

Abstract
Microglia, the brain’s resident macrophages, help to regulate brain function by removing dying neurons, pruning non-functional synapses, and producing ligands that support neuronal survival1. Here we show that microglia are also critical modulators of neuronal activity and associated behavioural responses in mice. Microglia respond to neuronal activation by suppressing neuronal activity, and ablation of microglia amplifies and synchronizes the activity of neurons, leading to seizures. Suppression of neuronal activation by microglia occurs in a highly region-specific fashion and depends on the ability of microglia to sense and catabolize extracellular ATP, which is released upon neuronal activation by neurons and astrocytes. ATP triggers the recruitment of microglial protrusions and is converted by the microglial ATP/ADP hydrolysing ectoenzyme CD39 into AMP; AMP is then converted into adenosine by CD73, which is expressed on microglia as well as other brain cells. Microglial sensing of ATP, the ensuing microglia-dependent production of adenosine, and the adenosine-mediated suppression of neuronal responses via the adenosine receptor A1R are essential for the regulation of neuronal activity and animal behaviour. Our findings suggest that this microglia-driven negative feedback mechanism operates similarly to inhibitory neurons and is essential for protecting the brain from excessive activation in health and disease.
Human and animal behaviour relies on the coordinated activity of excitatory and inhibitory neurons, which collectively define the output of distinct neuronal circuits and associated behaviours. Although the regulation of neuronal activity in the brain has long been viewed as an exclusive prerogative of neurons, recent findings have suggested that the brain’s immune cells – the microglia – might be involved in this process2–8. We found that, similar to inhibitory neurons, microglia sense neuronal activation and suppress excessive neuronal activity. Microglia respond to neuronal activation or inhibition with distinct changes in gene expression (Fig. 1a–c, Extended Data Fig. 1, Supplementary Tables 1, 2). The overall pattern of changes in microglial gene expression in the striatum in response to the selective activation of CamkII+ neurons in the mouse forebrain (Fig. 1b, c) indicates that neuronal activity is communicated to microglia and may alter microglia-neuron interactions. In particular, the upregulation of genes involved in chemotaxis (Ccl24, Ccl2, Ccl3) and actin filament polymerization (Kank2, Twf1) (Fig. 1b, Supplementary Table 1), and the downregulation of genes that govern baseline motility of microglia, such as the key regulator Kcnk13 (also known as THIK-1)9,10 (Fig. 1b, c, Supplementary Table 1), suggest that neuronal activation alters neuron-microglia interactions by affecting microglial process extension and motility11,12.
Figure 1: Microglia respond to neuronal activation and prevent excessive neurostimulation.
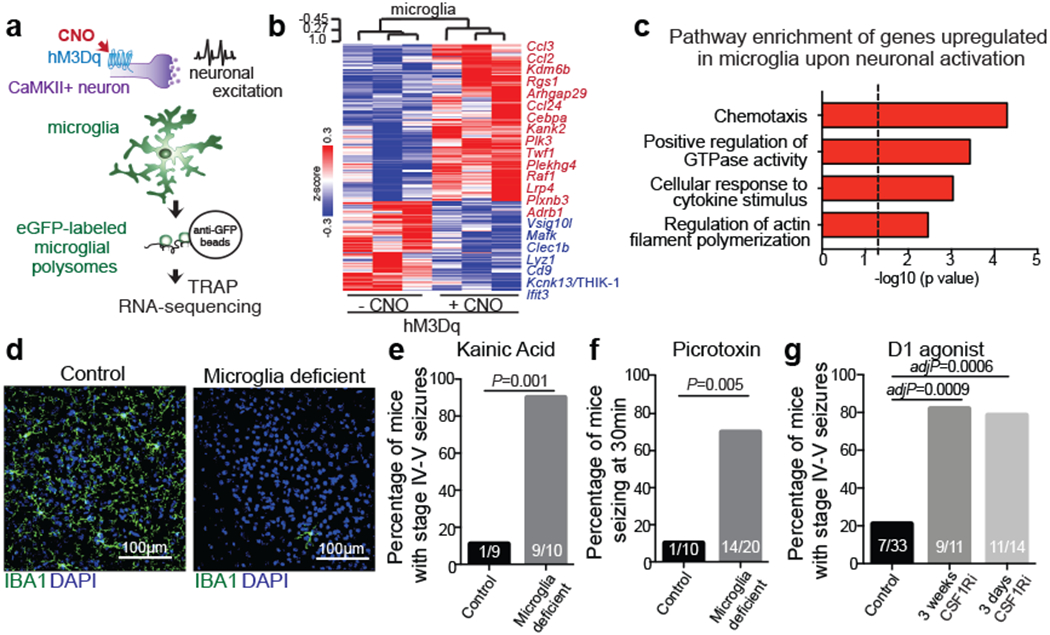
a, Neuronal activity-induced transcriptional changes in microglia. CaMKII-tTa;tetO-CHRM3 mice expressing the human M3 muscarinic (hM3Dq) receptor in CaMKII+ neurons were bred to Cx3cr1Cre Ert2/+(Litt);Eef1a1 LSL.eGFPL10a/+ mice. Neurons were activated by clozapine-N-oxide (CNO) followed by microglia-specific mRNA analysis using translating ribosome affinity purification (TRAP). b, Gene expression changes in striatal microglia following CNO-induced activation (z-scored log2(RPKM); n=3 mice) c, Selected gene ontology (GO) annotations (ENRICHR) for upregulated genes (DESeq2) in striatal microglia (dotted line, P=0.05). d, Microglia depletion by CSF1R inhibitor PLX5622. Representative images of sagittal striatal sections from control (left) or PLX5622 treated mice (right) show nucleated (DAPI+, blue) IBA1+ (green) microglia e-g, Percentage of mice exhibiting behavioral seizures in response to i.p. injections of e, kainic acid (18mg/kg, 1hr), f, picrotoxin (1mg/kg, 30min) and g, D1 agonist (SKF81297, 5mg/kg,1hr) (e: n=9 and 10 mice, Fisher’s exact test; f: n=10 and 20 mice, Fisher’s exact test; g: n=33, 11, and 14 mice, P<0.0001, Chi-squared test with Bonferroni adjustment). The experiments shown in d, e and g have been independently repeated with same results. RPKM: reads per kilobase of transcript per million mapped reads; data shown as mean ± s.e.m
We found that microglial response to neuronal activation is associated with a reciprocal microglia-mediated suppression of neuronal activity. Brain-wide ablation of microglia in adult mice by pharmacological inhibition of the microglia pro-survival receptor CSF1R13 (Fig. 1d, Extended Data Fig. 2a) had no major effect on animal behaviors at baseline13 (Extended Data Fig. 2b–e), but rendered animals hyper-responsive to neurostimulants at levels that normally do not cause excessive neuronal activation (Fig. 1e–g, Extended Data Fig. 2f–h). The intraperitoneal (i.p.) administration of a sub-threshold dose of kainic acid, a kainate receptor agonist that activated subtypes of glutamate receptors (kainate and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors) in the brain resulted in seizures (Racine stages above IV-V) in 90% of microglia-depleted mice compared with 11% of controls (Fig. 1e, Extended Data Fig. 2g). In line with this observation, we found that i.p. administration of picrotoxin–an inhibitor of GABAA (γ-aminobutyric acid A) receptors that enhances excitatory neuron activity by alleviating suppression by inhibitory neurons–resulted in significantly prolonged seizure responses in mice lacking microglia (Fig. 1f). Similar effects were seen upon stimulation of dopamine D1 receptor-expressing striatal neurons (D1 neurons). Activation of D1 receptors elicits a dose-dependent increase in motor activity and seizures in mice14. The pro-convulsive effect of the D1 agonist depends on the downstream PKA-DARPP32-ERK signaling pathway that increases neuronal firing frequency and is likely to involve recurrent activation of striato-thalamo-cortical neuronal circuits14. Both long-term sustained ablation of microglia (for more than three weeks) and their acute depletion (for three days) by CSF1R-inhibition triggered increased motor responses (Extended Data Fig. 2h) and seizures (Fig. 1g, Extended Data Fig. 2i) in response to i.p. administration of the D1 agonist SKF81297 at subthreshold doses.
The microglia-mediated suppression of stimulus-induced neuronal activation is executed by grey matter microglia in a highly region-specific fashion. The maintenance of microglia is controlled by interleukin-34 (IL34) and colony stimulating factor 1 (CSF1), both of which can activate CSF1R signaling and promote the survival of microglia15–17. In the striatum, the expression of Il34 and Csf1 is spatially separated; Il34 is expressed by D1 and D2 medium spiny neurons (MSNs) in the grey matter while Csf1 is expressed by oligodendrocytes and astrocytes within white matter regions16,18 (Fig. 2a, b, Extended Data Fig. 3a, b). Ablation of Il34 specifically in neuronal progenitor cells (Nestin-Cre), which give rise to neurons, astrocytes, and oligodendrocytes, resulted in a selective, gene dose-dependent loss of neuron-associated microglia in the grey matter areas of the striatum (Fig. 2c, d, Extended Data Fig. 3c–g). Conversely, ablation of Csf1 led to the loss of microglia predominantly in the white matter of the striatum (Fig. 2e, f, Extended Data Fig. 3h–j). Loss of Il34-maintained microglia in striatal grey matter, but not the ablation of Csf1-maintained microglia in white matter, resulted in an enhanced response to D1 agonist treatment as measured by the induction of seizures (Fig. 2g, h). Furthermore, the loss of striatal microglia has a selective impact on striatal neuron responses. Deletion of Il34 in either D1 or D2 neurons (Fig. 2i, Extended Data Fig. 4a–e), which causes a striatum-specific ~50% reduction in microglia numbers, led to exaggerated responses to D1 agonist treatment (Fig. 2j, Extended Data Fig. 4f, g). Notably, mice with striatum-specific ablation of microglia respond like wild-type mice to kainic acid and picrotoxin (Fig. 2k, Extended Data Fig. 4h), both of which trigger seizures via the activation of cortical and hippocampal neurons (Extended Data Fig. 2f). Our data show that microglia are critical for neurostimulant-induced neuronal activity regulation in a region-specific and microglia number-dependent fashion (Extended Data Fig. 4i). The increase in neuronal responses to D1 stimulation that was induced by microglia ablation occurred in the absence of detectable changes in striatal cellular composition or D1 and D2 neuron-specific phenotypes, including morphology, intrinsic excitability, and mRNA expression patterns (Extended Data Fig. 5, Supplementary Table 3). Thus, loss of Il34 has no overall effect on neuron or glia phenotypes (Extended Data Fig. 5j–l) in the striatum, and the abnormal hyper-responsiveness of neurons in the absence of local microglia is not a consequence of Il34 deficiency (Extended Data Fig. 5).
Figure 2: Spatial control of neuronal activity by microglia.
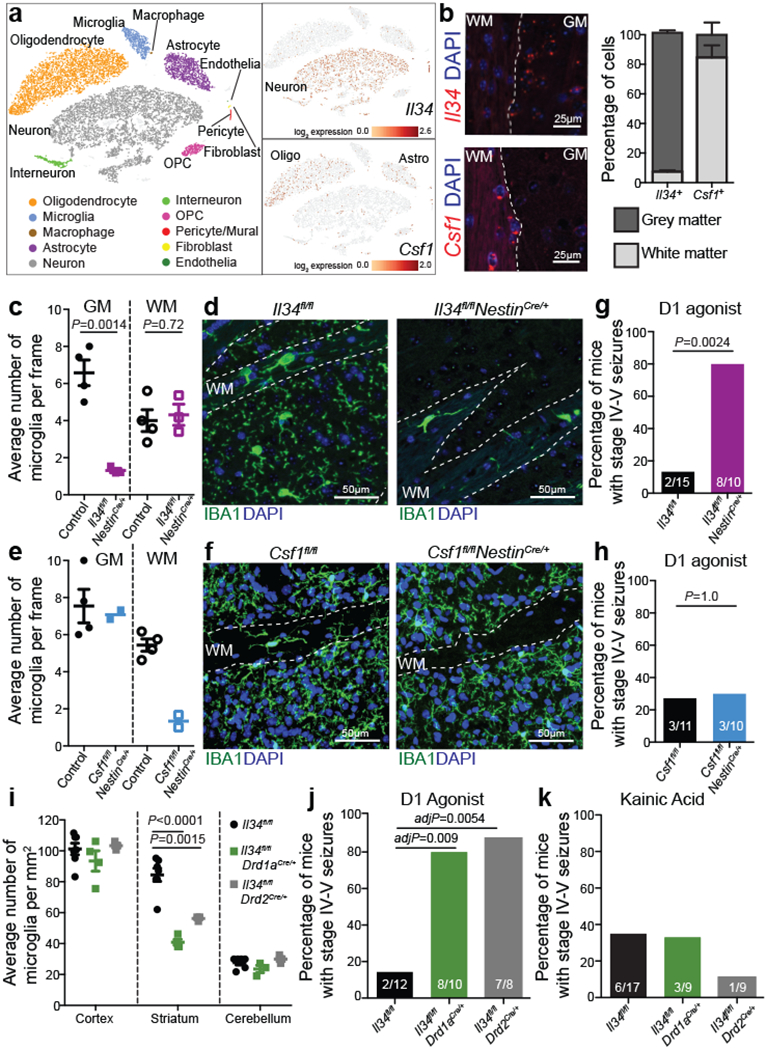
a, Left, cell populations in the mouse striatum (t-distributed stochastic neighbour embedding (t-SNE) plot) identified by single-nuclei RNA expression analysis (15,950 nuclei). Right, Il34 (top) or Csf1 (bottom) RNA-expressing cells. b, Il34+ (top left; red) and Csf1+ cells (bottom left; red) identified by RNA in situ hybridization, DAPI+ nuclei (blue), grey matter (GM), and white matter (WM). Right, distribution of Il34+ and Csf1+ cells in GM (Il34+, 93%; Csf1+, 15%) and WM (Il34+, 7%; Csf1+, 85%) in the striatum (n = 2 and 4 mice per group, respectively). c-h, Il34fl/fl and Csf1fl/fl mice were bred to NestinCre/+ mice to generate Il34fl/fl;NestinCre/+ (purple) and Csf1fl/fl;NestinCre/+ (blue) mice. c, e, Striatal microglia numbers in GM and WM in control and mutant mice shown per frame (c: n=4 and 3 mice; unpaired two-tailed t-test e: n=4 and 2 mice) d, f, Representative images of control and mutant striatum sections showing IBA1+ (green) nucleated (DAPI+, blue) microglia. g, h, Percentages of mice with seizures in response to D1 agonist (SKF81297, 5 mg/kg) (g: n=15 and 10 mice, P=0.0024, Fisher’s exact test; h: n=11 and 10 mice; P=1.0, Fisher’s exact test). i-k, Il34fl/fl mice were bred to Drd1aCre/+ or Drd2Cre/+ mice to generate Il34fl/fl;Drd1aCre/+ (green) and Il34fl/fl;Drd2Cre/+ (grey) mice. i, Number of microglia/mm2 in cortex, striatum, and cerebellum (n=7, 4, and 3 mice, cortex: P=0.38; striatum: P<0.0001; cerebellum: P=0.14, One-way ANOVA with Tukey’s post hoc test). j-k, Percentage of mice with seizures within 1hr in response to D1 agonist (j, SKF81297, 5mg/kg) or kainic acid (k, 18mg/kg). (j: n=12, 10, and 8 mice; P=0.0014; k: n=17, 9, and 9 mice; P=0.40, Chi-squared test with Bonferroni adjustment). The experiments in g have been independently repeated in a second cohort with identical results. Data shown as mean± s.e.m.
Using two-photon in vivo imaging of neuronal calcium responses in the dorsal striatum (Fig. 3a, Extended Data Fig. 6a, Supplementary Video 1), we found that ablation of microglia led to increased striatal neuron synchrony and increased the probability that striatal neurons would fire simultaneously (Fig. 3b, c, Extended Data Fig. 6b). Increased neuronal synchrony has been shown to underlie seizure progression19 and is likely to contribute to seizures in microglia-deficient mice. In addition, we found that microglia determine the threshold for striatal neuron activation in response to D1 agonist treatment (Fig. 3d, e, f). Acute treatment with a low dose of D1 agonist was sufficient to induce a substantial and prolonged increase in the frequency of neuronal events, with no difference in magnitude, in animals lacking microglia as compared to control mice (Fig. 3d–f, Extended Data Fig. 6c–g).
Figure 3. Microglia control striatal neuron synchrony and firing frequency in vivo.
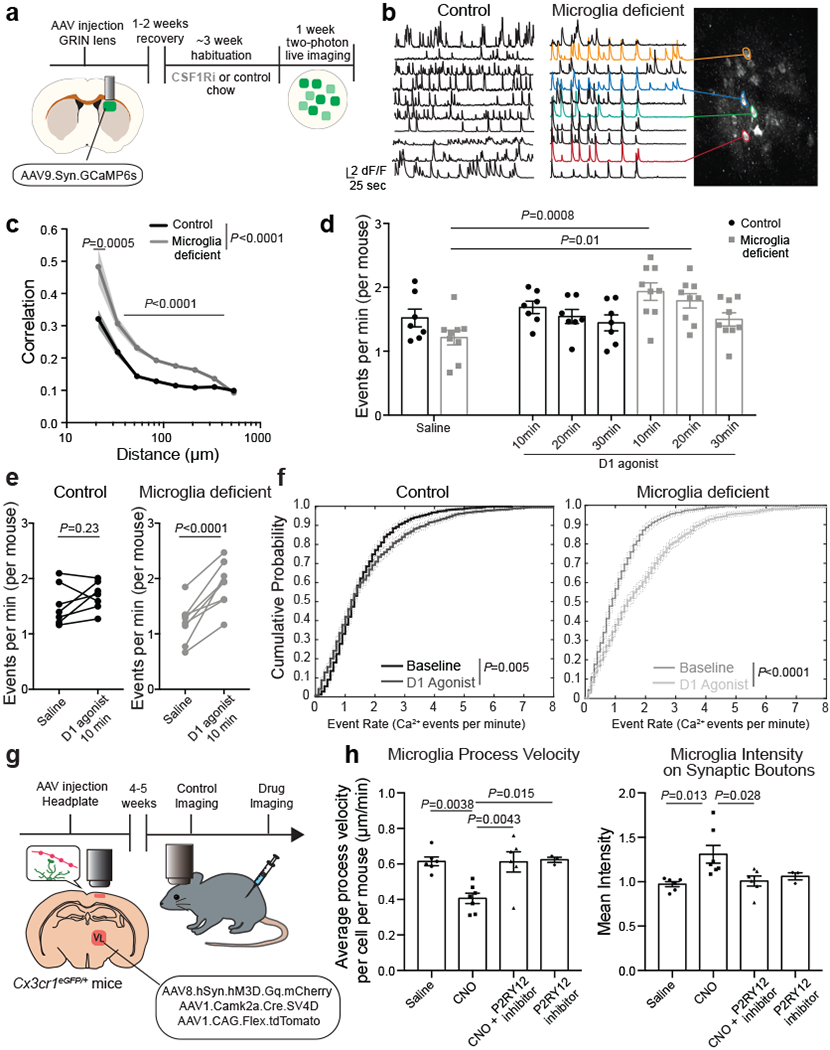
a, Two-photon calcium imaging of striatal neurons in control and microglia-deficient mice. b-c, Increased spatiotemporal coding of striatal neuronal activity in microglia-depleted mice at baseline. b, Representative neuron traces (n=10 neurons/mouse) and c, Correlation of spatiotemporal coding of striatal neuron activity (n=7 and 9 mice, treatment: P<0.0001, distance: P<0.0001, interaction: P<0.0001; Two-way ANOVA with Sidak’s multiple comparison test). d-e, Average frequency of Ca2+ events in control (black) and microglia-deficient (gray) mice at baseline and in response to D1 agonist (SKF81297, 3mg/kg)(n=7 and 9 mice; d, diet: P=0.48, treatment: P=0.0024, interaction: P=0.08, Two-way ANOVA with Tukey’s post hoc test. e, paired two-tailed t-test). f, Cumulative probability of Ca2+ events per neuron per minute in control (left) and microglia deficient (right) mice at baseline and in response to D1 agonist (3mg/kg) (n=7 and 9 mice, two-sample Kolmogorov-Smirnov test). g-h, Neuronal ATP-dependent changes in motility and localization of microglia protrusions. Using live two-photon imaging, eGFP+ microglia protrusions (Cx3cr1eGFP/+ mice) were tracked in proximity to virally-labeled tdTomato+ hM3Dq+ neurons. h, Microglial protrusion velocity per mouse (left) and mean intensity of microglial protrusions within 1μm2 of neuronal terminals per mouse (right) at baseline (saline) or upon neuronal activation (CNO, 5mg/kg, i.p.) in the absence or presence of P2RY12 antagonist (Clopidogrel, 100mg/kg, i.p.), (n= 6, 7, 6, and 3 mice, left: P=0.0012; right: P=0.01, One-way ANOVA with Tukey’s post hoc test). The data shown in c-f are data combined from two independent imaging cohorts of mice. Data shown as mean± s.e.m.
Neuronal activation is associated with the local release of ATP by neurons and astrocytes20–23. Microglia can detect synaptic ATP release through the surface-expressed purinergic receptor P2RY1224, which is highly expressed in forebrain microglia25 and controls ATP/ADP-dependent microglia chemotaxis and motility3,8,24,26,27. Although extracellular ATP levels in the striatum at baseline (3-5 nM)23 are far below concentrations that activate P2RY12 (>100nM24,26), local ATP release at the synapse of activated neurons can reach concentrations up to 5-500uM20, which is more than sufficient to trigger microglial P2RY12 signaling10,24. Using two-photon live imaging of eGFP-expressing microglia in the cortex, we tracked the positioning and velocity of microglia protrusions in vivo (Fig. 3g, h, Supplementary Video 2, Extended Data Fig. 6h–j). Depending on the level of neuronal activity, microglia can display different modes of motility: in the absence of strong neuronal activation, microglia motility is characterized by the constant extension and retraction of microglia protrusions, which has been defined as microglial baseline or surveillance activity9–12. The activation of neurons resulted in a decrease in microglial baseline motility indicated by reduced protrusion velocity (Fig. 3h) while enhancing microglial targeted motility (directional branch extension towards target sites3,9,10,27) leading to increased microglia–neuron proximity (Fig. 3h). Following neuronal activation, an increased number of microglia protrusions were recruited to the synaptic boutons of active thalamocortical projection neurons in an ATP-dependent manner (Fig. 3h). The pharmacological inhibition of microglial ATP/ADP-sensing by blocking P2RY12 activity both prevented the neuronal activity-induced synaptic recruitment of microglia protrusions and restored microglia baseline motility (Fig. 3h). Our findings suggest that activity-induced synaptic ATP release can act as a local chemoattractant that leads to the targeted recruitment of microglia protrusions to activated synapse.
In addition to its role as a chemoattractant, extracellular ATP can also serve as a substrate for the ATP/ADP-hydrolyzing ectoenzyme CD39 (encoded by Entpd1)28,29 the rate-limiting enzyme catalyzing ATP-AMP conversion; this is followed by conversion of AMP to adenosine (ADO)–a potent suppressor of neuronal activity30 –by CD73 (encoded by Nt5e)28 and/or the non-tissue-non-specific alkaline phosphatase TNAP31. ADO restrains neuronal activity by binding to pre- and postsynaptic Gi/o protein-coupled adenosine A1 receptors (A1R), the adenosine receptor subtype with the highest affinity for ADO in the brain (dissociation constant KD=2nM)30. Activation of A1R suppresses D1 neuron responses in the striatum both by limiting synaptic transmission via presynaptic neurotransmitter release22,32 and by suppressing the activation of postsynaptic D1 neuron signaling pathways via A1R-mediated inhibition of Protein Kinase A (PKA) activity33,34.
In support of the idea that ADO-A1R signalling mediates the suppressive effect of microglia on D1 neurons, we found that ex vivo isolated primary microglia could support the conversion of ATP to ADO (Fig. 4a). The production of ADO by microglia is suppressed by pharmacological inhibition of either CD39 (Fig 4a), which is primarily expressed by microglia18,29 (Extended Data Fig. 7a–e), or CD73 (Fig. 4a), which is expressed by striatal microglia, albeit at levels lower than in striatal neurons, as judged by CD73 mRNA and protein expression18,25,35 (Fig. 4b, Extended Data Fig. 7a, f). In line with these data, CD73 was expressed on the cell surface of CD39+ microglia isolated from the forebrains of adult wild-type mice at levels higher than on microglia from CD73-deficient mice, but lower than the CD73 expression levels on non-microglial CD39- cells (Fig. 4b). These findings suggest that microglia in the striatum in vivo can contribute to the production of ADO in either a cell autonomous fashion and/or by involving neighboring cells including neurons. Concurrently, microglia deficient mice display a significant decrease in extracellular ADO levels in the striatum (Fig. 4c). As expected to result from reduced ADO-mediated activation of A1R, a decrease in striatal microglia is associated with enhanced PKA activity in striatal D1 neurons as measured by increased phosphorylation of several PKA targets33,36 (Fig. 4d, e). These data show that microglia have a key role in the production of ADO and in ADO-mediated modulation of D1 neurons in the striatum.
Figure 4. Microglia suppress neuronal activation via an ATP/AMP/ADO/A1R- dependent feedback mechanism.
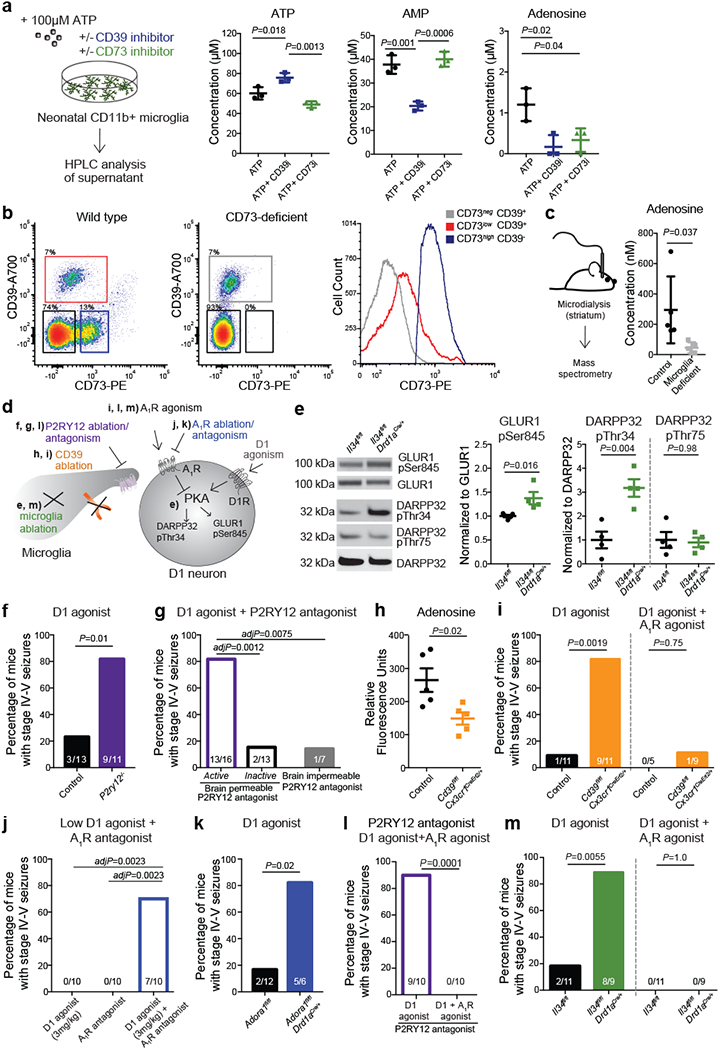
a, Levels of extracellular ATP, AMP, and adenosine (ADO) in primary microglia culture 60 min after addition of ATP (100μM) in the presence of CD39 inhibitor (ARL67156, 200μM) or CD73 inhibitor (APCP, 10μM) analyzed by high performance liquid chromatography (HPLC) (n=3 wells; ATP: P=0.0016; AMP: P=0.0005; ADO: P=0.017; One-way ANOVA with Tukey’s post hoc test). b, Left, surface expression of CD39 and CD73 in isolated forebrain cells from adult wild type (left) and CD73-deficient Nt5e−/− (right) mice by fluorescence-activated cell sorting (FACS). Right, expression of CD73 on CD39+ microglia (red) and CD39− non-microglia cells (blue) from wild-type mice as compared to CD39+CD73− microglia from Nt5e−/− mice (grey) (representative of three independent experiments). c, Extracellular ADO concentration in striatal dialysate from control and microglia deficient mice analyzed by mass spectrometry (n=5 mice, unpaired two-tailed t-test). d, Pharmacological and genetic dissection of the ATP-AMP-ADO-A1R circuit in microglia-dependent neuron regulation. e, PKA activity in striatal protein lysate from Il34fl/fl and Il34fl/fl;Drd1aCre/+ mice was measured by phosphorylation of GLUR1 at Ser845, DARPP32 at Thr34 (marker of D1 neuron activation36), and DARPP32 at Thr75 (marker of D2 neuron activation36) (Values normalized to total protein on the same blot, GLUR1Ser845: n=4 and 5 mice, two-tailed Mann-Whitney test; DARPP32Thr34: n=4, unpaired two-tailed t-test; DARPP32Thr75: n=4, unpaired two-tailed t-test). f-g, Percentage of mice showing seizures in response to D1 agonist (SKF81297, 5mg/kg) upon P2ry12 gene ablation (f, n=13 and 11 mice, Fisher’s exact test) or P2RY12 pharmacological inhibition (g, n= 16, 13, and 7 mice, P=0.0004, Chi-squared test with Bonferroni adjustment). h-i, Microglia-specific ablation of Entpd1/CD39. h, Striatal adenosine levels in control and Cd39fl/fl;Cx3cr1CreErt2/+ mice after D1 agonist (SKF81297, 5mg/kg) (n=5 mice, unpaired two-tailed t-test). i, Percentage of mice with seizures in response to D1 agonist alone (left) (SKF81297, 5mg/kg, n=11 mice, Fisher’s exact test) or in combination with A1R agonist (right) (CPA, 0.1mg/kg, i.p., n=5 and 9 mice, Fisher’s exact test with Yates correction). j, Percentage of mice with seizures in response to D1 agonist (SKF81297, 3mg/kg), A1R antagonist (DCPCX, 1mg/kg) or both (n=10 male mice; P=0.0018, Chi-squared test with Bonferroni adjustment). k, Percentage of mice with D1 neuron-specific ablation of A1R (Adora1fl/fl;Drd1aCre/+ mice) that seized in response to D1 agonist (SKF81297, 5mg/kg) (n=12 and 6 mice, Fisher’s exact test with Yates correction). l-m, Prevention of D1 agonist induced seizures by co-treatment with A1R agonist (CPA, 0.1mg/kg, i.p.) in mice following P2RY12 inhibition (l, n=10 mice, Fisher’s exact test) or striatum-specific microglia ablation (m, right, n=11 and 9 mice, D1 agonist: Fisher’s exact test; D1 agonist and A1R agonist: Fisher’s exact test). Experiments shown in f-g are combined from two independent cohorts of mice. For gel source data, see Supplementary Figure 1. All tests are two-tailed; data shown as mean± s.e.m.
We assessed the functional importance of the microglia-dependent ATP-AMP-ADO-A1R cascade in vivo by monitoring mice for seizures following interference with the individual components of the circuit (Fig. 4d). Blocking ATP/ADP-mediated microglia recruitment either by inactivating the P2ry12 gene (Fig. 4f) or in response to acute pharmacological inhibition of P2RY12 in the brain (Fig. 4g) triggered an increase in neuronal responses to D1 agonist treatment, supporting the idea that P2RY12 modulates neuronal activity and seizures3,7. The same effect was observed by rendering microglia unable to convert ATP to AMP/ADO. The same effect was observed when we rendered microglia unable to convert ATP to AMP/ADO. Microglia-specific deletion of Entpd1 (which encodes CD39) in adult mice (Extended Data Fig. 8a–c) is associated with an increase in striatal neuron PKA activity (Extended Data Fig. 8d), a significant decrease in striatal adenosine levels (Fig. 4h) and increased seizure susceptibility in response to D1 agonist treatment (Fig. 4i (left), Extended Data Fig. 8e). In addition, pharmacological inhibition of A1R activity (Fig. 4j) or D1 neuron-specific ablation of A1R expression in mice (Fig. 4k, Extended Data Fig. 8g) triggers an exaggerated D1 neuron response that recapitulates the effect of microglia ablation (Fig. 1g, 2j, 4m) or of rendering microglia unable to respond to and process ATP (Fig. 4f, g, i). Conversely, the alterations in striatal neuron activity and seizures in mice that lack microglia (Fig. 1g, 2j, 3c, d, 4m), the microglial P2RY12-mediated ATP response (Fig. 4f, g), or microglial CD39-mediated conversion of ATP to AMP (Fig. 4i) could be reversed by the administration of an A1R receptor agonist (Fig. 4i, l, m, Extended Data Fig. 6d–g, Extended Data Fig. 8i, j). The increased D1 agonist-induced seizure response in mice lacking A1R in D1 neurons was not prevented by pharmacological activation of A1R receptors on non-D1 neurons (Extended Data Fig. 8h), further supporting the highly localized nature of this mechanism. Collectively, these findings suggest that the ATP-AMP-ADO-A1R cascade has a critical role in local microglia-mediated suppression of D1 neurons in the striatum.
This novel microglia-controlled negative feedback mechanism is also likely to operate in other brain regions. Indeed, we found that microglia can reduce cortical neuron firing rates and seizures in response to glutamate receptor stimulation in a CD39- and A1R- dependent fashion in vitro (Extended Data Fig. 9) and in vivo (Extended Data Fig. 8f). Microglia-driven neurosuppression is likely to play a key role in constraining excessive neuronal activation that cannot be sufficiently suppressed by inhibitory neurons alone. This potent mechanism may also allow microglia to relay changes in the state of the local or peripheral environment to neurons and thereby to direct specific behavioral responses. Microglia can respond directly to pro-inflammatory signals that arrive from the periphery37,38 or are generated locally in the brain during neuro-inflammation or neurodegeneration39,40. It is conceivable that the downregulation in P2ry12 and Entpd1 expression in reactive microglia during various inflammatory24,40 and neurodegenerative diseases including Alzheimer’s and Huntington’s (summarized in Extended Data Fig. 10a–g)39,40 contributes to the pathological increases in neuron excitability and behavioural alterations that are associated with these disorders29,41,42 (Extended data Fig. 10h–j). It is further tempting to speculate that abnormal neuronal functions during sickness behavior or depression, which have been linked to aberrant activation of microglia43, might reflect changes in the ATP-AMP-ADO metabolic pathway or in neuronal A1R-mediated signaling responses.
Methods
Animals
Mice w ere housed two to five animals per cage with a 12-hour light/dark cycle (lights on from 0700 to 1900 hours) at constant temperature (23°C) and humidity (~50%) with ad libitum access to food and water. All animal protocols were approved by IACUC at Icahn School of Medicine at Mount Sinai and were performed in accordance with NIH guidelines.
For brain-wide microglia ablation, adult C57/Bl6 wild type mice (Jackson Laboratory, stock number 000664) between 8-16 weeks of age were treated with CSF1R inhibitor, PLX562244 (1200ppm chow, Plexxikon, Berkeley, CA), or control chow (same formula lacking only the inhibitor; Plexxikon, Berkeley, CA) for 3 weeks or 3 days as indicated. While a 3-week long PLX5622 treatment leads to a 99% microglia loss, around 80% of microglia are already lost after 3 days44.
For all pharmacological seizure experiments (those not involving genetic models), adult male C57/Bl6 wild type mice (Jackson Laboratory, stock number 000664) between 8-16 weeks of age were used.
To generate mice allowing for forebrain projection neuron-specific DREADD-mediated activation or inactivation, we bred transgenic CaMKII-tTa mice45 (Jackson Laboratory, stock number 003010) to transgenic TetO-CHRM3 mice46 (Jackson Laboratory, stock number 014093) or transgenic TetO-CHRM4 mice46 (Jackson Laboratory, stock number 024114). We then bred these DREADD mice to our microglia-specific TRAP (Cx3cr1CreErt2/+(Litt); Eef1a1LSL.eGFPL10a/+, both are knock-in lines to the endogenous locus) mice25,47,48, resulting in CaMKII-tTa;TetO-CHRM3;Cx3cr1CreErt2/+(Litt);Eef1a1LSL.eGFPL10a/+ (CHRM3) mice, CaMKII-tTa;TetO-CHRM4; Cx3cr1CreErt2/+(Litt);Eef1a1LSL.eGFPL10a/+ mice (CHRM4) and control Cx3cr1CreErt2/+(Litt); Eef1a1LSL.eGFPL10a/+ mice. To activate Tamoxifen-inducible CreErt2, all mice were gavaged at 4-6 weeks of age with five doses of 100 mg/kg of Tamoxifen (T5648, Sigma, St. Louis, MO) in corn oil (C8267, Sigma) with a separation of at least 48 hours between doses. For immunofluorescence, behavior, and gene expression, we induced neuronal excitation by intraperitoneal (i.p.) injection of 0.25 mg/kg clozapine N-oxide (CNO) and induced neuronal inhibition by i.p. injection of 1.0 mg/kg CNO 2 hours prior to tissue dissection.
To generate mice with forebrain and/or striatum neuron-specific conditional ablation of Il34, conditional Il34fl/fl mice49 in which exons 3-5 were targeted for deletion, were bred to transgenic NestinCre/+ mice50 (Jackson Laboratory, stock number 03771), BAC-transgenic Drd1aCre/+ mice (EY262, Gensat), BAC-transgenic Drd2Cre/+ mice (ER44, Gensat), BAC-transgenic Drd1aeGFPL10a/+ mice (CP73, Gensat), or BAC-transgenic Drd1aTdTomato/+ mice (Jackson Laboratory, 016204). For D1 neuron TRAP, Drd1aeGFPL10a/+ mice, which express an eGFP-tagged ribosome protein L10 under the Drd1 promoter, were bred to Il34fl/fl; Drd1aCre/+ mice generating Il34fl/fl;Drd1aCre/+;Drd1aeGFPL10a/+ mice. For identification of D1 neurons for electrophysiological recording, Il34fl/fl;Drd1aCre/+ mice were bred to Drd1aTdTomato/+ mice to identify D1 neurons by tdTomato expression or Il34fl/fl;Drd1aCre/+;Drd1aeGFPL10a/+ mice were used to identify D1 neurons by eGFP expression.
To generate mice with a brain-specific conditional ablation of Csf1, conditional Csf1fl/fl mice51 in which exons 4-6 were targeted for deletion, were bred to transgenic NestinCre/+ mice50 (Jackson Laboratory, stock number 03771).
To achieve conditional microglia-specific ablation of CD39, conditional Cd39fl/fl mice29,52 were bred to knock in Cx3cr1CreErt2/+(Jung) (Jackson Laboratory, stock number 020940)53 mice.
To generate mice with a D1 neuron-specific conditional ablation of Adora1, conditional Adora1fl/fl mice54 were bred to BAC-transgenic Drd1aCre/+ mice (EY262, Gensat).
For non-microglia cell-type specific TRAP, adult BAC-transgenic Aldh1l1eGFPL10a/+ mice (Jd130, Gensat) were used for astrocyte specific TRAP, BAC-transgenic Drd1aeGFPL10a/+ mice (Cp73, Gensat) were used for D1 neuron specific TRAP, and BAC-transgenic Drd2eGFPL10a/+ mice (Cp101, Gensat) were used for D2 neuron specific TRAP.
For mice with an ablation of CD73 (Nt5e), Nt5e−/− mice55 with a targeted deletion of Nt5e were purchased from Jackson Laboratory (stock number 018986).
For mice with an ablation of P2RY12, P2ry12−/− mice3,24,56 were obtained from Dr. Ukpong Eyo at the University of Virginia.
For use of an Alzheimer’s disease mouse model, 5xfAD57 mice were purchased from Jackson Laboratory/MMRRC (stock number 034840-JAX).
All mice used for experiments were backcrossed to the C57Bl/6J background for ≥ 5 generations. If not otherwise specified, Cre negative littermate controls were used as controls. Unless otherwise specified, male and female mice were used for all experiments (only male mice were used for social interaction behavior, live two-photon imaging of calcium transients in neurons, and live two-photon imaging of microglial process velocity and contact with boutons). Routine genotyping was performed by tail biopsy and PCR as previously described25,45–52,58.
RNA Isolation and Quantitative PCR (qPCR)
Mice were anesthetized with CO2 followed by decapitation. Brain regions of interest were rapidly dissected and frozen in liquid nitrogen and stored at −80°C until further processing. RNA extraction from frozen samples was performed using the TRIzol/Chloroform technique according to the manufacturer’s instructions (Invitrogen Corporation, Carlsbad, CA). After extraction, RNA was precipitated overnight at −80°C in isopropanol with 0.15M sodium acetate and Glycoblue (Life Technologies). The pellet was washed twice with 70% ethanol, air-dried, and resuspended in nuclease-free water. cDNA was prepared from total RNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems). Relative gene expression of the cDNA was assayed by qPCR (StepOne Software, ThermoFisher) using pre-designed recommended TaqMan gene expression assays from Applied Biosystems following manufacturers recommendations (Il34, Csf1, Gapdh, Entpd1, Adora1, Ccl3, Cd74, Kdm6b, Adrb1, Ccl24, Kckn13, Ikbkb). Cycle counts for mRNA quantification were normalized to Gapdh. Relative expression (ΔCT) and quantification (RQ=2-ΔΔCT) for each mRNA were calculated using ΔΔCT method as suggested.
Translating Ribosome Affinity Purification (TRAP)
This approach relies on the genetic labeling of the ribosomal protein L10a with the enhanced Green Fluorescent Protein (eGFP) in a cell type-specific fashion followed by eGFP-based immunoaffinity purification of the ribosome-associated mRNAs25,59,60. The microglia-specific TRAP approach allows to assess rapid changes in microglial ribosomal RNA-association in the absence of aberrant microglia activation following tissue dissection and cell isolation25. To assess changes in microglia in response to CaMKII+ neuron activation and inhibition using the DREADD approach, microglia-specific eGFP-L10a expression in CaMKII-tTa;tetO-CHRM3;Cx3cr1CreErt2/+(Litt);Eef1a1LSL.eGFPL10a/+ mice, CaMKII-tTa;tetO-CHRM4;Cx3cr1CreErt2/+(Litt); Eef1a1LSL.eGFPL10a/+ mice, and control Cx3cr1CreErt2/+(Litt);Eef1a1LSL.eGFPL10a/+ mice was induced at 4-6 weeks of age using 6 consecutive administrations of 100mg/kg Tamoxifen (T5648, Sigma) with a separation of at least 48 hours between doses. Neuronal excitation in CHRM3 mice was induced with 0.25 mg/kg CNO 2 hours prior to sacrificing the animals. Neuronal inhibition in CHRM4 mice was induced with 1 mg/kg CNO 2 hours prior to sacrificing the animals.
Mice were euthanized with CO2 and the striatum was rapidly dissected and ribosome-bound RNA was isolated as described25,60,61. Microglia, astrocyte, D1, and D2 TRAP experiments were performed using freshly isolated tissue. D1 TRAP on Il34fl/fl;Drd1aCre/+;Drd1aeGFPL10a/+ and littermate controls was performed on rapidly frozen tissue. Brain tissue was immediately homogenized with a motor-driven Teflon glass homogenizer in ice-cold polysome extraction buffer (10mM HEPES [pH 7.3], 150mM KCl, 5mM MgCl2 0.5mM dithiothreitol (Sigma), 100 μg/ml cyclohexamide (Sigma), EDTA-free protease inhibitor cocktail (Roche), 10μl/ml RNasin (Promega) and Superasin (Applied Biosystems)). Homogenates were centrifuged for 10min at 2000 x g, 4°C, to pellet large cell debris. NP-40 (EMD Biosciences, CA) and 1,2-Diheptanoyl-sn-Glycero-3-Phosphocholine (Avanti Polar Lipids, AL) were added to the supernatant as a final concentration of 1% and 30mM, respectively. After incubation on ice for 5 min, the lysate was centrifuged for 10min at 13000 x g to pellet insoluble material. Mouse monoclonal anti-GFP antibodies (clones 19C8 and 19F7, Antibody and Bioresource Core Facility, Memorial Sloan Kettering, NY) and biotinylated Protein L (GenScript, Piscataway, NJ)-coated Streptavidin MyOne T1 Dynabeads (Invitrogen) were added to the supernatant, and the mixture was incubated at 4°C with end-over-end rotation overnight. Beads were collected on a magnetic rack and washed four times with high-salt polysome wash buffer (10mM HEPES [pH 7.3], 350mM KCl, 5mM MgCl2, 1%NP-40, 0.5mM dithiothreitol (Sigma), and 100 μg/ml cyclohexamide (Sigma)). RNA was purified from beads directly using RNeasy Mini Kit (Qiagen) following manufacturer’s instructions.
RNA sequencing
RNA purification from TRAP samples and from 5% of their corresponding unbound fractions was performed using RNeasy Mini Kit (Qiagen) following manufacturer’s instructions and used for subsequent sequencing. RNA integrity was assayed by using an RNA Pico chip on Bioanalyzer 2100 using 2100 Expert Software (Agilent, Santa Clara, CA) and only samples with RIN>9 were considered for subsequent analysis. Double-stranded cDNA was generated from 1-5 ng RNA using Nugen Ovation V2 kit (NuGEN, San Carlos, CA) following manufacturer’s instructions. 500 ng of cDNA per sample was sonicated to obtain fragments of 200 base pairs using Covaris-S2 system (Duty cycle: 10%, Intensity: 5.0, Bursts per second: 200, Duration: 120 seconds, Mode: Frequency sweeping, Power: 23W, Temperature: 5.5°C - 6°C, Covaris Inc., Woburn, MA). These fragments were used to produce libraries for sequencing by TruSeq DNA Sample kit (Ilumina, San Diego, CA, USA) following manufacturer’s instructions. The quality of the libraries was assessed by the 2200 TapeStation (Agilent). Multiplexed libraries were directly loaded on NextSeq 500 (Ilumina) with High Output single read sequencing using 75 cycles. Raw sequencing data was processed with Illumina bcl2fastq2 Conversion Software v2.17.
Bioinformatic analysis of RNA sequencing data
Raw sequencing reads were mapped to the mouse genome (mm9) using the TopHat2 package (v2.1.0)62. Reads were counted using HTSeq-count (v0.6.0)63 against the Ensembl v67 annotation. The read alignment, read counting as well as quality assessment using metrics such as total mapping rate, mitochondrial and ribosomal mapping rates were done in parallel using an in-house workflow pipeline called SPEctRA64. The raw counts were processed through a variance stabilizing transformation (VST) procedure using the DESeq2 package65 to obtain transformed values that are more suitable than the raw read counts for certain data mining tasks. Principal component analysis (PCA) was performed on the top 500 most variable genes across all samples based on the VST data to visually assess if there were any outliers. Additionally, hierarchical clustering was used to assess the outliers once again to protect against false positives/negatives from PCA, and the outliers were further justified by the aforementioned quality control metrics as well as experimental metadata. After outlier removal, all pairwise comparisons were performed on the count data of entire gene transcripts using the DESeq2 package (v1.20.0)65.
For the analysis of microglia-specific TRAP in DREADD mice and in D1 neuron TRAP in Il34fl/fl;Drd1aCre/+;Drd1aeGFPL10a/+ mice and Cre negative controls, an enrichment cutoff of p-value < 0.05 and fold change > 2 for each TRAP group over its respective unbound fraction was applied. For microglia-specific TRAP in DREADD mice, a cutoff of p-value < 0.05 and mean expression > 30 (DESeq2; n=2-3 per group; 3-6-month-old males and females) was applied. For D1 neuron TRAP, a cutoff of p-value < 0.05, fold change >1.5, and mean expression > 30 (DESeq2; n=3-4 per group; 4-month-old males and females) was applied. All analyses were carried out with age- and sex-matched controls.
For analysis of glial activation in bulk RNAseq, astrocyte activation markers were selected from those identified by Liddelow et al66 and microglia sensome genes were selected from those identified in Hickman et al (92).
All of the MA plots and volcano plots were made using R (v3.1.1; https://www.R-project.org). For all heatmaps, expression of each gene in log2 reads per kilobase of transcript per million mapped reads, or log2 RPKM was normalized to the mean across all samples (z-scored). Heatmaps with hierarchical clustering were made on Multiple Experiment Viewer 4.8 (v.10.2; http://www.tm4.org) with Pearson Correlation by average link clustering67. Gene Ontology (GO) term enrichment analysis was performed using Enrichr68,69. ENRICHR FDR values were calculated using the Benjamini–Hochberg test in ENRICHR and P values were calculated using Fisher’s exact test in ENRICHR. Selected and significantly enriched (p-value < 0.05 with Benjamini-Hochberg correction) GO annotations for biological processes and are represented as bar graphs.
Striatal Nuclei Isolation
Striatal Nuclei Isolation was performed as previously described25. Mice were euthanized with CO2, and brain regions were quickly dissected and homogenized in 0.25 M sucrose, 150 mM KCl, 5 mM MgCl2, 20 mM Tricine pH 7.8 with a glass Dounce homogenizer (1984-10002, Kimble Chase, Vineland, NJ). All buffers were supplemented with 0.15 mM spermine, 0.5 mM spermidine, and EDTA-free protease inhibitor cocktail (11836170001, Roche). The homogenate was then spun through a 29% iodixanol cushion. The resulting nuclear pellet was resuspended in 0.25 M sucrose, 150 mM KCl, 5 mM MgCl2, 20 mM Tricine pH 7.8, supplemented with 10 μM DyeCycle Ruby (V10304, Invitrogen) and 10% donkey serum (017-000-121, Jackson Immunoresearch, West Grove, PA). Striatal nuclei were sorted in a BD FACSAria II cell sorter by gating for the lowest DyeCycle Ruby population, which indicates nuclei singlets.
Single Nuclei RNA Library Preparation and Sequencing
Single nuclei RNA-Seq (snRNA-seq) was performed on these samples using the Chromium platform (10x Genomics, Pleasanton, CA) with the 3’ gene expression (3’ GEX) V2 kit, using a targeted input of ~5,000 nuclei per sample. Briefly, Gel-Bead in Emulsions (GEMs) were generated on the sample chip in the Chromium controller. Barcoded cDNA was extracted from the GEMs by Post-GEM RT-cleanup and amplified for 12 cycles. Amplified cDNA was fragmented and subjected to end-repair, poly A-tailing, adapter ligation, and 10X-specific sample indexing following the manufacturer’s protocol. Libraries were quantified using Bioanalyzer (Agilent) and QuBit (Thermofisher) analysis and then sequenced in paired end mode on a HiSeq 2500 instrument (Illumina, San Diego, CA) targeting a depth of 50,000-100,000 reads per nuclei.
The raw read data were demultiplexed, aligned and analyzed by 10X Cell Ranger (v2.1.0). In order to capture unspliced pre-mRNA in the single nuclei RNA expression assay, intronic regions in 10X Cell Ranger mm10 v1.2.0 reference were marked as exonic as suggested by 10X for pre-mRNA reference generation. Data from two libraries were aggregated together with read depth normalized according to the number of mapped reads per nuclei of each library. A UMI threshold of 250 was manually selected to include the microglial population, resulting in a total of 15950 sequenced nuclei. The median number of UMIs detected per nuclei was 1707 UMI, with a median of 1075 genes per nuclei. Doublet populations were manually excluded from the analysis. Unique populations were manually annotated using known markers for striatal cell types70 using the 10X Loupe Cell Browser (v2.0.0) (10X Genomics). Expression of individual genes was visualized by t-SNE on the Loupe Browser, with zero UMIs in gray. Cluster-specific upregulated genes (P<0.05) were determined using the “Significant Genes” tool in the Loupe Browser. Normalized expression values (UMIs per 10^3 UMIs) for selected genes were calculated for each sequenced nucleus using R and graphed by cell type with the ggplot2 package
In situ hybridization
In situ hybridization was performed as previously described25. 3-6 month old male and female wild type mice were anesthetized with ketamine (120 mg/kg) and xylazine (24 mg/kg) and perfused transcardially with 10 ml PBS and 40ml of 10% neutral-buffered formalin, followed by removal and fixation of the brain in 10% neutral-buffered formalin. Fixed brains were embedded in paraffin and sliced in 5-μm-thick sections, baked for 1 hour at 60°C, and stored at room temperature until further processing. In situ hybridization was carried out using RNAScope custom-designed probes for Il34, Csf1, and Entpd1 in combination with the RNAScope 2.0 Red kit per manufacturer’s recommendation (Advanced Cell Diagnostics). After completing in situ hybridization, before colorimetric reaction, sections were rinsed with ddH20 and a gradient concentration of PBS (0.1X-1X), blocked in 2% normal goat serum in PBS for one hour at room temperature and incubated with IBA1 antibody (019-19741, 1:100, Wako) overnight at 4°C. IBA1 signal was amplified with anti-rabbit horseradish-peroxidase-conjugated secondary antibody and AlexaFluor 488 tyramide from Tyramide Signal Amplification Kit (Life Technologies) following manufacturer’s instructions. Sections were stained with DAPI (0.2 mg/ml), rinsed with a gradient concentration of PBS (1X-0.1X) and ddH20), subjected to colorimetric reaction, dried 15min at 60°C, mounted using EcoMount (EM897L, BioCare Medical), and dried overnight. Single plane images were taken on Zeiss LSM 780 confocal microscope and analyzed using Zen 2012 software. Cell counting was done using ImageJ cell counter tool. For localization of cells expressing Il34 or Csf1 in grey matter or white matter, single plane images (0.5mm x 0.5mm) that were on average 75% grey matter and 25% white matter were counted. 4-11 images were counted per mouse.
Microglia isolation
Microglia were isolated from neonatal (P5-P7) wild type mice for plating in Axion MEA plates, culture, and protein as previously described25,71. Mice were rapidly killed, brains were extracted, and meninges were removed prior to extracting forebrain. Tissue was mechanically dissociated in glass homogenizers and enrichment of live cells was achieved by centrifugation over 20% Percoll (17-0891-02, GE Healthcare). The microglia were selected by anti-CD11b-coated microbeads (130-093-636, Miltenyi) with the QuadroMACs separator following the manufacturer’s recommendations. Cells were manually counted with a hemocytometer using trypan exclusion staining. For co-culture with neurons in Axion MEA plates, 100,000 microglia were added per well. For HPLC, microglia were plated at 56,000/cm2 in a 12-well plate with DMEM supplemented with 10% FBS (F4135, Sigma) and 1% penicillin–streptomycin (15140, Gibco). Microglia purity was determined by FACS and immunostaining, cultured cells were also used for immunoblotting.
Microglia Flow Cytometry
FACS analysis was used to determine microglial CD39 and CD73 surface expression as well purity of CD11b+ bead-isolated microglia. For confirmation of the purity of CD11b+ bead-isolated microglia, single cell suspensions were prepared from neonatal pups (4-6 forebrains, age P5-P7). For analysis of CD39 and CD73 expression on adult microglia, single cell suspensions were prepared from the forebrain of wild type, Cx3cr1Cre Ert2/+(Litt) mice, which express cytosolic YFP under the microglial Cx3cr1 promoter, and Nt5e−/− (CD73 deficient) mice.
Tissue was rapidly dissected and mechanically dissociated in HBSS and centrifuged over 20% Percoll (GE Healthcare) and mononuclear cells were collected. Cells were resuspended in FACS buffer (500mL sterile PBS, .5% BSA, 2ml 0.5M EDTA), blocked in FC blocker (BioRad) as per manufacturer’s instructions and then stained on ice for 15 minutes with a combination of the following antibodies: CD73-PE (12-0731-82, ThermoFisher), CD39-Alexafluor700 (24DMS1, ThermoFisher). The samples were stained with DAPI on the final resuspension. The percentage of CD39+ and CD73+ cells was determined using fluorescence activated cell sorting (FACS) on the Attune NxT flow cytometer (Invitrogen) and data was analyzed with FCS Express Plus software (De Novo Software). Compensation was performed on single-stained samples of UltraComp eBeads (ThermoFisher), unstained beads, and YFP unstained brain (YFP only) and unstained control cells. Gates were determined using unstained samples, fluorescence minus one (FMO) controls, in which one antibody was omitted per sample, and appropriate isotype controls for the CD73 antibody. Forward and side scatter was used to gate on a defined population of cells to exclude debris and also select single cells. Live cells were determined as DAPI negative. In Cx3cr1Cre Ert2/+(Litt) mice, microglia were identified by YFP expression. FACS plots show gating on live cells that correspond to microglia, which made up 6-7% percentage of all cells in samples prepared from adult forebrain.
100% of YFP+ cells were CD39+. 0% of cells were YFP-/CD39+. Therefore, CD39-A700 was used to identify microglia in subsequent FACS experiments. In wild type and Nt5e−/− mice, single, live cells were identified by forward and side scatter gating as well as lack of DAPI- expression. Wild type and Nt5e−/− mice were stained with CD39-A700 to label microglia72 and CD73-PE to identify CD73+ cells. Nt5e−/− mice were used in parallel to determine specificity of the CD73-PE antibody. 7% of live cells were CD39+ microglia in both WT and Nt5e−/− mice. 0% of cells were CD73+ in Nt5e−/− mice. CD73+ cells were determined using gates created from Nt5e−/− mice, IgG isotype control samples, and unstained samples.
Immunofluorescence staining
Immunostaining was performed as previously described25,61. Male WT mice aged 3 months treated with control or PLX5622 diet for 3 weeks, 4-8 month old male and female CaMKII-tTa;tetO-CHRM3;Cx3cr1CreErt2/+(Litt);Eef1a1LSL.eGFPL10a/+ mice and CaMKII-tTa;tetO-CHRM4; Cx3cr1CreErt2/+(Litt);Eef1a1LSL.eGFPL10a/+ mice 2 hours after CNO, 3 month old Il34fl/fl;NestinCre/+, Il34fl/fl;Drd1aCre/+, Il34fl/fl;Drd2Cre/+, Csf1fl/fl;NestinCre/+, and Cd39fl/fl;Cx3cr1CrErt2/+(Litt) mice and littermate controls were anesthetized with ketamine (120 mg/kg) and xylazine (24 mg/kg) and perfused transcardially with 10 ml PBS and 40 ml 4% paraformaldehyde (Electron Microscopy Sciences). Fixed brains were removed and dehydrated in 5%, 15% and 30% sucrose in PBS. Following dehydration, brains were frozen in Neg-50 (Thermo Scientific) on dry ice and stored at −80 °C until further processing. Brains were cut using a cryostat and 25-μm sections were mounted on Superfrost Plus microscope slides (Fisher Scientific). Slides were stored at −80 °C until staining. Slides were washed with PBS, permeabilized with PBS + 0.2% Triton X-100 (PBST) and blocked with 2% normal goat serum in PBST for one hour at room temperature. Slides were incubated with primary antibodies (IBA1 (1:500) 019-19741, Wako; CD11b (1:500) MCA711GT, Biorad; GFP (1:2000) ab6556, Abcam; GFP (1:500) ab13970, Abcam; NeuN (1:500) MAB377, Millipore; NEUN (1:500) ABN91, Millipore, cFOS (1:1000) ab190289, Abcam; GFAP (1:500) G3893, Sigma; OLIG2 (1:500) sc-293163, Santa Cruz; P2RY12 (1:1000) AS-55043A, Anaspec;) in 2% normal goat serum in PBST overnight at 4 °C. Slides were washed in PBST and incubated in Alexa Fluor-conjugated secondary antibodies (Alexa Fluor 488-, 546-, and 568- and 647-labeled goat anti-mouse, goat anti-rat, goat anti-chicken, or goat anti-rabbit IgGs (H+L); 1:500, Thermo Scientific) in 2% normal goat serum in PBST for 1 h at RT. Slides were washed and coverslipped using Prolong Gold anti-fade with DAPI (Invitrogen) and dried overnight. Imaging was performed using a Zeiss LSM 780 Confocal Microscope (Zeiss, Oberkochen, DE). For z-stack images, 20-μm z-stack confocal images were acquired at 2-μm intervals, with 40×/1.3 oil objective at 0.6 or 1x zoom. For single plane images, 4.6 μm images were acquired at 20x/.8 or 40x/1.3 objectives 0.6 or 1x zoom. Image processing was performed using Zen 2012 software (Zeiss). Cell counting was done using ImageJ cell counter tool by counting cFOS+ cells or IBA1+ cells that overlapped with DAPI+ nuclei. Graphs show average number of cells (n=3-4 images) per mouse.
For identification of microglial localization (white matter or grey matter within the striatum), white matter was identified by the high levels of autofluorescence and lack of neuronal soma. The specificity of the approach was validated by immunostaining with tyrosine hydroxylase (TH, ab112, Abcam), which labels neuronal terminals in the striatum and is restricted to grey matter (data not shown). Graphs show average number of cells in WM and GM per mouse (n=2-4 mice, 4-11 images per mouse). Striatal images were taken at 20x as described above. ImageJ was used to calculate the percentage of the image that was white matter or gray matter.
Immunoblotting
Western blot analysis was performed as previously described25,61. Male and female age-matched mice were used. Mice were anesthetized with CO2 followed by decapitation, and the region of interest was rapidly dissected and frozen in liquid nitrogen and stored at −80 °C until further processing. For analysis of DARPP32 phosphorylation, mice were euthanized, brains were immediately frozen in liquid nitrogen, and frozen striatum punches were extracted. Samples were sonicated at 4 °C in 1% SDS solution supplemented with protease inhibitor (Roche, Switzerland) and PhosStop phosphatase inhibitor (Roche, Switzerland), and boiled for ten minutes. The protein concentration was determined using a BCA protein assay kit (ThermoFisherScientific, USA) according to the manufacturer’s instructions. Protein samples were diluted in equal volume of 2X LDS sample buffer (Invitrogen) and supplemented with DTT to a final concentration of 200 mM (Sigma).
20-40 μg of tissue protein samples were separated on 4-12% Bolt Bis-tris precast denaturing gels or 10% NuPAGE Bis-tris precast denaturing gels (Invitrogen, USA) and transferred onto PVDF membranes. Membranes were blocked for 1 hour and probed with primary antibodies diluted in 5% milk-TBST solution or 2% BSA overnight at 4 °C (DARPP32, (1:1000), Novus, Cat#NB300-304; DARPP32-THR34, (1:1000), Cell Signaling, Cat#12438; DARPP32-THR75, (1:1000), clone cc911 and DARPP32, (1:5000), clone 6a kindly provided by A Nairn and P Greengard; IL34, (1:1000), Abcam, Cat#AF5195; GLUR1-SER845, (1:1000), Cell Signaling, Cat#8084; GLUR1, (1:1000), Millipore, Cat#MAB2263, DRD1, (1:1000), Abcam, Cat#ab20066; CD39, (1:1000), R&D Systems, Cat#AF4398; CD73, (1:500), Cell Signaling, Cat#13160 H3, (1:5000), Abcam, Cat#ab1791; IBA1, (1:1000), Wako, Cat#016-20001; P2RY12, (1:1000), Anaspec, Cat#AS-55043A). Membranes were then washed and probed with horseradish-peroxidase conjugated anti-mouse (GE), anti-rabbit IgG (GE), anti-rat IgG (Invitrogen) or anti-sheep IgG (Jackson Immunoresearch, USA) secondary antibody (all 1:10000) for one hour at room temperature. Membranes were developed using enhanced chemiluminescence substrate (PerkinElmer, USA) and exposed on film. Exposed films were scanned, and protein bands were quantified using ImageJ Software (NIH, USA). All values were plotted relative to littermate control samples.
For immunoblotting of CD11b+ isolated microglia, cells were counted on a hemocytometer and centrifuged at 300g for 15 min to pellet cells. Cells were resuspended in 100uL of lysis buffer (see above), sonicated, and boiled for ten minutes. The lysed cells were concentrated by centrifugation (Protein Concentrator Tubes, 3K, Pierce) and diluted in equal volume of 2X LDS sample buffer (Invitrogen) supplemented with DTT (final concentration 200mM) to a final concentration of 50,000 cells/uL. Increasing amounts of cells were loaded along with whole tissue samples of control and CD73−/− striatal lysate (5ng). Samples were run, transferred, and incubated in primary and secondary antibody as described above. Membranes were developed using chemiluminescence substrate (PerkinElmer) (CD39, P2RY12, IBA1, H3) or with SuperSignal West Femto Maximum Sensitivity Substrate (ThermoScientific) for less abundant proteins (CD73).
Slice biotinylation assay of membrane bound proteins
Slice biotinylation was adapted from previously described protocols73,74. 10-week old male and female Il34fl/fl and Il34fl/fl;Drd1aCre/+ mice were anesthetized with isoflurane and the brain was rapidly removed and sliced (300uM) in prechilled, pre-saturated (95%/5% O2/CO2) 1X SACSF on a vibratome. Slices were recovered for 45 minutes at 31°C in oxygenated ACSF. Following recovery, slices were incubated with 1.0 mg/mL sulf-NHS-SS-biotin on ice for 45 minutes. Slices were washed 3x with ice cold ACSF and incubated for 10 minutes in ice cold ACSF. Slices were then washed 3x with ice cold quench buffer (ACSF, 100mM glycine and incubated twice for 25 minutes on ice to quench excess free biotin. Slices were then washed in ice cold ACSF three times and the striatum was micro-dissected. Tissue was pelleted by centrifuging at 200g for 1 min. Supernatant was removed and tissue was resuspended in 300uL ice cold RIPA buffer (Thermo Scientific) with protease inhibitors (Roche) and pipetted to break up tissues. To complete tissue lysis, samples were incubated for 30min at 4°C with end-over-end rotation. Debris was pelleted by spinning at 1800g for 15 minutes. Supernatant was collected and heated at 98°C for 10 minutes and sample concentration was assessed by BCA protein assay kit (ThermoFisher Scientific, USA) according to manufacturer’s instructions. A portion of protein was aliquoted and diluted in equal volume of 2X LDS sample buffer (Invitrogen) and supplemented with DTT to a final concentration of 200mM (Sigma) to serve as input. Streptavidin MyOne T1 Dynabeads (Invitrogen) were washed in RIPA/PI buffer and added to known concentration of protein lysis and were rotated at 4°C overnight. The following day, samples were spun down and biotinylated and unbound fractions were magnetically separated and collected. Biotinylated proteins were eluted off magnetic beads in 2X LDS sample buffer supplemented with DTT by boiling for 5 minutes at 95°C and separated on magnet. Equal amounts of protein (5ug-20ug) were loaded for immunoblotting.
Neuronal culture
Primary neuronal culture was performed as previously described75. Embryonic day 18 (E18) timed-pregnant female mice were anesthetized with CO2 and sacrificed by cervical dislocation. In a dissection hood, 10-12 embryos per experiment were collected through an incision of the mother’s abdomen, taken out of the amniotic sacs, and decapitated in ice-cold Hank’s Balanced Salt Solution (HBSS). Using fine scissors and forceps, brains were rapidly dissected and the cortex cleared from meninges and isolated under a dissection microscope. Cortices were collected in ice-cold HBSS and kept on ice until all embryos had been dissected. In a tissue culture hood, HBSS was removed and the cortex tissue was digested by 0.25% Trypsin-EDTA for 15 min at 37°C, followed by DNase1 treatment for 10 min at 37°C. The tissue was dissociated by serial trituration with a 25-ml serological pipette, followed by trituration with 10 and 5 ml serological pipettes. Cell suspension was washed once with DMEM medium, supplemented with 10% FBS and 1% penicillin/streptomycin, and passed through a 40μm cell strainer before being counted on a hemocytometer. Single cells were seeded on poly-d-lysine (0.1 mg ml−1)–coated wells at a density of 106 cells per well on a 12-well plate. Cells were grown in neurobasal medium, complemented with B27 supplement, N2 supplement, and 0.5 mM l-glutamine and maintained at 37°C in 5% CO2.
Axion recording
For multiple electron array (MEA) recordings, AccuSpot Classic MEA 48 plate with 16 microelectrodes per well were used (M768-KAP-48A, Axion). Plates were pre-incubated at 37 degrees Celsius for one hour with 5 microliters of 0.1% PEI (P3143, Sigma) in borate buffer (Boric acid (A73-500, Fisher), Sodium tetraborate (221732, Sigma). After incubation, plates were washed 4 times with 200 microliters dH20 and dried overnight in the hood with the lid cracked. The following day, neurons were cultured as described above, and the dried Axion plate was coated with 20 micrograms/ml laminin in neurobasal medium (L2020, Sigma) mid-way through the neuronal culture. 5 microliters of laminin were dropped into each well of the plate and incubated at 37 degrees Celsius for 1-2 hours without letting the plate dry. Laminin was removed directly before seeding the well with neurons. Neurons were diluted to 12 million cells/mL and 5 microliters were dropped onto each well to seed 60,000 neurons/well. The plate was incubated for 1 hour and then 250 microliters of DMEM medium, supplemented with 10% FBS and 1% penicillin/streptomycin was added to each well. The following day 250 microliters of neurobasal medium supplemented with B27 supplement, N2 supplement, and 0.5 mM l-glutamine was added to the wells. Half media change with neurobasal medium with supplements was performed every 3 days. Microglia were isolated using the Macs Miltenyi system as described in Ayata et al.25 and 100,000 isolated microglia were added to each well of the MEA plate on DIV12. Baseline Axion recordings were performed at DIV 14 on the Axion Maestro MEA reader and the electrical activity of the culture was recorded for 10 minutes and analyzed with the AxIS software. On DIV14 two hours after baseline recording, the MEA plate was treated with combinations of the following compounds: 10μM Glutamate (Sigma), 100nM A1R agonist (CPA, Tocris), 100nM A1R antagonist (DCPCX, Sigma), 10uM adenosine (Sigma) 200μM CD39 inhibitor (2 hour pretreatment, ARL 67156, Tocris). Recordings were performed 1 hour after treatment. For analysis, wells were excluded if the mean firing rate was <1Hz or if the well contained inactive electrodes (<16 active electrodes).
Adenosine Assay
Mice were anesthetized with isoflurane and striata were rapidly dissected, snap frozen, and stored at −80°C until processing. Weighed tissue was sonicated in 1XPBS and adenosine was measured by fluorometric assay (MET-5090, Cell BioLabs) following manufacturer’s instructions.
Two-photon imaging of neuronal activity
Surgery.
Male mice were anesthetized with isoflurane gas/carbogen mixture (5% for induction and 1.5% for maintenance during surgery) and carefully placed to a stereotaxic frame (David Kopf Instruments, CA, USA). Body temperature was maintained at 36–37 °C and ophthalmic ointment was applied to prevent eyes from drying. After shaving their hair and sterilizing the skin with chlorhexidine, a midline incision was made with a sterile scalpel. Then a surgical scissor was used to cut off extra skin and further expose the skull surface. The skull surface was wiped and cleaned with autoclaved cotton swabs. Bregma and lambda were identified and leveled to be on the same z-axis. Then a craniotomy hole was drilled over the striatum (antero-posterior: −0.1 mm, medio-lateral: - 1.5 mm relative to Bregma) with drill bits (#73 size, Kyocera). 600μl of AAV9 Syn-GCaMP6s (Addgene, #100843) were injected into the striatum (antero-posterior: −0.1 mm, medio-lateral: - 1.5 mm, dorso-ventral: −2.85 mm relative to Bregma) using a blunt 35-gauge microinjection needle within a 10μL microsyringe (NanoFil, World Precision Instruments, FL, USA), which was controlled by a microsyringe pump (UMP3, World Precision Instruments) and a controller (Micro4, World Precision Instruments). The AAV is injected at a rate of 50nl/min.
Following viral injection, a 23-gauge needle is mounted to a stereotaxic cannula holder (Doric lenses) and lowered to 0.5mm above the injection site (antero-posterior: −0.1 mm, medio-lateral: - 1.5 mm, dorso-ventral: −2.25 mm relative to Bregma). After holding the needle at that location for 5 minutes, it was retracted and lowered to coordinate mentioned above several times to create the path for GRIN Lens implantation. After the complete retraction of the 23-gauge needle, the GRIN Lens of 0.6mm diameter, 7.3mm length (INSCOPIX, INC, CA, USA) was assembled with a ferrule using super glue (Loctite) and mounted to a stereotaxic cannula holder (Doric lenses), then lowered to 0.15mm above the injection site (antero-posterior: −0.1 mm, medio-lateral: - 1.5 mm, dorso-ventral: −2.7 mm relative to Bregma). A layer of adhesive cement (C&B Metabond, Parkell Inc.) was applied to the skull surface to strongly hold the implanted ferrule. After adhesive cement was completely dried, a thick layer of dental cement (Lang Dental) was applied to build a head cap. Before the head cap fully solidified, we applied some super glue on the bottom surface of the head ring and place the head ring around the exposed ferrule. Then we wrapped the exposed upper surface of the lens with a small piece of parafilm, then add KWIK-SIL on top to protect the lens. Mice were given 1 mg/kg buprenorphine SR and 5 mg/kg ketoprofen s.c. intraoperatively and received 30 mg/kg ibuprofen in their home cage water for seven days post-surgery.
Habituation to head-fixation and intraperitoneal (i.p.) injection.
After a 1-2 week recovery from the surgery, the mice were randomly divided into 2 groups, one group received PLX5622 and one group received control chow (lacking inhibitor).
The mice were later put under water restriction (1.5 ml/day) and were handled and habituated daily to head-fixation and immobilization for ~2 weeks. They were immobilized in a polyethylene tube and head-fixed in the future recording environment under 2-photon microscope. During this habituation period, we increased the head-fixation period from 3 minutes to 40 minutes gradually. After they showed no signs of stress and drank water provided randomly during the 40-minute session, we switched to no-water-provided head fixation habituation for future recording.
After habituation to head-fixation, we also performed daily habituation to i.p. injection before the head-fixation session. Mice were injected i.p. with a microlitre volume equivalent to 10× the body weight in grams (10 × BW μl) of saline, matching the volume to be injected in future imaging sessions, and then performed head-fixation. This habituation was performed for ~1 week until the mice showed a reduction in clear signs of stress upon handling and i.p. injection.
In vivo two-photon imaging.
We use a customized 2-Photon microscope with galvo-galvo scanner for imaging. The setting we used through the session below is 4Hz scan rate at 194 by 194 pixels. The imaging protocol lasted for 7 days and included 3 sessions.
Session 1: 3 days of baseline recording, we i.p. injected 10XBW μl of saline and started the recording 10 minutes after the injection and image for 10 minutes. For analysis of baseline neuronal activity, data was pooled from three days of 10 minute baseline (saline injection) recording.
Session 2: 3 days of recording after SKF81297 injection, we i.p. injected 10XBW μl of SKF81297 solution (3mg/kg, diluted in saline) and started the recording 10 minutes after injection. A low dose of SKF81297 was used to avoid confounds induced by seizure activity. The imaging lasted for 30 minutes, analyzed in 10-minute bins, to capture the whole post-injection dynamics. For the analysis of neuronal activity in response to SKF81297, data was pooled across three days of imaging for each 10-minute bin starting 10 minutes post injection (first bin: 10-20 minutes after injection, second bin: 20-30 minutes after injection, and third: 30-40 minutes after injection).
Session 3: 1 day of recording after i.p. injection of mixed solution of SKF81297 (3 mg/kg, diluted in saline) and CPA (0.1 mg/kg, diluted in saline). The recording was started 10 minutes after injection and lasted for 30 minutes. For the analysis of neuronal activity in response to co-administration of SKF81297 and CPA, data was pooled from the 30 minute imaging session.
Calcium Imaging Data Analysis.
The calcium imaging data were originally acquired as DAT file in our customized system and later processed into TIF files. The data were firstly rigid motion corrected using the custom script adapted from NoRMCorre (https://github.com/flatironinstitute/NoRMCorre), the technical details could be found in the relevant paper76.
The motion-corrected data was later ran through pipeline adapted from Suite2P (https://github.com/cortex-lab/Suite2P)77 for automatic ROI detection and spike deconvolution. To account for out-of-focus contamination from background signals Fbg, a fraction d = 0.7, of the background was subtracted from the raw fluorescence Fraw. The relative change in fluorescence was calculated as:
Where F is the background corrected fluorescence F = Fraw − d · Fbg and F0 is the median of the F distribution.
To identify significant calcium events, we use a peak detection algorithm that identifies maxima in the derivative of the dF/F signal implemented in Matlab97. The identified maxima must be above a threshold, defined as the mean plus three standard deviations of the dF/F distribution.
To understand how the similarity between calcium traces of neurons varies as a function of their distance between each other, we utilized a method described before78 which involves calculating the Pearson’s correlation coefficient (PCC) between two time series traces x and y, were:
Where E[·] is the expectation operator and μ and σ denote the mean and standard deviation, respectively. The PCC was calculated for pairs of neurons where the distance between them was calculated as the Euclidean distance between the centers of their somas.
Synchrony was calculated as previously defined in Barbera et al., 201679. In brief, onsets of calcium transients were identified with a threshold crossing of 2 times the standard deviation of the calcium trace baseline. In order to account for uncertainty associated with threshold detection, each event is represented as a 750 ms (3 frame) pulse centered at the onset of the calcium trace. We defined two events to be synchronous if they overlap at least at one time instant. The binary matrix was then used to calculate synchrony between two cells as the average of the ratio of the number of times both cells were simultaneously active to the total activations of each cell.
Two-photon imaging of microglial protrusion and synaptic terminals
For imaging of microglia and synaptic terminals, male Cx3cr1eGFP/+ mice (8-10 weeks) derived from the C57BL/6J strain, in which enhanced green fluorescent protein (eGFP) expresses under microglial Cx3cr1 promoter were used. For Ca2+ imaging of synaptic terminals, male C57BL/6J mice (6-10 weeks) were used.
Surgery and virus injection.
Mice were anesthetized by mixture of ketamine (74 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). The skull was exposed and disinfected, and a custom-made head plate was firmly attached using dental cement (Fujiryu-to BC; GC, Tokyo, Japan, Bistite II; Tokuyama Dental, Tokyo, Japan) onto the skull. Mice were allowed to recover for 1 day before the following craniotomy and viral injection.
For virus injection, a circular craniotomy (2 mm diameter) was performed over the left primary motor cortex (centered at 0.2 mm anterior and 1 mm lateral from bregma) under isoflurane (1%) anesthesia. For imaging of microglia and synaptic terminals, mixed AAV (Addgene; AAV8.hSyn.hM3D(Gq).mCherry: 5 × 1012 vector genomes/ml, UPenn Vector Core; AAV2/1-CAG-FLEX-Tdtomato: 7.6 × 1012 vector genomes/ml, and AAV2/1-CaMKII-Cre: 2.94 × 1013 vector genomes/ml (1:10,000) diluted in saline) solution was injected into the ventral lateral nucleus of the thalamus (1 mm anterior and 1 mm lateral from bregma, 3,600 μm deep). For Ca2+ imaging of synaptic terminals, AAV2/1-Syn-GCaMP6s: 2.7 ×1013 vector genomes/mL (1:2 diluted in saline) and AAV8.hSyn.hM3D.Gq.mCherry was injected into ventral lateral nucleus of the thalamus (1 mm anterior and 1 mm lateral from bregma, 3,600 μm deep). To exclude the effect of CNO, control mice with the same AAV2/1-CAG-FLEX-Tdtomato and AAV2/1-CaMKII-Cre cocktail but omitting AAV8.hSyn.hM3D(Gq).mCherry were used. Virus was filled in glass capillary with filament (GDC-1; Narishige, Tokyo, Japan) and injected into stereotaxic coordinates over 5 minutes. After the injection, a double glass window comprised of 2 and 4.5 mm glass slips (Matsunami Glass, Osaka, Japan) jointed together with an ultraviolet curable adhesive (NOR-61, Norland) was implanted over the craniotomy.
Two-photon imaging.
All the imaging was conducted 5 weeks after the virus injection to acquire sufficient virus expression and brain tissue recovery to prevent microglial activation80.
The two-photon microscopy setup was composed of a laser scanning system (NIS-Elements; Nikon Instech Co., Ltd., Tokyo, Japan), a mode-locked Ti:Sapphire Chameleon Ultra II laser (Coherent, Santa Clara, CA) set at 950-nm and a water-immersion objective lens (25×, N.A. 1.10; Nikon Instech Co., Ltd.). XYZT imaging was conducted over the primary motor cortex, and the imaging plane was within 100-150μm from the surface. 1024×1024 pixels imaging field was 129.96μm × 129.96μm with a pixel size of 0.1269μm. Z-step was 1μm, each XYZ frame duration was 1 minute and whole imaging session was 3 hours long. For DREADDs excitation or microglial P2Y12R inhibition, Clozapine-N-oxide (CNO; Tocris Bioscience, Bristol, UK; 5 mg/kg) or Clopidogrel (Sanofi-Aventis; 100 mg/kg) was dissolved in saline and freshly prepared before every injection. CNO, Clopidogrel and CNO + Clopidogrel imaging sessions were started immediately after the intravenous administration of CNO to mice. Note that the mice used for each experiment were totally naive for all drugs. In control imaging, mice were treated with saline injection. Mice were imaged for 3 hours immediately after injection.
Two-photon image analysis.
Analysis was performed using ImageJ (1.52v; NIH). All images were corrected for focal plane and depth direction displacements using HyperStackReg (Ved Sharma, 2015-2016). For quantification of interactions between microglia and pre-synapses, 2 μm diameter ROIs were manually defined around axonal boutons, and mean intensity in green channel (microglia) within each ROI was measured for all frames. To normalize value, mean intensity was divided by the average intensity of 5 control frames within the same ROI, which were recorded before drug stimulation. Microglial contact onto boutons was further demonstrated by measuring the Pearson’s correlation coefficient value of red and green channels with ImageJ Coloc 2 plugin. For the assessment of microglial motility in time-lapse images, ImageJ built-in tool Manual Tracking was used to track tips of primary processes in each frame. Microglial tips were identified by the eGFP expression (Cx3cr1eGFP/+ mice). 7-9 microglia tips per microglia were tracked for 2-4 microglia per mouse. We measured the trajectory of primary processes that were defined as branches emanating directly from the cell body. To track the process tips, we first identified the tips by projecting 20 × 1 μm z slices and the farthest point of each primary processes were plotted with ImageJ software. The ImageJ built-in tool Manual Tracking was used to track tips of processes for each frame.
Confirmation of viral targeting and CNO-mediated activation of neurons
Two-photon Ca2+ imaging.
All the imaging was conducted 5 weeks after the virus injection to acquire sufficient virus expression and brain tissue recovery to prevent microglial activation.
The two-photon microscopy setup was composed of a laser scanning system (NIS-Elements; Nikon Instech Co., Ltd., Tokyo, Japan), a mode-locked Ti:Sapphire Chameleon Ultra II laser (Coherent, Santa Clara, CA) set at 950nm and a water-immersion objective lens (25X, N.A. 1.10; Nikon Instech Co., Ltd.). XYT imaging was conducted over the primary motor cortex with axonal terminals expressing GCaMP6s via viral transfection in ventral lateral nucleus. Imaging plane was within 100-150μm from the cortical surface. 512×512 pixels imaging field was 130.02 μm × 130.02 μm with a pixel size of 0.2539 μm. 1,000 XYT frame was taken in each imaging at 2 fps. Ca2+ responses under three different conditions were conducted on the same day in each mouse, in the order of pre-injection, saline injection, and CNO injection. For DREADDs excitation of neurons, Clozapine-N-oxide (CNO; Tocris Bioscience, Bristol, UK; 5 mg/kg) was dissolved in saline and freshly prepared before every experiment. Saline and CNO imaging sessions were started 30 minutes after the intravenous administration of drug or the same amount of saline to mice.
Ca2+ imaging analysis.
Analysis was performed using ImageJ (1.52v; NIH) and MATLAB (2019b; The Mathworks, Natick, MA). All images were corrected for focal plane using TurboReg81. Axonal terminal identification, Ca2+ activity extraction and quantification of Area Under the Curve (AUC) were conducted using the customized MATLAB script.
For electrophysiological recordings of sEPSC
Brain slice preparation for physiology recordings.
8- to 12-week old male and female Il34fl/fl; Drd1aCre/+; Drd1atdTomato mice were used for all electrophysiology experiments. Mice were anesthetized with isoflurane followed by transcardial perfusion with oxygenated (95% O2/5% CO2) N-methyl-D-glucamine (NMDG) HEPES solution (in mM: 92 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na+ ascorbate, 2 thiourea, 3 Na+ pyruvate, 10 MgSO4 7H2O, 0.5 CaCl2 2 H2O, with pH adjusted to 7.3– 7.4, 300 –310 mOsm). The brain was quickly removed into ice-cold NMDG HEPES solution for 1 min. Two hundred μm-thick coronal slices containing striatum were cut with a vibratome (Leica VT1200S, Germany). Slices were then moved into a pre-warmed (32°C) recovery chamber and carried out the stepwise Na+ spike-in procedure82, then kept at room temperature for at least 1 h, in the following solution: 95% O2/5% CO2-equilibrated HEPES-holding-solution containing the following (in mM) 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na+ ascorbate, 2 thiourea, 3 Na+ pyruvate, 2 MgSO4 7H2O, and 2 CaCl2, 2 H2O.
Voltage-clamp electrophysiology.
Recordings were made under an upright microscope (Scientifica SliceScope Pro 2000, Scientifica, UK) equipped with infrared differential interference contrast optics for visualization. Slices were transferred to a recording chamber superfused with standard recording ACSF containing (in mM) 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 12.5 glucose, 2 MgSO4 7H2O and 2 CaCl2 2H2O, adjusted to pH 7.3–7.4, 295 – 305 mOsm. Recordings were performed at 32°C. Patch pipettes were made from borosilicate glass capillary tubing (1B150F-4; World Precision Instruments) using a micropipette puller (PC-10; Narishige, Japan).
For spontaneous EPSCs (sEPSCs) measurements in the striatum, the internal recording pipette solution was potassium-based contained the following (in mM): K+ gluconate 130, KCl 4, EGTA 0.3, HEPES 10, MgATP 4, Na2GTP 0.3, Phosphocreatine 10; pH adjusted to 7.3 with KOH while the external solution was ACSF + 100 μM picrotoxin. sEPSCs were recorded from the red fluorescent protein (RFP) positive dorsal striatum neurons using a Multiclamp 700B amplifier (Molecular Devices), filtered at 3 kHz, amplified five times, and then digitized at 10 kHz with a Digidata 1550 analog-to-digital converter (Molecular Devices). Voltage was held at −80 mV (Vhold = −80 mV). Neurons were allowed to stabilize after rapturing membrane for at least 10 min and sEPSCs were recorded. For sEPSCs event analysis, the baseline was adjusted to 0 pA and the events threshold was set at 5 pA, and then sequential events from >60 sec analysis window were individually identified using Clampfit 10.3 software (Molecular Devices). Experiment and analysis were performed without knowledge of genotype.
Ex vivo slices for electrophysiology
Mice were anesthetized with ketamine (100 mg/kg)/xylazine (7 mg/kg), transcardially perfused with ice cold modified artificial cerebrospinal fluid (aCSF; containing in mM: 124.0 NaCl, 3.0 KCl, 1.0 CaCl2, 2.0 MgCl2, 26 NaHCO3, 1.0 NaH2PO4, and 16.66 glucose), and sagittal brain slices (275 μm thick) were sectioned using a vibratome (Leica Biosystems). Ex vivo brain slices entailing the dorsolateral striatum were transferred to a holding chamber (34°C) with aCSF (containing in mM: 124.0 NaCl, 3.0 KCl, 2.0 CaCl2, 1.0 MgCl2, 26 NaHCO3, 1.0 NaH2PO4, and 16.66 glucose) for a recovery period of 30-40 minutes prior to experimentation; all solutions were pH 7.4, 310-320 mOsm and continually bubbled with 95% O2/5% CO2.
Two-photon laser scanning microscopy (2PLSM) and electrophysiology.
Slices were placed in a recording chamber and direct spiny projection neurons (dSPNs) visualized by ribosomal eGFP expressed in dSPNs with a laser scanning microscope system (Bruker) and two-photon laser (Coherent, Inc.); 810 nm excitation was used throughout. Identified SPNs were patched under an Olympus 60X/0.9NA lens using glass pipettes (3–4.5MΩ resistance); patch pipettes were filled with internal recording solution containing (in mM): 135.0 KMeSO4, 5.0 KCL, 10.0 HEPES, 2.0 Mg-ATP, 0.5 Na-GTP, 5 phosphocreatine-Tris, 5.0 phospocreatine-Na, 0.1 spermine; pH 7.25–7.30 and 270–280 mOsm. EGTA was omitted and replaced with the calcium sensitive dye Fluo-4 (200μM); to visualize dendrites the anatomical dye Alexa 568 (50μM) was included. Whole-cell patch clamp recordings were acquired at 32-34 degrees Celsius using a Multiclamp 700B amplifier as previously described83,84. Intrinsic excitability was tested in current clamp with 500 ms somatic current injections (25 pA steps). Dendritic excitability was assayed by somatic current injection to evoke back propagating action potentials (bAPs). bAP associated Fluo-4 fluorescent signals were measured in proximal (30-60 μm from the soma) and distal (>80 μm from the soma) dendrites and spines using 2PLSM.84 Data are presented as the dendritic index which is calculated by dividing distal by proximal fluorescence; ‘fluorescence’ measurements refers to area under the curve of the fluorescence trace normalized to the fluorescence intensity of the anatomical dye (AlexaFluor 568). This method of analysis was used to correct for differences in dye loading, laser power and optical path. Whole cell z-series were acquired as previously described83,84 and dendritic arborization analyzed using Neurolucida software (MBF Bioscience).
Microdialysis
C57/Bl6 male mice were put on PLX5622 or control chow (n=5 PLX5622 diet, n=5 control diet). for one week. Dialysis probes were implanted and mice recovered for one week before commencing microdialysis collection. Microdialysis guide cannulae (Harvard Apparatus, Holliston, MA) were stereotaxically implanted in the striatum (A/P: +1.4 mm; M/L: −1.0 mm; D/V: −3.8 mm from skull). Microdialysis experiments were conducted following a one-week recovery period following guide cannula implantation. Dialysis tubing was flushed prior to initial use with 70% EtOH for 5 minutes, followed by dH2O syringe pump (Model R99-E, Razel Scientific Instruments, Saint Albans, VT) at a flow rate of 1 μL/min. The tubing was then attached to the microdialysis probe (Cuprophane (6kD), membrane length 1mm; Harvard Apparatus, Holliston, MA), which was primed by placing the probe in artificial cerebrospinal fluid (aCSF; pH 7.4: 148 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 0.85 mM MgCl2) and running aCSF through the microdialysis tubing and probe at a 0.8 μL/min rate. Microdialysis experiments were done in anesthetized animals. Briefly, mice were placed into a stereotaxic frame under isoflurane anesthesia (4% induction,1.75% sustained). The probe was inserted via the guide cannula and allowed to equilibrate. Dialysate was collected after 20 minutes. All collections were frozen at −80 degrees Celsius immediately following collection completion.
Liquid Chromatography/Mass Spectrometry
Adenosine was quantified using ultra pressure liquid chromatography/mass spectrometry (UPLC/MS) in the Vanderbilt University Neurochemistry Core. Analytes in 5μL of microdialysis fluid were derivatized by sequential dilution with 10uL 100mM NaCO3 (aq) and 2% benzoyl chloride (BZC) in acetonitrile. Following a two minute incubation, the reaction was stopped by the addition of 10uL internal standard solution (in 20% acetonitrile containing 3% sulfuric acid). The adenosine internal standard was prepared by derivatization with 13C6-benzoyl chloride as described. Standard stocks were frozen at −80 °C in aliquots to prevent multiple freeze/thaw cycles. A single internal standard stock aliquot was thawed the day of use, diluted 100-fold in 20% (v/v) acetonitrile containing 3% (v/v) sulfuric acid. BZC and NaCO3 solutions were made fresh daily.
LC was performed on a 2.0 x 50 mm, 1.7mm particle Acquity BEH C18 column (Waters Corporation, Milford, MA, USA) using a Waters Acquity UPLC attached to a Waters Xevo TQ-XS triple quadrupole mass spectrometer. Mobile phase A was 15% aqueous formic acid and mobile phase B was acetonitrile. The flow rate was 200uL/min and the elution gradient was as follows: initial, 1% B; 0.1 min, 7% B; 0.5 min, 15% B; 14 min, 55% B; 14.5 min, 70% B; 18 min, 99% B; 19 min, 99% B; and 19.1 min, 1% B. An additional 5 min of equilibration time was required for reproducible chromatography.
The adenosine internal standard was prepared by derivatization with 13C6-benzoyl chloride as described85. Standard stocks were frozen at−80 °C in aliquots to prevent multiple freeze/thaw cycles. A single internal standard stock aliquot was thawed the day of use, diluted 100-fold in 20% (v/v) acetonitrile containing 3% (v/v) sulfuric acid.
High performance liquid chromatography (HPLC)
Microglia were isolated from neonatal pups as described above. Cultured microglia were pre-treated with CD39 inhibitor (ARL67156, 200uM), CD73 inhibitor (APCP, 10uM) or control (.04% DMSO) 30 minutes prior to treatment with 100uM ATP. 60min after ATP treatment, 200uL of media was removed and chilled on ice for 10 minutes. Samples were spun at 400g for 10minutes at 4°C. The supernatant was transferred to a new tube and treated with 7.5uL of 8M perchloric acid (PCA), vortexed, flash frozen and stored at −80°C until processing. PCA-treated samples were neutralized with 0.4 M K2HPO4 (Sigma-Aldrich, St. Louis, MO) and analyzed with an Agilent 1260 Infinity HPLC system (Agilent Technologies, Santa Clara, CA) equipped with a G1312B binary pump, a G1367C high performance autosampler and a G1314C Variable Wavelength Detector VL+ set at 254 nm. Nucleotides were separated by ion-pair reversed-phase chromatography using an Atlantis dC18 column (3 mm x 150 mm, particle size 3 μm; Waters Corporation, Milford, MA). The samples were loaded on the column equilibrated with buffer A (0.1 M KH2PO4, 4 mM tetrabutylammonium hydrogen sulfate, Sigma-Aldrich; pH 6). The mobile phase developed linearly from 0 to 100% buffer B (70% buffer A/30% methanol) during the first 13 min and remained isocratic at 100% buffer B for 12 min. Subsequently, the column was re-equilibrated with buffer A for 7 minutes. The flow rate was 0.5 ml/min. ATP, ADP, AMP, and adenosine were identified by their retention times and concentrations were calculated using known standards run in parallel.
Telemetric EEG and EMG recording
10-week old male mice were implanted with a wireless EEG device (HD-X02, Data Sciences International) into the subcutaneous pocket posterior to the neck and continuously recorded in the home cage. EEG leads were placed between the dura and skull, with one lead above the frontal cortex and one lead over the contralateral parietal cortex. EMG leads were placed in the neck muscle. Meloxicam (1 mg/kg) and Ampicillin (20 mg/kg) was administered both during surgery and for 2 days after surgery. Data were acquired with a telemetry system (Data Sciences International) running Ponemah v6 software and analyzed using Neuroscore v3.2.9 software (Data Sciences International). For comparison of control and microglia deficient mice, transmitters were implanted and mice were placed on PLX or control chow and allowed to recover for at least one week. Seizures were visually identified behaviorally and from the EEG and EMG.
Behavior
All behavior analyses were performed during the 0700–1900 light cycle. When possible, experimenters were blinded to the genotypes of the animals and genotypes were decoded after data was processed and analyzed. All subjects correspond to data points within two standard deviations from the sample mean and no subjects were excluded from the behavioral analyses. For all behavioral experiments, sex and age-matched corresponding littermate control animals were used. No randomization protocol was used. Animals were allocated to treatment groups to ensure uniform distribution of ages and sexes in each group. All procedures were conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the IACUC at Icahn School of Medicine at Mount Sinai.
Seizure Induction and Monitoring.
For all seizure experiments, 14-20 week old mice were used. Mice were injected intraperitoneally (i.p.) with the microliter volume equivalent to 10x the body weight (g). D1 receptor agonist (SKF 81297, Tocris) was diluted from stock (50mg/ml in DMSO) with 0.9% saline to indicated concentration to induce seizures86. Kainic acid (Sigma, K0250) was dissolved in saline just before use to indicated concentration for i.p. injections to induce seizures87. Picrotoxin (Tocris) was diluted from stock (10 mg/mL in ethanol) with 0.9% saline to administer at 1 mg/kg to induce seizures88. A1R-agonist (N6-Cyclopentyladenosine, CPA, Tocris) was diluted from stock (50 mg/mL in DMSO) in 0.9% saline to 0.1 mg/kg. A1R antagonist (8-Cyclopentyl-1,3-dipropylxanthine, DPCPX, Sigma-Aldrich) was diluted from stock (10 mg/mL in DMSO) to inject at 1 mg/mL. A2A agonist (CGS 21680, Sigma) was diluted from stock (10 mg/mL in DMSO) in 0.9% saline to inject at a final dose of 0.1 mg/kg. Clopidogrel (Sanofi-Aventis, kindly provided by J.J. Badimon, as administered in Sipe et al89) was diluted from stock (50 mg/ml in DMSO) with 0.9% saline to inject at 100 mg/kg. Ticagrelor (Astra Zeneca, kindly provided by J.J. Badimon, as administered in Sipe et al.89) was dissolved in DMSO:PEG400:Triton80 5:35:10 and then diluted in saline. To assess the effects of inflammation on seizure susceptibility, mice were pretreated with lipopolysaccharide (LPS, Sigma, 0.2 mg/kg or 2 mg/kg) 24 hours prior to D1 agonist administration.
Animals were observed for the time of onset of seizures and scored on the Racine scale90. Seizure behavior was monitored under a modified Racine scale as follows: (1) normal behavior; (2) rigid posture with raised tail; (3) continuous head bobbing and forepaws shaking; (4) rearing, falling, and jumping; (5) loss of posture and generalized convulsion activity90.
Open field analysis.
Locomotion and exploratory behavior was measured using the open field analysis in a new environment (clear plexiglass 40 × 40 × 30 cm open field arena) as previously described25,61,75. Activity in the open-field was quantified with Fusion Software (v5.0) (Omnitech Electronics). The distance traveled was recorded for each mouse. Data were collected at 5-10 min intervals over 30-90 minute test sessions.
Accelerating Rotarod.
The motor function and balance of mice was analyzed using the standard accelerated rotarod test (4-40 rpm/5 min, Med Associates, St. Albans, VT) as previously described25,61. The time taken for the mice to fall from the rod was measured in seconds. If a mouse, clinging on to the rod, completed two full passive rotations, the mouse was removed from the trial and the time was recorded as fallen from the rod. If a mouse stayed on the rod until the end of the 5-minute trial, a time of 300 seconds was recorded. After one training trial, mice were subjected to three consecutive trials/day with 5 min inter-trial intervals for three consecutive days and measurements were taken from each trial.
Elevated Plus Maze.
The elevated plus maze test was used to determine the unconditioned response to a potentially dangerous environment. Anxiety-related behavior was measured by the degree to which the rodent avoided the open arms of the maze. The mice were placed at the junction of the elevated 4-arm maze in which 2 arms are open and 2 are enclosed. The number of times the animal entered each of the arms and the time spent in each arm was recorded for 5 min by the EthoVision video-tracking system (Noldus Information Technology Inc., Leesburg, VA). Total arm entries, percentage of open arm entries, and percentage of time spent in the open arms were calculated as previously described25.
Olfaction Test (Sniff test).
Sniff test was conducted as previously described75. Olfaction was tested by exposing mice (n = 13-21) to a small amount of palatable food (Cinnamon Toast Crunch cereal; General Mills) once per day for 2 days. Mice were deprived of food overnight before the test. A clean cage was filled with roughly 3 inches of fresh bedding, and the stimulus food was buried in the bedding until it was not visible. Mice were then placed in the cage one at a time and allowed to freely explore. The latency to localize and retrieve the food was measured. All mice retrieved the food within 1 min. Bedding was mixed in between trials and tested mice were placed in a new holding cage until all cage-mates had been tested. After this, all mice were returned to their original cage and ad libitum food access was restored.
Three chamber social compatibility.
Social preference and social memory91 was performed as described75 by using a Plexiglas chamber divided into three compartments. The two edge compartments contain an empty wire cup. Mice were habituated to the testing room for at least 1 hour before the experiment. Stimulus mice were age- and sex- matched C67Bl/6 male mice that were housed in separate areas of the animal facility and had no prior contact with the test mice. Stimulus mice were habituated to the wire cup before testing. For the sociability test, the test mouse was introduced to the middle chamber and allowed to freely move and habituate to all three compartments for 10 min. Then, the mouse was restricted to the middle chamber using the dividers, while a novel object (Lego) was placed under the wire cup in one chamber and an unfamiliar mouse in the other. The test mouse was then allowed to investigate the whole apparatus for 10 min. After, the mouse was again restricted to the middle chamber while the object was replaced by a second, unfamiliar mouse. The test mouse was allowed 10 min to investigate. Data were acquired using the Ethovision system (Noldus) to automatically track motion while manual scoring was used to quantify time spent sniffing the stimuli. Counterbalancing was used to control for potential left-right preferences.
Statistical Analysis
Statistics were analyzed using GraphPad Prism v7.0a, and significance was determined at p-value < 0.05. All statistical analyses were two-tailed. Normal distribution was assessed by the Shapiro-Wilk normality test. Grubbs’ test was used to identify and remove outliers. For two-group comparisons with equal variance as determined by the F-test, an unpaired two-tailed t-test was used. Normally distributed data with unequal variance was analyzed with Welch’s correction. Not normally distributed data was analyzed by Mann-Whitney U test. For analysis across three groups or more, a One-way ANOVA was used. For the analysis of seizure susceptibility, Fisher’s exact test was used for comparison between two groups. For the analysis of seizure susceptibility across three groups, a Chi-squared test with Bonferroni post hoc adjustment was used to calculate adjusted P values to maintain an alpha of 0.05. For the analysis of accelerated rotarod a two-way ANOVA with repeated measures was used. For Gene Ontology analysis using ENRICHR, FDR values were calculated using the Benjamini–Hochberg test in ENRICHR and P values were calculated using Fisher’s exact test in ENRICHR68,69. All data is shown is represented as vertical dot plots, vertical bar graphs, or XY plots with mean ± standard error of mean (SEM). Statistical methods were not used to predetermine the sample size, but our sample sizes are similar to those generally employed in the field. All samples were randomly allocated into treatment groups. Experimenters were blind to genotype whenever possible.
Data Availability
The accession number for gene expression analysis in the paper is available at NCBI Gene Expression Omnibus (GEO) under the accession number GSE149897.
Code Availability
The code used for analysis of calcium transience in neurons to analyze event rates, magnitude, spatial correlation and synchrony can be found at https://github.com/GradinaruLab/striatum2P
Extended Data
Extended Data Figure 1. DREADD-based mouse models to study microglia responses to neuronal activation and inhibition reveals distinct microglia responses.
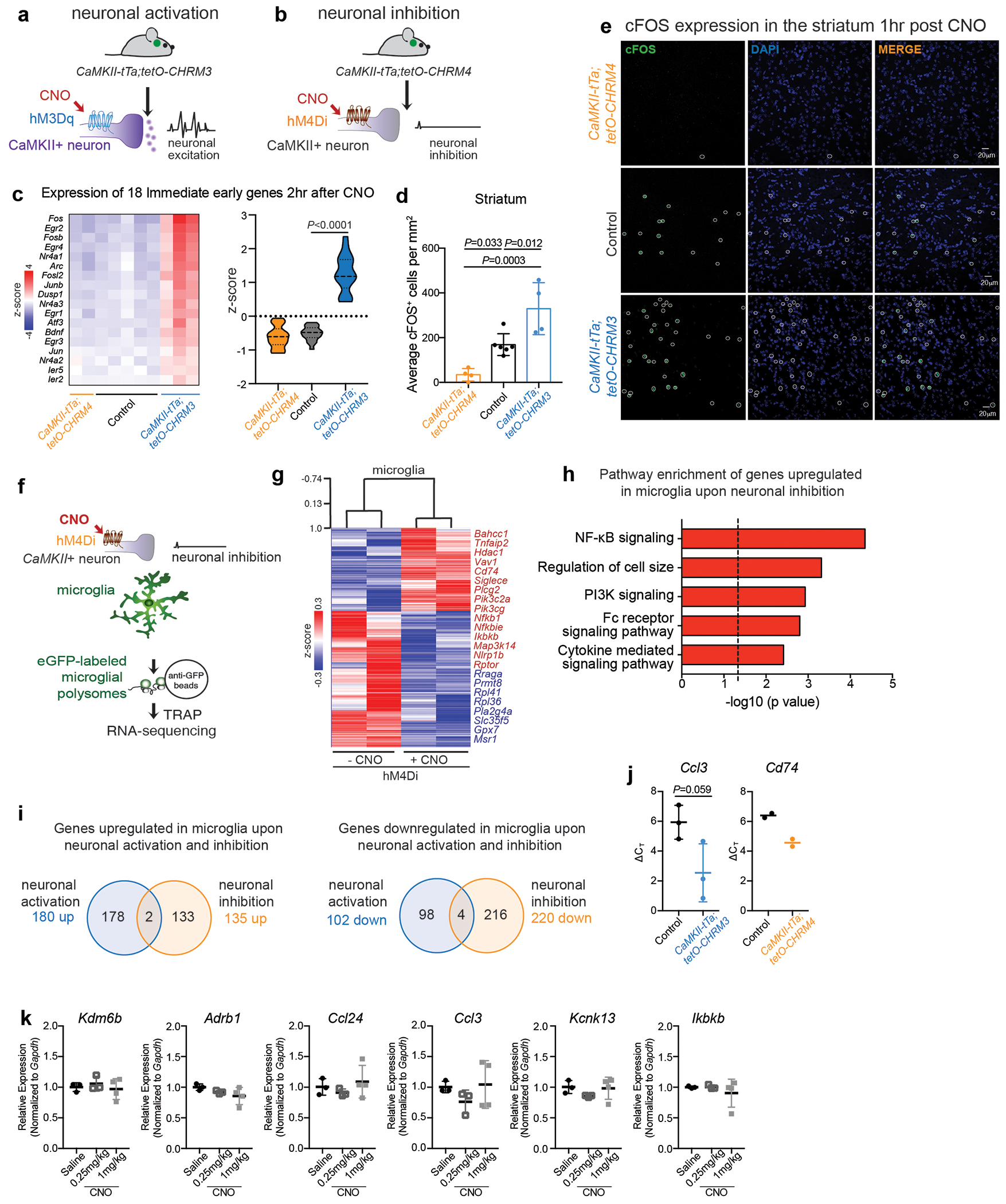
a, b, Neuron-specific activation (a) and inhibition (b) has been achieved by the expression of the Gq-coupled (activating) hM3Dq or Gi-coupled (inhibiting) hM4Di in CaMKII+ forebrain neurons. The CaMKII-tTa mice were bred to either tetO-CHRM3 or tetO-CHRM4 mice to generate CaMKII-tTa;tetO-CHRM3 or CaMKII-tTa;tetO-CHRM4 mice. hM3Dq or hM4Di were activated by i.p. injection of clozapine-N-oxide (CNO) to activate (0.25mg/kg) or inhibit (1mg/kg) CaMKII+ neuronal activity, respectively. c-e, Validation of CNO-mediated neuronal activation and inhibition: c, Heatmap (left) and violin plot (right) show RNA expression levels of 18 immediate early genes in total striatum 2 hours after CNO-mediated neuronal inhibition (orange) or neuronal activation (blue) as compared with controls (n=2 CaMKII-tTa;tetO-CHRM4, n=5 control, and n=3 CaMKII-tTa;tetO-CHRM3 mice) (right, P=0.0001, One-way ANOVA (Kruskal–Wallis test) with Dunn’s multiple comparison test). d, Dot plot showing quantification of the average number of cFOS+ cells in the dorsal striatum of CaMKII-tTa;tetO-CHRM4 (orange, n=4 mice), control (black, n=6 mice), and CaMKII-tTa;tetO-CHRM3 (blue, n=4 mice) mice one hour after treatment with CNO (P=0.0004, One-way ANOVA with Tukey’s post hoc test). e, Representative images showing cFOS+ cells (green) in the striatum of CaMKII-tTa;tetO-CHRM4 (top), control (middle), and CaMKII-tTa;tetO-CHRM3 (bottom) mice in response to CNO, DAPI (blue) (image are representative of two independent cohorts of mice). f, To allow for the microglia-specific analysis of changes in ribosome-associated RNA levels following neuron inhibition, the CaMKII-tTa;tetO-CHRM4 mice were bred to Cx3cr1CreErt2/+(Litt); Eef1a1LSL.eGFPL10a/+ mice followed by tamoxifen-induced Cre-mediated L10a-eGFP expression in microglia. g, Changes in ribosome-bound mRNA levels in striatal microglia were determined using the TRAP-sequencing approach. The heatmap shows the variation in the expression levels of 135 upregulated and 220 downregulated genes (z-scored log2(RPKM) at 2 hours following CNO-mediated neuronal inhibition. h, Selected gene ontology (GO) annotations for upregulated genes (DESeq2) in striatal microglia in response to neuronal inhibition, GO analysis was performed using ENRICHR analysis68,69 (dotted line, P=0.05). i, Venn diagrams comparing microglial genes up- and down-regulated following CaMKII+ neuronal activation and inhibition reveals highly differential microglia response. j, qPCR confirmation of increased mRNA expression (lower ΔCT, normalized to Gapdh) in microglia upon neuronal activation (Ccl3, left, n=3 mice, P=0.059, unpaired two-tailed t-test) and neuronal inhibition (Cd74, right, n=2 mice). k, Dot plots show lack of expression changes in selected genes in the striatum of wild type mice 2 hrs after saline, 0.25mg/kg CNO injection, or 1mg/kg CNO injection (n=3, 3, and 3 mice; Kdm6b: P=0.70 Adrb1: P=0.22, Ccl24: P=0.54, Ccl3: P=0.43, Kcnk13: P=0.37, Ikbkb, P=0.62, One-way ANOVA with Tukey’s post hoc test). RPKM: reads per kilobase of transcript per million mapped reads, TRAP: translating ribosome affinity purification; Data shown as mean± s.e.m.
Extended Data Figure 2. Microglia deficient mice show normal baseline behaviors but exaggerated responses to neurostimulants.
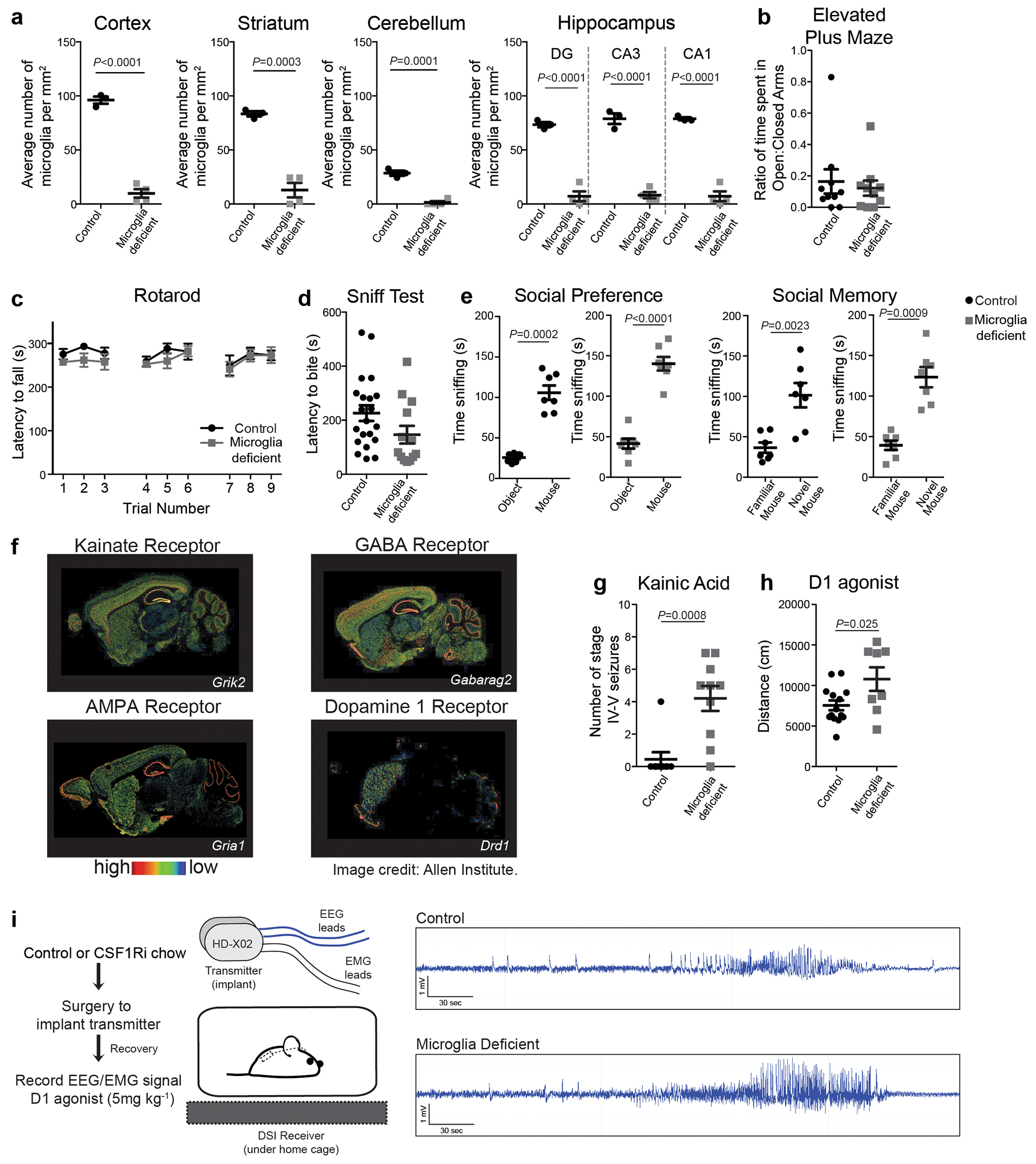
a, Dot plots show the average number of microglia per mm2 in cortex, striatum, cerebellum and hippocampus in control and microglia deficient mice (n=3 and 4 mice, cortex: P<0.0001, striatum: P=0.0003, cerebellum: P=0.0001, DG: P<0.0001, CA3: P<0.0001, CA1: P<0.0001, unpaired two-tailed t-test). b-e, Behavioral characteristics of microglia deficient mice. b, Anxiety-like behavior was measured by the ratio of time spent in the open arms/closed arms in the elevated plus maze (n=10 mice, P=0.65, unpaired two-tailed t-test). c, Motor coordination was measured by latency to fall from the accelerating rotarod (n=8 and12 mice, interaction: P=0.89, time: P=0.13, treatment: P=0.36; subjects: P<0.0001, Two-way repeated measures ANOVA). d, Olfactory behavior was measured by the sniff test (n=21 and 13 mice, P=0.09, unpaired two-tailed t-test). e, Social behavior was measured by using the classic three-chamber sociability task (Social preference: mouse preference for sniffing another mouse over object, Control: n=7 mice; P=0.0002, microglia deficient: n=9 mice, P<0.0001; Social Memory: mouse preference for sniffing novel mouse over familiar mouse, Control n=7 mice, P=0.0023, microglia deficient: n=7 mice, P=0.0009; paired two-tailed t-test). f, Representative images show brain-wide gene expression patterns of receptors targeted by kainic acid (kainate and AMPA receptor), picrotoxin (GABAA receptor), and SKF81297 (D1 receptor) (Allen Institute). g, Number of stage IV-V seizures (Racine scale90) per mouse visually recorded within one hour in response to kainic acid (18mg/kg, i.p.) are shown as a dot plot (n=9 and 10 mice, P=0.0008, unpaired two-tailed t-test). h, Dot plot showing distance traveled in response to D1 agonist in one hour in the open field (SKF81297, 3mg/kg, i.p.)(n=14 and 8 mice, P=0.025, unpaired two-tailed t-test). i, Representative cortical EEG traces during a tonic-clonic seizure event in response to D1 agonist treatment (SKF81297, 5mg/kg i.p.) in control (top) and microglia deficient (bottom) mice showing high amplitude and rhythmic discharges followed by EEG depression. DG: dentate gyrus; Data shown as mean± s.e.m.
Extended Data Figure 3. Generation and characterization of Il34-deficient and Csf1-deficient mice.
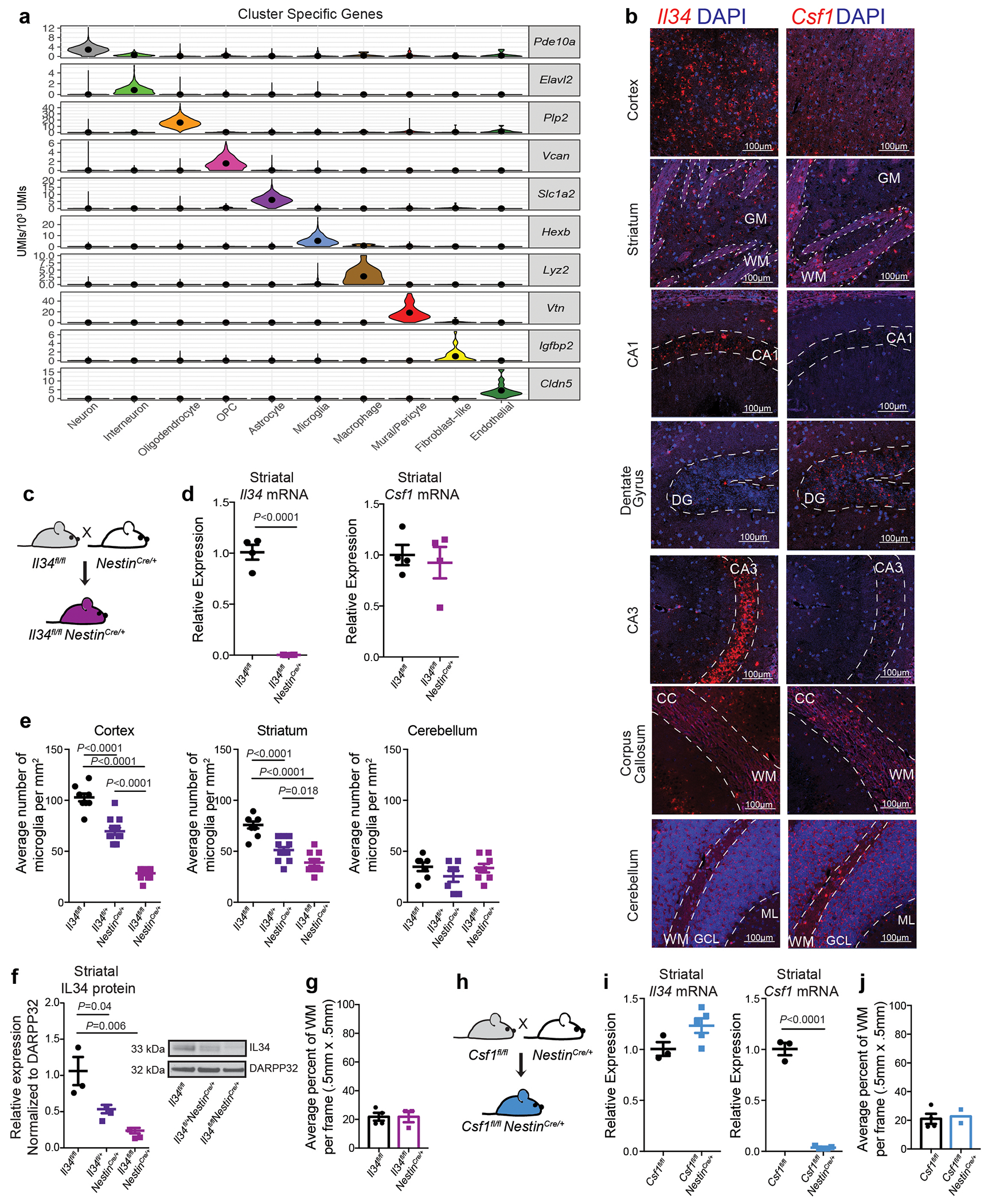
a, Violin plots show the expression levels of cell-type specific representative marker genes across the 10 identified cell types from striatum single nuclei RNA-seq (snRNA-seq) data analysis. Black dots indicate mean expression of selected gene per cell type. b, In situ hybridization for Il34 (left) and Csf1 (right) mRNAs show differential, region-specific expression in cortex, striatum, CA1, dentate gyrus (DG), CA3, corpus callosum (CC), and cerebellum of wild-type mice (WM: white matter, GM: grey matter, ML: molecular layer, GCL: granule cell layer, scale bar=100μm). c, h, The striatal grey matter-specific or white matter-specific microglia depletion was achieved by breeding NestinCre/+ mice to Il34fl/fl mice or Csf1fl/fl mice, respectively, to generate Il34fl/fl;NestinCre/+ (purple, c) and Csf1fl/fl;NestinCre/+ mice (blue, h). d, i, Dot plots showing relative expression levels of Il34 and Csf1 mRNA normalized to Gapdh in the striatum of Il34fl/fl;NestinCre/+ mice (d) or Csf1fl/fl;NestinCre/+ mice (i) compared with littermate controls (d, n=4 mice each, Il34 P<0.0001, Csf1 P =0.69; i, n=3 and 5 mice, Il34 P=0.07, Csf1 p<0.0001, unpaired two-tailed t-test). e, Dot plots show the average microglia density per mm2 per mouse in cortex, striatum, cerebellum (cortex: n=9, 12, and 10 mice, P<0.0001, striatum: n=9, 13, and 10 mice, P<0.0001, cerebellum: n=7, 7, and 8 mice, P=0.34, One-way ANOVA with Tukey’s post hoc test). f, left, Dot plot shows levels of IL34 protein as determined by Western blot analysis of striatal protein lysate from Il34fl/fl, Il34fl/+;NestinCre/+ or Il34fl/fl;NestinCre/+ mice normalized to DARPP32 expression (n=3 mice, P=0.0077, One-way ANOVA with Tukey’s post hoc test). g, j, Bar graphs show the average percentage of white matter regions in striatal images (0.5mm x 0.5mm) used to count WM and GM microglia in control and mutant mice for the data shown in Fig. 2c and e. (g, P = 0.99, n = 4 and 3 mice, unpaired two-tailed t-test; j, n = 4 and 2 a mice). For gel source data, see Supplementary Figure 1. Data shown as mean± s.e.m.
Extended Data Figure 4. Generation of mice with striatum-specific microglia depletion.
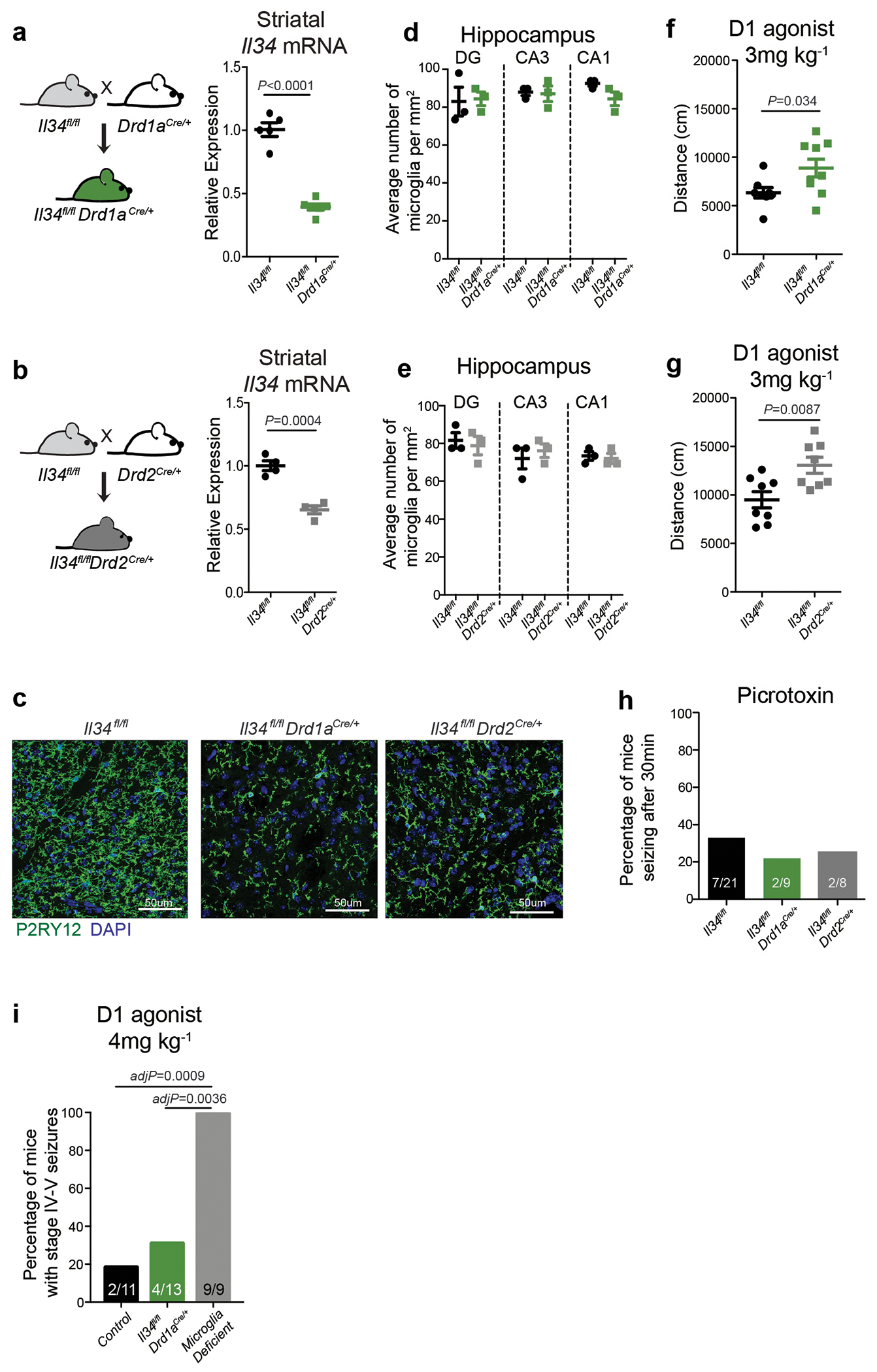
a, b, (left), The striatum-specific microglia depletion was achieved by breeding Il34fl/fl mice to Drd1aCre/+ or Drd2Cre/+ mice to generate Il34fl/fl;Drd1aCre/+ (a, green) and Il34fl/fl;Drd2Cre/+ mice (b, grey). Right, dot plots show relative expression of Il34 mRNA in the striatum normalized to Gapdh (a, n=6 and 7 mice, P<0.0001; b, n=4 mice, P=0.0004, unpaired two-tailed t-test). c, Representative striatal images of sagittal brain slices from Il34fl/fl, Il34fl/fl;Drd1aCre/+ and Il34fl/fl;Drd2Cre/+ mice following immunofluorescent staining for P2RY12 (microglia, green) and DAPI (nuclei, blue) (scale bar = 50μm). d, e, Dot plots show the average microglia density per mm2 per mouse per specific region in the hippocampus of Il34fl/fl;Drd1aCre/+ (d) and Il34fl/fl;Drd2Cre/+ mice (e) compared to littermate controls (d, n=3 mice, DG: P=0.88, CA3: P=0.85, CA1: P=0.1; e, n=3 mice, DG: P=0.69, CA3: P=0.56, CA1: P=0.72; unpaired two-tailed t-test). f, g, Dot plots showing total distance traveled in response to D1 agonist (SKF81297, 3mg/kg, i.p.) in one hour in the open field for Il34fl/fl;Drd1aCre/+ (f) and Il34fl/fl;Drd2Cre/+ mice (g) compared with littermate controls (f: n=8 and 9 mice, P=0.034 g: n=8 mice, P=0.0087, unpaired two-tailed t-test). h, Percentage of mice seizing 30 minutes after administration of picrotoxin (i.p., 1mg/kg) shown as a bar graph (n=21, 9, and 8 mice; P=0.80, Chi-squared test). DG: dentate gyrus. i, Microglia-neuron ratio defines the threshold of D1 neuron activation by D1 agonist. Bar graph shows the percentage of mice with stage IV-V seizures in response to D1 agonist (4mg/kg, i.p.) in control, Il34fl/fl;Drd1aCre/+, and microglia deficient mice (n=11, 13, and 9 mice; right, P=0.0005, Chi-squared test). While all mice display an increased seizure response to 5mg/kg D1 agonist treatment, only microglia deficient (99% reduction of microglia), but not Il34fl/fl;Drd1aCre/+ (60% reduction of microglia in the striatum) display an increased seizure response at 4mg/kg D1 agonist treatment. Data shown as mean± s.e.m.
Extended Data Figure 5. Striatum-specific microglia reduction has no overall effects on striatal cellular composition, D1/D2 neuronal morphology, D1/D2 MSN characteristic electrophysiological and molecular phenotypes, and glial phenotypes.
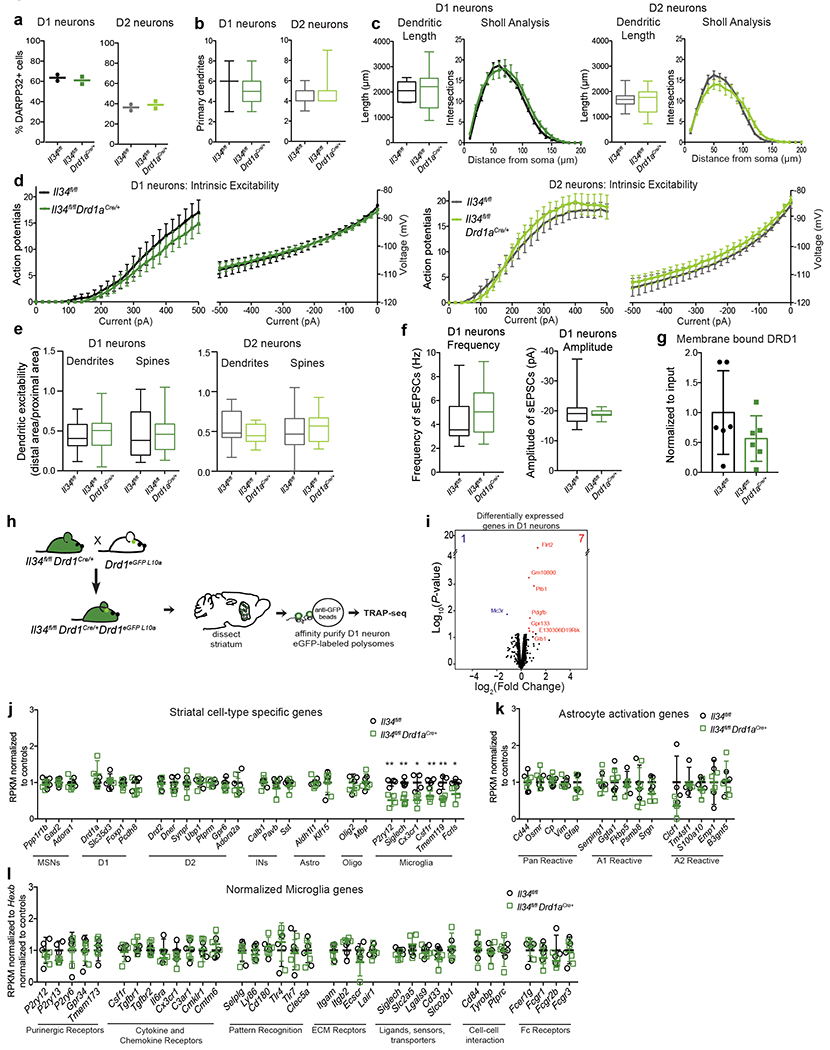
a, Dot plots show average number of D1 neurons (left, dark green, GFP+, DARPP32+) and D2 neurons (right, light green, GFP-, DARPP32+) per mouse in the striatum of Il34fl/fl;Drd1aeGFPL10a and Il34fl/fl;Drd1aeGFPL10a;Drd1aCre/+ mice. Mice expressing eGFP-tagged ribosomal subunit L10a under the Drd1a promoter were used to identify GFP+ D1 neurons and GFP− D2 neurons in control Il34fl/fl;Drd1aeGFPL10a and mutant Il34fl/fl;Drd1aeGFPL10a;Drd1aCre/+ (n=2 mice). b, c, D1 or D2 neuron cell morphology was determined by the number of primary dendrites (b), total dendritic length (c, left), and sholl analysis (c, right) (b, D1 neurons: n=11 and 15 D1 neurons, P=0.33; D2 neurons: n=15 and 11 D2 neurons, P=0.59; unpaired two-tailed t-test; c, D1 neurons, n=11 and 15 D1 neurons, dendritic length: P=0.83, unpaired two-tailed t-test; sholl, interaction: P=0.99; genotype: P=0.069; distance: P<0.0001, two-way ANOVA; D2 neurons, n=15 and 10 D2 neurons, dendritic length: P=0.80, unpaired two-tailed t-test; sholl: interaction: P=0.051; genotype: P=0.67; distance: P<0.0001, two-way ANOVA). d, Intrinsic excitability of D1 neurons (left) and D2 neurons (right) in ex vivo slices as measured by current-evoked action potentials (AP, left) and equilibrium potentials as voltage-current (VC) plots (right) (D1: n=11 and 15 D1 neurons, AP: interaction: P=1.0; genotype: P=0.98; pA: P<0.0001, subjects: P<0.0001; VC: interaction: P=1.0; genotype: P=0.48; distance: P<0.0001, subjects: P<0.0001; D2: n=16 and 10 D2 neurons; AP: interaction: P=1.0; genotype: P=0.5; distance: P<0.0001, subjects: P<0.0001; VC: interaction: P=0.99; genotype: P=0.7; distance: P<0.0001, subjects: P<0.0001; two-way ANOVA). e, Dendritic excitability of D1 neurons (left) and D2 neurons (right) in ex vivo slices as determined by back-propagating action potentials as measured by Ca2+-sensitive fluorescence (D1: n=12 and 15 D1 neurons, dendrites: P=0.90, spines: P=0.85; D2: n=16 and 10 D2 neurons, dendrites, P=0.27, spines, P=0.61; two-way ANOVA). f, Frequency (Hz) and amplitude (pA) of sEPSPs in D1 neurons from ex vivo slices shown as box and whisker plots (Frequency: n=19 cells from 5 mice and 16 cells from 5 mice, P=0.23, unpaired two-tailed t-test; amplitude: n=19 cells from 5 mice and 16 cells from 5 mice, P=0.796, unpaired two-tailed t-test with Welch’s correction). g, Membrane bound DRD1 protein expression normalized to total DRD1 expression as determined by ex vivo brain slice biotinylation assay shown as a dot plot (n=6 mice, P=0.21). h, Generation of Il34fl/fl;Drd1aCre/+;Drd1eGFP-L10a for D1 neuron specific TRAP sequencing analysis. i, Volcano plot shows lack of any major gene expression changes in D1 neurons in 3 month old Il34fl/fl;Drd1aCre/+;Drd1eGFP-L10a mice and littermate controls as determined by differential expression analysis (DESeq2, n=3 mice each, P<0.05, fold change>1.5, red: upregulated, blue: downregulated). j-k, Total striatal RNA expression analysis from control and Il34fl/fl;Drd1aCre/+ mice reveals unperturbed striatum cell-type specific gene expression pattern except the expected ~50% reduction in the expression of microglia-enriched genes. j, RPKM, normalized to controls, showing pan-medium spiny neuron (MSN), D1 neuron (D1), D2 neuron (D2), interneuron (IN), astrocyte (astro), oligodendrocyte (oligo), and microglia specific genes in Il34fl/fl;Drd1aeGFPL10a and Il34fl/fl;Drd1aCre/+;Drd1aeGFPL10a mice (n=4 mice each, P2ry12: P=0.003, Siglech: P=0.001, Cx3cr1: P=0.01, Csf1r: P=0.007, Tmem119: P=0.005, Fcrls: P=0.03, unpaired two-tailed t-test). k, RPKM, normalized to controls, showing unperturbed expression of astrocyte-specific activation markers66 (n=4 mice each, unpaired two-tailed t-test). l, Microglia show wild-type like expression of selected microglia sensome genes92, RPKMs of selected genes have been normalized to Hexb RPKM, (n=4 mice each, unpaired two-tailed t-test). The experiments shown in h-k have been independently repeated in a second cohort (n=3 mice) with identical results. For gel source data, see Supplementary Figure 1. Box and whisker plots in b, c, e, and f are shown with arithmetic median (middle line), box shows upper and lower quartile, whiskers show min-max range. Data shown as mean± s.e.m.
Extended Data Figure 6. Microglia regulate striatal neuron synchrony and responses to D1 agonist treatment in an ADO/A1R dependent fashion.
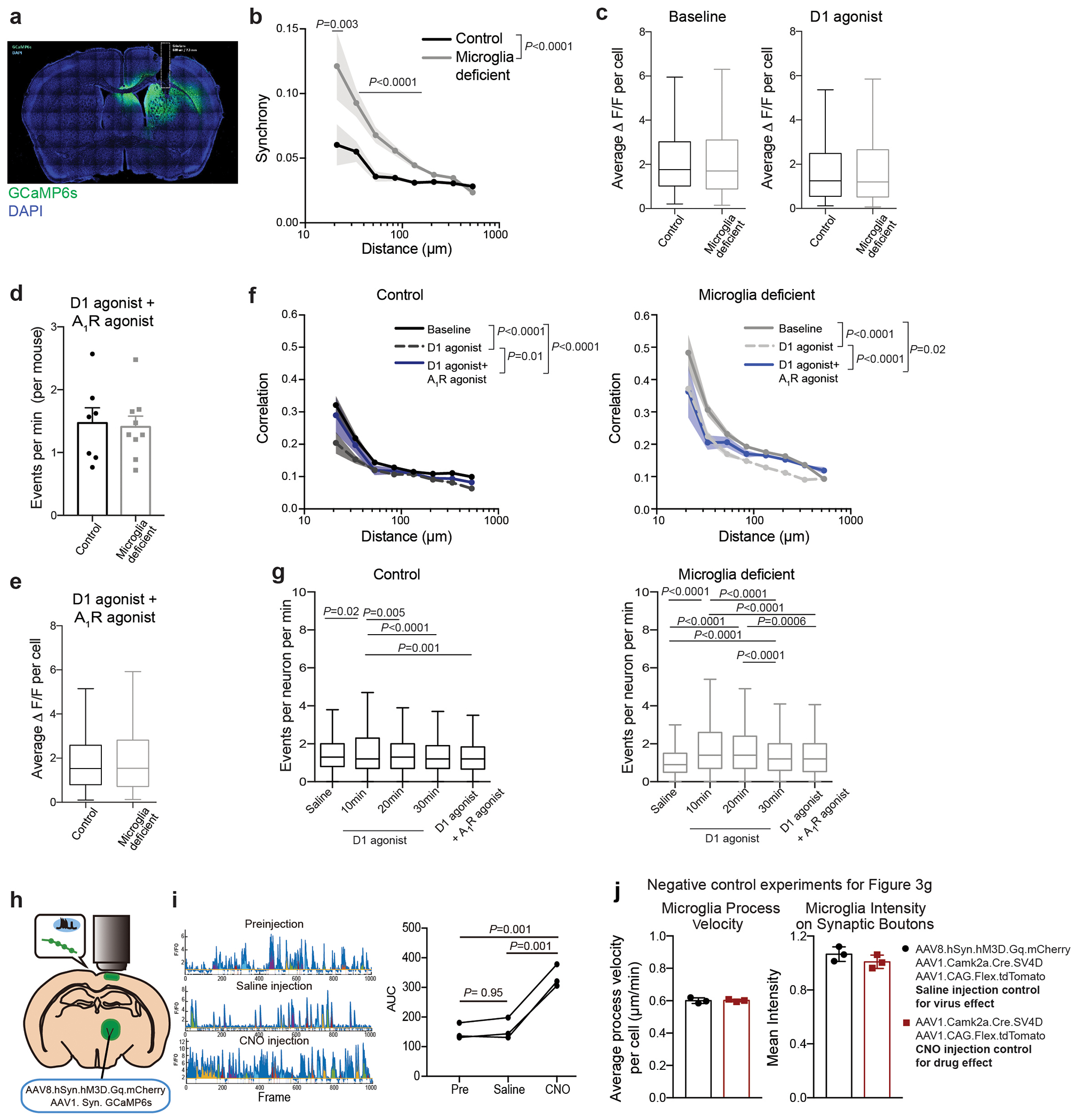
a, Representative tile scan of coronal brain slice showing implantation of GRIN lens and AAV9.hSyn.GCaMP6s expression in the dorsal striatum. b, Increased synchrony in the dorsal medial striatum of microglia deficient mice (n=9 mice) at baseline compared with controls (n=7 mice) (treatment: P<0.0001, distance: P<0.0001, interaction: P<0.0001; Two-way ANOVA with Sidak’s multiple comparisons test). c, Bar graphs show magnitude of Ca2+ events (ΔF/F) recorded in control (black) and microglia deficient mice (grey) at baseline (left) and in response to D1 agonist (SKF81297, 3mg/kg, right) (baseline: control, n=824 cells from 7 mice; microglia deficient, n=775 cells from 9 mice, P=0.87; D1 agonist: control, n=995 cells from 7 mice; microglia deficient, n=1021 cells from 9 mice; P=0.89, unpaired two-tailed t-test). d, e, Co-administration of A1R agonist (CPA, 0.1mg/kg) with D1 agonist (SKF81297, 3mg/kg) normalizes increased neuronal activity in microglia deficient mice. Bar graphs show wild type-like frequency (per mouse, d) and magnitude (ΔF/F, e) of Ca2+ events per neuron per minute in control (black) and microglia deficient (grey) (d, control, n= 7 mice; microglia deficient, n= 9 mice, P=0.82, unpaired two-tailed t-test; e, control, n=387 cells from 7 mice; microglia deficient, n=305 cells from 9 mice; P=0.69, unpaired two-tailed t-test). f, Spatiotemporal coding of neuronal activity (baseline shown in Fig. 3c) is disrupted by D1 agonist administration (dotted line) and largely normalized by co-administration with an A1R agonist (blue line) in control (top, n=7 mice) and microglia deficient mice (left, n=9 mice). For better visualization, the distance axis was logarithmically scaled. (Control, n=7 mice: interaction: P=0.0012, distance: P<0.0001, treatment: P<0.0001; Microglia deficient, n=9 mice: interaction: P=0.0014, distance: P<0.0001, treatment: P<0.0001; Two-way ANOVA with Sidak’s multiple comparisons test). g, Bar graphs show the frequency of Ca2+ events per neuron per minute in control (left) and microglia deficient (right) mice at baseline, in response to D1 agonist (SKF81297, 3mg/kg, i.p.) alone, or in response to D1 agonist and A1R agonist treatment (CPA, 0.1mg/kg, i.p.) treatment (Control: n=332-995 cells from 7 mice, P<0.0001; Microglia deficient: n=243-1021 cells from 9 mice, P<0.0001; One-way ANOVA with Bonferroni post hoc test). h, Confirmation of CNO-mediated neuronal activation for data shown in Fig. 3h. The neuron-specific expression of GCaMP6s and hM3Dq was achieved by injecting the indicated viruses. Virally labeled thalamocortical projection neurons were identified (mCherry expression) and calcium transients were recorded at baseline, after saline injection, and after CNO injection. i, Representative traces (left) and quantification of the area under the curve (AUC) (right) of calcium transients per mouse in virally labeled neurons pre-injection, after saline injection, and after CNO injection (n=3 mice, P=0.0009, One-way ANOVA with Tukey’s post hoc test). j, Microglia baseline process velocity (left) and contact with synaptic boutons (right) is not affected by either the expression of the DREADD virus (red bars) or by CNO injection (5mg/kg, black bars) alone (n=3 mice, left: P=0.96, right, P=0.25, unpaired two-tailed t-test). The experiments shown in a-g are data combined from two independent imaging cohorts of mice. Box and whisker plots in c, e, and g are shown with arithmetic median (middle line), box shows upper and lower quartile, whiskers show 1.5x interquartile range. CNO: clozapine-N-oxide; Data shown as mean± s.e.m.
Extended Data Figure 7. Microglial expression of Entpd1/CD39 and Nt5e/CD73 in vitro and in vivo.
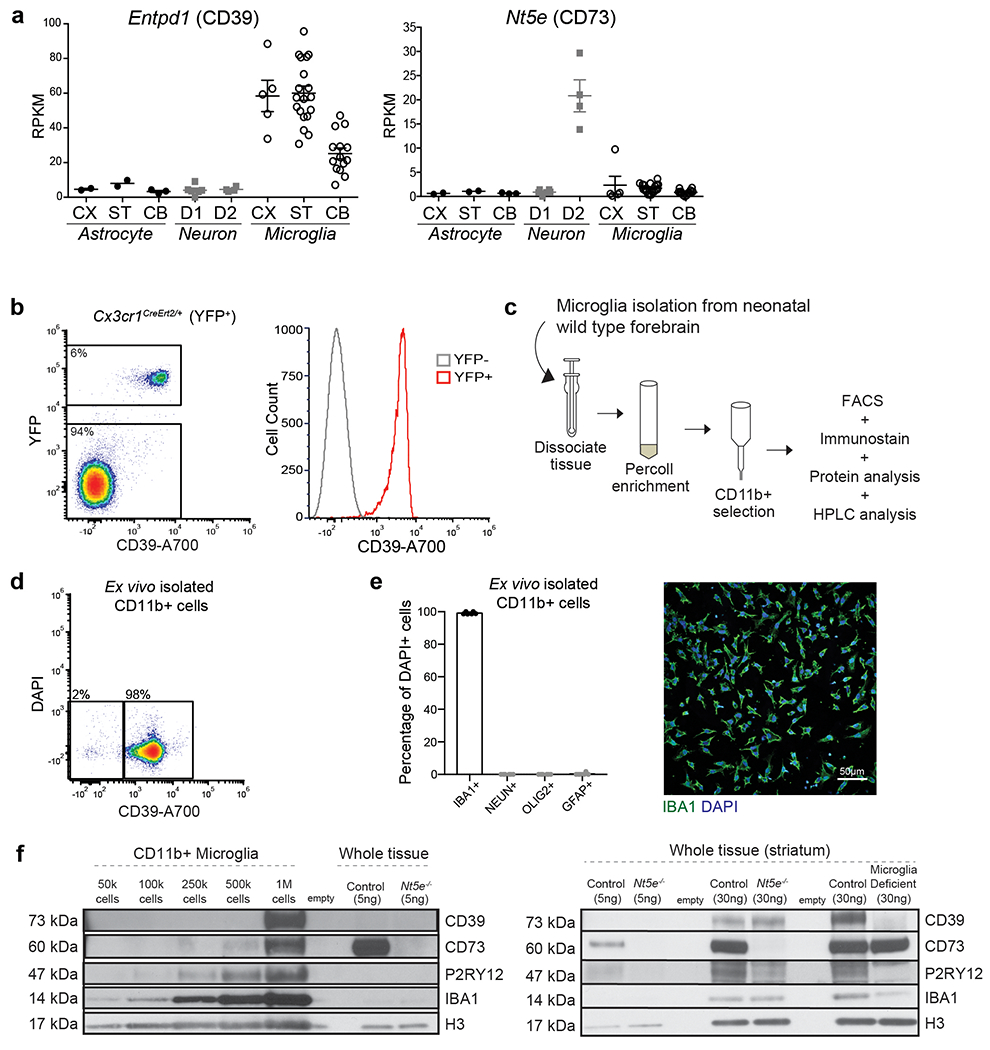
a, Dot plots show normalized, ribosome-associated mRNA levels (RPKM) for Entpd1/CD39 (left) and Nt5e/CD73 (right) in astrocytes, neurons, and microglia from distinct brain regions of adult mice using cell-type specific TRAP sequencing (n=2, 2, 3, 6, 4, 5, 19, and 15 mice). b, CD39 surface expression on ex vivo isolated forebrain cells of Cx3cr1CreErt2/+(Litt) mice (mice express cytosolic YFP in Cx3cr1+ microglia). Percoll-purified cells were incubated with anti-CD39-AlexaFluor700 followed by FACS analysis. The histogram shows expression levels of CD39, which is almost exclusively restricted to YFP+ microglia (red) and is not found on YFP- non-microglia cells (gray) as shown previously72 (data is representative of three independent experiments). c, Scheme shows ex vivo isolation procedure of CD11b+ microglia following neonatal mouse forebrain tissue dissociation and Percoll enrichment for live cells. d, e, Ex vivo CD11b+ microglia isolation procedure from neonatal pups yields highly pure microglia population. d, Microglia were positively selected for by using CD11b+ magnetic bead purification and were incubated with anti-CD39-AlexaFluor700 followed by FACS analysis to assess the purity of the population. The numbers show the percentage of live (DAPI-) cells with distinct pattern of CD39 expression levels (>98% CD39+; data is representative of two independent experiments). e, Immunofluorescent analysis of purity of CD11b+ microglia isolation. Left, cells were plated on cover slips and stained for cell-type specific protein expression using antibodies specific for IBA1 (microglia), GFAP (astrocyte), OLIG2 (oligodendrocytes) or NEUN (neurons) to identify and quantify different cells within the populations in order to assess microglia purity (n=6 GFAP/IBA1 images and 6 OLIG2/NEUN/IBA1 images). Right, representative image of cover slip containing 99% pure microglia following CD11b+ isolation procedure is shown (IBA1, green; DAPI, blue). f, left, Cell lysates of increasing numbers of CD11b+ bead-purified microglia cells have been analyzed for CD39, CD73, P2RY12, and IBA1 protein expression by Western Blot analysis as indicated, 5ng of total striatal lysate from control or Nt5e−/− (CD73-deficient) mice have been used to verify CD73 antibody specificity, H3 protein expression has been used as a loading control (k= thousand, M=million; SuperSignal ECL substrate was used to visualize CD73 expression, regular ECL was used for all other proteins) Right, Whole striatal tissue lysates of control and Nt5e−/− (CD73-deficient) striatal tissue were loaded at low (5ng) and high (30ng) concentrations and analyzed for microglia-specific protein expression (CD39, CD73, P2RY12, and IBA1) by Western Blot analysis as indicated. Whole striatal tissue lysates of control and microglia deficient mice have been used to verify antibody specificity. H3 protein expression has been used as a loading control. (SuperSignal ECL substrate was used to visualize P2RY12 expression, regular ECL was used for all other proteins). Blots are representative from two independent experiments. For gel source data, see Supplementary Figure 1. Data shown as mean± s.e.m.
Extended Data Figure 8. Microglia suppress neuronal activation via an ATP/AMP/ADO/A1R- dependent feedback mechanism.
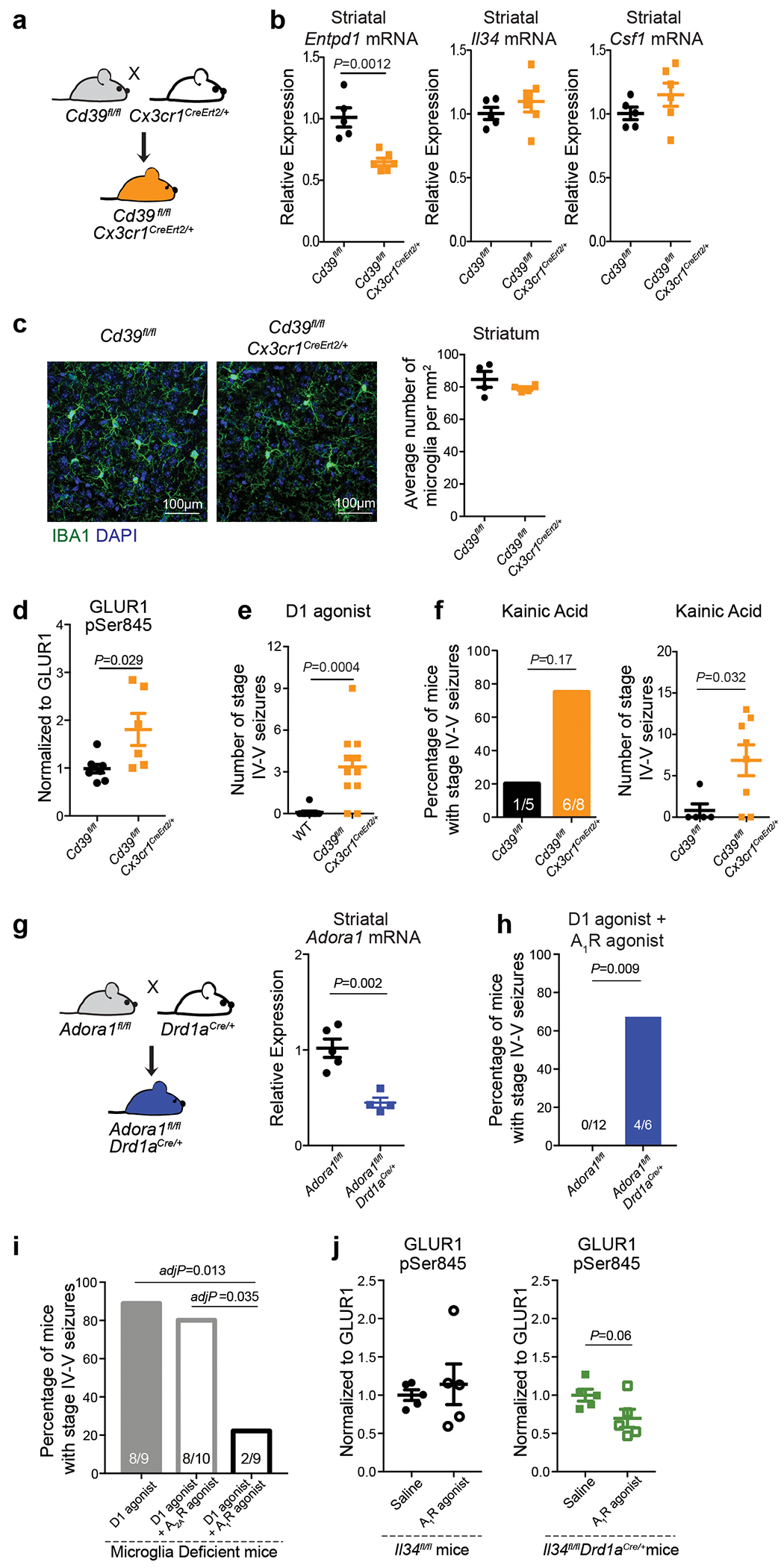
a, Scheme for generation of mice with microglia-specific CD39 depletion by breeding Cd39fl/fl mice to Cd39fl/fl;Cx3cr1CreErt2/+(Jung) mice followed by tamoxifen-mediated Cre induction at 4-6 weeks of age. b, Dot plots show relative expression of Entpd1, Il34, and Csf1 mRNA in the striatum of Cd39fl/fl;Cx3cr1CreErt2/+ mice and littermate controls normalized to Gapdh (n=5 and 6 mice, Entpd1: P=0.0012, Il34: P=0.38, Csf1: P=0.22, unpaired two-tailed t-test). c, left, Representative images of striatal sections from Cd39fl/fl and Cd39fl/fl;Cx3cr1CreErt2/+ mice stained for IBA1 (microglia, green) and DAPI (nuclei, blue) (scale bar:100μm); right, dot plots show the average number of microglia per mm2 per mouse in the striatum of Cd39fl/fl and Cd39fl/fl;Cx3cr1CreErt2/+ mice (n=4 mice, P=0.33, unpaired two-tailed t-test with Welch’s correction for variance). d, Microglia-specific CD39 ablation leads to increased levels of neuronal PKA activity in the striatum as measured by phosphorylation levels of GLUR1 at Ser845 in striatal protein lysate from Cd39fl/fl;Cx3cr1CreErt2/+ and littermate controls, pGLUR1 levels have been normalized to total GLUR1 in each sample, (n=8 and 6 mice, P=0.029, two-tailed Mann-Whitney Test). e, f, Increased seizure response in Cd39fl/fl;Cx3cr1CreErt2/+: e, Dot plot shows number of stage IV-V seizures recorded within one hour in response to D1 agonist (SKF81297, 5mg/kg) (n=11 mice each, P=0.0004; unpaired two-tailed t-test). f, Bar graph showing percentage of mice (left) and dot plot showing number (right) of stage IV-V seizures in response to kainic acid (15mg/kg) in Cd39fl/fl;Cx3cr1CreErt2/+ mice as compared to littermate controls (n=5 and 8 mice; left, P=0.17, Fisher’s exact test with Yates correction, right, P=0.032, unpaired two-tailed t-test). g, left, Scheme for the generation of mice with a D1 neuron-specific Adora1 depletion by breeding Adora1fl/fl mice to Drd1aCre/+ mice; right, dot plots show relative expression of Adora1 mRNA in the striatum of Adora1fl/fl;Drd1aCre/+ mice and littermate controls normalized to Gapdh (n=5 and 4 mice, P=0.002, unpaired two-tailed t-test). h, Co-administration of A1R agonist (CPA, 0.1mg/kg) and D1 agonist (SKF81297, 5mg/kg) does not prevent the increased seizure susceptibility in Adora1fl/fl;Drd1aCre/+ mice (n=12 and 6 mice, P=0.009, Fisher’s exact test with Yates correction). i, Bar graph shows percentage of microglia deficient mice with seizures in response to D1 agonist alone (SKF81297, 5mg/kg, i.p.) or co-administered with an A2AR agonist (CGS21680, 0.1mg/kg, i.p.) or an A1R agonist (CPA, 0.1m/kg, i.p.) (n=9-10 mice, P=0.005, Chi-squared test with Bonferroni post hoc adjustment). j, A1R agonist administration (CPA, 0.1mg/kg) normalizes increased PKA activity in Il34fl/fl;Drd1aCre/+ mice but does not affect PKA activity in control Il34fl/fl mice as measure by phosphorylation levels of GLUR1 at Ser845 in striatal protein lysate, pGLUR1 levels have been normalized to total GLUR1 expression in each sample (Il34fl/fl mice, n=5 mice, P= 0.62, Il34fl/fl;Drd1aCre/+ mice, n=5 mice, P=0.06, unpaired two-tailed t-test). All statistical tests are two-tailed; Data shown as mean± s.e.m.
Extended Data Figure 9. Microglia can suppress glutamate-induced cortical neuron activation in a CD39/ADO/A1R-dependent fashion in vitro.
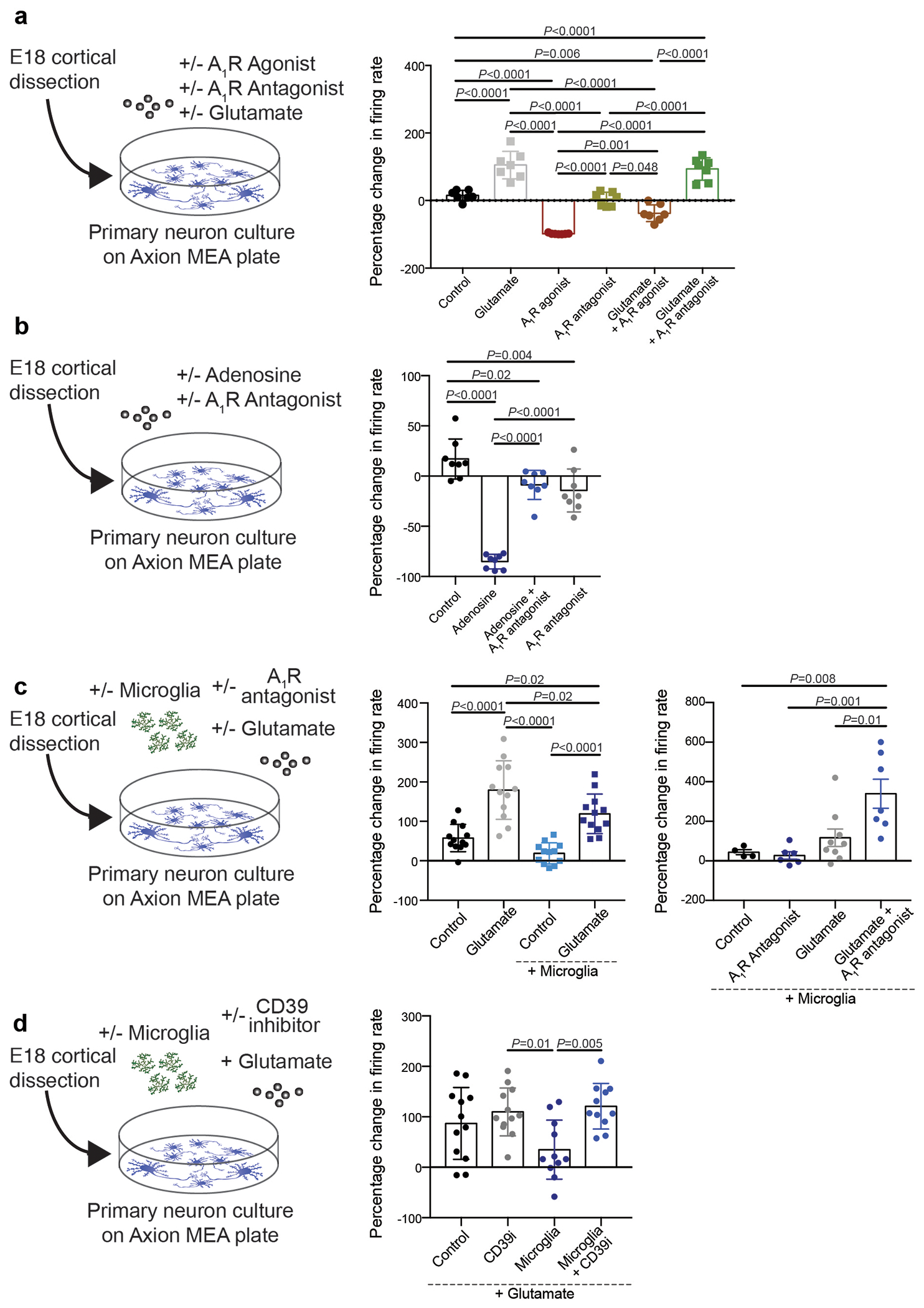
a-d, Experimental approaches for the assessment of adenosine-mediated regulation of cortical neuron activity in vitro. Embryonic cortical neurons were cultured on Axion microelectrode array (MEA) plates which allows for continuous electrical field recordings. a, A1Rs modulate cortical neuronal activity at baseline and in response to glutamate. On day in vitro (DIV) 14, neuronal cultures were treated with vehicle, glutamate (10μM), A1R agonist (CPA, 100nM), A1R antagonist (DCPCX, 100nM), glutamate and A1R agonist, or glutamate and A1R antagonist. Dot plot shows the percentage change in mean firing rate of neurons 1 hour after treatment compared to their baseline prior to drug treatment. (n=7 wells, P<0.0001, One-way ANOVA with Tukey’s post hoc test). b, Adenosine suppresses neuronal activity via A1R activation. On DIV14, cultures were treated with vehicle, adenosine (10μM), A1R antagonist (DCPCX, 100nM), or co-treated with adenosine and A1R antagonist. Dot plot shows percentage change in mean firing rate of neurons 1 hour after treatment compared to their baseline prior to drug treatment. (n=8 wells, P<0.0001, One-way ANOVA with Tukey’s post hoc test). c, Microglia suppress neuronal activity in response to glutamate-induced activation in an A1R-dependent manner. Microglia were isolated from neonatal pups, plated onto the neuronal culture on DIV 14, and allowed to settle for 48 hrs. Mixed cultures were treated with vehicle and/or glutamate (10μM) and/or A1R antagonist (100nM) on DIV 16. Dot plot shows percentage change in mean firing rate of neurons 1 hour after treatment compared to their baseline prior to drug treatment. (left, n=12 wells, P<0.0001, right, n=4, 6, 9, and 7 wells, P=0.001, One-way ANOVA with Tukey’s post hoc test). d, Microglia suppress neuronal activity in a CD39-dependent manner in response to glutamate-induced activation. Microglia were isolated from neonatal pups, plated onto the neuronal culture on DIV 14, and allowed to settle for 48 hrs. Mixed cultures were pretreated with CD39 inhibitor (ARL67156, 200μM) or vehicle (30 min) and then treated with glutamate (10μM). Dot plot shows percentage change in mean firing rate of neurons 1 hour after treatment compared to the corresponding baseline neuronal activity levels prior to their baseline prior to drug treatment. (n=12, 12, 11, and 11 wells, P=0.0045, One-way ANOVA with Tukey’s post hoc test). Data shown as mean ± s.e.m. and representative of 2-3 independent experiments.
Extended Data Figure 10. Reactive microglia in different neuroinflammatory and neurodegenerative conditions show a reduction in Entpd1 and P2ry12 expression that is associated with an A1R-dependent increase in D1 neuron responses.
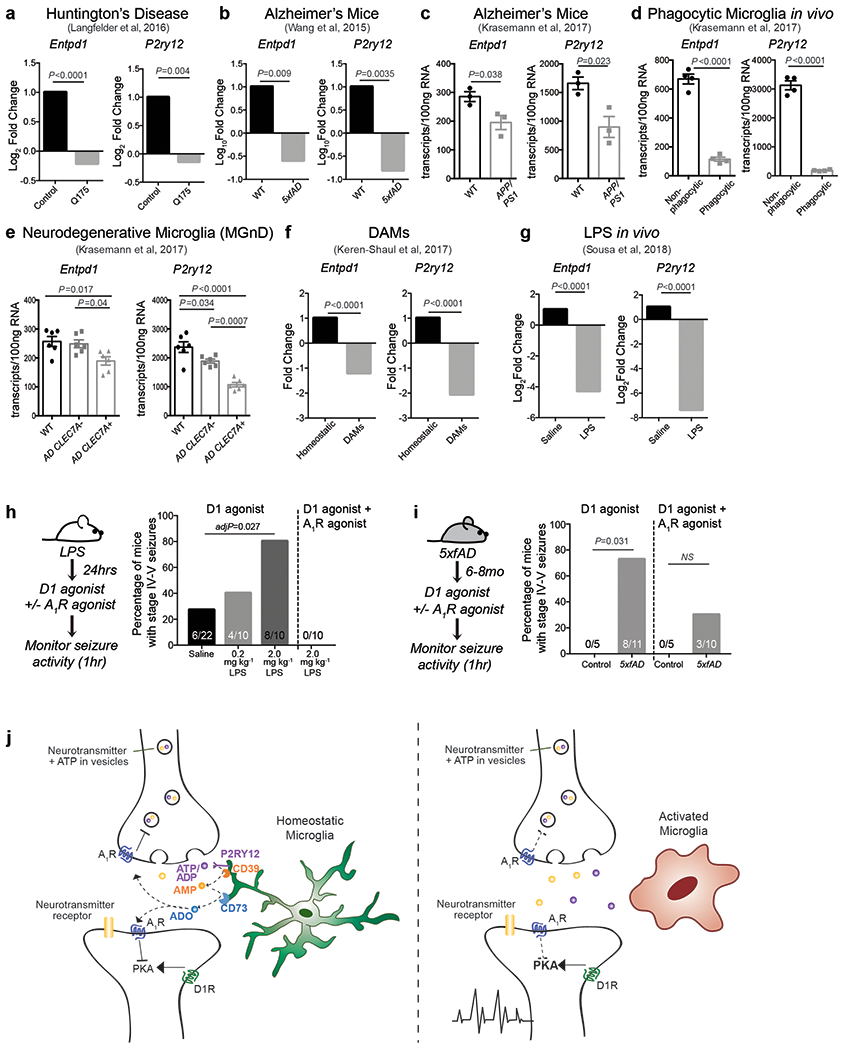
a-g, Changes in Entpd1 and P2ry12 gene expression are shown in: a, RNA extracted from whole striatum of 6-month old control mice and Q175 (Huntington’s disease) mice93 (Entpd1: P=0.0001; P2ry12: P=0.004; n=8 mice, fold change and P-value provided in publication). b, RNA from FACS-sorted CD11b+/F4/80+ cortical and hippocampal microglia from 8.5-month old control and 5xfAD mouse model of Alzheimer’s Disease94 (Entpd1: P=0.009; P2ry12: P=0.0035; n=5 mice, fold change and P-value provided in publication). c, RNA from FACS-sorted forebrain microglia from 10-month old control and APP/PS1 Alzheimer’s disease mouse model39 (n=3 mice, Entpd1: P=0.038; P2ry12: P=0.023, unpaired two-tailed t-test). d, RNA from FACS-sorted FCRLS+ phagocytic and non-phagocytic microglia isolated after stereotaxic injection of apoptotic neurons39 (n=4 mice, Entpd1: P<0.0001; P2ry12: P<0.0001, unpaired two-tailed t-test). e, FACS-sorted FCRLS+ microglia in 24-month old control mice or APP/PS1 Alzheimer’s disease mouse model. Plaque associated microglia were identified and sorted based on CLEC7A expression39 (n=6 mice, Entpd1: P=0.01; P2ry12: P<0.0001, One-way ANOVA with Tukey’s post hoc test). f, Massively parallel single-cell RNA-seq (MARS-seq) from isolated homeostatic microglia and disease associated microglia (DAM) in 5xfAD mice Alzheimer’s disease mouse model95 (Entpd1: P<0.0001; P2ry12: P<0.0001; n=8,016 cells from 3 WT and 3 AD mice, fold change and P-value provided in publication). g, FACS-sorted CD11b+CD45int single microglia in control and LPS-injected mice (4mg/kg)96 (Entpd1: P<0.0001; P2ry12: P<0.0001; n=477 microglia from saline injected mice and 770 microglia from LPS injected mice, fold change and P-value provided in publication). h, i, Bar graphs show increased seizure susceptibility to D1 agonist administration (SKF81297, 5mg/kg, i.p.) in LPS-injected (indicated doses, i.p.) (h) and 6-8-month old 5xfAD Alzheimer’s mice (i) that is prevented by co-administration of an A1R agonist (CPA, 0.1mg/kg, i.p.) (h, n=10-22 male mice, P=0.032, Chi-squared test; i, n=5-10 mice per genotype, left, P=0.031, Fisher’s exact test with Yates correction; right, P=0.49, Fisher’s exact test with Yates correction). j, Scheme illustrating the model of microglia-mediated adenosine-controlled regulation of D1 neuron responses in the healthy striatum (left) and its potential dysfunction upon microglia activation during inflammatory and/or neurodegenerative diseases (right). All statistical tests are two-tailed; Data shown as mean± s.e.m.
Supplementary Material
Acknowledgements
We thank P. Greengard and A. Nairn for sharing the DARPP32 antibodies; J. J. Badimon for the Ticagrelor and Clopidogrel; R. Greene for the Adora1fl/fl mice; M. Merad and F. Desland for the Csf1fl/fl ;NestinCre mice, the MSSM FACS facility and J. Ochando, C. Bare, and G. Viavattene for assistance with flow cytometry analysis; A. Lopez and A. Watters for assistance with microdialysis experiments; G. Milne and the Vanderbilt University Neurochemistry Core for LC-MS analysis; D. Wagenaar and CalTech Neurotechnology Laboratory for help with construction of the two-photon system; and all Schaefer laboratory members and A. Tarakhovsky for discussions and critical comments on the manuscript. This work was supported by the National Institutes of Health (NIH) Director New Innovator Award DP2 MH100012-01 (A.S.), NIH grants R01NS091574 (A.S.), R01MH118329 (A.S.), DA047233 (A.S.), R01NS106721 (A.S.) and U01AG058635 (A.S.), a Robin Chemers Neustein Award (P.A.), NIH grant RO1AG045040 (J.X.J.), Welch Foundation Grant AQ-1507 (J.X.J.), NARSAD Young Investigator Award no. 25065 (P.A.), NIH grants T32AG049688 (A.B.), T32AI078892 (A.T.C.), 1K99NS114111 (M.A.W.), T32CA207201 (M.A.W.), R01NS102807 (F.J.Q.), R01AI126880 (F.J.Q.), and R01ES025530 (F.J.Q.), a TCCI Chen Graduate Fellowship (X.C.), an A*STAR National Science Scholarship (A.N.), the CZI Neurodegeneration Challenge Network (V.G.), NIH BRAIN grant RF1MH117069 (V.G.), NIH grants HL107152 (S.C.R.), HL094400 (S.C.R.), AI066331 (S.C.R.), GM-136429 (W.G.J.), GM-51477 (W.G.J.), GM-116162 (W.G.J.), HD-098363 (W.G.J.), DA042111 (E.S.C.), DA048931 (E.S.C.), funds from a VUMC Faculty Research Scholar Award (M.G.K.), the Brain and Behavior Research Foundation (M.G.K. and E.S.C), the Whitehall Foundation (E.S.C.), and the Edward Mallinckrodt Jr. Foundation (E.S.C.). The Vanderbilt University Neurochemistry Core is supported by the Vanderbilt Brain Institute and the Vanderbilt Kennedy Center (EKS NICHD of NIH Award U54HD083211).
Footnotes
Competing Interests
The authors declare no competing interests.
Main References
- 1.Werneburg S, Feinberg PA, Johnson KM & Schafer DP A microglia-cytokine axis to modulate synaptic connectivity and function. Current Opinion in Neurobiology (2017). doi: 10.1016/j.conb.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Du XF, Liu CS, Wen ZL & Du JL Reciprocal Regulation between Resting Microglial Dynamics and Neuronal Activity In Vivo. Dev. Cell (2012). doi: 10.1016/j.devcel.2012.10.027 [DOI] [PubMed] [Google Scholar]
- 3.Eyo UB et al. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci. (2014). doi: 10.1523/JNEUROSCI.0416-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akiyoshi R et al. Microglia enhance synapse activity to promote local network synchronization. eNeuro 5, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kato G et al. Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity. eNeuro (2016). doi: 10.1523/ENEURO.0004-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wake H, Moorhouse AJ, Jinno S, Kohsaka S & Nabekura J Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci (2009). doi: 10.1523/JNEUROSCI.4363-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng J et al. Microglial P2Y12 receptor regulates ventral hippocampal CA1 neuronal excitability and innate fear in mice. Mol. Brain (2019). doi: 10.1186/s13041-019-0492-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cserép C et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science (80-. ). (2020). doi: 10.1126/science.aax6752 [DOI] [PubMed] [Google Scholar]
- 9.Bernier LP et al. Nanoscale Surveillance of the Brain by Microglia via cAMP-Regulated Filopodia. Cell Rep. 27, 2895–2908.e4 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Madry C et al. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K+ Channel THIK-1. Neuron (2018). doi: 10.1016/j.neuron.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu YU et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci (2019). doi: 10.1038/s41593-019-0511-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stowell RD et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat. Neurosci (2019). doi: 10.1038/s41593-019-0514-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elmore MRP et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014). doi: 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bozzi Y & Borrelli E The role of dopamine signaling in epileptogenesis. Frontiers in Cellular Neuroscience (2013). doi: 10.3389/fncel.2013.00157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chitu V, Gokhan Ş, Nandi S, Mehler MF & Stanley ER Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends in Neurosciences (2016). doi: 10.1016/j.tins.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kana V et al. CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J. Exp. Med (2019). doi: 10.1084/jem.20182037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Easley-Neal C, Foreman O, Sharma N, Zarrin AA & Weimer RM CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia Development and Maintenance in White and Gray Matter Brain Regions. Front. Immunol 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saunders A et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell (2018). doi: 10.1016/j.cell.2018.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenzel M, Hamm JP, Peterka DS & Yuste R Acute focal seizures start as local synchronizations of neuronal ensembles. J. Neurosci 3176–18 (2019). doi: 10.1523/jneurosci.3176-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pankratov Y, Lalo U, Verkhratsky A & North RA Vesicular release of ATP at central synapses. Pflugers Archiv European Journal of Physiology (2006). doi: 10.1007/s00424-006-0061-x [DOI] [PubMed] [Google Scholar]
- 21.Pascual O et al. Neurobiology: Astrocytic purinergic signaling coordinates synaptic networks. Science (80-. ). (2005). doi: 10.1126/science.1116916 [DOI] [PubMed] [Google Scholar]
- 22.Corkrum M et al. Dopamine-Evoked Synaptic Regulation in the Nucleus Accumbens Requires Astrocyte Activity. Neuron (2020). doi: 10.1016/j.neuron.2019.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beamer E, Conte G & Engel T ATP release during seizures - A critical evaluation of the evidence. Brain Res. Bull 151, 65–73 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Haynes SE et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci (2006). doi: 10.1038/nn1805 [DOI] [PubMed] [Google Scholar]
- 25.Ayata P et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci (2018). doi: 10.1038/s41593-018-0192-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madry C et al. Effects of the ecto-ATPase apyrase on microglial ramification and surveillance reflect cell depolarization, not ATP depletion. Proc. Natl. Acad. Sci. U. S. A (2018). doi: 10.1073/pnas.1715354115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dissing-Olesen L et al. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci (2014). doi: 10.1523/JNEUROSCI.0405-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robson SC, Sévigny J & Zimmermann H The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal. (2006). doi: 10.1007/s11302-006-9003-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanser AJ et al. Disruption of the ATP/adenosine balance in CD39−/− mice is associated with handling-induced seizures. Immunology (2017). doi: 10.1111/imm.12798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunwiddie TV & Masino SA The Role and Regulation of Adenosine in the Central Nervous System. Annu. Rev. Neurosci (2001). doi: 10.1146/annurev.neuro.24.1.31 [DOI] [PubMed] [Google Scholar]
- 31.Zimmermann H, Zebisch M & Sträter N Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. (2012). doi: 10.1007/s11302-012-9309-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flagmeyer I, Haas HL & Stevens DR Adenosine A1 receptor-mediated depression of corticostriatal and thalamostriatal glutamatergic synaptic potentials in vitro. Brain Res. (1997). doi: 10.1016/S0006-8993(97)01060-3 [DOI] [PubMed] [Google Scholar]
- 33.Yabuuchi K et al. Role of adenosine A1 receptors in the modulation of dopamine D1 and adenosine A2A receptor signaling in the neostriatum. Neuroscience (2006). doi: 10.1016/j.neuroscience.2006.04.047 [DOI] [PubMed] [Google Scholar]
- 34.Trusel M et al. Coordinated Regulation of Synaptic Plasticity at Striatopallidal and Striatonigral Neurons Orchestrates Motor Control. Cell Rep. (2015). doi: 10.1016/j.celrep.2015.10.009 [DOI] [PubMed] [Google Scholar]
- 35.Zhou S et al. Pro-inflammatory effect of downregulated CD73 expression in EAE astrocytes. Front. Cell. Neurosci (2019). doi: 10.3389/fncel.2019.00233 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Bateup HS et al. Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat. Neurosci 11, 932–939 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wendeln AC et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature (2018). doi: 10.1038/s41586-018-0023-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Süß P et al. Chronic Peripheral Inflammation Causes a Region-Specific Myeloid Response in the Central Nervous System. Cell Rep. (2020). doi: 10.1016/j.celrep.2020.02.109 [DOI] [PubMed] [Google Scholar]
- 39.Krasemann S et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity (2017). doi: 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mildner A, Huang H, Radke J, Stenzel W & Priller J P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia (2017). doi: 10.1002/glia.23097 [DOI] [PubMed] [Google Scholar]
- 41.Palop JJ et al. Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer’s Disease. Neuron 55, 697–711 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam AD et al. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. (2017). doi: 10.1038/nm.4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wohleb ES, Franklin T, Iwata M & Duman RS Integrating neuroimmune systems in the neurobiology of depression. Nature Reviews Neuroscience (2016). doi: 10.1038/nrn.2016.69 [DOI] [PubMed] [Google Scholar]
Extended References
- 44.Spangenberg EE et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain (2016). doi: 10.1093/brain/aww016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bejar R, Yasuda R, Krugers H, Hood K & Mayford M Transgenic calmodulin-dependent protein kinase II activation: Dose-dependent effects on synaptic plasticity, learning, and memory. J. Neurosci (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alexander GM et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stanley S et al. Profiling of glucose-sensing neurons reveals that ghrh neurons are activated by hypoglycemia. Cell Metab. (2013). doi: 10.1016/j.cmet.2013.09.002 [DOI] [PubMed] [Google Scholar]
- 48.Parkhurst CN et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol (2012). doi: 10.1038/ni.2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tronche F et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet (1999). doi: 10.1038/12703 [DOI] [PubMed] [Google Scholar]
- 51.Harris SE et al. Meox2Cre-mediated disruption of CSF-1 leads to osteopetrosis and osteocyte defects. Bone (2012). doi: 10.1016/j.bone.2011.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rothweiler S et al. Selective deletion of ENTPD1/CD39 in macrophages exacerbates biliary fibrosis in a mouse model of sclerosing cholangitis. Purinergic Signal. (2019). doi: 10.1007/s11302-019-09664-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yona S et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity (2013). doi: 10.1016/j.immuni.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scammell TE et al. Focal deletion of the adenosine A1 receptor in adult mice using an adeno-associated viral vector. J. Neurosci 23, 5762–5770 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson LF et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med (2004). doi: 10.1084/jem.20040915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.André P et al. P2Y12 regulates platelet adhesion/activation, thrombus growth, and thrombus stability in injured arteries. J. Clin. Invest (2003). doi: 10.1172/JCI17864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oakley H et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci (2006). doi: 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casanova E et al. A CamKIIα iCre BAC allows brain-specific gene inactivation. Genesis (2001). doi: 10.1002/gene.1078 [DOI] [PubMed] [Google Scholar]
- 59.Doyle JP et al. Application of a Translational Profiling Approach for the Comparative Analysis of CNS Cell Types. Cell (2008). doi: 10.1016/j.cell.2008.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heiman M et al. A Translational Profiling Approach for the Molecular Characterization of CNS Cell Types. Cell (2008). doi: 10.1016/j.cell.2008.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Von Schimmelmann M et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci (2016). doi: 10.1038/nn.4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim D & Salzberg SL TopHat-Fusion: An algorithm for discovery of novel fusion transcripts. Genome Biol. (2011). doi: 10.1186/gb-2011-12-8-r72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anders S, Pyl PT & Huber W HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics (2015). doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Purushothaman I & Shen L SPEctRA: A Scalable Pipeline for RNA-seq Analysis. (2016). doi: 10.5281/ZENODO.60547 [DOI] [Google Scholar]
- 65.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014). doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017). doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Howe EA, Sinha R, Schlauch D & Quackenbush J RNA-Seq analysis in MeV. Bioinformatics (2011). doi: 10.1093/bioinformatics/btr490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen EY et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics (2013). doi: 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. (2016). doi: 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gokce O et al. Cellular Taxonomy of the Mouse Striatum as Revealed by Single-Cell RNA-Seq. Cell Rep. (2016). doi: 10.1016/j.celrep.2016.06.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bohlen CJ, Bennett FC & Bennett ML Isolation and Culture of Microglia. Curr. Protoc. Immunol (2019). doi: 10.1002/cpim.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Butovsky O et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci (2014). doi: 10.1038/nn.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gabriel LR, Wu S & Melikian HE Brain slice biotinylation: An ex vivo approach to measure region-specific plasma membrane protein trafficking in adult neurons. J. Vis. Exp (2014). doi: 10.3791/51240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crupi MJF, Richardson DS & Mulligan LM Cell surface biotinylation of receptor tyrosine kinases to investigate intracellular trafficking. Methods Mol. Biol (2015). doi: 10.1007/978-1-4939-1789-1_9 [DOI] [PubMed] [Google Scholar]
- 75.Sullivan JM et al. Autism-like syndrome is induced by pharmacological suppression of BET proteins in young mice. J. Exp. Med (2015). doi: 10.1084/jem.20151271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pnevmatikakis EA & Giovannucci A NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods (2017). doi: 10.1016/j.jneumeth.2017.07.031 [DOI] [PubMed] [Google Scholar]
- 77.Pachitariu M et al. Suite2p: beyond 10,000 neurons with standard two-photon microscopy. bioRxiv (2016). doi: 10.1101/061507 [DOI] [Google Scholar]
- 78.Klaus A et al. The Spatiotemporal Organization of the Striatum Encodes Action Space. Neuron 95, 1171–1180.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barbera G et al. Spatially Compact Neural Clusters in the Dorsal Striatum Encode Locomotion Relevant Information. Neuron (2016). doi: 10.1016/j.neuron.2016.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kato D et al. In Vivo Two-Photon Imaging of Microglial Synapse Contacts. in Methods in Molecular Biology (2019). doi: 10.1007/978-1-4939-9658-2_20 [DOI] [PubMed] [Google Scholar]
- 81.Thévenaz P, Ruttimann UE & Unser M A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. (1998). doi: 10.1109/83.650848 [DOI] [PubMed] [Google Scholar]
- 82.Ting JT et al. Preparation of Acute Brain Slices Using an Optimized N-Methyl-D-glucamine Protective Recovery Method. J. Vis. Exp (2018). doi: 10.3791/53825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fieblinger T et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat. Commun (2014). doi: 10.1038/ncomms6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Graves SM & Surmeier DJ Delayed spine pruning of direct pathway spiny projection neurons in a mouse model of parkinson’s disease. Front. Cell. Neurosci (2019). doi: 10.3389/fncel.2019.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wong JMT et al. Benzoyl chloride derivatization with liquid chromatography-mass spectrometry for targeted metabolomics of neurochemicals in biological samples. J. Chromatogr. A (2016). doi: 10.1016/j.chroma.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gangarossa G et al. Convulsant doses of a dopamine D1 receptor agonist result in erk-dependent increases in Zif268 and Arc/Arg3.1 expression in mouse dentate gyrus. PLoS One (2011). doi: 10.1371/journal.pone.0019415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bunch L & Krogsgaard-Larsen P Subtype selective kainic acid receptor agonists: Discovery and approaches to rational design. Medicinal Research Reviews (2009). doi: 10.1002/med.20133 [DOI] [PubMed] [Google Scholar]
- 88.Willoughby JO, Mackenzie L, Medvedev A & Hiscock JJ Distribution of Fos-positive neurons in cortical and subcortical structures after picrotoxin-induced convulsions varies with seizure type. Brain Res. (1995). doi: 10.1016/0006-8993(95)00366-X [DOI] [PubMed] [Google Scholar]
- 89.Sipe GO et al. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun (2016). doi: 10.1038/ncomms10905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Racine RJ Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol (1972). doi: 10.1016/0013-4694(72)90177–0 [DOI] [PubMed] [Google Scholar]
- 91.Silverman JL, Yang M, Lord C & Crawley JN Behavioural phenotyping assays for mouse models of autism. Nature Reviews Neuroscience (2010). doi: 10.1038/nrn2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hickman SE et al. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci (2013). doi: 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Langfelder P et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci (2016). doi: 10.1038/nn.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell (2015). doi: 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell (2017). doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 96.Sousa C et al. Single‐cell transcriptomics reveals distinct inflammation‐induced microglia signatures. EMBO Rep. (2018). doi: 10.15252/embr.201846171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yoder NC peakfinder(x0, sel, thresh, extrema, includeEndpoints, interpolate). https://www.mathworks.com/matlabcentral/fileexchange/25500-peakfinder-x0-sel-thresh-extrema-includeendpoints-interpolate (Matlab Central File Exchange, 2016).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for gene expression analysis in the paper is available at NCBI Gene Expression Omnibus (GEO) under the accession number GSE149897.