

Summary
Metabolites are the essential substrates for epigenetic modification enzymes to write or erase the epigenetic blueprint in cells. Hence, the availability of nutrients and activity of metabolic pathways strongly influence the enzymatic function. Recent studies have shed light on the choreography between metabolome and epigenome in the control of immune cell differentiation and function, with a major focus on histone modifications. Yet, despite its importance in gene regulation, DNA methylation and its relationship with metabolism is relatively unclear. In this review, we will describe how the metabolic flux can influence epigenetic networks in innate and adaptive immune cells, with a focus on the DNA methylation cycle and the metabolites S‐adenosylmethionine and α‐ketoglutarate. Future directions will be discussed for this rapidly emerging field.
Keywords: 5‐hydroxymethylcytosine, B cells, DNA methylation, DNA methyltransferases, epigenetics, immunometabolism, Krebs cycle, macrophages, mitochondria, one‐carbon metabolism, T cells, ten–eleven translocation
The interconnected metabolic and DNA methylation cycles.
Abbreviations
- 2‐HG
2‐hydroxyglutarate
- 2OGDD
2OG‐dependent dioxygenases
- 5hmC
5‐hydroxymethylcytosine
- 5mC
5‐methylcytosine
- αKG
α‐ketoglutarate
- αKGDH
α‐ketoglutarate dehydrogenase
- CoA
coenzyme A
- DNMT
DNA methyltransferases
- EAE
experimental autoimmune encephalomyelitis
- IDH
isocitrate dehydrogenase
- IFN
interferon
- IL‐1β
interleukin‐1β
- ImmGen
Immunological Genome Project
- JmjC
Jumonji C
- JMJD
lysine (and/or arginine) demethylases
- LCMV
lymphocytic choriomeningitis virus
- NADH
nicotinamide adenine dinucleotide
- oxi‐MC
oxidized methylcytosine
- PD‐1
programmed cell death protein 1
- PHD
prolyl‐hydroxylase domain
- SAH
S‐adenosylhomocysteine
- SAM
S‐adenosylmethionine
- TET
Ten‐Eleven Translocation
- Th1
T helper type 1
- Treg
regulatory T
Introduction
Metabolism is the constellation of chemical reactions in cells. These reactions are catalyzed by specific metabolic enzymes to support and maintain cellular and tissue homeostasis. In immune cells, cell differentiation, effector function, and cell proliferation are accompanied by a meticulous metabolic reprogramming. Therefore, the imbalance of intrinsic and/or extrinsic metabolites can strongly influence the differentiation and function of immune cells and could attribute to immune‐related diseases. 1 , 2
Epigenetics describes the inheritable traits without changes in the DNA sequence. In 1957, Conrad Waddington introduced the concept of the ‘Epigenetics Landscape’ to describe the decision‐making process during cellular development. 3 In the model, developing cells are marbles on top of a hill among the landscape of choices. Despite having the same genetic material, each cell can ‘roll down’ and adopt one of the genetically pre‐defined paths and differentiate into various cell lineages. Many factors can influence the decision‐making and impact the outcome of cells, and studies have implicated cell‐intrinsic metabolic activity and cell‐extrinsic nutrient availability. Epigenetic processes are known to contribute significantly to immune cell development, activation, and function. 4 , 5 , 6 Increasing evidence has suggested that epigenetic modification enzymes can modulate the epigenetic patterns by integrating metabolic cues from metabolic pathways, including one‐carbon metabolism and the Krebs cycle. More specifically, the level of intermediary metabolites strongly affects the activity of most epigenetic enzymes, 7 which ultimately influences gene expression in immune cells in response to tissue homeostasis and inflammation.
In this review article, we first introduce the metabolism of S‐adenosylmethionine (SAM) and α‐ketoglutarate (αKG), followed by the roles of the DNA‐modifying enzymes DNA methyltransferases (DNMT) and Ten–Eleven Translocation (TET) in controlling immune cell fate. We will discuss the interconnection between metabolism, epigenome, and immunity.
One‐carbon metabolism and SAM
One‐carbon metabolism provides one‐carbon units required for the synthesis of essential metabolites, including amino acids, nucleotides, nicotinamide adenine dinucleotide (NADH), and SAM. 8 It is a crucial and complex metabolic network involving the folate and methionine cycles (Fig. 1). Notably, the one‐carbon metabolic process is largely derived from the non‐essential amino acids serine and glycine, which can be acquired from the extracellular environment or by de novo synthesis. The de novo serine biosynthesis starts from the 3‐phosphoglycerate of glycolysis, followed by serial conversions through phosphoglycerate dehydrogenase, phosphoserine aminotransferase 1, and phosphoserine phosphatase (Fig. 1). Deprivation of serine was showed to induce cell cycle arrest in proliferating lymphocytes 9 , 10 and tumor cells. 11
Figure 1.
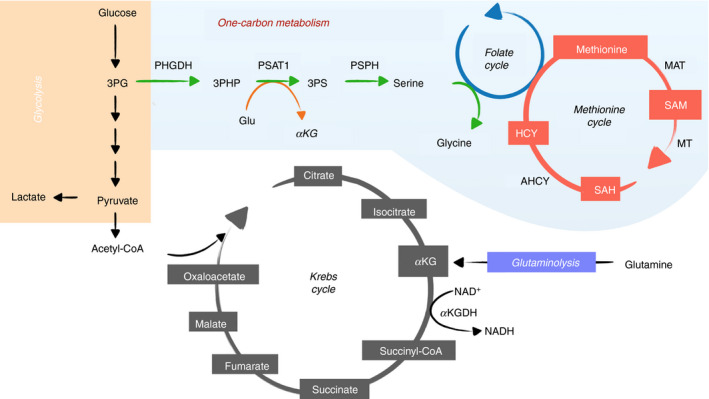
Metabolic pathways that provide metabolic substrates for epigenetic cycle. Glycolysis (orange), involves the enzymatic catabolism of glucose to pyruvate and lactate in the cytoplasm. Pyruvate can be converted to acetyl‐CoA in mitochondria and shuttled through several enzymatic reactions of the Krebs cycle (gray) to generate metabolic intermediates. Glutamine through glutaminolysis (purple) can be metabolized to α‐ketoglutarate in the Krebs cycle. Intermediates from glucose catabolism during glycolysis can branch out through the serine one‐carbon metabolism (blue) to generate amino acids of serine and glycine fueling into the folate and methionine cycle to generate S‐adenosyl‐methionine. 3PG, 3‐phosphoglycerate; 3PHP, 3‐phosphohydroxypyruvate; 3PS, 3‐phosphoserine; PHGDH, phosphoglycerate dehydrogenase; PSAT1, phosphoserine aminotransferase 1; PSPH, phosphoserine phosphatase; MAT, methionine adenosyltransferase; SAM, S‐adenosyl‐methionine; MT, methyltransferase; SAH, S‐adenosylhomocysteine; HCY, homocysteine; AHCY, S‐adenosylhomocysteine hydrolase; αKG, α‐ketoglutarate; KGDH, α‐ketoglutarate dehydrogenase
One‐carbon metabolism is essential for immune function. For instance, T‐cell expansion requires serine to fuel the de novo nucleotide biosynthesis. 10 , 12 Whereas most serine is acquired extracellularly, 10 cell‐intrinsic serine synthesis is also required as effector T‐cell proliferation was impaired when the rate‐limiting enzyme phosphoglycerate dehydrogenase was inhibited. 13 In addition, compared with young mice, naive CD4 T cells from aged mice were decreased in percentage and showed a proliferation defect that was linked to impaired mitochondrial respiration and one‐carbon metabolism. 14 In myeloid cells, lipopolysaccharide stimulation enhanced serine one‐carbon metabolism to support the production of interleukin‐1β (IL‐1β) in inflammatory macrophages. 15 , 16 Concordantly, inhibition of de novo serine synthesis protected mice from endotoxemia. 15 However, in a mouse model of Pasteurella multocida infection, exogenous administration of serine abated macrophage pro‐inflammatory response, decreased bacterial colonization, and increased animal survival. 17 While serine one‐carbon metabolism is clearly essential for T cells, its role in regulating myeloid function remains to be determined.
Methionine is an intermediate of the methionine cycle in one‐carbon metabolism (Fig. 1) and can be adenylated by methionine adenosyltransferase to serve as the substrate for the production of SAM, the primary methyl donor in cellular methylation. 18 It has been shown that the activity of methionine adenosyltransferase for SAM synthesis is essential for the function of T cells 19 and macrophages. 16 During methyl transfer, SAM is catalyzed by cytosolic or nuclear methyltransferases to form the methylated substrate and S‐adenosylhomocysteine (SAH). SAH is hydrolyzed back to homocysteine and adenosine through a reversible reaction catalyzed by S‐adenosylhomocysteine hydrolase to complete the methionine cycle (Fig. 1). As the increased level of homocysteine can have a negative effect on the activity of methyltransferases, it is efficiently remethylated back to methionine. 18
Methionine metabolic regulation is crucial for the differentiation and function of CD4 T cells and B cells. 20 , 21 , 22 Restriction of methionine availability in activated T cells decreased the intracellular levels of SAM and histone methylation (active histone mark H3K4me3), impaired cell proliferation and cytokine production, and reduced the severity of experimental autoimmune encephalomyelitis (EAE) in mice. 21 In human B cells, extracellular methionine is required for BLIMP1‐dependent plasmablast differentiation. 22 BACH2 represses the expression of PRDM1, which encodes BLIMP1. Methionine induced the H3K27 methyltransferase EZH2, which catalyzed the repressive mark H3K27me3 at the BACH2 locus and decreased the gene expression. 22 Whether methionine affects SAM level was not addressed. In myeloid cells, Toll‐like‐receptor‐4‐stimulated macrophages require both exogenous serine and methionine to sustain the methionine cycle for generating SAM, which supports methylation reactions including the H3K36 trimethylation at the gene body of Il1b. 16 . However, oral SAM supplementation inhibited inflammatory response and fibrosis in a chronic asthma model. 23 As in the case of dietary serine supplementation, the discrepancy between in vitro and in vivo data is likely due to the pleiotropic effect of amino acids on whole animals.
Krebs cycle and αKG
Krebs cycle, also known as the tricarboxylic acid cycle, is the central hub of the metabolic network and provides essential metabolites and substrates in the mitochondrial matrix to maintain cellular homeostasis and function (Fig. 1). 24 This cycle contains a series of reactions to convert intermediates such as citrate, isocitrate, αKG, succinate, fumarate, malate, and oxaloacetate. Primarily, the Krebs cycle is supplied with new substrate in the form of acetyl‐CoA, which is generated from glucose‐derived pyruvate, fatty acid oxidation, or amino acid catabolism. Additionally, glutamine‐derived glutamate can be catabolized by glutamate dehydrogenase to generate αKG, sending the carbon backbone towards the cycle anaplerosis. The completion of the Krebs cycle produces NADH and flavin adenine dinucleotide that fuel into the complex I and complex II of the electron transport chain to generate electrons to support oxidative phosphorylation for cellular energy production in the mitochondria.
α‐Ketoglutarate dehydrogenase (αKGDH) is a rate‐limiting metabolic enzyme in the flux of the Krebs cycle. 25 αKGDH is a multiprotein complex that reacts with acetyl‐coenzyme A (CoA) and NAD+ to decarboxylate αKG to succinyl‐CoA, which is further metabolized to become succinate. An increase of cytosolic calcium leads to rapid mitochondrial acidification and promotes αKGDH activity, thereby boosting NADH production and oxidative metabolism. 26 , 27 In addition, αKGDH is responsive and sensitive to the levels of reactive oxygen species, and oxidative stress‐induced reactive oxygen species impair αKGDH function. 28 It has been reported that αKGDH reaction is essential for neuronal viability, and deficiency in the αKGDH activity appears to be associated with the chronic aberrant inflammation of neurodegenerative disorders, including Alzheimer's disease. 29 , 30 Moreover, Toll‐like receptor 4 stimulation in pro‐inflammatory macrophages, but not in the alternatively activated macrophage, markedly induced the expression of αKGDH, which promotes the conversion of αKG to succinate and limits the production of anti‐inflammatory cytokine IL‐10. 31
Chemical modifications of nucleosome and DNA play a central role in the development and effector function of immune cells. 4 , 5 , 6 The epigenetic enzymes responsible for these modifications often require the intermediary metabolites from the Krebs cycle. Among metabolites, αKG (also known as 2‐oxoglutarate or 2OG), is a crucial molecule involved in multiple metabolic and cellular pathways. αKG is the essential co‐factor for the reaction of 2OG‐dependent dioxygenases (2OGDD), a diverse superfamily of Fe(II)‐dependent, oxygen‐consuming enzymes that are crucial for cell development and diseases. 32 The 2OGDD family includes the prolyl‐hydroxylase domain (PHD) ‐containing proteins, Jumonji C (JmjC) domain‐containing proteins, and TET proteins. These enzymes use αKG and O2 to hydroxylate various types of substrates (including proteins, nucleic acids, and lipids) and produce succinate and CO2. Therefore, the activities of 2OGDD positively correlate with the intracellular ratio of αKG to succinate and fumarate, another Krebs cycle downstream metabolite; elevated levels of succinate or fumarate will therefore inhibit 2OGDD function. In addition, 2OGDD can be inhibited by 2‐hydroxyglutarate (2‐HG), a metabolite structurally similar to αKG (discussed below). 33
One branch of the 2OGDD family, the PHD proteins, is critical for hypoxia response. In normoxia (sufficient oxygen level), PHDs hydroxylate the prolines of the transcription factor hypoxia inducible factor‐1α (HIF) that becomes the target for proteasome degradation. Under hypoxia, or reduced αKG level, the activity of PHDs is abolished, resulting in the stabilization and activation of hypoxia inducible factor‐1α, which in turn induces gene expression related to metabolism and modulates immune cell function. 24 , 34 The other two branches of the 2OGDD family, JmjC and TET proteins, are essential epigenetic erasers, the activity of which is similarly regulated by αKG/succinate/fumarate. 35 Most JmjC proteins are lysine (and/or arginine) demethylases (JMJD) that target histones and other proteins. In macrophages, the increased αKG from glutaminolysis contributed the alternative activation of macrophages upon IL‐4 stimulation. αKG suppresses IκB kinase/nuclear factor‐κB‐dependent pro‐inflammatory effects, and modulates the activity of the histone demethylase JMJD3, thus favoring the acquisition of an anti‐inflammatory phenotype. 31
DNA methyltransferases
DNA methylation is the earliest known epigenetic modification. 36 Although DNA methylation was once thought to be a static repressive mark, recent studies have shown that DNA methylation is a dynamically regulated process and could have various functions depending on the protein binders and genomic locations. 37 In mammals, most DNA methylation occurs at the cytosines of CG motifs and is catalyzed by one of the DNA methyltransferases: DNMT1, DNMT3A, and DNMT3B. DNMT transfers the methyl group from SAM to the fifth carbon of cytosine on DNA, producing 5‐methylcytosine and SAH (Figs 2, 3). The de novo methyltransferases DNMT3A and DNMT3B methylate the unmodified cytosine to establish the methylation pattern. During DNA replication, methylation patterns at CG motifs are replicated onto the newly synthesized DNA by the maintenance methyltransferase DNMT1 complex. Since the discovery of TET enzymes, it is now known that cytosine can go through the methylation cycle, which describes the intermediates between methylation and demethylation (see below and Fig. 2).
Figure 2.
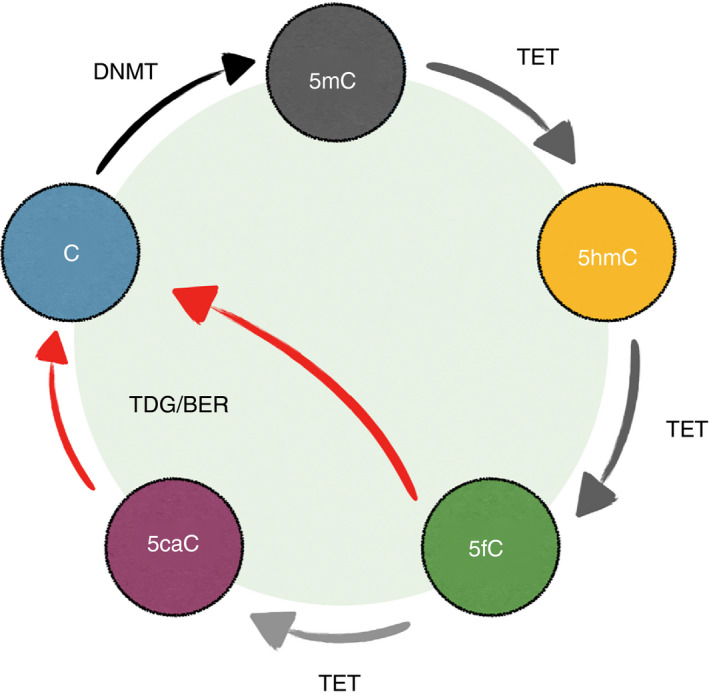
The DNA methylation cycle. In the mammalian genome, the majority of the cytosines at CG motifs are methylated. DNA methyltransferases (DNMTs) catalyze the addition of a methyl group to the fifth carbon of cytosine (C), generating 5‐methylcytosine (5mC). TETs then convert 5mC into oxidized methylcytosines (oxi‐mCs): 5‐hydroxymethylcytosine (5hmC), 5‐formylcytosine (5fC), and 5‐carboxylcytosine (5caC). While TETs are capable of the complete oxidization of 5mC to 5caC in vitro, the majority of oxi‐mCs in the cells are 5hmC. 5hmC is stable and a potential epigenetic mark. 5fC and 5caC are unstable and are removed by TDG (thymine DNA glycosylase) with the base‐excision repair. The base removal process (red arrows) constitutes ‘active DNA demethylation’. During DNA replication, the pairing between newly synthesized DNA with the original modified CpG motif creates the hemi‐modified CpG. The maintenance DNA methyltransferase complex DNMT1/UHRF1 recognizes the hemi‐methylated CpG and methylates the unmodified cytosine on the new DNA. However, DNMT1/UHRF1 cannot recognize the hemi‐methylated CpG containing oxi‐mCs, preventing the methylation of the newly synthesized DNA. Therefore, the methylation pattern will be erased after rounds of DNA replication
Figure 3.
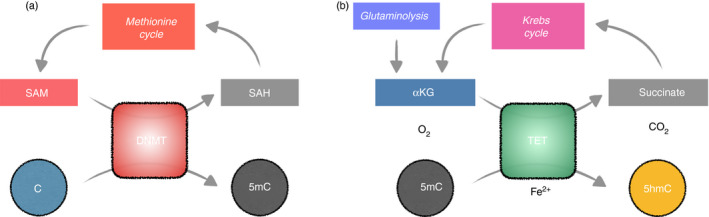
The intersection between metabolic cycles and DNA methylation cycle. (a) DNMT and methionine cycle. To methylate cytosine, DNMT uses S‐adenosyl methionine (SAM) as the methyl group donor, producing S‐adenosylhomocysteine (SAH) as a result. SAH is then recycled back to the methionine cycle (Fig. 1), regenerating SAM for additional methylation. (b) TET and Krebs cycle. With reduced iron (Fe2+) as a co‐factor, TET converts the substrates 5mC, α‐ketoglutarate (αKG), and oxygen into the products 5hmC, succinate, and carbon dioxide. TET can further oxidize 5hmC into 5fC and 5caC (not depicted). Succinate can be shuttled back to the Krebs cycle and regenerating αKG. Additional αKG can be derived from glutamine via glutaminolysis (Fig. 1).
Maintenance methyltransferase DNMT1
In the immune system, DNMT1 is highly expressed in CD4– CD8– double‐negative and Pro‐B cell stages in developing T and B cells, respectively (Immunological Genome Project; ImmGen). Consistently, conditional deletions of Dnmt1 using Lck‐Cre in early T cells 38 and Mb1‐Cre in early B cells 39 resulted in the corresponding developmental blockades. Whereas T cells were able to develop when Dnmt1 was deleted at the CD4+ CD8+ double‐positive stage using Cd4‐Cre, 38 studies have shown that DNMT1 is required to repress the ectopic expression of IL‐4 40 and Foxp3 41 in CD4 and CD8 T cells.
Foxp3 is the essential transcription factor for regulatory T (Treg) cells. 83 Although loss of DNMT1 de‐represses Foxp3, DNMT1 is required to maintain the lineage stability of Treg cells and prevents autoimmune diseases as shown by Foxp3‐Cre Dnmt1‐deficient mice. 42 In CD8 T cells, when Dnmt1 is conditionally deleted with granzymeB (GzmB)‐Cre, antigen‐specific CD8 T cells failed to differentiate into effector and memory cells. 43 Similarly, peripheral B cells from a mouse expressing a DNMT1 hypomorphic mutant failed to differentiate into germinal center B cells in response to immunization. 44 In myeloid cells, the deletion of Dnmt1 in macrophage enhanced the alternative polarization (M2) and increased M2 adipose tissue macrophages. 45 . Similarly, treatment with the DNMT inhibitor 5‐Aza‐2′‐deoxycytidine potentiated macrophage M2 polarization in a dose‐dependent manner. 45 As DNMT1 is essential for the maintenance of DNA methylation during DNA replication, pharmacological or metabolic inhibition (low SAM/SAH ratio) of DNMT will likely result in a global loss of DNA methylation and lead to global ectopic gene expression and genomic instability. 46
De novo methyltransferases DNMT3A and DNMT3B
During acute infection with lymphocytic choriomeningitis virus (LCMV), antigen‐specific CD8 T cells remodel their methylome when differentiating from naive to effector or memory cells. 47 In the absence of DNMT3A, CD8 T cells preferentially differentiate into memory cells. 48 , 49 TCF‐1 (encoded by Tcf7) is the transcription factor that is important for CD8 memory cell differentiation. In early effector cells, DNMT3A methylates the regulatory elements of Tcf7, repressing TCF‐1 expression and memory differentiation. 48 , 49 Therefore, DNMT3A promotes terminal effector differentiation and restricts memory precursor in CD8 T cells. Besides effector differentiation, de novo DNA methylation is required for terminal T‐cell exhaustion, a dysfunctional state describing the loss of T‐cell effector function. 50 The inhibitory receptor programmed cell death protein‐1 (PD‐1; encoded by Pdcd1) is essential for T‐cell exhaustion and antibody blockage of PD‐1 has been the key to recent success in cancer immunotherapy. In CD8 T cells, inhibition of de novo DNA methylation, either by deletion of Dnmt3a or by pharmacological inhibition, potentiated the effect of anti‐PD‐1‐mediated reversal of exhaustion that is induced by chronic LCMV infection and tumor. 50 Consistent with the acute LCMV infection models, 48 , 49 DNMT3A methylates the elements at Tcf7 and Ifng, both of which contribute to the tumor clearance. Intriguingly, in mouse CD8 T cells, two conserved regions of Pdcd1 are methylated in naive, demethylated in effector, and remethylated in memory CD8 T cells. 51 In exhausted CD8 T cells, these regions were fully demethylated and correlated with PD‐1 expression. The role of DNMT3 and TET enzymes in PD‐1 regulation remains to be established. In summary, dependent on the context, de novo DNA methylation facilitates effector differentiation and exhaustion while limiting memory formation in CD8 T cells.
De novo DNA methylation is important for lineage restriction in CD4 T cells. Although only wild‐type T helper 1 (Th1) cells express Ifng, CD4 T cells from Cd4‐Cre Dnmt3afl/fl mice fail to silence Ifng after differentiation into Th2, Th17, and induced Treg cells. 52 , 53 Although DNMT1 is required for Treg lineage stability, the deletion of Dnmt3a with Foxp3‐Cre has no effect in the steady state. 42 Interestingly, in an EAE model, most Treg cells express BLIMP1 at the sites of tissue inflammation. BLIMP1 protects the lineage stability by inhibiting the expression of Dnmt3a, which would otherwise be induced by IL‐6. In the absence of BLIMP1, DNMT3A methylates Foxp3 CNS2, decreasing Foxp3 expression and compromising Treg cell identity. 54
In B cells, DNMT3A and DNMT3B are required for the B‐lineage commitment from hematopoietic stem cells in vitro. 55 Once the B‐cell progenitor has committed, de novo DNA methylation is not required for the major checkpoints during B‐cell development as demonstrated in the Mb1‐Cre Dnmt3a/b‐deficient mice. 56 , 57 However, the use of light‐chain Vκ genes was skewed, 56 potentially due to altered CTCF binding and long‐range chromatin interactions. 58 In the periphery, antigen‐stimulated B cells remodel their methylome during differentiation into germinal center B cells, plasma, and memory cells. 59 , 60 Similar to the Mb1‐Cre model, the deletion of Dnmt3a/b with Cd19‐Cre has no discernable phenotype in bone marrow development. However, Dnmt3a/b deficiency in B cells increased germinal center B cells and plasma cells in response to immunization. 57 Consistent with its opposite role as an eraser for DNA methylation, Tet2 deficiency inhibits plasma cell differentiation, potentially through a lack of demethylation at Prdm1/BLIMP1, a transcription factor essential for plasma cells. 61 However, both TET2 and DNMT3A/B function to limit the expansion of germinal center B cells, a similar overlapping function was also observed in hematopoietic malignancies. 62 How these enzymes with seemingly opposite functions have a similar role in repressing cell proliferation remains to be addressed.
In macrophages, DNMT3B represses M2 differentiation, and the deficiency of Dnmt3b increased the expression of IL‐4‐induced M2 genes in bone‐marrow‐derived mouse macrophage. 63 Mechanistically, DNMT3B methylates the promoter of peroxisome proliferator‐activated receptor‐γ (PPAR), a key transcription factor for promoting macrophage alternative M2 polarization. 63 In addition, peritoneal macrophages from Lyz2‐Cre Dnmt3a‐deficient mice were defective in the production of type I interferon (IFN‐I), and animals were more susceptible to the infection of vesicular stomatitis virus. 64 DNMT3A regulates interferon induction indirectly by maintaining the expression of HDAC9, which in turns deacetylates TBK1, the key kinase for innate immune sensing. 64 Therefore, in macrophages, both DNMT1 and DNMT3B are required for the restricting alternative M2 activation, while DNMT3A is important for IFN‐I production.
TET methylcytosine oxidases
TET enzymes (TET1, TET2, TET3) are 2OGDD that are essential for cell differentiation and functions. 65 , 66 The function of TET is to oxidize and demethylate cytosine on DNA to regulate gene expression and other undefined processes. Similar to other 2OGDD, TETs use αKG and oxygen to hydroxylate 5‐methylcytosine (5mC) into 5‐hydroxymethylcytosine (5hmC), a stable epigenetic mark and one of the oxidized methylcytosines (Fig. 2). Further reaction can produce the other two oxidized methylcytosines: 5‐carboxylcytosine and 5‐formylcytosine, both of which are removed by base‐excision DNA repair and are about 10‐fold and 100‐fold lower in abundance compared with 5hmC, respectively. These oxidized methylcytosines serve as the key intermediates for passive (replication‐dependent) or active (replication‐independent) DNA demethylation. 65 The three TET family members regulate distinct regions in the genome: TET1 preferentially regulates promoters, while TET2 and TET3 regulate enhancers. Therefore, 5hmC is enriched at enhancers, promoters, and gene bodies, with the level of enhancer 5hmC positively correlating with enhancer activity (marked by H3K27Ac) and gene body 5hmC with transcriptional activity. The current model suggests that TET enzymes (especially TET2 and TET3) regulate the activity of lineage‐specific super‐enhancers, 67 in part by facilitating the enhancer accessibility. 68 , 69 , 70 As 5hmC is stable on the DNA, the pleiotropic phenotypes after TET deletion are often observed at delayed times and are usually manifested after rounds of cell proliferation. Mice with germline deletion of Tet1 and Tet2 are largely viable, 71 , 72 , 73 but the Tet3 homozygous mutation results in embryonic or perinatal lethality. 73 , 74
In humans, TET2 is one of the most recurring loss‐of‐function mutations in hematopoietic malignancies, implying a role of TET in immune cell differentiation and function (TET in hematopoietic cancers was recently reviewed 75 ). In mouse blood cells, TET1 is preferentially expressed in hematopoietic stem cells, developing B and T lymphocytes, and naive T cells (data from ImmGen); TET2 and TET3 are expressed ubiquitously. The genome‐wide 5hmC undergoes dynamic changes often around the key lineage genes during T‐cell and B‐cell differentiation. 67 , 76 , 77 , 78 As TET2 and TET3 function redundantly, no significant B‐cell or T‐cell developmental phenotype was observed in either germline or conditional Tet2‐single‐deficient models. 68 , 69 , 70 , 71 , 79 , 80 However, when both Tet2 and Tet3 (Tet2/3‐double knockout) were both deleted in developing B cells using Mb1‐Cre, bone marrow B‐cell development was blocked at the pro‐B to pre‐B cell stage transition due to a deficiency in the rearrangement of immunoglobulin light chain. 68 , 70 In T cells, while conventional CD4 and CD8 T cells were able to develop, Cd4‐Cre Tet2/3‐double knockout developed a massive lymphoproliferation caused by the expansion of self‐reactive RORγt+ IL‐17+ natural killer T cells. 69
TET enzymes regulate a diverse array of immune cell functions. In CD4 T cells, TET2 is required for the production of IFN‐γ and IL‐17 by Th1 and Th17 in vitro, respecitvely. 79 However, Tet2 deficiency in T cells exacerbated the IL‐17‐dependent pathology in two EAE models, potentially due to decreased IL‐10 production. 79 Unlike CD4 T cells, CD8 T cells from the Cd4‐Cre Tet2fl/fl mice produced more IFN‐γ in response to the acute LCMV infection, with increased differentiation of memory precursor cells and decreased short‐lived effector cells. 81 Consistent with mouse CD8 T cells, TET2 deficiency in human CD8 CAR‐T cells resulted in the differentiation of central memory cells with enhanced tumor clearance. 82 Hence, although promoting pro‐inflammatory cytokines in vitro, TET enzymes function to suppress T cell immune response in vivo.
TET2 and TET3 are required for the demethylation of Foxp3 CNS2, an intronic enhancer known to be demethylated in Treg cells. Deletion of Tet2 and Tet3 either using Cd4‐Cre or Foxp3‐Cre increased the DNA methylation at CNS2, resulting in decreased Foxp3 expression, lineage instability, and the unleashing of the effector potentials of these Treg cells. 84 , 85 , 86 Interestingly, Treg‐specific deletion of Uqcrsf1, the gene encoding the essential mitochondrial complex III protein Rieske iron‐sulfur protein, resulted in an autoimmune phenotype caused by impaired suppressive function of Treg cells. Loss of complex III resulted in increased levels of the 2‐HG and succinate, both of which are TET inhibitors, and so destabilized Treg cell lineage identity. 87 Therefore, existing data suggest that TET enzymes are required for Treg cell function.
In B cells, loss of TET enzymes resulted in impaired class switch recombination, a process by which antibody switch from IgM to other isotypes. 61 , 67 , 88 Mechanistically, TET2 and TET3 cooperate with transcription factor Basic leucine zipper transcription factor (BATF) to promote the expression of activation‐induced cytidine deaminase, the key enzyme for antibody maturation. 67 Tet2 deficiency resulted in increased germinal center B cells and decreased plasma cell differentiation after immunization. 61 Therefore, TET enzymes have both positive and negative roles in B‐cell responses.
In myeloid cells, one of the functions of TET2 is to repress pro‐inflammatory cytokines, including IL‐1β 89 , 90 and IL‐6. 91 , 92 Consistent with the anti‐inflammatory role of TET2, loss of Tet2 facilitates atherosclerosis in mice, 90 a phenotype reminiscent of humans with clonal hematopoiesis caused by TET2 mutation. 93 Most importantly, Tet2‐deficient macrophages (Lyz2‐Cre) exhibited a pro‐inflammatory phenotype in the immunosuppressive tumor microenvironment and delayed melanoma growth in vivo. 94
Links between metabolism, epigenome, and immune function
Epigenetic modifications are essentially biochemical reactions catalyzed by enzymes. Therefore, the concentrations of metabolic substrates, co‐factors, and products would dictate the reaction rate. For instance, 2OGDDs including TET and JmjC histone demethylases require αKG, the availability of which directly affects the epigenome. Indeed, in activated T cells, the IL‐2‐sensitive differentiation programs depend on the level of αKG and glutamine. 95 High levels of IL‐2 favor effector differentiation; whereas low levels of IL‐2 favor memory or follicular T helper cells. High IL‐2 induces the accumulation of glycolysis and glutaminolysis metabolites including αKG. The level of αKG appears to be instructive in gene expression: the addition of cell‐permeable αKG can mimic a high‐IL‐2‐like gene expression profile even when cultured in low IL‐2. Mechanistically, αKG likely promotes the enzymatic reactions by JmjC proteins and TETs to promote histone and DNA demethylation, respectively. DNA demethylation at CG‐containing CTCF motifs permits CTCF binding, facilitating the genome reorganization and gene expression. 95 Another example of metabolism affecting the epigenome is a study of methionine metabolism in T cells (discussed above). In vitro activated T helper cells actively uptake extracellular methionine, from which the majority of the SAM pool is derived. Activated CD8 T cells cultured in methionine‐restricted conditions had dramatically decreased the SAM level and global H3K4me3 level, whereas the global H3K4me3 level was slightly decreased in Th1 and Th17 cells. Nonetheless, Th17 cells cultured in low methionine had decreased expression of Il17a, Batf, Cd5l, and cell cycle‐related genes accompanied by decreased H3K4me3 at the corresponding promoters. A low‐methionine diet also ameliorates the severity of the Th17‐driven disease EAE, at least in part by limiting the proliferation of Th17 cells.
2‐HG was first identified as the ‘oncometabolite’ produced by the isocitrate dehydrogenase 1 and 2 (IDH1/2) mutant in glioma cells. 33 Structurally similar to αKG, 2‐HG can inhibit the activity of 2OGDD enzymes. 2‐HG exists as two enantiomers that differ in their ability to inhibit 2OGDD enzymes; with (S)‐2HG (also known as l‐2HG) being the more potent inhibitor than (R)‐2HG (or known as d‐2HG). Notably, it has been reported that 2‐HG can be generated endogenously in normal cells. For instance, in CD8 T cells, T‐cell receptor stimulation induces the generation of l‐2HG as early as 2 days post‐activation. 96 Similar to Tet2 deficiency, treatment with l‐2HG induced higher levels of Eomes and CD62L, resembling central memory cells. Interestingly, OT‐I T‐cell‐receptor‐transgenic CD8 T cells treated with L‐2HG in vitro appear to have increased survival and tumor clearance after in vivo transfer to recipients bearing tumors expressing ovalbumin, 96 suggesting that the effect of l‐2HG is likely via epigenome. Therefore, these recent examples showcased the link between metabolism and epigenome and their effect on immune responses.
Concluding remarks
The DNA methylation program mediated by DNMT and TET is essential for regulating cell homeostasis and effector function in immune cells. Most studies of the metabolome–epigenome focus on the global level of histone modifications, but the effect of metabolites on the DNA methylome should warrant more attention. The perturbation of epigenetic enzymes may not affect the abundance of a given epigenetic mark globally, 21 , 67 thus genome‐wide epigenome profiling will likely be required to pinpoint the local changes underlying the phenotype. In addition, recent advances in single‐cell technologies in epigenome sequencing 97 and mass spectrometry 98 will greatly improve the capacity of analysis in the often small immune populations isolated in vivo, including tumor‐infiltrating and tissue‐resident immune cells.
The studies discussed above have demonstrated promising therapeutic interventions to achieve the desired immune response by modulating the cellular metabolism and/or epigenome. However, it has yet to explore the broad effects of metabolite levels on global epigenetic landscapes of immune cells. Similar to the ImmGen consortium for genomics and epigenomics, it would be beneficial to expand the scope to include metabolomic profiles for individual immune cell types. Understanding the differential metabolic circuits in each cell type may allow cell‐specific rewiring of the metabolome and potentially the epigenome.
Disclosures
The authors declare no conflict of interests.
Acknowledgements
SCCH was supported by the Cancer Research Institute CLIP Grant and Case Comprehensive Cancer Center American Cancer Society Pilot Grants (IRG‐91‐022‐19, IRG‐16‐186‐21). CWJL was supported by the Independent Investigator Fund (La Jolla Institute/Kyowa Kirin) and National Cancer Institute K22 Career Transition Award (K22CA241290).
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Metabolic mediators: how immunometabolism directs the immune response to infection. Immunology 2020, 161: 163‐164.
The role of O‐GlcNAcylation in immunity against infections. Immunology 2020, 161: 175‐185.
The battle for iron in enteric infections. Immunology 2020, 161: 186‐199.
Implications of cellular metabolism for immune cell migration. Immunology 2020, 161: 200‐208.
Contributor Information
Chan‐Wang Jerry Lio, Email: lio.4@osu.edu.
Stanley Ching‐Cheng Huang, Email: stan.huang@case.edu.
References
- 1. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell 2017; 169:570–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li X, Wenes M, Romero P, Huang SC‐C, Fendt S‐M, Ho P‐C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol (R Coll Radiol) 2019; 16:425–41. [DOI] [PubMed] [Google Scholar]
- 3. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell 2007; 128:635–8. [DOI] [PubMed] [Google Scholar]
- 4. Álvarez‐Errico D, Vento‐Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol 2015; 15:7–17. [DOI] [PubMed] [Google Scholar]
- 5. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8+ T cell differentiation. Nat Rev Immunol 2018; 18:340–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol 2019; 19:417–32. [DOI] [PubMed] [Google Scholar]
- 7. Etchegaray J‐P, Mostoslavsky R. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol Cell 2016; 62:695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ducker GS, Rabinowitz JD. One‐carbon metabolism in health and disease. Cell Metab 2017; 25:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allen RW, Moskowitz M. Arrest of cell growth in the G1 phase of the cell cycle by serine deprivation. Exp Cell Res 1978; 116:127–37. [DOI] [PubMed] [Google Scholar]
- 10. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B et al Serine is an essential metabolite for effector T cell expansion. Cell Metab 2017; 25:345–57. [DOI] [PubMed] [Google Scholar]
- 11. Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E et al Serine starvation induces stress and p53‐dependent metabolic remodelling in cancer cells. Nature 2013; 493:542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ron‐Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB et al Mitochondrial biogenesis and proteome remodeling promote one‐carbon metabolism for T cell activation. Cell Metab 2016; 24:104–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S et al Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 2019; 51:856–870.e5. [DOI] [PubMed] [Google Scholar]
- 14. Ron‐Harel N, Notarangelo G, Ghergurovich JM, Paulo JA, Sage PT, Santos D et al Defective respiration and one‐carbon metabolism contribute to impaired naïve T cell activation in aged mice. Proc Natl Acad Sci USA 2018; 115:13347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V et al Serine metabolism supports macrophage IL‐1β production. Cell Metab 2019; 29:1003–1011. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu W, Wang Z, Zhang K, Chi Z, Xu T, Jiang D et al One‐carbon metabolism supports S‐adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol Cell 2019; 75:1147–1160.e5. [DOI] [PubMed] [Google Scholar]
- 17. He F, Yin Z, Wu C, Xia Y, Wu M, Li P et al l‐Serine lowers the inflammatory responses during Pasteurella multocida infection. Infect Immun 2019; 87:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clare CE, Brassington AH, Kwong WY, Sinclair KD. One‐carbon metabolism: linking nutritional biochemistry to epigenetic programming of long‐term development. Annual Rev Anim Biosci 2019; 7:263–87. [DOI] [PubMed] [Google Scholar]
- 19. LeGros HL, Geller AM, Kotb M. Differential regulation of methionine adenosyltransferase in superantigen and mitogen stimulated human T lymphocytes. J Biol Chem 1997; 272:16040–7. [DOI] [PubMed] [Google Scholar]
- 20. Sinclair LV, Howden AJ, Brenes A, Spinelli L, Hukelmann JL, Macintyre AN et al Antigen receptor control of methionine metabolism in T cells. eLife 2019; 8:1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roy DG, Chen J, Mamane V, Ma EH, Muhire BM, Sheldon RD et al Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming. Cell Metab 2020; 31:250–266.e9. [DOI] [PubMed] [Google Scholar]
- 22. Zhang M, Iwata S, Hajime M, Ohkubo N, Todoroki Y, Miyata H et al Methionine commits cells to differentiate into plasmablasts through epigenetic regulation of BTB and CNC homolog 2 by the methyltransferase enhancer of zeste homolog 2. Arth Rheumatol 2020. [DOI] [PubMed] [Google Scholar]
- 23. Yoon S‐Y, Hong GH, Kwon H‐S, Park S, Park SY, Shin B et al S‐adenosylmethionine reduces airway inflammation and fibrosis in a murine model of chronic severe asthma via suppression of oxidative stress. Exp Mol Med 2016; 48:e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martínez‐Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 2020; 11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sheu KF, Blass JP. The α‐ketoglutarate dehydrogenase complex. Ann N Y Acad Sci 1999; 893:61–78. [DOI] [PubMed] [Google Scholar]
- 26. Tretter L, Adam‐Vizi V. Alpha‐ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci 2005; 360:2335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochem Biophys Acta 2009; 1787:1324–33. [DOI] [PubMed] [Google Scholar]
- 28. McLain AL, Szweda PA, Szweda LI. α‐Ketoglutarate dehydrogenase: a mitochondrial redox sensor. Free Radical Res 2011; 45:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age‐associated neurodegenerative diseases. Biochem Biophys Acta 2010; 1802:122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Starkov AA. An update on the role of mitochondrial α‐ketoglutarate dehydrogenase in oxidative stress. Mol Cell Neurosci 2013; 55:13–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S et al α‐Ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017; 18:985–94. [DOI] [PubMed] [Google Scholar]
- 32. McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2‐oxoglutarate dependent oxygenases. Curr Opin Struct Biol 2010; 20:659–72. [DOI] [PubMed] [Google Scholar]
- 33. Dang L, Su S‐SM. Isocitrate dehydrogenase mutation and (R)‐2‐hydroxyglutarate: From basic discovery to therapeutics development. Annu Rev Biochem 2017; 86:305–31. [DOI] [PubMed] [Google Scholar]
- 34. Bargiela D, Burr SP, Chinnery PF. Mitochondria and hypoxia: metabolic crosstalk in cell‐fate decisions. Trends Endocrinol Metab 2018; 29:249–59. [DOI] [PubMed] [Google Scholar]
- 35. Kaelin WG, McKnight SL. Influence of metabolism on epigenetics and disease. Cell 2013; 153:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010; 11:204–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schübeler D. Function and information content of DNA methylation. Nature 2015; 517:321–6. [DOI] [PubMed] [Google Scholar]
- 38. Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW et al A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001; 15:763–74. [DOI] [PubMed] [Google Scholar]
- 39. Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R et al Testing gene function early in the B cell lineage in mb1‐cre mice. Proc Natl Acad Sci U S A 2006; 103:13789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Makar KW, Pérez‐Melgosa M, Shnyreva M, Weaver WM, Fitzpatrick DR, Wilson CB. Active recruitment of DNA methyltransferases regulates interleukin 4 in thymocytes and T cells. Nat Immunol 2003; 4:1183–90. [DOI] [PubMed] [Google Scholar]
- 41. Josefowicz SZ, Wilson CB, Rudensky AY. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J Immunol 2009; 182:6648–52. [DOI] [PubMed] [Google Scholar]
- 42. Wang L, Liu Y, Beier UH, Han R, Bhatti TR, Akimova T et al Foxp3+ T‐regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood 2013; 121:3631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chappell C, Beard C, Altman J, Jaenisch R, Jacob J. DNA methylation by DNA methyltransferase 1 is critical for effector CD8 T cell expansion. J Immunol 2006; 176:4562–72. [DOI] [PubMed] [Google Scholar]
- 44. Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME et al DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B‐cell differentiation. Blood 2011; 118:3559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang X, Cao Q, Yu L, Shi H, Xue B, Shi H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016; 1:e87748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karpf AR, Matsui S‐i. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Can Res 2005; 65:8635–9. [DOI] [PubMed] [Google Scholar]
- 47. Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol 2013; 191:3419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ladle BH, Li KP, Phillips MJ, Pucsek AB, Haile A, Powell JD et al De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T‐cell fate decisions following activation. Proc Natl Acad Sci U S A 2016; 113:10631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A et al Effector CD8 T cells dedifferentiate into long‐lived memory cells. Nature 2017; 552:404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P et al De novo epigenetic programs inhibit PD‐1 blockade‐mediated T cell rejuvenation. Cell 2017; 170:142–157.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE et al Chronic virus infection enforces demethylation of the locus that encodes PD‐1 in antigen‐specific CD8+ T cells. Immunity 2011; 35:400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thomas RM, Gamper CJ, Ladle BH, Powell JD, Wells AD. De novo DNA methylation is required to restrict T helper lineage plasticity. J Biol Chem 2012; 287:22900–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA methyltransferase 3a as a T cell receptor‐induced regulator of Th1 and Th2 differentiation. J Immunol (Baltimore, Md. 1950); 2009:2267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garg G, Muschaweckh A, Moreno H, Vasanthakumar A, Floess S, Lepennetier G et al Blimp1 prevents methylation of Foxp3 and loss of regulatory T cell identity at sites of inflammation. Cell Rep 2019; 26:1854–1868.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B et al Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014; 15:350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Manoharan A, Du Roure C, Rolink AG, Matthias P. De novo DNA Methyltransferases Dnmt3a and Dnmt3b regulate the onset of Igκ light chain rearrangement during early B‐cell development. Eur J Immunol 2015; 45:2343–55. [DOI] [PubMed] [Google Scholar]
- 57. Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, Haines RR et al B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun 2018; 9:1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kenter AL, Feeney AJ. New insights emerge as antibody repertoire diversification meets chromosome conformation. F1000Research 2019; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS et al DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet 2016; 48:253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barwick BG, Scharer CD, Bally AP, Boss JM. Plasma cell differentiation is coupled to division‐dependent DNA hypomethylation and gene regulation. Nat Immunol 2016; 17:1216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L et al TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B‐cell lymphomagenesis. Cancer Discov 2018; 8:1632–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shih AH, Abdel‐Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 2012; 12:599–612. [DOI] [PubMed] [Google Scholar]
- 63. Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol 2014; 28:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K et al Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat Immunol 2016; 17:806–15. [DOI] [PubMed] [Google Scholar]
- 65. Lio CJ, Yue X, Lopez‐Moyado IF, Tahiliani M, Aravind L, Rao A. TET methylcytosine oxidases: new insights from a decade of research. J Biosci 2020; 45. [PMC free article] [PubMed] [Google Scholar]
- 66. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lio CJ, Shukla V, Samaniego‐Castruita D, Gonzalez‐Avalos E, Chakraborty A, Yue X et al TET enzymes augment activation‐induced deaminase (AID) expression via 5‐hydroxymethylcytosine modifications at the Aicda superenhancer. Sci Immunol 2019;4: eaau7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lio CW, Zhang J, Gonzalez‐Avalos E, Hogan PG, Chang X, Rao A. Tet2 and Tet3 cooperate with B‐lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife 2016;5: e18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tsagaratou A, Gonzalez‐Avalos E, Rautio S, Scott‐Browne JP, Togher S, Pastor WA et al TET proteins regulate the lineage specification and TCR‐mediated expansion of iNKT cells. Nat Immunol 2017; 18:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Orlanski S, Labi V, Reizel Y, Spiro A, Lichtenstein M, Levin‐Klein R et al Tissue‐specific DNA demethylation is required for proper B‐cell differentiation and function. Proc Natl Acad Sci U S A 2016; 113:5018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R et al Ten‐Eleven‐Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A 2011; 108:14566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW et al Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011; 9:166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kang J, Lienhard M, Pastor WA, Chawla A, Novotny M, Tsagaratou A et al Simultaneous deletion of the methylcytosine oxidases Tet1 and Tet3 increases transcriptome variability in early embryogenesis. Proc Natl Acad Sci U S A 2015; 112:E4236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W et al The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011; 477:606–10. [DOI] [PubMed] [Google Scholar]
- 75. Lio CJ, Yuita H, Rao A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood 2019; 134:1487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nestor CE, Lentini A, Hagg Nilsson C, Gawel DR, Gustafsson M, Mattson L et al 5‐Hydroxymethylcytosine remodeling precedes lineage specification during differentiation of human CD4+ T cells. Cell Rep 2016; 16:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tsagaratou A, Aijo T, Lio CW, Yue X, Huang Y, Jacobsen SE et al Dissecting the dynamic changes of 5‐hydroxymethylcytosine in T‐cell development and differentiation. Proc Natl Acad Sci U S A 2014; 111:E3306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Caron G, Hussein M, Kulis M, Delaloy C, Chatonnet F, Pignarre A et al Cell‐cycle‐dependent reconfiguration of the DNA methylome during terminal differentiation of human B cells into plasma cells. Cell Rep 2015; 13:1059–71. [DOI] [PubMed] [Google Scholar]
- 79. Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S et al The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015; 42:613–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E et al The loss of TET2 promotes CD8+ T cell memory differentiation. J Immunol 2018; 200:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E et al The loss of TET2 promotes CD8+ T cell memory differentiation. J Immunol 2017200: 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ et al Disruption of TET2 promotes the therapeutic efficacy of CD19‐targeted T cells. Nature 2018; 558:307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol 2012; 12:157–67. [DOI] [PubMed] [Google Scholar]
- 84. Yue X, Lio CJ, Samaniego‐Castruita D, Li X, Rao A. Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nat Commun 2019; 10:2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yue X, Trifari S, Aijo T, Tsagaratou A, Pastor WA, Zepeda‐Martinez JA et al Control of Foxp3 stability through modulation of TET activity. J Exp Med 2016; 213:377–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nakatsukasa H, Oda M, Yin J, Chikuma S, Ito M, Koga‐Iizuka M et al Loss of TET proteins in regulatory T cells promotes abnormal proliferation, Foxp3 destabilization and IL‐17 expression. Int Immunol 2019; 31:335–47. [DOI] [PubMed] [Google Scholar]
- 87. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez‐Reyes I et al Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019; 565:495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schoeler K, Aufschnaiter A, Messner S, Derudder E, Herzog S, Villunger A et al TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. FEBS J 2019; 286:3566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Neves‐Costa A, Moita LF. TET1 is a negative transcriptional regulator of IL‐1β in the THP‐1 cell line. Mol Immunol 2013; 54:264–70. [DOI] [PubMed] [Google Scholar]
- 90. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R et al Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017; 355:842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol 2017; 55:56–70. e13. [DOI] [PubMed] [Google Scholar]
- 92. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X et al Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL‐6. Nature 2015; 525:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018; 22:157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pan W, Zhu S, Qu K, Meeth K, Cheng J, He K et al The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor‐infiltrating myeloid cells to promote melanoma progression. Immunity 2017; 47:284–297.e5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chisolm DA, Savic D, Moore AJ, Ballesteros‐Tato A, León B, Crossman DK et al CCCTC‐Binding factor translates interleukin 2‐ and α‐ketoglutarate‐sensitive metabolic changes in T cells into context‐dependent gene programs. Immunity 2017; 47:251–267.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P et al S‐2‐hydroxyglutarate regulates CD8+ T‐lymphocyte fate. Nature 2016; 540:236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shema E, Bernstein BE, Buenrostro JD. Single‐cell and single‐molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat Genet 2019; 51:19–25. [DOI] [PubMed] [Google Scholar]
- 98. Duncan KD, Fyrestam J, Lanekoff I. Advances in mass spectrometry based single‐cell metabolomics. Analyst 2019; 144:782–93. [DOI] [PubMed] [Google Scholar]