

ABSTRACT
Treatment of ocular disease is hindered by the presence of the blood-retinal barrier, which restricts access of systemic drugs to the eye. Intravitreal injections bypass this barrier, delivering high concentrations of drug to the targeted tissue. However, the recommended dosing interval for approved biologics is typically 6–12 weeks, and frequent travel to the physician’s office poses a substantial burden for elderly patients with poor vision. Real-world data suggest that many patients are under-treated. Here, we investigate IgMs as a novel platform for treating ocular disease. We show that IgMs are well-suited to ocular administration due to moderate viscosity, long ocular exposure, and rapid systemic clearance. The complement-dependent cytotoxicity of IgMs can be readily removed with a P436G mutation, reducing safety liabilities. Furthermore, dodecavalent binding of IgM hexamers can potently activate pathways implicated in the treatment of progressive blindness, including the Tie2 receptor tyrosine kinase signaling pathway for the treatment of diabetic macular edema, or the death receptor 4 tumor necrosis family receptor pathway for the treatment of wet age-related macular degeneration. Collectively, these data demonstrate the promise of IgMs as therapeutic agonists for treating progressive blindness.
KEYWORDS: IgM, antibody engineering, ocular therapeutics, agonism, long-acting delivery
Introduction
Retinal diseases, including age-related macular degeneration (AMD), geographic atrophy, and diabetic macular edema (DME), affect the vision of approximately 6 million Americans.1 Vitreally administered anti-vascular endothelial growth factor (VEGF) therapies have become the standard of care for both wet AMD (wAMD) and DME, allowing widespread stabilization and even improvement in patients’ vision.2–4 Still, approximately 40% of patients treated under optimal conditions lose sight and could benefit from additional therapies. Furthermore, the high treatment burden posed by frequent administration leads to poor compliance, which can result in permanently reduced vision.5
Biologics intended to treat ocular diseases are typically injected into the vitreous humor. This site-specific injection provides a safety window (i.e., high concentrations at the site of action, and relatively little systemic drug), but the procedure is invasive and less frequent administrations are preferred.6 Notably, the vitreous is largely devoid of receptors that drive systemic clearance (e.g., asialoglycoprotein receptors or mannose receptors).7 Instead, clearance from the vitreous seems to be driven primarily by diffusion into the aqueous humor and ultimately into the blood; molecules containing larger hydrodynamic radii exit more slowly.8,9 Thus, if they could be generated and safely administered, larger molecules present a theoretical advantage for long-acting delivery. In this context we investigated immunoglobulin Ms (IgMs) as a potential platform for treating ocular disease.
IgMs are megadalton-sized protein complexes of 21 or 24 protein subunits. By molecular weight they are 6–7x larger than an IgG and approximately 20x larger than an antigen-binding fragment (Fab). The ‘monomeric’ component of an IgM consists of two light chains (LCs), each containing two Ig-domains, and two heavy chains (HCs), containing five Ig-domains and a short C-terminal tail piece (CTP, Figure 1a).10 These four chains assemble to form a homodimer of HC-LC heterodimers. Subsequently, disulfide formation and folding of the C-terminal tail-piece into beta-strands drives formation of pentamers containing five homodimers and a J-chain (JC) or hexamers with six homodimers. In humans, pentamers are the dominant species, accounting for ~95−98% of the IgMs isolated from serum.11 These four chains assemble to form a homodimer of HC-LC heterodimers. Subsequently, disulfide formation and folding of the C-terminal tail-piece into beta-strands drives formation of pentamers containing five homodimers and a J-chain (JC) or hexamers with six homodimers. In humans, pentamers are the dominant species, accounting for ~95−98% of the IgMs isolated from serum.,11–13 It is believed that avid binding of 10 or 12 variable fragments (Fvs) enables IgMs to bind targets without substantial affinity maturation, and thus to serve as sentinel adaptive immune receptors.12,13
Figure 1.
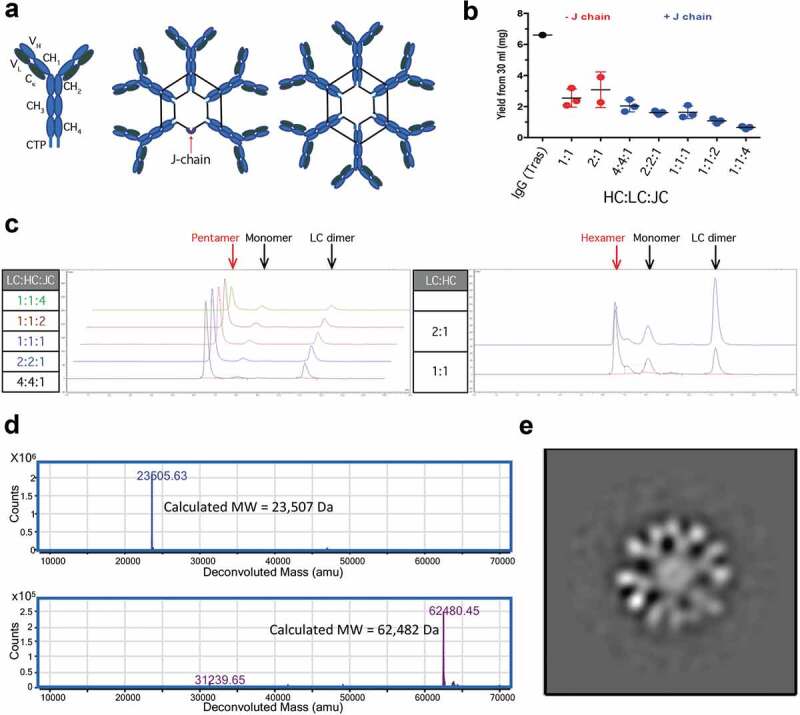
Characterization of recombinant IgMs. Structural representations of IgM ‘monomers’, pentamers, and hexamers. Black lines joining HC represent disulfide bonds (a). 30 ml of Expi293 cells were transiently transfected with various ratios of hIgM HC:LC:JC incubated for 7 d and partially purified using CaptoL resin. Samples were analyzed for total protein by A280 using trastuzumab (Tras) as a positive control (b) and for homogeneity using SEC (c). Human IgM hexamers were purified via two-step purifications. Purified hexamer was reduced and deglycosylated prior to analysis by LC-MS/MS. Extracted ion chromatograms are shown for LC (top) and heavy chain (bottom) (d). Negative Staining TEM characterization of hIgM hexamer reveals the anticipated six-fold symmetry (e).
Despite the potential potency advantages resulting from high avidity, immunoglobulin Gs (IgGs) rather than IgMs have emerged as the preferred immunoglobulin therapeutic format. IgGs have increased systemic exposure due to binding to the neonatal Fc receptor (FcRn) and potentially reduced lectin-mediated clearance.14 They have interactions with a variety of Fc-gamma receptors (FcγRs), which can contribute to therapeutic efficacy,15 and the smaller size of IgGs results in reduced manufacturing and analytical complexity. Several monoclonal IgMs have entered clinical trials, with one molecule advancing as far as Phase 3 trials in the United States and a short-lived approval in Europe.16–19 However, the combination of manufacturing difficulties, poor pharmacokinetics, and lack of efficacy has thus far contributed to an absence of marketed IgM therapeutics.
While IgGs remain the therapeutic standard, recent work shows that multivalent agonism enables activities not achievable with simple bivalent engagement, for example in the activation of trimeric tumor necrosis factor (TNF) superfamily receptors or in mimicking cellular synapses for immuno-oncology applications.20,21 In the ocular space, death receptor 4 (DR4), and Tie-2 have both been implicated in retinal disease and require multivalent engagement to signal.22,23 DR4 is a TNF superfamily member whose oligomerization drives caspase-8 activation and initiation of apoptosis.24 The minimal clustering unit of DR4 contains three subunits. Direct activation of DR4 on choroidal neovasculature may atrophy leaky vessels that are a key driver of neovascular AMD. Tie-2 is a receptor tyrosine kinase implicated in reducing vessel leakiness. Clusters of angiopoetin-1 (Ang-1) bind to and oligomerize Tie-2, driving tightening of cell-cell junctions and promoting cell survival.25 Overexpression of angiopoietin-2 (Ang-2) antagonizes Ang-1 signaling and is thought to drive ocular edema in DME.26 Two recent trials found that Anti-Ang-2 therapeutics drove partial reductions in ocular edema,23,27 but only one of the two trials demonstrated improvements in visual acuity. Direct agonism may more potently activate Tie-2 signaling, driving improvement in vision in cases where Ang-1, is scarce or absent.28
In addition to opportunities for novel activities, IgMs may provide additional safety advantages suited to the unique environment of the eye. The vitreous has ~1% of the serum levels of both IgMs and IgGs,29 which compete with therapeutics for binding to FcγRs or C1q. The risk of potent effector function driving toxicity can be addressed by removing the Fc (e.g., ranibizumab (Lucentis®)) or by mutating the FcγR and complement binding sites to reduce effector function and the FcRn binding sites to reduce exposure (e.g., faricimab (anti-Ang-2/VEGF)).30,31 These solutions aren’t without risk; the small size of the Fabs can drive more rapid clearance from the vitreous (t1/2 in humans of ~7 days vs. ~10 days for IgGs),32 and mutations introduce foreign sequence into therapeutics. IgMs do not bind to FcγRs or FcRn, but they do potently activate complement, a feature that may need to be abrogated for safe intravitreal administration.
To assess the ocular applicability of IgMs, we demonstrate recombinant expression of murine, rabbit, and human IgMs, and facile conversion between pentameric and hexameric forms by inclusion/omission of the J-chain. IgMs are shown to have acceptable viscosities, extended vitreal exposure following intravitreal injection, and rapid systemic clearance, features consistent with a high therapeutic index and reduced frequency of injection. Homology-guided mutagenesis is shown to remove complement activity from IgMs, thus minimizing safety risks. Finally, IgMs are shown to activate two signaling pathways implicated in ocular disease that are not amenable to direct engagement with IgGs.
Results
Expression and characterization of IgMs
To investigate the feasibility of applying IgMs to ocular targets we first sought to optimize their expression and purification. Expi293 cells were transiently transfected with various amounts of DNA encoding human IgM LC and HC with or without JC, supernatants were harvested, and LC-containing species were isolated by single-step purification off of a Capto L column. Isolated material was quantified and analyzed by size-exclusion chromatography (SEC) to estimate the various species present in the samples. Hexamer optimization proceeded by transfecting cells with plasmids containing LC and HC at a 1:1 or 2:1 ratio of plasmid DNA. Each ratio resulted in similar total protein yields (2.5–3 mg from a 30 ml expression) (Figure 1b), but product quality appeared improved at a 1:1 ratio, with reduced amounts of LC-dimer present (Figure 1c). Expression of putative pentamers was conducted by including J-chain in the transfection. The optimal ratio of 4:4:1 LC:HC:JC, maximized both expression and product quality (Figure 1b-c). Notably, the high level of LC and HC DNA relative to JC in the optimized conditions approximates the 10:10:1 chain ratio in the final IgM pentamer.
Having optimized chain ratios for expression of putative pentameric and hexameric IgMs, we scaled up expression and purification of human and murine hexameric and pentameric IgMs. Approximately 20 mg/L of purified hexameric or pentameric IgM was isolated via two-step purification from Chinese hamster ovary (CHO) cells. Though the transient yields are 3- to 4-fold lower than typical for IgGs expressed under this process, the relatively facile purification combined with the low material requirements for ocular drugs (typically 0.3–20 mg/eye) should enable manufacturing feasibility. Chain composition of the human IgMs was confirmed by SDS-PAGE and LC-MS of the reduced and deglycosylated species showing the presence of LC, HC, and JC if present (Figure 1d and Supplementary Figure 1–2). SEC showed monodisperse peaks for pentameric and hexameric murine IgM and for hexameric human IgM. Pentameric human IgM eluted from the SEC in two closely related peaks, both containing J-chains and with high apparent MW by SEC (Supplementary Figure 3). Assessment by light scattering found hydrodynamic radii (Rh) of approximately 12 nm with predicted molecular weights for the hexamers (~1050 kDa) slightly exceeding those for the pentamers (~950 kDa) (Supplementary Table 1). The ratio of radius of gyration to hydrodynamic radius (Rg/Rh) provides some insight into molecular shape, with spherical molecules averaging ~0.775, and more elongated species trending higher. Monodisperse IgMs ranged from 0.85 to 0.91, consistent with the anticipated structures. To further assess the polymeric identity of the IgMs we utilized negative stain transmission electron microscopy (TEM) and reference-free 2D classification to describe the architecture of IgM particles, which appear to match their anticipated geometries (Supplementary Figs 4 & 5). No obvious difference was observed between the two peaks corresponding to putative human pentamers. The human IgM hexamer peak was selected for further investigation. We manually collected 110 TEM micrographs, which resulted in a total of 1500 particles. Following three rounds of reference-free 2D classification using the Relion suite,34 we found that the majority of the particles organized into a hexameric structure (Figure 1e). A small number of particles (<2%) refined into pentamers (Supplementary Figure 6). While each Fab is clearly visible in the structure, the CH3 and CH4 domains are visible only as an overlapping central circle. This is consistent with previous atomic force microscopy investigations into IgM pentamers35 in which the CH3 and CH4 domains form a stem-like domain that is orthogonal to the plane of the image, though modestly differentiated from the more planar structure revealed by high-resolution cryo-electron microscopy of the IgM pentamer (CH3-4 only) in complex with the secretory chain.36 It’s unclear whether these differences are artifacts of the lower resolution techniques or if removal of the Fabs and CH2 and/or interaction with the secretory chain impact IgM structure. While pairs of Fabs attached to the same Fc appear coplanar in our image, there is some indication that Fabs on adjacent Fcs are offset for the hexamer. A partial offset may allow the hexamer to accommodate all chains in a sterically crowded environment. Human hexameric IgMs were selected as the preferred therapeutic scaffold due to the improved purity of production and the absence of JC, which both simplifies manufacturing and obviates the need to consider polymeric-Ig receptor-mediated binding in vivo.
Characterization of IgMs for ocular administration
Next, we investigated the suitability of IgMs for ocular applications using New Zealand White Rabbits as a model system. The rabbit eye is of sufficient size to enable relatively facile intravitreal administration, and pharmacokinetic parameters in rabbits have previously been correlated to the results in cynomolgus monkeys and humans.29,30 To minimize the risk of anti-drug antibodies interfering with the pharmacokinetic evaluation of IgMs, we generated a fully rabbit IgM hexamer (rabbit IgM Fc with rabbit Fvs targeting an intracellular epitope). Consistent with data for both human and murine IgM hexamers, elution off a Capto L column followed by SEC provided a single major peak of high molecular weight, which was confirmed to contain bands of ~75 and ~25 kDa by reducing SDS-PAGE (data not shown). Notably, intravitreal administration to humans poses a series of technical challenges. Needles are usually limited to 30 G, and injection volumes are limited to 50 or rarely 100 μL.37 To avoid frequent repeated injections, the amount and therefore the concentration of protein injected is maximized. Under this set of conditions, protein viscosity is of considerable interest. We evaluated the viscosity of our IgM hexamer versus historical data surveying a range of IgG viscosities (Figure 2).38 Viscometer readings for IgMs show the expected log-linear response. Under viscosities conducive to intravitreal administration without patient pain or undue backpressure on the syringe (~10-50 cP), the median reported IgGs were limited to ~120-150 mg/ml, while the IgM was limited to 100–130 mg/ml. Thus, IgMs can be administered with modest reductions in total protein versus IgGs.
Figure 2.
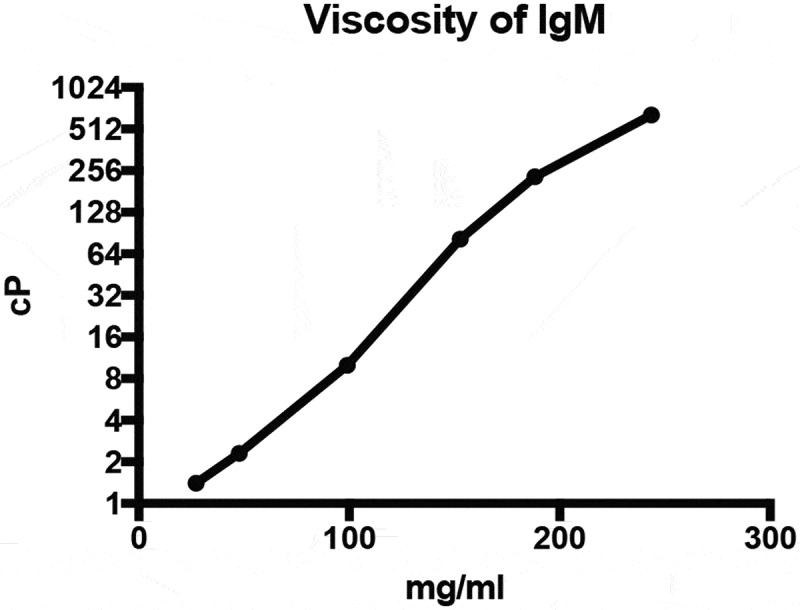
Rheology measurements of IgMs. Viscosity of a non-targeting rabbit IgM was assessed by rotary rheology.
While viscosity limits the amount of therapeutic that can be administered, the frequency of administration is influenced by vitreal half-life. The primary means of biotherapeutic escape from the vitreous is diffusion into the anterior segment followed by removal through aqueous humor outflow.33 The rate of anterior diffusion has been shown to be inversely proportional to the molecule’s hydrodynamic radius.8 Application of SEC-quasi-elastic light scattering (QELS) analysis to the rabbit IgM found a hydrodynamic radius of 12.9 ± 1.1 nm, considerably larger than the ~2.5–2.7 nm radius previously reported for this Fab.8 To assess the functional consequence of this increase in molecular size, a rabbit Fab and IgM were labeled with Alexa Fluor 488, and 0.5 mg or 1.2 mg, respectively, were intravitreally administered to New Zealand White rabbits. In-life quantitative fluorescent measurements taken over 28 days revealed the durability of each of the two therapeutics (Figure 3). In this experiment, the rabbit Fab showed a half-life of 3.5 days, comparable to previous reports of 3.2 days and typical Fab half-lives of ~3.4 days in rabbit vitreous.39 The IgM showed substantially extended vitreal exposure of 7.8 days, consistent with predictions for its larger size.39
Figure 3.

Vitreal pharmacokinetic analysis of IgMs. A non-binding Fab and IgM were nonspecifically labeled with Alexa Fluor-488. Following administration of 0.5 or 1.2 mg respectively, vitreal concentrations were assessed via in vivo fluorophotometric measurements as previously described.33 Data is shown as observed concentrations (symbols) and noncompartmental projections (lines) in vitreous humor following intravitreal injection of untargeted Fab or untargeted IgM in New Zealand White rabbits.
Systemic exposure of IgMs was also of interest, as rapid systemic clearance helps restrict activity of ocular therapeutics to the eye. Interestingly, while previous investigations of IgMs isolated from serum or ascites fluid have reported half-lives of a few days,40,41 recombinantly produced IgM typically clears with half-lives of hours.18,42,43 To confirm these findings we compared the pharmacokinetics of non-binding recombinant human IgM pentamer and hexamer dosed at 5 mg/kg versus IgM isolated from human serum at both 1 and 5 mg/kg (Figure 4a). Following intravenous injection into SCID mice, recombinant IgM pentamers and hexamers were rapidly cleared (t1/2 of 0.25 and 0.50 days, respectively), while isolated IgM showed prolonged and dose-proportional exposure (t1/2 = 2.1–2.9 d). We hypothesized that rapid clearance may be due to differential glycan occupancy or composition of the IgMs. There are up to 51 N-linked glycans per human pentamer and 60 per human hexamer. In support of this theory, glycan analysis showed 75% of glycans on IgMs purified from human serum contained at least one sialic acid versus 24% and 29% of recombinant hexamers and pentamers, respectively (Figure 4b) consistent with asialoglycoprotein-mediated clearance.
Figure 4.
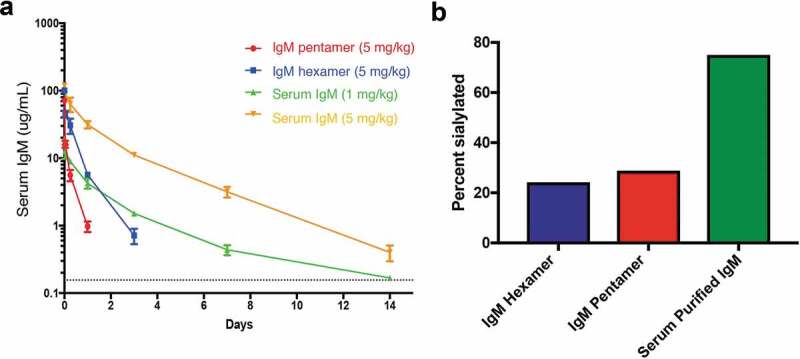
Systemic pharmacokinetic analysis of IgMs. Non-binding recombinant hIgM pentamers and hexamers along with IgM isolated from human serum injected intravenously into female SCID mice (a). At indicated time points, serum samples were taken and analyzed for the presence of human IgM. The dotted line is the minimum quantifiable of IgM (0.156 µg/ml). Glycans were removed from the IgMs using PNGase F, and global N-linked glycan profiles were assessed using LC-MS (b).
Based on the hypothesis that removal of glycans might drive extended systemic exposure, additional glycan characterization was undertaken. Consistent with previous reports,44 glyco-peptide analysis showed both serum and recombinant IgMs had full glycan occupancy at N171, N332, N395 and N402, while N563 was 92% occupied for serum IgM and 46% for the recombinant pentamer (Supplementary Table 2). Glycan composition was also similar to the previous report,44 with sialic acid present at N171, N332, but not N563. N395 and N402 were not separable in our analysis, though at least one of the two glycans contained sialic acid the majority of the time. Various asparagine to glutamine mutations were made to assess the feasibility of removing sites of N-linked glycosylation. All sites except N402 could be individually removed without substantial impact on transiently expressed product quality or yield (Supplementary Figure 7). Combinations of these mutations, however, substantially reduced product yield and/or quality potentially due to reduced solubility of the HC, and this approach was abandoned.
Protein engineering to minimize complement activity
IgMs have previously been reported to potently recruit C1q, and induce target cell-killing via complement-dependent cytotoxicity (CDC).45,46 While this represents an alternative potential mechanism of action for therapeutics inducing cell death (e.g., anti-DR4 IgMs), it can be undesired for therapeutics signaling through other pathways. In addition to this theoretical risk, mutations in multiple steps of the complement pathway are associated with increased risk for AMD, further highlighting the risk of activating complement in the eye.47 Thus, we sought to reduce the CDC activity of IgMs.
We chose to evaluate CDC using antibodies targeting CD20 on B-cells.48 Rituximab (anti-CD20) is known to have potent CDC activity as an IgG, providing a stringent test for complement attenuation. We generated targeted IgM hexamers and IgG1s (rituximab) and evaluated their activity on Wil-2S cells expressing CD20 in the presence of 20% human complement. Following a two-hour incubation, cells were assessed using an Alamar Blue cell viability assay.49 Both IgG and IgM showed potent CDC (Figure 5a), with IgM showing ~6x greater potency on a molar basis. Prior work on IgGs identified P327 and P329 as important residues to facilitate complement binding and determined that the P329G mutation could inactivate complement activity while maintaining IgG expression and stability.50,51 Alignment of the human IgG1-Fc with the human IgM-Fc (Figure 5c) revealed that this mutation mapped to a site known to influence complement binding in mouse IgM.52 Thus, we generated the P436G mutant and evaluated its impact on CDC (Figure 5b). Consistent with our expectations, introduction of the P436G mutation removed all detectable complement activity, thus enabling the application of IgM hexamers in the ocular space.
Figure 5.
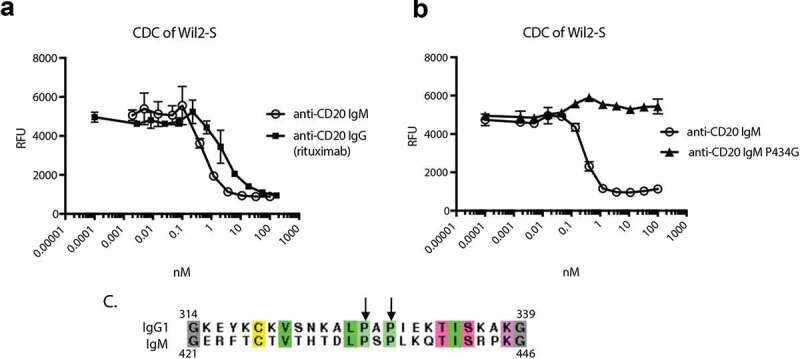
Characterization and attenuation of hIgM CDC activity. Rituximab and the corresponding anti-CD20 hIgM hexamer were incubated with Wil-2S cells in the presence of 20% human serum for 2 h. Extent of cell death was evaluated by addition of Alamar blue (a). The P436G mutant shows attenuated CDC compared to the anti-CD20 IgM hexamer (b). Alignment of hIgG1 with hIgM highlighting the prolines critical for effective engagement with complement (c).
IgMs cluster receptors to drive downstream signaling
Having characterized the suitability of IgMs to ocular administration, we next turned to probing their ability to activate relevant signaling pathways. Tie-2 is a receptor tyrosine kinase that is broadly expressed on endothelial cells, including retinal vasculature, and is thought to regulate tight junction strength and vessel leakiness.53–55 Natural activation of Tie-2 is regulated by expression levels of Ang-1, a full agonist, and Ang-2, a competitive binder and partial agonist. Ang-1 associates multivalently on the cell surface, and in the absence of competition from Ang-2, binds to and activates Tie-2. Overexpression of Ang-2 during DME is thought to prevent Ang-1 engagement and full Tie-2 activation.26 As a result, Tie-2 mediated stabilization of vessels and reduction in vascular leakage does not occur. Two recent clinical trials investigated the impact of Ang2 inhibition when combined with standard of care VEGF-inhibition.23,27 The trials anticipated that endogenous Ang-1 levels would be sufficient to drive Tie-2 signaling once inhibitory Ang-2 was removed. Both trials showed inhibition of the Ang-2 pathway led to significant reductions in ocular edema above VEGF-inhibition alone, suggesting that Ang-2 inhibition led to a pharmacodynamic effect; however, only one of the trials showed significant improvements in vision. Direct activation via targeted Tie-2 clustering could prove to be a more effective therapeutic strategy.28
Tie-2 targeted IgM hexamers were expressed and analyzed for their ability to drive phospho-AKT signaling downstream of Tie-2. Rat aortic endothelial cells were treated with increasing doses of either endothelial-targeted anti-Tie-2 human IgM hexamers or untargeted anti-CD20 human IgM hexamers for 15 minutes, lysed, and intracellular phospho-AKT levels were evaluated using homogeneous time-resolved fluorescence (Figure 6). Increases in cellular phospho-AKT levels were seen only for the targeted IgM. No activity was seen for the corresponding IgG in the absence of cross-linking (data not shown). Interestingly, while the anti-Tie-2 IgM was active at picomolar concentrations, pAKT production was partially suppressed at high concentrations, perhaps due to receptor internalization or to super-saturation of IgM on the cell-surface reducing the extent of Tie-2 clustering and downstream signaling. Still, at 100 nM IgM (~100 µg/ml) approximately 30% of the maximum activity was attained. These data suggest that anti-Tie-2 IgMs are likely to drive Tie-2 signaling over at least three orders of magnitude in protein concentration.
Figure 6.
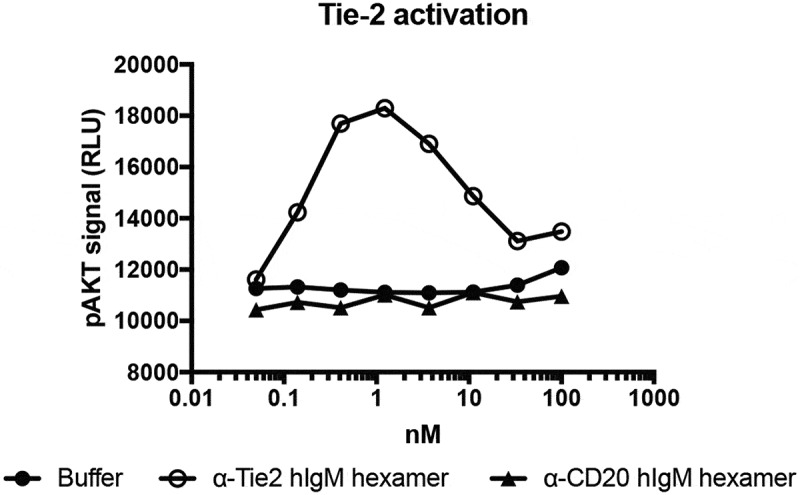
Phospho-(p)AKT signaling downstream of Tie-2 clustering. Serial dilutions of Tie-2 or CD20 targeted IgMs were incubated with rat-aortic endothelial cells for 15 minutes prior to lysis and analysis of pAKT levels using the CisBio Ser473 pAKT kit.
As an alternative example of ocular agonism, we examined IgM activation of DR4. DR4 is a TNF-superfamily member agonized by TNF-related ligand/Apo2 ligand (TRAIL/Apo2L).24 Binding drives activation of the death-inducing signaling complex and caspase-8, and induces apoptosis. DR4 is expressed on developing blood vessels, and activation of DR4 on these cells is anticipated to reduce choroidal neovascularization and the corresponding vascular leak in wAMD. Similar to other TNF-superfamily members, DR4 is a trimeric receptor and TRAIL is a trimeric ligand.56 Bivalent engagement with IgGs is typically insufficient to drive activation.57 To investigate the role of multivalent engagement in DR4 signaling, we generated anti-DR4 IgMs based on a previously published IgG (4H6)57 and compared them to a variety of other published DR4-, DR5-, or DR4/5-targeted therapeutics. Addition of these molecules to COLO 205 cells over 24 hours drove both activation of Caspase-8 and induction of cell-death (Figure 7). 4H6 showed no activity as a IgG alone (closed blue circles), requiring cross-linking with an anti-human IgG1 secondary antibody to drive partial killing and caspase activation (open blue circles). By contrast, the same variable regions attached to an IgM-scaffold drove nearly complete killing with an EC50 ~ 100-fold lower than the crosslinked IgG (open blue squares). We also evaluated Apomab (an antibody known to agonize a related death receptor (DR5))58 and Apo2L-FLAG (the native DR4 and DR5 ligand), with and without cross-linking. Apo2L-FLAG is known to spontaneously trimerize,59 whereas Apomab has previously been shown to partially activate DR5 signaling in the absence of cross-linking antibody. Both trimeric Apo2L-FLAG (closed red triangles), and Apomab (closed green squares) showed intrinsic death receptor signaling that is enhanced by Fc- or FLAG-crosslinking (open red triangles and open green squares, respectively). Despite binding to only DR4, the IgM showed substantial increases in killing and caspase-8 activation versus uncrosslinked ligands, and slight increases versus ligands cross-linked by IgGs. Collectively, these data speak to the ability of IgMs to potently activate TNF-receptors.
Figure 7.
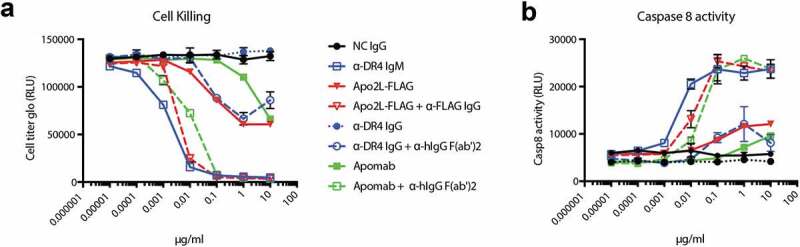
Death receptor activation downstream of clustering. Serial dilutions of death receptor targeted therapeutics were added to COLO 205 cells in the presence or absence of cross-linker. Viability was determined by Cell-titer glo after 24 h (a). Caspase-8 activity was determined by Caspase-glo 8 4 h following addition of therapeutics (b). The legend is shared for both graphs.
Discussion
To close the gap between optimal ocular therapy as conducted in clinical trials and real-world use, multiple approaches are being pursued to extend ocular therapeutic dosing intervals and reduce patient burden. The small size and low viscosity of brolocizumab, a recently approved anti-VEGF single-chain variable domain, enables concentration up to 120 mg/ml, and the resulting high molar dose of drug extends the approved dosing interval to 3 months.60,61 Kodiak Sciences has pursued an alternate approach where conjugation of a ~ 800 kDa biocompatible polymer to an anti-VEGF IgG results in a nearly megadalton conjugate with slow diffusion out of the eye resulting in a retinal half-life of 10.5 days in rabbits and durability of up to 6 months in a Phase 1 clinical trial (NCT03790852).62 Finally, surgical implantation of a port delivery system has allowed ranibizumab to be dosed with a median durability of up to 15 months.63 Additional depot approaches have been described, but are not yet validated in clinical trials.64 Each of the described approaches has limitations. The three-month durability with brolocizumab is not markedly distinguished from existing therapies, and given the relatively short vitreal half-lives of small proteins, increasing the durability further would require a substantially higher concentration of the drug. Kodiak Sciences’ KSI-301 is a biopolymer antibody conjugate, which is approximately 80% biopolymer by mass. To achieve reported dosing of 5 mg of antibody per eye, they inject ~25 mg of conjugate in 100 uL of solution (the limit of clinical acceptability). The decision to implement this high-volume dosing, typically performed as two separate 50 uL injections administered at least 15 minutes apart to allow re-equilibrium of the vitreal pressure, speaks to an inability to further concentrate the molecule.
In our hands, the tested IgM could be concentrated to >100 mg/ml, enabling a 5 mg protein within the target 50 uL injection. Human IgM viscosity has been assessed more broadly in the context of patients with Waldenstrom’s Macroglobulinemia, a lymphoma resulting in extremely high levels of monoclonal IgMs.65 While the variable domains do influence viscosity, most patients with circulating IgM levels between 40–85 mg/ml showed serum viscosities between 3–12 cP, suggesting that many IgMs may have appropriate viscosities for high-dose intravitreal injection. Finally, while the durability achieved with the port delivery system is unlikely to be matched without assistance of a device, it does require surgery performed at a specialty eye clinic, a procedure that is not desirable for all patients. IgMs, which are one megadalton in size, can be intravitreally injected. Comparing across studies, the vitreal exposure of IgMs is modestly reduced compared to the reported retinal exposure of KSI-301 (half-life in rabbits of 7.9 vs. 10.5 days).63 Notably, these studies analyzed different tissues using different drug quantification methods, resulting in uncertainty in the comparability. In contrast to KSI-301, IgMs are simply fermented, without additional polymer generation or conjugation required. Furthermore, IgMs have 12 Fabs per megadalton as opposed to two for KSI-301, potentially allowing for higher initial dose-equivalents.
Beyond durability, IgMs appear to demonstrate a suite of properties conducive to ocular therapy and distinct from other long-acting-delivery technologies. IgMs targeting DR4 and Tie-2 agonized receptors not addressable by uncrosslinked IgGs. This extends the subset of druggable targets in a way not readily achievable with other approaches targeting ocular durability. From a manufacturing perspective, IgMs were recombinantly expressed and purified with acceptable yields. Selection of an IgM hexamer without a J-chain as the preferred ocular format both simplifies production and characterization of the final material (one fewer chain) and removes polymeric-Ig-receptor (pIgR) binding and corresponding risk of transcytosis. In addition, they do not require extensive conjugation or assembly post-purification, providing advantages over other alternative complex formats. A key manufacturing risk associated with IgMs is the large number of glycosylation sites. This does not appear to drive rapid clearance from the vitreous, and thus the risk profile for ocular therapeutics is lower than it would be for systemic medicines, but demonstrating consistent and homogeneous production could be a substantial challenge. IgMs also provide potential safety advantages for ocular applications. By agonizing targets in the absence of FcγR crosslinking, they eliminate FcγR effector function-associated safety risks. Given the tight genetic correlation between activation of complement and risk of retinal disease,48 we further extended the safety profile by removing CDC via P436G mutation. While modification of effector function has previously been explored for murine IgMs,45,46 to the extent of our knowledge these findings have not previously been extended to the human Fc.
Consistent with predictions based on previously investigated PEGylated Fabs,8 the large size of the IgM results in durable protein exposure. Notably, half-life extension is quantitatively aligned with previous predictions,39 indicating that these models accurately estimate both globular proteins and protein conjugates, along with the irregular mushroom shape of an IgM. Finally, extensive glycosylation of IgMs (up to 60 N-linked glycans in a hexamer), which presumably drives rapid systemic clearance of recombinant material in vivo,18 does not result in rapid ocular clearance. This combination of durable ocular exposure coupled to rapid systemic clearance results in a wide exposure differential that should enable application of IgM application to targets with high potential systemic toxicity. Collectively, we believe these data highlight the potential of IgMs as ocular agonists.
Materials and Methods
Antibody construct design and synthesis
All antibodies in this work are numbered using EU numbering systems for constant domains.66 Antibody constructs were generated by gene synthesis (GeneWiz) or through mutagenesis using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs). DNAs encoding IgG-HC, IgM-HC, Fab-HC, LC, or JC were subcloned into the expression plasmid pRK5.67 IgM-HCs were constructed with the variant P436G as noted.
Antibody expression and purification
Plasmids encoding IgGs were transiently transfected into Expi293F cells (ThermoFisher) as recommended by the manufacturer and grown for 7 d prior to harvesting and purification. Harvested media was loaded onto 5 ml MabSelect SuRe column (GE Healthcare). The loaded column was washed with 10 column volumes (CV) of Tris buffer (25 mM Tris pH 7.0, 150 mM NaCl, 5 mM EDTA, 2 mM EDTA), 5 CV of Triton X-114 buffer (25 mM Tris pH 7.0, 150 mM NaCl, 5 mM EDTA, 0.1% Triton X-114) 10 CV of Tris buffer, 2 CV of KP buffer (0.4 M potassium phosphate pH 7.0, 5 mM EDTA, 0.02% PS20) and finally 10 CV of Tris buffer. Protein was eluted with 5 CV of elution buffer (50 mM sodium citrate pH 3.0, 150 mM NaCl) and immediately neutralized with 1 M Tris pH 8.0. The neutralized elution was concentrated and purified over a HiLoad 16/600 Superdex 200 SEC column (GE Healthcare) with arginine buffer (200 mM arginine, 137 mM succinic acid) as the mobile phase. Eluted fractions were collected in 1 mL fractions increments and analyzed by SDS-PAGE. The final eluant pool was concentrated and formulated into 20 mM histidine acetate (HisOAc) pH 5.5, 240 mM sucrose, 0.02% PS20. Optimization of IgM expression and small-scale IgM expression for in vitro assays was conducted in 30 ml cultures of Expi293F cells transfected with a total of 30 µg of plasmid DNA. Cultures were grown for 7 d prior to harvesting and purification. Large-scale IgM expression for detailed biochemical and structural characterization along with in vivo studies was conducted by transiently transfecting CHO-DKO cells and growing cells for 14 d prior to harvesting purification. IgMs were purified via one-step purification (for expression optimization) or two-step purification (other studies). Harvested media was loaded onto CaptoL or Kappa Select columns (GE healthcare) and washed and eluted as recommended by the manufacturer. IgMs undergoing two-step purifications were passed over a HiLoad 16/600 Superdex 200 SEC column (GE Healthcare) with arginine buffer (200 mM arginine, 137 mM succinic acid) as the mobile phase. Eluted fractions were collected in 1 mL fractions increments and analyzed SDS-PAGE. Pooled fractions were concentrated and analyzed by analytical SEC on an XBridge Protein BEH SEC 450 Å column (Waters). Human serum IgM (Sigma-Aldrich, cat# I8260) was purchased commercially and dialyzed into arginine buffer (200 mM arginine, 137 mM succinic acid, pH 5.0).
RP-LCMS
Data were acquired as described previously9 using an Agilent 1290 Infinity UPLC in tandem with an Agilent 6230 electrospray ionization time-of-flight mass spectrometer, operating in positive ion mode. Protein was loaded onto a reverse-phase (RP) PLRP-S column (Agilent) with dimension of 4.6 × 50 mm. Mobile phase A consisted of 0.05% trifluoroacetic acid (TFA) while mobile phase B consisted of 0.05% TFA and 80% acetonitrile, and was used for the gradient between 20% and 90% solvent B.
Size-exclusion chromatography-multiangle light scattering-quasi-elastic light scattering
Hydrodynamic radii of IgMs were experimentally determined by SEC-multiangle light scattering (MALS)-quasi-elastic light scattering (QELS). An Agilent 1200 HPLC (Agilent Technologies, Inc., Santa Clara, CA) equipped with a binary pump, diode-array detector, Optilab T-rEX refractive index (RI) detector, and HELEOS-II MALS detector (Wyatt Technology Corp., Santa Barbara, CA) with a QELS module was fitted with an XBridge Protein BEH SEC 450 Å, 3.5 µm, 7.8 × 300 mm column (Waters Corp., Milford, MA) maintained at 25 °C. Mobile phase of phosphate-buffered saline (PBS) pH 7.4 + 0.01% sodium azide was used to elute samples under isocratic conditions at 0.5 mL/min after injections of 100 µg per sample. ASTRA software (Wyatt Technology Corp., Santa Barbara, CA) was used to process MALS and QELS data using differential refractive index as a concentration source.
Electron microscopy
Purified recombinant hexameric or pentameric IgMs were diluted to a final concentration of 0.08 mg/ml and deposited onto a 400-mesh copper grid (Electron Microscopy Sciences) previously glow discharged using a GloQube glow discharge system (Quorum Technologies). After incubating the samples for 30 seconds the grids were blotted with Whatman filter paper, and negatively stained by dipping 3 times into 30 µL distilled water and then twice into 30 µL of 0.22 μm filtered solution of 2% uranyl acetate. Grids were imaged using a JOEL 1400 equipped with a 2Kx2K Ultrascan 1000 (Gatan). Particles were selected and extracted using the EMAN2 e2boxer.py algorithm.68 Reference-free 2D classes were generated and refined using Relion.34
Rheology
Viscosity measurements were made using the Anton Paar Physica MCR 501 rotary rheometer, with the CP20-0.5° cone and plate configuration. The CP20-0.5 geometry has a diameter of 20-mm and an angle of with 0.5°. The measurements were performed at 25°C using the temperature controller of the rheometer (Peltier plate with circulating fluid from water bath). Approximately 20 μL of each sample was loaded onto the bottom plate for measurement, after which the cone was lowered slowly to the desired gap width. The measured torque determined the shear stress, from which the viscosity was calculated, as previously described.69
Assessment of vitreal pharmacokinetics
IgMs or Fabs (Genentech, South San Francisco, CA, USA) in 0.1 M sodium bicarbonate were labeled with of AF488 NHS ester (product number A-10235; Life Technologies) according to the manufacturer’s instructions to create an approximate 1.34:1 labeling ratio for the Fab and an 8.03:1 labeling ratio for the IgM. The excess dye was removed from AF488-ranibizumab using S200 (GE Healthcare, London, UK) SEC. The eluted peak was pooled, concentrated, dialyzed into PBS, and sterile filtered. Endotoxin levels were assessed using the Pierce LAL chromogenic endotoxin quantitation kit (Life Technologies, Foster City, CA, USA). Endotoxin levels were less than 0.05 EU/mg.
Vitreal pharmacokinetic (PK) data were determined following intravitreal injection of labeled IgMs or Fabs in New Zealand white rabbits. All animal studies were conducted in accordance with ethical standards of the Genentech institutional animal care and use committee guidelines and in agreement with the Association for Research in Vision and Ophthalmology’s Statement for the Use of Animals in Ophthalmic and Vision Research. Animal studies were conducted at the laboratory animal resource facility at Genentech. In all rabbit studies described herein, test articles were administered by a board-certified veterinary ophthalmologist. Test articles typically were formulated in sterile PBS (pH 7.4), typically at protein concentrations of 10 mg/mL (Fab) or 24 mg/ml (IgM) such that a 50 µL injection delivered 0.5 or 1.2 mg/eye dose. Assessment of labeled proteins within the vitreous by use of fluorophotometry was conducted as previously described.70 PK parameters were determined by noncompartmental analysis with nominal time and dose (Phoenix WinNonlin, Certara, Inc., Mountain View, CA).
Assessment of systemic pharmacokinetics
All animal work performed was reviewed and approved by Genentech’s Institutional Animal Care and Use Committee. Female SCID mice (6–8 weeks old) were obtained by Charles River Laboratories. Upon arrival, all mice were maintained in a pathogen-free animal facility under a standard 12 h light/12 h dark cycle at 21°C room temperature (RT) with access to food and water ad libitum. All mice received a single intravenous injection of IgM hexamer, pentamer, or mixture of serum purified hexamer and pentamer. Blood samples (150–200 μL) were collected via either retro-orbital sinus or cardiac puncture under isoflurane anesthesia at various times post injection. Samples were collected into serum separator tubes. The blood was allowed to clot at ambient temperature for at least 20 min. Clotted samples were maintained at RT until centrifuged, commencing within 1 h of the collection time. Each sample was centrifuged at a relative centrifugal force of 1500–2000 x g for 5 min at 2–8°C. The serum was separated from the blood sample within 20 min after centrifugation and transferred into labeled 2.0-mL polypropylene, conical-bottom microcentrifuge tubes. Only animals that appeared to be healthy and that were free of obvious abnormalities were used for the study.
IgM antibody levels were measured by sandwich ELISA. Wells of 384-microtiter plates were coated overnight at 4°C with 0.5 μg/ml of goat anti-human IgM antibody (Jackson ImmunoResearch, Cat#109-005-129) in 25 μL of PBS, pH 7.4, followed by blocking with 50 μL of Blocker Casein in PBS (Thermo Fisher Scientific, cat#37528) for 1 h at RT. Samples (25 μl) diluted (a minimum of 1:100) in sample buffer (PBS, pH 7.4, 0.5% bovine serum albumin (BSA), 0.35 M NaCl, 0.05% Tween20, 0.25% CHAPS, 5 mM EDTA) were then added to the blocked plates and incubated for 1.5 h at RT. After incubation, 25 μL of horseradish peroxidase-conjugated donkey anti-human IgM (Jackson ImmunoResearch, Cat# 709–035-073) were added and incubated for 1 h at RT. The plates were then incubated with 25 μL of 3,3ʹ,5,5ʹ-tetramethylbenzidine (Kirkegaard & Perry Laboratories, cat#50-76-00) for 10 min, and the reaction was stopped with 25 μL 1 M phosphoric acid. Absorbance was measured at 450 nm with a reduction at 630 nm using a plate reader. Between steps, plates were washed six times with 100 μL of washing buffer (0.05% Tween-20 in PBS). As a reference for quantification, a standard curve was established using serially diluted stock material (200 ng/ml-1.56 ng/ml) for each IgM molecule. The IgM ELISA tolerates 1% mouse serum.
Global N-linked glycan composition (LC-MS analysis)
IgM samples (10 µg) were denatured with 8 M guanidine HCl at 1:1 volume ratio and reduced with 100 mM dithiothreitol (DTT) for 10 min at 95°C. Samples were diluted with 100 mM Tris HCl, pH 7.5, to a final concentration of 2 M guanidine HCl, followed by overnight N-linked deglycosylation at 37°C with 2 μL of P0705S PNGase F (New England BioLabs). After deglycosylation, 150 ng of each sample were injected onto an Agilent 1260 Infinity LC system and eluted by an isocratic gradient of 2% to 32% solvent B (solvent A: 99.88% water containing 0.1% formic acid and 0.02% TFA; solvent B: 90% acetonitrile containing 9.88% water plus 0.1% formic acid and 0.02% TFA). The HPLC system was coupled via an Agilent G4240A Chip Cube MS system to a G6520B Q-TOF mass spectrometer. The samples were glycan enriched and separated using porous-graphitized carbon columns built within a G4240-64025 mAb-Glyco chip in the Chip Cube MS system. Data acquisition: 1.9 kV spray voltage; 325°C gas temperature; 5 l/min drying gas flow; 160 V fragmentor voltage; 65 V skimmer voltage; 750 V oct 1 RF Vpp voltage; 400 to 3000 m/z scan range; positive polarity; MS1 centroid data acquisition using extended dynamic range (2 GHz) instrument mode; 333.3 ms/spectrum and 3243 transients/spectrum. Acquired mass spectral data were searched against a glycan library in the Agilent MassHunter Qualitative Analysis software utilizing a combination of accurate mass with a mass tolerance of 10 ppm and expected retention time for glycan identification. N-linked glycans were label-free quantified relative to all identified N-linked glycans within each sample based on the area under the curve (AUC) in the extracted compound chromatogram of each glycan.
Glycopeptide analysis (LC-MS/MS analysis)
Ten μg of proteins were reduced with 20 mM DTT at 37°C for 30 min followed by alkylation with 40 mM iodoacetamide at room temperature for 30 min. Proteins were digested with trypsin (1:25 enzyme:substrate ratio) in 50 mM Tris, pH 7.5 overnight at 37°C. Digests were quenched with 0.1% TFA and subjected to C18 stage-tip clean up with a 50% acetonitrile +0.1% TFA elution step. After C18 clean up, peptides were lyophilized and reconstituted in 50 μl 0.1% TFA, where 1 μl was injected onto a NanoAcquity UPLC system (Waters Corp.) via an autosampler. Injected samples were separated on a 45°C heated Waters Acquity M-Class BEH C18 column (75 µm × 100 mm, 1.7 μm resin) at a flow rate of 1 µL/min. A gradient from 98% solvent A (water +0.1% formic acid) to 80% solvent B (acetonitrile +0.08% formic acid) was applied over 40 min. Samples were analyzed on-line via nanospray ionization into a hybrid LTQ-Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Data were collected in data-dependent mode with the parent ion being analyzed in the Fourier Transform Ion Cyclotron Resonance Mass Spectrometer and the top 15 most abundant ions being selected for fragmentation and analysis in the LTQ. Data were searched using Byonic™ software (Protein Metrics Inc.) with the following parameters: 20 ppm precursor mass tolerance with 0.5 Da fragment mass tolerance; fully specific digestion specificity with up to 1 missed cleavage; static carbamidomethylation on cysteine; variable oxidation on methionine; and variable N-glycosylation on N-linked motifs. Searched data from Byonic™ was imported and analyzed in Byologic® software (Protein Metrics Inc.). Peptide identification was confirmed by MS/MS fragmentation. Glycosylated peptides were label-free quantified relative to its unmodified and modified forms by AUC integration of their extracted ion chromatograms in Byologic®.
Characterization of complement activity
Complement activity was assessed essentially as previously described.48 Briefly, 3-fold serial dilutions of IgMs were added to 50,000 WIL2S cells in the presence of a 1/5 human complement dilution (final volume of 100 µL) and the mixture was incubated for 2 h at 37°C. Alamar Blue (Aldrich) 50 µL was added to the plates, and they were incubated for an additional 5 h at 37°C. The plates were cooled to RT for 10 min, and fluorescence was read using a 96-well fluorometer with excitation at 530 nm and emission at 590 nm.
Characterization of pAKT induction downstream of Tie-2 agonism
Rat Aortic Endothelial Cells (RAECs) were purchased from VEC Technologies and cultured in growth medium (VEC Technologies, Inc., CAT# MCDB-131 10). RAECs were seeded at a density of 12,000 cells/well in 96-well cell culture plate and cultured in 100 µl EGM2 MV medium overnight at 37°C with 5% CO2. After overnight culture, cells were starved in EBM2 basal medium with 0.1% BSA for 3 h prior to incubation with the Tie-2 agonists. For evaluation of the activity of Tie-2 agonists, the molecules were diluted to 100 nM in media followed by 3-fold serial dilution. These dilutions (50 µL) were added to each well after removal of serum starvation media, and the plates were incubated at 37°C, 5% CO2, for 15 min. The solution was removed and 50 µL of lysis buffer containing blocking buffer from pAKT Ser473 Kit (Cisbio) was added to cells. The plates were incubated at RT for 45 min with gentle shaking and kept in −80°C freezer until used. Cell lysate (15 µL) was thawed on ice then mixed with 5 µL of 1:40 dilution of each Phospho-AKT d2 antibody and Phospho-AKT Cryptate antibody from pAKT Ser473 Kit in 384-well microplate (Greiner Bio-One North America, Inc) and the plates were incubated at RT for 4 h or at 4°C overnight and read at 620 nm and 665 nm on CLARIOstar (BMG LABTECH, software version:5.01 R2). The data were calculated as the ratio of the acceptor and donor emission signals times 104 for each individual well.
Characterization of cell death and caspase-8 activity downstream of death receptor agonism
COLO 205 cell death assays were performed as previously reported.20 COLO 205 cells were maintained in RPMI medium supplemented with L-glutamine (L-glut) and 10% heat-inactivated fetal bovine serum (Rockland Immunochemicals Inc., Limerick, PA, USA) under 5% CO2 at 37°C. COLO 205 cells were plated at 10,000 cells per well of a 96-well white-walled plate (Corning) in 50 µL of growth media and allowed to adhere overnight. Dilutions of antibodies were prepared in growth media, and 50 µL of each antibody sample was added to each well. For caspase-8 activity, cells were incubated with antibody for 4 h prior to the addition of Caspase-Glo 8 (Promega). For cell viability, cells were incubated with antibody for 24 h prior to the addition of CellTiter-Glo (Promega). Experiments were done in triplicate and luminescence was read using Envision (PerkinElmer). Apo2L-FLAG was obtained from internal repository and crosslinked with anti-FLAG M2 antibody (Sigma, F3165). For extrinsic crosslinking of IgG, polyclonal goat anti-human Fc fragment specific F(ab’)2 (Jackson Immunochemicals, 109–006-008) was used at a 1:1 molar ratio.
Supplementary Material
Acknowledgments
The authors would like to thank the members of the Genentech Ophthalmology Platform Team and Christoph Spiess for their insight and critical perspective. Additionally, we would like to thank Daniela Yadav, the Antibody Production and Automation Technologies Group, and the Research Materials Group for key technical support.
Abbreviations
- AMD
age-related macular degeneration
- Ang
angiopoetin
- AUC
area under the curve
- BSA
bovine serum albumin
- CHO
Chinese hamster ovary
- CV
column volumes
- CDC
complement-dependent cytotoxicity
- CTP
C-terminal tail piece
- DME
diabetic macular edema
- DR4
death receptor 4
- DTT
dithiothreitol
- Fab
antigen-binding fragment
- FcγR
Fc-gamma receptor
- FcRn
neonatal Fc receptor
- Fv
variable fragment
- HC
heavy chain
- IgG
immunoglobulin G
- IgM
immunoglobulin M
- JC
J-chain
- LC
light chain
- pIgR
polymeric-Ig receptor
- MALS
multiangle light scattering
- PBS
phosphate-buffered saline
- QELS
quasi-eleastic light scattering
- RP
reversed phase
- Rg
radius of gyration
- Rh
hydrodynamic radius
- RT
room temperature
- SEC
size-exclusion chromatography
- TEM
transmission electron microscopy
- TNF
tumor necrosis factor
- TNF
related apoptosis inducing ligand/Apo2 ligand, TRAIL/Apo2L
- VEGF
vascular endothelial growth factor
- wAMD
wet AMD
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Congdon N, O’Colmain B, Klaver CCW, Klein R, Muñoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P, Eye Diseases Prevalence Research Group . Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–13. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, Gibson A, Sy J, Rundle AC, Hopkins JJ, et al. Ranibizumab for diabetic macular edema. Ophthalmology. 2012;119:789–801. doi: 10.1016/j.ophtha.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 3.Brown DM, Nguyen QD, Marcus DM, Boyer DS, Patel S, Feiner L, Schlottmann PG, Rundle AC, Zhang J, Rubio RG, et al. Long-term outcomes of ranibizumab therapy for diabetic macular edema: the 36-month results from two phase III trials. Ophthalmology. 2013;120:2013–22. doi: 10.1016/j.ophtha.2013.02.034. [DOI] [PubMed] [Google Scholar]
- 4.Korobelnik J-F, Do DV, Schmidt-Erfurth U, Boyer DS, Holz FG, Heier JS, Midena E, Kaiser PK, Terasaki H, Marcus DM, et al. Intravitreal aflibercept for diabetic macular edema. Ophthalmology. 2014;121:2247–54. doi: 10.1016/j.ophtha.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Chong V. Ranibizumab for the treatment of wet AMD: a summary of real-world studies. Eye (Lond). 2016;30:270–86. doi: 10.1038/eye.2015.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.National Academies of Sciences, Engineering, and Medicine, Health and Medicine Division, Board on Population Health and Public Health Practice, Committee on Public Health Approaches to Reduce Vision Impairment and Promote Eye Health. Welp A, Woodbury RB, McCoy MA, Teutsch SM. Making eye health a population health imperative: vision for tomorrow. Washington (DC): National Academies Press (US); 2016. [PubMed] [Google Scholar]
- 7.Murthy KR, Goel R, Subbannayya Y, Jacob HK, Murthy PR, Manda SS, Patil AH, Sharma R, Sahasrabuddhe NA, Parashar A, et al. Proteomic analysis of human vitreous humor. Clin. Proteom. 2014;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shatz W, Hass PE, Mathieu M, Kim HS, Leach K, Zhou M, Crawford Y, Shen A, Wang K, Chang DP, et al. Contribution of antibody hydrodynamic size to vitreal clearance revealed through rabbit studies using a species-matched Fab. Mol Pharmaceutics. 2016;13:2996–3003. doi: 10.1021/acs.molpharmaceut.6b00345. [DOI] [PubMed] [Google Scholar]
- 9.Shatz W, Hass PE, Peer N, Paluch MT, Blanchette C, Han G, Sandoval W, Morando A, Loyet KM, Bantseev V, et al. Identification and characterization of an octameric PEG-protein conjugate system for intravitreal long-acting delivery to the back of the eye. PLoS ONE. 2019;14:e0218613–20. doi: 10.1371/journal.pone.0218613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brewer JW, Randall TD, Parkhouse RM, Corley RB. Mechanism and subcellular localization of secretory IgM polymer assembly. J Bio Chem. 1994;269:17338–48. [PubMed] [Google Scholar]
- 11.Petrušić V, Živković I, Stojanović M, Stojićević I, Marinković E, Dimitrijević L. Hexameric immunoglobulin M in humans: desired or unwanted? Med Hypotheses. 2011;77:959–61. doi: 10.1016/j.mehy.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 12.Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J, Critical A. Role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. 1998;188:2381–86. doi: 10.1084/jem.188.12.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boes M. Role of natural and immune IgM antibodies in immune responses. Mol Immunol. 2000;37:1141–49. doi: 10.1016/S0161-5890(01)00025-6. [DOI] [PubMed] [Google Scholar]
- 14.Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6:576–88. doi: 10.1002/psp4.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–90. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 16.Greenman RL, Schein RM, Martin MA, Wenzel RP, MacIntyre NR, Emmanuel G, Chmel H, Kohler RB, McCarthy M, Plouffe J. A controlled clinical trial of E5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. The XOMA sepsis study group. JAMA. 1991;266:1097–102. doi: 10.1001/jama.1991.03470080067031. [DOI] [PubMed] [Google Scholar]
- 17.Beutner U, Lorenz U, Illert B, Rott L, Timmermann W, Vollmers HP, Müller-Hermelink HK, Thiede A, Ulrichs K. Neoadjuvant therapy of gastric cancer with the human monoclonal IgM antibody SC-1: impact on the immune system. Oncol Rep. 2008;19:761–69. [PubMed] [Google Scholar]
- 18.Rasche L, Duell J, Castro IC, Dubljevic V, Chatterjee M, Knop S, Hensel F, Rosenwald A, Einsele H, Topp MS, et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma - results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica. 2015;100:377–84. doi: 10.3324/haematol.2014.117945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marks L. The birth pangs of monoclonal antibody therapeutics: the failure and legacy of Centoxin. mAbs. 2012;4:403–12. doi: 10.4161/mabs.19909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Yeh SH, Madireddi S, Matochko WL, Gu C, Sanchez PP, Ultsch M, de Leon Boenig G, Harris SF, Leonard B, et al. Tetravalent biepitopic targeting enables intrinsic antibody agonism of tumor necrosis factor receptor superfamily members. mAbs. 2019;11:996–1011. doi: 10.1080/19420862.2019.1625662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overdijk MB, Strumane K, Ortiz Buijsse A, Vermot-Desroches C, Kroesm T, De Jong B, Hoevenaars N, Beurskens FJ, de Jong RN, Lingnau A, et al. DR5 agonist activity of HexaBody-DR5/DR5 (GEN1029) is potentiated by C1q and independent of Fc-gamma receptor binding in preclinical tumor models. Canc Res. 2019;79(Supplement). [Google Scholar]
- 22.Arakawa S, Takahashi A, Ashikawa K, Hosono N, Aoi T, Yasuda M, Oshima Y, Yoshida S, Enaida H, Tsuchihashi T, et al. Genome-wide association study identifies two susceptibility loci for exudative age-related macular degeneration in the Japanese population. Nat Genet. 2011;43:1001–04. doi: 10.1038/ng.938. [DOI] [PubMed] [Google Scholar]
- 23.Sahni J, Patel SS, Dugel PU, Khanani AM, Jhaveri CD, Wykoff CC, Hershberger VS, Pauly-Evers M, Sadikhov S, Szczesny P, et al. Simultaneous inhibition of angiopoietin-2 and vascular endothelial growth factor-A with faricimab in diabetic macular edema: BOULEVARD phase 2 randomized trial. Ophthalmology. 2019;126:1155–70. doi: 10.1016/j.ophtha.2019.03.023. [DOI] [PubMed] [Google Scholar]
- 24.Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–13. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 25.Kim K-T, Choi -H-H, Steinmetz MO, Maco B, Kammerer RA, Ahn SY, Kim H-Z, Lee GM, Koh GY. Oligomerization and multimerization are critical for angiopoietin-1 to bind and phosphorylate tie2. J Biol Chem. 2005;280:20126–31. doi: 10.1074/jbc.M500292200. [DOI] [PubMed] [Google Scholar]
- 26.Klaassen I, Van Noorden CJF, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res. 2013;34:19–48. doi: 10.1016/j.preteyeres.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Ho AC. Combination therapy with intravitreal nesvacumab and aflibercept for neovascular age-related macular degeneration. Paper Presented at: Angiogenesis, Exudation and Degeneration, Miami (FL), February 10, 2018. [Google Scholar]
- 28.David S, Ghosh CC, Kümpers P, Shushakova N, Van Slyke P, Khankin EV, Karumanchi SA, Dumont D, Parikh SM. Effects of a synthetic PEG-ylated Tie-2 agonist peptide on endotoxemic lung injury and mortality. Am J Physiol-Lung Cell Mol Physiol. 2011;300:L851–62. doi: 10.1152/ajplung.00459.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose GE, Billington BM, Chignell AH. Immunoglobulins in paired specimens of vitreous and subretinal fluids from patients with rhegmatogenous retinal detachment. Br J Ophthalmol. 1990;74:160–62. doi: 10.1136/bjo.74.3.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Wiesmann C, Fuh G, Li B, Christinger HW, McKay P, de Vos AM, Lowman HB. Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J Mol Biol. 1999;293:865–81. doi: 10.1006/jmbi.1999.3192. [DOI] [PubMed] [Google Scholar]
- 31.Regula JT, Lundh von Leithner P, Foxton R, Barathi VA, Cheung CMG, Bo Tun SB, Wey YS, Iwata D, Dostalek M, Moelleken J, et al. Targeting key angiogenic pathways with a bispecific cross MAb optimized for neovascular eye diseases. EMBO Mol Med. 2016;8:1265–88. doi: 10.15252/emmm.201505889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krohne TU, Liu Z, Holz FG, Meyer CH. Intraocular pharmacokinetics of ranibizumab following a single intravitreal injection in humans. Am J Ophthalmol. 2012;154:682–2. doi: 10.1016/j.ajo.2012.03.047. [DOI] [PubMed] [Google Scholar]
- 33.Urtti A. Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv Drug Deliv Rev. 2006;58:1131–35. doi: 10.1016/j.addr.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 34.Scheres SHW. Semi-automated selection of cryo-EM particles in RELION-1.3. J Struct Biol. 2015;189:114–22. doi: 10.1016/j.jsb.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czajkowsky DM, Shao Z. The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. Proc Natl Acad Sci USA. 2009;106:14960–65. doi: 10.1073/pnas.0903805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Wang G, Li N, Wang Y, Zhu Q, Chu H, Wu W, Tan Y, Su X-D, Gao N, et al. Structural insights into immunoglobulin M. Science. 2020;367:1014–17. doi: 10.1126/science.aaz5425. [DOI] [PubMed] [Google Scholar]
- 37.Mandal A, Pal D, Agrahari V, Trinh HM, Joseph M, Mitra AK. Ocular delivery of proteins and peptides: challenges and novel formulation approaches. Adv Drug Deliv Rev. 2018;126:67–95. doi: 10.1016/j.addr.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav S, Liu J, Scherer TM, Gokarn Y, Demeule B, Kanai S, Andya JD, Shire SJ. Assessment and significance of protein–protein interactions during development of protein biopharmaceuticals. Biophys Rev. 2013;5:121–36. doi: 10.1007/s12551-013-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crowell SR, Wang K, Famili A, Shatz W, Loyet KM, Chang V, Liu Y, Prabhu S, Kamath AV, Kelley RF. Influence of charge, hydrophobicity, and size on vitreous pharmacokinetics of large molecules. Trans Vis Sci Tech. 2019;8:1–9. doi: 10.1167/tvst.8.6.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maiorella BL, Winkelhake J, Young J, Moyer B, Bauer R, Hora M, Andya J, Thomson J, Patel T, Parekh R. Effect of culture conditions on IgM antibody structure, pharmacokinetics and activity. Biotechnology (NY). 1993;11:387–92. doi: 10.1038/nbt0393-387. [DOI] [PubMed] [Google Scholar]
- 41.Challacombe SJ, Russell MW. Estimation of the intravascular half-lives of normal rhesus minkey IgG, IgA, and IgM. Immunology. 1979;36:331–38. [PMC free article] [PubMed] [Google Scholar]
- 42.Ditzel H, Rasmussen JW, Erb K, Jensenius JC. Tumor detection with 131I-labeledHuman monoclonal antibody COU-1 in patients with suspected colorectal carcinoma1. Cancer Res. 1993;53:5920–28. [PubMed] [Google Scholar]
- 43.Irie RF, Ollila DW, O’Day S, Morton DL . Phase I pilot clinical trial of human IgM monoclonal antibody to ganglioside GM3 in patients with metastatic melanoma. Cancer Immunol Immunother. 2004;53:110–17. doi: 10.1007/s00262-003-0436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moh ESX, Lin C-H, Thaysen-Andersen M, Packer NH. Site-specific N-glycosylation of recombinant pentameric and hexameric human IgM. J Am Soc Mass Spectr. 2016;27:1143–55. doi: 10.1007/s13361-016-1378-0. [DOI] [PubMed] [Google Scholar]
- 45.Shulman MJ, Pennell N, Collins C, Hozumi N. Activation of complement by immunoglobulin M is impaired by the substitution serine-406 asparagine in the immunoglobulin mu heavy chain. Proc Natl Acad Sci USA. 1986;83:7678–82. doi: 10.1073/pnas.83.20.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wright JF, Shulman MJ, Isenman DE, Painter RH. C1 binding by mouse IgM. J Biol Chem. 1990;265:10506–13. [PubMed] [Google Scholar]
- 47.van Lookeren Campagne M, Strauss EC, Yaspan BL. Age-related macular degeneration: complement in action. Immunobiology. 2016;221:733–39. doi: 10.1016/j.imbio.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 48.Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47:115–23. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gazzano-Santoro H, Ralph P, Ryskamp TC, Chen AB, Mukku VR. A non-radioactive complement-dependent cytotoxicity assay for anti-CD20 monoclonal antibody. J Immunol Methods. 1997;202:163–71. doi: 10.1016/S0022-1759(97)00002-1. [DOI] [PubMed] [Google Scholar]
- 50.Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, Mulkerrin MG. Engineered antibodies with increased activity to recruit complement. J Immunol. 2001;166:2571–75. doi: 10.4049/jimmunol.166.4.2571. [DOI] [PubMed] [Google Scholar]
- 51.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes J-M, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJY, et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Bio Chem. 2017;292:3900–08. doi: 10.1074/jbc.M116.767749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright JF, Shulman MJ, Isenman DE, Painter RH. C1 binding by murine IgM. J Biol Chem. 1988;263:11221–26. [PubMed] [Google Scholar]
- 53.Hackett SF, Wiegand S, Yancopoulos G, Campochiaro PA. Angiopoietin-2 plays an important role in retinal angiogenesis. J Cell Physiol. 2002;192:182–87. doi: 10.1002/jcp.10128. [DOI] [PubMed] [Google Scholar]
- 54.Khalaf N, Helmy H, Labib H, Fahmy I, Abd El Hamid M, Moemen L. Role of angiopoietins and Tie-2 in diabetic retinopathy. Electron Physician. 2017;9:5031–35. doi: 10.19082/5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 56.Ramamurthy V, Yamniuk AP, Lawrence EJ, Yong W, Schneeweis LA, Cheng L, Murdock M, Corbett MJ, Doyle ML, Sheriff S. The structure of the death receptor 4-TNF-related apoptosis-inducing ligand (DR4-TRAIL) complex. Acta Cryst. 2015;F71:1273–81. doi: 10.1107/S2053230X15016416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chuntharapai A, Dodge K, Grimmer K, Schroeder K, Marsters SA, Koeppen H, Ashkenazi A, Kim KJ. Isotype-dependent inhibition of tumor growth in vivo by monoclonal antibodies to death receptor 4. J Immunol. 2001;166:4891–98. doi: 10.4049/jimmunol.166.8.4891. [DOI] [PubMed] [Google Scholar]
- 58.Adams C, Totpal K, Lawrence D, Marsters S, Pitti R, Yee S, Ross S, DeForge L, Koeppen H, Sagolla M, et al. Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ. 2008;15:751–61. doi: 10.1038/sj.cdd.4402306. [DOI] [PubMed] [Google Scholar]
- 59.Kelley RF, Totpal K, Lindstrom SH, Mathieu M, Billeci K, DeForge L, Pai R, Hymowitz SG, Ashkenazi A. Receptor-selective mutants of apoptosis-inducing ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J Bio Chem. 2005;280:2205–12. doi: 10.1074/jbc.M410660200. [DOI] [PubMed] [Google Scholar]
- 60.Dugel PU, Koh A, Ogura Y, Jaffe GJ, Schmidt-Erfurth U, Brown DM, Gomes AV, Warburton J, Weichselberger A, Holz FG. HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology. 2020;127:72–84. doi: 10.1016/j.ophtha.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 61.Nguyen QD, Das A, Do DV, Dugel P, Gomes A, Holz FG, Koh A, Pan C, Sepah YJ, Patel N, et al. Brolucizumab: evolution through preclinical and clinical studies and the implications for the management of neovascular age-related macular degeneration. Ophthalmology. 2020;127:1–14. [DOI] [PubMed] [Google Scholar]
- 62.Do DV. Extended durability in exudative retinal diseases using the novel intravitreal anti-VEGF antibody biopolymer conjugate KSI-301. Paper presented at: Angiogenesis, Exudation and Degeneration, Miami (FL), February 8, 2020. [Google Scholar]
- 63.Campochiaro PA, Marcus DM, Awh CC, Regillo C, Adamis AP, Bantseev V, Chiang Y, Ehrlich JS, Erickson S, Hanley WD, et al. The port delivery system with ranibizumab for neovascular age-related macular degeneration. Ophthalmology. 2019;126:1141–54. doi: 10.1016/j.ophtha.2019.03.036. [DOI] [PubMed] [Google Scholar]
- 64.Rau H, Sprogoe K. Transcon technology for sustained delivery of proteins, peptides, and small molecules. Paper presented at: Boulder Peptide Symposium, Denver (CO), September 26, 2018. [Google Scholar]
- 65.MacKenzie MR, Lee TK. Blood viscosity in Waldenstrom macroglobulinemia. Blood. 1977;49:507–10. doi: 10.1182/blood.V49.4.507.507. [DOI] [PubMed] [Google Scholar]
- 66.Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc National Acad Sci. 1969;63:78–85. doi: 10.1073/pnas.63.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eaton DL, Wood WI, Eaton D, Hass PE, Hollingshead P, Wion K, Mather J, Lawn RM, Vehar GA, Gorman C. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry. 1986;25:8343–47. doi: 10.1021/bi00374a001. [DOI] [PubMed] [Google Scholar]
- 68.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 69.Zarzar J, Shatz W, Peer N, Taing R, McGarry B, Liu Y, Greene DG, Zarraga IE. Impact of polymer geometry on the interactions of protein-PEG conjugates. Biophys Chem. 2018;236:22–30. doi: 10.1016/j.bpc.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 70.Dickmann LJ, Yip V, Li C, Abundes J Jr, Maia M, Young C, Stainton S, Hass PE, Joseph SB, Prabhu S, et al. Evaluation of fluorophotometry to assess the vitreal pharmacokinetics of protein therapeutics. Invest Ophthalmol Vis Sci. 2015;56:6991–99. doi: 10.1167/iovs.15-17457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.