

Altered metabolism and genome instability are hallmarks of cancer. A mechanism now explains how three small molecules that accumulate in tumours connect abnormal metabolism to genomic problems by hindering DNA repair.
See p.586
Towards the end of the nineteenth century, chromosomal abnormalities detected under the light microscope revealed that a type of massive genome instability resulting in an abnormal number of chromosomes occurs in certain types of cancer. Not long after, the biochemist Otto Warburg observed that tumour cells tend to use pathways of glucose and energy metabolism that are distinct from those used by normal cells. We now know that genome instability and altered metabolism are two common characteristics of most tumour cells. Genome instability has been investigated continuously since its discovery; altered metabolism was rediscovered as a research area only recently. But not much crosstalk between these two processes in cancer has been reported so far. Sulkowski et al.1 reveal on page 586 how several metabolites that accumulate to high levels in tumour cells suppress DNA repair, thus revealing a direct link between altered metabolism and genome instability caused by DNA damage.
Mutations targeting the genes encoding the enzymes isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) result in cells accumulating high levels of the metabolite 2-hydroxyglutarate (2-HG). Mutations in the genes encoding the enzymes fumarate hydratase and succinate dehydrogenase cause cells to accumulate high levels of the molecules fumarate and succinate, respectively. These three small molecules are often referred to as oncometabolites because their accumulation boosts tumour development2,3, and they are structurally similar to the molecule α-ketoglutarate (α-KG). This is an intermediate in the Krebs-cycle pathway that also serves as a component, called a co-substrate, needed for the function of a family of enzymes called α-KG/Fe(II)-dependent dioxygenases.
This enzyme family, which comprises 65 members in humans4, catalyses a diverse range of oxidation reactions in proteins, DNA, RNA and lipids. In these reactions, α-KG binds to the active site of the enzyme to aid catalysis. However, 2-HG, succinate and fumarate can compete with α-KG for binding to this catalytic site and thus inhibit these enzymes. One such enzyme is lysine histone demethylase (KDM), which modifies chromatin - the complex of DNA and proteins of which chromosomes are made5–7.
Two closely related KDMs, called KDM4A and KDM4B, catalyse the removal of a methyl group (demethylation) from a lysine amino-acid residue (termed K9) in the DNA-binding histone 3 (H3) proteins in chromatin. The methylation of H3K9 is linked to a pathway called the homology-dependent repair (HDR) pathway, which mends double-strand breaks (DSBs) in DNA8. DSBs are the most dangerous type of DNA damage. If left unrepaired, they can cause chromosome breakage and genomic instability that might promote tumour growth or lead to cell death.
Sulkowski and colleagues investigated HDR in human cancer cells grown in vitro. They found that, at a DSB site, the local addition of three methyl groups to H3K9 to generate trimethylated H3K9me3 residues has a key role in the initiation of HDR. In tumour cells that have mutations in the genes encoding IDH1, IDH2, fumarate hydratase or succinate dehydrogenase, the authors report that high levels of oncometabolites inhibit KDM4B. This inhibition of demethylation results in a widespread hypermethylation of H3K9 that masks the specific local appearance of H3K9me3 marks and impairs the recruitment of factors needed for HDR and DSB repair (Fig. 1).
Figure 1 |. How molecules in cancer cells inhibit the repair of DNA damage.
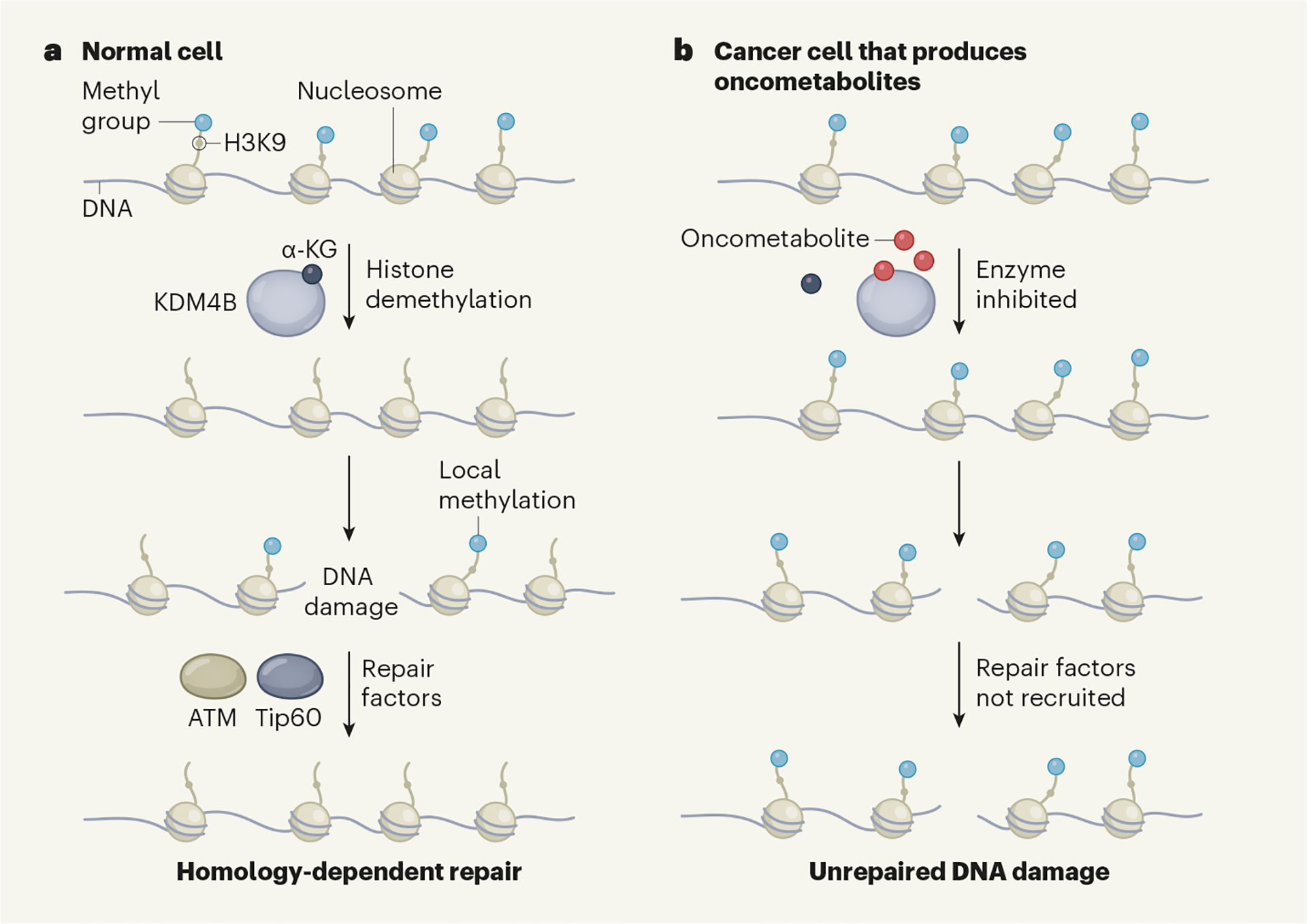
a, DNA wraps around histone proteins to form a structure called a nucleosome. In normal cells, the enzyme KDM4B catalyses the removal of methyl groups from the lysine 9 (K9) amino-acid residue of the protein histone 3 (H3) in the nucleosome. This H3K9 demethylation activity requires the small molecule α-ketoglutarate (α-KG). If a double-strand break in DNA occurs, H3K9 is methylated at the damage site and this local methylation signal recruits DNA-repair factors that include the proteins Tip60 and ATM. These fix the damage through a process called homology-dependent repair. b, As a result of certain mutations, some cancer cells accumulate small molecules termed oncometabolites that promote tumour growth. Sulkowski et al.1 have revealed a mechanism that underlies this phenomenon. Oncometabolites compete with α-KG for binding to KDM4B and thus inhibit the enzyme’s function. This results in H3K9 methylation across the genome. This global hypermethylation masks a local spike in H3K9 methylation occurring after DNA damage, and hinders the recruitment of DNA-repair factors. Unrepaired DNA damage can lead to genome instability and thus boost tumour growth.
A link between oncometabolites and DNA-repair defects was previously suggested by the clinical finding that people who have a type of cancer called glioma with mutations in the IDH1 or IDH2 genes benefited from a combination of chemotherapy and radiation therapy, both of which induce DNA damage9. That finding indicates that tumours that accumulate high levels of oncometabolites are vulnerable to therapy that causes DNA damage. Moreover, a genomic analysis of different types of cancer ranked IDH1 as being the fifth most frequently mutated human gene that is connected to DNA repair10.
Two mechanisms have previously been proposed to explain how the 2-HG that accumulates when IDH1 or IDH2 are mutated causes DNA-repair defects. One idea is that 2-HG directly inhibits the enzymes ALKBH2 and ALKBH3, which repair methylation-induced single-strand DNA damage11. Another suggestion is that 2-HG inhibits H3K9 demethylases and thereby causes a reduction in the expression of ATM, a key protein required for DNA repair12.
Sulkowski and colleagues had previously found that oncometabolites suppressed the HDR pathway and had identified KDM4A and KDM4B as being important for DSB repair11. The authors therefore explored possible connections between these processes. HDR is a complex event that involves the sequential recruitment of multiple repair factors to DSB sites, with the protein Tip60 being among the first to arrive at the damaged region8. Sulkowski et al. used a system in which human cells grown in vitro were engineered to allow the precise initiation of DSB and monitoring of the repair process.
The authors found that in control cells that did not have high levels of oncometabolites, a rapid spike of H3K9me3 modifications occurred locally in chromatin in the vicinity of the DSB within 30 minutes of the DSB being induced. This was followed by the coordinated recruitment of factors needed for HDR. However, in cancer cells with high levels of oncometabolites, H3K9me3 was elevated throughout the genome before DSB induction, and the subsequent recruitment of the factors needed for HDR was substantially impaired compared with that in the control cells. These defects in repair-factor recruitment could be prevented by deleting the mutant version of IDH1 or by treatment with a pharmacological inhibitor of mutant IDH1 protein to block 2-HG production. These results establish a causal relationship between the presence of oncometabolites and impaired DSB repair.
How might KDM4B inhibition by oncometabolites impair HDR? Local H3K9 methylation activates Tip60, which in turn activates ATM, a key enzyme needed for HDR. Results from a series of experiments support the authors’ model that a sudden increase in H3K9me3 modifications at a DSB site serves as a key signal to recruit repair factors. Blocking the accumulation of oncometabolites, adding α-KG, or engineering cells to express KDM4A or KDM4B (but not other KDMs or ALKBH2 or ALKBH3), resulted in a decrease in global genomic H3K9me3 modifications and restored both the recruitment of repair factors and DSB repair at an engineered DNA-damage site, compared with the effects seen in cells that did not receive such treatment. If cells producing oncometabolites were engineered to have a mutant version of a histone that sequesters H3K9 methyltransferase enzymes and thus reduces the genomic level of H3K9me3 modifications, the cells displayed an H3K9me3 spike on DSB formation that led to Tip60 recruitment and repair of DNA damage.
Sulkowski and colleagues’ findings expand the known roles of oncometabolites and raise several interesting questions. How does the rapid spike in H3K9me3 at a DSB site result in the coordinated recruitment of repair proteins, and what factor(s) might recognize such a modification of a DSB site? Around the DSB site, does hypermethylation of H3K9, which is known to recruit repressive factors that drive the formation of a condensed form of chromatin called heterochromatin, prevent the binding of factors needed for HDR? Questions also remain about whether the roles of KDM4A and KDM4B differ in HDR. Both enzymes catalyse the same type of H3K9 demethylation, and boosting their expression can overcome inhibition by oncometabolites and prevent HDR defects. Yet the authors report that the depletion only of KDM4B impairs HDR.
The enzyme PARP promotes the repair of single-strand DNA breaks, and inhibitors that block PARP are used to treat certain types of cancer. Tumour cells that produce 2-HG are particularly prone to death if treated with PARP inhibitors11. The findings by Sulkowski et al. might lead to new therapeutic strategies that exploit the therapeutic opportunities arising from oncometabolite accumulation, given that we now have a clearer picture of how such cancer cells are vulnerable if DNA-repair processes are targeted.
References
- 1.Sulkowski PL et al. Nature 582, 586–591 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King A, Selak MA & Gottlieb E Oncogene 25, 4675–4682 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Ye D, Guan K-L & Xiong Y Trends Cancer 4, 151–165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rose NR, McDonough MA, King ONF, Kawamura A & Schofield CJ Chem. Soc. Rev 40, 4364–4397 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Chowdhury R et al. EMBO Rep. 12, 463–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu W et al. Cancer Cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao M et al. Genes Dev. 26, 1326–1338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y et al. Nature Cell Biol. 11, 1376–1382 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cairncross JG et al. J. Clin. Oncol 32, 783–790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knijnenburg TA et al. Cell Rep. 23, 239–254.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sulkowski PL et al. Sci. Transl. Med 9, eaal2463 (2017).28148839 [Google Scholar]
- 12.Inoue S et al. Cancer Cell 30, 337–348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]