

Summary
Cell migration is an essential, energetically demanding process in immunity. Immune cells navigate the body via chemokines and other immune mediators, which are altered under inflammatory conditions of injury or infection. Several factors determine the migratory abilities of different types of immune cells in diverse contexts, including the precise co‐ordination of cytoskeletal remodelling, the expression of specific chemokine receptors and integrins, and environmental conditions. In this review, we present an overview of recent advances in our understanding of the relationship of each of these factors with cellular metabolism, with a focus on the spatial organization of glycolysis and mitochondria, reciprocal regulation of chemokine receptors and the influence of environmental changes.
Keywords: immunology, metabolism, migration
Migration is an essential function of immune cells in diverse contexts of homeostasis and inflammation. This review presents an overview of how cell motility is regulated by the integration of cellular metabolism with cytoskeletal remodelling and signalling. The effects of the microenvironment and host metabolism on immune cell migration are also discussed.

Abbreviations
- AMPK
5’ adenosine monophosphate‐activated protein kinase
- CXCL
C‐X‐C motif ligand
- CCL
C‐C motif ligand
- CCR
C‐C motif receptor
- DRP1
dynamin‐related protein 1
- ECM
extracellular matrix
- ETC
electron transport chain
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- HIF‐1ɑ
hypoxia inducible factor‐1ɑ
- LDHA
lactate dehydrogenase
- LDL
low‐density lipoprotein
- HDL
high‐density lipoprotein
- MIRO‐1
mitochondrial Rho GTPase 1
- MLC
myosin light chain
- mTOR
mammalian target of rapamycin
- OXPHOS
oxidative phosphorylation
- PDK1
pyruvate dehydrogenase kinase 1
- PFKFB3
6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase 3
- PFK
phosphofructokinase
- PI3K
phosphoinositide 3‐kinase
- PKM2
pyruvate kinase M2
- ROS
reactive oxygen species
- TCA
tricarboxylic
- Treg cells
regulatory T cells
INTRODUCTION
Migration of immune cells is a fundamental process during both homeostasis and response to infection, governing the recruitment of these cells to where they are needed to carry out their functions. Immune cells are trafficked via the circulatory and lymphatic systems, and some become resident to various tissues throughout the body. Certain cell types, such as neutrophils and monocytes, patrol and circulate in the blood, ready to be rapidly recruited to sites of inflammation. Naive T cells also recirculate and repeatedly home to secondary lymphoid organs to search for their cognate antigen. Injury, inflammation and infection stimulate the recruitment of immune cells into tissues. As well, activated dendritic cells (DCs) migrate from the inflammatory site to the tissue‐draining lymph node to instruct T‐cell differentiation and proliferation. Spatial positioning of cells within the lymph node also facilitates interactions between specific immune cell subsets, promoting the development of a context‐specific adaptive immune response.
In each of these situations, the migratory capacity of the immune cell depends on the combination of chemokine receptors and integrins expressed, the ability to dynamically remodel the cytoskeleton and sufficient metabolic activity to meet the bioenergetic demands of cell motility. The interplay between cellular metabolism, signalling and cell motility is highly integrated and co‐ordinated to enable rapid migration of immune cells in diverse contexts. In this review, we present an overview of recent advances in our understanding of the spatial relationship between the cytoskeletal and metabolic pathways, how chemotaxis depends on glycolytic metabolism and the impact of environmental conditions on cellular metabolism. The potential implications for immune cell migratory capacity and function during infection and in certain disease contexts are discussed.
OVERVIEW OF IMMUNOMETABOLISM
Cellular metabolism is a central determinant of immune cell function, activation and differentiation. Immune cells have diverse fates and functions that require specific metabolic activity to support their bioenergetic and biosynthetic needs. ATP is the main energetic currency of the cell and is generated by glycolysis and oxidative phosphorylation (OXPHOS). Glycolysis occurs in the cytoplasm of the cell, breaking down glucose into pyruvate, which can be converted into lactic acid, or shuttled into the mitochondria to enter the tricarboxylic acid (TCA) cycle (Figure 1). The TCA cycle produces reducing agents that donate electrons to the electron transport chain (ETC) during OXPHOS. The transfer of electrons creates a proton gradient across the inner mitochondrial membrane, with the final electron acceptor being oxygen. The proton gradient powers ATP synthesis via proton flow through ATP synthase, coupling energy generation with oxygen consumption. The cell also consumes oxygen outside of energy production, such as for making reactive oxygen species (ROS), which are by‐products of oxidative metabolism, but are also produced by some innate cells as an antimicrobial defence mechanism. In addition to energy production, both glycolysis and OXPHOS involve metabolic intermediates that can be points of entry into these pathways or generated to fuel other pathways in a complex metabolic network for building nucleotides, lipids and proteins. Nutrients in the environment affect immune cell function by shaping metabolic programming. Proteins such as mTOR, AMPK and HIF‐1ɑ are involved in sensing the signals immune cells receive and coordinating the metabolic network to respond accordingly. Several reviews highlight details of the rapidly expanding field of immunometabolism. 1 , 2 , 3
Figure 1.
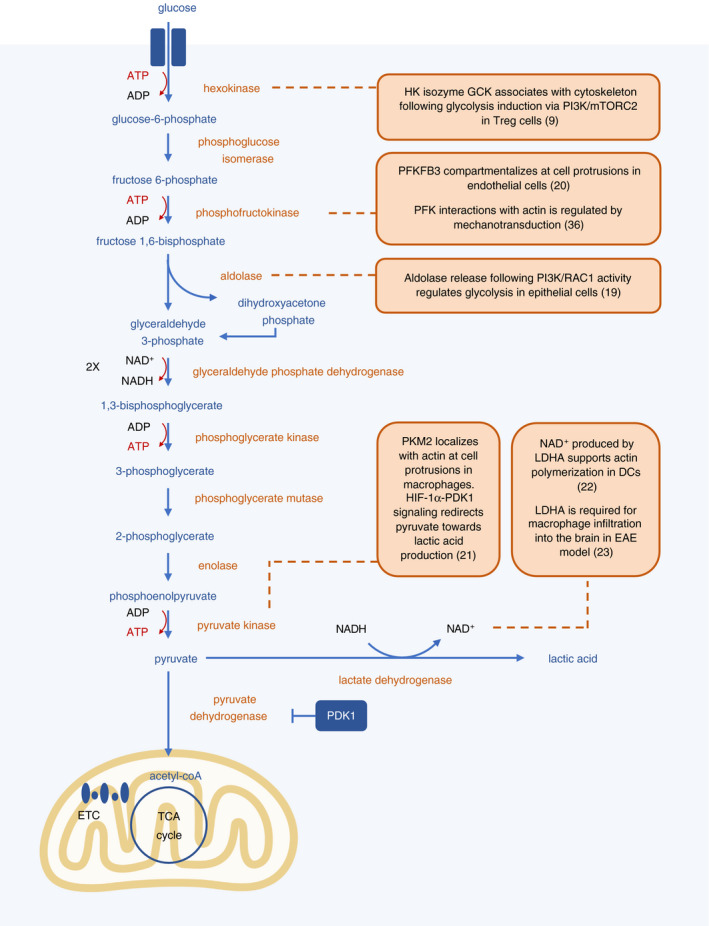
Several glycolytic enzymes localize at the cytoskeleton. During glycolysis, glucose is broken down to pyruvate in a series of enzymatic reactions that take place in the cytosol, resulting in a net output of 2 molecules each of ATP and NADH. Pyruvate can be converted to lactic acid, or transported into the mitochondria and converted into acetyl‐coA. Several enzymes involved in the glycolysis pathway have been shown to be intimately linked with local energy generation and cytoskeletal rearrangements in both immune and non‐immune cells. Scaffolding to cytoskeletal components also increases the efficiency of enzymatic reactions.
COMPARTMENTALIZED METABOLISM IN CELL ADHESION AND MOTILITY
Immune cells largely engage in amoeboid movements, with leading edge protrusion and contraction of the cell body. 4 This mode of migration differs from modes that are used by other cell types during development and tissue remodelling. The weaker adhesive interactions of immune cells allow for more rapid movement and probing of their extracellular environment. 4 Two‐dimensional migration, such as crawling along or crossing endothelial or epithelial linings, is integrin‐dependent for immune cells. 5 During three‐dimensional migration, such as movement within tissues, immune cells are capable of integrin‐dependent and integrin‐independent movement. 5 , 6 Once the immune cell is within the tissue, the cell moves by protrusive flow of actin. When the environment is especially dense, contraction of the trailing edge is mediated by myosin II in order to push along the relatively inflexible nuclei. These observations were established in DCs, T cells, B cells and granulocytes, suggesting these are common migratory mechanisms among migrating immune cells. 5 , 7
The dynamic remodelling of the cytoskeleton that occurs during cell motility is energetically demanding, most notably for the use of ATP in actin polymerization and GTP by the Rho family of GTPases and microtubules. Although OXPHOS is more efficient at generating ATP, the requirement of glycolysis for cell motility has been observed in multiple systems for over 40 years. 8 , 9 , 10 , 11 Unlike the enzymes involved in OXPHOS, glycolytic enzymes are not partitioned by membrane‐bound organelles. However, co‐ordination of cell movement demands local energy production furnished through compartmentalization of glycolysis. Indeed, it has long been observed that many glycolytic enzymes are associated with the cytoskeleton. 12 , 13 , 14 The anchoring of glycolytic enzymes at the cytoskeleton also promotes the efficiency of glycolysis by bringing the enzymes in close proximity. 15
More recently, specific compartmentalization of glycolytic enzymes has been shown to be important for cytoskeletal remodelling required for cell motility (Figure 1). The phosphoinositide 3‐kinase (PI3 K)‐AKT pathway links several types of cell surface receptors—including growth factor receptors, T‐cell receptors, B‐cell receptors, Toll‐like receptors and integrins—to metabolic changes, primarily promoting glycolysis. 16 , 17 PI3 K signalling substantially colocalizes with F‐actin bundles at membrane protrusions and participates in a positive feedback loop of actin polymerization. 18 Aldolase, a glycolytic enzyme that converts fructose‐1,6‐bisphosphate to glyceraldehyde 3‐phosphate and dihydroxyacetone phosphate, is associated with F‐actin. 19 Upon actin remodelling co‐ordinated by PI3 K and RAC1, aldolase is released, activating its activity in the glycolytic pathway. 19 These studies suggest that PI3 K signalling is a mechanism that spatially regulates glycolysis at areas of active cytoskeletal remodelling. Another enzyme that localizes at the cytoskeleton is 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase 3 (PFKFB3), which converts fructose‐6‐phosphate to fructose‐2,6‐bisP and increases glycolytic activity. 20 PFKFB3 compartmentalizes at cell protrusions through actin interactions in endothelial cells to promote their migration during vessel formation. 20 In T regulatory (Treg) cells, glucokinase, a hexokinase isozyme, inducibly associates with the cytoskeleton following PI3 K‐mTORC2 signalling to generate energy to fuel Treg cell migration. 9
Compartmentalized metabolism may also help direct the flux of metabolites to specific pathways. For example, pyruvate kinase M2 (PKM2) was found to be co‐localized with F‐actin at cell protrusions in macrophages. 21 Under hypoxic conditions, macrophages have enhanced migratory capacity, attributed in part to HIF‐1ɑ‐pyruvate dehydrogenase kinase 1 (PDK1) signalling that redirects flux of pyruvate away from the TCA cycle and instead into lactate production, leading to localized energy generation. 21 In DCs, NAD+ produced by lactate dehydrogenase (LDHA) has been shown to support actin polymerization and thus migration. 22 Generation of NAD+ can help sustain glycolytic flux, as glycolytic enzyme glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) requires NAD+ for its enzymatic reaction. Inflammatory macrophages in the encephalomyelitis mouse model of multiple sclerosis likely rely on a similar mechanism, as they also require LDHA as well as lactate transporter MCT‐4 for infiltration into the brain. 23 In addition, nucleoside diphosphate kinases, which can generate GTP from local ATP, can bind the membrane remodeler dynamin and microtubule filament tubulin, channelling GTP to these proteins locally and further supporting cytoskeletal rearrangements. 15
Subcellular localization of mitochondria also plays an important role in immune cell migration (Figure 2). Mitochondria have been shown to accumulate at the rear (uropod) of migrating T cells in response to C‐X‐C motif ligand 12 (CXCL12) or C‐C motif ligand 21 (CCL21). 24 Mitochondrial Rho GTPase 1 (MIRO‐1) contributes to mitochondrial redistribution by coupling mitochondria to microtubule dynein motors, thereby facilitating their positioning to the cell uropod. 25 This redistribution of mitochondria requires mitochondrial fission, which is regulated by the GTPase dynamin‐related protein 1 (DRP1), and DRP1 deficiency results in impaired T‐cell transmigration across endothelial cells as well as infiltration into tumours. 26 Localized mitochondrial ATP is necessary for the ATP‐dependent phosphorylation of myosin light chain (MLC), which activates the ATPase activity of myosin II that is required for contraction of the actin cytoskeleton at the cell rear. 24 , 26 While most mitochondria localize to the cell rear, a smaller portion of mitochondria accumulate near the front of the migrating T cell. 27 These mitochondria provide ATP that is released and acts on the cell's purinergic P2X4 receptor, which promotes the formation of actin protrusions. 27 Neutrophils, despite generating energy primarily through glycolysis, 28 have also been shown to exhibit this differential activation of mitochondria in response to a chemoattractant. 29 The minor portion of mitochondria that localizes to the front of the cell displays higher membrane potential and is enhanced by local mTOR signalling. 29 In addition, disruption of components of the ETC and antioxidant proteins in neutrophils of zebrafish hampers their migratory abilities, demonstrating the importance of the ETC and redox status, respectively, for neutrophil motility. 30 Together, these studies provide evidence that mitochondria with contrasting metabolic properties localize to either ends of a polarized immune cell, and that both groups significantly support migration.
Figure 2.
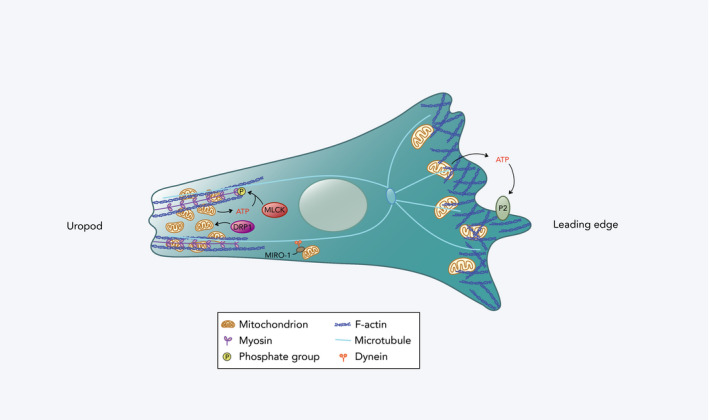
Mitochondrial redistribution in a migrating immune cell. At the leading edge, a small proportion of mitochondria with relatively high membrane potential accumulates. ATP derived from these mitochondria is released extracellularly and acts on the cell's purinergic P2 receptors (P2X4 and P2Y2 receptors in T cells and neutrophils, respectively), promoting the formation of actin protrusions. At the cell rear (uropod), there is localization of a larger fraction of mitochondria with lower membrane potential that are regulated by DRP1. These mitochondria are redistributed by coupling to dynein motor proteins on microtubules by MIRO‐1. They facilitate contraction of the uropod by providing ATP for myosin light chain kinase (MLCK) phosphorylation, and thus, MLC activation in T cells.
Cellular metabolism can also be affected by interactions with the extracellular matrix (ECM). Integrins couple the cytoskeleton and the ECM to generate traction along a surface during cell migration. Integrins are also chief mediators of mechanotransduction, a process by which mechanical stimuli such as stiffness are translated to biochemical signals, which impact cellular functions. 6 , 31 , 32 Integrin signalling engages signalling cascades that promote cell spreading and survival, and stimulates pathways such as PI3 K/AKT known to affect cellular metabolism. 33 Although there is a well‐established link between cytoskeletal rearrangements, integrin signalling and mechanotransduction in immune cells, 32 , 34 how these processes directly affect cellular metabolism and migratory capacity of immune cells is currently not well understood. However, a number of studies in other cell types suggest that integrin engagement and signalling result in changes in metabolism. 35 For example, a recent study establishes a link between integrins, mechanotransduction and glycolysis. 36 Cells can sense the stiffness of their environment through engagement of integrins. Under low adhesion and reduced integrin signalling (low stiffness), E3 ubiquitin ligase TRIM21 can be released from the actin cytoskeleton to target rate‐limiting enzyme phosphofructokinase (PFK) for degradation, resulting in downregulation of glycolysis. 36 Another study shows the integrin coreceptor SLC3A2 (CD98hc) regulates integrin mechanosensing via sphingolipid metabolism, potentially by altering membrane composition. 37 As integrin engagement is often localized to establish cell polarity in migrating cells, it is likely a mechanism that triggers localized glycolysis at areas of active cytoskeletal rearrangements. 38 While integrins are undoubtedly crucial for an effective immune system, 39 , 40 determining the impact of mechanosensing through integrin signalling on metabolic activity in immune cells requires further investigation.
RECIPROCAL RELATIONSHIP BETWEEN METABOLISM AND CHEMOTAXIS
Migration of immune cells is often directed by chemokine gradients and their corresponding receptors. C‐C motif chemokine receptor 7 (CCR7), a chemokine receptor expressed by several cell types, has been the most studied within the context of metabolism. CCR7 detects chemokines CCL19 and CCL21, which are expressed by lymphatic endothelial cells and high endothelial venules, resulting in migration into the lymph node. 41 Within the lymph node, these chemokines are produced by a network of follicular reticular cells to assist with positioning of CCR7‐expressing DCs and T cells to promote efficient interactions. 42 Cells residing in peripheral tissues, such as DCs, upregulate CCR7 expression upon pathogen encounter, enabling their migration to lymph nodes. 43 Glycolytic metabolism has been shown to be necessary for upregulation of CCR7 expression as well as CCR7 oligomerization that is required for optimal signalling. 8 , 44 , 45 HIF‐1ɑ, which promotes expression of glycolytic machinery, 46 is essential for myeloid cell motility. 47 Accordingly, HIF‐1ɑ deficiency results in less CCR7 expression and decreased migration towards CCL19 in bone marrow‐derived DCs under hypoxic conditions. 48
However, the relationship observed between glycolysis and chemokine receptors is not universal. In HIF1ɑ‐deficient DCs, migration towards CXCR4 ligand CXCL12 was unaffected, indicating differing metabolic regulation between chemokine receptors. 48 CD62L, like CCR7, promotes trafficking to secondary lymphoid organs and is normally expressed at lower levels in cytotoxic T lymphocytes (CTLs) relative to naive and memory T cells. In contrast to HIF‐1 deficiency in DCs, CTLs deficient in HIF‐1β, which dimerizes with HIF‐1ɑ to form active HIF‐1, have higher expression of CCR7 and CD62L compared to wild‐type CTLs. 49 The observed increase in CD62L expression was shown to be due to the loss of HIF‐1β‐regulated maintenance of glucose uptake, rather than direct transcriptional regulation by HIF‐1β. 49
There is also evidence that chemokine receptors can modulate metabolism via intracellular signalling. In DCs, CCR7‐mediated suppression of HIF‐1ɑ expression by the long non‐coding RNA lnc‐Dpf3 is an important regulatory mechanism in the later stages of an immune response to decrease glycolysis and prevent aberrant inflammation. 22 In Treg cells, engagement of CCL22 with CCR4 also increases glycolysis, but does not affect oxygen consumption. 9 Furthermore, signalling through CCR5 by inflammatory chemokine CCL5 promotes AMPK signalling and glucose uptake by human T cells, and blocking glucose metabolism impairs chemotaxis. 50
Collectively, these studies demonstrate that there are reciprocal and context‐dependent interactions between cellular metabolism and chemokine receptors. Targeting these mechanisms may be a way for pathogens to evade the immune system. Many intracellular pathogens reprogramme host cell metabolism to obtain nutrients and energy for replication. 51 For example, HIV‐1 can use CCR5 as a coreceptor to enter T cells and preferentially infects CD4+ T‐cell subsets that have high rates of glycolysis and oxidative phosphorylation. 52 Glucose metabolism is required to support the metabolic needs of the virus; however, infection also inhibits T‐cell migration. 52 , 53 , 54 Several mechanisms mediating this inhibition, including CCR7 downregulation 55 and inhibition of cytoskeletal remodelling by cofilin inactivation, 56 have been described. Since infected T cells display enhanced metabolic activity compared to non‐infected cells, it has been proposed that binding of CCR5 by HIV‐1 may boost glycolysis similarly to CCL5 engagement. 50 Reliance on the host cell's glycolytic machinery is common for many intracellular pathogens, 57 , 58 and whether diversion of the glycolytic pathway to support viral functions also contributes to migration defects should be examined.
CONSEQUENCES OF HYPOXIC MICROENVIRONMENTS ON CELL MIGRATION
Immune cells can encounter varying levels of O2 under physiological conditions, such as in circulation or within different tissues. O2 levels can also differ in pathological contexts, such as at a site of infection or inflammation, or within tumours. Hypoxia leads to HIF‐1ɑ stabilization and increased glycolysis that can result in elevated microenvironmental lactate levels. 59 Extracellular lactate can impair motility and IL‐17 production in CD4+ T cells. 60 Synovial fluid from the joints of rheumatoid arthritis patients contains higher levels of lactate compared to those with a non‐inflammatory type of arthritis, and this is thought to drive chronic inflammation by inhibiting migration and, therefore, entrapping Th17 cells. 60 The tumour microenvironment can also have high lactate levels due to the strikingly glycolytic nature of many tumour types. Deleting LDHA in melanoma cells in combination with anti‐PD1 therapy improves infiltration and anti‐tumour functions of CTLs, 61 further highlighting the inhibitory effect of lactate on T‐cell motility.
The capacity of immune cells to adapt to fluctuating levels in O2 varies among different immune cell types. In the context of glioblastoma, hypoxic regions correlate with tumour malignancy, 62 and Treg cells promote tumour growth by aiding in immunosuppression. Treg cells have previously been shown to predominantly require oxidative metabolism for their suppressive functions, while glycolysis promotes their migratory capacity. 9 In line with these findings, Treg cells that are present in the tumour microenvironment of brain tumours rely on a HIF‐1ɑ‐mediated metabolic shift to glycolysis for enhanced infiltration into the tumours, and lipid oxidation for their suppressive functions. 63 Under homeostatic conditions, circulating T cells face regular changes in oxygen availability. Circulating memory T cells possess greater spare respiratory capacity compared to naive T cells, ensuring sufficient ATP levels under hypoxic conditions to bioenergetically support their migration. 64
In certain pathological contexts, neutrophils, along with some other myeloid cell types, can create hypoxic regions themselves by rapidly releasing reactive oxygen species (ROS), generated by NADPH oxidase in a process known as respiratory burst. In a murine model of acute colitis, neutrophils that migrate into the intestinal tissues consume microenvironmental O2 to support respiratory burst. 65 This depletion of O2 stabilizes intestinal epithelial cell HIF, inducing the expression of genes that are protective to the barrier, thus inhibiting further infiltration and promoting resolution of inflammation. Neutrophils lacking NADPH oxidase cannot induce hypoxia in the intestinal epithelium and, as a result, display defective inflammatory resolution. 65 Another setting where neutrophil infiltration creates a hypoxic microenvironment is during the development of herpes stromal keratitis, a chronic inflammatory condition of the cornea due to recurrent herpes simplex virus‐1 infection. 66 However, in this case, hypoxia partly contributes to the progression of these lesions, with HIF‐2ɑ becoming stabilized in the epithelial cells of these lesions, while HIF‐1ɑ is stabilized in the infiltrating immune cells. Microenvironmental O2 depletion during disease settings that involve heavy neutrophil infiltration is not always as a result of neutrophil respiratory burst, however. During intestinal infection by Shigella flexneri, aerobic respiration by the bacteria, rather than the recruited neutrophils, is mainly responsible for the observed O2 depletion and formation of hypoxic foci of infection. 67 Local O2 depletion by pathogenic bacteria may affect O2‐dependent antimicrobial functions of neutrophils, such as respiratory burst, due to competition.
DYSLIPIDEMIA AND IMMUNE CELL MOTILITY
While cellular metabolism certainly impacts immune cell migration, host metabolism and metabolic alterations can significantly contribute to migration defects. One example of this is the impact of abnormal levels of lipids in the blood—referred to as dyslipidemia—on immune cell migration. 68 Lipids are well appreciated to have many effects on immune cells, including roles in generating energy and membrane components, and in activation and differentiation. 69 In the extracellular environment, excessive levels of saturated fatty acids promote pro‐inflammatory phenotypes of macrophages and T cells, while polyunsaturated fatty acids have generally been described to have anti‐inflammatory properties. 70 Lipids such as cholesterol and triglycerides are transported in the blood in association with lipoproteins, with low‐density lipoproteins (LDL) delivering lipids to tissues from the liver, while high‐density lipoproteins (HDL) transport lipids from tissues back to the liver. Abnormal levels of lipids in the blood are strongly associated with an increased risk of developing cardiovascular disease. 71 Dyslipidemia occurs in obesity and diabetes, following excessive consumption of dietary fats, and is also observed in several chronic inflammatory disorders including rheumatoid arthritis, psoriasis and infections like HIV. 71 Obesity and diabetes are known to increase the risk of mortality and severe complications resulting from respiratory infection, 72 , 73 , 74 including the novel coronavirus SARS‐CoV‐2. 75 Epidemiological evidence suggests dyslipidemia itself—both abnormally low or high cholesterol levels—is associated with heightened susceptibility to acute infection as well, although causality has not yet been proven clinically. 76 , 77 Mouse models of spontaneous hypercholesterolaemia have demonstrated that dyslipidemia increases susceptibility to infections by pathogens such as Mycobacterium tuberculosis, 78 Listeria monocytogenes 79 and Leishmania major. 80
While dyslipidemia and associated diseases can affect immune cell function in many ways, there are increasing associations with a direct affect on cell migration and motility. For example, in macrophages, cholesterol accumulation can inhibit their migration. 81 , 82 ATP‐binding cassette transporters ABCA1 and ABCG1 promote cholesterol efflux to apolipoproteins, and HDL, respectively. 83 Macrophages deficient in both Abca1 and Abcg1 have increased plasma membrane cholesterol due to reduced movement of cholesterol across the plasma membrane bilayer. 83 This results in excessive and dysregulated activated RAC1 and increased actin polymerization, but a defective migratory response. Treatment of macrophages containing excessive cholesterol with apolipoprotein A1 (apo A1;a major component of HDL), or HDL, restores migration of control macrophages, but not of macrophages deficient in Abca1 and Abcg1, confirming the involvement of these transporters in cholesterol accumulation and macrophage migration. 81 , 83 In monocytes and macrophages that do not contain excessive cholesterol, apoA1 has inhibitory effects on chemotaxis and recruitment by disrupting membrane lipid rafts, which then attenuates PI3 K/AKT signalling involved in cytoskeletal remodelling. 84 Acute exposure to apoA1 is sufficient to inhibit monocyte and macrophage migration, while chronic exposure to apoA1 in other studies leads to additional effects on the cells, including decreased expression of chemokine receptors. 85 These studies indicate that the level of cholesterol in the cell requires precise regulation for optimal cell motility. Furthermore, very low‐ and low‐density lipoproteins (VLDL and LDL, respectively) cause neutral lipid accumulation in human monocytes and mouse peritoneal macrophages, impairing their migratory abilities due to blunting of RHOA activation, and consequently of cytoskeletal rearrangements. 86 Oxidized LDL inhibits macrophage migration by interacting with the scavenger receptor CD36, which triggers a pathway that results in RAC1 activation and RAC‐mediated inhibition of MLC kinase. This leads to MLC dephosphorylation and retraction of the front end lamellipodia, thus resulting in loss of cell polarity. 87 Oxidized LDL has been shown to impair DC trafficking as well. 88 These studies mainly describe mechanisms by which dyslipidemia can affect immune cell migration via effects on the cytoskeleton; other reviews have comprehensively covered further ways dyslipidemia can affect immune cell trafficking, including the effects on lymphatic vessels and on immune cell retention in atherosclerotic plaques. 89 , 90 , 91
PERSPECTIVES
A vital function of immune cells is migration during development, inflammation and homeostasis. Migration is an energetically demanding process requiring significant metabolic activity, which is co‐ordinated through cytoskeletal interactions to provide local, efficient energy production. Enzymes in the TCA cycle and ETC are compartmentalized in the mitochondrion for optimal enzymatic activity, whereas the glycolytic pathway is compartmentalized by anchoring to the cytoskeleton. However, how this compartmentalization is important for local production of other metabolites or redox factors, and their impact on cellular function, requires further research. Integrated networks among metabolic pathways and other signalling pathways through engagement of chemokine receptors and integrins are evident; however, it is important to uncover other pathways necessary for immune function that are also intimately linked to cellular metabolism. Further, a better understanding of the extent to which metabolic pathways beyond glycolysis and OXPHOS are compartmentalized to support local cellular needs will shed light on the metabolic flexibility of cells and their ability to function in different environments.
Nutrient availability in microenvironments—whether in primary or secondary lymphoid tissues or in inflamed peripheral tissues—will impact the metabolic pathways used by cells to support their bioenergetic and biosynthetic needs. 92 How local nutrient availability affects immune homeostasis and response to infection adds another dimension to our understanding of host immunity. For example, the increased metabolic demands of both tumour cells and activated immune cells lead to competition for nutrients in the tumour microenvironment. Anti‐tumour immunity can be influenced by nutrient depletion in the tumour microenvironment, such as glucose restriction by tumours impairing T‐cell effector function, 93 and glutamine depletion promoting the differentiation of T cells into Treg cells. 94 Nutrient competition in the dense lymph node environment likely also occurs, potentially impacting both spatial organization of immune cells and egress to the periphery. Studies examining systemic altering of nutrient availability, largely through diet manipulations, are demonstrating profound effects on immune function. Dietary restriction of methionine reduces disease severity in the experimental autoimmune encephalomyelitis model of multiple sclerosis by limiting the expansion of Th17 cells. 95 Furthermore, mice fed a diet lacking non‐essential amino acids serine and glycine exhibit a reduced number of antigen‐specific CD8+ T cells when challenged with Listeria monocytogenes infection. 96 A ketogenic diet is protective in mice during an influenza A virus infection by promoting the expansion of γδ T cells in the lung. 97
While it is clear that diet has considerable effects on immune function and response to infection, the specific effects on the ability of immune cells to patrol the host and migrate into inflamed tissues is not known. Further, whether nutrient depletion in a highly inflammatory microenvironment serves as a feedback mechanism to reduce inflammatory migration and resolve inflammation needs to be examined. Unravelling the molecular basis of the reciprocal relationship between cellular metabolism and immune cell migration will expand our understanding of host immunity. The impact of systemic nutrient alteration through diet or disease will shed light on whether specific aspects of host nutrition can be targeted to increase immunity or decrease inflammation.
CONFLICT OF INTEREST
The authors declare no competing interests.
ACKNOWLEDGMENTS
We thank members of the Krawczyk laboratory for thoughtful discussions and advice. We thank Drs. N. Truong, E. Ma, C. Scullion for feedback and editing. H.G. was supported by a FRQS Doctoral Award.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Metabolic mediators: how immunometabolism directs the immune response to infection. Immunology 2020, 161: 163‐164.
Circles of Life: linking metabolic and epigenetic cycles to immunity. Immunology 2020, 161: 165‐174.
The role of O‐GlcNAcylation in immunity against infections. Immunology 2020, 161: 175‐185.
The battle for iron in enteric infections. Immunology 2020, 161: 186‐199.
DATA AVAILABILITY STATEMENT
No new data were generated for this manuscript.
REFERENCES
- 1. Buck M.D., Sowell R.T., Kaech S.M., Pearce E.L.. Metabolic instruction of immunity. Cell 2017; 169(4):570–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ganeshan K., Chawla A.. Metabolic regulation of immune responses. Annu Rev Immunol. 2014; 32:609–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O’Neill LAJ, Kishton R.J., Rathmell J.. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016; 16(9):553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Friedl P., Weigelin B.. Interstitial leukocyte migration and immune function. Nat Immunol. 2008; 9(9):960–9. [DOI] [PubMed] [Google Scholar]
- 5. Lämmermann T., Bader B.L., Monkley S.J., Worbs T., Wedlich‐Söldner R., Hirsch K. et al. Rapid leukocyte migration by integrin‐independent flowing and squeezing. Nature 2008; 453(7191):51–5. [DOI] [PubMed] [Google Scholar]
- 6. Doyle A.D., Carvajal N., Jin A., Matsumoto K., Yamada K.M.. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility‐dependent adhesions. Nat Commun. 2015; 6:8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Friedl P., Entschladen F., Conrad C., Niggemann B., Zänker K.S.. CD4+ T lymphocytes migrating in three‐dimensional collagen lattices lack focal adhesions and utilize β1 integrin‐independent strategies for polarization, interaction with collagen fibers and locomotion. Eur J Immunol. 1998; 28(8):2331–43. [DOI] [PubMed] [Google Scholar]
- 8. Guak H., Al Habyan S., Ma E.H., Aldossary H., Al‐Masri M., Won S.Y. et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun. 2018; 9(1):2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kishore M., Cheung KCP, Fu H., Bonacina F., Wang G., Coe D. et al. Regulatory T cell migration is dependent on glucokinase‐mediated glycolysis. Immunity 2018; 48(4):831–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shiraishi T., Verdone J.E., Huang J., Kahlert U.D., Hernandez J.R., Torga G. et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget. 2015; 6(1):130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D’Silva H., Gothoskar B.P., Jain V.K., Advani S.H.. Cell migration and energy generating metabolic activities. Eur J Cancer. 1978; 14(11):1243–8. [DOI] [PubMed] [Google Scholar]
- 12. Masters C.. Interactions between glycolytic enzymes and components of the cytomatrix. J Cell Biol. 1984; 99(1 Pt 2):222s–225s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arnold H., Pette D.. Binding of glycolytic enzymes to structure proteins of the muscle. Eur J Biochem. 1968; 6(2):163–71. [DOI] [PubMed] [Google Scholar]
- 14. Srere P.A.. Complexes of sequential metabolic enzymes. Annu Rev Biochem. 1987; 56:89–124. [DOI] [PubMed] [Google Scholar]
- 15. Zala D., Schlattner U., Desvignes T., Bobe J., Roux A., Chavrier P. et al. The advantage of channeling nucleotides for very processive functions. F1000Res. 2017; 6:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoxhaj G., Manning B.D.. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2019; 20(2):74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. So L., Fruman D.A.. PI3K signalling in B‐ and T‐lymphocytes: new developments and therapeutic advances. Biochem J. 2012; 442(3):465–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson H.E., King S.J., Asokan S.B., Rotty J.D., Bear J.E., Haugh J.M.. F‐actin bundles direct the initiation and orientation of lamellipodia through adhesion‐based signaling. J Cell Biol. 2015; 208(4):443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu H., Juvekar A., Lyssiotis C.A., Lien E.C., Albeck J.G., Oh D. et al. Phosphoinositide 3‐kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 2016; 164(3):433–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B.W., Cantelmo A.R. et al. Role of PFKFB3‐driven glycolysis in vessel sprouting. Cell 2013; 154(3):651–63. [DOI] [PubMed] [Google Scholar]
- 21. Semba H., Takeda N., Isagawa T., Sugiura Y., Honda K., Wake M. et al. HIF‐1α‐PDK1 axis‐induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016; 7:11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J., Zhang X., Chen K., Cheng Y., Liu S., Xia M. et al. CCR7 Chemokine Receptor‐inducible lnc‐Dpf3 restrains dendritic cell migration by inhibiting HIF‐1α‐mediated glycolysis. Immunity 2019; 50(3):600–15.e15. [DOI] [PubMed] [Google Scholar]
- 23. Kaushik D.K., Bhattacharya A., Mirzaei R., Rawji K.S., Ahn Y., Rho J.M. et al. Enhanced glycolytic metabolism supports transmigration of brain‐infiltrating macrophages in multiple sclerosis. J Clin Invest. 2019; 129(8):3277–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Campello S., Lacalle R.A., Bettella M., Mañes S., Scorrano L., Viola A.. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006; 203(13):2879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morlino G., Barreiro O., Baixauli F., Robles‐Valero J., Gonzalez‐Granado J.M., Villa‐Bellosta R. et al. Miro‐1 links mitochondria and microtubule dynein motors to control lymphocyte migration and polarity. Mol Cell Biol 2014; 34(8):1412–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simula L., Pacella I., Colamatteo A., Procaccini C., Cancila V., Bordi M. et al. Drp1 controls effective T cell immune‐surveillance by regulating T cell migration, proliferation, and cMyc‐dependent metabolic reprogramming. Cell Rep. 2018; 25(11):3059–73.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ledderose C., Liu K., Kondo Y., Slubowski C.J., Dertnig T., Denicoló S. et al. Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J Clin Invest. 2018; 128(8):3583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maianski N.A., Geissler J., Srinivasula S.M., Alnemri E.S., Roos D., Kuijpers T.W.. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004; 11(2):143–53. [DOI] [PubMed] [Google Scholar]
- 29. Bao Y., Ledderose C., Graf A.F., Brix B., Birsak T., Lee A. et al. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol. 2015; 210(7):1153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou W., Cao L., Jeffries J., Zhu X., Staiger C.J., Deng Q.. Neutrophil‐specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. Dis Model Mech. 2018; 11(3):dmm033027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jaalouk D.E., Lammerding J.. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009; 10(1):63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rossy J., Laufer J.M., Legler D.F.. Role of Mechanotransduction and Tension in T Cell Function. Front Immunol. 2018; 9:2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geiger B., Bershadsky A., Pankov R., Yamada K.M.. Transmembrane crosstalk between the extracellular matrix and the cytoskeleton. Nat Rev Mol Cell Biol. 2001; 2(11):793–805. [DOI] [PubMed] [Google Scholar]
- 34. Upadhyaya A.. Mechanosensing in the immune response. Semin Cell Dev Biol. 2017; 71:137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ata R., Antonescu C.N.. Integrins and cell metabolism: an intimate relationship impacting cancer. Int J Mol Sci. 2017; 18(1):189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park J.S., Burckhardt C.J., Lazcano R., Solis L.M., Isogai T., Li L. et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020; 578(7796):621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boulter E., Estrach S., Tissot F.S., Hennrich M.L., Tosello L., Cailleteau L. et al. Cell metabolism regulates integrin mechanosensing via an SLC3A2‐dependent sphingolipid biosynthesis pathway. Nat Commun. 2018; 9(1):4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huttenlocher A., Horwitz A.R.. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011; 3(9):a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gunawan M., Venkatesan N., Loh J.T., Wong J.F., Berger H., Neo W.H. et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat Immunol 2015; 16:505–16. [DOI] [PubMed] [Google Scholar]
- 40. Morrison V.L., James M.J., Grzes K., Cook P., Glass D.G., Savinko T. et al. Loss of beta2‐integrin‐mediated cytoskeletal linkage reprogrammes dendritic cells to a mature migratory phenotype. Nat Commun. 2014; 5:5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Griffith J.W., Sokol C.L., Luster A.D.. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014; 32:659–702. [DOI] [PubMed] [Google Scholar]
- 42. Eisenbarth S.C.. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. 2019; 19(2):89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Worbs T., Hammerschmidt S.I., Förster R.. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017; 17(1):30–48. [DOI] [PubMed] [Google Scholar]
- 44. Hauser M.A., Schaeuble K., Kindinger I., Impellizzieri D., Krueger W.A., Hauck C.R. et al. Inflammation‐induced CCR7 oligomers form scaffolds to integrate distinct signaling pathways for efficient cell migration. Immunity 2016; 44(1):59–72. [DOI] [PubMed] [Google Scholar]
- 45. Everts B., Amiel E., Huang SC.‐C., Smith A.M., Chang C.‐H., Lam W.Y. et al. TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKɛ supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014; 15(4):323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Corcoran S.E., O’Neill LAJ. HIF1α and metabolic reprogramming in inflammation. J Clin Invest. 2016; 126(10):3699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cramer T., Yamanishi Y., Clausen B.E., Förster I., Pawlinski R., Mackman N. et al. HIF‐1α Is essential for myeloid cell‐mediated inflammation. Cell 2003; 112(5):645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Köhler T., Reizis B., Johnson R.S., Weighardt H., Förster I.. Influence of hypoxia‐inducible factor 1α on dendritic cell differentiation and migration. Eur J Immunol. 2012; 42(5):1226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Finlay D.K., Rosenzweig E., Sinclair L.V., Feijoo‐Carnero C., Hukelmann J.L., Rolf J. et al. PDK1 regulation of mTOR and hypoxia‐inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012; 209(13):2441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chan O., Burke J.D., Gao D.F., Fish E.N.. The chemokine CCL5 regulates glucose uptake and AMP kinase signaling in activated T cells to facilitate chemotaxis. J Biol Chem. 2012; 287(35):29406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Eisenreich W., Rudel T., Heesemann J., Goebel W.. How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front Cell Infect Microbiol. 2019; 9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valle‐Casuso J.C., Angin M., Volant S., Passaes C., Monceaux V., Mikhailova A. et al. Cellular metabolism is a major determinant of HIV‐1 reservoir seeding in CD4+ T cells and offers an opportunity to tackle infection. Cell Metab. 2019; 29(3):611–26.e5. [DOI] [PubMed] [Google Scholar]
- 53. Kavanagh Williamson M., Coombes N., Juszczak F., Athanasopoulos M., Khan M.B., Eykyn T.R. et al. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV‐1. Viruses 2018; 10(3):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vérollet C., Le Cabec V., Maridonneau‐Parini I.. HIV‐1 infection of T lymphocytes and macrophages affects their migration via Nef. Front Immunol 2015; 6:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ramirez P.W., Famiglietti M., Sowrirajan B., DePaula‐Silva A.B., Rodesch C., Barker E. et al. Downmodulation of CCR7 by HIV‐1 Vpu results in impaired migration and chemotactic signaling within CD4+ T cells. Cell Rep. 2014; 7(6):2019–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stolp B., Reichman‐Fried M., Abraham L., Pan X., Giese S.I., Hannemann S. et al. HIV‐1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe. 2009; 6(2):174–86. [DOI] [PubMed] [Google Scholar]
- 57. Thaker S.K., Ch’ng J., Christofk H.R.. Viral hijacking of cellular metabolism. BMC Biol. 2019; 17(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Escoll P., Buchrieser C.. Metabolic reprogramming of host cells upon bacterial infection: Why shift to a Warburg‐like metabolism? FEBS J. 2018; 285(12):2146–60. [DOI] [PubMed] [Google Scholar]
- 59. Taylor C.T., Colgan S.P.. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. 2017; 17(12):774–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Haas R., Smith J., Rocher‐Ros V., Nadkarni S., Montero‐Melendez T., D’Acquisto F. et al. Lactate regulates metabolic and pro‐inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015; 13(7):e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Daneshmandi S., Wegiel B., Seth P.. Blockade of lactate dehydrogenase‐A (LDH‐A) improves efficacy of anti‐programmed cell death‐1 (PD‐1) therapy in melanoma. Cancers. 2019; 11(4):450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Søndergaard K.L., Hilton D.A., Penney M., Ollerenshaw M., Demaine A.G.. Expression of hypoxia‐inducible factor 1α in tumours of patients with glioblastoma. Neuropathol Appl Neurobiol. 2002; 28(3):210–7. [DOI] [PubMed] [Google Scholar]
- 63. Miska J., Lee‐Chang C., Rashidi A., Muroski M.E., Chang A.L., Lopez‐Rosas A. et al. HIF‐1α Is a Metabolic switch between glycolytic‐driven migration and oxidative phosphorylation‐driven immunosuppression of Tregs in glioblastoma. Cell Rep 2019; 27:226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dimeloe S., Mehling M., Frick C., Loeliger J., Bantug G.R., Sauder U. et al. The immune‐metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J Immunol. 2016; 196(1):106–14. [DOI] [PubMed] [Google Scholar]
- 65. Campbell E.L., Bruyninckx W.J., Kelly C.J., Glover L.E., McNamee E.N., Bowers B.E. et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014; 40:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rao P., Suvas S.. Development of inflammatory hypoxia and prevalence of glycolytic metabolism in progressing herpes stromal keratitis lesions. J Immunol. 2019; 202(2):514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tinevez J.‐Y., Arena E.T., Anderson M., Nigro G., Injarabian L., André A. et al. Shigella‐mediated oxygen depletion is essential for intestinal mucosa colonization. Nat Microbiol. 2019; 4(11):2001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rahman M.S., Murphy A.J., Woollard K.J.. Effects of dyslipidaemia on monocyte production and function in cardiovascular disease. Nat Rev Cardiol. 2017; 14(7):387–400. [DOI] [PubMed] [Google Scholar]
- 69. Cucchi D., Camacho‐Muñoz D., Certo M., Pucino V., Nicolaou A., Mauro C.. Fatty acids – from energy substrates to key regulators of cell survival, proliferation and effector function. Cell Stress. 2020; 4:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hubler M.J., Kennedy A.J.. Role of lipids in the metabolism and activation of immune cells. J Nutr Biochem. 2016; 34:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Feingold K.R., Grunfeld C.. The Effect of Inflammation and Infection on Lipids and Lipoproteins In: Feingold K.R., Anawalt B., Boyce A., Chrousos G., Dungan K., Grossman A., eds. Endotext. South Dartmouth (MA): MDText.com, Inc., 2019. [Google Scholar]
- 72. Sun Y., Wang Q., Yang G., Lin C., Zhang Y., Yang P.. Weight and prognosis for influenza A(H1N1)pdm09 infection during the pandemic period between 2009 and 2011: a systematic review of observational studies with meta‐analysis. Infect Dis. 2016; 48(11–12):813–22. [DOI] [PubMed] [Google Scholar]
- 73. Allard R., Leclerc P., Tremblay C., Tannenbaum T.‐N.. Diabetes and the severity of pandemic influenza A (H1N1) infection. Diabetes Care 2010; 33(7):1491–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Badawi A., Ryoo S.G.. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus (MERS‐CoV): a systematic review and meta‐analysis. Int J Infect Dis. 2016; 49:129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Erener S.. Diabetes, infection risk and COVID‐19. Mol Metab. 2020; 39:101044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Delgado‐Rodríguez M., Medina‐Cuadros M., Martínez‐Gallego G., Sillero‐Arenas M.. Total cholesterol, HDL‐cholesterol, and risk of nosocomial infection: a prospective study in surgical patients. Infect Control Hosp Epidemiol. 1997; 18(1):9–18. [DOI] [PubMed] [Google Scholar]
- 77. Madsen C.M., Varbo A., Tybjærg‐Hansen A., Frikke‐Schmidt R., Nordestgaard B.G.. U‐shaped relationship of HDL and risk of infectious disease: two prospective population‐based cohort studies. Eur Heart J. 2018; 39(14):1181–1190. [DOI] [PubMed] [Google Scholar]
- 78. Martens G.W., Arikan M.C., Lee J., Ren F., Vallerskog T., Kornfeld H.. Hypercholesterolemia impairs immunity to tuberculosis. Infect Immun. 2008; 76(8):3464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Roselaar S.E., Daugherty A.. Apolipoprotein E‐deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J Lipid Res. 1998; 39(9):1740–3. [PubMed] [Google Scholar]
- 80. Shamshiev A.T., Ampenberger F., Ernst B., Rohrer L., Marsland B.J., Kopf M.. Dyslipidemia inhibits Toll‐like receptor–induced activation of CD8α‐negative dendritic cells and protective Th1 type immunity. J Exp Med. 2007; 204(2):441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Adorni M.P., Favari E., Ronda N., Granata A., Bellosta S., Arnaboldi L. et al. Free cholesterol alters macrophage morphology and mobility by an ABCA1 dependent mechanism. Atherosclerosis 2011; 215(1):70–6. [DOI] [PubMed] [Google Scholar]
- 82. Nagao T., Qin C., Grosheva I., Maxfield F.R., Pierini L.M.. Elevated cholesterol levels in the plasma membranes of macrophages inhibit migration by disrupting RhoA regulation. Arterioscler Thromb Vasc Biol. 2007; 27(7):1596–602. [DOI] [PubMed] [Google Scholar]
- 83. Pagler T.A., Wang M., Mondal M., Murphy A.J., Westerterp M., Moore K.J. et al. Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ Res. 2011; 108(2):194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Iqbal A.J., Barrett T.J., Taylor L., McNeill E., Manmadhan A., Recio C. et al. Acute exposure to apolipoprotein A1 inhibits macrophage chemotaxis in vitro and monocyte recruitment in vivo. Elife. 2016; 5:e15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bursill C.A., Castro M.L., Beattie D.T., Nakhla S., van der Vorst E., Heather A.K. et al. High‐density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2010; 30(9):1773–8. [DOI] [PubMed] [Google Scholar]
- 86. Jackson W.D., Weinrich T.W., Woollard K.J.. Very‐low and low‐density lipoproteins induce neutral lipid accumulation and impair migration in monocyte subsets. Sci Rep. 2016; 6:20038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Park Y.M., Drazba J.A., Vasanji A., Egelhoff T., Febbraio M., Silverstein R.L.. Oxidized LDL/CD36 interaction induces loss of cell polarity and inhibits macrophage locomotion. Mol Biol Cell. 2012; 23(16):3057–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Angeli V., Llodrá J., Rong J.X., Satoh K., Ishii S., Shimizu T. et al. Dyslipidemia associated with atherosclerotic disease systemically alters dendritic cell mobilization. Immunity 2004; 21(4):561–74. [DOI] [PubMed] [Google Scholar]
- 89. Kataru R.P., Park H.J., Baik J.E., Li C., Shin J., Mehrara B.J.. Regulation of lymphatic function in obesity. Front Physiol. 2020; 11:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tall A.R., Yvan‐Charvet L.. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015; 15(2):104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Getz G.S., Reardon C.A.. The mutual interplay of lipid metabolism and the cells of the immune system in relation to atherosclerosis. Clin Lipidol. 2014; 9:657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kedia‐Mehta N., Finlay D.K.. Competition for nutrients and its role in controlling immune responses. Nat Commun. 2019; 10(1):2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chang C.‐H., Qiu J., O’Sullivan D., Buck M.D., Noguchi T., Curtis J.D. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015; 162(6):1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Klysz D., Tai X., Robert P.A., Craveiro M., Cretenet G., Oburoglu L. et al. Glutamine‐dependent α‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal. 2015; 8(396):ra97. [DOI] [PubMed] [Google Scholar]
- 95. Roy D.G., Chen J., Mamane V., Ma E.H., Muhire B.M., Sheldon R.D. et al. Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming. Cell Metab. 2020; 31(2):250–66.e9. [DOI] [PubMed] [Google Scholar]
- 96. Ma E.H., Bantug G., Griss T., Condotta S., Johnson R.M., Samborska B. et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017; 25(2):482. [DOI] [PubMed] [Google Scholar]
- 97. Goldberg E.L., Molony R.D., Kudo E., Sidorov S., Kong Y., Dixit V.D. et al. Ketogenic diet activates protective γδ T cell responses against influenza virus infection. Sci Immunol. 2019; 4(41):eaav2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated for this manuscript.