

Abstract
Following its introduction as an antituberculosis agent close to 75 years ago, the use of para‐aminosalicylic acid (PAS) has been limited by gastrointestinal intolerance and multiple formulations were produced in attempts to reduce its occurrence. More recently, an enteric‐coated, granular, slow‐release PAS formulation (PASER) was introduced and is now in wide‐spread use for the treatment of drug‐resistant tuberculosis. The current PASER dosing regimen is based on recommendations derived from older studies using a variety of different PAS formulations and relegate PAS to a role as an exclusively bacteriostatic agent. However, there is ample evidence that if sufficiently high serum concentrations are reached, PAS can be bactericidal and that intolerance following once daily dosing, that aids the achievement of such concentrations, is no worse than that following intermittent daily dosing. In particular, prevention of resistance to companion drugs appears to be dependent on the size of the single dose, and hence the peak concentrations, and not on maintaining serum levels consistently above minimum inhibitory concentration. We present a narrative review of the development of PAS formulations, dosing practices, and published data regarding pharmacokinetics and pharmacodynamics and the relationship of PAS dosage to intolerance and efficacy. Our conclusions suggests that we are at present not using PAS to its maximum ability to contribute to regimen efficacy and protect companion drugs.
Keywords: efficacy, intolerance, para‐aminosalicylic acid, pharmacokinetics
1. INTRODUCTION
In 1943, Jorgen Lehmann proposed that para‐aminosalicylic acid (PAS) might have antituberculosis activity 1 , 2 , 3 , 4 ; and, by 1944, PAS was successfully used in Sweden to treat pulmonary tuberculosis (PTB) patients. 5
At almost the same time, streptomycin (SM) was discovered and its activity studied in the USA by Schatz, Bugie and Waksman and other clinicians 6 , 7 , 8 in parallel to a series of randomized controlled studies conducted by the British Medical Research Council (BMRC) between 1948 and 1952. 9 , 10 , 11 , 12 Although initially significant improvement in the condition of PTB patients was documented following SM monotherapy, SM resistance emerged within the first months of treatment, and, by 3 months, 85% (35/41) of the patients remaining sputum culture‐positive were producing viable bacilli resistant to SM. 13 The combination of sodium PAS (NaPAS) with SM and, later isoniazid (INH), 9 , 11 , 12 , 14 , 15 inhibited the emergence of resistance to SM and INH and created an efficacious antituberculosis regimen that was the backbone of TB chemotherapy for approximately 30 years. PAS disappeared from many pharmacies after ethambutol was found to be effective and better tolerated. 16 , 17
The emergence of human immunodeficiency virus infection, however, and the concomitant multidrug‐resistant (MDR) and extensively drug‐resistant (XDR) TB epidemics, led to a renewed interest for PAS to protect companion drugs within regimens comprised of second‐ and third‐line agents. Amongst the formulations introduced in response to this need was an enteric‐coated PAS formulation, PASER (Jacobus Pharmaceuticals, Princeton, NJ, USA). Assuming that PAS is essentially a bacteriostatic agent, PASER was designed to provide a slow‐release of PAS allowing a prolonged period of PAS concentrations above the minimum inhibitory concentration (MIC) of Mycobacterium tuberculosis of 1–2 μg/mL when given in divided daily doses, 18 , 19 , 20 , 21 , 22 , 23 and with less intolerance. This formulation is now widely used to manage certain forms of drug‐resistant TB. 21 , 22 , 24 , 25
Other older antituberculosis drugs such as rifampicin are now undergoing re‐evaluation using modern techniques and parent molecules 26 , 27 ; the recommendations for PAS dosing are seldom questioned and have not been reassessed by methodologies that might be now considered routine, such as determination of early bactericidal activity (EBA), evaluation of the maximum tolerated dose or the relationship between dosage, pharmacokinetics (PK), activity and intolerance. The knowledge from older studies regarding these aspects of PAS use is commonly ignored as is the fact that the introduction of PASER requires old dogmas to be challenged, because its PK is very different from the formulations used when the principles of PAS usage were established. Irrespective of the formulation used, it could be argued that PAS therapy with PASER should aim at providing exposures similar to those achieved by the most successful NaPAS treatments used in the BMRC studies. 9 , 11 , 12 , 14 , 15
In 2018, the World Health Organization revised its treatment recommendation on MDR‐TB, partly based on a recent meta‐analysis investigating the relationship of individual antituberculosis agents with treatment success and death. 28 , 29 , 30 This meta‐analysis found PAS to contribute little or nothing to treatment success in patients with MDR‐TB susceptible to PAS, 28 but it should be noted that those patients with MDR‐TB isolates resistant to PAS had worse outcome. 28 Other reports, however, do provide a more positive picture of the value of PAS in managing drug‐resistant TB patients. 31 This emphasizes the need to re‐examine all existing data on PAS to determine if we are currently using PASER optimally.
In this paper, we expand our previous review of the development of PAS regarding formulations, dosing practices and their relationship to the prevention of resistance in companion drugs and intolerance 22 and review the PK of PAS in greater depth and in particular regarding the lack of relationship of PAS concentrations to intolerance. We also present previously unappreciated evidence that PAS may well have some bactericidal efficacy.
2. METHODS
We conducted a literature search in PubMed to identify articles on the PK, pharmacodynamics (PD), safety, and tolerability of PAS. The search terms used in various combinations were: “Aminosalicylic Acid”[MeSH], “para‐aminosalicylic acid”, “PAS”, “efficacy”, “intolerance”, “dosage”, “dose”, “intravenous”, “PK”, “PD”, “PK PD”, “Pharmacokinetic*”, “Pharmacokinetics”[MeSH], “Pharmacodynamic*”, “Pharmacology”[MeSH], “Tuberculosis”[MeSH], “Tubercul*”. The identified articles were screened by title and abstract. Additional articles were identified from referenced articles and related citations in PubMed. Data on PAS PK/PD including intolerance were assessed with particular emphasis on comparison between single‐daily doses vs the same dose but in divided smaller doses; we also noted the PK and intolerance related to intravenous administration of NaPAS. R version 3.5.1 and WebPlotDigitizer version 4.2 were used to reconstruct figures from 2 studies. 32 , 33 , 34 , 35
3. RESULTS AND DISCUSSION
3.1. PAS formulations
In the face of severe intolerance noted soon after its introduction various formulations of PAS were manufactured, including PAS acid and various PAS salts such as NaPAS, potassium PAS (KPAS), and calcium PAS (CaPAS). 22 , 36 Several granular and enteric coated formulations also became available in an attempt to reduce the gastrointestinal intolerance to PAS but were often associated with lower peak concentrations (Cmax) and total exposure (area under the curve, AUC) compared to PAS salts. 22 , 37 , 38 , 39 Similarly, the PASER formulation of PAS was designed to be better tolerated than earlier formulations and does appear to cause less gastrointestinal intolerance. 20 , 21 , 22 , 23 , 40 , 41 Multiple other PAS formulations have been reported in the literature, few of which have less gastrointestinal intolerance when compared to NaPAS. 22 , 37 , 38 , 39 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51
3.2. The dose of PAS
Several PAS dosing regimens use oral and intravenous routes of administration, ranging from once daily to multiple daily doses.
3.2.1. Intravenous dosing regimens
As early as the 1950s, intravenous NaPAS at 24–25 g was utilized in patient care. 3 , 52
3.2.2. Oral dosing regimens
PAS in multiple daily doses has been in use from its first introduction into clinical use. 5 , 9 , 11 , 14 , 15 , 53 The dosage of NaPAS in the earliest studies was 20 g/dadministered orally in 4 divided 5‐g doses. 9 , 11 Subsequent studies evaluated lower doses including NaPAS dosages of 5 g, and 10 g/d administered orally in 4 divided 1.25‐ and 2.5‐g doses, respectively. 11 In a later BMRC study, NaPAS dose of 10 g/d was administered orally but in 2 divided 5‐g doses. 14 Other PAS regimens investigated in clinical studies are 3.3 g twice or thrice daily or 6.6 g twice daily, and 4 g PASER twice daily. 18 , 21 , 40 , 54
Various researchers have studied single daily PAS administration. The doses include PASER at 6 g single dose and 8 g once daily 21 , 23 , 41 ; NaPAS at 15 and 17 g once daily 39 , 55 ; and, Neopasalate 12 g once daily. 47
For use in the clinic, current recommendation is for PAS to be used as 4 g twice or thrice daily. 24 , 56 Similarly, the World Health Organization recommends 8–12 g/d in 2 or 3 divided doses. 30
It is important to note that 1 g of PAS acid is equivalent to 1.19 g of NaPAS, 1.37 g of crystalline NaPAS and 1.12 g of CaPAS. 3 To complicate matters, these conversion factors could vary between formulations of the same generic. For example, 1 g of PAS acid is equivalent to 1.43–1.7 g of NaPAS, 44 , 45 1.12–1.54 g of CaPAS and 1.54 g of KPAS. 3 , 44
3.3. PAS PK
The absorption of PAS salts including NaPAS and KPAS is rapid and complete following oral administration, which usually produces higher PAS concentrations than a PAS acid formulation. 44 PAS acid is poorly soluble in acidic environments, tends to be slowly released while still in the stomach, and is therefore readily acetylated during first‐pass metabolism. 22 Compared to the PAS acid, NaPAS, KPAS and CaPAS formulations are more water soluble, and more easily absorbed and more easily saturate the N‐acetyltransferase‐1 (NAT1) acetylation capacity of the gut and liver. 22 , 44 , 51
PASER administration with food results in 1.5 and 1.7‐fold higher PAS Cmax and AUC from time zero to infinity, respectively, compared to its administration when fasting. 41 In addition to the better absorption when given with food, intolerance to PASER might be less. 41 The plasma protein binding of PAS ranges between 50 and 73%. 2 , 22 , 57 PAS distribution to various sites of disease was shown in animal studies to depend on how high the concentrations were in the blood. 51 , 57
The elimination half‐life of PAS varies from about 0.5 to 2.5 hours depending on the PAS formulation and administration with or without food or antacid. 21 , 24 , 41 , 45 Following oral administration, PAS is metabolised in the gut to acetyl‐PAS, and in the liver to both acetyl‐PAS and glycine‐PAS. 18 , 22 , 51 About 80–90% of an administered dose is excreted in urine following glomerular filtration and tubular secretion as PAS, glycine‐PAS and acetyl‐PAS. 2 , 18 , 22 , 44 , 45 , 51 , 58 Although, previously considered monomorphic, NAT1 is now proven to be polymorphic just like N‐acetyl transferase‐2 (NAT2). 23 , 59 , 60 A South African study in patients with MDR‐ or XDR‐TB, found NAT1*4/*10 genotype in 44%, NAT1*14A genotype in 6%, NAT1*10/*3 genotype in 3%, and heterozygotes or homozygotes NAT2*5 genotype in 37% of the study population, all of which are slow acetylators. Whereas, 66% of the study population were heterozygotes or homozygotes for the rapid acetylator NAT2*4 genotype. 23 A population PK modelling that included NAT1 and NAT2 alleles as covariates, found NAT1*3, NAT1*14 and NAT2*5 alleles to result in significant reduction in the oral clearance of PAS by 17, 14 and 27%, respectively. 23
It is important to note that more PAS is inactivated when administered in repeated small doses in comparison to same daily dose administered as a single large dose. Lehmann in a 1969 paper, reported the PK of a microgranulate PAS formulation (PASolac, A/B Ferrosan) that was designed to be rapidly absorbed. 51 The microgranulate was administered orally either as 4 g thrice daily or as a single 12‐g dose. 51 This study showed the excretion of acetyl‐PAS was larger with repeated small doses (4 g × 3) than with a single large dose (12 g × 1). 51 This is probably because at high PAS concentrations, acetyl coenzyme A depletion contributes to the suppressed transformation to acetyl‐PAS. 22 , 38 , 51
Some PK parameters of various PAS formulations are provided in Table 1 and it is clear that the plasma PAS concentrations achieved following administration of different PAS formulations vary greatly. Furthermore, the considerable interindividual and interoccasion variability found with different PAS formulations contributes to the challenges of using PAS. 21 , 23 , 37 , 43 , 44 , 45
TABLE 1.
Para‐aminosalicylic acid pharmacokinetics
| Study | PAS formulation | Route of administration | Dose/dosing frequency | Cmax (μg/mL) | AUC (h*μg/mL) | Tmax (h) | Half‐life (h) |
|---|---|---|---|---|---|---|---|
| Hollander 1955 46 | PAS‐resin complex | Oral | 8 g thrice daily | 78–85 | |||
| Hollander 1955 46 | PAS‐resin complex | Oral | 16 g# | 180 | |||
| Riska 1959 61 | NaPAS granules | Oral | 19 g once daily | 277 | |||
| Riska 1959 61 | NaPAS granules | Oral | 22 g once daily | 284 | |||
| Riska 1959 61 | NaPAS granules | Intravenous | 24 g once daily | 323 | |||
| Riska 1962 43 | NaPAS granules | Oral | 16‐g single‐dose | 240 | |||
| Riska 1962 43 | NaPAS granules | Oral | 4 g 4 times daily | 60–80 | |||
| Riska 1962 43 | PAS (free acid) | Intravenous | 16‐g single‐dose over 1 h infusion | 450 | |||
| Riska 1962 43 | PAS (free acid) | Intravenous | 16‐g single‐dose over 2 h infusion | 350 | |||
| Riska 1962 43 | PAS (free acid) | Intravenous | 16‐g single‐dose over 3 h infusion | 290 | |||
| Yue and Cohen 1966 47 | Neopasalate | Oral | 6 g once daily | 108.9 | 2 | ||
| Yue and Cohen 1966 47 | NaPAS | Oral | 6 g once daily | 136.2 | 2 | ||
| Yue and Cohen 1966 47 | Neopasalate | Oral | 12 g once daily | 169.8 | 2 | ||
| Yue and Cohen 1966 47 | NaPAS | Oral | 12 g once daily | 153.3 | 2 | ||
| Wan et al. 1974 44 | PAS acid | Oral | 4‐g single‐dose | 50 | 209 | 3.54 | 0.94 |
| Wan et al. 1974 44 | NaPAS | Oral | 4‐g single‐dose | 155 | 313 | 0.83 | 0.91 |
| Wan et al. 1974 44 | CaPAS | Oral | 4‐g single‐dose | 140 | 327 | 1.02 | 0.91 |
| Wan et al. 1974 44 | KPAS | Oral | 4‐g single‐dose | 121 | 313 | 1.1 | 0.96 |
| Peloquin et al. 1994 18 | PASER | Oral | 4‐g single‐dose | 20.23 (8.8) a | 107.92 (52.45) a , b | 7.95 (6.12) a | 1.62 (0.85) a |
| Peloquin et al. 2001 41 | PASER | Oral, fasting | 6‐g single‐dose | 21.4 (11.4–79.3) c | 140 (20.8–255) b , c | 4.43 (2.09–6.64) c | 1.88 (1.21–3.91) c |
| Peloquin et al. 2001 41 | PASER | Oral, food | 6‐g single‐dose | 32.5 (6.83–72.3)c | 240 (71.1–472) b , c | 6.56 (3.57–8.97) c | 1.85 (1.10–6.0) c |
| Peloquin et al. 2001 41 | PASER | Oral, orange juice | 6‐g single‐dose | 24.7 (6.93–36.4) c | 175 (29.2–295) b , c | 4.57 (3.43–11.1) c | 2.53 (0.95–4.09) c |
| Peloquin et al. 2001 41 | PASER | Oral, antacids | 6‐g single‐dose | 18.4 (5.22–46.0) c | 124 (55.9–227) b , c | 5.25 (2.7–13.0) c | 2.1 (0.94–3.39) c |
| Liwa et al. 2013 40 | PASER | Oral, acidic food or beverage | 4 g twice daily | 51.3 (20.0) a | 368 (194.0) a , d | 5.2 (2.04)a | |
| Sy et al. 2015 23 | PASER | Oral, acidic food or beverage | 4 g twice daily | 61 (10–112) c | 428 (119–934) c , d | 4.0 (0.0–12.0)c | |
| Sy et al. 2015 23 | PASER | Oral, acidic food or beverage | 8 g once daily | 80 (21–135) c | 652 (161–1055)c,d | 8 (3.0–121.1)c |
All pharmacokinetics parameter values are given as mean unless otherwise stated. Para‐aminosalicylic acid (PAS), sodium PAS (NaPAS), calcium PAS (CaPAS), potassium PAS (KPAS), para‐aminosalicylic acid buffer complex (Neopasalate), granular slow‐release PAS formulation (PASER), peak plasma concentration (Cmax), time to Cmax (Tmax)
mean (standard deviation).
area under the curve from time zero to infinity.
median (range).
area under the curve from 0 to 12 hours.
Dosing frequency is unclear.
PAS can be a subject of drug interactions at both PK and PD levels. PAS absorption could be interfered with by antacid such as aluminium hydroxide because of its adsorption effect, 58 a later study, however, showed negligible effect on overall PAS exposure when PASER formulation was administered with a combination of aluminium hydroxide and magnesium hydroxide/simethicone. 41 In addition, digoxin could reduce PAS absorption and vice versa. 62 , 63 PAS, by competing with INH for acetylation possibly through depletion of coenzyme A, could result in elevated INH concentrations. 51 , 58 The oral clearance of PAS is increased by efavirenz. 21 , 23 A recent in vitro study reported PAS to be a substrate of multiple organic and cation transporters. 64 In this study, nonsteroidal anti‐inflammatory drugs such as diclofenac and indomethacin, and proton pump inhibitors such omeprazole inhibit PAS uptake through OAT1 and OAT3 inhibition. 64 Similarly, in vitro study showed metformin inhibits OCT1 and OCT2 mediated PAS uptake. At the PD level, PAS combination with ethionamide/prothionamide could increase the risk of reversible thyroid dysfunction. 62 , 65
3.4. Efficacy
PAS is structurally related to para‐aminobenzoic acid, a substrate for dihydropteroate synthase. 2 , 64 PAS mechanism of action is thought to involve PAS incorporation into the folate biosynthetic pathway by dihydropteroate synthase and dihydrofolate synthase to generate a hydroxyl dihydrofolate antimetabolite that in turn inhibits dihydrofolate reductase. 2 , 64 In addition, PAS is hypothesized to also inhibit synthesis of mycobactin, a mycobacterial cell wall component. 24
3.4.1. Early studies of PAS in the prevention of resistance in companion drugs
There is in vitro evidence of a concentration‐related bacteriostatic effect of PAS; an early study by Singh and Mitchison demonstrated the failure of PAS in combination with either SM or INH to protect these companion drugs against resistance emergence at low PAS concentrations of 10 μg/mL, but that PAS concentrations of 100 μg/mL were effective in preventing emergence of resistant organisms. 66 Mitchison later described this effect attained at higher PAS dosages as bactericidal. 67 Other in vitro studies demonstrated a concentration‐related bacteriostatic effect of PAS. 36 , 51 , 68 , 69 In addition, in vivo guinea pig and mouse TB models also showed a dose and concentration‐related bacteriostatic effect of PAS. 68
In early clinical studies of PAS, SM and INH, all the drugs were given in divided daily dosages as was then the practice with infectious diseases, it being considered necessary to maintain the constant presence of inhibitory concentration of the relevant drug. 9 , 11 , 14 , 15 , 53 The dosage of NaPAS in the earliest studies was 20 g/d but administered in 5‐g doses. 9 , 11 In later studies, NaPAS dosages of 5, 10 and 20 g were evaluated, and a NaPAS dosage of 20 g (5 g × 4) used in combination with SM was more efficacious in the prevention of resistance to SM than 10 g (2.5 g × 4) or 5 g (1.25 g × 4) daily. 11 When used combined with INH, efficacy in prevention of INH resistance was similar whether a daily PAS dosage of 10 g (5 g × 2) or 20 g (5 g × 4) was used. 14 This suggests a dose‐related gradient for PAS efficacy in the prevention of resistance developing in companion drugs, with a 5‐g NaPAS single dose being more efficacious than 1.25‐ or 2.5‐g doses. When discussing these studies Singh and Mitchison suggested that it was possible the doses used rather than the total daily dosage that was responsible for the improved prevention of resistance in companion drugs that accompanied the use of 5 g single doses of PAS. In other words, it is likely that the PAS Cmax rather than just AUC or percentage of time above MIC (%T > MIC) was responsible for suppression of resistance in companion drugs.
3.4.2. Studies documenting the efficacy of PAS monotherapy in reducing sputum acid‐fast bacilli counts
It is important to note that a neglected, blinded randomized, placebo‐controlled early study also evaluated the efficacy of PAS monotherapy in causing a fall in the percentage of patients coughing sputum containing acid‐fast bacilli (AFB) seen on microscopy. In total, 176 patients were randomized to receive either placebo (n = 82) or an enteric‐coated PAS granulate (n = 94). 34 The enteric‐coated PAS granulate was given in 4 daily doses of 5, 2, 2 and 5 g. 34 In comparison to placebo, the percentage of PTB patients with sputum smears positive for AFB fell significantly from 62.2% (standard error ±5.35) to 18.9% (±3.27) during the first 8 weeks of PAS monotherapy; no similar response occurred in patients receiving placebo (Figure 1). 34
FIGURE 1.
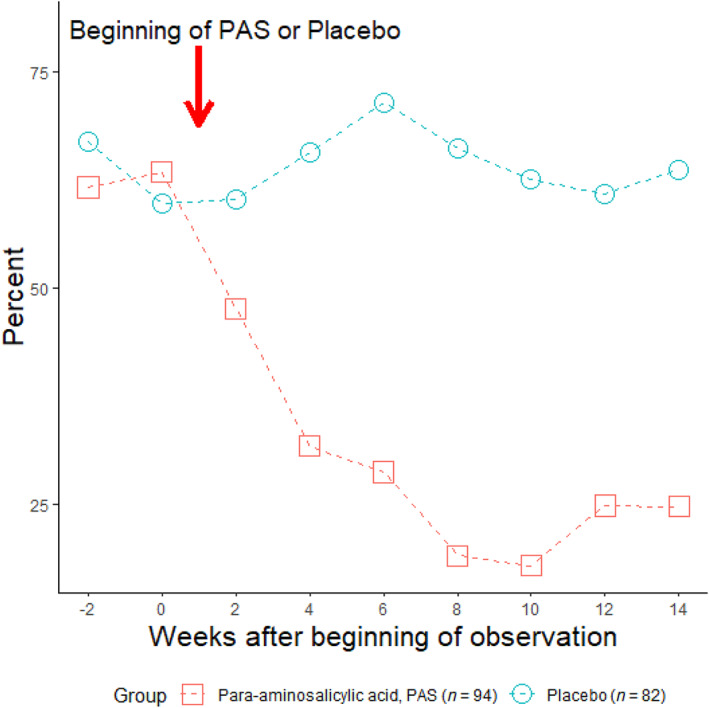
Percentage of patients with sputum smears positive for acid‐fast bacilli after receiving a placebo or enteric‐coated para‐aminosalicylic acid (PAS) granulate given in 4 daily doses of 5, 2, 2, and 5 g (redrawn from 34 )
In 1969, the results of 2 studies were published during which a research group in Berlin assessed the efficacy of the new potential antituberculosis agents, thiocarlide and morphazinamide, in comparison to the established agents such as PAS, isoniazid, rifampicin and ethambutol by measuring the fall in counts of AFB/mL of sputum quantified by Gaffky counts, a recognized manner of quantifying AFB/mL of sputum. 35 The response to treatment was thus assessed in a manner similar to that of today's EBA studies. In the first study, the activity of thiocarlide was compared to that of PAS at a NaPAS dosage of 12 g given orally 3 times daily in 4 g doses. During the second, similar study, morphazinamide, was assessed in comparison to INH, ethionamide and cycloserine and PAS given intravenously 35 and the results are illustrated in Figure 2.
FIGURE 2.
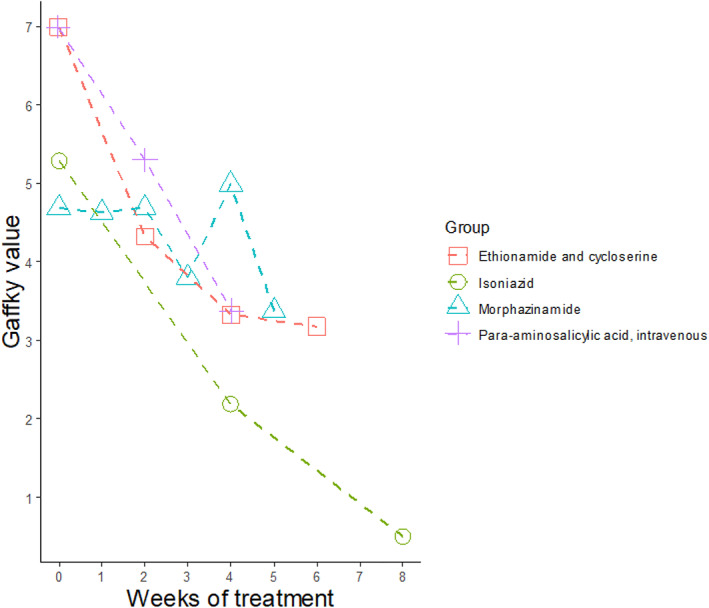
Behaviour of the Gaffky values in response to the treatment of cavitary pulmonary tuberculosis with different therapeutic modalities in the lung clinic Heckeshorn (redrawn from 35 )
Although PAS serum concentrations were not determined in either of the studies, the mean peak concentrations reached in patients during the first study (4 g × 3) would probably have been 50–100 μg/mL. 44 , 45 During both studies, a significant fall in log counts of AFB/mL of sputum in the patients receiving PAS was documented. 35 Remarkably, the fall in AFB in the second study after intravenous PAS, following which PAS concentrations of well over 200–300 μg/mL would probably be reached, matches the fall in AFB counts associated with INH the most bactericidal of our currently available antituberculosis agents.
During the well‐known, first comprehensive study of the EBA of antituberculosis agents carried out in Nairobi by Jindani et al. in 1980, a small group of patients received NaPAS 15 g once daily for 14 days. 55 During the first 2 days (2‐day EBA), a fall in log10 counts of viable colony forming units of 0.259 per mL of sputum per day was found, a value similar to that of 10 mg/kg rifampicin in this same study. 55 Plasma PAS concentrations were not measured in this study; but the mean Cmax achieved was probably close to 190–240 μg/mL, based on the results of investigation by Frostad in few patients that were administered 12 g PAS formulation equivalent to 17 g NaPAS as a single dose. 39
A study by Reisner over a 6‐month period showed similar PAS efficacy was achieved irrespective of PAS administration as a single daily dose of 6 g, or as a 12‐g daily dose (4 g × 3), in combination with INH. 70 Efficacy in this study was defined as improvement in chest X‐ray abnormalities, cavity closure, and the disappearance of tubercle bacilli from previously positive sputum. 70 Even though plasma PAS concentrations were not measured in this study, one could assume that the Cmax achieved with 6 g is probably higher than with 4 g PAS doses. Indeed, early researchers and clinicians, including Jorgen Lehmann, the discoverer of PAS recommended single daily dosing of PAS, if this could be tolerated. 39 , 43 , 61 , 71
The usual PAS recommendation for divided daily doses is mainly to ensure that its plasma concentrations between dosing intervals are above the MIC of 1–2 μg/mL, 18 , 19 , 20 , 21 , 22 , 54 and to ensure that less intolerance is experienced. This is in keeping with the understanding that %T > MIC is the most important PAS PK/PD parameter of efficacy. 52 However, Charles, in 1955, suggested PAS Cmax and/or its overall systemic exposure were more important determinants of efficacy, and not necessarily %T > MIC. 52 In his study, 25 g NaPAS solution administered as intravenous infusions 3 times a week, produced at least 10‐fold higher PAS blood concentrations compared to oral PAS administration, and he claimed, with a better treatment outcome in the former. 52
Furthermore, the designs of the aforementioned BMRC studies 9 , 11 , 14 suggest that individual PAS doses and, by extension Cmax, might be linked to suppression of resistance emergence. Although, no PAS PK data are available from the studies, 9 , 11 , 14 Citron and Kuper later reported that a mean Cmax of approximately 100 μg/mL was achieved following 5 g oral doses of NaPAS, this being the dose used in the most successful arms of the BMRC studies with the lowest Cmax being 50 μg/mL. 22 , 37 It is reasonable to propose a clinical equipoise; that PAS Cmax of at least 100 μg/mL may be desirable to achieve suppression of resistance‐emergence in companion drugs, and that at high doses and by implication high PAS concentrations, bactericidal activity may be achieved as can be inferred from the study by Jindani et al. 55
Studies by Peloquin et al.,18,41 Liwa et al. 40 and Sy et al. 23 provided evidence that much lower plasma PAS Cmax is achieved with PASER (Table 1). Indeed, we found that despite increasing PASER dose to 8 g once daily, the median Cmax was 80 μg/mL. 23 PASER at the currently recommended dosing regimen of 8–12 g/d given in 2 or 3 divided doses of 4 g provides Cmax values considerably lower than those reached with the NaPAS formulation used during the BMRC studies. There is, therefore, an urgent need to reconsider the current dosing of PASER, and to perform a prospective evaluation of its PK/PD using the current methodologies. We propose evaluating the PK/PD of high, once daily administration of PASER, as this facilitates use and simplifies supervision of drug intake.
3.5. PAS intolerance
It is essential to distinguish between the toxicity and gastrointestinal intolerance of PAS. A major problem with intolerance is that it carries the liability of nonadherence and a risk of not completing treatment and following these, treatment failure, relapse and further development of drug resistance.
The frequency and severity of gastrointestinal intolerance including nausea, vomiting and diarrhoea vary with different PAS formulations, dose, route of administration and administration as a single daily dose or in divided daily doses, and, possibly, when taken with or without food. 3 , 9 , 11 , 41 , 47 , 52 In the BMRC studies, the gastrointestinal intolerance to NaPAS administered orally in divided daily doses was reported to occur in 12–58% of patients, with higher NaPAS doses associated with more frequent intolerance. 9 , 11 , 22 By contrast, intolerance to PASER formulation seems to be low following its administration at dosing regimens of 8 g once daily or at 4 g once, twice, or thrice daily. 18 , 23 , 72 Peloquin et al. found that <4% of patients reporting intolerance with this formulation. 18 Other studies also reported PASER to be well tolerated by patients. 23 , 72 The better tolerability to PASER could be because of PAS release in the small intestine rather than in the stomach and the reduced production of meta‐aminophenol. 20 , 24 Furthermore, the enteric‐coating of PASER probably prevents direct gastric irritation by PAS. 20
Yue and Cohen reported fewer patients having gastrointestinal symptoms when a Neopasalate PAS formulation was administered orally as a 12‐g single daily dosage than following the same total daily dosage, but in divided doses. 47 Gastrointestinal symptoms were reported in 6.9% (5/72), 10.7% (3/28) and 4.5% (1/22) of patients administered oral Neopasalate at 4 g thrice daily, 6 g twice daily and 12 g once daily, respectively. 47 Similarly, gastrointestinal symptoms were reported in 14.8% (4/27), 12.5% (3/24), and 10.5% (2/19) of patients administered oral NaPAS at 4 g thrice daily, 6 g twice daily and 12 g once daily, respectively. 47 Importantly, a single daily dose of 12 g Neopasalate/NaPAS compared to the same total daily dosage in 2 or 3 divided doses did result in similar sputum culture conversion rate, despite failure to maintain PAS concentrations above 1–2 μg/mL throughout the dosing interval in those receiving single daily doses. 47 Similar observations were made by other investigators using various PAS formulations. 23 , 73
Of further interest, the Yue and Cohen study provided evidence that intolerance to PAS is formulation related. Gastrointestinal symptoms were seen in 7.4% (9/112) of patients on Neopasalate, 12.3% (18/146) on Rezipas, 19.3% (23/119) on Phenyl PAS, 20.8% (35/168) on NaPAS, 24.8% (25/101) on CaPAS and in 50.9% (27/53) of patients on KPAS. 47
Also in the study by Yue and Cohen, a mean Cmax of 169.8 μg/mL was reached in patients administered 12 g once daily Neopasalate compared to a mean Cmax of 40.6 μg/mL and 108.9 μg/mL in those administered 4 g thrice daily, and 6 g twice daily, respectively. 47 In spite of the higher Cmax achieved with 12 g once daily, intolerance was lowest at 4.5% (1/22) compared to 6.9% (5/72) and 10.7% (3/28) in the 4 g × 3 (12 g/d) and 6 g × 2 (12 g/d) regimens, respectively. 47 Similarly, Riska also found NaPAS orally administered as a single daily dose rather than divided daily doses was associated with fewer side‐effects and better tolerability, in spite of achieving very high PAS blood concentrations. 43 , 61 Earlier mentioned BMRC studies found an association between PAS dosage and intolerance. However, gastrointestinal intolerance to PAS seems to be independent of its plasma concentrations, as demonstrated in several PAS studies. 3 , 21 , 23 , 40 , 47 , 69 More recently, no association was found between PAS concentration and intolerance when PASER was given as 4 g twice daily or 8 g once daily. 21 , 23 , 40 Adams et al. found no relationship between plasma PAS, acetyl‐PAS or glycine‐PAS concentrations and gastrointestinal intolerance, irrespective of PASER administration as 4 g twice daily or 8 g once daily. 74
In addition, Jones administered 4.8–5.0% solution of crystalline NaPAS (equivalent to 24‐25 g NaPAS), intravenously to TB patients and found it well tolerated with only mild gastrointestinal symptoms seen in about 7.4% (2/27) of patients, despite very high plasma PAS concentrations. 3 Charles, in his 1955 paper, found no gastrointestinal intolerance in any of the 50 patients administered 25 g NaPAS intravenously. 52 In this study, each patient received on average, 50 infusions over a 4‐month period. PAS concentrations reached were not provided, but based on similar dose and formulation used by Riska, the mean (range) PAS Cmax would probably be about 323 μg/mL (190–570). 52 , 61 Therefore, PAS plasma concentrations probably play little role in gastrointestinal intolerance to PAS. Data from intravenous PAS administration show that if gastrointestinal tract is bypassed, intolerance to PAS is minimized; because gastrointestinal intolerance requires the presence of PAS in the gastrointestinal tract. 74
3.6. PAS toxicity
The overall incidence of adverse events (AEs) ascribed to PAS varies from 10 to 30%, 2 and several studies reported PAS to have minimal toxicity even at very high doses, with the most pressing concern being gastrointestinal intolerance. 3 , 21 , 40 , 41 , 42 , 43 , 51 , 61 PAS is associated with reversible hypothyroidism that is amplified when used at high doses and when combined with ethionamide or prothionamide. 3 , 75 Other potentially serious AEs associated with PAS include hepatitis, haemolytic anaemia, granulocytopenia, polyneuritis, psychosis and angioedema. 3 Furthermore, hypersensitivity reactions to PAS are experienced in about 5–10% of patients, but can be managed and PAS can usually be reintroduced. 2
It is important to note that PAS overall appears to be safer than several other antituberculosis agents used in treatment of MDR‐ or XDR‐TB. Potentially serious AEs such as central nervous system and haematologic disorders have been associated with other agents. 75 , 76 Moreover, serious toxicities such as peripheral neuropathy, optic neuritis, and hearing loss following the use of other agents can be irreversible. 75 Thus, if tolerated, PAS appears to have a safety profile comparable, if not better, than that of several other agents used in MDR‐ or XDR‐TB treatment.
4. CONCLUSION
In summary, available evidence summarized above suggests that PAS Cmax, AUC and, by extension, Cmax/MIC and/or AUC/MIC rather than %T > MIC are the more important determinants of efficacy and suppression of resistance development in companion drugs. Data obtained from studies using PASER in healthy volunteers and TB patients suggest that PAS exposures with currently used dosing regimens are lower than those expected to have been achieved in the BMRC studies of the 1950s and that we are not using the PASER formulation optimally. 18 , 21 , 40 , 41 Importantly it appears that PAS at higher dosages that achieve higher blood concentrations has a hitherto unappreciated bactericidal effect that is not currently exploited.
Future PAS investigations should focus on exploring the relationships between PAS dose, PK and pharmacogenetics and the efficacy and tolerability of PAS in its different formulations. The slow‐release formulation of PASER may well offer an acceptable compromise between intolerance and efficacy if dosed once daily. The demonstration of bactericidal activity by PAS, as is suggested by some of the studies summarised above and better knowledge of the relationship of PAS intolerance and efficacy to pharmacokinetic factors and dose might enable the far greater rational use of PAS in community‐based programmes for patients otherwise therapeutically destitute. Resistance to several of the newly introduced drugs for the management of MDR‐ and XDR‐TB has already been reported 77 , 78 ; toxicity to several of the currently recommended agents for managing MDR‐ and XDR‐TB patients is also not infrequent. 75 , 76 PAS remains an attractive orally administered, alternative agent for these patients, but we urgently need to be better informed regarding the most efficacious manner in which to administer PAS and the appropriate doses to use. Should further studies confirm that PAS once daily is acceptable, both as regards efficacy and intolerance, one might envisage a possible regimen where PAS is given once daily in the evening with the evening meal.
4.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
COMPETING INTERESTS
All authors have completed the unified competing interest form, and declare no support from any organisation for the submitted work, and no financial interest to declare. The European and Developing Countries Clinical Trials Partnership (EDCTP) support A.A.A., grant number TRIA.2015.1102. There was no specific funding source for this manuscript.
CONTRIBUTORS
P.R.D., A.H.D. and H.R. developed the study concept. A.A.A., E.M.S., A.H.D. and H.R. designed the study. A.A.A., P.R.D. and K.A. conducted electronic searches and selections of studies. A.A.A., P.R.D. and K.A. conducted data extraction. A.H.D., A.A.A., P.R.D., E.M.S. and H.R. were involved in interpretation of the results; A.A.A. and P.R.D. wrote the first draft of the manuscript. All authors were involved in critical review and revision of the manuscript. All authors approved the final version of the manuscript.
Abulfathi AA, Donald PR, Adams K, Svensson EM, Diacon AH, Reuter H. The pharmacokinetics of para‐aminosalicylic acid and its relationship to efficacy and intolerance. Br J Clin Pharmacol. 2020;86:2123–2132. 10.1111/bcp.14395
REFERENCES
- 1. Vallentin G, Törnell E, Beskow A, et al. PAS in pulmonary tuberculosis. Tubercle. 1950;31(1):2‐10. [DOI] [PubMed] [Google Scholar]
- 2. Brunton LL, Knollmann BC, Hilal‐Dandan R. Goodman & Gilman's the Pharmacological Basis of Therapeutics , 13th Ed. McGraw Hill Medical; 2018. [Google Scholar]
- 3. Jones JS. Intravenous P.a.S. in relation to pulmonary tuberculotherapy, with an account of 1,145 transfusions. Br J Tuberc Dis Chest. 1954;48(4):286‐297. [DOI] [PubMed] [Google Scholar]
- 4. Dubovsky H. Correspondence with a pioneer, Jürgen Lehmann (1898‐1989), producer of the first effective antituberculosis specific. S Afr Med J. 1991;79(1):48‐50. [PubMed] [Google Scholar]
- 5. Lehmann J. Para‐aminosalicylic acid in the treatment of tuberculosis. Lancet. 1946;247(6384):15‐16. [DOI] [PubMed] [Google Scholar]
- 6. Feldman W, Hinshaw H, Mann F. Streptomycin in experimental tuberculosis. Am Rev Tuberc. 1945;52:269‐298. [Google Scholar]
- 7. Schatz A, Bugie E, Waksman S. Streptomycin, a substance exhibiting antibiotic activity against gram‐positive and gram‐negative bacteria. Proc Soc Exp Biol Med. 1944;55(1):66‐69. [DOI] [PubMed] [Google Scholar]
- 8. Hinshaw H, Feldman W, Pfuetze K. Treatment of tuberculosis with streptomycin; a summary of observations on one hundred cases. JAMA. 1946;132(13):778–782. [DOI] [PubMed] [Google Scholar]
- 9. Treatment of pulmonary tuberculosis with streptomycin and Para‐aminosalicylic acid; a Medical Research Council investigation. Br Med J. 1950;2(4688):1073‐1085. [PMC free article] [PubMed] [Google Scholar]
- 10. The chemotherapy of tuberculosis. Br Med J. 1950;2(4688):1102‐1103. [PMC free article] [PubMed] [Google Scholar]
- 11. The prevention of streptomycin resistance by combined chemotherapy; a Medical Research Council investigation. Br Med J. 1952;1:1157‐1162. [PMC free article] [PubMed] [Google Scholar]
- 12. Various combinations of isoniazid with streptomycin or with P.a.S. in the treatment of pulmonary tuberculosis; seventh report to the Medical Research Council by their tuberculosis chemotherapy trials committee. Br Med J. 1955;1(4911):435‐445. [PMC free article] [PubMed] [Google Scholar]
- 13. Streptomycin treatment of pulmonary tuberculosis. Br Med J. 1948;2(4582):769‐782. [PMC free article] [PubMed] [Google Scholar]
- 14. Isoniazid in combination with streptomycin or with P.a.S. in the treatment of pulmonary tuberculosis; fifth report to the Medical Research Council by their tuberculosis chemotherapy trials committee. Br Med J. 1953;2(4844):1005‐1014. [PMC free article] [PubMed] [Google Scholar]
- 15. Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946‐1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999;3(10 Suppl 2):S231‐S279. [PubMed] [Google Scholar]
- 16. Murray JF, Schraufnagel DE, Hopewell PC. Treatment of tuberculosis. A historical perspective. Ann am Thorac Soc. 2015;12(12):1749‐1759. [DOI] [PubMed] [Google Scholar]
- 17. Iseman MD. Tuberculosis therapy: past, present and future. Eur Respir J Suppl. 2002;36(36 suppl):87s‐94s. [DOI] [PubMed] [Google Scholar]
- 18. Peloquin CA, Henshaw TL, Huitt GA, Berning SE, Nitta AT, James GT. Pharmacokinetic evaluation of Para‐aminosalicylic acid granules. Pharmacother J Hum Pharmacol Drug Ther. 1994;14(1):40‐46. [DOI] [PubMed] [Google Scholar]
- 19. Heifets LB. Antituberculosis Drugs: Antimicrobial Activity in Vitro In: Heifets LB, ed. Drug Susceptibility in the Chemotherapy of Mycobacterial Infections; 1991:13‐58. [Google Scholar]
- 20. Kibleur Y, Brochart H, Schaaf HS, Diacon AH, Donald PR. Dose regimen of Para‐aminosalicylic acid gastro‐resistant formulation (PAS‐GR) in multidrug‐resistant tuberculosis. Clin Drug Investig. 2014;34(4):269‐276. [DOI] [PubMed] [Google Scholar]
- 21. de Kock L, Sy SKB, Rosenkranz B, et al. Pharmacokinetics of Para‐aminosalicylic acid in HIV‐uninfected and HIV‐coinfected tuberculosis patients receiving antiretroviral therapy, managed for multidrug‐resistant and extensively drug‐resistant tuberculosis. Antimicrob Agents Chemother. 2014;58(10):6242‐6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Donald PR, Diacon AH. Para‐aminosalicylic acid: the return of an old friend. Lancet Infect Dis. 2015;15(9):1091‐1099. [DOI] [PubMed] [Google Scholar]
- 23. Sy SKB, de Kock L, Diacon AH, et al. N‐acetyltransferase genotypes and the pharmacokinetics and tolerability of Para‐aminosalicylic acid in patients with drug‐resistant pulmonary tuberculosis. Antimicrob Agents Chemother. 2015;59(7):4129‐4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paser (Jacobus Pharmaceutical Company, Inc.) . https://medlibrary.org/lib/rx/meds/paser/. 2010.
- 25. WHO . Companion handbook to the WHO guidelines for the programmatic management of drug‐resistant tuberculosis In: Companion Handb to WHO Guidel Program Manag Drug‐Resistant Tuberc; 2014:1‐20. [PubMed] [Google Scholar]
- 26. Boeree MJ, Diacon AH, Dawson R, et al. A dose‐ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am J Respir Crit Care Med. 2015;191(9):1058‐1065. [DOI] [PubMed] [Google Scholar]
- 27. Diacon AH, Patientia RF, Venter A, et al. Early bactericidal activity of high‐dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob Agents Chemother. 2007;51(8):2994‐2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahmad N, Ahuja SD, Akkerman OW, et al. Treatment correlates of successful outcomes in pulmonary multidrug‐resistant tuberculosis: an individual patient data meta‐analysis. Lancet. 2018;392(10150):821‐834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. WHO . WHO consolidated guidelines on drug‐resistant tuberculosis treatment. Geneva: World Health Organization; World Health Organization; 2019. License: CC BY‐NC‐SA 3.0 IGO. [PubMed] [Google Scholar]
- 30. WHO . WHO consolidated guidelines on drug‐resistant tuberculosis treatment. Geneva: World Health Organization; 2019. Licence: CC BY‐NC‐SA 3.0 IGO. [PubMed] [Google Scholar]
- 31. Desai U, Joshi J. Utility of Para‐aminosalicylic acid in drug‐resistant tuberculosis: should it be classified as group D3 or group C? Lung India. 2018;35(6):488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. R: The R project for statistical computing . https://www.r-project.org/
- 33. Rohatgi A. WebPlotDigitizer version 4.2. https://apps.automeris.io/wpd/. 2019.
- 34. Para‐aminosalicylic acid treatment in pulmonary tuberculosis. Am Rev Tuberc. 1950;61(5):597‐612. [PubMed] [Google Scholar]
- 35. Cooperative controlled trial of thiocarlide (DATC), PAS and bedrest alone in short‐term single‐drug treatment in retreated cavitary pulmonary tuberculosis. Beitr Klin Erforsch Tuberk Lungenkr. 1969;139(2):115‐139. [PubMed] [Google Scholar]
- 36. Bogen E, Loomis RN, Will DW. Para‐aminosalicylic acid treatment of tuberculosis; a review. Am Rev Tuberc. 1950;61(2):226‐246. [DOI] [PubMed] [Google Scholar]
- 37. Citron KM, Kuper SW. Para‐aminosalicylic‐acid (PAS) concentrations in the serum during treatment with various PAS preparations. Tubercle. 1959;40(6):443‐452. [DOI] [PubMed] [Google Scholar]
- 38. Bang HO, Jacobsen LK, Strandgaard E, Yde H. Metabolism of isoniazid and Para‐amino salicylic acid (PAS) in the organism and its therapeutic significance. Acta Tuberc Pneumol Scand. 1962;41:237‐251. [PubMed] [Google Scholar]
- 39. Frostad S. Continued studies in concentrations of Para‐amino‐salicylic acid (PAS) in the blood. Acta Tuberc Pneumol Scand. 1961;41:68‐82. [PubMed] [Google Scholar]
- 40. Liwa AC, Schaaf HS, Rosenkranz B, Seifart HI, Diacon AH, Donald PR. Para‐aminosalicylic acid plasma concentrations in children in comparison with adults after receiving a granular slow‐release preparation. J Trop Pediatr. 2013;59(2):90‐94. [DOI] [PubMed] [Google Scholar]
- 41. Peloquin CA, Zhu M, Adam RD, Singleton MD, Nix DE. Pharmacokinetics of Para‐aminosalicylic acid granules under four dosing conditions. Ann Pharmacother. 2001;35(11):1332‐1338. [DOI] [PubMed] [Google Scholar]
- 42. Riska N. Variations in the PAS concentration in the blood and their influence on treatment of tuberculosis. Acta Tuberc Scand. 1954;30(1–2):144‐163. [PubMed] [Google Scholar]
- 43. Riska N, Tennberg C. Optimal PAS dosage. Am Rev Respir Dis. 1962;86:430‐433. [DOI] [PubMed] [Google Scholar]
- 44. Wan SH, Pentikainen PJ, Azarnoff DL. Bioavailability of aminosalicylic acid and its various salts in humans III: absorption from tablets. J Pharm Sci. 1974;63(5):708‐711. [DOI] [PubMed] [Google Scholar]
- 45. Pentikäinen PJ, Wan SH, Azarnoff DL. Bioavailability of aminosalicylic acid and its various salts in humans IV: comparison of four brands of the sodium salt. J Pharm Sci. 1974;63(9):1431‐1434. [DOI] [PubMed] [Google Scholar]
- 46. Hollander AG. Para‐aminosalicylic acid‐resin complex: studies in absorption, serum electrolytes, and tolerance. Am Rev Tuberc. 1955;72(4):548‐551. [DOI] [PubMed] [Google Scholar]
- 47. Yue WY, Cohen SS. Toleration and absorption of sodium Para‐aminosalicylate and Para‐aminosalicylic acid (neopasalate). Comparison with other forms of Para‐aminosalicylic acid. Dis Chest. 1966;49(2):165‐174. [DOI] [PubMed] [Google Scholar]
- 48. Jeker K, Lauener H, Regli J, Friedrich T. The tolerability of calcium benzoyl‐PAS and calcium PAS; a double blind study. Am Rev Tuberc. 1959;79(3):351‐356. [DOI] [PubMed] [Google Scholar]
- 49. Duncan DK, Carr DT, Pfuetze KH, Power MH. Serum concentrations of Para‐aminosalicylic acid (PAS) produced by various forms of PAS. Dis Chest. 1951;19(2):138‐148. [DOI] [PubMed] [Google Scholar]
- 50. Lewis DO. Drug resistance in pulmonary tuberculosis treated with calcium B‐PAS. Tubercle. 1958;39(4):247‐250. [DOI] [PubMed] [Google Scholar]
- 51. Lehmann J. The role of the metabolism of p‐aminosalicylic acid (PAS) in the treatment of tuberculosis. Interaction with the metabolism of isonicotinic acid hydrazide (INH) and the synthesis of cholesterol. Scand J Respir Dis. 1969;50(3):169‐185. [PubMed] [Google Scholar]
- 52. Charles F. The treatment of pulmonary tuberculosis with intravenous PAS‐infusions. Tubercle. 1955;36(2):40‐44. [DOI] [PubMed] [Google Scholar]
- 53. Daniels M, Hill AB. Chemotherapy of pulmonary tuberculosis in young adults; an analysis of the combined results of three Medical Research Council trials. Br Med J. 1952;1(4769):1162‐1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang MJ, Jin B, Chae J, et al. Population pharmacokinetics of moxifloxacin, cycloserine, p ‐aminosalicylic acid and kanamycin for the treatment of multi‐drug‐resistant tuberculosis. Int J Antimicrob Agents. 2017;49(6):677‐687. [DOI] [PubMed] [Google Scholar]
- 55. Jindani A, Aber VR, Edwards EA, Mitchison DA. The early bactericidal activity of drugs in patients with pulmonary tuberculosis. Am Rev Respir Dis. 1980;121(6):939‐949. [DOI] [PubMed] [Google Scholar]
- 56. Nahid P, Mase SR, Migliori GB, et al. Treatment of drug‐resistant tuberculosis. An official ATS/CDC/ERS/IDSA clinical practice guideline. Am J Respir Crit Care Med. 2019;200(10):e93‐e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Way EL, Smith PK, Howie DL, Weiss R, Swanson R. The absorption, distribution, excretion and fate of Para‐aminosalicylic acid. J Pharmacol Exp Ther. 1948;93(3):368‐382. [PubMed] [Google Scholar]
- 58. Mitchell RS, Bell JC. Clinical implications of isoniazid blood levels in pulmonary tuberculosis. N Engl J Med. 1957;257(22):1066‐1070. [DOI] [PubMed] [Google Scholar]
- 59. Vatsis KP, Weber WW. Structural heterogeneity of Caucasian N‐acetyltransferase at the NAT1 gene locus. Arch Biochem Biophys. 1993;301(1):71‐76. [DOI] [PubMed] [Google Scholar]
- 60. Walker K, Ginsberg G, Hattis D, Johns DO, Guyton KZ, Sonawane B. Genetic polymorphism in N‐acetyltransferase (NAT): population distribution of NAT1 and NAT2 activity. J Toxicol Environ Heal Part B. 2009;12(5–6):440‐472. [DOI] [PubMed] [Google Scholar]
- 61. Riska N. PAS therapy with a daily unfractionated dose. Acta Tuberc Scand. 1959;37:104‐111. [PubMed] [Google Scholar]
- 62. Arbex MA, Varella M. de CL, Siqueira HR de, Mello FAF de. Drogas antituberculose: interações medicamentosas, efeitos adversos e utilização em situações especiais ‐ parte 2: fármacos de segunda linha. J Bras Pneumol. 2010;36(5):641‐656.21085831 [Google Scholar]
- 63. Rodin SM, Johnson BF. Pharmacokinetic interactions with digoxin. Clin Pharmacokinet. 1988;15(4):227‐244. [DOI] [PubMed] [Google Scholar]
- 64. Parvez M. Masud, Shin Ho Jung, Jung Jin Ah, Shin Jae‐Gook. Evaluation of para‐Aminosalicylic Acid as a Substrate of Multiple Solute Carrier Uptake Transporters and Possible Drug Interactions with Nonsteroidal Anti‐inflammatory Drugs In Vitro. Antimicrobial Agents and Chemotherapy. 2017;61(5):e02392‐16. 10.1128/aac.02392-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guidelines for the programmatic management of drug‐resistant tuberculosis 2011 update.
- 66. Singh B., Mitchison D. A.. The Bactericidal Activities of Combinations of Streptomycin, Isoniazid, p‐Aminosalicylic Acid (PAS), Oxytetracycline (Terramycin) and Viomycin against Mycobacterium tuberculosis. Journal of General Microbiology. 1955;13(1):176–184. 10.1099/00221287-13-1-176 [DOI] [PubMed] [Google Scholar]
- 67. Mitchison DA. Problems of drug resistance. Br Med Bull. 1954;10(2):115‐124. [DOI] [PubMed] [Google Scholar]
- 68. Bartmann K(K). Antituberculosis Drugs. Berlin Heidelberg: Springer; 1988. 10.1007/978-3-642-72873-0 [DOI] [Google Scholar]
- 69. Naito M, Tokushima K. The intravenous administration of a high concentration of Para‐aminosalicylic acid (PAS). Acta Tuberc Jpn. 1957;7(1–2):1‐11. [PubMed] [Google Scholar]
- 70. Reisner D. Comparison of single and divided daily dosages of isoniazid and PAS in the treatment of pulmonary tuberculosis. XV. A report of the veterans administration‐armed forces cooperative. Study on the chemotherapy of tuberculosis. Am Rev Respir Dis. 1966;94(6):849‐857. [DOI] [PubMed] [Google Scholar]
- 71. Bang HO, Strandgaard E. Continued studies on the problems of PAS dosage. Acta Tuberc Scand. 1960;39:81‐96. [PubMed] [Google Scholar]
- 72. Peloquin CA, Berning SE, Huitt GA, Childs JM, Singleton MD, James GT. Once‐daily and twice‐daily dosing of p‐aminosalicylic acid granules. Am J Respir Crit Care Med. 1999;159(3):932‐934. [DOI] [PubMed] [Google Scholar]
- 73. Wilson JL, Lampe WT. Single daily dose regimen of isoniazid and PAS in the treatment of pulmonary tuberculosis. Am Rev Respir Dis. 1964;89:756‐759. [DOI] [PubMed] [Google Scholar]
- 74. Adams KT, Donald PR, Abulfathi AA, Diacon AH, Stander MA, Reuter H. Pharmacokinetics of Para‐Aminosalicylic acid and its 2 major metabolites: a potential relationship to the development of gastrointestinal intolerance. J Clin Pharmacol. 2020;60(4):489‐494. [DOI] [PubMed] [Google Scholar]
- 75. WHO treatment guidelines for drug‐ resistant tuberculosis October 2016 Revision.
- 76. Zhang Y, Wu S, Xia Y, et al. Adverse events associated with treatment of multidrug‐resistant tuberculosis in China: an ambispective cohort study. Med Sci Monit. 2017;23:2348‐2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hoffmann H, Kohl TA, Hofmann‐Thiel S, et al. Delamanid and bedaquiline resistance in mycobacterium tuberculosis ancestral Beijing genotype causing extensively drug‐resistant tuberculosis in a Tibetan refugee. Am J Respir Crit Care Med. 2016;193(3):337‐340. [DOI] [PubMed] [Google Scholar]
- 78. Polsfuss S, Hofmann‐Thiel S, Merker M, et al. Emergence of low‐level delamanid and bedaquiline resistance during extremely drug‐resistant tuberculosis treatment. Clin Infect Dis. 2019;69(7):1229‐1231. [DOI] [PubMed] [Google Scholar]