

Abstract
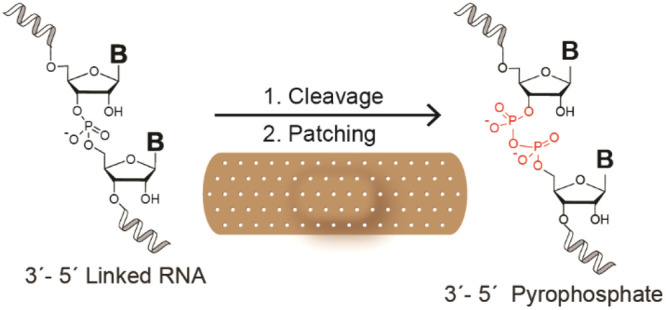
Achieving multiple cycles of RNA replication within a model protocell would be a critical step toward demonstrating a path from prebiotic chemistry to cellular biology. Any model for early life based on an “RNA world” must account for RNA strand cleavage and hydrolysis, which would degrade primitive genetic information and lead to an accumulation of truncated, phosphate-terminated strands. We show here that cleavage of the phosphodiester backbone is not an end point for RNA replication. Instead, 3′-phosphate-terminated RNA strands can participate in template-directed copying reactions with activated ribonucleotide monomers. These reactions form a pyrophosphate linkage, the stability of which we have characterized in the context of RNA copying chemistry. The presence of free magnesium cations results in cleavage of the pyrophosphate bond within minutes. However, we found that the pyrophosphate bond is relatively stable within an RNA duplex and in the presence of chelated magnesium. We show that, under these conditions, pyrophosphate-linked RNA can act as a template for the polymerization of ribonucleotides into canonical 3′–5′ phosphodiester-linked RNA. We suggest that primer extension of 3′-phosphate-terminated RNA followed by template-directed copying represents a plausible nonenzymatic pathway for the salvage and recovery of genetic information following strand cleavage.
Introduction
The RNA world hypothesis proposes an early stage in the evolution of life in which RNA was responsible for both catalysis and genetic inheritance.1−4 In existing biology, ribonucleotide monomers are joined to form polynucleotides in a template-directed process by the enzyme-catalyzed reaction of 3′-hydroxyl groups with nucleoside 5′-triphosphates, forming a 3′–5′ phosphodiester-linked backbone. Before the evolution of protein enzymes, a replicase made of RNA may have catalyzed RNA synthesis, a possibility explored by the in vitro selection and evolution of ribozymes with ligase5 and polymerase6−8 activity. A more challenging problem is how these ribozymes could have emerged from a nonenzymatic process of chemical RNA replication. Chemical RNA replication starts with the binding of energy-rich activated ribonucleotides to an RNA template through Watson–Crick base pairing. The ribonucleotide monomers react with each other to form phosphodiester bonds, leading to a double-stranded RNA duplex; subsequent amplification requires separation of the strands, so that they can act as templates for further cycles of copying. Mechanistic studies which revealed the critical role of 5′–5′ imidazolium-bridged dinucleotides as the active species in primer extension,9 and the discovery of 2-aminoimidazole as a superior 5′-phosphate activating group,10,11 have recently enabled the copying of short mixed sequence RNA templates within vesicle models of early cells.12 Despite this progress, the chemical copying of RNA is not currently able to produce RNA products of the length and complexity necessary to sustain ribozyme evolution.13,14
Any model proposing RNA or RNA-like polymers as early carriers of genetic information must also account for cleavage of the phosphodiester backbone,15 which competes with copying chemistry. The cleavage of RNA strands results in a mixture of 2′ and 3′ phosphate-terminated strands via an initial transesterification reaction followed by hydrolysis of a cyclic phosphate intermediate (Figure 1a). Cleavage of RNA strands is greatly accelerated by divalent metal cations, such as magnesium, that are also required for the copying chemistry to proceed at a reasonable rate. This degradation pathway constrains plausible rates of nonenzymatic RNA synthesis and would result in a net loss of material and thus information from a primordial genetic system if there were no mechanism for recycling of the phosphate-terminated strands.15 2′,3′-Cyclic phosphate-terminated strands can participate in templated ligation reactions,16,17 albeit with slow rates and poor regiospecificity, largely yielding 2′–5′-linked products. The Sutherland lab18 has demonstrated a prebiotically plausible cycle of backbone repair that converts 2′–5′ to 3′–5′ phosphodiester linkages via iterative cycles of strand cleavage and repair in which a 3′ phosphate plays a central role as a substrate for ligation chemistry. Identification of complementary pathways for RNA salvage or repair following strand cleavage18,19 or the discovery of additional functions for phosphate-terminated strands20 could further strengthen the case for RNA as the earliest carrier of genetic information.
Figure 1.
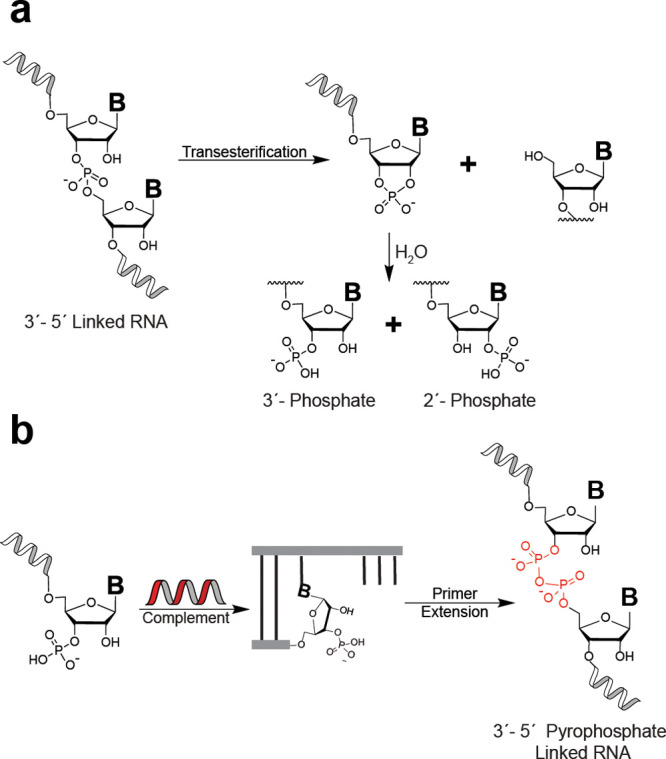
RNA strand cleavage and possible recovery of genetic information via pyrophosphate bond formation. (a) Cleavage of 3′–5′ phosphodiester-linked RNA leads to a mixture of 2′ and 3′ monophosphate-terminated strands via a cyclic phosphate intermediate. (b) Nonenzymatic primer extension starting from a 3′-monophosphate leads to 3′–5′ pyrophosphate-linked RNA, a possible mechanism for salvage of truncated RNA strands.
Consideration of potentially prebiotic nucleoside phosphorylation reactions provides further motivation for studying terminally phosphorylated RNA. Phosphorylation of nucleosides typically gives varying mixtures of 5′-, 3′-, and 2′-phosphorylated products as well as the 2′,3′-cyclic phosphate. Although regioselective 5′-phosphorylation has been demonstrated, specific conditions such as the addition of borate minerals21 or the use of gas-phase reactions are required.22 The diversity of pathways to nucleoside phosphorylation suggests that a mixture of phosphorylation states may have been likely at the monomer level. Heterogeneity of the phosphorylation state at the 2′ and 3′ position of nucleotide monomers could therefore be incorporated into RNA strands via templated copying or nontemplated reactions in addition to the hydrolytic pathway.
Considering this implied presence of terminal phosphates in any “RNA world” scenario and bearing in mind the greater nucleophilicity of phosphate relative to hydroxyl groups, we became interested in exploring whether phosphate-terminated RNA primers could participate in RNA copying chemistry. We hypothesized that the reaction of terminally phosphorylated primers with incoming nucleotides could lead to a mechanism for preservation of genetic information via formation of a pyrophosphate linkage (Figure 1b). Early work from Schwartz and Orgel demonstrated that 3′-phosphorylated, 2′-deoxy, imidazole-activated nucleotides efficiently polymerize on DNA templates to form 3′–5′ pyrophosphate-linked oligomers.23 In turn, the 3′–5′ pyrophosphate-linked oligomers could be used as templates for further synthesis, again employing activated 3′-phosphate, 2′-deoxy monomers, indicating that a pyrophosphate-linked backbone may not preclude cycles of nonenzymatic replication.24 Unfortunately, these early results were never extended to RNA-like systems.
Here, we report the results of our initial investigations into the behavior of phosphate-terminated RNA primers in nonenzymatic primer extension. We confirm that terminally phosphorylated RNA primers participate in primer extension reactions, leading to RNA polymers containing a pyrophosphate linkage. We have undertaken a thorough study of the stability of a single pyrophosphate linkage embedded within an RNA strand, which revealed pronounced lability toward cleavage reactions in the presence of magnesium ions. However, similarly to “native” RNA, the pyrophosphate linkage is protected from strand cleavage in the context of a duplex and in the presence of magnesium chelators. These observations enabled us to survey the kinetic parameters of primer extension from phosphate-terminated primers and following incorporation of a pyrophosphate bond. We further demonstrate that pyrophosphate-linked RNA can function as a template to direct the polymerization of canonical ribonucleotides, pointing toward a possible nonenzymatic salvage pathway for primordial RNA.
Results
We first synthesized RNA primers terminated with either a 2′ or a 3′ monophosphate using a solid-phase approach (SI Figure 1a). Using strong anion-exchange chromatography18 we confirmed the regioisomeric purity of the individual primers; no contamination with the alternative terminal phosphate was observed in either case (SI Figure 1b). We therefore tested the 2′ and 3′ phosphorylated primers in a primer-extension assay (Figure 2), employing a templating region with the sequence 3′-CCCA-5′, which can be extended efficiently by addition of activated guanosine and uridine ribonucleotide monomers. Citrate-chelated magnesium was used in these assays as it enables RNA copying chemistry to proceed within vesicles composed of fatty acids,25 which are the most likely candidates for protocell membranes but are destabilized by Mg2+ at low millimolar concentrations.26 The two phosphorylated primers and a nonphosphorylated control were annealed separately with the template, incubated with 50 mM Mg2+, 200 mM citrate, and a 10 mM concentration of both guanosine 2-aminophosphorimidazolide (2AImpG) and uridine-2-aminophosphorimidazolide (2AImpU), and monitored over the course of 24 h. All three primer–template duplexes were extended, although extension in the case of the 2′-phosphate-terminated primer was very poor. Surprisingly, the 3′ phosphate-terminated primer gave comparable extension to that obtained in the 3′-hydroxyl system with 81% of +3 products after 24 h (cf. 87% of +3 product for 3′-hydroxyl).
Figure 2.
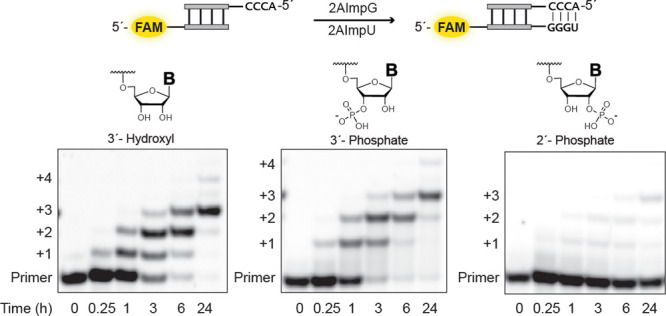
Efficient nonenzymatic copying of RNA commences from a primer with a terminal 3′-monophosphate. Time course of primer extension with guanosine 2-aminophosphorimidazolide (2AImpG) and uridine-2-aminophosphorimidazolide (2AImp-U) monomers was monitored using polyacrylamide gel electrophoresis (PAGE). All reactions were performed at pH 8.0, 200 mM HEPES, 50 mM Mg2+, and 200 mM citrate with 10 mM each of guanosine 2-aminophosphorimidazolide and uridine-2-aminophosphorimidazolide.
We hypothesized that extension of the phosphate-terminated primers could be due to formation of a pyrophosphate linkage by reaction of the 2′ or 3′ terminal phosphate with the activated 5′-phosphate of the incoming monomer. However, we aimed to exclude an alternate possibility in which the reaction proceeds through the nonphosphorylated hydroxyl, as chemical primer extension can proceed via either the 2′ or the 3′ hydroxyl group.27 To confirm the presence of a pyrophosphate linkage in the reaction products derived from the 3′-phosphorylated primer, we made use of the fact that RNase A is known to cleave pyrophosphate bonds28 while leaving 2′–5′ phosphodiester linkages intact27 (Figure 3a). The primer utilized was designed so that it is cleaved by RNase A at a single site, after the first bond-forming step of the primer extension reaction. Thus, for the 3′ terminal phosphate reaction we expected cleavage if the addition of the first monomer occurs through the 3′ phosphate and no cleavage if the reaction instead proceeded through the 2′ hydroxyl. Treating a primer extension reaction mixture with increasing concentrations of RNase A resulted in cleavage of the +1 to +3 bands and the appearance of a band with the same gel mobility as the original primer. As RNase A requires a free 2′-OH for cleavage and should lead to regeneration of the 3′- monophosphate, this result rules out formation of a 2′–5 phosphodiester bond during primer extension and strongly suggests the presence of a 3′–5′ pyrophosphate linkage.
Figure 3.
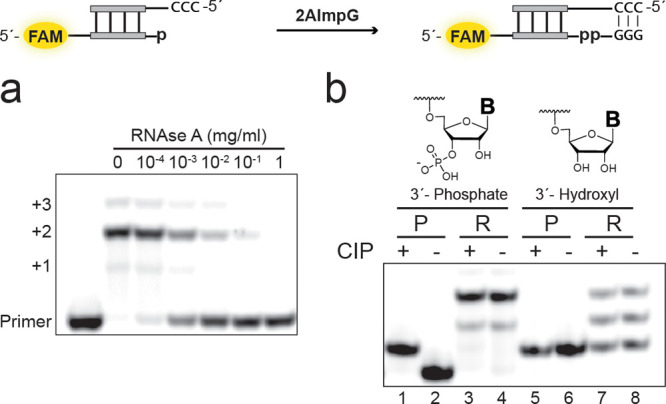
Confirmation of 3′–5′ pyrophosphate linkage by enzymatic digestion assays. (a) PAGE analysis of RNase treatment. RNase A treatment of a primer extension reaction commenced from a primer bearing a terminal 3′-monophosphate leads to regeneration of the primer. (b) PAGE analysis of calf intestinal phosphatase (CIP) treatment of primer extension reaction mixtures. “P” refers to primer, and “R” refers to reaction mixture derived from primer extension. Lanes 1 and 2: 3′-phosphorylated primer standard. Lanes 3 and 4: Reaction mixture from extension of the 3′-phosphorylated primer with (Lane 3) and without (Lane 4) CIP added. Lanes 5 and 6: Nonphosphorylated RNA primer. Lanes 7 and 8: Reaction mixture from extension of the same RNA primer.
If primer extension proceeded from the free terminal hydroxyl and not from the terminal phosphate group, a phosphate monoester internal to the RNA chain would remain in the +1 and higher extension products. To test this hypothesis, we treated the reaction mixtures with calf intestinal phosphatase (CIP), which has been shown to cleave internal phosphate monoesters29 (Figure 3b). For reaction mixtures derived from the 3′-phosphorylated primer, gel bands corresponding to extended products were not affected by CIP treatment. Taken together, these results obtained from enzymatic digestion support formation of a pyrophosphate linkage during primer extension initiated from a terminal phosphate and rule out the alternative possibility of a 2′–5′ phosphodiester-linked structure.
To further investigate the properties of pyrophosphate-linked RNA, we required a method to generate pure, single-stranded RNA containing site-specific pyrophosphate linkages for use both as primers and as templates in downstream studies. Notably, there exist no synthetic or enzymatic routes to produce RNA containing defined 3′–5′ pyrophosphate linkages. We thus devised a strategy that enabled us to generate pure (>95% by PAGE analysis) RNA strands containing a single pyrophosphate linkage (Figure 4). Our approach begins with a primer extension reaction using a 3′-phosphate-terminated primer and a mixed sequence template which allows us to control the number and identity of added nucleotides simply by adjusting the reaction times or adding/removing nucleotide phosphorimidazolides from the reaction mixture. Critically, we use a DNA–RNA hybrid template in which the region to be copied is RNA but the remainder of the template is DNA to allow for DNase digestion of the template which facilitates recovery of the modified primer. Incubation of the primer template duplexes with 2-aminoimidazole-activated monomers leads to robust conversion of the primer to extended products in which the +1 nucleotide is connected to the RNA primer by a pyrophosphate linkage. Following desalting to remove unreacted monomer, a 5′-phosphorylated ligator RNA oligonucleotide is annealed and ligated to the pyrophosphate-containing primer by T4 RNI2 ligase. The ligation step combined with design of the primer and template sequence allows for the generation of essentially any RNA sequence in the final product. After ligation, treatment of the reaction mixture with DNase followed by gel purification affords reasonable yields (typically 20–30%, based on known input of primer) of high-purity (>95% by PAGE analysis), single-stranded “pyrophosphate-containing RNA”. To produce ssRNA species containing a terminal pyrophosphate (Figure 4), a template containing only a single binding site for the activated G-G dinucleotide intermediate is employed using a 5′-CC-3′ templating region and the ligation step is omitted. The +1 primer extension product, bearing a terminal pyrophosphate linkage, can be isolated directly following DNase digestion. Although a recent report detailed the synthesis of pyrophosphate-linked DNA via a solid-phase synthesis approach,30 our method represents the first route to RNA containing defined pyrophosphate bonds.
Figure 4.
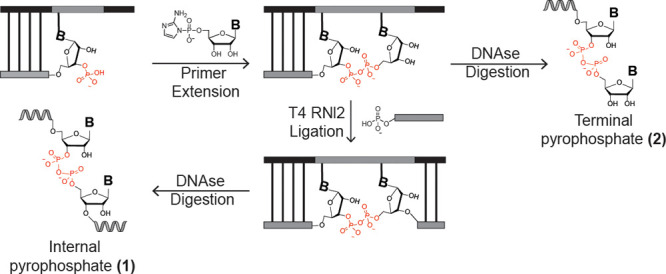
Synthetic strategy to access RNAs containing a single, defined pyrophosphate linkage. RNA primer containing a terminal 3′-phosphate monoester is annealed to a chimeric DNA/RNA template. Primer extension with activated guanosine monomers generates a 3′–5′ pyrophosphate linkage. Digestion of the DNA residues of the template with Turbo DNase generates a ssRNA containing a terminal pyrophosphate linkage. Alternatively, T4 RNA ligase 2 can be used to ligate an RNA oligonucleotide downstream in excellent yields (>95%). The same DNase treatment can then be used to generate ssRNA containing an internal pyrophosphate linkage. Gray: RNA. Black: DNA. See the Supporting Information for detailed experimental procedures.
We were interested in exploring the relative stability of the pyrophosphate linkage within RNA to determine whether it could support the chemical replication of genetic information and whether it could in principle enable ribozyme function. We prepared two single-stranded RNAs containing either an internal (AAGGGAAGAAGC-pp-GGGCUAGCAUGAC 1) or a terminal pyrophosphate linkage (AAGGGAAGAAGC-pp-G 2) using the strategies outlined above. The pyrophosphate–RNAs were then incubated at 22 °C in a pH 8.0 solution (conditions typical for primer extension reactions) in the presence or absence of magnesium, which was included as either the free cation or in citrate-chelated form (Table 1). We included citrate as it protects both RNA and fatty acid vesicles from magnesium-induced degradation while still allowing RNA copying reactions to proceed, albeit at a reduced rate.25 In the presence of 1 mM EDTA, which should chelate trace divalent cations, the pyrophosphate linkage was relatively stable with a half-life of 16 days. In contrast, incubation with free magnesium (50 mM) led to rapid cleavage of the pyrophosphate bond with a half-life measured to be on the order of minutes for both internal and terminal pyrophosphate linkages. Even at low millimolar concentrations of magnesium ions, the half-life of the pyrophosphate bond was only extended slightly (SI Table 1). The product of strand cleavage was determined by LC-MS (SI Figure 2a) and CIP digestion (SI Figure 2b) to be the 2′–3′-linked cyclic phosphate, consistent with a mechanism in which the α-phosphorus atom is subject to nucleophilic attack by the adjacent 2′-hydroxyl group. Incubating the pyrophosphate–RNA with magnesium citrate afforded modest protection, extending the half-life of the linkage from minutes to hours. Notably, the reduction in the rate of cleavage afforded by citrate chelation is much larger than the effect of chelation on the rate of the copying chemistry.25
Table 1. Hydrolytic Half-Life of a Single 3′–5′ Pyrophosphate Linkage within RNAa.
| ssRNA | dsRNA | stabilization | |
|---|---|---|---|
| internal pyrophosphate | |||
| Mg2+ | 0.12 ± 0.01 | 690 ± 20 | 5500 ± 200 |
| Mg–citrate | 3.7 ± 0.1 | ||
| citrate | 30 ± 1 | ||
| EDTA | 390 ± 20 | ||
| terminal pyrophosphate | |||
| Mg2+ | 0.20 ± 0.01 | 26 ± 1 | 130 ± 10 |
| Mg–citrate | 3.7 ± 0.2 | ||
The half-life values are presented with the standard error of the mean and reported in hours.
Duplex formation has been shown to protect 3′–5′-linked phosphodiester bonds in RNA from strand cleavage while promoting cleavage in the case of 2′–5′ bonds.31,32 This observation has been used to explain the ultimate “selection” of the canonical 3′–5′ linkage in extant RNA. We therefore examined the effect of duplex formation on the rate of cleavage of the pyrophosphate linkage to determine whether pyrophosphate bonds, once formed, could be retained in RNA duplex structures and ultimately serve as components of templates for further copying cycles. For the RNA 25mer 1 containing an internal pyrophosphate, addition of the complementary strand “buries” the pyrophosphate bond in an extensive duplex region. Incubating this duplex with free Mg2+ revealed a significant stabilization effect with the half-life of the pyrophosphate bond increased from minutes to 28 days: a stabilization factor of 5500. Protection in the case of the terminal pyrophosphate was far more modest, consistent with reduced helicity at the primer terminus.
Together, our degradation studies constrain the conditions under which the pyrophosphate linkage could support the storage and propagation of early genetic information. Pyrophosphate retention is possible within a duplex but is strongly disfavored in single-stranded RNA in the presence of Mg2+. The fate of a pyrophosphate bond at the terminal end of a duplex will depend on the relative rates of downstream extension or ligation reactions and strand cleavage and is thus expected to also depend on the RNA sequence (see Discussion below).
A critical factor determining the proportion of pyrophosphate-containing RNAs within a prebiotic population of polynucleotides would be the relative rates of reaction of phosphate and hydroxyl-terminated primers with activated nucleotides. We were therefore interested in comparing the kinetics of primer extension for the initial reaction step. We measured the rates of primer extension on a template designed to provide a single binding site for C*C dimer (5′–5′ aminoimidazolium-bridged cytidine dinucleotide), the reactive species in primer extension using 2-aminoimidazole activated cytidine ribonucleotides (Figure 5). Using varying concentrations of C*C, we obtained Michaelis–Menten parameters for reactions using primers terminated in either a hydroxyl or a monophosphate group at the 3′ position. We performed these experiments with magnesium–citrate, which protects against pyrophosphate degradation on the time-scales examined. Notably, we observed both higher vmax (14.1 vs 4.4 h–1 obtained for hydroxyl) and KM values (62.6 vs 8.4 mM for hydroxyl) for the phosphate-terminated primer (Figure 5). The observed binding defect could be due to either charge repulsion between the negatively charged phosphate and the dimer species or steric hindrance. The rate enhancement may be rationalized by consideration of the active nucleophile in each case. The actual nucleophilic species in primer extension with “native” RNA is likely a Mg-bound alkoxide, which is poorly populated at pH 8.13 The nucleophile for the 3′-phosphorylated primer is most likely a phosphate monoester dianion, which is more highly populated at pH 8.33 The difference in the population of these two nucleophiles under primer extension conditions may therefore explain the observed rate difference.
Figure 5.
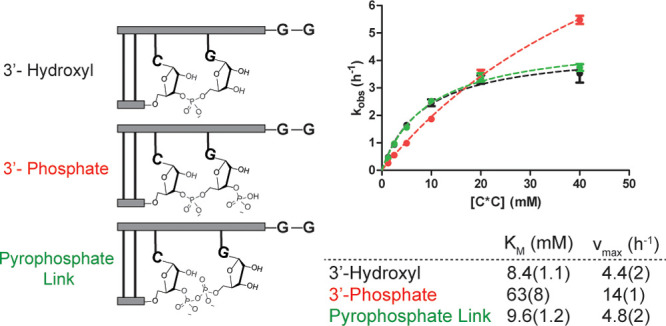
Michaelis–Menten analysis of nonenzymatic copying from 3′-hydroxyl- and 3′-monophosphate-terminated primers and for a 3′-hydroxylated terminus following a pyrophosphate bond. (Left) Schematic representation of the primer–template duplexes analyzed. (Right) Michaelis–Menten curves and obtained kinetic parameters. Plotted is kobs (h–1) against the concentration of C*C dimer. All reactions were performed at pH 8.0, 200 mM HEPES, 50 mM Mg2+, and 200 mM citrate.
Previous reports have demonstrated a stalling effect on primer extension following the incorporation of mismatched bases34 and certain noncanonical nucleotides.35 Our examination of the stability of the +1 extended product indicates that downstream extension is required to afford protection against cleavage of the newly formed pyrophosphate bond. We therefore sought to quantify the relative rates of reaction for primers in which the terminal 3′ nucleotide is joined by either a pyrophosphate or a phosphodiester bond (Figure 5). The kinetic parameters for incorporation of the next nucleotide were almost identical for the pyrophosphate- and phosphodiester-linked systems (vmax of 4.8 h–1 following a pyrophosphate linkage vs 4.4 h–1 obtained for phosphodiester linked). Importantly, the defect observed for binding of the imidazolium-bridged dimer intermediate C*C to the 3′ phosphate-terminated primer was almost completely ameliorated (KM 9.6 vs 8.4 mM for phosphodiester linked). This result implies that after the initial pyrophosphate linkage formation, the downstream steps in primer extension proceed essentially as if the primer contains only native RNA.
To better understand how RNA can accommodate the 3′–5′ pyrophosphate linkage during primer extension, we turned to crystallography. A 14mer self-complementary RNA strand containing a terminal 3′-monophosphate (5′-mCmCmCGACUUAAGUCGp-3′, italic: locked methylcytidine nucleotides, Figure 6a) was incubated with 2-AImpG under primer extension conditions (10 mM MgCl2, 10 mM Tris pH 8.0, 8 h). The extension product was crystallized and the complex structure determined to 1.6 Å resolution. The duplexes are slip stacked with one another in an end-to-end fashion, as observed in our previous structure of RNA–GMP complexes36 (Figure 6b). The crystal structure clearly indicates extension of the 3′-phosphorylated RNA by one guanosine nucleotide via pyrophosphate bond formation at both ends of the duplex (Figure 6c). The newly incorporated guanosine and the pyrophosphate linkage are well ordered. Canonical Watson–Crick base pairing is observed between the pyrophosphate-linked terminal 3′-guanosine and the templating methylcytidine and the guanosine sugar pucker is in the 3′-endo conformation. Both canonical base pairing and the 3′-endo conformation are favored for nonenzymatic primer extension, suggesting a structural rationale for the correspondence of reaction rates we observed for extension of phosphodiester-linked and pyrophosphate-linked RNA (Figure 5). To afford a more detailed comparison with phosphodiester-linked RNA, we superimposed the pyrophosphate–RNA structure over the native RNA–GMP structure (PDB 5L00) we obtained previously (Figure 6d). The two structures possess strikingly similar geometry, again suggesting that the pyrophosphate group does not cause significant structural perturbation to the reaction center. Due to the flexibility of the phosphodiester backbone and the accommodating capacity of the RNA major groove, the pyrophosphate moiety is accommodated with only a slight structural shift (Figure 6e).
Figure 6.

(a–e) Crystal structure of RNA containing a pyrophosphate linkage. (a) Schematic of the RNA–monomer complex used for crystallization. (Green) RNA/LNA duplex. Italics represent LNA. (Orange) Pyrophosphate linkage connecting the +1 G nucleobase and G monomer at +2 position. (b) Overall structure of the RNA–monomer complex. (c) Local structure of the pyrophosphate linkage and the extended guanosine, which forms a Watson–Crick pair with the template methylcytidine. 2Fo – Fc omit map (counter level of 1.0 σ) indicates the pyrophosphate linkage and guanosine are ordered. (d) Superimposed structures of the pyrophosphate-containing RNA and the native RNA–GMP complex (PDB 5L00). (Red) Pyrophosphate–RNA. (Black) Native RNA. (e) Local structural comparison of the pyrophosphate linkage and the phosphodiester linkage.
If pyrophosphate-linked RNA can act as a template for primer extension with canonical, 3′-hydroxyl-containing ribonucleotide monomers, a mechanism for re-enrichment of phosphodiester-linked RNA could operate over cycles of replication as the altered backbone structure will not be passed on to daughter strands. We were therefore curious whether a single 3′–5′ pyrophosphate linkage internal to an RNA template influences the ability of the template to direct monomer incorporation. For these experiments, we prepared 20mer ssRNA 3 (AUCGAAGGGppGGCAACACGAC), which contains a single pyrophosphate linkage as the template strand. The template was designed to contain a stretch of guanosine residues 5′-GppGGG-3′ such that we could directly compare the kinetic parameters of template copying across different systems using the same C*C dimer employed in our previous kinetic experiments. We examined three cases that differ only in the position of the primer strand relative to the pyrophosphate bond within the template (Figure 7). In the first case, the pyrophosphate bond joins the two nucleotides of the dimer binding site (Figure 7a). In the second case, the pyrophosphate bond is located after the primer annealing site, such that the expected distance between the primer 3′-hydroxyl and the 5′-phosphate of the incoming dimer is greater than in the case of phosphodiester-linked RNA (Figure 7b). Finally, in the third case examined the primer is “clamped” over the pyrophosphate linkage, rendering it internal to the primer–template duplex structure (Figure 7c). Using varying concentrations of C*C we evaluated initial rates of primer extension and determined Michaelis–Menten parameters for the three cases using hydroxyl-terminated primers and either the pyrophosphate-containing template or a fully RNA template of the same sequence.
Figure 7.

(a–c) Michaelis–Menten analysis of nonenzymatic copying across 3′–5′ pyrophosphate-linked template 3 compared with phosphodiester-linked control. (Top) Schematic representation of the primer–template duplexes analyzed, showing the binding site for the C*C dimer in each case. (Bottom) Michaelis–Menten curves and obtained kinetic parameters. Plotted is kobs (h–1) against the concentration of C*C dimer. All reactions were performed at pH 8.0, 200 mM HEPES, 50 mM Mg2+, and 200 mM citrate. Further experimental details and data analysis procedures can be found in the Supporting Information.
For all three situations, extension could be observed and rates determined for concentrations of imidazolium-bridged intermediate as low as 2 mM. This result indicates that pyrophosphate-linked RNA can indeed act as template for the incorporation of canonical ribonucleotide monomers (Figure 7). Binding of the imidazolium-bridged C*C dimer to the template was only strongly affected when the pyrophosphate linkage directly connects the two bases of the binding site (∼7-fold increase in KM observed, Figure 6a). However, in all three cases the maximum rate (vmax) values obtained were lower for the pyrophosphate-linked templates.
When comparing the three cases, the observed rates of copying across the pyrophosphate template were higher and the difference in the maximum rate between RNA and pyrophosphate templates was less pronounced when the primer “clamps” over the pyrophosphate linkage (Figure 6c). This may imply that conformational distortion in the ternary complex of primer:template:bound dimer due to the pyrophosphate bond becomes less significant as the linkage is buried within an RNA duplex.
Discussion
We found that RNA strand cleavage and hydrolysis is not a dead end for primitive, RNA-based genetic systems. Instead, the 3′-phosphorylated RNAs that result are able to participate in nonenzymatic primer extension when supplied with activated nucleotides. Upon reaction of phosphate-terminated primers with incoming nucleotides, a 3′–5′ pyrophosphate linkage is formed. By surveying the kinetic parameters for both the synthesis and degradation of this linkage, we have been able to place reasonable constraints on the likelihood of pyrophosphate-linked RNA playing a role in early systems of genetic inheritance.
The kinetic stability of the phosphodiester linkage is a critical feature enabling the storage and use of genetic information.37 In an RNA world dependent on ribozymes for catalysis, the rate of cleavage of the phosphodiester bond should also determine the lifetime and thus biosynthesis requirements of the functional machinery of the protocell.15 In the transition from prebiotic chemistry to the first genetic polymers, the phosphodiester linkage may have been “selected” from a range of alternative backbones that were feasible from the standpoint of chemical reactivity but were not stable enough or for other structural or kinetic reasons were unable to support the long-term storage and transmission of genetic information or the functioning of ribozymes. Using only canonical ribonucleotide monomers as precursors to primitive RNA oligonucleotides, four principal backbone linkages can be considered which are derived from either templated copying reactions or the cleavage pathways discussed in this manuscript: 3′–5′ and 2′–5′ phosphodiester linkages and the 3′–5′ and the 2′–5′ pyrophosphate linkages explored here. Other linkages, such as 5′–5′ pyrophosphate linkages,38,39 have been observed in nonenzymatic polymerization of activated nucleotides. These linkages can disrupt information transfer by interfering with the directionality of copying chemistry. Our initial primer extension results suggest that 2′–5′ pyrophosphate linkages were unlikely to play a significant role in early forms of RNA-based genetics, as initiation of copying chemistry from a 2′-phosphate is strongly disfavored. In contrast, copying from 3′-phosphate-terminated primers is robust and comparable to extension from 3′-hydroxyl groups. When considering degradation reactions, the rate of cleavage of a 3′–5′ pyrophosphate linkage within single-stranded RNA in the presence of magnesium ions is orders of magnitude greater than that of either a 2′–5′ or a 3′–5′ phosphodiester linkage.31,32 However, duplex formation protects 3′–5′ phosphodiester-linked bonds from cleavage by constraining RNA in a helical conformation that disfavors attack on the phosphodiester unit by the adjacent 2′-hydroxyl. We observed a similar effect for 3′–5′ pyrophosphate linkages, suggesting a similar mechanism of protection. In the case of 2′–5′ phosphodiester linkages, duplex formation has the opposite effect, promoting attack by the 3′-hydroxyl and leading to more rapid cleavage. We expect a similar effect to operate for 2′–5′-linked pyrophosphate bonds, although the inefficiency of primer extension from 2′ phosphates has prevented us from directly testing this hypothesis. The observation of enhanced stability for the 3′–5′ phosphodiester linkage in duplex structures has previously been used to support the idea of “selection” over time for the more stable 3′–5′ linkage in the context of genetic inheritance.31 Our results suggest that susceptibility to strand cleavage in the presence of magnesium ions, which are required for RNA folding and catalysis, could explain why biology does not currently employ 3′–5′ pyrophosphate linkages in the RNA backbone. However, before the emergence of enzymatic mechanisms for synthesis and proof-reading of genetic polymers, pyrophosphate linkages could have been tolerated as part of duplex structures and thus have contributed to the transmission of primitive genetic information.
We have shown that pyrophosphate linkages in template strands do not block template-directed primer extension. This result suggests pyrophosphate linkages may have been tolerated in primordial RNA-based genetics and raises the prospect of a nonenzymatic “salvage” and proof-reading cycle for cleaved RNA (Figure 8). Following a cleavage (which could take place in a single strand or within a duplex structure), a cyclic phosphate terminus results and is further hydrolyzed to a mixture of 2′ and 3′ terminal phosphates with the latter being slightly favored.40 If a 3′-phosphorylated RNA is bound by a template strand, reaction with activated monomers could “patch” the cleaved strand via formation of a pyrophosphate linkage. This would result in a duplex structure bearing a single pyrophosphate linkage, the lifetime of which depends on the relative rate of pyrophosphate cleavage compared to primer extension or ligation reactions. Cleaved pyrophosphate bonds can simply re-enter the cycle, while those sequences able to extend efficiently (or undergo rapid ligation reactions) will “bury” the labile pyrophosphate bond within a duplex structure, affording some protection from cleavage. 2′-Phosphorylated RNA will not form pyrophosphate linkages in appreciable quantities but can re-enter the cycle as a 2′,3′ cyclic phosphate upon activation and cyclization.18
Figure 8.

Nonenzymatic pathway for the recovery of genetic information via a pyrophosphate “patch”. (From top left) Following a cleavage event, a cyclic phosphate terminus is formed which hydrolyzes to a mixture of 3′ and 2′ monophosphate-terminated strands. Primer extension from a 3′-monophosphate terminus leads to duplex RNA containing a single 3′–5′ pyrophosphate linkage. Strand displacement and templated copying with canonical ribonucleotides leads to re-enrichment of “native”, 3′–5′ phosphodiester-linked RNA over multiple cycles of replication. Unreactive 2′ monophosphate-terminated strands can be reactivated to the cyclic phosphate and re-enter the cycle.
For regeneration of the original phosphodiester-linked RNA strand, strand separation and copying of the pyrophosphate-linked RNA (now acting as template) are required. If only canonical ribonucleotide monomers are available, we have demonstrated that copying over pyrophosphate linkages is feasible, albeit slower than for purely phosphodiester-linked RNA. Nonenzymatic separation of RNA strands and multiple cycles of copying (replication) have yet to be demonstrated experimentally.14,41 Assuming such a cycle, phosphodiester bond formation over a pyrophosphate template would re-enrich canonical RNA in a crude, nonenzymatic form of backbone proof-reading. The extreme susceptibility of pyrophosphate linkages to metal-ion-catalyzed cleavage in single strands renders this pathway unlikely under a model of nonenzymatic replication that requires temperature cycling, as this promotes a fully single-stranded state under conditions likely to promote cleavage.
However, if a strand displacement synthesis is possible, temperature cycling is not required and the salvage pathway may be feasible. Mariani et al.18 demonstrated a different cycle of backbone repair that converts 2′–5′ to 3′–5′ phosphodiester linkages via iterative cycles of strand cleavage, 2′-acetylation, and nonenzymatic RNA ligation.19 Such a cycle provides a pathway for the repair of genetic information that also relies on the formation of a 3′-phosphate as the critical intermediate. In conditions that promote the chemical activation of phosphate groups, ligation with a 5′-hydroxyl is thus a complementary pathway to that described in this manuscript. Under such activating conditions, if a 5′-phosphate is present on a bound ligator, pyrophosphate formation should also occur. Hydrolysis of a 3′–5′ pyrophosphate linkage produces such a 5′- phosphate on the downstream cleaved fragment, which could feed back into the system as an input for repeated ligation with a 3′-phosphate or ligation to an oligo bearing a 3′-hydroxyl. A network of interconnecting salvage pathways for RNA may therefore operate under nonenzymatic conditions, all of which depend on the unique reactivity of 3′-phosphates generated upon hydrolytic cleavage.
Acetylation of the vicinal 2′-hydroxyl group of a terminal 3′-phosphate before primer extension18,19 should render the resultant pyrophosphate linkage stable to strand cleavage as the 2′-hydroxyl would be incapable of transesterification. Such prebiotically plausible “protection” could greatly enhance the retention of pyrophosphate linkages within a population of RNAs and bolster the chances of repair by enhancing the kinetic stability of pyrophosphate-containing templates. As a proof of principle that protecting the 2′-hydroxyl would not inhibit primer extension from a terminal 3′-monophosphate while reducing the problem of strand cleavage, we synthesized a primer with a 2′-OMe, 3′-phosphorylated terminus and compared its activity with our original 2′-OH, 3′-phosphorylated primer in a primer extension assay with free magnesium cations (SI Figure 3). Over 18 h, both primers were extended up to +6 products with a slightly greater yield of +6 products observed for the 2′-O-methylated primer. Pleasingly, minimal remaining primer (5%) was observed for the 2′-O-methylated system during the course of the assay. In contrast, the reaction with a free 2′-hydroxyl yielded 22% of a product that comigrates with the original primer band, consistent with cleavage leading to a cyclic phosphate-terminated strand (evidence for the comigration of our 3′-phosphorylated primer and its derived cyclic phosphate is presented in SI Figure 2b). The fraction of remaining primer observed for the 2′-O-methylated system correlates with that observed in identical reactions conducted with nonphosphorylated RNA in which native phosphodiester bonds are formed that do not appreciably cleave on this time scale. This result suggests that prebiotically plausible 2′-OH protection could indeed facilitate the increased retention of pyrophosphate bonds in early RNA polymers and perhaps expand potential roles for such linkages in the RNA world. Work toward this end is currently underway in our laboratory.
This study examined primer extension using only canonical ribonucleotides reacting with phosphate groups introduced to the 3′ ends of primers by solid-phase synthesis. A remaining question is the efficiency of primer extension using 2′- or 3′-phosphorylated monomers. Schwartz and Orgel have shown that 3′-monophosphate, 2′-deoxy-modified monomers can polymerize on DNA templates to form 3′–5′ pyrophosphate-linked oligomers.23 Isolated pyrophosphate-linked oligonucleotides could also be used as templates for further polymerization, employing the same activated monomers.24 These results imply that a full cycle of nonenzymatic replication may be possible in a system composed of phosphorylated monomers and pyrophosphate-linked genetic polymers. Here, we examined the consequences of a single pyrophosphate linkage in the context of RNA copying. Primer extension using 3′-phosphorylated ribonucleotide monomers or in a system containing monomers with a mixture of phosphorylation states should introduce multiple pyrophosphate linkages. The implications of multiple pyrophosphate bonds for the function of RNA, as both genetic material and catalyst, remain to be explored.
Acknowledgments
J.W.S is an investigator of the Howard Hughes Medical Institute. We thank Dr. Li Li for helpful discussions. X-ray diffraction data were collected at the Advanced Light Source (ALS) SIBYLS beamlines 821 and 822, a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. This work was supported in part by a grant (290363FY18) from the Simons Foundation to J.W.S., a grant from the NSF (CHE-1607034) to J.W.S., and a grant from NASA (NNX15AL18G) to J.W.S. D.K.O. is a recipient of a Postdoctoral Research Scholarship (B3) from the Fonds de Recherche du Quebec–Nature et Technologies (FRQNT), Quebec, Canada, and a Postdoctoral Fellowship from Canadian Institutes of Health Research (CIHR) from Canada.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08237.
Methods and materials; synthesis and characterization of terminally phosphorylated primers; characterization of the product of magnesium-catalyzed cleavage of ssRNA 25mer 1 containing a single pyrophosphate linkage; 2′-OMe modification of a 3′-phosphorylated primer eliminates cleavage of the pyrophosphate linkage during primer extension; magnesium dependence of the hydrolytic half-life of 3′–5′ pyrophosphate-linked RNA; data collection statistics; structure refinement statistics; sequences of oligonucleotides used in this study; additional references (PDF)
Author Present Address
‡ Alnylam Pharmaceuticals, 300 Third Street, Cambridge, Massachusetts 02142, United States.
Author Contributions
† T.H.W. and C.G.: These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Crick F. H. C. The Origin of the Genetic Code. J. Mol. Biol. 1968, 38, 367–379. 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- Orgel L. E. Evolution of the Genetic Apparatus. J. Mol. Biol. 1968, 38 (3), 381–393. 10.1016/0022-2836(68)90393-8. [DOI] [PubMed] [Google Scholar]
- Gilbert W. Origin of Life: The RNA World. Nature 1986, 319 (6055), 618. 10.1038/319618a0. [DOI] [Google Scholar]
- Joyce G. F.; Szostak J. W. Protocells and RNA Self-Replication. Cold Spring Harbor Perspect. Biol. 2018, 10 (9), a034801. 10.1101/cshperspect.a034801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P.; Szostak J. W. Isolation of New Ribozymes from a Large Pool of Random Sequences. Science 1993, 261 (5127), 1411–1418. 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- Johnston W. K.; Unrau P. J.; Lawrence M. S.; Glasner M. E.; Bartel D. P. RNA-Catalyzed RNA Polymerization: Accurate and General RNA-Templated Primer Extension. Science 2001, 292 (5520), 1319–1325. 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- Horning D. P.; Joyce G. F. Amplification of RNA by an RNA Polymerase Ribozyme. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (35), 9786–9791. 10.1073/pnas.1610103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwater J.; Raguram A.; Morgunov A. S.; Gianni E.; Holliger P. Ribozyme-Catalysed RNA Synthesis Using Triplet Building Blocks. eLife 2018, 7, e35255 10.7554/eLife.35255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton T.; Szostak J. W. A Highly Reactive Imidazolium-Bridged Dinucleotide Intermediate in Nonenzymatic RNA Primer Extension. J. Am. Chem. Soc. 2016, 138 (36), 11996–12002. 10.1021/jacs.6b07977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenbach A. C.; Giurgiu C.; Tam C. P.; Li L.; Hongo Y.; Aono M.; Szostak J. W. Common and Potentially Prebiotic Origin for Precursors of Nucleotide Synthesis and Activation. J. Am. Chem. Soc. 2017, 139 (26), 8780–8783. 10.1021/jacs.7b01562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Prywes N.; Tam C. P.; O’Flaherty D. K.; Lelyveld V. S.; Izgu E. C.; Pal A.; Szostak J. W. Enhanced Nonenzymatic RNA Copying with 2-Aminoimidazole Activated Nucleotides. J. Am. Chem. Soc. 2017, 139 (5), 1810–1813. 10.1021/jacs.6b13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flaherty D. K.; Kamat N. P.; Mirza F. N.; Li L.; Prywes N.; Szostak J. W. Copying of Mixed-Sequence RNA Templates inside Model Protocells. J. Am. Chem. Soc. 2018, 140 (15), 5171–5178. 10.1021/jacs.8b00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgiu C.; Wright T. H.; O’Flaherty D. K.; Szostak J. W. A Fluorescent G-Quadruplex Sensor for Chemical RNA Copying. Angew. Chem., Int. Ed. 2018, 57 (31), 9844–9848. 10.1002/anie.201805785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak J. W. The Eightfold Path to Non-Enzymatic RNA Replication. J. Syst. Chem. 2012, 3 (1), 2. 10.1186/1759-2208-3-2. [DOI] [Google Scholar]
- Lazcano A.; Miller S. L. The Origin and Early Evolution of Life: Prebiotic Chemistry, the Pre-RNA World, and Time. Cell 1996, 85 (6), 793–798. 10.1016/S0092-8674(00)81263-5. [DOI] [PubMed] [Google Scholar]
- Usher D. A.; McHale A. H. Nonenzymic Joining of Oligoadenylates on a Polyuridylic Acid Template. Science 1976, 192, 53–54. 10.1126/science.1257755. [DOI] [PubMed] [Google Scholar]
- Lutay A. V.; Zenkova M. A.; Vlassov V. V. Nonenzymatic Recombination of RNA: Possible Mechanism for the Formation of Novel Sequences. Chem. Biodiversity 2007, 4, 762–767. 10.1002/cbdv.200790062. [DOI] [PubMed] [Google Scholar]
- Mariani A.; Sutherland J. D. Non-Enzymatic RNA Backbone Proofreading through Energy-Dissipative Recycling. Angew. Chem., Int. Ed. 2017, 56 (23), 6563–6566. 10.1002/anie.201703169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler F. R.; Chan C. K. W.; Duffy C. D.; Gerland B. B. B. B.; Islam S.; Powner M. W.; Sutherland J. D.; Xu J. Prebiotically Plausible Oligoribonucleotide Ligation Facilitated by Chemoselective Acetylation. Nat. Chem. 2013, 5 (5), 383–389. 10.1038/nchem.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron J.-P.; Parkes A. L.; Pascal R.; Sutherland J. D. Expeditious, Potentially Primordial, Aminoacylation of Nucleotides. Angew. Chem., Int. Ed. 2005, 44 (41), 6731–6734. 10.1002/anie.200501591. [DOI] [PubMed] [Google Scholar]
- Kim H.-J.; Furukawa Y.; Kakegawa T.; Bita A.; Scorei R.; Benner S. A. Evaporite Borate-Containing Mineral Ensembles Make Phosphate Available and Regiospecifically Phosphorylate Ribonucleosides: Borate as a Multifaceted Problem Solver in Prebiotic Chemistry. Angew. Chem., Int. Ed. 2016, 55, 15816. 10.1002/anie.201608001. [DOI] [PubMed] [Google Scholar]
- Hu J.; Lei W.; Wang J.; Chen H.-Y.; Xu J.-J. Regioselective 5′-Position Phosphorylation of Ribose and Ribonucleosides: Phosphate Transfer in the Activated Pyrophosphate Complex in the Gas Phase. Chem. Commun. 2019, 55 (3), 310–313. 10.1039/C8CC08510B. [DOI] [PubMed] [Google Scholar]
- Schwartz A. W.; Orgel L. E. Template-Directed Synthesis of Novel, Nucleic Acid-Like Structures. Science 1985, 228 (4699), 585–587. 10.1126/science.228.4699.585. [DOI] [PubMed] [Google Scholar]
- Visscher J.; Bakker C. G.; van der Woerd R.; Schwartz A. W. Template-Directed Oligomerization Catalyzed by a Polynucleotide Analog. Science 1989, 244 (4902), 329–331. 10.1126/science.2540527. [DOI] [PubMed] [Google Scholar]
- Adamala K.; Szostak J. W. Nonenzymatic Template-Directed RNA Synthesis Inside Model Protocells. Science 2013, 342, 1098–1100. 10.1126/science.1241888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. A.; Salehi-Ashtiani K.; Szostak J. W. RNA Catalysis in Model Protocell Vesicles. J. Am. Chem. Soc. 2005, 127 (38), 13213–13219. 10.1021/ja051784p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgiu C.; Li L.; O’Flaherty D. K.; Tam C. P.; Szostak J. W. A Mechanistic Explanation for the Regioselectivity of Nonenzymatic RNA Primer Extension. J. Am. Chem. Soc. 2017, 139 (46), 16741–16747. 10.1021/jacs.7b08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine A. M.; Leonidas D. D.; Jenkins J. L.; Park C.; Raines R. T.; Acharya K. R.; Shapiro R. Cleavage of 3′,5′-Pyrophosphate-Linked Dinucleotides by Ribonuclease A and Angiogenin. Biochemistry 2001, 40 (34), 10262–10272. 10.1021/bi010888j. [DOI] [PubMed] [Google Scholar]
- Konarska M.; Filipowicz W.; Domdey H.; Gross H. J. Formation of a 2′-Phosphomonoester, 3′,5′-Phosphodiester Linkage by a Novel RNA Ligase in Wheat Germ. Nature 1981, 293 (5828), 112–116. 10.1038/293112a0. [DOI] [PubMed] [Google Scholar]
- Anderson B. A.; Krishnamurthy R. Heterogeneous Pyrophosphate-Linked DNA–Oligonucleotides: Aversion to DNA but Affinity for RNA. Chem. - Eur. J. 2018, 24 (26), 6837–6842. 10.1002/chem.201800538. [DOI] [PubMed] [Google Scholar]
- Usher D. A.; McHale A. H. Hydrolytic Stability of Helical RNA: A Selective Advantage for the Natural 3′,5′-Bond. Proc. Natl. Acad. Sci. U. S. A. 1976, 73 (4), 1149–1153. 10.1073/pnas.73.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R.; Bartel D. P.; Szostak J. W. Nonenzymatic, Template-Directed Ligation of Oligoribonucleotides Is Highly Regioselective for the Formation of 3 ′ - 5 ′ Phosphodiester Bonds. J. Am. Chem. Soc. 1996, 118 (9), 3340–3344. 10.1021/ja9537134. [DOI] [PubMed] [Google Scholar]
- Phillips R.; Eisenberg P.; George P.; Rutman R. J. Thermodynamic Data for the Secondary Phosphate Ionizations of Adenosine, Guanosine, Inosine, Cytidine, and Uridine Nucleotides and Triphosphate. J. Biol. Chem. 1965, 240 (11), 4393–4397. [PubMed] [Google Scholar]
- Rajamani S.; Ichida J. K.; Antal T.; Treco D. A.; Leu K.; Nowak M. A.; Szostak J. W.; Chen I. A. Effect of Stalling after Mismatches on the Error Catastrophe in Nonenzymatic Nucleic Acid Replication. J. Am. Chem. Soc. 2010, 132 (16), 5880–5885. 10.1021/ja100780p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. C.; O’Flaherty D. K.; Zhou L.; Lelyveld V. S.; Szostak J. W. Inosine, but None of the 8-Oxo-Purines, Is a Plausible Component of a Primordial Version of RNA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (52), 13318–13323. 10.1073/pnas.1814367115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Tam C. P.; Wang J.; Szostak J. W. Unusual Base-Pairing Interactions in Monomer-Template Complexes. ACS Cent. Sci. 2016, 2, 916–926. 10.1021/acscentsci.6b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westheimer F. H. Why Nature Chose Phosphates. Science 1987, 235 (4793), 1173–1178. 10.1126/science.2434996. [DOI] [PubMed] [Google Scholar]
- Sulston J.; Lohrmann R.; Orgel L. E.; Miles H. T. Nonenzymatic Synthesis of Oligoadenylates on a Polyuridylic Acid Template. Proc. Natl. Acad. Sci. U. S. A. 1968, 59, 726–733. 10.1073/pnas.59.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez L.; Orgel L. E. Pyrophosphate Formation as the Most Efficient Condensation Reaction of Activated Nucleotides. J. Mol. Evol. 1991, 32, 101–104. 10.1007/BF02515382. [DOI] [PubMed] [Google Scholar]
- Oivanen M.; Kuusela S.; Lonnberg H. Kinetics and Mechanisms for the Cleavage and Isomerization of the Phosphodiester Bonds of RNA by Bronsted Acids and Bases. Chem. Rev. 1998, 98 (3), 961–990. 10.1021/cr960425x. [DOI] [PubMed] [Google Scholar]
- Mariani A.; Bonfio C.; Johnson C. M.; Sutherland J. D. PH-Driven RNA Strand Separation under Prebiotically Plausible Conditions. Biochemistry 2018, 57 (45), 6382–6386. 10.1021/acs.biochem.8b01080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.