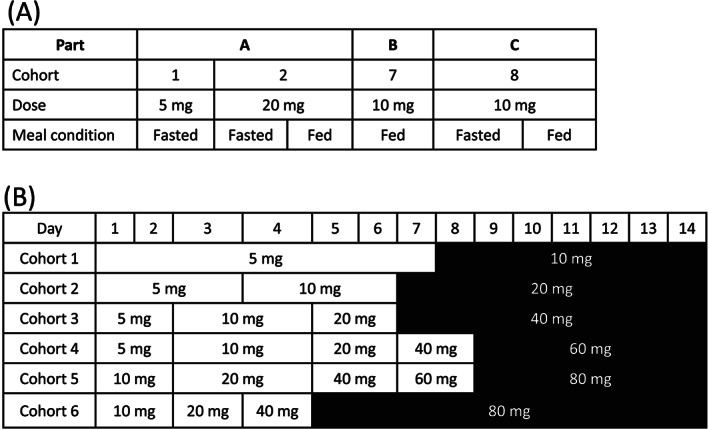
Study designs: (A) single‐ascending dose study; (B) multiple‐ascending dose study. Note (B): titration period presented without shading and maintenance period with shading
3.3. Subjects
Eligible subjects included healthy males and females who were not pregnant or breastfeeding, aged 18–55 years inclusive and body weight ≥ 45 kg, with a body mass index ≥18 and ≤ 30 kg m−2. Exclusion criteria included, but were not limited to: clinically significant abnormal cardiac examinations, haematology, biochemistry or urinalysis; significant history or presence of hepatic, renal, cardiovascular or pulmonary disease; history or presence of psychiatric or neurological disease or condition. TS‐134 and placebo were administered orally as a solution on Day 1 in the SAD study and on Day 1 through Day 14 in the MAD study. TS‐134 drug product (MGS0274 besylate) and placebo solutions were prepared extemporaneously at the clinical site by an unblinded pharmacist. The dosages were calculated as free base (MGS0274). Subjects remained on site from Day −1 until 48 hours following the last drug administration.
3.4. Blood, CSF and urine sampling
In the SAD study, blood samples for the determination of MGS0274 and MGS0008 concentrations in plasma were collected at predose, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, and 48 hours postdose. For the CSF sample collection, an indwelling epidural catheter was inserted into the lumbar spine at least 1 hour predose. CSF was collected at predose, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12 and 24 hours postdose. Urine samples were collected at predose, and 0–6, 6–12, 12–24, 24–36 and 36–48 hours postdose.
In the MAD study, Cohort 1 blood samples were collected intensively on the first and last days of TP and MP (Days 1, 7, 8, 14: predose, 0.25, 0.5, 1, 2, 3, 4, 5, 6, 8 and 12 hours postdose), predose only on Days 2–6 and Days 9–13, and at 24, 36 and 48 hours after the final dose on Day 14. Blood sample collection times in Cohorts 2 to 6 were similar to Cohort 1, except there were no intense samplings during TP and only predose samples were collected. Urine samples were collected for 0–24 hours postdose on Days 1, 7, 8, and 14 in Cohort 1, or MP Day 1 and Day 14 in Cohorts 2 to 6, with additional samples at 24–48 hours postdose on Day 14.
3.5. Bioanalytical methods
Plasma, urine and CSF samples were analysed at CMIC Pharma Science Co., Ltd. (Hyogo, Japan). The samples were quantified for MGS0274 and MGS0008 in plasma and urine, and for MGS0008 in CSF, using validated high‐performance liquid chromatography/tandem mass spectrometry methods. The lower limit of quantitation was 0.1 ng mL−1 and the upper limit of quantitation (ULOQ) was 100 ng mL−1 for both MGS0008 and MGS0274 in the plasma and CSF samples. In the urine samples, the lower limit of quantitation was 1 ng mL−1 and the ULOQ was 1000 ng mL−1 for both MGS0008 and MGS0274. Samples for which the concentrations could potentially exceed the ULOQ, or for which the assayed values exceeded the ULOQ, were diluted to yield concentrations within the calibration range. All the analytical runs met the acceptance criteria: the accuracy for quality control samples ranged from 90.4 to 106.1% (plasma), 89.4 to 103.3% (urine) and 95.3 to 102.0% (CSF) for MGS0008, and 86.4 to 114.5% (plasma) and 92.4 to 111.7% (urine) for MGS0274.
3.6. Statistics
Placebo treated subjects from different cohorts were pooled as control groups by condition (fasted or fed) for the SAD study and as 1 control group for the MAD study in summary statistics.
3.6.1. Sample size
The sample sizes for both studies were customary for Phase 1 studies evaluating safety and PK and were not based on statistical power calculations.
3.6.2. PK
PK parameters were calculated by noncompartmental analysis methods from the concentration–time data using Phoenix WinNonlin version 6.3 or later. Dose proportionality and food effects were analysed using SAS version 9.2 or later.
Dose proportionality of plasma MGS0274 and MGS0008 PK variables Cmax and AUC(0‐tau) over the dose range tested in the MAD study were investigated using the following power model: log (Parameter) = a + b * log (dose), where a is the intercept and b is the slope. Linearity was assessed based on a 90% confidence interval (CI) of the slope, and those that included 1.0 were interpreted as the absence of significant evidence of nonproportionality.
The food effect was evaluated in Cohorts 2 and 8 in the SAD study by performing an analysis of variance, with terms for treatment effects, on the log‐transformed values of Cmax and AUC(0‐inf). Subject was treated as a random effect and treatment was considered as a fixed effect. From these analyses, least‐squares (LS) means, LS treatment differences and 90% CI for the treatment differences on log‐scale were obtained. The results were transformed back to the original scale by exponentiation to provide treatment geometric LS means, point estimates of the geometric test (fed)/reference (fasted) LS mean ratios and 90% CI for these ratios.
3.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
4. RESULTS
4.1. Preclinical pharmacology profiles of MGS0008
The present study demonstrated that MGS0008 normalizes aberrant brain activity induced by hypofunction of NMDA receptor because it attenuated memantine‐increased 2‐deoxy‐D‐glucose uptake (measured as 2‐deoxyglucose 6‐phosphate formed in the cells) in the prefrontal cortex in mice (Figure 2A). Accordingly, MGS0008 significantly attenuated not only methamphetamine‐ but also phencyclidine‐induced locomotor hyperactivity, and significantly reversed MK‐801‐induced impaired social memory in rats (Figures 2B, C and D), indicating that MGS0008 attenuates behavioural abnormalities induced by NMDA receptor blockade. Notably, MGS0008 also significantly reduced spontaneous locomotor activity in rats at doses similar to those which block psychostimulants‐induced hyperlocomotion (Figure S1a). In contrast, MGS0008 did not induce catalepsy in rats, even at doses 3–10 times higher than those showing the antipsychotic‐like effects (Figure S1b), suggesting that MGS0008 may be devoid of extrapyramidal side effects.
FIGURE 2.
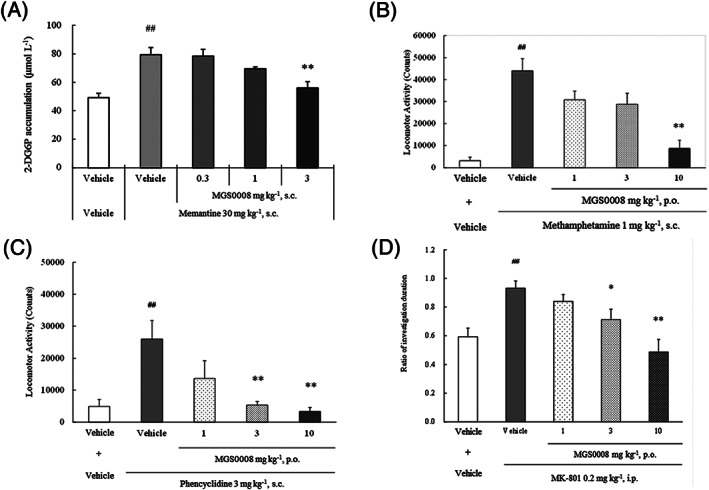
Pharmacological profiles of MGS0008: Effects of MGS0008 on (A) memantine‐increased 2‐deoxyglucose 6‐phosphate (2‐DG6P) accumulation in the mouse prefrontal cortex; (B) methamphetamine‐induced locomotor hyperactivity in rats; (C) phencyclidine‐induced locomotor hyperactivity in rats, and (D) MK‐801‐induced social recognition deficits in rats. Data represent mean ± standard error of the mean (A‐B: n = 8, C: n = 10, D: n = 12–14 animals per each group). ##P < .01 vs the vehicle + vehicle‐treated group (Student's t‐test or Welch's test), *P < .05, **P < .01 vs the vehicle + memantine‐treated group, the vehicle + methamphetamine‐treated group, the vehicle + phencyclidine‐treated group or the vehicle + MK‐801‐treated group (Dunnett's test or Steel's test). s.c., subcutaneous; p.o, per os, orally; i.p., intraperitoneal injection
4.2. Subject disposition and demographics
In the SAD study, a total of 33 subjects were enrolled (27 active and 6 placebo) and 28 subjects (84.85%) completed the study per protocol. Two fasted subjects (20 mg TS‐134) were discontinued due to TEAEs of severe vomiting and/or severe nausea. In the MAD study, 59 subjects were enrolled (47 active and 12 placebo) in 6 treatment cohorts. Forty‐four subjects who were administered TS‐134 and 12 who were administered placebo completed the study per protocol. There were no discontinuations due to TEAEs. Subject disposition and demographics are summarized in Tables 1 and 2, respectively.
TABLE 1.
Subject disposition
| SAD study | MAD study | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parts A and C | Part B | All parts | ||||||||||||||
| Cohort | 1–2, 8 | 2, 8 | 1 | 8 | 2 | 7 | 1–2, 7–8 | 1–6 | 1 | 2 | 3 | 4 | 5 | 6 | ||
| Dose (mg) | PBO | PBO | 5 | 10 | 20 | 10 | PBO | 5–10 | 5–20 | 5–40 | 5–60 | 10–80 | 10–80 | |||
| Food condition | fasted | fed | fasted | fasted | fed | fasted | fed | fed | fed | fed | fed | fed | fed | fed | fed | |
| Randomized | 6 | 4 | 6 | 6 | 5 a | 7 b | 3 c | 8 | 33 | 12 | 8 | 8 | 8 | 8 | 8 | 7 |
| Completed | 6 | 4 | 6 | 5 | 5 | 3 | 3 | 8 | 28 | 12 | 8 | 8 | 8 | 7 | 7 | 6 |
| Withdrawn | 0 | 0 | 0 | 1 | 0 | 4 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 1 | 1 | 1 |
| TEAE | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Subject withdrew | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Lost to follow‐up | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Other | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Safety population | 6 | 4 | 6 | 6 | 5 | 7 | 3 | 8 | 33 | 12 | 8 | 8 | 8 | 8 | 8 | 7 |
| PK population | 0 | 0 | 6 | 4 | 3 | 3 | 3 | 7 | 20 | 0 | 8 | 8 | 8 | 8 | 8 | 7 |
Placebo subjects were pooled. SAD, single‐ascending dose; MAD, multiple‐ascending dose; PBO, placebo; TEAE, treatment‐emergent adverse event; PK, pharmacokinetics.
One subject who completed the 10 mg TS‐134 fasted period was withdrawn from the study before initiating dosing in the 10 mg TS‐134 fed period due to a positive urine alcohol result at the admission visit for fed treatment.
One withdrawn subject in the 20 mg TS‐134 fasted period was replaced for compensating sufficient number of subjects to evaluate food effect on PK adequately.
Two subjects who vomited with severe intensity during 20 mg TS‐134 fasted period were withdrawn from the study and 2 other subjects who vomited during 20 mg TS‐134 fasted period were not allowed to enter the fed period due to the Principal Investigator's decision.
TABLE 2.
Subject demographics
| SAD study | MAD study | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parts A and C | Part B | All parts | |||||||||||||
| Cohort | PBO | 1 | 8 | 2 | 7 | PBO | 1 | 2 | 3 | 4 | 5 | 6 | All cohorts | ||
| Sex | Male | 1 | 2 | 4 | 3 | 3 | 13 | 10 | 5 | 7 | 8 | 4 | 6 | 4 | 44 |
| Female | 5 | 4 | 2 | 4 | 5 | 20 | 2 | 3 | 1 | 0 | 4 | 2 | 3 | 15 | |
| Age (y) | Mean ± SD | 40.2± 5.88 | 36.5± 6.89 | 31.8± 9.66 | 29.7± 6.65 | 40.6± 11.3 | 35.9± 9.16 | 35.8± 7.30 | 32.5± 5.93 | 27.6± 6.35 | 36.0± 9.87 | 33.0± 7.98 | 37.3± 11.7 | 37.6± 7.39 | 34.3± 8.43 |
| Height (cm) | Mean ± SD | 162± 3.01 | 164± 8.75 | 173± 13.2 | 169± 8.42 | 168± 7.83 | 167± 8.99 | 174± 6.78 | 170± 7.41 | 172± 7.98 | 172± 9.87 | 165± 9.97 | 174± 9.58 | 177± 6.37 | 172± 8.53 |
| Weight (kg) | Mean ± SD | 66.4± 3.00 | 68.6± 8.60 | 72.8± 11.6 | 71.7± 10.6 | 73.4± 5.74 | 70.8± 8.35 | 76.8± 10.9 | 72.1± 10.5 | 76.5± 8.08 | 69.7± 10.8 | 67.1± 8.65 | 77.5± 13.6 | 82.8± 7.01 | 74.6± 10.8 |
| BMI (kg m−2) | Mean ± SD | 25.5± 1.28 | 25.4± 1.77 | 24.2± 1.68 | 25.4± 4.22 | 26.2± 1.88 | 25.4± 2.40 | 25.4± 2.64 | 24.8± 2.02 | 25.8± 2.30 | 23.4± 2.49 | 24.6± 1.82 | 25.6± 2.30 | 26.6± 2.61 | 25.2± 2.40 |
| Race | WH | 3 | 2 | 2 | 2 | 6 | 15 | 8 | 6 | 6 | 4 | 7 | 4 | 6 | 41 |
| AS | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 3 | 1 | 0 | 0 | 5 | |
| BL | 1 | 2 | 4 | 5 | 0 | 12 | 3 | 2 | 1 | 1 | 0 | 3 | 1 | 11 | |
| AM | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MU | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Other | 1 | 0 | 0 | 0 | 2 | 3 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 2 | |
| Ethnicity | Hispanic or Latino | 2 | 2 | 0 | 2 | 4 | 10 | 4 | 2 | 4 | 1 | 6 | 3 | 1 | 21 |
| Not Hispanic or Latino | 4 | 4 | 6 | 5 | 4 | 23 | 8 | 6 | 4 | 7 | 2 | 5 | 6 | 38 | |
Placebo subjects were pooled. SAD, single‐ascending dose; MAD, multiple‐ascending dose; PBO, placebo; BMI, body mass index; SD, standard deviation; WH, white; AS, Asian; BL, Black or African American; AM, American Indian or Alaska Native; MU, multiple.
4.3. Safety
TS‐134 was safe and generally well‐tolerated when administered orally to healthy subjects as single doses up to 10 mg under both fasted or fed conditions, and up to 80 mg when administered daily under fed condition using the multiple dose titration schemes for 14 days. There were no deaths or serious adverse events in both studies. The summary of TEAEs by treatment (SAD study) and by cohort (MAD study) is presented in Table 3. There were no apparent treatment‐ or dose‐related trends in clinical laboratory test results, vital sign measurements, 12‐lead electrocardiography measurements or physical examination findings.
TABLE 3.
Summary of treatment‐emergent adverse events occurring in 2 or more subjects in any dosing group by treatment (SAD study) and cohort (MAD study)
| SAD study | MAD study | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parts A and C | Part B | |||||||||||||||||
| MedDRA Preferred Term | 5 mg fasted (n = 6) | 10 mg fasted (n = 6) | 10 mg fed (n = 5) | 20 mg fasted (n = 7) | 20 mg fed (n = 3) | Total TS‐134 fasted (n = 19) | Total TS‐134 fed (n = 8) | Total placebo fasted (n = 6) | Total placebo fed (n = 4) | 10 mg fed (n = 8) | Cohort 1 TS‐134 (n = 8) | Cohort 2 TS‐134 (n = 8) | Cohort 3 TS‐134 (n = 8) | Cohort 4 TS‐134 (n = 8) | Cohort 5 TS‐134 (n = 8) | Cohort 6 TS‐134 (n = 7) | Total TS‐134 (n = 47) | Pooled placebo (n = 12) |
| Total number of TEAEs | 2 | 6 | 4 | 11 | 3 | 19 | 7 | 0 | 0 | 10 | 11 | 17 | 16 | 26 | 30 | 25 | 125 | 9 |
| Number of subjects with at least 1 TEAE | 2 (33.3) | 4 (66.7) | 3 (60.0) | 5 (71.4) | 1 (33.3) | 11(57.9) | 4 (50.0) | 0 | 0 | 6 (75.0) | 5 (62.5) | 5 (62.5) | 5 (62.5) | 8 (100) | 7 (87.5) | 6 (85.7) | 36(76.6) | 3 (25.0) |
| Nausea | 0 | 1 (16.7) | 1 (20.0) | 3 (42.9) | 1 (33.3) | 4 (21.1) | 2 (25.0) | 0 | 0 | 0 | 2 (25.0) | 3 (37.5) | 2 (25.0) | 3 (37.5) | 3 (37.5) | 2 (28.6) | 15 (31.9) | 1 (8.3) |
| Vomiting | 0 | 2 (33.3) | 1 (20.0) | 4 (57.1) | 0 | 6 (31.6) | 1 (12.5) | 0 | 0 | 2 (25.0) | 0 | 0 | 1 (12.5) | 2 (25.0) | 0 | 0 | 3 (6.4) | 0 |
| Dizziness | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 3 (37.5) | 1 (12.5) | 0 | 3 (42.9) | 8 (17.0) | 0 |
| Headache | 1 (16.7) | 0 | 1 (20.0) | 1 (14.3) | 1 (33.3) | 2 (10.5) | 2 (25.0) | 0 | 0 | 2 (25.0) | 1 (12.5) | 3 (37.5) | 3 (37.5) | 5 (62.5) | 3 (37.5) | 1 (14.3) | 16 (34.0) | 1 (8.3) |
| Somnolence | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (5.3) | 0 | 0 | 0 | 2 (25.0) | 3 (37.5) | 1 (12.5) | 1 (12.5) | 1 (12.5) | 1 (12.5) | 2 (28.6) | 9 (19.1) | 1 (8.3) |
| Abnormal dreams | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25.0) | 0 | 0 | 2 (4.3) | 1 (8.3) |
| Insomnia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (37.5) | 0 | 1 (14.3) | 4 (8.5) | 1 (8.3) |
| Pruritus | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 | 3 (42.9) | 4 (8.5) | 0 |
| Post lumbar puncture syndrome | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (37.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Number of subjects (%) are presented. SAD, single‐ascending dose; MAD, multiple‐ascending dose; MedDRA, Medical Dictionary for Regulatory Activities; TEAE, treatment‐emergent adverse event.
In the SAD study, the most frequent TEAEs reported in Parts A and C included vomiting (31.6%) and nausea (21.1%) in fasted conditions, and nausea (25.0%) and headache (25.0%) in fed conditions. There were dose‐related increases in the incidences of vomiting and nausea. For food effect cohorts, there were no apparent differences in the incidences of TEAEs between fasted and fed conditions, except for vomiting, which was decreased under fed conditions. In Part B, during fed 10 mg TS‐134 treatment, the most frequent TEAEs included post lumbar puncture syndrome (37.5%), vomiting (25.0%), headache (25.0%) and somnolence (25.0%). All TEAEs were considered to be mild or moderate in severity; except for 2 reports of severe vomiting and 1 report of severe nausea at 20 mg in the fasted treatment group (one subject experienced both severe vomiting and severe nausea).
In the MAD study, the most frequent TEAEs in the total TS‐134 group included headache (34.0%), nausea (31.9%), somnolence (19.1%) and dizziness (17.0%). All TEAEs were considered to be mild or moderate in severity, except for 1 report of severe fatigue and 1 report of severe anxiety, both of which were reported by the same subject at the 80 mg MP in Cohort 5 (data not shown). Unlike the SAD study, where TEAEs resulted in early termination of 2 subjects, there were no TEAEs leading to subject withdrawal in the MAD study.
Employment of the multiple‐dose titration schemes starting from 5 or 10 mg under fed conditions improved the tolerability of TS‐134 administered orally in healthy subjects from 10 to 80 mg.
4.4. PK
Mean plasma concentration–time profiles of MGS0008 and MGS0274 on Day 1 in the SAD study (fasted condition) and on Day 14 in the MAD study (fed condition) are presented in Figure 3. The PK parameters and urinary excretion data are shown in Tables 4 and 5, respectively. In both studies, MGS0274 was rapidly absorbed, reached Cmax within 1 hour postdose and then rapidly decreased. Plasma MGS0008 (molecular weight: 203.17) was detected at the first sampling time point (15 min postdose), reached a Cmax approximately 6‐fold higher than that of MGS0274 (molecular weight: 429.49) in a molar ratio within 4 hours postdose and decreased slower than MGS0274, with a t1/2 of around 10 hours (range: 6.9–10.9 h). In the MAD study, plasma trough concentrations of MGS0008 appeared to reach steady state by 2 days after initiating multiple doses of target dose levels (data not shown), and dose proportional increases of steady‐state Cmax and AUC of both MGS0274 and MGS0008 were observed over the dose range tested (5–80 mg; Table S1). The plasma AUC of MGS0008 was approximately 30‐fold higher than that of MGS0274 in a molar ratio in all the treatment groups with or without food. These results demonstrated rapid and sufficient conversion of MGS0274 into MGS0008 in humans. In the SAD study, food effects on the PK of MGS0008 and MGS0274 were investigated at 10 and 20 mg TS‐134, and the Cmax and AUC of MGS0008 were reduced by up to 57 and 25%, respectively, in the fed group. The Cmax of MGS0274 was reduced up to 45% in the fed group, while almost no food effect was observed in AUC of MGS0274 (Table S2).
FIGURE 3.
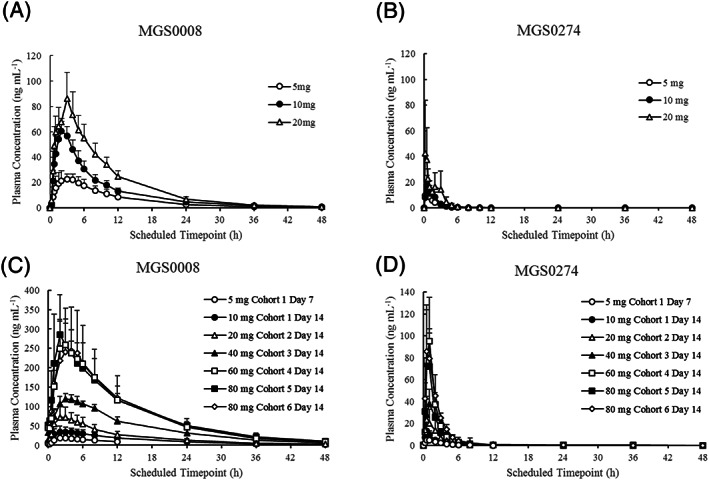
Mean plasma concentration–time profiles of MGS0008 and MGS0274 following single (SAD) and multiple (MAD) dose titrations of TS‐134: (A) MGS0008 in SAD study; (B) MGS0274 in SAD study; (C) MGS0008 on Day 14 in MAD study; (D) MGS0274 on Day 14 in MAD study. Cohort 1, 5 mg in (C) and (D) represent results on Day 7. Figures are presented as the mean + standard deviation
TABLE 4.
Plasma and CSF pharmacokinetic parameters of MGS0008 and MGS0274 by treatment
| (a) SAD study | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter (unit) | MGS0008 | MGS0274 | |||||||||||
| Plasma | Plasma | CSF | Plasma | ||||||||||
| Parts A and C | Part B | Parts A and C | Part B | ||||||||||
| 5 mg fasted (n = 6) | 10 mg fasted (n = 4) | 10 mg fed (n = 3) | 20 mg fasted (n = 3) | 20 mg fed (n = 3) | 10 mg fed (n = 7) | 5 mg fasted (n = 6) | 10 mg fasted (n = 4) | 10 mg fed (n = 3) | 20 mg fasted (n = 3) | 20 mg fed (n = 3) | 10 mg fed (n = 7) | ||
| C max (ng mL−1) a | 25.92 (22.65) | 60.60 (12.41) | 28.13 (43.31) | 89.40 (17.15) | 60.23 (12.80) | 33.96 (29.52) | 1.244 (38.39) | 13.43 (55.44) | 17.88 (40.87) | 8.137 (16.40) | 49.07 (66.56) | 33.27 (45.09) | 13.00 (82.69) |
| AUC 0‐inf (h•ng mL−1) a | 277.6 (18.69) | 535.1 (11.33) | 390.4 (24.70) | 839.5 (19.89) | 676.6 (5.83) | 381.9 f (17.02) | 18.85 f (33.61) | 23.94 c (30.64) | 29.33 (31.92) | 22.69 (8.60) | 72.09 (50.40) | 84.30 d (16.80) | 36.47 e (61.58) |
| t max (h) b | 2.0 | 2.0 | 4.0 | 3.0 | 2.0 | 4.0 | 8.0 | 0.5 | 1.0 | 0.25 | 0.25 | 0.5 | 0.5 |
| t 1/2 (h) a | 7.747 (7.62) | 8.407 (19.40) | 8.699 (20.55) | 6.963 (20.66) | 6.909 (19.91) | 8.492 (27.56) | 16.33 g (NC) | 1.013 c (35.22) | 0.8765 (17.66) | 1.505 (7.14) | 1.477 (14.30) | 1.669 d (7.05) | 1.377 e (26.45) |
| (b) MAD study (Day 14 in plasma only) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter (unit) | MGS0008 | MGS0274 | ||||||||||||
| 5 mg (n = 8) | 10 mg (n = 8) | 20 mg (n = 8) | 40 mg (n = 8) | 60 mg (n = 8) | 80 mg h (n = 7) | 80 mg i (n = 6) | 5 mg (n = 8) | 10 mg (n = 8) | 20 mg (n = 8) | 40 mg (n = 8) | 60 mg (n = 8) | 80 mg h (n = 7) | 80 mg i (n = 6) | |
| C max (ng mL−1) a | 19.91 (37.61) | 37.70 (31.89) | 77.76 (37.74) | 124.0 (9.46) | 262.6 (25.09) | 297.6 (30.13) | 255.5 (43.42) | 6.090 (79.22) | 13.91 (51.77) | 26.92 (66.42) | 40.38 (29.69) | 95.14 (42.43) | 85.27 (43.68) | 98.55 (37.09) |
| AUC 0‐tau (h•ng mL−1) a | 233.1 (27.58) | 475.0 (26.29) | 816.8 (28.39) | 1,644 (11.45) | 3,097 (24.63) | 3,197 (12.21) | 3,137 (42.99) | 14.90 (37.92) | 29.40 (23.81) | 56.36 (48.86) | 122.0 (29.47) | 170.0 (23.85) | 142.0 (19.40) | 203.9 (24.30) |
| t max (h) b | 2.0 | 2.5 | 3.0 | 3.5 | 3.0 | 2.0 | 3.5 | 1.0 | 0.5 | 0.5 | 1.0 | 1.0 | 1.0 | 0.5 |
| t 1/2 (h) a | 9.680 (17.24) | 10.91 (11.66) | 10.40 (12.97) | 10.28 (19.16) | 9.431 (8.94) | 10.11 (7.45) | 9.705 (16.70) | 1.247 (40.98) | 1.212 (35.09) | 1.567 (39.16) | 4.711 (52.68) | 7.549 (93.72) | 10.52 (57.99) | 6.810 (23.98) |
CSF, cerebrospinal fluid; SAD, single‐ascending dose; MAD, multiple‐ascending dose; CV, coefficient of variation; Cmax, maximum concentration; AUC0‐inf, area under the concentration–time curve extrapolated to infinity; AUC0‐tau, area under the concentration–time curve over a dosing interval; tmax, time to maximum observed concentration; t1/2, terminal half‐life.
Mean (CV[%]) values are presented;
median values are presented;
n = 5 due to 1 subject who failed to meet minimum λz requirements for the regression;
n = 2 due to 1 subject who failed to meet minimum λz requirements for the regression;
n = 6 due to 1 subject who failed to meet minimum λz requirements for the regression;
data represent AUC0‐24h;
n = 1 due to 6 subjects who failed to meet minimum λz requirements for the regression;
Cohort 5;
Cohort 6.
TABLE 5.
Urine pharmacokinetic parameters of MGS0008 by treatment: (a) SAD study; (b) MAD study (Day 14)
| (a) SAD study | ||||||
|---|---|---|---|---|---|---|
| Parameter (unit) | TS‐134 treatment | |||||
| Parts A and C | Part B | |||||
| 5 mg fasted (n = 6) | 10 mg fasted (n = 4) | 10 mg fed (n = 3) | 20 mg fasted (n = 3) | 20 mg fed (n = 3) | 10 mg fed (n = 7) | |
| Fe (0–48) (%) | 69.01 (10.23) | 60.61 (36.10) | 49.65 (38.35) | 38.55 (65.52) | 29.38 (52.53) | 55.77 (15.99) |
| CLr (L h−1) | 6.059 (13.09) | 5.390 (32.75) | 6.164 (32.20) | 4.436 (59.49) | 4.238 (55.34) | 6.101 (12.04) |
| (b) MAD study | |||||||
|---|---|---|---|---|---|---|---|
| Parameter (unit) | TS‐134 treatment | ||||||
| 5 mg (n = 8) | 10 mg (n = 8) | 20 mg (n = 8) | 40 mg (n = 8) | 60 mg (n = 8) | 80 mg a (n = 7) | 80 mg b (n = 6) | |
| Fe (0–24) (%) | 48.03 (36.10) | 51.79 (24.95) | 50.42 (20.58) | 48.06 (13.48) | 50.48 (24.14) | 43.69 (36.76) | 37.21 (35.47) |
| CLr (L h−1) | 5.110 (40.10) | 5.291 (23.53) | 6.180 (27.68) | 5.596 (19.06) | 4.722 (24.02) | 5.225 (37.51) | 4.831 (37.14) |
Mean (CV[%]) values are presented. SAD, single‐ascending dose; MAD, multiple‐ascending dose; CV(%), coefficient of variation; Fe(0‐x), percentage of the amount of drug excreted in urine between time 0 and X hours; CLr, renal clearance.
Cohort 5;
Cohort 6.
In the SAD study at 10 mg TS‐134, the CSF concentration of MGS0008 which is proposed to be a surrogate for a concentration in brain interstitial fluid contributing to actions on the mGlu2/3 receptors, showed gradual increase, reaching a Cmax of 1.244 ng mL−1 (coefficient of variation: 38.4%) at 8 hours postdose, and then decreased slower than from plasma, with a t1/2 of 16.33 hours (Figure 4 and Table 4). The CSF‐to‐plasma exposure ratios of MGS0008 were calculated to be 3.66% (Cmax) and 4.94% (AUC), respectively, which is higher than the CSF‐to‐plasma AUC ratio observed in rats (2.8%). 26
FIGURE 4.
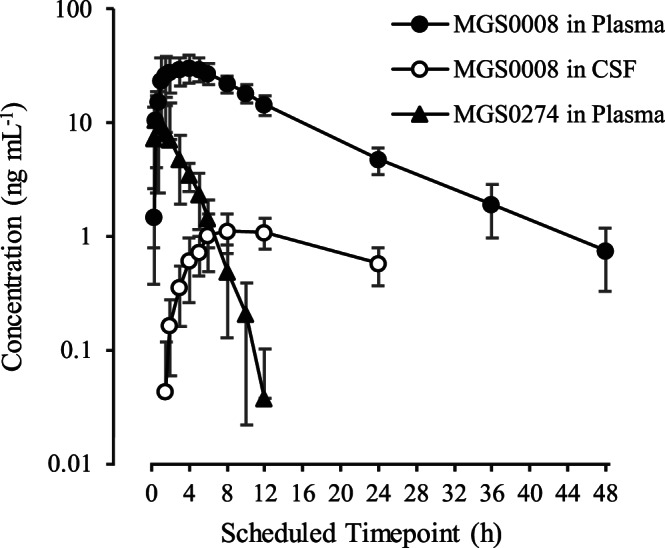
Plasma concentration–time profiles of MGS0008 and MGS0274, and cerebrospinal fluid (CSF) concentration–time profiles of MGS0008 on semi‐log scale following a single oral administration of 10 mg TS‐134 in the fed condition. Figure is presented as the mean ± standard deviation
In the SAD and MAD studies, the urinary excretion rate of MGS0008 ranged from 29.38 to 69.01% of the administered doses across the dose range of 5–80 mg (Table 5), while the urinary excretion rate of MGS0274 was around 1% of the administered doses (data not shown). The renal clearance of MGS0008 ranged from 4.238–6.180 L h−1, which was approximately equal to or less than the human glomerular filtration rate (5.88–7.86 L h−1), 28 suggesting that renal MGS0008 elimination is less likely to be by tubular secretion or reabsorption in humans.
5. DISCUSSION
In this Phase 1 study, we demonstrate that (i) TS‐134 is orally bioavailable in healthy subjects and is converted rapidly and extensively into the active compound (MGS0008) after oral administration; (ii) MGS0008 exhibits good CSF penetration to exert its effect; and (iii) TS‐134 is safe and generally well‐tolerated. These results support that TS‐134 can be an excellent tool to explore therapeutic values in patients in which the hyperglutamatergic state is involved, such as schizophrenia.
We previously reported that MGS0008 is a potent and selective agonist for mGlu2/3 receptor. 24 In the present preclinical study, we first confirmed if MGS0008 improves hyperactivity of the frontal cortex which has been considered as a pathophysiological feature of schizophrenia, 3 , 4 and then we extended therapeutic profiles of MGS0008 for schizophrenia in rodent models. Our preclinical studies indicated that MGS0008 ameliorates abnormalities of the frontal cortical activity caused by NMDA receptor hypofunction in mice. Moreover, our studies showed that MGS0008 exerts antipsychotic‐like effects and improves cognitive impairment in rats at doses that normalize the frontal cortical abnormalities. Thus, MGS0008 exerts the effects by normalizing abnormal activity in the frontal cortex caused by NMDA receptor hypofunction, the pathophysiological conditions mimicking schizophrenia.
MGS0008 is a rigid glutamate analogue and was predicted to exhibit poor oral bioavailability in humans due to its highly hydrophilic property, like other orthosteric mGlu2/3 receptor agonists such as LY354740 and LY404039. 18 , 29 Eli Lilly previously designed amino acid prodrugs to be absorbed via intestinal peptide transporter 1 for two mGlu2/3 receptor agonists (LY544344 for LY354740, and LY2140023 for LY404039), 18 , 29 and both prodrugs did indeed improve oral bioavailability of their active compounds in humans. However, as to LY2140023, the plasma exposure of the prodrug was found to be approximately 30% of that of LY404039. 18 Since higher exposure of prodrugs causes insufficient exposure of active compounds and might trigger unexpected toxic effects, we synthesized MGS0274 besylate, an ester‐based lipophilic prodrug, not only to achieve good gastrointestinal absorption but also to further reduce the systemic exposure to the prodrug itself in humans. 26
In this study, we confirmed that MGS0274 is rapidly absorbed and extensively converted into MGS0008 after dosing of TS‐134 in humans, and that the Cmax and AUC of MGS0008 at steady state increased dose‐proportionally over the dose range tested (5–80 mg). The plasma exposure (AUC) of MGS0274 after single and multiple oral administrations of TS‐134 was much lower than that of MGS0008 in all the treatment groups, approximately 3% of MGS0008 AUC in a molar ratio; that is, almost all the systemic exposure can be accounted for by MGS0008. Note that comparison of exposure was made based on molar concentrations of MGS0008 and MGS0274, of which molecular weights are 203.17 and 429.49, respectively. The urinary excretion of MGS0008 was ranging from 29.38–69.01% of the administered dose without relation to the dose and dosing days (1% for MGS0274), demonstrating that gastrointestinal absorption rate of the prodrug was approximately ≥30%. These results showed that MGS0274 besylate has a desirable characteristic as a prodrug, that is, it is orally well‐absorbed and hydrolysed rapidly and sufficiently to MGS0008. Notably, our prodrug strategy is better than that used for another mGlu2/3 receptor agonist, LY2140023, in terms of avoiding unnecessary exposure of prodrug to patients. For reference, plasma levels of LY2979165, an amino acid prodrug of the mGlu2 receptor‐preferring agonist LY2812223, 30 were reported to be <1% of its active compound. 31
The mean CSF Cmax of MGS0008 after administration of 10 mg TS‐134 under fed condition was 1.244 ng mL−1 (6.12 nM), which is approximately 1/5 of the in vitro EC50 value for mGlu2 receptor (29.4 nM) and 1/7 of that of mGlu3 receptor (45.4 nM). 24 Due to lack of an appropriate tracer for positron emission tomography to determine receptor occupancy, the concentration of the active compound in the CSF would give important information for dose determination for mGlu2/3 receptor agonists. Indeed, it has been presumed that CSF exposure achieving the EC50 value for the mGlu2 receptor would be needed to exert their effects in the case of LY544344 for generalized anxiety disorder 32 and LY2140023 for schizophrenia. 18 Considering that the EC50 value was higher in our assay system (EC50 of LY354740 for mGlu2 receptor is 18.3 nM) 24 than that used by Eli Lilly (EC50 of LY354740 is 5.1 nM), 33 CSF exposure estimated in this study may approximate the EC50 value of MGS0008 for the mGlu2 receptor, which may achieve sufficient activation of the mGlu2/3 receptor in the brain. Notably, although CSF concentrations of MGS0008 were measured only at the 10‐mg dose level in humans, the dose‐proportional increase in CSF exposure of MGS0008 was confirmed in rats within the plasma MGS0008 exposure levels that were observed in this clinical study (data not shown); therefore, MGS0008 CSF concentrations in humans are also expected to be dose proportional.
In the SAD study, the most significant and frequent TEAEs limiting the tolerability of oral administrations of TS‐134 under fasted conditions were vomiting and nausea. Tolerability of a single dose TS‐134 solution was observed up to 10 mg. In the 14‐day MAD study, the tolerability was observed up to 80 mg by employing the multiple dose titration schemes under fed conditions, and the incidence of vomiting in the total TS‐134 group decreased from 31.6% (SAD, Parts A and C, fasted) to 6.4% (MAD). It is important to note that all vomiting and nausea in the MAD study were mild in intensity and there was no apparent dose‐related increase in these TEAEs as was seen in the SAD study. The mechanism of vomiting and nausea that occurred with TS‐134 is currently not clear. Overcoming TEAEs related to initial exposure to the drug seem to be the key to improve tolerance of TS‐134, as the dose increases by titrations in the MAD study showed no dose‐dependent increases in incidents of nausea and vomiting, even when titrated up to 80 mg. The results of these studies showed that implementation of careful titration schemes and dose administrations under fed conditions allow better management of vomiting and nausea; thus, improving the tolerability profile of TS‐134.
In conclusion, oral administrations of TS‐134, prodrug MGS0274 besylate, up to 10 mg as single dose under fasted conditions and up to 80 mg as multiple dose titration under fed conditions were safe and well‐tolerated. Oral administration of TS‐134 in these studies proved a successful prodrug strategy in humans, where the prodrug, MGS0274, was rapidly converted into its active compound, MGS0008, which in turn showed good CSF penetration to act on presumed drug targets.
CLINICAL TRIAL REGISTRATION
Clinical Trials.gov, NCT03746067, NCT03742791.
COMPETING INTERESTS
M. W. and B. M. are employees of the study sponsor, Taisho Pharmaceutical R&D Inc. K. K., M. F., H. H., S. C., S. O., S. Y. are employees of Taisho Pharmaceutical Co., Ltd. Hakop Gevorkyan is an employee of California Clinical Trials Medical Group and was Principal Investigator of the clinical studies.
CONTRIBUTORS
Clinical study design: M.W., B.M., S.Y. Clinical study conduct: M.W., B.M., H.G. Pharmacology study: H.H., S.C., S.O. Pharmacokinetic study: K.K., M.F. Drafting manuscript: M.W., K.K., S.C., S.Y. Editing manuscript: B.M., M.F., H.H., S.O.
Supporting information
FIGURE S1 Effect of MGS0008 on (A) spontaneous locomotor activity and (B) the duration of catalepsy in rats. Data represent mean ± standard error of the mean, n = 8 animals per each group. **P < .01 vs the Vehicle‐ or Vehicle B‐treated group (Steel's test or Wilcoxon's test). Vehicle A: distilled water, Vehicle B: 0.5 w/v% methylcellulose.
TABLE S1 Assessment of dose proportionality on plasma MGS0008 and MGS0274 at steady state in the multiple‐ascending dose study
TABLE S2 Assessment of the food effect on plasma MGS0008 and MGS0274 in the single‐ascending dose study
ACKNOWLEDGEMENTS
The authors wish to thank all subjects who participated in the studies, and internal and external project teams for their services. The nonclinical studies were funded by Taisho Pharmaceutical Co., Ltd. and the clinical studies were funded by Taisho Pharmaceutical R&D Inc.
Watanabe M, Marcy B, Kinoshita K, et al. Safety and pharmacokinetic profiles of MGS0274 besylate (TS‐134), a novel metabotropic glutamate 2/3 receptor agonist prodrug, in healthy subjects. Br J Clin Pharmacol. 2020;86:2286–2301. 10.1111/bcp.14331
The authors confirm that the PI for this paper is Hakop Gevorkyan and that he had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Li CT, Yang KC, Lin WC. Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies. Front Psych. 2019;9:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7(5):426‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lewis DA, González‐Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33(1):141‐165. [DOI] [PubMed] [Google Scholar]
- 4. Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35(1):57‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abi‐Saab WM, D'Souza DC, Moghaddam B, Krystal JH. The NMDA antagonist model for schizophrenia: promise and pitfalls. Pharmacopsychiatry. 1998;31(Suppl 2):104‐109. [DOI] [PubMed] [Google Scholar]
- 6. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301‐1308. [DOI] [PubMed] [Google Scholar]
- 7. Balu DT, Coyle JT. The NMDA receptor 'glycine modulatory site' in schizophrenia: D‐serine, glycine, and beyond. Curr Opin Pharmacol. 2015;20:109‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krystal JH, D'Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl). 2003;169(3‐4):215‐233. [DOI] [PubMed] [Google Scholar]
- 9. Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258(5082):597‐603. [DOI] [PubMed] [Google Scholar]
- 10. Chaki S. Group II metabotropic glutamate receptor agonists as a potential drug for schizophrenia. Eur J Pharmacol. 2010;639(1‐3):59‐66. [DOI] [PubMed] [Google Scholar]
- 11. Chaki S, Hikichi H. Targeting of metabotropic glutamate receptors for the treatment of schizophrenia. Curr Pharm des. 2011;17(2):94‐102. [DOI] [PubMed] [Google Scholar]
- 12. Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N‐methyl‐D‐aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117(3):697‐706. [DOI] [PubMed] [Google Scholar]
- 13. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349‐1352. [DOI] [PubMed] [Google Scholar]
- 14. Gozzi A, Large CH, Schwarz A, Bertani S, Crestan V, Bifone A. Differential effects of antipsychotic and glutamatergic agents on the phMRI response to phencyclidine. Neuropsychopharmacology. 2008;33(7):1690‐1703. [DOI] [PubMed] [Google Scholar]
- 15. Fell MJ, McKinzie DL, Monn JA, Svensson KA. Group II metabotropic glutamate receptor agonists and positive allosteric modulators as novel treatments for schizophrenia. Neuropharmacology. 2012;62(3):1473‐1483. [DOI] [PubMed] [Google Scholar]
- 16. Hikichi H, Kaku A, Karasawa J, Chaki S. Stimulation of metabotropic glutamate (mGlu) 2 receptor and blockade of mGlu1 receptor improve social memory impairment elicited by MK‐801 in rats. J Pharmacol Sci. 2013;122(1):10‐16. [DOI] [PubMed] [Google Scholar]
- 17. Rorick‐Kehn LM, Johnson BG, Burkey JL, et al. Pharmacological and pharmacokinetic properties of a structurally novel, potent, and selective metabotropic glutamate 2/3 receptor agonist: in vitro characterization of agonist (−)‐(1R,4S,5S,6S)‐4‐amino‐2‐sulfonylbicyclo[3.1.0]‐hexane‐4,6‐dicarboxylic acid (LY404039). J Pharmacol Exp Ther. 2007;321(1):308‐317. [DOI] [PubMed] [Google Scholar]
- 18. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med. 2007;13(9):1102‐1107. [DOI] [PubMed] [Google Scholar]
- 19. Downing AM, Kinon BJ, Millen BA, et al. A double‐blind, placebo‐controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry. 2014;14:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kinon BJ, Zhang L, Millen BA, et al. A multicenter, inpatient, phase 2, double‐blind, placebo‐controlled dose‐ranging study of LY2140023 monohydrate in patients with DSM‐IV schizophrenia. J Clin Psychopharmacol. 2011;31(3):349‐355. [DOI] [PubMed] [Google Scholar]
- 21. Stauffer VL, Millen BA, Andersen S, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr Res. 2013;150(2‐3):434‐441. [DOI] [PubMed] [Google Scholar]
- 22. Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry. 2015;78(11):754‐762. [DOI] [PubMed] [Google Scholar]
- 23. Mehta MA, Schmechtig A, Kotoula V, et al. Group II metabotropic glutamate receptor agonist prodrugs LY2979165 and LY2140023 attenuate the functional imaging response to ketamine in healthy subjects. Psychopharmacology (Berl). 2018;235(7):1875‐1886. [DOI] [PubMed] [Google Scholar]
- 24. Nakazato A, Kumagai T, Sakagami K, et al. Synthesis, SARs, and pharmacological characterization of 2‐amino‐3 or 6‐fluorobicyclo[3.1.0]hexane‐2,6‐dicarboxylic acid derivatives as potent, selective, and orally active group II metabotropic glutamate receptor agonists. J Med Chem. 2000;43(25):4893‐4909. [DOI] [PubMed] [Google Scholar]
- 25. Takamori K, Hirota S, Chaki S, Tanaka M. Antipsychotic action of selective group II metabotropic glutamate receptor agonist MGS0008 and MGS0028 on conditioned avoidance responses in the rat. Life Sci. 2003;73(13):1721‐1728. [DOI] [PubMed] [Google Scholar]
- 26. Kinoshita K, Ochi M, Iwata K, Fukasawa M, Yamaguchi J. Preclinical disposition of MGS0274 besylate, a prodrug of a potent group II metabotropic glutamate receptor agonist MGS0008 for the treatment of schizophrenia. Pharmacol Res Perspect. 2019;7(5):e00520 10.1002/prp2.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexander SP, Christopoulos A, Davenport AP, et al. The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol. 2017;174:S17‐S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koopman MG, Koomen GCM, van Acker BAC, Arisz L. Circadian rhythm in glomerular transport of macromolecules through large pores and shunt pathway. Kidney Int. 1996;49(5):1242‐1249. [DOI] [PubMed] [Google Scholar]
- 29. Bueno AB, Collado I, de Dios A, et al. Dipeptides as effective prodrugs of the unnatural amino acid (+)‐2‐aminobicyclo[3.1.0]hexane‐2,6‐dicarboxylic acid (LY354740), a selective group II metabotropic glutamate receptor agonist. J Med Chem. 2005;48(16):5305‐5320. [DOI] [PubMed] [Google Scholar]
- 30. Monn JA, Prieto L, Taboada L, et al. Synthesis and pharmacological characterization of C4‐(thiotriazolyl)‐substituted‐2‐aminobicyclo[3.1.0] hexane‐2,6‐dicarboxylates. Identification of (1R,2S,4R,5R,6R)‐2‐amino‐4‐(1H‐1,2,4‐ triazol‐3‐ylsulfanyl)bicyclo[3.1.0]hexane‐2,6‐dicarboxylic acid (LY2812223), a highly potent, functionally selective mGlu2 receptor agonist. J Med Chem. 2015;58:7526‐7548. [DOI] [PubMed] [Google Scholar]
- 31. McColm J, Brittain C, Suriyapperuma S, et al. Evaluation of single and multiple doses of a novel mGlu2 agonist, a potential antipsychotic therapy, in healthy subjects. Br J Clin Pharmacol. 2017;83(8):1654‐1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD. Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology. 2008;33(7):1603‐1610. [DOI] [PubMed] [Google Scholar]
- 33. Schoepp DD, Johnson BG, Wright RA, et al. LY354740 is a potent and highly selective group II metabotropic glutamate receptor agonist in cells expressing human glutamate receptors. Neuropharmacology. 1997;36(1):1‐11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Effect of MGS0008 on (A) spontaneous locomotor activity and (B) the duration of catalepsy in rats. Data represent mean ± standard error of the mean, n = 8 animals per each group. **P < .01 vs the Vehicle‐ or Vehicle B‐treated group (Steel's test or Wilcoxon's test). Vehicle A: distilled water, Vehicle B: 0.5 w/v% methylcellulose.
TABLE S1 Assessment of dose proportionality on plasma MGS0008 and MGS0274 at steady state in the multiple‐ascending dose study
TABLE S2 Assessment of the food effect on plasma MGS0008 and MGS0274 in the single‐ascending dose study
Data Availability Statement
Research data are not shared.