

Abstract
Malignant gliomas are the most common primary central nervous system tumors and their prognosis is very poor. In recent years, ion channels have been demonstrated to play important roles in tumor pathophysiology such as regulation of gene expression, cell migration, and cell proliferation. In this review, we summarize the current knowledge on the role of ion channels on the development and progression of gliomas. Cell volume changes through the regulation of ion flux, accompanied by water flux, is essential for migration and invasion. Signaling pathways affected by ion channel activity play roles in cell survival and cell proliferation. Moreover, ion channels are involved in glioma-related seizures, sensitivity to chemotherapy, and tumor metabolism. Ion channels are potential targets for the treatment of these lethal tumors. Despite our increased understanding of the contributions of ion channels to glioma biology, this field remains poorly studied. This review summarizes the current literature on this important topic.
Keywords: Ion channels, glioma, invasion, proliferation, treatment
Introduction
Gliomas account for ~80% of malignant brain tumors. Gliomas are divided into four grades (I-IV) by the World Health Organization classification according to the relative malignancy. The high-grade (III-IV) gliomas include glioblastoma (GBM), anaplastic astrocytoma, and anaplastic oligodendroglioma(1). GBM is a WHO grade IV tumor and the most common and aggressive type of glioma in adults. The prognosis of glioblastoma is very poor with a median survival less than two years, with current standard treatments including maximum safe resection followed by chemoradiation and adjuvant temozolomide(2). One of the main challenges in the treatment of GBM is its highly invasive and rapidly proliferative character. Recently, the role of ion channels in glioma invasion and proliferation has been the focus of several articles(3-5). A better understanding of the involvement of ion channels in glioma biology will help uncover potential mechanisms of cell invasion, proliferation, apoptosis, and treatment resistance. In this article, we reviewed the current literature on ion channels and their involvement in the pathophysiology of gliomas. We searched PubMed for articles published after 2015, using the following keywords: “Ion channel glioma”, “Ion channel glioblastoma”, “Ion channel glioma proliferation”, “Ion channel glioma invasion”, “Ion channel glioma epilepsy”, and “Ion channel glioma seizure”. We limited our search to full-text English articles with available access from our institution and other relevant literature from the reference lists of the identified articles. We also discussed ion channels as potential targets in the personalized treatment of gliomas.
Ion Channels
Ion channels are membrane proteins that regulate the flux of ions such as sodium (Na+), potassium (K+), calcium (Ca2+), and chloride (Cl−) into and out of cells. Previous studies have demonstrated that ion channel activity is important for cell migration, proliferation, and regulation of gene expression in cancer cells(6). For example, cell volume changes during cell migration/invasion are regulated by the flux of Cl− and K+ together with water movement. According to the HUGO Gene Nomenclature Committee (HGNC), there are 328 ion channel genes in the human genome (https://www.genenames.org/data/genegroup/#!/group/177). These pore-forming proteins play an essential role in cellular physiology including maintenance of the resting membrane potential, regulation of cell volume, differentiation, proliferation, activation of signaling pathways, and apoptosis. However, alterations in ion channel function is still a poorly understood aspect of the biology of cancer cells.
Ion Channels and Glioma Invasion/Migration
During the migration of glioma cells along blood vessels and white matter tracts, cell volume changes are required to navigate narrow interstitial spaces. Cell migration involves protrusion in the migrating edge and retraction at the rear-end of the cell body, and these steps depend on the activity of multiple ion channels (Figure 1)(3,4). Progressive cell migration and invasion into surrounding normal brain parenchyma is one of the defining characteristics of malignant gliomas and makes complete surgical tumor resection virtually impossible. It is conceivable that alterations in ion channel function contribute to the invasiveness of glioma cells. Ion channels reported to be involved in glioma cell migration and invasion are summarized in Table 1.
Figure 1.
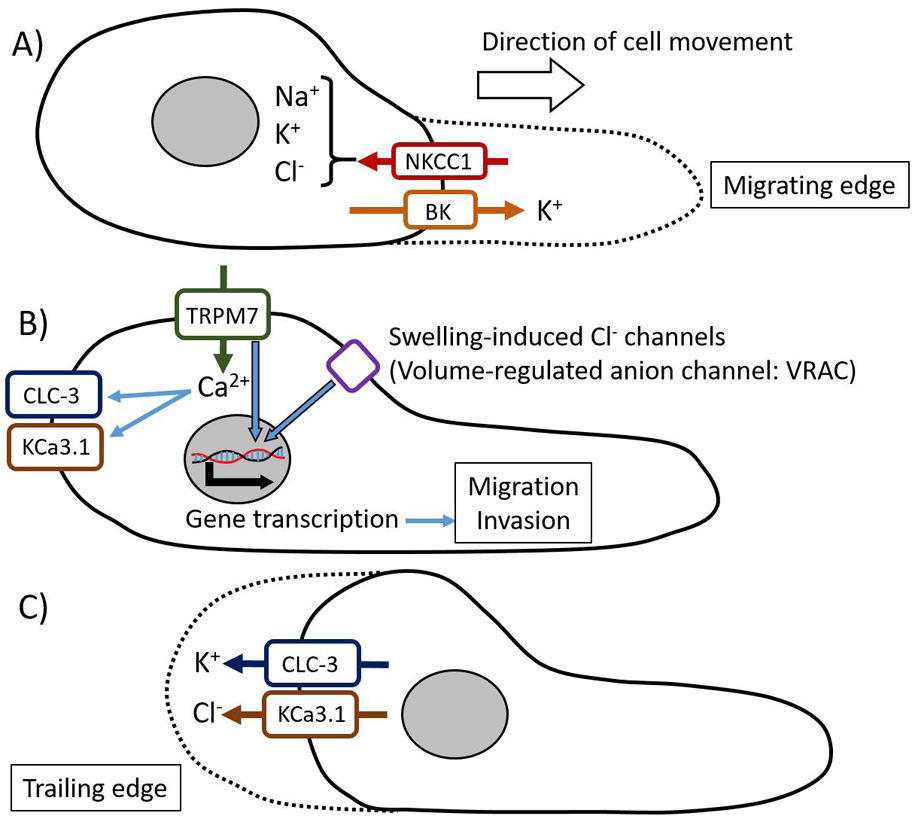
Cell volume changing model during cell migration. Cell migration is thought to be modeled as a continuous cycle of protrusion of the migrating cell front and retraction of the trailing end. A) In the migrating edge, ion channels and transporters, such as NKCC1, cause ion influx leading to an osmotic gradient, which results in water flowing into the cell. B) TRP channels allow the influx of Ca2+, which activates ion channels in the trailing edge. TRPM7 and swelling-induced Cl− channels are also thought to activate signaling pathways, such as MAPK/ERK signaling and PI3K/Akt pathways, leading to alterations in gene expression which enhance glioma cell migration and invasion. C) In the rear-end of the cell body, activated ion channels, such as CLC-3 and KCa3.1, induce ion and water efflux that causes cell shrinking only in the trailing end and results in cell retraction.
Table 1.
Ion Channels and Glioma Invasion/Migration
| Ion channel | Ions | Mechanisms/Effects on Migration/Invasion |
|---|---|---|
| KCa3.1 (Ca2+ activated K+ channel) | K+ | K+ efflux in the trailing edge |
| BK (or KCa1.1) (Ca2+ activated K+ channel) | K+ | K+ efflux in the migrating edge |
| CLC-3 (voltage-gated Cl− channel 3) | Cl− | Cl− efflux in the trailing edge |
| NKCC1 (Na+K+Cl− cotransporter 1) | Na+, K+, Cl− | Na+, K+, Cl− influx in the migrating edge |
| Kir4.1 (Inward rectifier K+ channel) | K+ | Inhibition of this channel enhances cell migration |
| Kir4.2 | K+ | α9β1 integrin-mediated migration |
| Swelling-induced Cl− channels (VRAC) | Cl− | Promotes gene transcription through modulation of signaling pathways |
| ANO1 (Anoctamin-1) Ca2+ activated Cl− channel (TMEM16A) | Cl− | Activation of NF-κB signaling pathway |
| KCC3 (K+Cl− cotransporter) | K+, Cl− | Unclear |
| TRPM7 | Ca2+ | Ca2+ influx and activating Ca2+-dependent pathways |
| TRPV4 | Ca2+ | Activation of Rac1 by Akt phosphorylation |
| TRPC1 | Ca2+ | Localizes in the migrating edge, Involved in chemotaxis towards EGF |
| Calcium Release Channel IP3R | Ca2+ | Ca2+ release from intracellular Ca2+ stores |
| Cav3.1, Cav3.2 (voltage-gated Ca2+ channels) | Ca2+ | Ca2+ influx and activating Ca2+-dependent pathways |
| ASIC1 and ENaC (Epithelial sodium channel) | Na+ | Involved in lamellipodium expansion |
| Na+/K+-ATPase adhesion molecule on glia (AMOG) | Na+, K+ | Regulating cytoskeletal remodeling, cell polarity, and lamellipodia formation |
| Hv1 (voltage-gated proton channels) | H+ | Regulating intracellular pH |
The KCa3.1 channel (KCNN4), is a member of the Ca2+-activated K+ channel family and is one of the most studied channels in the context of glioma invasion(5,7-11). Expression of KCa3.1 in the normal brain has been found to be limited to microglia, vascular endothelial cells, and the nodes of Ranvier of cerebellar Purkinje neurons, however, glioma cells show high expression(9,11). The REepository of Molecular BRAin Neoplasia DaTa (REMBRANDT) database showed overexpression of KCa3.1 in 32% of all types of gliomas, and it correlated with significantly poor survival(7). However, the interpretation of this study should be done with caution, since the glioma subtype composition of each group was unclear and various types of gliomas have different biology and prognosis. Synchronous activation of KCa3.1 and the Cl− channel CLC-3 was shown in the invading processes of glioma cells. These channels regulate the flux of K+ and Cl− ions causing changes in cell volume required for cell migration(10). In addition, a recent study indicates that KCa3.1 promotes GBM cell migration by inducing or modulating Ca2+ oscillations, a recurrent cycle reinforcing the Ca2+ signal(9). In addition, a combination of KCa3.1 inhibition and temozolomide reduced migration and invasion of glioma cells and increased cytotoxicity in-vitro(11). Taken together, these studies indicate KCa3.1 is involved in glioma invasion and is a potential target for treatment.
Another well studied K+ channel that is dysregulated in GBM and is associated with invasion, is the large-conductance, Ca2+-activated K+ channel (BK or KCa1.1), encoded by the KCNMA1 gene. BK channels promote ion and water fluxes, working in combination with Cl− channels. At the invading extensions of glioma cells, finger-like microprojections such as filopodia and microvilli express BK channels. When the BK channels are activated, the K+ leaves the cell and Na+ enters, while Cl− anions and water follow Na+ into the cell through channels/exchangers. This process enables the filopodia to swell(3,12-15). Ionizing radiation induces SDF-1 (stromal cell-derived factor-1, CXCL12) signaling, altering Ca2+ signaling, which contributes to BK channel activation through channel phosphorylation by Ca2+/calmodulin-dependent kinase II (CaMKII). Inhibition of the BK channel mitigates the infiltration of glioblastoma cells in-vitro and in-vivo(12,13). Although the detailed mechanisms of BK channel activity in irradiated glioblastoma cell migration remain unclear, the BK channel may be a therapeutic target, particularly in the prevention of radiation-induced glioma cell spreading.
In addition, K+ selective ion channels known as inwardly rectifying K+ (or Kir or IRK) channels are involved in glioma cell migration. Kir channels in glial cells maintain ionic homeostasis through potassium spatial buffering. Kir4.1 (KCNJ10), is predominantly expressed in glial cells. A study showed that miR-5096 significantly inhibited Kir4.1 expression in glioblastoma cells; treatment with miR5096 or a Kir4.1 blocker, increased the release of extracellular vesicles and filopodia outgrowth, enhancing cell motility(16). These findings are consistent with other studies showing KCNJ10 expression levels were negatively correlated with glioma grade(17,18). Another study also presented α9β1 integrin-mediated glioma cell migration related to Kir4.2(19). In contrast to Kir4.1, inhibition or silencing of Kir4.2 reduced glioma migration.
During cell volume changes, the coordinated release of K+ and Cl−, along with osmotically obligated water, is thought to be essential. Thus, Cl− ions also play an important role in glioma cell migration and invasion. One of the voltage-gated chloride channels, CLC-3, encoded by the CLCN3 gene, also has been recognized as a crucial regulator of glioma cell migration and invasion by modulating cellular shape and volume(10,20-22). CLC-3 is highly expressed on the plasma membrane of glioma cells compared to the normal brain. Both, inhibition by a non-specific chloride channel blocker, 5-nitro-2-(3-phenylpropylamino)-benzoate (NPPB)(23), and CLC-3 silencing with siRNA, suppressed glioma cell migration(20). CLC-3 activity is regulated by phosphorylation via CaMKII. Also, inhibition of CaMKII and direct inhibition of CLC-3 reduced CLC-3-expressing glioma cell invasion(21). Together with KCa3.1, CLC-3 localizes to the invading processes of glioma cells, and contribute to cell volume change by facilitating the flux of Cl− along with water(10). Temozolomide conjugated with NPPB has been shown to suppress both, glioma cell proliferation and migration, in-vitro(22). A recent study reported that CLC-3 expression was positively correlated with histological grade of gliomas; glioma patients with high CLC-3 expression had a significantly shorter survival. This study also indicated that CLC-3 promotes glioma cell invasion partially through the nuclear factor-κB (NF-κB) signaling pathway(24). Thus, CLC-3 is also a possible therapeutic target and biomarker for gliomas. However, the expression of CLC-3 in many different types of tissues is an important consideration regarding the selective targeting of tumor cells.
The volume-regulated anion channel (VRAC) is also involved in sustaining the shape and cell volume changes needed for cell migration and proliferation(4,25,26). VRAC is highly expressed in GBM cells and mediates the swelling-induced chloride current (ICl, swell). Both, acute hypoxia and exposure to serum, occur in a typical GBM microenvironment and will stimulate cell migration and evasion from cell death by activating VRAC(4,25). GBM often induces the breakdown of the blood brain barrier (BBB) and increases vascular permeability, which causes serum leakage into the tissue around a tumor. DCPIB, a blocker of ICl, swell, has been shown to reduce proliferation, migration, and invasion of glioblastoma cells, by potentially suppressing the JAK2/STAT3 and PI3K/Akt signaling pathways(26). Novel compounds inhibiting ICl, swell need to be explored because DCPIB does not cross the BBB.
Anoctamin-1 (ANO1), also known as TMEM16A (transmembrane protein with unknown function 16A), acts as a Ca2+-activated Cl− channel. TMEM16A is overexpressed in gliomas with higher abundance in higher-grade gliomas. The knockdown of TMEM16A significantly decreased proliferation, migration, and invasion of glioma cells. Furthermore, the NF-κB signaling was activated by overexpressed TMEM16A, which promoted proliferation, migration, and invasion(27). A recent study revealed that ANO1 expression was enhanced by 14-3-3γ, a protein having the ability to bind other signaling proteins. Gene silencing of ANO1 or 14-3-3γ suppressed migration and invasion of glioblastoma cells(28). Further in-vivo experiments and clinical trials are needed to conclusively clarify whether ANO1 can be a therapeutic target of glioma.
The sodium-potassium-chloride co-transporter isoform 1 (NKCC1) also regulates cell volume and intracellular Cl− accumulation, and thus it is involved in glioma cell motility, though it is not an ion channel but an ion co-transporter. NKCC1 mediates the movement of Na+, K+, and Cl− across the plasma membrane using the inward gradient for Na+, generated by the Na+/K+ ATPase. Cl− concentration in glioma cells is higher than in mature neurons and normal astrocytes, and this gradient of Cl− provides a driving force for cell shrinkage as the cells invade surrounding tissues(29,30). NKCC1 expression levels were significantly higher in high-grade glioma than in grade II glioma and normal brain, and its expression was mostly localized to the extending processes of migrating cells. Pharmacological inhibition of NKCC1 or shRNA knockdowns of NKCC1 disrupted Cl− uptake and inhibited glioma cell invasion in-vitro and in-vivo. Furthermore, NKCC1 modulated glioma cell invasion through the regulation of focal adhesion dynamics and cell contractility. NKCC1 knockdown displayed larger focal adhesions and smaller projected cell area, accompanied by a decrease in contractile forces(29). A recent study reported that NKCC1 interacts with Cofilin-1, an actin-regulating protein involved in actin polymerization/depolymerization and cell migration. The study suggests that NKCC1 polarizes to the cell edge, regulates the localization of Cofilin-1, and can act as an actin anchor at the leading edge during cell migration(30). These suggest that targeting NKCC1 has great therapeutic potential to reduce glioma cell invasion.
Only a few studies talk about, Na+-independent, K+-Cl− co-transporter in gliomas. One study showed that inhibition of the K+-Cl− co-transporters (KCCs) contributed to more invasive behavior of glioblastoma cells(29), however, another study indicates that KCC3 inhibition reduced glioma cell motility(31). Given the conflicting results, further studies are needed to determine how KCCs are involved in glioma cell migration.
TRP (transient receptor potential) channels are cation-selective channels that are important for cellular calcium homeostasis and Ca2+-dependent processes such as neurotransmitter release, proliferation, gene transcription, and apoptosis. TRP channels are also thought to influence cancer and stromal cell migration through growth factors, cytokines, and cytoskeletal remodeling. A study showed that TPRC1, TPRC6, TRPM2, TRPM3, TRPM7, TRPM8, TRPV1, and TRPV2 were expressed at significantly higher levels in glioblastoma(32). Among them, one of the best-studied channels is TRPM7(33-35). TRPM7 is Ca2+ and Mg2+ permeable, and also has serine/threonine kinase function. TRPM7 activates JAK2/STAT3 and/or Notch signaling pathways and induces cell proliferation and migration(33). Suppression of TRPM7 by siRNA, or inhibition, significantly impaired migration and invasion of a glioma cell line. Also, it decreased matrix metalloproteinase-2 (MMP-2) expression; a protein capable of degrading components of the basement membrane and extracellular matrix to assist in the invasion process. Moreover, inhibiting TRPM7 showed attenuation of PI3K/Akt and MEK/MAPK/ERK signaling pathways(34). In contrast, potentiating TRPM7 with activators (e.g., naltriben) enhanced migration and invasion of glioblastoma cells. Activated TRPM7 upregulated MAPK/ERK signaling pathway, but not the PI3K/Akt pathway, suggesting that the MAPK/ERK signaling pathway plays a more significant contribution to glioma invasiveness(35). Carvacrol, a Food and Drug Administration (FDA) approved food additive and TRPM7 inhibitor is able to cross the BBB; a clinical trial is needed to evaluate the efficacy of this compound in glioma patients.
TRPC1 has also been reported to play a role in glioma migration. Stimulation of glioma cells with epidermal growth factor (EGF) showed TRPC1 localization to the invading edge of migrating cells. In addition, inhibition of TRPC1 caused a loss of chemotaxis toward EGF(37). A recent study indicates that PDGF induced TRPC1 translocation from inside of the cell to the front of migration and activated sphingosine kinase that formed sphingosine-1-P (S1P), a potential activator of TRPC1 activity(38).
TRPV4, another TRP channel involved in glioma migration, has been shown to be upregulated in gliomas and its higher expression is associated with a worse prognosis. Activating TRPV4 promoted the activation of Rac1 (Ras-related C3 botulinum toxin substrate 1) by Akt phosphorylation, which enhanced glioma migration and invasion. Also, inhibition of this channel has been shown to suppress glioma migration(39) and colon cancer development(40), supporting a potential role of TRPV4 in carcinogenesis.
Ca2+ signaling is important for the motility of glioblastoma cells. Inositol 1,4,5-triphosphate receptor (IP3R) is a Ca2+ release channel located in intracellular Ca2+ stores such as endoplasmic reticulum. Inhibiting IP3R subtype 3 by caffeine reduces glioblastoma invasion and is associated with extended survival in-vivo(41). The FDA-approved antipsychotic drug, trifluoperazine (TFP)(42), also suppressed proliferation and invasion of glioblastoma cells. TFP interacts at the TFP-binding site of calmodulin subtype 2 (CaM2), causing dissociation of CaM2 from IP3R, the opening of IP3R, and triggering the massive release of Ca2+ from intracellular stores(43). The authors propose that an aberrant increase in Ca2+ signaling could also inhibit the invasiveness of glioblastoma cells, however, the mechanism of how inhibition of intracellular Ca2+ increases by caffeine and increased intracellular Ca2+ by TFP inhibit GBM invasion needs further investigation.
Ca2+ influx is also mediated by voltage-gated Ca2+ channels (VGCCs) located at the plasma membrane. There are five types: L-type, P-type, N-type, R-type, and T-type. When VGCCs are open, Ca2+ enters the cytoplasm and acts as a secondary messenger. Among the VGCCs, T-type Ca2+ channels (Cav3.2) are thought to play a key role in regulating intracellular Ca2+ levels and tumor progression(44). Endostatin (ES), a c-terminal proteolytic fragment of collagen XVIII, inhibited T-type Ca2+ channel currents. ES and the specific T-type Ca2+ channel blocker, mibefradil, inhibit proliferation and migration of glioblastoma cells(45). Mibefradil was FDA-approved for the treatment of hypertension and angina, but was withdrawn from the market after approval due to potential health hazards. However, there are other FDA-approved T-type Ca2+ channels on the market that could be explored as therapeutic strategies for gliomas (e.g., ethosuximide, trimethadione, amlodipine)(46).
High-grade gliomas express epithelial sodium channel (ENaC)/Degenerin family and the ENaC/Degenerin family also includes acid-sensing ion channels (ASICs). It is reported that ASIC1, αENaC, and γENaC are overexpressed in a glioblastoma cell line, interact with each other, and form an amiloride-sensitive nonselective cation channel. The knockdown of any one of these channels inhibited cell migration possibly because of the decreased Na+ influx inhibited the cell swelling required for lamellipodium expansion(47). Moreover, a physical and functional interaction between these channels and integrin-β1 has been postulated. The knockdown of integrin-β1 caused a loss of expression of ASIC1 and attenuated the amiloride-sensitive current. The link between the amiloride-sensitive channel and integrin-β1 was mediated by α-actinin. Downregulation of α-actinin-1 or −4 also attenuated the amiloride-sensitive current and migration due to loss of surface expression of ASIC1(48). Although these studies strongly indicated that ENac/ASICs are involved in GBM migration, they have the limitation of being in-vitro studies. Further investigation with animal models will help elucidate the therapeutic importance of these channels.
The Na+/K+-ATPase has the essential function in maintaining cell homeostases, such as ion translocation and maintenance of pH balance, but also has a role in cellular adhesion and migration through the regulation of cytoskeletal remodeling, cell polarity, and lamellipodia formation. The adhesion molecule on glia (AMOG) is an isoform of the β2-subunit of Na+/K+-ATPase, and it is highly expressed in the CNS. A study showed that most GBMs have a dramatic loss of AMOG expression, and AMOG expression in glioblastoma inhibited its invasion. In normal astrocytes, the downregulation of AMOG causes increased invasiveness, which suggests that loss of AMOG may be one of the mechanisms involved in glioblastoma cell invasion(49). Another study showed that AMOG and neural cell adhesion molecule L1 (L1CAM) interdependently regulate their expression. Both molecules are involved in regulating apoptosis through Akt and Erk activation. The authors also found that the reduction of AMOG expression delays senescence and correlates with decreased cell death(50). These studies suggest that strategies to increase levels or activity of AMOG could be potential avenues for the treatment of gliomas.
Hypoxia can increase tumor aggressiveness and cause resistance to radiation and chemotherapy. The hypoxic microenvironment, which commonly occurs in glioblastoma, induces tumor cells to have high glycolytic activity and produce acidic metabolites. As a result, cells need to extrude more protons to overcome intracellular acidification. A study showed that the voltage gated proton channel (Hv1) are expressed in glioblastoma cell lines and regulate the intracellular pH (pHi). Blocking Hv1 channels with ZnCl2 significantly reduced pHi, cell survival, and migration(51). Although Hv1 channels could be a therapeutic target for glioblastoma, expression in normal tissues may potentially lead to side effects.
Ion Channels and Glioma Proliferation
Ion channels also play a key role in the proliferation of gliomas, mainly through the activation of intracellular signaling pathways (Figure 2) (Table 2)(3,4). Ca2+ channels are involved in glioma development since the higher expression of Ca2+ channels on the plasma membrane increases Ca2+ influx which promotes Ca2+ dependent proliferative signaling pathways. TRP channels are important for cellular calcium homeostasis and cell proliferation, as we described above in the invasion/migration section. TRPM7 activates the JAK2/STAT3 and/or Notch signaling pathways and induces cell proliferation and migration. Activated STAT3 also upregulates aldehyde dehydrogenase1 (ALDH1) which is recognized as a novel stem cell marker in glioblastoma, and ALDH1 is thought to have important roles of self-protection, differentiation, expansion, and proliferation(33). Suppression of TRPM7 with siRNA, or by pharmacological inhibition, reduced not only migration and invasion but also the proliferation of glioblastoma cells, probably through inhibiting PI3K/Akt and MEK/MAPK/ERK signaling pathways(34). A study showed that midazolam(36), a short-acting benzodiazepine with the ability to cross the BBB, inhibited TRPM7 currents and calcium influx causing G0/G1 cell cycle arrest and a decrease in cell proliferation in glioblastoma cells(52).
Figure 2.

Representative contributions of ion channels to glioma cell proliferation. TRPM7 may activate proliferation through the Notch, PI3K/mTOR, and MAPK pathways. The nuclear localization sequence (NLS) on hEAG/hERG causes perinuclear localization of these channels and activates the MAPK pathway. Increased hEAG/hERG expression and VGCC activity results in an increased influx of Ca2+ ions which causes increased cell cycle transition and cell proliferation through calmodulin activation. ANO1 overexpression activates NF-κB. Interestingly, Kv2.1 overexpression increases autophagy through phosphorylated ERK1/2, which results in increased apoptosis and attenuates proliferation. *; Postulated mechanism.
Table 2.
Ion Channels and Glioma Proliferation/Glioma-related Epilepsy
| Ion Channels and Glioma Proliferation | ||
|---|---|---|
| Ion channel | Ions | Mechanisms/Effects on Proliferation |
| TRPM7 | Ca2+ | Ca2+ influx and activating Ca2+-dependent pathways |
| TRPC6 | Ca2+ | Regulation of cell cycle (G2/M) progression |
| TRPML2 | Ca2+ | Association with Akt/PKB and Erk1/2 pathways |
| Cav3.1, Cav3.2 (voltage-gated Ca2+ channels) | Ca2+ | Ca2+ influx and activation of Ca2+-dependent pathways |
| Kv10.1 (Eag1, Ether-a-go-go-1) | K+ | Involved in modulation of signaling pathways |
| Ether-a-go-go-Related Gene (hERG) | K+ | Involved in modulation of signaling pathways |
| Kv1.5 | K+ | Inversely correlated with increasing glioma grade |
| Kv2.1 | K+ | Regulation of ERK pathway |
| Swelling-induced Cl− channels (VRAC) | Cl− | Promoting gene transcription through signaling pathways |
| CLIC1 (Chloride Intracellular Channel-1) | Cl− | Unclear |
| ANO1 (Anoctamin-1) Ca2+ activated Cl− channel (TMEM16A) | Cl− | Activation of NF-κB signaling pathway |
| ASIC1 and ENaC (Epithelial sodium channel) | Na+ | Regulation of ERK pathway through integrin-β1 |
| Ion Channels and Glioma-related Epilepsy | ||
| Ion channel | Ions/Molecule | Mechanisms/Effect on glioma-related Epilepsy |
| Kir4.1 | K+ | Decreased expression impairs K+ buffering and increase propensity for seizures |
| KCC2 (K+-Cl− cotransporter 2) | K+, Cl− | Altered Cl− homeostasis causes GABAergic disinhibition |
| AQP-4 | H2O | Regulating extracellular potassium concentration and efflux of glutamate |
TRPC6 has also been shown to be overexpressed in glioma cells(32,53). Inhibition of TRPC6 expression suppressed glioma growth both in-vitro and in-vivo. TRPC6 affected glioma growth through the regulation of G2 to M phase cell cycle progression. The inhibition of TRPC6 induces G2 cell cycle arrest and enhances radiosensitivity(53). Among the TRP family, mucolipins (TRPML1, −2, and −3) represent a distinct subfamily of endosome/lysosome Ca2+ channel proteins. A study showed higher expression of TRPML2 in gliomas and silencing this channel reduced viability and proliferation of glioblastoma cells, associated with abrogation of Akt/PKB and Erk1/2 pathways(54).
Another Ca2+ channel that has been the focus of recent studies is the T-type voltage-gated Ca2+ channel (Cav3.2), as mentioned in the invasion/migration section. Cav3.2 is highly expressed in glioblastoma cells and its higher expression might correlate with poor prognosis. Cav3.2 blockade inhibited tumor growth of glioblastoma cell lines and enhanced the effects of temozolomide on glioblastoma cells and mouse glioma models(44,45). A phase I clinical trial evaluating the T-type Ca2+ channel blocker, mibefradil, was performed(55) and demonstrated safety and some responses. However, given the withdrawal of mibefradil, additional studies with other T-type Ca2+ channel blockers should be performed.
K+ channels also play an important role in controlling the proliferation and apoptosis of glioma cells. The human ether-à-go-go potassium channel (hEAG1, Kv10.1, encoded by KCNH1) and the human ether-à-go-go-related channel (hERG, Kv11.1, encoded by KCNH2) are voltage-dependent K+ channels, overexpressed in GBM, and are reported to be involved in signaling pathways promoting proliferation and inhibiting apoptosis(56-59). Overexpression of miR-296-3p suppressed cell proliferation and modulated sensitivity to anticancer drugs by reducing hEAG(56). Arsenic trioxide, which elevated the level of miR-133b, or overexpression of miR-133b, also inhibited proliferation and induced apoptosis in GBM cells through targeting hERG(57). A recent study showed that GBM patients with high expression of hERG had a worse prognosis, and the hERG blocker was beneficial in improving survival(58). Since this study supports the efficacy of hERG blockers in a xenograft model and in glioblastoma patients, this therapeutic approach is promising. However, prospective clinical trials are needed to validate hERG inhibitor as a novel therapy.
Voltage-gated K+ channels are involved in proliferation and apoptosis as well. Quinidine, a commonly used voltage-gated K+ channel blocker, inhibited voltage-gated K+ channel currents, decreased GBM cell proliferation, and induced apoptosis, partly by regulating miRNA expression(60). Immunohistochemical studies in infiltrating astrocytomas of various grades showed that Kv1.5 expression inversely correlated with the increasing histologic tumor grade. Increased Kv1.5 expression in GBM patients was associated with a favorable outcome, but it was not significant(61). However, this study did not mention the IDH mutational status of GBM cases. Comparing Kv1.5 expression between IDH-mutant and IDH-wildtype glioma of various grades is critical. A recent study investigated that Kv2.1 (KCNB1), was associated with malignant progression and outcome in gliomas; high Kv2.1 expression correlated with significantly longer survival. The same study also suggested that Kv2.1 influenced the induction of autophagy, accompanied by increased apoptosis and reduced proliferation and invasion, by regulation of the ERK pathway(62). However, the group of patients in this study contained both grade III and IV gliomas. As KCNB1 expression was negatively correlated with malignant progression, grade differences might influence survival and should have been taken into consideration for the analysis. Also, IDH mutation status and MGMT promoter methylation status should be included as independent variables in multivariate analysis. This study also postulates an interesting role for Kv2.1 in non-canonical ERK signaling, contributing to anti-tumor effects via activation of autophagy.
The VRAC that mediates the swelling-induced chloride current (ICl, swell), is also involved in proliferation(4,25,26). Acute hypoxia following cell swelling activates VRAC, resulting in the efflux of Cl−, together with K+. This is followed by osmotic water loss and reestablishment of the original cell volume, a process called regulatory volume decrease (RVD). RVD is thought to limit cell swelling and prevent necrotic death under hypoxic conditions, contributing to glioma cell survival(25). The Ca2+-activated Cl− channel, Anoctamin-1 (ANO1), is also involved in proliferation through activation of the NF-κB signaling pathway(27). Another Cl− channel involved in proliferation is the first member of the Chloride Intracellular Channel family, CLIC1(63,64). CLIC1 was shown to be overexpressed in GBM compared to normal tissues, and its expression was associated with a worse prognosis. Moreover, CLIC1 exists also as a circulating protein, secreted in extracellular vesicles (EVs). Treatment of GBM cells with EVs derived from CLIC1-overexpressing GBM cells strongly induced proliferation both, in-vitro and in-vivo(64). These studies indicate that CLIC1 is a strong prognostic marker in GBM and a potential therapeutic target.
Lack of expression of ASIC1 reduced phosphorylation of ERK, resulting in decreased proliferation. The amiloride-sensitive channel was composed of ASIC1, αENaC, and γENaC was associated with integrin-β1 and was dependent on the presence of α-actinin. Interaction of these proteins with integrins formed a multiprotein signaling complex that regulates cell migration and proliferation. In contrast, loss of this channel surface localization can reduce cell migration and proliferation(48). Recent research indicates that ASIC1a and ASIC3 expression in primary GBM stem cell lines, and states that these channels might endow glioblastoma cells with the capacity to sense extracellular pH. They also described that the expression of either of these channels was associated with a significant survival benefit in glioma patients. However, the patient group included a mixture of GBM, oligodendrogliomas, and astrocytomas, which can confound the interpretation of the results(65).
Ion channels in glioma stem-like cells
GBM is highly refractory to therapy, and such resistance is due in part to glioma stem-like cells (GSCs). Ion channels have important functions also in GSCs. Blocking KCa3.1 channels with a specific inhibitor, TRAM-34(66), reduces migration and motility of GSCs(7,8). GSCs expressed higher levels of BK channels, compared to normally cultured cells, which appears to be essential to their increased migratory ability. It has also been suggested that BK channels might have an important role in maintaining the stemness of GSCs(14). AMOG expressions at both the protein and mRNA levels were increased in GSCs compared with primary differentiated GBM cells, which suggests that the loss of AMOG represents an important step in GSCs differentiation(49). In addition, Cav3.2 is highly expressed in GSC and Cav3.2 inhibition reduced the growth of GSCs, induced GSCs differentiation, and enhanced the effects of temozolomide on GSCs(67). CLIC1 is functionally active as a chloride channel in GSCs and CLIC1 inhibition regulated GBM progression by targeting GSCs(63).
Ion channels and Glioma-related Epilepsy
Epilepsy is one of the most typical presenting symptoms in glioma patients. Recent studies revealed that IDH1 mutant gliomas are more likely to develop seizures than IDH-wildtype tumors. The D2-HG (D-2-hydroxyglutarate), a product of the mutant IDH1 enzyme, has a similar chemical structure to glutamate, which is an excitatory neurotransmitter. Therefore, it is thought that D2-HG increases neuronal activity by mimicking glutamate(68). However, the pathophysiology of glioma-related seizures is likely to be multifactorial(69). Alterations in ion channels are potential factors contributing to epileptogenesis and understanding their biological role in the context of glioma cells, is critical for the development of antiepileptics, especially in IDH-wildtype gliomas in which D2-HG is not expected to play a major role.
There are a few studies discussing ion channels and glioma related epilepsy (Table 2). One of these postulates a significantly lower inwardly rectifying potassium channel 4.1 (Kir4.1) expression in glioma patients with epilepsy, as well as, higher levels of the inflammatory cytokine IL-1β. Decreased expression of Kir4.1 may contribute to impaired K+ buffering and an increased propensity for seizures(70). Increased IL-1β may also be involved in enhancing neuronal excitability in the peritumoral area. For example, glutamate reuptake inhibited by cytokines can lead to increased extracellular glutamate concentrations.
Aquaporin-4 (AQP-4) is widely expressed, and it is the most important water channel in the brain. It has been reported that AQP-4 expression was more frequently detected on the GBM-cell membranes from patients with seizures than without seizures. AQP-4 forms a complex or functional coupling with K+ channels, Cl− channels, and K+-Cl− cotransporter. Thus, AQP-4 is involved in the control of cell volume through regulation of extracellular space volume, extracellular potassium concentration, and efflux of glutamate. The net effect of increased extracellular potassium and glutamate concentration is a depolarization of neuronal membrane potential with increased excitability(71). Unfortunately, these two studies did not include data on the IDH status of the gliomas(70,71).
Another study implicates GABAergic disinhibition and decreased KCC2, K+-Cl− cotransporter in glioma-related epilepsy. Loss of peritumoral GABAergic inhibitory interneurons and significantly decreased KCC2 membrane expression in peritumoral neurons were observed in tissue from a glioma-related seizure mouse model. In the normal brain, GABAergic interneurons are essential to counteract excitatory glutamatergic activity and maintain a delicate excitation-inhibition balance. This typical inhibitory response of GABA is conferred by a low intracellular Cl− established by the KCC2 cotransporter, which exports Cl−. Peritumoral neurons exhibit depolarizing GABAergic responses as a result of altered Cl− homeostasis, which was accompanied by impaired KCC2 functional expression. This means that the excitation-inhibition balance shifted towards hyperexcitability. The study also stated that both, GABAergic disinhibition and increased SXC (cysteine-glutamate transporter) expression, is required for epileptogenesis(72). There is strong evidence to support the importance of tumor-derived excitatory glutamate release mediated by SXC in epileptogenesis(73).
Ion channels and Sensitivity to Chemotherapy
There are some reports linking ion channels to sensitivity to chemotherapeutic agents (Supplementary Table S1). NKCC1 plays an important role in a regulatory volume increase in response to hypertonic and isotonic cell shrinkage. Loss of cell volume and lowering of intracellular K+ and Cl− is a hallmark of apoptosis. Inhibition of NKCC1 augmented temozolomide-induced apoptosis due to failure of the compensatory influx of K+ and Cl− (74). Suppression of CLC-3 caused the inhibition of Akt and autophagy, and sensitized the apoptosis-resistant glioblastoma cell line to cisplatin-mediated cell death. It has been studied that Akt phosphorylation is involved in the resistance of tumor cells to cisplatin and autophagy has a protective role against cisplatin in glioblastoma cells(75). Another study from the same group showed that CLC-3 suppression caused lysosomal dysfunction involved in cisplatin sensitivity(76). Na+/K+-ATPase blocker induced cell death with features of both apoptosis and necrosis, and effectively sensitized drug-resistant glioblastoma cells to temozolomide. The increases in intracellular Na+ and Ca2+ were associated with necrosis and K+ depletion was linked to apoptosis. Since the Na+/K+-ATPase α2/α3 subunits were highly expressed in drug-resistant glioblastoma cells, Na+/K+-ATPase might be a therapeutic target for the treatment of glioblastoma(77), although achieving selectivity for tumor cells could be a limitation of this strategy.
Regulation of ion channels by chemotherapy agents has also been studied. Oxaliplatin and temozolomide affect KCa3.1 expression in glioma cells. In contrast, neither the activity of BK channels or Kir channels was affected by either oxaliplatin and temozolomide(78,79). However, another study showed that temozolomide induced increased expression of glioma BK ion channel (gBK) and downregulated fascin-1. Fascin-1 controls cytoskeletal elements that stabilize actin-rich membranous projections such as microvilli and filopodia. Temozolomide increased HLA-A2 and gBK tumor antigen production. Moreover, temozolomide increased cytolytic T-lymphocyte mediated cytolysis of glioma cells due to the loss of their defensive membrane protrusions supported by fascin-1(15). The different responses of BK channels by temozolomide may be caused by different cell lines used in these studies.
Ion channels and Glioma Metabolism
Tumor metabolism has been the focused of several recent studies and modulating tumor metabolism has been postulated as a possible strategy for cancer treatment. Recent studies have also shown an association between glioma cell metabolism and ion channels (Supplementary Table S1). TRPC6 channels control the stability of HIF-1α which is a key transcription factor involved in adaptation to a hypoxic microenvironment. Inhibition of TRPC6 promoted HIF-1α hydroxylation and degradation to suppress HIF-1α accumulation. Moreover, TRPC6 regulated glucose transporter 1 (GLUT1) mRNA and protein levels through HIF-1α to affect glucose uptake under hypoxia(80). The voltage-dependent anion channel 1 (VDAC1) is a mitochondrial protein controlling cell energy and metabolic homeostasis. VDAC1 also plays a key role in apoptosis. Depletion of VDAC1 in glioblastoma mouse models resulted in inhibited cell growth, associated with marked decreases in GLUT1, hexokinase type I (HK-I), GAPDH, LDH, and enzymes of the Krebs cycle. Depletion of VDAC1 reversed oncogenic properties such as reprogrammed metabolism, stemness, angiogenesis, epithelial-mesenchymal transition, and invasiveness(81). Inhibiting T-type voltage-gated Ca2+ or KCa channels induced selective cell death of glioma-initiating cells (GIC) and increased host survival in a glioma mouse model. Blocking these channels resulted in plasma membrane depolarization and the resulting elevated intracellular Na+ compromised Na+-dependent nutrient transport. The resulting nutrient deficit triggered resulted in starvation and GIC cell death(82). The role of ion channels in the metabolism of cancer cells is a promising and uninvestigated area of cancer biology.
Ion channels and Other Biological Processes
Tumor-associated microglia/macrophages are polarized toward an anti-inflammatory, pro-tumor phenotype induced by interaction with cytokines and factors released by glioma cells, to allow glioma cells to invade the brain parenchyma. Anti-inflammatory microglia/macrophages have a higher expression of KCa3.1. Preventing KCa3.1 activation in microglia/macrophages switches them toward a pro-inflammatory, antitumor phenotype(83). Whether modulation of ion channels could be an effective way of influencing the response of GBM to immunotherapy, is something that warrants further investigation.
A study reported a tumor-suppressive role of voltage-gated Ca2+ channel subunit alpha-2/delta-3 (CACNA2D3). Downregulation of CACNA2D3 was a common feature of glioma tissue, correlated with poor survival, and was accompanied by increased methylation. Overexpression of CACNA2D3 increased intracellular Ca2+, which induced mitochondrial-mediated apoptosis, activated Nemo-like kinase (NLK) via the Wnt/Ca2+ non-canonical Wnt pathway, and inhibited the epithelial-mesenchymal transition. Silencing of NLK increased Wnt/β-catenin signaling and cell motility, confirming that NLK is a negative regulator of the canonical Wnt/β-catenin pathway(84).
Connexins are plasma membrane proteins and the key structural component of gap junctions. Also, connexin hemichannels facilitate paracrine communication between the cytosol and extracellular space. The opening of Cx43 hemichannels results in the release of molecules such as ATP, glutamate, and Ca2+ ions. Connexin43 (Cx43) is the commonest connexin and thought to play an important role in tumor invasion, progression, and temozolomide resistance in glioblastoma cells(85,86). A study reported that Cx43 levels were inversely correlated with temozolomide sensitivity and patient survival. Administration of the C-terminal peptide mimetic αCT1, a selective inhibitor of Cx43 hemichannels, sensitized temozolomide-resistant glioblastoma cells to temozolomide, by inhibition of Akt/mTOR signaling(87). This study shows the therapeutic potential of combining Cx43 inhibitors and temozolomide for the treatment of GBMs.
Pannexins are members of gap junction proteins. Most past studies about glioma and pannexins are related to gap junction, and there is limited literature about the hemichannel activity of pannexins and gliomas. However, a study showed that pannexin-1 plays an important role in the release of IL-6, IL-8, and glutamate from a glioblastoma cell line(88). Further studies are needed to investigate the association between glioma and pannexin hemichannels.
Ion Channel Expression and Glioma Prognosis
In recent years, several studies have postulated ion channel signatures that correlate with the prognosis of glioblastoma patients. A study identified 18 ion channel genes that are differentially expressed in high-grade gliomas compared to lower-grade gliomas. Among these, only two ion channel genes, CLIC1 and CLIC4 were up-regulated in high-grade gliomas. The remaining 16 ion channel genes (16/18), including KCNB1, KCNJ10, and KCNMA1, were down-regulated in high-grade gliomas. A gene signature based on these ion channels predicted glioma outcomes in three independent validation cohorts. Moreover, this ion channel gene signature was the most significant factor in the multivariate model including clinical factors such as age, performance status, and IDH1 mutation status (17). Wang HY, et al. presented a molecular signature with tree ion channel genes, KCNN4, KCNB1, and KCNJ10, that significantly associated with survival of glioblastoma patients. They also demonstrated that high-risk patients of unfavorable outcomes with this signature were sensitive to chemotherapy(89). The same group reported on another study an ion channel signature comprising 47 ion channel genes, including the three genes in the initial study. The risk score based on that signature predicted the prognosis of glioma patients. In addition, the expression of these signatures was preferentially seen in the mesenchymal subtype and IDH-wild-type gliomas. Gene ontology analysis and gene set variation analysis revealed that high-risk score patients tended to have reduced expression of proteins associated with the regulation of apoptosis and cell adhesion, and the increased expression of proteins associated with cell cycle and proliferation(18). Ion channels deregulated or activated, among these ion channel signature studies and other studies in the above sections, are listed in Supplementary Table S2. Another study identified a signature composed of 25 ion channel genes enriched in GSCs, including SCN8A, KCNB1, and GRIA3. Expression of these ion channels correlated with survival in glioblastoma databases such as The Cancer Genome Atlas (TCGA). Genetic knockdown or pharmacological inhibition of these ion channels attenuated the growth of GSCs compared to normal neural stem cells. This suggests that ion channels that are highly expressed in GSCs could be treatment candidates for targeting GSCs(90).
Conclusion
Ion channels and transporters play a significant role in glioma biology and influence the tumor microenvironment. The regulation of ion fluxes, associated with water flux, is essential for cell migration and invasion. Intracellular signaling pathways affected by the activity of ion channels influence cell proliferation and cell survival. Moreover, recent studies indicated that ion channels are involved in glioma-related seizures, sensitivity to chemotherapy, and tumor metabolism. Gene expression signatures based on ion channel genes have been shown to correlate with patient survival, indicating that the biological effects of ion channels have a significant impact on the behavior of glioma cells. A better comprehension of ion channels in gliomas will improve our understanding of gliomagenesis and glioma-related epileptogenesis. Moreover, the existing literature supports the notion that ion channels are potential therapeutic targets that might be beneficial in the treatment of these lethal tumors.
Supplementary Material
Acknowledgments
Financial support: This study was supported in part by the Japan-U.S. Brain Research Cooperation Program (T.T.).
Abbreviations
- siRNA
small interfering RNA
- shRNA
small hairpin RNA
- JAK
Janus kinase
- STAT
signal transducer and activator of transcription
- MEK
MAPK/ERK Kinase
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- PI3K
phosphatidylinositol-3-kinase
- PKB
protein kinase B
- GABA
gamma-aminobutyric acid
- IDH1
isocitrate dehydrogenase 1
- ATP
adenosine triphosphate
- HIF-1
hypoxia-inducible factor-1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- LDH
lactate dehydrogenase
- MGMT
O6-methylguanine-DNA methyltransferase
Footnotes
A conflict of interest disclosure statement: The authors declare no potential conflicts of interest.
References
- 1.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 2.Roger S, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 3.Simon OJ, Müntefering T, Grauer OM, Meuth SG. The role of ion channels in malignant brain tumors. J Neurooncol 2015;125:225–35. [DOI] [PubMed] [Google Scholar]
- 4.Caramia M, Sforna L, Franciolini F, Catacuzzeno L. The volume-regulated anion channel in glioblastoma. Cancers (Basel) 2019;11:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Qu C, Wu J, An L-J, Zou W. Potassium Channel and Glioma. Biomed J Sci Tech Res 2019;16:12179–84. [Google Scholar]
- 6.Litan A, Langhans SA. Cancer as a channelopathy: ion channels and pumps in tumor development and progression. Front Cell Neurosci 2015;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner KL, Honasoge A, Robert SM, McFerrin MM, Sontheimer H. A pro-invasive role for the Ca2+-activated K+ channel KCa3.1 in malignant glioma. Glia 2014;62:971–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruggieri P, Mangino G, Fioretti B, Catacuzzeno L, Puca R, Ponti D, et al. The inhibition of KCa3.1 channels activity reduces cell motility in glioblastoma derived cancer stem cells. PLoS One 2012;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catacuzzeno L, Franciolini F. Role of KCa3.1 channels in modulating Ca2+ oscillations during glioblastoma cell migration and invasion. Int J Mol Sci 2018;19:E2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuddapah VA, Turner KL, Seifert S, Sontheimer H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J Neurosci 2013;33:1427–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Alessandro G, Grimaldi A, Chece G, Porzia A, Esposito V, Santoro A, et al. KCa3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 2016;7:30781–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinle M, Palme D, Misovic M, Rudner J, Dittmann K, Lukowski R, et al. Ionizing radiation induces migration of glioblastoma cells by activating BK K+ channels. Radiother Oncol 2011;101:122–6. [DOI] [PubMed] [Google Scholar]
- 13.Edalat L, Stegen B, Klumpp L, Haehl E, Schilbach K, Lukowski R, et al. BK K+ channel blockade inhibits radiation-induced migration/brain infiltration of glioblastoma cells. Oncotarget 2016;7:14259–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosa P, Sforna L, Carlomagno S, Mangino G, Miscusi M, Pessia M, et al. Overexpression of large-conductance calcium-activated potassium channels in human glioblastoma stem-like cells and their role in cell migration. J Cell Physiol 2017;232:2478–88. [DOI] [PubMed] [Google Scholar]
- 15.Hoa NT, Ge L, Martini F, Chau V, Ahluwalia A, Kruse CA, et al. Temozolomide induces the expression of the glioma Big Potassium (gBK) ion channel, while inhibiting fascin-1 expression: possible targets for glioma therapy. Expert Opin Ther Targets 2016;20:1155–67. [DOI] [PubMed] [Google Scholar]
- 16.Thuringer D, Chanteloup G, Boucher J, Pernet N, Boudesco C, Jego G, et al. Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget 2017;8:37681–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang R, Gurguis CI, Gu W, Ko EA, Lim I, Bang H, et al. Ion channel gene expression predicts survival in glioma patients. Sci Rep 2015;5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu F fei Wang HY, He X zheng Liang TY, Wang W Hu HM, et al. Prognostic value of ion channel genes in Chinese patients with gliomas based on mRNA expression profiling. J Neurooncol 2017;134:397–405. [DOI] [PubMed] [Google Scholar]
- 19.Veeravalli KK, Ponnala S, Chetty C, Tsung AJ, Gujrati M, Rao JS. Integrin α9β1-mediated cell migration in glioblastoma via SSAT and Kir4.2 potassium channel pathway. Cell Signal 2012;24:272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lui VCH, Lung SSS, Pu JKS, Hung KN, Leung GKK. Invasion of human glioma cells is regulated by multiple chloride channels including ClC-3. Anticancer Res 2010;30:4515–24. [PubMed] [Google Scholar]
- 21.Cuddapah VA, Sontheimer H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J Biol Chem 2010;285:11188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park M, Song C, Yoon H, Choi KH. Double blockade of glioma cell proliferation and migration by temozolomide conjugated with NPPB, a chloride channel blocker. ACS Chem Neurosci 2016;7:275–85. [DOI] [PubMed] [Google Scholar]
- 23.Walsh KB, Long KJ, Shen X. Structural and ionic determinants of 5-nitro-2-(3-phenylpropylamino)-benzoic acid block of the CFTR chloride channel. Br J Pharmacol 1999;127:369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang B, Xie J, He HY, Huang EW, Cao QH, Luo L, et al. Suppression of CLC-3 chloride channel reduces the aggressiveness of glioma through inhibiting nuclear factor-κB pathway. Oncotarget 2017;8:63788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sforna L, Cenciarini M, Belia S, Michelucci A, Pessia M, Franciolini F, et al. Hypoxia modulates the swelling-activated Cl current in human glioblastoma cells: Role in volume regulation and cell survival. J Cell Physiol 2017;232:91–100. [DOI] [PubMed] [Google Scholar]
- 26.Wong R, Chen W, Zhong X, Rutka JT, Feng ZP, Sun HS. Swelling-induced chloride current in glioblastoma proliferation, migration, and invasion. J Cell Physiol 2018;233:363–70. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Liu Y, Ren Y, Kang L, Zhang L. Transmembrane protein with unknown function 16A overexpression promotes glioma formation through the nuclear factor-κB signaling pathway. Mol Med Rep 2014;9:1068–74. [DOI] [PubMed] [Google Scholar]
- 28.Lee YS, Lee JK, Bae Y, Lee BS, Kim E, Cho CH, et al. Suppression of 14-3-3γ-mediated surface expression of ANO1 inhibits cancer progression of glioblastoma cells. Sci Rep 2016;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garzon-Muvdi T, Schiapparelli P, ap Rhys C, Guerrero-Cazares H, Smith C, Kim DH, et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol 2012;10:e1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiapparelli P, Guerrero-Cazares H, Magaña-Maldonado R, Hamilla SM, Ganaha S, Goulin Lippi Fernandes E, et al. NKCC1 regulates migration ability of glioblastoma cells by modulation of actin dynamics and interacting with cofilin. EBioMedicine 2017;21:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gagnon KB. High-grade glioma motility reduced by genetic knockdown of KCC3. Cell Physiol Biochem 2012;30:466–76. [DOI] [PubMed] [Google Scholar]
- 32.Alptekin M, Eroglu S, Tutar E, Sencan S, Geyik MA, Ulasli M, et al. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumor Biol 2015;36:9209–13. [DOI] [PubMed] [Google Scholar]
- 33.Liu M, Inoue K, Leng T, Guo S, Xiong Z. TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell Signal 2014;26:2773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W-L, Barszczyk A, Turlova E, Deurloo M, Liu B, Yang BB, et al. Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget 2015;6:16321–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong R, Turlova E, Feng Z-P, Rutka JT, Sun H-S. Activation of TRPM7 by naltriben enhances migration and invasion of glioblastoma cells. Oncotarget 2017;8:11239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerecke M Chemical structure and properties of midazolam compared with other benzodiazepines. Br J Clin Pharmacol 1983;16:11S–16S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bomben VC, Turner KL, Barclay TTC, Sontheimer H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J Cell Physiol 2011;226:1879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lepannetier S, Zanou N, Yerna X, Emeriau N, Dufour I, Masquelier J, et al. Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium 2016;60:373–83. [DOI] [PubMed] [Google Scholar]
- 39.Ou-yang Q, Li B, Xu M, Liang H. TRPV4 promotes the migration and invasion of glioma cells via AKT/Rac1 signaling. Biochem Biophys Res Commun 2018;503:876–81. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Zhang P, Xie C, Sham KWY, Ng SSM, Chen Y, et al. Activation of PTEN by inhibition of TRPV4 suppresses colon cancer development. Cell Death Dis 2019;10:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res 2010;70:1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, et al. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 2003;4:463–76. [DOI] [PubMed] [Google Scholar]
- 43.Kang S, Hong J, Lee JM, Moon HE, Jeon B, Choi J, et al. Trifluoperazine, a well-known antipsychotic, inhibits glioblastoma invasion by binding to calmodulin and disinhibiting calcium release channel IP3R. Mol Cancer Ther 2017;16:217–27. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Wang H, Qian Z, Feng B, Zhao X, Jiang X, et al. Low-voltage-activated T-type Ca2+ channel inhibitors as new tools in the treatment of glioblastoma: The role of endostatin. Pflugers Arch Eur J Physiol 2014;466:811–8. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Zhang J, Jiang D, Zhang D, Qian Z, Liu C, et al. Inhibition of T-type Ca2+ channels by endostatin attenuates human glioblastoma cell proliferation and migration. Br J Pharmacol 2012;166:1247–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kopecky BJ, Liang R, Bao J. T-type calcium channel blockers as neuroprotective agents. Pflugers Arch Eur J Physiol 2014;466:757–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapoor N, Bartoszewski R, Qadri YJ, Bebok Z, Bublen JK, Fuller CM, et al. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J Biol Chem 2009;284:24526–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rooj AK, Liu Z, McNicholas CM, Fuller CM. Physical and functional interactions between a glioma cation channel and integrin-β1 require α-actinin. Am J Physiol Physiol 2015;309:C308–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun MZ, Kim JM, Oh MC, Safaee M, Kaur G, Clark AJ, et al. Na+/K+-ATPase β2-subunit (AMOG) expression abrogates invasion of glioblastoma-derived brain tumor-initiating cells. Neuro Oncol 2013;15:1518–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang Q, Xie Q, Hu C, Yang Z, Huang P, Shen H, et al. Glioma malignancy is linked to interdependent and inverse AMOG and L1 adhesion molecule expression. BMC Cancer 2019;19:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ribeiro-Silva L, Queiroz FO, Da Silva AMB, Hirata AE, Arcisio-Miranda M. Voltage-gated proton channel in human glioblastoma multiforme cells. ACS Chem Neurosci 2016;7:864–9. [DOI] [PubMed] [Google Scholar]
- 52.Chen J, Dou Y, Zheng X, Leng T, Lu X, Ouyang Y, et al. TRPM7 channel inhibition mediates midazolam-induced proliferation loss in human malignant glioma. Tumor Biol 2016;37:14721–31. [DOI] [PubMed] [Google Scholar]
- 53.Ding X, He Z, Zhou K, Cheng J, Yao H, Lu D, et al. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J Natl Cancer Inst 2010;102:1052–68. [DOI] [PubMed] [Google Scholar]
- 54.Morelli MB, Nabissi M, Amantini C, Tomassoni D, Rossi F, Cardinali C, et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: role in tumor growth and progression. Oncotarget 2016;7:43654–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holdhoff M, Ye X, Supko JG, Nabors LB, Desai AS, Walbert T, et al. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro Oncol 2017;19:845–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bai Y, Liao H, Liu T, Zeng X, Xiao F, Luo L, et al. MiR-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-à-go-go (EAG1). Eur J Cancer 2013;49:710–24. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Li Y, Jiang C. MiR-133b Contributes to Arsenic-induced apoptosis in U251 glioma cells by targeting the hERG channel. J Mol Neurosci 2015;55:985–94. [DOI] [PubMed] [Google Scholar]
- 58.Pointer KB, Clark PA, Eliceiri KW, Salamat MS, Robertson GA, Kuo JS. Administration of non-torsadogenic human Ether-à-go-go-related gene inhibitors is associated with better survival for high hERG-expressing glioblastoma patients. Clin Cancer Res 2017;23:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martínez R, Stühmer W, Martin S, Schell J, Reichmann A, Rohde V, et al. Analysis of the expression of Kv10.1 potassium channel in patients with brain metastases and glioblastoma multiforme: Impact on survival. BMC Cancer 2015;15:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ru Q, Tian X, Pi M-S, Chen L, Yue K, Xiong Q, et al. Voltage-gated K+ channel blocker quinidine inhibits proliferation and induces apoptosis by regulating expression of microRNAs in human glioma U87-MG cells. Int J Oncol 2014; 833–40. [DOI] [PubMed] [Google Scholar]
- 61.Arvind S, Arivazhagan A, Santosh V, Chandramouli BA. Differential expression of a novel voltage gated potassium channel Kv 1.5 in astrocytomas and its impact on prognosis in glioblastoma. Br J Neurosurg 2012;26:16–20. [DOI] [PubMed] [Google Scholar]
- 62.Wang HY, Wang W, Liu YW, Li MY, Liang TY, Li JY, et al. Role of KCNB1 in the prognosis of gliomas and autophagy modulation. Sci Rep 2017;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Setti M, Savalli N, Osti D, Richichi C, Angelini M, Brescia P, et al. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J Natl Cancer Inst 2013;105:1644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Setti M, Osti D, Richichi C, Ortensi B, Del Bene M, Fornasari L, et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015;6:31413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tian Y, Bresenitz P, Reska A, El Moussaoui L, Beier CP, Gründer S. Glioblastoma cancer stem cell lines express functional acid sensing ion channels ASIC1a and ASIC3. Sci Rep 2017;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown BM, Pressley B, Wulff H. KCa3.1 channel modulators as potential therapeutic compounds for glioblastoma. Curr Neuropharmacol 2017;15:618–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Cruickshanks N, Yuan F, Wang B, Pahuski M, Wulfkuhle J, et al. Targetable T-type calcium channels drive glioblastoma. Cancer Res 2017;77:3479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen H, Judkins J, Thomas C, Wu M, Khoury L, Benjamin CG, et al. Mutant IDH1 and seizures in patients with glioma. Neurology 2017;88:1805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tandon N, Esquenazi Y. Resection strategies in tumoral epilepsy: Is a lesionectomy enough? Epilepsia 2013;54(Suppl. 9):72–8. [DOI] [PubMed] [Google Scholar]
- 70.Zurolo E, de Groot M, Iyer A, Anink J, van Vliet EA, Heimans JJ, et al. Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: A role for interleukin-1 β. J Neuroinflammation Journal of Neuroinflammation; 2012;9:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Isoardo G, Morra I, Chiarle G, Audrito V, Deaglio S, Melcarne A, et al. Different Aquaporin-4 expression in glioblastoma multiforme patients with and without seizures. Mol Med 2012;18:1147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Campbell SL, Robel S, Cuddapah VA, Robert S, Buckingham SC, Kahle KT, et al. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 2015;63:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sørensen MF, Heimisdóttir SB, Sørensen MD, Mellegaard CS, Wohlleben H, Kristensen BW, et al. High expression of cystine–glutamate antiporter xCT (SLC7A11) is an independent biomarker for epileptic seizures at diagnosis in glioma. J Neurooncol 2018;138:49–53. [DOI] [PubMed] [Google Scholar]
- 74.Algharabli J, Kintner DB, Wang Q, Begum G, Clark PA, Yang S-S, et al. Inhibition of Na+-K+-2Cl− cotransporter isorom 1 accelerates temozolomide-mediated apoptosis in glioblastoma cancer cells. Cell Physiol Biochem 2012;30:33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Su J, Xu Y, Zhou L, Yu H-M, Kang J-S, Liu N, et al. Suppression of chloride channel 3 expression facilitates sensitivity of human glioma U251 cells to cisplatin through concomitant inhibition of Akt and autophagy. Anat Rec 2013;296:595–603. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y, Zhou L, Zhang J, Zhang L, Yan X, Su J. Suppression of chloride voltage-gated channel 3 expression increases sensitivity of human glioma U251 cells to cisplatin through lysosomal dysfunction. Oncol Lett 2018;16:835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen D, Song M, Mohamad O, Yu SP. Inhibition of Na+/K+-ATPase induces hybrid cell death and enhanced sensitivity to chemotherapy in human glioblastoma cells. BMC Cancer 2014;14:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang MH, Huang YM, Wu SN. The inhibition by oxaliplatin, a platinum-based anti-neoplastic agent, of the activity of intermediate-conductance Ca2+-Activated K+ channels in human glioma cells. Cell Physiol Biochem 2015;37:1390–406. [DOI] [PubMed] [Google Scholar]
- 79.Yeh PS, Wu SJ, Hung TY, Huang YM, Hsu CW, Sze CI, et al. Evidence for the inhibition by temozolomide, an imidazotetrazine family alkylator, of intermediate-conductance Ca2+-Activated K+ channels in glioma cells. Cell Physiol Biochem 2016;38:1727–42. [DOI] [PubMed] [Google Scholar]
- 80.Li S, Wang J, Wei Y, Liu Y, Ding X, Dong B, et al. Crucial role of TRPC6 in maintaining the stability of HIF-1 in glioma cells under hypoxia. J Cell Sci 2015; 3317–29. [DOI] [PubMed] [Google Scholar]
- 81.Arif T, Krelin Y, Nakdimon I, Benharroch D, Paul A, Dadon-Klein D, et al. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol 2017;19:951–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Niklasson M, Maddalo I, Sramkova Z, Mutlu E, Wee S, Sekyrova P, et al. Membrane-depolarizing channel blockers induce selective glioma cell death by impairing nutrient transport and unfolded protein/amino acid responses. Cancer Res 2017;77:1741–52. [DOI] [PubMed] [Google Scholar]
- 83.Grimaldi A, D’Alessandro G, Golia MT, Grössinger EM, Di Angelantonio S, Ragozzino D, et al. KCa3.1 inhibition switches the phenotype of glioma-infiltrating microglia/macrophages. Cell Death Dis 2016;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jin Y, Cui D, Ren J, Wang K, Zeng T, Gao L. CACNA2D3 is downregulated in gliomas and functions as a tumor suppressor. Mol Carcinog 2017;56:945–59. [DOI] [PubMed] [Google Scholar]
- 85.Aasen T, Leithe E, Graham SV, Kameritsch P, Mayán MD, Mesnil M, et al. Connexins in cancer: bridging the gap to the clinic. Oncogene 2019;38:4429–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xing LY, Yang T, Sen Cui S, Chen G. Connexin hemichannels in astrocytes: Role in CNS disorders. Front Mol Neurosci 2019;12:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murphy SF, Varghese RT, Lamouille S, Guo S, Pridham KJ, Kanabur P, et al. Connexin 43 inhibition sensitizes chemoresistant glioblastoma cells to temozolomide. Cancer Res 2016;76:139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei L, Sheng H, Chen L, Hao B, Shi X, Chen Y. Effect of pannexin-1 on the release of glutamate and cytokines in astrocytes. J Clin Neurosci 2016;23:135–41. [DOI] [PubMed] [Google Scholar]
- 89.Wang HY, Li JY, Liu X, Yan XY, Wang W, Wu F, et al. A three ion channel genes-based signature predicts prognosis of primary glioblastoma patients and reveals a chemotherapy sensitive subtype. Oncotarget 2016;7:74895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pollak J, Rai KG, Funk CC, Arora S, Lee E, Zhu J, et al. Ion channel expression patterns in glioblastoma stem cells with functional and therapeutic implications for malignancy. PLoS One 2017;12:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.