

 This work is licensed under a
This work is licensed under a Abstract
Summary
Familial hypocalciuric hypercalcaemia (FHH) is a dominantly inherited, lifelong benign disorder characterised by asymptomatic hypercalcaemia, relative hypocalciuria and variable parathyroid hormone levels. It is caused by loss-of-function pathogenic variants in the calcium-sensing receptor (CASR) gene. Primary hyperparathyroidism (PHPT) is characterised by variable hypercalcaemia in the context of non-suppressed parathyroid hormone levels. Unlike patients with FHH, patients with severe hypercalcaemia due to PHPT are usually symptomatic and are at risk of end-organ damage affecting the kidneys, bone, heart, gastrointestinal system and CNS. Surgical resection of the offending parathyroid gland(s) is the treatment of choice for PHPT, while dietary adjustment and reassurance is the mainstay of management for patients with FHH. The occurrence of both FHH and primary hyperparathyroidism (PHPT) in the same patient has been described. We report an interesting case of FHH due to a novel CASR variant confirmed in a mother and her two daughters and the possible coexistence of FHH and PHPT in the mother, highlighting the challenges involved in diagnosis and management.
Learning points:
Familial hypocalciuric hypercalcaemia (FHH) and primary hyperparathyroidism (PHPT) can coexist in the same patient.
Urinary calcium creatinine clearance ratio can play a role in distinguishing between PHPT and FHH.
Genetic testing should be considered in managing patients with PHPT and FHH where the benefit may extend to the wider family.
Family segregation studies can play an important role in the reclassification of variants of uncertain significance.
Parathyroidectomy has no benefit in patients with FHH and therefore, it is important to exclude FHH prior to considering surgery.
For patients with coexisting FHH and PHPT, parathyroidectomy will reduce the risk of complications from the severe hypercalcaemia associated with PHPT.
Patient Demographics: Adult, Female, White, United Kingdom
Clinical Overview: Parathyroid, Neuroendocrinology
Related Disciplines: Genetics
Publication Details: New disease or syndrome: presentations/diagnosis/management, September, 2020
Background
Familial hypocalciuric hypercalcaemia (FHH) and primary hyperparathyroidism (PHPT) are both causes of hypercalcaemia. FHH is an autosomal dominant, lifelong, generally benign disorder that does not require any treatment or surgery. Most patients with FHH are asymptomatic, but chondrocalcinosis and acute pancreatitis have occasionally been observed. Patients with FHH have often been misdiagnosed as having PHPT as both disorders can have elevated or inappropriately normal parathyroid hormone and elevated serum calcium. As such, the prevalence of FHH is difficult to estimate. Previous population-based studies have suggested 1:10 000 to 1:100 000 (1). Affected patients usually have mild hypercalcaemia, normal or mildly elevated parathyroid hormone levels, and low urine calcium. The urinary calcium creatinine clearance ratio (CCCR) is a useful biochemical test to differentiate this condition from PHPT. Its hallmark feature is an inappropriately low CCCR (typically <0.01), but 20% of FHH patients may have a CCCR >0.01, and therefore be indistinguishable from individuals with PHPT (2). Therefore, genetic testing is the confirmatory test for FHH (1).
FHH is most commonly caused by inactivating pathogenic variants in the calcium-sensing receptor gene CASR (leading to FHH1) or rarely by inactivating variants in the G-protein subunit alpha 11 gene GNA11 (leading to FHH2) and missense variants at the Arg15 residue of the adaptor protein 2 sigma subunit AP2S1 gene (leading to FHH3). All forms of hereditary FHH are inherited in an autosomal dominant fashion (2).
PHPT is suspected when parathyroid hormone (PTH) values are higher than characteristic for FHH with increasing serum urinary calcium levels. Imaging can help detect an abnormally enlarged parathyroid gland which is more characteristic for PHPT. PHPT affects up to 3 in 1000 people with the highest incidence observed in the elderly female population (3). PHPT is usually sporadic but can be hereditary in a minority of cases (~10%) and can present as part of a syndrome such as the multiple endocrine neoplasia (MEN1, MEN2, MEN4) and CDC73-related disorders (4). Surgical intervention is recommended in PHPT for both stabilisation of parathyroid function and to prevent long term complications of chronic hypercalcaemia and hypercalciuria (5).
The occasional coexistence of FHH and PHPT in the same patient has been reported and this should be considered in patients with hypercalcaemia, hypophosphatemia, mildly elevated parathyroid hormone levels and inappropriate hypocalciuria. Although surgical intervention for the parathyroid over-activity may not resolve the hypercalcaemia completely, it will alleviate the symptoms and prevent potential complications of the hypercalcaemia secondary to PHPT. We report an interesting case of FHH1 found in a mother and her two daughters and the likely coexistence of FHH and PHPT in the mother, highlighting the challenges involved in diagnosis and management and clarification through genetic testing.
Case presentation
Case 1
Medical history
A 37-year-old Caucasian woman was found to have mildly raised calcium levels with PTH at the upper end of normal range when she presented to her GP with fatigue, sweats and palpitations. Investigations by her local endocrine unit confirmed hypercalcaemia of 2.63–2.71 mmol/L with a slightly raised PTH of 7.9 pmol/L. She had no significant past medical history. General examination revealed no abnormal clinical findings.
Investigations
Blood investigations revealed mild hypercalcaemia (adjusted calcium of 2.63–2.71 mmol/L) with a PTH of 7.9 pmol/L (Table 1). The calculated CCCR was 0.0096 (Table 2). Imaging of neck with US and Sestamibi reported no lesions. A 24 h urine metanephrines assay was normal. A bone density scan revealed normal bone density. Sequencing of the CASR gene detected a heterozygous novel, missense variant in CASR exon 7 – NM_000388.4:c.2414A>G; p.(Lys805Arg). This is located in the cytoplasmic domain between transmembrane domains 5 and 6. The p.Lys805Arg variant has not previously been reported in the ExAC or gnomAD databases. The CASR gene has a low rate of benign missense variation as evidenced by a significant (z = 3.52) ExAC constraint score. The p.Lys805 residue is conserved across 18 species (to Lamprey) and has a conservation (ConSurf) score of 8 and the p.Lys805Arg variant is predicted by SIFT and PolyPhen to have a deleterious effect on protein function. At this stage, it was classified as a variant of unknown significance (VUS).
Table 1.
Results of biochemical blood tests at presentation.
| Blood parameters | Normal ranges | Case 1 | Case 2 | Case 3 |
|---|---|---|---|---|
| Sodium (mmol/L) | 132–145 | 141 | 142 | 135 |
| Potassium (mmol/L) | 3.4–5.1 | 4.0 | 4.1 | 4.5 |
| Creatinine (µmol/L) | 45–84 | 73 | 77 | 106 |
| 25-hydroxy vitamin D (nmol/L) | >50 | – | 34 | 12 |
| Adjusted calcium (mmol/L) | 2.2–2.6 | 2.71 | 2.65 | 3.15 |
| Phosphate (mmol/L) | 0.8–1.5 | 1.05 | 0.75 | 1.23 |
| Magnesium (mmol/L) | 0.7–1.0 | – | 0.97 | 0.83 |
| Alkaline phosphatase (U/L) | 30–130 | 93 | 124 | 111 |
| Parathyroid hormone (pmol/L) | 1.4–6.2 | 7.9 | 3.7 | 11.9 |
| Thyroid-stimulating hormone (mU/L) | 0.3–4.2 | 2.98 | 3.38 | 11.2 |
Table 2.
Results of urine analysis at presentations.
| Urine parameters | Normal ranges | Case 1 | Case 2 | Case 3 |
|---|---|---|---|---|
| Calcium creatinine clearance ratio (%) | >0.01 | 0.0096 | 0.003 | 0.0052 |
| 24-h calcium excretion (mmol/24 h) | 2.5–7.5 | 2.09 | 2.9 | 2.2 |
Treatment
The patient was reassured and advised to continue adequate fluid intake.
Outcome and follow-up
The patient remained well with a mildly raised serum calcium level (2.6–2.7 mmol/L) and a normal serum parathyroid hormone level.
Case 2
Medical history and demographics
A 44-year-old woman (sister of Case 1) was found to have mild hypercalcaemia while being investigated for tiredness. She had a past medical history of a renal stones and mildly raised serum calcium (2.63 mmol/L) 4 years prior to this presentation. Current medication consisted of the oral contraceptive pill. General examination revealed a patient with normal weight (BMI: 24 kg/m2) with no abnormal clinical findings.
Investigation
Blood results demonstrated mild hypercalcaemia with a normal parathyroid hormone level (Table 1). A 24-h urine collection demonstrated a calcium excretion at the lower end of the normal range, and CCCR was calculated at 0.003. Further results revealed vitamin D deficiency. Renal function, fasting gut hormones and pituitary hormones were all normal. A bone density scan revealed osteopenia at the hip region (T score of −1.5 s.d., 0.765 g/cm2). Ultrasonography of the kidneys was normal. An ultrasonography and nuclear medicine scan (sestamibi) of the parathyroid glands were also normal (no adenoma detected). She was tested for the familial CASR VUS and was found to also be heterozygous for this variant.
Treatment
The patient was reassured and given vitamin D replacement. She was advised to continue adequate fluid intake and regular weight-bearing exercises.
Outcome and follow-up
The patient remained well with a mildly raised serum calcium level (2.5–2.7 mmol/L) and a normal serum parathyroid hormone level.
Case 3
Medical history and demographics
A 71-year-old female (mother of Cases 1 and 2) was referred for evaluation of asymptomatic hypercalcaemia. The hypercalcaemia was discovered during routine screening carried out because she had a family history of hypercalcaemia in her two children. The patient had a past medical history of mild chronic kidney disease, hypothyroidism, and hypertension. Current medication included Atenolol, Doxazosin, Ramipril, Amitriptyline, and Simvastatin. General examination revealed an overweight patient (BMI: 34 kg/m2) with no abnormal clinical findings.
Investigation
Blood results demonstrated severe hypercalcaemia (3.15–3.25 mmol/L) with a significantly raised parathyroid hormone level of 11.9 pmol/L (reference: 1.4–6.2) which were not characteristic of FHH (Table 1). However, a 24-h urine collection demonstrated a calcium excretion of 2.2 mmol in 24 h, which was surprisingly low for that amount of hypercalcaemia. Furthermore, her CCCR was significantly low at 0.0052 when checked at a later date. Further results revealed Vitamin D deficiency. Renal function test revealed stable chronic kidney disease. Fasting gut hormone and pituitary hormone profiles were all normal. She had a raised thyroid-stimulating hormone level indicating hypothyroidism. A bone density scan was normal. Ultrasonography demonstrated a 26 mm hypoechoic solid mass lesion posterior to the lower pole of the right thyroid lobe (Fig. 1A). This was collaborated by a similar finding on a parathyroid nuclear medicine scan (sestamibi) and appearances were consistent with a parathyroid adenoma (Fig. 1B). The above results raised the question of a possible coexistence of a FHH and PHPT in this patient.
Figure 1.
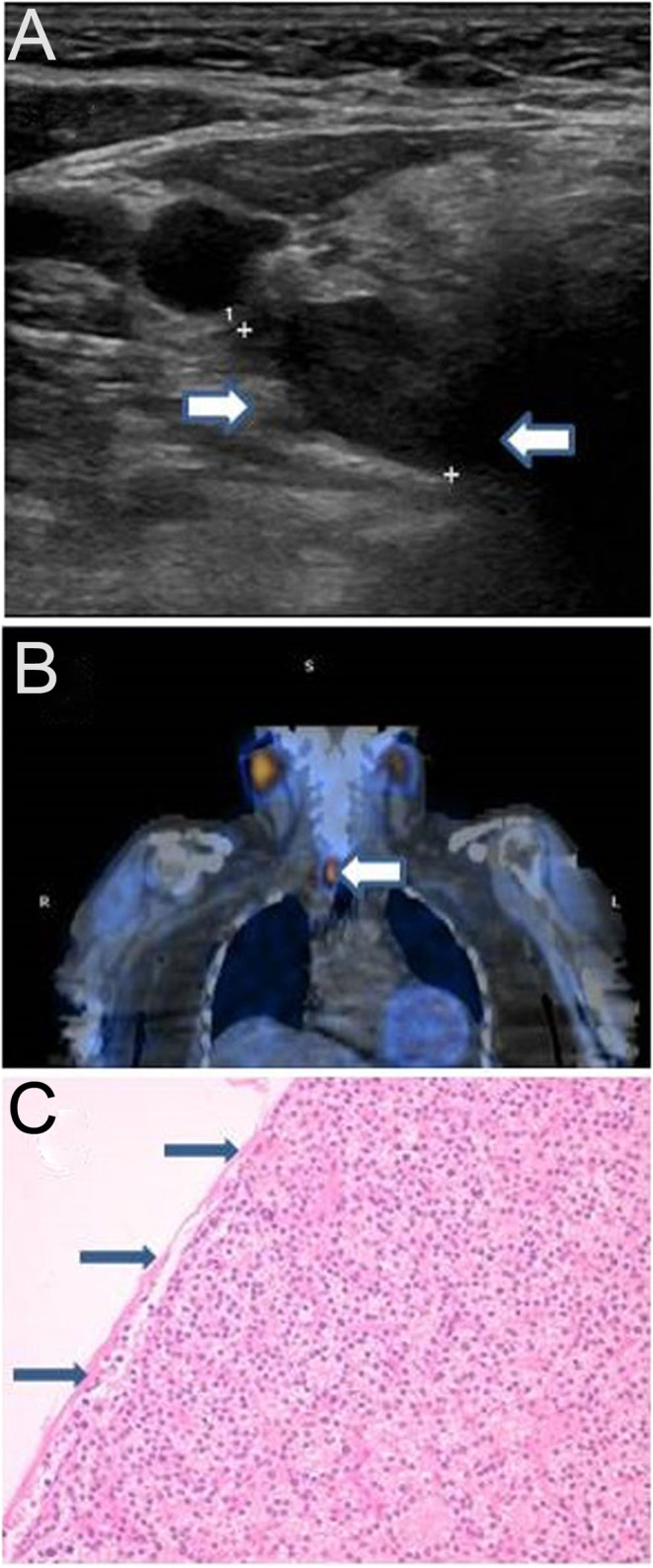
(A) Enlarged parathyroid gland measuring 26 mm on ultrasound scan (arrows). (B) Enlarged parathyroid gland on 99mTc-sestamibi scan (arrow). (C) Histology specimen of the resected parathyroid adenoma with thin capsule (arrows).
Treatment
After rehydration and intravenous bisphosphonate therapy, the serum calcium level gradually reduced to 2.96 mmol/L. The patient was also started on levothyroxine 50 µg daily. A right parathyroidectomy was performed and histopathological examination confirmed the presence of a thinly encapsulated parathyroid adenoma (Fig. 1C). The patient was also tested for the familial CASR VUS detected in her daughters and confirmed as also being heterozygous for this variant.
Outcome and follow-up
During postoperative follow-up the patient remained clinically well. Despite parathyroidectomy, her serum calcium level remained slightly elevated (2.64–2.77 mmol/L) thereafter. The serum parathyroid hormone level remained in the normal range. In addition, her calcium creatinine clearance ratio remained low (<0.01) on two more tests done a year apart, indicating that the chronic mild hypercalcaemia was due to the FHH.
Discussion
We have described a family with two sisters (Cases 1 and 2) and their mother (Case 3) who all presented with hypercalcaemia, elevated PTH and CCCR < 0.01. The mother also had evidence of a parathyroid adenoma which was felt to be contributory to her more extreme hypercalcaemia as post-operatively, the calcium levels reduced to mildly elevated levels. As a result of Case 3 requiring urgent parathyroidectomy, an estimate of her urinary CCCR was not performed before the surgery. However, a 24-h urine collection done prior to the surgery did indicate an inappropriately low calcium excretion for that amount of hypercalcaemia. Additionally, mild hypercalcaemia with normal parathyroid hormone levels remained after surgery for this patient and subsequent follow-up demonstrated low CCCR <0.01 suggesting underlying FHH.
A novel missense variant in the CASR gene was first detected in the index sister, which was initially classified as a VUS. However, there were several features upon computational analysis, including in silico analysis, suggesting possible pathogenicity. Family segregation studies in this situation with multiple affected members are valuable as they can provide further evidence for pathogenicity. We demonstrated that the CASR variant in this family segregates with all three relatives with a biochemical phenotype consistent with FHH with a significantly low CCCR of <0.01. Therefore, based on the pathogenicity evidence given under the ACMG standards and guidelines and applying the January 2018 ClinGen SVI Bayesian classification framework, giving a posterior probability of 0.900 (using tools on DECIPHER https://decipher.sanger.ac.uk/), this CASR variant can now be classified as likely pathogenic and a diagnosis of likely FHH1 in this family. Unlike other genetic disorders, such as developmental paediatric disorders, VUSs detected in endocrine patients are more likely to be re-classified, based on computational analysis supported by careful phenotyping using biochemistry, family history and family segregation studies. This re-classification allows for appropriate management of disorders in the affected patients and their families.
Calcium-sensing receptor (CASR) protein is a large glycoprotein which belongs to family C or family 3 of the superfamily of G protein-coupled receptors. Structurally it is composed of 1078 amino acids with a large extracellular domain, seven-transmembrane spanning regions and an intracellular tail. It is encoded by the CASR gene which is located on chromosome 3q13.3–q21.1. This receptor is expressed predominantly in parathyroid glands but also in the kidneys, gut, pancreas and bone. The receptor enables CASR expressing cells to detect alterations in the serum calcium levels and when a certain threshold is reached the receptor is activated to normalise calcium levels by regulating parathyroid hormone secretion and urinary calcium excretion. Inactivating pathogenic variant in CASR decreases the receptor’s sensitivity to extracellular calcium leading to persistent hypercalcaemia and in the kidneys it leads to increased renal tubular reabsorption of calcium resulting in hypocalciuria (6).
In heterozygous individuals, loss of function mutations of CASR gene manifest as FHH1 or Marx–Auerbach syndrome. In 20% of FHH cases, the underlying genetic cause is yet to be discovered. Furthermore, hypocalciuric hypercalcaemia can be autoimmune in nature and acquired via anti-CASR blocking antibodies. FHH1 is transmitted as an autosomal dominant trait. The treatment of FHH1 involves close observation with dietary and medication modifications. Surgical approach has proven non-beneficial and hence not recommended (6). Bi-allelic loss-of-function variants in the CASR gene can lead to neonatal severe primary hyperparathyroidism (NSHPT), where severe PHPT can lead to calcium levels of 4–8 mmol/L associated with severe parathyroid hyperplasia which is often lethal without urgent total parathyroidectomy (1).
PHPT is characterised by mild to severe hypercalcaemia in the context of non-suppressed parathyroid hormone levels. Eighty percent of cases are due to a parathyroid adenoma, 10% of the cases have more than one adenoma and less than 10% have hyperplasia. PHPT can occur in isolation or as part of familial endocrinopathies such as multiple endocrine neoplasia (MEN1, MEN2A, MEN4) and CDC73-related disorders. Hypercalcaemia in PHPT is usually symptomatic and patients are at increased risk of renal stones, cortical bone loss and fractures. Patients may also develop gastrointestinal, neuropsychiatric, and cardiovascular manifestations. The diagnostic workup of PHPT includes biochemical tests (serum calcium, PTH, eGFR, 25-hydroxy vitamin D and 24-h urine calcium excretion), renal ultrasound examination, bone density scan and ultrasonic and nuclear medicine imaging of the parathyroid glands. Surgery remains the definitive treatment leading to resolution of symptoms and normalisation of calcium and PTH levels. Guidelines suggest surgical intervention is recommended for patients with symptoms of hypercalcaemia (thirst, frequent or excessive urination) or end-organ disease (renal stones, fragility fractures or osteoporosis) or adjusted serum calcium level of 2.85 mmol/L or above. Patients who are unsuitable for surgery or surgery has been unsuccessful are offered medical treatment with calcimimetics (5).
The occasional coexistence of FHH and PHPT in the same patient has been reported and can be a diagnostic challenge. For example, one such patient was found to have a missense mutation in the exon 4 of the CASR gene (7). Another patient was found with the same mutation in the exon 4 of the CASR gene (8). One patient was identified with a C562Y variant commonly found in Norwegian FHH families (9). One patient of Japanese origin was found to have a novel heterozygous 3193delA variant (10). Another patient of German origin was found to have a novel c.1651A>G p.(Arg551Gly) variant (11). More recently, one patient was found to have a c.2208G>C, p.(Gly670Arg) variant in the CASR gene (12). A common feature to all these cases is the fact they all presented with hypercalcaemia along with overlapping features common to both FHH and PHPT. In about 50% of reports FHH was diagnosed prior to parathyroidectomy while in the rest FHH was diagnosed after parathyroidectomy. All patients had a parathyroid adenoma on pathologic examination and excision resulted in normal serum parathyroid hormone levels with reduced but still slightly elevated calcium levels, indicating that the FHH was still present. Our patient with coexisting FHH and PHPT was found to have a novel c.2414A>G, p.(Lys805Arg) missense variant: the serum calcium levels remained slightly elevated while the CCCR remained significantly low after parathyroidectomy.
A German series highlighted the coexistence of PHPT and FHH in 4 out of 139 patients with hypercalcaemia and suggested a pathogenic role of CASR variant in PHPT (13). The authors speculated that some CASR pathogenic variants play a role in parathyroid cell proliferation, therefore leading to hypertrophy and adenoma formation and thus cause PHPT.
In conclusion, we report a family with a mother and two daughters presenting with hypercalcaemia, elevated PTH, critically low CCCR where PHPT was found to coexist in the mother. Genetic testing detected a novel missense VUS in the index case which upon testing the other affected relatives confirmed segregation with the appropriate phenotype. Following the family segregation studies and careful phenotyping of the affected individuals, we were able to reclassify the CASR VUS to a likely pathogenic variant suggesting that the type 1 FHH is the probable diagnosis. Therefore, we highlight the importance of a thorough diagnostic work-up, including calculation of a CCCR and genetic testing in managing patients suspected of having PHPT. We also stress the value of family segregation studies in the re-classification of VUS’s. We hope that this case not only adds to the existing literature but also provides insight into an expanding clinical spectrum of CASR gene mutations and the possible link to parathyroid adenomas.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Informed and written consent has been obtained from all three patients and these are available for review if needed.
Author contribution statement
S V S, H J, S O O and S-M P wrote the first draft of this manuscript. All authors were involved in the writing and in the final review of the manuscript. S-M P is the named clinical geneticist for the patients. J T and T E contributed to the genetic testing and analysis performed and the ensuing diagnosis. S V S, A S and F J S H were the named physicians for each case. N A B was the named surgeon for the surgical case.
References
- 1.Lee JY & Shoback DM. Familial hypocalciuric hypercalcaemia and related disorders. Best Practice and Research: Clinical Endocrinology and Metabolism 2018. 32 609–619. ( 10.1016/j.beem.2018.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nesbit MA, Hannan FM, Howles SA, Babinsky VN, Head RA, Cranston T, Rust N, Hobbs MR, Heath 3rd H & Thakker RV. Mutations affecting G-protein subunit α11 in hypercalcemia and hypocalcemia. New England Journal of Medicine 2013. 368 2476–2486. ( 10.1056/NEJMoa1300253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adami S, Marcocci C & Gatti D. Epidemiology of primary hyperparathyroidism in Europe. Journal of Bone and Mineral Research 2002. 17 (Supplement 2) N18–N23. [PubMed] [Google Scholar]
- 4.Thakker RV. Genetics of parathyroid tumours. Journal of Internal Medicine 2016. 280 574–583. ( 10.1111/joim.12523) [DOI] [PubMed] [Google Scholar]
- 5.Hyperparathyroidism (primary): diagnosis, assessment and initial management. NICE guideline 2019 [NG132]. (available at: https://www.nice.org.uk/guidance/ng132/chapter/Recommendations). Accessed on 9 February 2020. [Google Scholar]
- 6.Vahe C, Benomar K, Espiard S, Coppin L, Jannin A, Odou MF & Vantyghem MC. Diseases associated with calcium-sensing receptor. Orphanet Journal of Rare Diseases 2017. 12 19. ( 10.1186/s13023-017-0570-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brachet C, Boros E, Tenoutasse S, Lissens W, Andry G, Martin P, Bergmann P & Heinrichs C. Association of parathyroid adenoma and familial hypocalciuric hypercalcaemia in a teenager. European Journal of Endocrinology 2009. 161 207–210. ( 10.1530/EJE-09-0257) [DOI] [PubMed] [Google Scholar]
- 8.Forde HE, Hill AD & Smith D. Parathyroid adenoma in a patient with familial hypocalciuric hypercalcaemia. BMJ Case Reports 2014. 2014 bcr2014206473 ( 10.1136/bcr-2014-206473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burski K, Torjussen B, Paulsen AQ, Boman H & Bollerslev J. Parathyroid adenoma in a subject with familial hypocalciuric hypercalcaemia: coincidence or causality? Journal of Clinical Endocrinology and Metabolism 2002. 87 1015–1016. ( 10.1210/jcem.87.3.8304) [DOI] [PubMed] [Google Scholar]
- 10.Yabuta T, Miyauchi A, Inoue H, Yoshida H, Hirokawa M & Amino N. A patient with primary hyperparathyroidism associated with familial hypocalciuric hypercalcaemia induced by a novel germline CaSR gene mutation. Asian Journal of Surgery 2009. 32 118–122. ( 10.1016/S1015-9584(09)60022-1) [DOI] [PubMed] [Google Scholar]
- 11.Papadakis M, Meurer N, Margariti T, Meyer A, Weyerbrock N & Dotzenrath C. A novel mutation of the calcium-sensing receptor gene in a German subject with familial hypocalciuric hypercalcemia and primary hyperparathyroidism. Hormones 2016. 15 557–559. ( 10.14310/horm.2002.1711) [DOI] [PubMed] [Google Scholar]
- 12.Kay S, Piltin M, Loseva V, Sinnot B, Brenna JR, Mehrotra S & Terris D. Collision diagnoses: primary hyperparathyroidism layered on familial hypocalciuric hypercalcaemia. AACE Clinical Case Reports 2018. 4 e362–e366. ( 10.4158/ACCR-2018-0005) [DOI] [Google Scholar]
- 13.Frank-Raue K, Leidig-Bruckner G, Haag C, Schulze E, Lorenz A, Schmitz-Winnenthal H & Raue F. Inactivating calcium-sensing receptor mutations in patients with primary hyperparathyroidism. Clinical Endocrinology 2011. 75 50–55. ( 10.1111/j.1365-2265.2011.04059.x) [DOI] [PubMed] [Google Scholar]