

 This work is licensed under a
This work is licensed under a Abstract
Summary
Maturity-onset diabetes of the young (MODY) is a form of monogenic diabetes mellitus characterised by early onset and dominant inheritance. Delayed diagnosis or misdiagnosis as type 1 or type 2 diabetes mellitus is common. Definitive genetic diagnosis is essential for appropriate treatment of patients with MODY. The hepatocyte nuclear factor 1-beta (HNF1B) gene is responsible for MODY type 5 (MODY5), which has distinctive clinical features including renal disease. MODY5 should always be considered by clinicians in patients with early onset diabetes and renal anomalies. We report a case of a 30-year-old Japanese male with early-onset diabetes mellitus, renal anomalies and family history of diabetes that was suggestive of MODY5. Renal histology showed no evidence of diabetic nephropathy. Genetic testing revealed a novel heterozygous splice-site mutation of the HNF1B gene in the family members. It was strongly suggested that the mutation could underlie our patient’s MODY5.
Learning points:
Genetic diagnosis of MODY is relevant for appropriate treatment.
Dominantly inherited early-onset diabetes mellitus with renal cysts suggests MODY5.
Scanning the non-coding regions is important for not missing a mutation in HNF1B.
Patient Demographics: Adult, Male, Asian - Japanese, Japan
Clinical Overview: Pancreas, Diabetes
Publication Details: Insight into disease pathogenesis or mechanism of therapy, September, 2020
Background
Maturity-onset diabetes of the young (MODY) is a type of diabetes mellitus (DM) characterised by early onset, typically before 25 years of age, in nonobese individuals with an absence of pancreatic beta cell autoimmunity markers. The frequency of MODY among patients with DM is estimated to be 1–2% (1). To date, 14 different causative genes have been identified for MODY. The most common forms of the disease are MODY2 and MODY3, while MODY5 and MODY6 are relatively rare. Generally, MODY represents the insufficient insulin secretion with a lean phenotype, which is common to type 2 diabetes mellitus (T2DM) in Japanese patients (2). HNF1B mutation manifesting as MODY5 was first described in 1997 (3). In fact, MODY5 is characterised by a variety of extrapancreatic phenotypes including renal cystic disease, genital malformation, hepatobiliary malformation, hypomagnesaemia, hyperuricaemia and early-onset gout. Here we report a Japanese case of MODY5 with early-onset DM and renal cystic disease. Genetic testing revealed a novel heterozygous splice-site mutation of the HNF1B gene.
Case report
A 30-year-old, nonobese male was referred to our hospital because of poor glycaemic control. He was diagnosed with DM at the age of 15 and was initially treated with intensified insulin for 1.5 years. His glycaemic control then got better and insulin therapy was discontinued. At the age of 18 he discontinued follow-ups. He restarted his treatment at age 25. He was treated with 5 mg of linagliptin, 1000 mg of metformin, 50 mg of ipragliflozin and 22 U/day of insulin glargine, however, his HbA1c remained above 10% (NGSP). He did not regularly monitor his blood glucose or take medications. At his first visit to our hospital, his HbA1c was 12.2% and he was admitted. His BMI was 23.3 kg/m2. He had a family history of early-onset diabetes; his younger sister (III-3) was diagnosed at age 14, his father (II-2) at age 40 and his paternal grandmother (I-2) at age 50. Both genetic and environmental factor might be related to an earlier age of onset in successive generations. The pedigree chart of the family is shown in Fig. 1. III-3 was treated with intensified insulin therapy from the onset of insulin-dependent DM. Her abdominal ultrasound showed pancreatic agenesis and renal cysts without renal dysfunction.
Figure 1.

The pedigree chart of the family and the characteristics of affected family. The proband is indicated by an arrow. Subjects with diabetes mellitus (DM) are shaded.
Investigation
Laboratory evaluation showed that fasting glucose was 96 mg/dL, serum C-peptide was 0.28 ng/mL, and urinary C-peptide in 24 h was 38.4 μg/day. His serum BUN and creatinine were 33.0 and 1.52 mg/dL, respectively, uric acid was 8.56 mg/dL and magnesium was 1.5 mg/dL. His urinary albumin excretion was 85.3 mg/day and serum amylase and lipase levels were normal. Other biochemical findings were within normal limits. Glutamic acid decarboxylase (GAD) antibodies, insulinoma-associated antigen-2 (IA-2) antibody, and insulin autoantibody were negative. Abdominal ultrasound showed bilateral hyperechogenic kidneys with a few cysts, and the pancreas was intact (Fig. 2). No other dysmorphic features were evident. A kidney biopsy obtained for histological analysis to identify the aetiology of the renal disease revealed enlarged tubular structures. Transmission electron microscopy findings were most likely within normal limits. Arteriolo- or glomerular sclerosis were not seen, suggesting that there was no evidence of diabetic nephropathy (Fig. 3).
Figure 2.
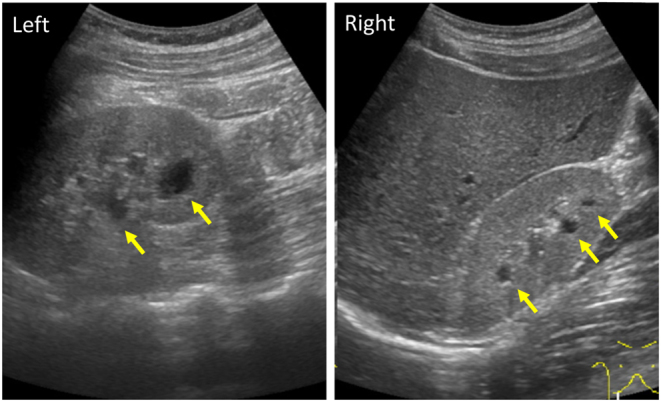
The ultrasonographic images of proband’s kidneys. A normal-sized hyperechoic kidney, and a few cysts are observed (arrows).
Figure 3.
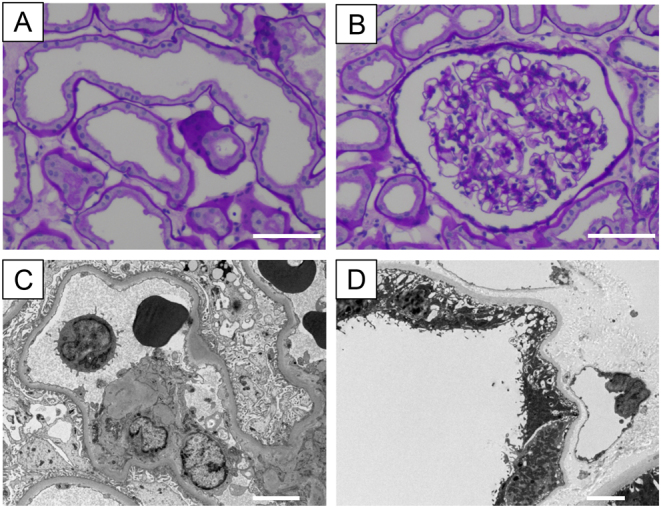
Histopathology of proband’s kidney biopsy. (A and B) Stained with periodic acid-Schiff. Dilated proximal tubules and a glomerulus within normal limits. Scale bar: 40 μm. (C and D) Transmission electron microscopy images. A glomerulus was most likely within normal limits (no diabetic nephropathy). A partial thinning of the tubular basement membrane was observed. Scale bar: 5 μm.
Based on the presence of early-onset DM without autoantibodies, renal abnormalities and family history, he was suspected to have MODY. His genomic DNA was purified from peripheral blood after obtaining written informed consent. The coding exons and intron–exon boundaries of the HNF1A, HNF1B, and HNF4A genes were then sequenced directly using Sanger sequencing. As a result, a novel heterozygous mutation, NC_000017.11:g.37731831C>G, in intron 3 of the HNF1B gene was identified (Fig. 4). This mutation could not be found in the SNP database (dbSNP) of the National Center for Biotechnology Information or in the Human Genetic Variation Database (4). The mutation was located in the splice acceptor site at the boundary between intron 3 and exon 4 of the HNF1B gene. The mutation altered the conserved AG-dinucleotide at the splice acceptor site to AC. The effect of this mutation was analysed using SpliceAI, which is a deep neural network precisely modelling mRNA splicing from a genomic sequence (5). Loss of splicing acceptor at the canonical splice site of intron 3 was predicted with high delta score of 0.97. In addition, gain of splicing acceptor at 5-nucleotide downstream from the canonical acceptor site was also predicted with delta score of 0.65. This result indicates, NC_000017.11:g.37731831C>G disrupts a normal splicing of intron 3 leading to 5-nucleotide intron 3 inclusion. Together with a possibility of exon skipping, either splicing at the potential gain site or exon skipping results in a frameshift and premature termination codon, which is consistent that this novel mutation can be pathogenic. Genomic DNA samples from the patient’s father and sister with young-onset DM were also obtained, and revealed that both of them shared the same, unusual splice-site mutation of the HNF1B gene.
Figure 4.

The gene structure of HNF1B is shown with its corresponding functional protein domains. Sequence analysis revealed a novel heterozygous mutation in intron 3, NC_000017.11:g.37731831C>G. The mutation located in the conserved AG-dinucleotide at the splice acceptor site.
Treatment
Because his insulin secretory capacity was severely impaired, multiple daily insulin injections with a total daily insulin dose of 0.32 units/kg was started and his glycaemic control improved. He had no signs of diabetic retinopathy, diabetic neuropathy or macroangiopathy.
Outcome and follow-up
Based on the above findings, his phenotype was compatible with MODY5. It is likely that his novel mutation of the HNF1B gene was inherited in an autosomal dominant manner in the family. The appropriate genetic diagnosis has empowered him in his care and improved his adherence to treatment. His HbA1c level was lowered to 8% after hospitalisation. The renal function and microalbuminuria have remained stable, so far. The most recent creatinine level was 1.33 mg/dL and urine albumin-to-creatinine ratio was 96.7 mg/g. Hyperuricaemia improved after initial treatment. Hypomagnesaemia remained at a similar level with no need of replacement therapy.
Discussion
Here, we report a case of MODY5 with a previously unidentified mutation in the splicing acceptor site of the HNF1B gene. MODYs are frequently misdiagnosed as other types of diabetes such as type 1 DM or type 2 DM. Glycaemic control, in this case, was insufficient for the long term with treatment following the standard for type 2 DM. In this case, an accurate genetic diagnosis significantly improved the management of both the patient and the family.
The autoantibody test and early-onset DM with a strong family history were strongly suggestive of MODY, which was true in this case. In addition, extrapancreatic features disclosed by a blood test or images is useful for some MODYs. Early-onset DM cases with renal abnormalities strongly suggest a mutation in the HNF1B gene. Moreover, the pathophysiology of DM in patients with HNF1B mutations is beta-cell dysfunction and reduced insulin secretion as well as reduced insulin sensitivity to endogenous glucose production (6). The majority of MODY5 patients require early treatment with insulin since they respond poorly to sulphonylureas. The HNF1B gene is located on chromosome 17q12, a Pit-1/Oct-1/Unc-86 (POU) transcription factor that is important for embryonic development. To date, 290 mutations have been reported within the HNF1B gene in The Human Gene Mutation Database. Mutations predominantly cluster in the first four exons, which encode dimerisation and DNA-binding domains. Most of them can explain a transcriptional impairment of the HNF1B protein (6). Here, sequencing of coding exons and intron–exon boundaries of the candidate genes allowed us to detect a mutation in an intron. Analysis of exons, which comprise ~2% of the genomic sequence, has been successful in many cases. However, ~98% of the genome remains in the non-coding regions, which can contain a significant variant affecting splicing or regulatory elements (7). An appropriate sequence technique should be chosen so as not to miss an accurate diagnosis. The presently identified point mutation at the splice acceptor site in intron 3 of the HNF1B gene is predicted to affect splicing, which might cause premature termination of the HNF1B by in silico analysis of splicing. According to the American College of Medical Genetics standards and guidelines (8), the mutation is classified into a pathogenic variant with one very strong, one moderate, and one supporting evidence of pathogenicity.
Previous studies have shown that mutations or deletions of HNF1B cause multi-system disease mediated by haploinsufficiency (9). HNF1B mutations characterise a variety of phenotypes, the most common being renal disease (multi-cystic dysplastic kidneys, renal dysplasia, renal dysfunction, etc) (9). In many cases, renal cysts are seen on an ultrasound, and MODY5 cases with renal cysts are referred to as renal cysts and diabetes (RCAD). Renal function ranges from normal to chronic kidney failure, including cases requiring dialysis. There are some cases without renal dysfunction, and the phenotypes vary between individuals and within families (10). When biopsies are performed, considerable variation is observed in the histological diagnosis, diabetic nephropathy is deniable, but the enlargement of tubular structures observed in this case is generally observed in glomerulocystic kidney disease, and is not specific for MODY5 (9). To facilitate diagnosis, ultrasonography readily evaluates renal anomaly as well as associated extra-renal abnormalities, and a precise family history is useful, especially for renal disease and DM. After these evaluations, the genetic test should be applied for a conclusive diagnosis.
In conclusion, we report a case of MODY5 with a novel splice acceptor site mutation in the HNF1B gene with diverse clinical characteristics who experienced a good clinical course by diagnosis. Although MODY identification remains challenging, understanding the phenotypic variation of MODY is essential for diagnosis, especially for cases with renal abnormalities. Genetic testing for successful identification of the disease results in better management for both the patient and the family.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research was partially funded by JSPS KAKENHI 18K08509.
Patient consent
Written informed consent was obtained from the patient prior to publication.
Author contribution statement
Y F, D T, and Y Y prepared the manuscript. D T, H T performed the genetic test on the patient and relatives. T K is responsible for renal biopsy and the histopathological assessment. M M, T H, Y H, N I, and Y S contributed to the discussion of content and reviewed the paper before submission.
Acknowledgement
The authors thank Dalmen Mayer for his help in the preparation of this manuscript.
References
- 1.Fajans SS & Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011. 34 1878–1884. ( 10.2337/dc11-0035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horikawa Y. Maturity-onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. Journal of Diabetes Investigation 2018. 9 704–712. ( 10.1111/jdi.12812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O, et al Mutation in hepatocyte nuclear factor-1β gene (TCF2) associated with MODY. Nature Genetics 1997. 17 384–38. ( 10.1038/ng1297-384) [DOI] [PubMed] [Google Scholar]
- 4.Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, Doi K, Shimizu M, Nakabayashi K, Aoki Y, et al Human genetic variation database, a reference database of genetic variations in the Japanese population. Journal of Human Genetics 2016. 61 547–5. ( 10.1038/jhg.2016.12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al Predicting splicing from primary sequence with deep learning. Cell 2019. 176 535, .e24–548.e24. ( 10.1016/j.cell.2018.12.015) [DOI] [PubMed] [Google Scholar]
- 6.El-Khairi R & Vallier L. The role of hepatocyte nuclear factor 1β in disease and development. Diabetes, Obesity and Metabolism 2016. 18 (Supplement 1) 23–32. ( 10.1111/dom.12715) [DOI] [PubMed] [Google Scholar]
- 7.De Franco E. From biology to genes and back again: gene discovery for monogenic forms of beta-cell dysfunction in diabetes. Journal of Molecular Biology 2020. 432 1535–1550. ( 10.1016/j.jmb.2019.08.016) [DOI] [PubMed] [Google Scholar]
- 8.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015. 17 405–4. ( 10.1038/gim.2015.30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clissold RL, Hamilton AJ, Hattersley AT, Ellard S & Bingham C. HNF1B-associated renal and extra-renal disease – an expanding clinical spectrum. Nature Reviews: Nephrology 2015. 11 102–1. ( 10.1038/nrneph.2014.232) [DOI] [PubMed] [Google Scholar]
- 10.Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, Shishido S, Hasegawa Y & Nakahata T. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1β gene due to germline mosaicism. Journal of Clinical Endocrinology and Metabolism 2004. 89 2905–290. ( 10.1210/jc.2003-031828) [DOI] [PubMed] [Google Scholar]